

Abstract
Several promising antimalarial drugs are currently being tested in human trials, such as artefenomel, cipargamin, ferroquine and ganaplacide. Many of these compounds were identified using high throughput screens against a single species of human malaria, Plasmodium falciparum, under the assumption that effectiveness against all malaria species will be similar, as has been observed for other antimalarial drugs. However, using our in vitro adapted line, we demonstrated recently that P. knowlesi is significantly less susceptible than P. falciparum to some new antimalarial drugs (e.g., cipargamin and DSM265), and more susceptible to others (e.g., ganaplacide). There is, therefore, an urgent need to determine the susceptibility profile of all human malaria species to the current generation of antimalarial compounds. We obtained ex vivo malaria samples from travellers returning to the United Kingdom and, using the [3H]hypoxanthine incorporation method, compared susceptibility to select established and experimental antimalarial agents among all major human infective Plasmodium species. We demonstrate that P. malariae and P. ovale spp. are significantly less susceptible than P. falciparum to cipargamin, DSM265 and AN13762, but are more susceptible to ganaplacide. Preliminary ex vivo data from single isolates of P. knowlesi and P. vivax demonstrate a similar profile. Our findings highlight the need to ensure cross species susceptibility profiles are determined early in the drug development pipeline. Our data can also be used to inform further drug development, and illustrate the utility of the P. knowlesi in vitro model as a scalable approach for predicting the drug susceptibility of non-falciparum malaria species in general.
Keywords: Plasmodium falciparum, Plasmodium knowlesi, Drug susceptibility, ex vivo
Graphical abstract

Highlights
-
•
P. knowlesi less susceptible than P. falciparum to some new antimalarials in vitro.
-
•
ex vivo malaria samples used to test susceptibility of other human malaria species.
-
•
P. malariae and P. ovale show similar drug susceptibility profile to P. knowlesi.
-
•
ex vivo drug screens from all malaria species can inform new drug development.
1. Introduction
Almost all human malaria infections are caused by six Plasmodium species: P. falciparum, P. vivax, P. malariae, P. ovale curtisi, P. ovale wallikeri, and the zoonotic P. knowlesi (Singh et al., 2004; Sutherland et al., 2010). However, with advances in genomics it is becoming evident that this number could be increased to include other primate malaria species now known to cause some human infections, including P. simium (Brasil et al., 2017), P. brasilianum (Lalremruata et al., 2015) and P. cynomolgi (Raja et al., 2020; Ta et al., 2014). While P. falciparum continues to exert the greatest burden of morbidity and mortality world-wide, P. vivax infections make up 75 % of the cases detected in the WHO Region of the Americas and 50 % of the cases detected in WHO South-East Asia Region (World Health Organization, 2020). In Malaysia, both P. falciparum and P. vivax infections have almost been eliminated while the zoonotic P. knowlesi infections have increased from 1600 cases in 2016 to over 4000 cases in 2018, and despite a small decline in 2019, nonetheless resulted in 12 deaths in 2018–19 (World Health Organization, 2020).
Since the turn of the century new drug development has increasingly been driven by high-throughput screens of large libraries of compounds against whole parasites, and thousands of new active chemical scaffolds have been identified (Gamo et al., 2010; Guiguemde et al., 2010; Plouffe et al., 2008). However, these screens have only been performed using P. falciparum parasites, as this is historically the only human malaria species that could be cultured in vitro continuously (Trager and Jensen, 1976), and in large numbers. However, the recent adaptation of the zoonotic parasite P. knowlesi to grow continuously in human red blood cells (Gruring et al., 2014; Lim et al., 2013; Moon et al., 2013) has enabled detailed exploration of in vitro drug susceptibility in a non-falciparum parasite species. Furthermore, both species can be cultivated in vitro under identical conditions allowing for a direct comparison of drug susceptibility (van Schalkwyk et al., 2017). Using the P. knowlesi in vitro model we have shown recently that some existing and experimental antimalarial agents exhibit large and significant species differences in susceptibility (van Schalkwyk et al., 2017, 2019). It remains an open question as to whether the other human-infecting malaria species (e.g., P. vivax, P. malariae or P. ovale spp.) are more like P. falciparum or P. knowlesi in their drug susceptibility profiles. In the absence of in vitro continuous cultivation methods for these other human malaria species, it is necessary to use ex vivo drug assays to obtain drug susceptibility data.
For this study, we developed the following two hypotheses: (a) that there are significant species differences in drug susceptibility, particularly to some of the newer antimalarials under development, and (b) that the drug susceptibility profiles of P. vivax, P. malariae and P. ovale spp. will be more similar to P. knowlesi than to P. falciparum, given their phylogenetic relatedness (Rutledge et al., 2017). In order to test these hypotheses we obtained infected whole blood samples representing the six common human-infecting malaria species from the Public Health England Malaria Reference Laboratory (MRL), United Kingdom. We then performed ex vivo drug susceptibility testing on short term cultures to compare the activity of a select panel of established and experimental antimalarial agents.
2. Materials and methods
2.1. Compounds
Established antimalarial drugs and investigational compounds were supplied by the Medicines for Malaria Venture, Geneva, Switzerland. Chloroquine stocks were prepared in sterile distilled water and all other compounds were dissolved in DMSO. [3H]hypoxanthine monohydrochloride [1 mCi (37 MBq)] was purchased from PerkinElmer (Catalogue number: NET177001MC).
2.2. ex vivo parasite sample acquisition
Whole blood samples from all malaria species were obtained from the Malaria Reference Laboratory (MRL) at the London School of Hygiene & Tropical Medicine. All patient identifiers were removed and a code assigned to each sample. Samples received were typically between 500 μL and 2500 μL and species were confirmed by microscopy before use. Only mono-infections (i.e., no mixed species infections) were used in this study. Approval for the study was obtained by the Research Ethics Committee of the London School of Hygiene & Tropical Medicine (Application reference: 14710) and the National Health Service (NHS), London-Chelsea Research Ethics Committee (Application reference: 18/LO/0738).
2.3. In vitro parasite cultivation
P. knowlesi (clone A1-H.1) and P. falciparum (clone 3D7) were maintained in culture as described previously (van Schalkwyk et al., 2019).
2.4. Sample preparation
Whole blood (ex vivo) samples were first transferred into microcentrifuge tubes. The samples were then centrifuged at 7000×g for 2 min after which the plasma and buffy coat was removed by aspiration. The infected erythrocytes were then washed twice with RPMI 1640 (Sigma product R5886). Parasites received, particularly non-falciparum species, are often present at low parasitaemia (<1 %). Therefore, for the drug susceptibility assays, parasite stocks were prepared at 2 % haematocrit without diluting the parasitaemia.
Comparison assays were set up with the in vitro P. knowlesi (clone A1-H.1) and P. falciparum (clone 3D7) lines. These drug susceptibility assays were initiated using stocks of unsynchronised parasites with the parasitaemia set to 0.1 % and haematocrit set to 2 %.
2.5. Drug susceptibility assays
All drug susceptibility assays (ex vivo and in vitro parasites samples) were set up in media composed of RPMI 1640 supplemented with 25 mM Na2HCO3, 25 mM HEPES, 10 mM D-glucose, 2 mM L-glutamine, 25 mg/mL gentamicin sulphate and 5 g/L AlbuMAX II.
Drug susceptibility assays were set up in 96-well, flat-bottom microplates in a final volume of 200 μL, as described previously (van Schalkwyk et al., 2017). Typically, five drugs were screened in duplicate per plate. Tritiated hypoxanthine ([3H]hypoxanthine (0.25 μCi/well; 110 nM)) was added to each well at the beginning of the experiment.
The parasites were exposed to the drugs for one complete life cycle: 24 h for P. knowlesi (ex vivo), 27 h for P. knowlesi (in vitro), 48 h for P. vivax, P. falciparum, P. ovale spp., and 72 h for P. malariae. The plates were incubated at 37 °C in a modular incubation chamber (Billups-Rothenburg Inc.) under a gas mixture of 96 % N2, 3 % CO2 and 1 % O2. Upon termination of the assay, the plates were stored at −80 °C overnight.
On the following day the 96-well plates were thawed at 37 °C for 1 h, and the contents harvested onto a glass fibre printed Filtermat A (PerkinElmer product 1450-421), using a 96-well cell harvester (Tomtec, Hamden, Conn.). Once dried, the filter is placed on a heating plate. A solid scintillator (Meltilex A, PerkinElmer product 1450-441) is placed over the filter and heated to 90 °C. Once the scintillator has melted into the filter, it is removed from the heating plate and allowed to cool and set at room temperature. The filter mat is then placed into a sample bag (PerkinElmer product 1450-432) and sealed. A Wallac Trilux 1450 Microbeta scintillation β-counter (PerkinElmer) is used to read the amount of radioactivity (in counts per minute) from the [3H]hypoxanthine incorporated into parasite material trapped in the filter mats.
2.6. Parasite DNA isolation
Genomic DNA was extracted from 200 μL of whole blood from the EDTA specimen tube using the QIAamp DNA Blood Mini Kit (Qiagen, UK) as per the manufacturer's instructions. The DNA was stored at −80 °C.
2.7. pfcrt genotyping
The Plasmodium falciparum chloroquine resistance transporter (pfcrt) genotype was determined by multiplex real-time PCR. A small segment around codons 72–76 was amplified using a Rotorgene Q thermocycler (QIAGEN, Germany) in the presence of three dual-labelled probes complementary to CVMNK (FAM fluorophore), CVIET (JOE fluorophore) and SVMNT (ROX fluorophore), as described previously (Sutherland et al., 2007; van Schalkwyk et al., 2013). For data analysis, a threshold for each probe was set manually using positive and negative controls.
2.8. Plasmodium ovale species discrimination
Discrimination between Plasmodium ovale curtisi and Plasmodium ovale wallikeri was achieved by quantitative real-time PCR (qPCR) of the porbp2 locus, as described previously (Oguike et al., 2011).
2.9. Statistics
P values for comparisons between EC50 values were calculated using Student's two-tailed t-test for unpaired samples.
3. Results
3.1. Sample acquisition
We obtained 43 non-falciparum whole blood samples from the Malaria Reference Laboratory for our drug screens over the period from June 2018 to December 2019. Most of the samples were Plasmodium ovale spp. (21), followed by P. vivax (12) and P. malariae (9) (Supplementary Table 1). Only a single P. knowlesi sample was received for testing. In addition, we included seven P. falciparum samples for comparison of ex vivo drug susceptibility among human malaria species. Of the fifty whole blood samples received in total, not all harboured viable parasites that could be successfully assayed for drug susceptibility. P. falciparum and P. malariae samples were most likely to yield viable parasites for successful drug assays with 86 % (6/7 samples) and 78 % (7/9 samples), respectively (Supplementary Table 1). About a third of P. ovale assays (8/21 samples; 38 %) produced viable parasites for drug screening. P. vivax was the species least likely to yield viable parasites for drug assays with only a single sample producing data out of twelve received. The single P. knowlesi sample we received was viable. With the exception of the P. knowlesi (Malaysia) and P. vivax (Afghanistan) sample, all other samples were from patients that reported travel to countries in Africa (Supplementary Table 1).
3.2. Testing in vitro parasite lines under ex vivo assay conditions
Before screening the ex vivo samples, we tested the viability of using the [3H]hypoxanthine method on our in vitro P. knowlesi (A1-H.1) and P. falciparum (3D7) clones. In order to mimic the likely conditions for the ex vivo samples, asynchronous in vitro parasite lines were diluted to a low starting parasitaemia (0.1 %). After adding the parasites to the drug dilutions in the 96-well plate, the [3H]hypoxanthine was added at the start of the experiment and drug + hypoxanthine exposure continued for a complete life cycle duration (27 h for P. knowlesi and 48 h for P. falciparum). The [3H]hypoxanthine assay provided a sufficient signal to allow for growth inhibition to be measured in spite of the low starting parasitaemia of 0.1 %.
Table 1 reports the results of the drug susceptibility tests. By using asynchronous parasite populations, and considering also the different life cycle lengths of P. knowlesi versus P. falciparum, we expect natural variability in our EC50 determinations. Consequently, we regard only differences in EC50 values greater than 3-fold between species as potentially important. In Table 1 we show that chloroquine, pyronaridine and artefenomel are uniformly potent (EC50 values < 8.8 nM) and exhibit a less than 3-fold difference between species. For the other compounds there was a greater than 4-fold difference in EC50 values between species, with an almost 55-fold difference between species for compound AN13762. AN13762 was also the least potent of the drugs screened (Table 1). Ganaplacide and dihydroartemisinin provide two examples where P. knowlesi is more susceptible than P. falciparum. On the contrary, we demonstrate that P. knowlesi is less susceptible to cipargamin, DSM265 and AN13762 than P. falciparum (Table 1). For comparison, we performed the same drug screens on synchronised ring stage cultures of both species (Supplementary Table 2), which are expected to mimic P. falciparum ex vivo conditions. The EC50 data for synchronised ring cultures (Supplementary Table 2) were very similar to those of asynchronous parasite cultures presented in Table 1.
Table 1.
Comparison of the susceptibility of the in vitro culture adapted P. knowlesi (A1-H.1 clone) and P. falciparum (3D7 clone) under ex vivo conditions. Asynchronized parasites set to a low starting parasitaemia of 0.1 % were exposed to drug dilutions for a complete life cycle, and [3H]hypoxanthine was added at the beginning of the experiment (see methods section for more details).
| Compound Screened | EC50 values (nM)a |
Fold Difference (Pk/Pf) | P-valueb | |
|---|---|---|---|---|
|
P. knowlesi (A1-H.1 clone) 27 h exposure |
P. falciparum (3D7 clone) 48 h exposure |
|||
| Chloroquine | 8.83 ± 0.65 | 5.38 ± 0.42 | 1.64 | 0.0046 |
| Dihydroartemisinin | 0.48 ± 0.04 | 2.00 ± 0.39 | 0.24 | 0.0080 |
| Pyronaridine | 0.38 ± 0.03 | 0.40 ± 0.08 | 0.95 | 0.8369 |
| Artefenomel | 1.05 ± 0.10 | 2.70 ± 0.34 | 0.39 | 0.0014 |
| Cipargamin | 5.20 ± 0.66 | 1.03 ± 0.15 | 5.05 | 0.0003 |
| Ganaplacide | 2.98 ± 0.42 | 12.3 ± 1.72 | 0.24 | 0.0004 |
| DSM265 | 44.9 ± 9.49 | 5.93 ± 0.78 | 7.57 | 0.0064 |
| AN13762 | 3763 ± 798 | 69.1 ± 3.65 | 54.46 | 0.0006 |
EC50 values report the mean ± SEM from at least 4 experiments, and some up to six repeats, each performed in duplicate.
p values are calculated by comparing EC50 values for P. falciparum versus P. knowlesi using Student's two-tailed unpaired t-test.
Apart from dihydroartemisinin, these differences between species are very similar to those we reported previously using different metholodogy for measuring drug susceptibility (SYBR green I readout previously (van Schalkwyk et al., 2019; van Schalkwyk et al., 2017) versus [3H]hypoxanthine used here, and 1 % starting parasitaemia versus 0.1 % used here). Some EC50 values reported in Table 1 are lower than those we reported previously (e.g., chloroquine, pyronaridine, DSM265) (van Schalkwyk et al., 2017). This is likely due the lower starting parasitaemia (inoculum effect) and/or to the absence of 10 % serum in the growth media (serum binding) used in this study. Apart from pyronaridine (p = 0.8369), all the EC50 differences between species were significantly different (p < 0.0080). These data confirm our previous findings that P. knowlesi is less susceptible than P. falciparum to DSM265, cipargamin and AN13762, but is more susceptible to ganaplacide and dihydroartemisinin (van Schalkwyk et al., 2017, 2019).
3.3. ex vivo species differences
In Fig. 1 we provide a graphic demonstration of the drug susceptibility data from the viable ex vivo samples received in the course of our study. For each drug, a single point in the graph represents the EC50 value averaged from a distinct, individual ex vivo sample assayed in duplicate. Not all drugs could be tested on all ex vivo parasites because of the small and variable volumes of sample received within the study. Also shown in Fig. 1 are the median EC50 values, which are then reported in Table 2 for a more detailed comparison. Statistical analyses were limited to comparisons with P. falciparum, P. malariae and P. ovale species and were not possible for P. knowlesi and P. vivax as only one sample was available for analysis in each case.
Fig. 1.
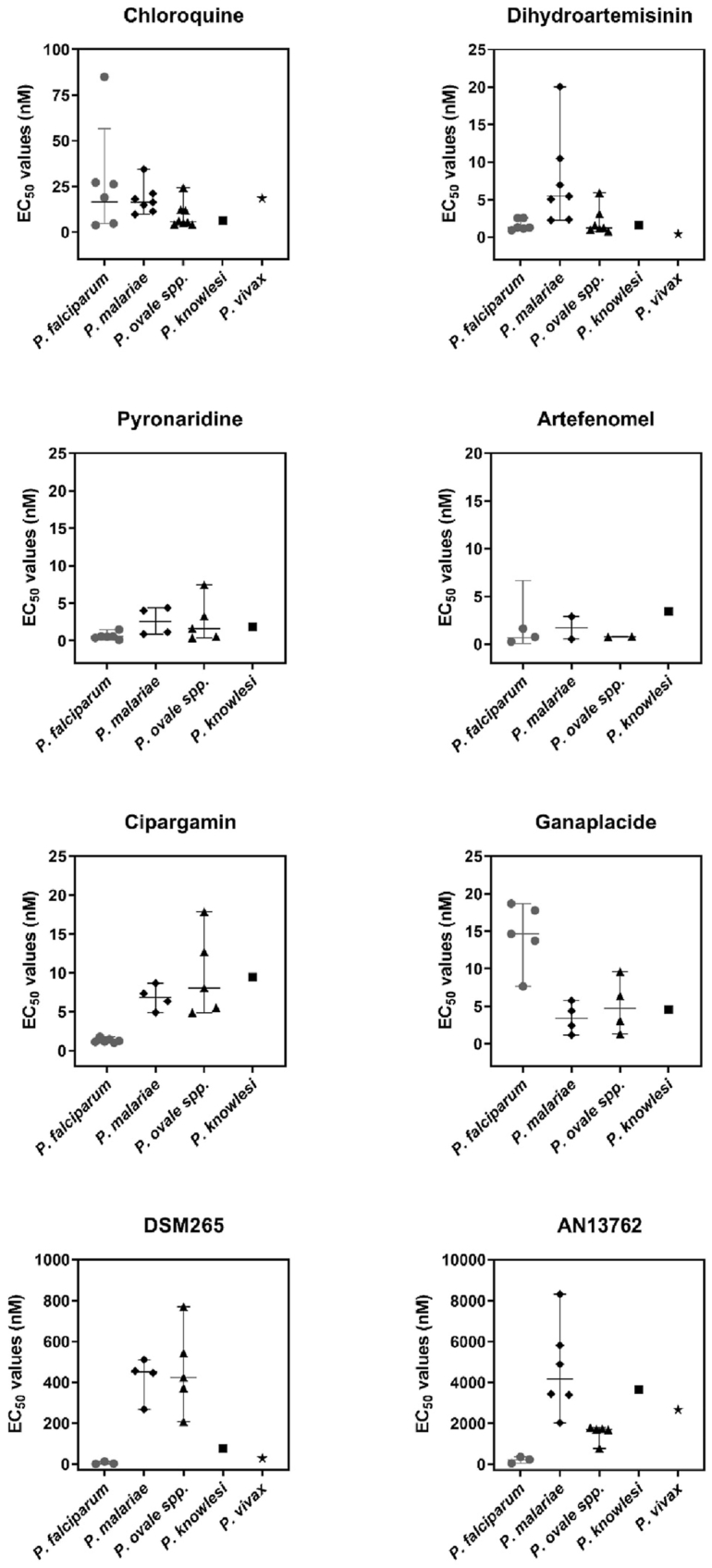
Susceptibility of human malaria species, obtained ex vivo through the Malaria Reference Laboratory (UK), to established and experimental antimalarial drugs. Each data point for a particular drug shows the average EC50 value from duplicate assays on a single, distinct ex vivo sample (i.e., no repeats). Median EC50 values and the 95 % confidence value are shown for P. falciparum, P. malariae and P. ovale species. For P. knowlesi and P. vivax only one ex vivo sample was successfully screened for EC50 data. Not all drugs could be tested against all ex vivo parasites because of small and variable volumes of sample received within the study.
Table 2.
Comparison of the susceptibility of the ex vivo Plasmodium species. Parasites were exposed to serial drug dilutions for a complete life cycle, and [3H]hypoxanthine was added at the beginning of the experiment (see methods section for more details).
| Compound screened | Median EC50 values (nM)a (range) |
Fold Difference (p-value) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| P. falciparum | P. malariae | P. ovale spp. | P. knowlesi | P. vivax | Pm/Pf | Po/Pf | Pk/Pfb | Pv/Pfb | |
| Chloroquine | 22.61 (3.85–85.0) | 16.38 (9.77–34.4) | 5.69 (4.16–24.3) | 6.18 | 18.46 | 0.72 (0.4247) | 0.25 (0.1130) | 0.27 | 0.82 |
| Dihydroartemisinin | 1.30 (0.94–2.57) | 5.45 (2.27–20.0) | 1.23 (0.77–5.92) | 1.64 | 0.42 | 4.19 (0.0420) | 0.95 (0.5639) | 1.26 | 0.32 |
| Pyronaridine | 0.57 (0.11–1.49) | 2.57 (0.88–4.41) | 1.63 (0.39–7.47) | 1.86 | ND | 4.51 (0.0332) | 2.86 (0.1223) | 3.26 | ND |
| Artefenomel | 0.76 (0.26–1.63) | 1.72 (0.55–2.89) | 0.78 (0.76–0.80) | 3.43 | ND | 2.26 (0.4715) | 1.03 (0.8504) | 4.51 | ND |
| Cipargamin | 1.21 (1.02–1.79) | 6.85 (4.92–8.68) | 8.06 (4.90–17.85) | 9.46 | ND | 5.66 (<0.0001) | 6.66 (0.0038) | 7.82 | ND |
| Ganaplacide | 15.76 (7.65–18.69) | 3.40 (1.18–5.76) | 4.72 (1.30–9.60) | 4.57 | ND | 0.22 (0.0066) | 0.30 (0.0236) | 0.29 | ND |
| DSM265 | 2.51 (0.23–12.18) | 450 (268–511) | 463 (207–769) | 76.4 | 28.2 | 179.3 (0.0064) | 184.5 (0.0105) | 30.44 | 11.24 |
| AN13762 | 226 (45.2–354) | 4166 (2022–8317) | 1708 (776–1789) | 3644 | 2664 | 18.43 (0.0126) | 7.56 (0.0023) | 16.12 | 11.79 |
ND = Not done. No P. vivax EC50 data are available for these compounds.
Each EC50 value is determined from the average of a duplicate experiment performed on a single ex vivo sample.
No p-value could be calculated as only a single EC50 value was obtained.
The results in Fig. 1 and Table 2 are summarised through the following key findings:
For chloroquine, across all species, most of the EC50 values are below 35 nM indicating that most ex vivo parasites remain susceptible to this antimalarial drug (Fig. 1). The one exception is a P. falciparum sample with a EC50 value of 85 nM. We suspected this parasite sample would harbour chloroquine-resistant parasites. Genotyping of archived DNA extracted from this sample confirmed the presence of only the chloroquine-resistant CVIET genotype, for amino acids 72–76 of PfCRT.
For dihydroartemisinin, pyronaridine and artefenomel, most EC50 values were below 10 nM and showed little or no significant difference when comparing P. falciparum to P. malariae or P. falciparum to P. ovale (Table 2; p ≥ 0.0332).
Cipargamin, DSM265 and AN13762 were more potent (≥5.66 fold) against P. falciparum than all the other species tested. For these compounds, the difference in susceptibility between P. falciparum and P. malariae, or P. falciparum and P. ovale, were all significant (Table 2; p ≤ 0.0126).
Ganaplacide was the only new antimalarial compound with greater potency against non-falciparum species. P. malariae and P. ovale (median EC50 values < 5 nM; Table 2) were significantly more susceptible to ganaplacide that P. falciparum (median EC50 value = 15.76 nM; P < 0.0236).
4. Discussion
We have reported previously on species differences in the susceptibility of Plasmodium falciparum and Plasmodium knowlesi to antimalarial drugs, such as cipargamin, DSM265, ganaplacide and proguanil (van Schalkwyk et al., 2017, 2019, 2020). These data have been generated using in vitro culture-adapted lines. Whether these susceptibility differences exist in the other species, and whether these different susceptibilities are more comparable to the P. knowlesi phenotype or P. falciparum phenotype, is challenging to assess in the absence of routine in vitro culture systems for these other species. One way of assessing the in vitro drug susceptibility of parasite species lacking representative culture-adapted lines is through using an ex vivo platform, where infected erythrocytes are removed from the patient and exposed to serial dilutions of drugs in a 96-well plate. These parasites are then allowed to grow for a period of time (up to one complete life cycle usually, as they do not re-invade if not culture-adapted) and the survivors can be quantified using either microscopy or measuring [3H]hypoxanthine incorporation (Chaorattanakawee et al., 2017; Fatih et al., 2013; Kaewpongsri et al., 2011; Mbaye et al., 2017; Rangel et al., 2018). Obtaining ex vivo non-falciparum parasites is a challenge, especially in a non-endemic setting such as the UK. Some of these challenges include: obtaining large numbers of samples, getting hold of viable parasites for drug assays in a timely manner, and obtaining mono-infections (as mixed infections are often present). We made use of the Malaria Reference Laboratory (MRL) as our source of ex vivo parasites.
The MRL is an essential resource of reference-level expertise for the surveillance of malaria infections within the United Kingdom, providing also diagnostic and advisory services. Suspected or confirmed malaria specimens are sent to the MRL by post or courier. Two thick and two thin films are requested from each sending laboratory together with a recommended minimum of 3.0 mL of EDTA-treated blood. The MRL confirms the diagnosis of malaria infection by microscopy and extracts DNA from the blood sample for additional PCR confirmation. Some blood is also archived in case further analyses are required. Despite the length of time over which the study was run, only a small number of viable samples were obtained for our drug screens (Supplementary Table 1). Among the reasons for the limited number of samples of non-falciparum species were: (a) blood sample volume received was too small to provide excess for our drug screening above that required for characterization and archiving by the MRL, (b) the sample received through the post was too old to yield viable parasites, (c) samples contained mixed species infections, and (d) parasitaemia was too low to generate a signal with the [3H]hypoxanthine method.
We employed the tritiated hypoxanthine ([3H]hypoxanthine) method (Desjardins et al., 1979) to measure drug susceptibility of our ex vivo samples. Live parasites take up [3H]hypoxanthine and incorporate it into their nucleic acids. Thus, using the [3H]hypoxanthine method to measure ex vivo growth eliminates the need to subjectively assess schizont maturation which is the method more widely used in other ex vivo studies in endemic countries (Chaorattanakawee et al., 2017; Russell et al., 2003, 2012). Using our approach, the [3H]hypoxanthine was added at the beginning of each experiment, and surviving parasites allowed to incorporate the DNA precursor for the entire length of the drug exposure, an approach previously adopted by Arnold et al. for testing P. knowlesi in vitro (Arnold et al., 2016). In our assays no unlabelled hypoxanthine was added so that parasites were forced to incorporate only the [3H]hypoxanthine, thereby ensuring the signal generated was as large as possible even with low parasitaemia samples. Traditionally, in vitro drug assays are initiated with parasites growing in a small amount of unlabelled hypoxanthine (i.e., 2 μM), with the tritiated hypoxanthine added 24 h before the end of the experiment to allow for the radiolabel to be incorporated into DNA. Furthermore, most ex vivo drug assays on P. vivax use large amounts of human serum (20 %) in their growth media. Human serum contains hypoxanthine which may also compete for uptake into DNA by parasites (Psychogios et al., 2011). We observed that both unlabelled hypoxanthine and high serum media reduced the incorporation of [3H]hypoxanthine, thereby reducing the signal window and the Zˊ value (a measure of the robustness of the assay) (Supplementary Figure 1). Since the in vitro parasites were able to grow successfully in the presence of only radiolabeled hypoxanthine, our ex vivo experiments were run without unlabelled hypoxanthine and high serum. Serum has previously been shown to be important in supporting P. vivax ex vivo growth (Brockelman et al., 1985; Chotivanich et al., 2001), and thus its absence here may have contributed to the low success rate of our P. vivax and P. ovale assays. However, the conditions adopted were able to support growth of the in vitro lines as well as the ex vivo P. knowlesi sample, most P. malariae and P. falciparum samples, and several P. ovale samples. Future work could focus on identifying optimal conditions for assaying P. vivax and P. ovale species ex vivo in non-endemic settings that allow for improved [3H]hypoxanthine incorporation so as to maintain a good signal window and assay quality.
Another reason for the small number of viable P. vivax and P. ovale samples could be the use of anticoagulant used during blood collection. Samples to the MRL were all received from hospitals using the recommended purple-top Vacutainer® tubes containing EDTA (1.8 mg/mL) as anticoagulant. EDTA is reported to have a toxic effect on parasite viability (Russell et al., 2012). This study used passive sample collection so we were unable to alter the method for blood collection. Interestingly, we have collected several ex vivo P. falciparum samples by similar methodology since 2012 and showed a high (>90 %) success rate for culture adaptation (e.g. (van Schalkwyk et al., 2013)). Also, most P. falciparum and P. malariae samples, and the single P. knowlesi sample used in this study were successfully assayed. This suggests that, if inhibition of growth by EDTA is a factor, some species (e.g., P. vivax) may be more susceptible to this toxicity than others.
Despite these limitations, we were able to obtain a small number of P. malariae, P. ovale and P. falciparum parasites to assay successfully for drug susceptibility. We limited our drug screen to only seven P. falciparum samples as these would provide a similar number of comparator samples to the P. malariae and P. ovale spp.
In the course of the study we obtained only one P. knowlesi sample – this species is rarely detected among malaria cases in the UK - but the sample volume and excellent viability allowed us to screen all our drugs of interest. The P. knowlesi ex vivo EC50 data (Table 2) showed remarkable congruence with the EC50 data reported in Table 1 for the in vitro A1-H.1 clone, tested under similar ex vivo conditions. For P. falciparum, there was also good agreement between ex vivo and in vitro data observed for most drugs. This provides us with confidence in the methodology used to screen the other species for drug susceptibility.
The ex vivo drug susceptibility data reported here for P. malariae and P. ovale spp., and compared with P. falciparum, confirm our hypotheses that there are species differences in drug susceptibility, and that these susceptibility profiles are more similar to the zoonotic P. knowlesi than to P. falciparum (Table 2). With a few exceptions, there were generally small differences in median EC50 values for chloroquine, pyronaridine, dihydroartemisinin and artefenomel for P. malariae or P. ovale when compared with P. falciparum (Table 2). Most comparisons between species for these compounds were not significantly different or were only marginally so (Table 2).
For the new antimalarial agents cipargamin, DSM265 and AN13762, the differences in median EC50 values between P. malariae or P. ovale spp. compared with P. falciparum were large (>5.6 fold; and up to 184-fold for DSM265) and were, in many cases, highly significant (Table 2). The reduced susceptibility of P. malariae or P. ovale spp. compared with P. falciparum to these three antimalarial agents match the susceptibility profiles obtained in comparisons of P. knowlesi with P. falciparum either in vitro (Table 1) or ex vivo (Table 2). Our ex vivo cipargamin susceptibility data are similar to those reported previously for ten P. falciparum and ten P. vivax isolates from Thailand (median EC50 values < 10 nM). However, there was no species difference in drug susceptibility reported between P. vivax and P. falciparum in that study (Rottmann et al., 2010). Of note, different methodologies were employed in those ex vivo drug screens. Cipargamin activity against either rings stages or trophozoite stages was determined using a modified WHO schizont maturation assay and the growth media used was McCoy's 5A medium supplemented with 20 % AB+ human serum as compared to our RPMI-based media lacking serum (Russell et al., 2003; Sharrock et al., 2008). Nevertheless, without more P. vivax data in our study, it is impossible to draw further conclusions about potential species differences with P. falciparum for comparison to that study. For DSM265, ex vivo drug assays performed by others revealed a 5-fold lower susceptibility to P. vivax versus P. falciparum in isolates sampled from Papua, Indonesia (Phillips et al., 2016). Coupled with our ex vivo data reported here, this confirms that DSM265 is more potent against P. falciparum than against any of the other malaria species.
Conversely, P. malariae and P. ovale spp. are more susceptible to ganaplacide than P. falciparum. Again, this greater susceptibility to ganaplacide is more similar to the profile of P. knowlesi than to that of P. falciparum (Table 1, Table 2). Our data are in close agreement with a recent study comparing the ex vivo activity of ganaplacide (KAF156) against P. falciparum and P. vivax. Using parasites obtained from Thailand and Indonesia, P. falciparum samples were shown to be less susceptible to ganaplacide (median EC50 value = 12.6 nM) than P. vivax samples (median EC50 value = 5.5 nM) (Kuhen et al., 2014).
We were only able to obtain data from a single P. vivax ex vivo sample, and could only screen four drugs with the limited sample size (Table 2). The reason for this high attrition rate for this species (only 1/12 successful assays, Supplementary Table 1) compared with other species tested, is unclear. As mentioned above, it could be related to the absence of serum in our assays or the use of EDTA vials for blood collection. However, the P. vivax drug susceptibility data we obtained are similar to the data reported for the other non-falciparum malaria species. The EC50 data for chloroquine and dihydroartemisinin against our non-falciparum species were less than 4-fold different from the median P. falciparum EC50 values. However, DSM265 and AN13762 were 11-fold less potent against P. vivax than P. falciparum, confirming trends observed for the other non-falciparum species.
It is interesting to note how similar the species differences were for the non-falciparum species compared to P. falciparum for cipargamin (5.66–7.82 fold), ganaplacide (0.22–0.30 fold) and AN13762 (7.56–18.43 fold; Table 2). However, there were large differences observed for DSM265, ranging from around 11-fold for P. vivax to 184-fold for P. ovale spp. when compared to P. falciparum (Table 2). Further studies are needed to confirm these data on more P. vivax and P. knowlesi samples, and to investigate this remarkable diversity in susceptibility to DSM265, an inhibitor of the enzyme dihydroorotate dehydrogenase (DHODH) (Phillips et al., 2015).
It should be noted that cipargamin remains highly potent against all species in spite of the lower susceptibility of non-falciparum species reported here (Table 2). Also, ganaplacide is revealed to be more potent against non-falciparum species which is good news. However, the observations of significantly lower potency of DSM265 and AN13762 against non-falciparum species are potentially of critical importance to future drug development (Table 2). We cannot directly predict the clinical relevance of our in vitro findings in the absence of in vivo data from non-falciparum malaria patients, but it would be remarkable if the large species-specific differences observed here were not reflected in different clinical outcomes in treated infections. For example, a recent clinical trial in Peru showed that DSM265 was highly effective at clearing most P. falciparum infections while none of the P. vivax infections were cleared, even at higher doses than the ones used to treat P. falciparum infections (Llanos-Cuentas et al., 2018). Therefore, based on our ex vivo drug susceptibility data presented here, we might predict that DSM265 treatment would also be insufficient to clear either P. malariae and P. ovale spp. infections, given that these species are less susceptible to DSM265 than P. vivax.
5. Conclusion
In this study we sought to test two hypotheses: (a) that there are species differences in drug susceptibility, and (b) that the other non-falciparum malaria species (P. vivax, P. malariae and the P. ovale species) will be more similar to P. knowlesi in their drug susceptibility profiles than to P. falciparum. Using a small panel of ex vivo P. malariae, P. ovale, and P. falciparum samples, and single P. vivax and P. knowlesi samples, we demonstrate that there are indeed species differences in drug susceptibility, particularly to the experimental antimalarial agents cipargamin, ganaplacide, DSM265 and AN13762. Furthermore, the susceptibility profiles of P. malariae and P. ovale spp. are more similar to the susceptibility profile of P. knowlesi (in vitro and ex vivo) than of P. falciparum. This reinforces the value of complementing in vitro drug discovery approaches using P. falciparum with concurrent screening against P. knowlesi.
Author contributions
CJS, DAvS, RM conceived and designed the study. DAvS and CJS performed the experiments. DAvS, RM, MD, DL and CJS analysed the data. DAvS and CJS wrote the paper. All authors read and approved the final manuscript.
Funding
This project was funded by the Medicines for Malaria Venture, grant MMV RD/15/0017 awarded to DAvS. RWM is supported by the UK Medical Research Council (MRC) Career Development Award (MR/M021157/1) jointly funded by the UK MRC and the UK Department for International Development. CJS is supported by Public Health England and the UK Medical Research Council.
Declaration of competing interest
MD and DL are employees of the funder, the Medicines for Malaria Venture. All other authors: no competing interests to declare.
Acknowledgements
The authors extend their deepest gratitude to the staff of the Malaria Reference Laboratory for their support through supplying the ex vivo parasite samples. We also wish to thank Lindsay Stewart for genotyping the P. falciparum isolate.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ijpddr.2021.07.002.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- Arnold M.S., Engel J.A., Chua M.J., Fisher G.M., Skinner-Adams T.S., Andrews K.T. Adaptation of the [3H]hypoxanthine uptake assay for in vitro-cultured Plasmodium knowlesi malaria parasites. Antimicrob. Agents Chemother. 2016;60:4361–4363. doi: 10.1128/AAC.02948-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasil P., Zalis M.G., de Pina-Costa A., Siqueira A.M., Junior C.B., Silva S., Areas A.L.L., Pelajo-Machado M., de Alvarenga D.A.M., da Silva Santelli A.C.F., Albuquerque H.G., Cravo P., Santos de Abreu F.V., Peterka C.L., Zanini G.M., Suarez Mutis M.C., Pissinatti A., Lourenco-de-Oliveira R., de Brito C.F.A., de Fatima Ferreira-da-Cruz M., Culleton R., Daniel-Ribeiro C.T. Outbreak of human malaria caused by Plasmodium simium in the Atlantic Forest in Rio de Janeiro: a molecular epidemiological investigation. Lancet Glob Health. 2017;5:e1038–e1046. doi: 10.1016/S2214-109X(17)30333-9. [DOI] [PubMed] [Google Scholar]
- Brockelman C.R., Tan-Ariya P., Laovanitch R. Observation on complete schizogony of Plasmodium vivax in vitro. J. Protozool. 1985;32:76–80. doi: 10.1111/j.1550-7408.1985.tb03016.x. [DOI] [PubMed] [Google Scholar]
- Chaorattanakawee S., Lon C., Chann S., Thay K.H., Kong N., You Y., Sundrakes S., Thamnurak C., Chattrakarn S., Praditpol C., Yingyuen K., Wojnarski M., Huy R., Spring M.D., Walsh D.S., Patel J.C., Lin J., Juliano J.J., Lanteri C.A., Saunders D.L. Measuring ex vivo drug susceptibility in Plasmodium vivax isolates from Cambodia. Malar. J. 2017;16:392. doi: 10.1186/s12936-017-2034-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chotivanich K., Silamut K., Udomsangpetch R., Stepniewska K.A., Pukrittayakamee S., Looareesuwan S., White N.J. Ex-vivo short-term culture and developmental assessment of Plasmodium vivax. Trans. R. Soc. Trop. Med. Hyg. 2001;95:677–680. doi: 10.1016/s0035-9203(01)90113-0. [DOI] [PubMed] [Google Scholar]
- Desjardins R.E., Canfield C.J., Haynes J.D., Chulay J.D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979;16:710–718. doi: 10.1128/aac.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatih F.A., Staines H.M., Siner A., Ahmed M.A., Woon L.C., Pasini E.M., Kocken C.H., Singh B., Cox-Singh J., Krishna S. Susceptibility of human Plasmodium knowlesi infections to anti-malarials. Malar. J. 2013;12:425. doi: 10.1186/1475-2875-12-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F.J., Sanz L.M., Vidal J., de Cozar C., Alvarez E., Lavandera J.L., Vanderwall D.E., Green D.V.S., Kumar V., Hasan S., Brown J.R., Peishoff C.E., Cardon L.R., Garcia-Bustos J.F. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- Gruring C., Moon R.W., Lim C., Holder A.A., Blackman M.J., Duraisingh M.T. Human red blood cell-adapted Plasmodium knowlesi parasites: a new model system for malaria research. Cell Microbiol. 2014;16:612–620. doi: 10.1111/cmi.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiguemde W.A., Shelat A.A., Bouck D., Duffy S., Crowther G.J., Davis P.H., Smithson D.C., Connelly M., Clark J., Zhu F., Jimenez-Diaz M.B., Martinez M.S., Wilson E.B., Tripathi A.K., Gut J., Sharlow E.R., Bathurst I., El Mazouni F., Fowble J.W., Forquer I., McGinley P.L., Castro S., Angulo-Barturen I., Ferrer S., Rosenthal P.J., Derisi J.L., Sullivan D.J., Lazo J.S., Roos D.S., Riscoe M.K., Phillips M.A., Rathod P.K., Van Voorhis W.C., Avery V.M., Guy R.K. Chemical genetics of Plasmodium falciparum. Nature. 2010;465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaewpongsri S., Sriprawat K., Suwanarusk R., Kyle D.E., Lek-Uthai U., Leimanis M., Lwin K.M., Phyo A.P., Zwang J., Russell B., Nosten F., Renia L. The presence of leukocytes in ex vivo assays significantly increases the 50-percent inhibitory concentrations of artesunate and chloroquine against Plasmodium vivax and Plasmodium falciparum. Antimicrob. Agents Chemother. 2011;55:1300–1304. doi: 10.1128/AAC.01103-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhen K.L., Chatterjee A.K., Rottmann M., Gagaring K., Borboa R., Buenviaje J., Chen Z., Francek C., Wu T., Nagle A., Barnes S.W., Plouffe D., Lee M.C., Fidock D.A., Graumans W., van de Vegte-Bolmer M., van Gemert G.J., Wirjanata G., Sebayang B., Marfurt J., Russell B., Suwanarusk R., Price R.N., Nosten F., Tungtaeng A., Gettayacamin M., Sattabongkot J., Taylor J., Walker J.R., Tully D., Patra K.P., Flannery E.L., Vinetz J.M., Renia L., Sauerwein R.W., Winzeler E.A., Glynne R.J., Diagana T.T. KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment, and prevention of disease transmission. Antimicrob. Agents Chemother. 2014;58:5060–5067. doi: 10.1128/AAC.02727-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalremruata A., Magris M., Vivas-Martinez S., Koehler M., Esen M., Kempaiah P., Jeyaraj S., Perkins D.J., Mordmuller B., Metzger W.G. Natural infection of Plasmodium brasilianum in humans: man and monkey share quartan malaria parasites in the Venezuelan Amazon. EBioMedicine. 2015;2:1186–1192. doi: 10.1016/j.ebiom.2015.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C., Hansen E., DeSimone T.M., Moreno Y., Junker K., Bei A., Brugnara C., Buckee C.O., Duraisingh M.T. Expansion of host cellular niche can drive adaptation of a zoonotic malaria parasite to humans. Nat. Commun. 2013;4:1638. doi: 10.1038/ncomms2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llanos-Cuentas A., Casapia M., Chuquiyauri R., Hinojosa J.C., Kerr N., Rosario M., Toovey S., Arch R.H., Phillips M.A., Rozenberg F.D., Bath J., Ng C.L., Cowell A.N., Winzeler E.A., Fidock D.A., Baker M., Mohrle J.J., Hooft van Huijsduijnen R., Gobeau N., Araeipour N., Andenmatten N., Ruckle T., Duparc S. Antimalarial activity of single-dose DSM265, a novel plasmodium dihydroorotate dehydrogenase inhibitor, in patients with uncomplicated Plasmodium falciparum or Plasmodium vivax malaria infection: a proof-of-concept, open-label, phase 2a study. Lancet Infect. Dis. 2018;18:874–883. doi: 10.1016/S1473-3099(18)30309-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbaye A., Gaye A., Dieye B., Ndiaye Y.D., Bei A.K., Affara M., Deme A.B., Yade M.S., Diongue K., Ndiaye I.M., Ndiaye T., Sy M., Sy N., Koita O., Krogstad D.J., Volkman S., Nwakanma D., Ndiaye D. Ex vivo susceptibility and genotyping of Plasmodium falciparum isolates from Pikine, Senegal. Malar. J. 2017;16:250. doi: 10.1186/s12936-017-1897-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon R.W., Hall J., Rangkuti F., Ho Y.S., Almond N., Mitchell G.H., Pain A., Holder A.A., Blackman M.J. Adaptation of the genetically tractable malaria pathogen Plasmodium knowlesi to continuous culture in human erythrocytes. Proc. Natl. Acad. Sci. U. S. A. 2013;110:531–536. doi: 10.1073/pnas.1216457110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguike M.C., Betson M., Burke M., Nolder D., Stothard J.R., Kleinschmidt I., Proietti C., Bousema T., Ndounga M., Tanabe K., Ntege E., Culleton R., Sutherland C.J. Plasmodium ovale curtisi and Plasmodium ovale wallikeri circulate simultaneously in African communities. Int. J. Parasitol. 2011;41:677–683. doi: 10.1016/j.ijpara.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M.A., Lotharius J., Marsh K., White J., Dayan A., White K.L., Njoroge J.W., El Mazouni F., Lao Y., Kokkonda S., Tomchick D.R., Deng X., Laird T., Bhatia S.N., March S., Ng C.L., Fidock D.A., Wittlin S., Lafuente-Monasterio M., Benito F.J., Alonso L.M., Martinez M.S., Jimenez-Diaz M.B., Bazaga S.F., Angulo-Barturen I., Haselden J.N., Louttit J., Cui Y., Sridhar A., Zeeman A.M., Kocken C., Sauerwein R., Dechering K., Avery V.M., Duffy S., Delves M., Sinden R., Ruecker A., Wickham K.S., Rochford R., Gahagen J., Iyer L., Riccio E., Mirsalis J., Bathhurst I., Rueckle T., Ding X., Campo B., Leroy D., Rogers M.J., Rathod P.K., Burrows J.N., Charman S.A. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci. Transl. Med. 2015;7:296ra111. doi: 10.1126/scitranslmed.aaa6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M.A., White K.L., Kokkonda S., Deng X., White J., El Mazouni F., Marsh K., Tomchick D.R., Manjalanagara K., Rudra K.R., Wirjanata G., Noviyanti R., Price R.N., Marfurt J., Shackleford D.M., Chiu F.C., Campbell M., Jimenez-Diaz M.B., Bazaga S.F., Angulo-Barturen I., Martinez M.S., Lafuente-Monasterio M., Kaminsky W., Silue K., Zeeman A.M., Kocken C., Leroy D., Blasco B., Rossignol E., Rueckle T., Matthews D., Burrows J.N., Waterson D., Palmer M.J., Rathod P.K., Charman S.A. A triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with improved drug-like properties for treatment and prevention of malaria. ACS Infect. Dis. 2016;2(12):945–957. doi: 10.1021/acsinfecdis.6b00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe D., Brinker A., McNamara C., Henson K., Kato N., Kuhen K., Nagle A., Adrian F., Matzen J.T., Anderson P., Nam T.G., Gray N.S., Chatterjee A., Janes J., Yan S.F., Trager R., Caldwell J.S., Schultz P.G., Zhou Y., Winzeler E.A. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. U. S. A. 2008;105:9059–9064. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psychogios N., Hau D.D., Peng J., Guo A.C., Mandal R., Bouatra S., Sinelnikov I., Krishnamurthy R., Eisner R., Gautam B., Young N., Xia J., Knox C., Dong E., Huang P., Hollander Z., Pedersen T.L., Smith S.R., Bamforth F., Greiner R., McManus B., Newman J.W., Goodfriend T., Wishart D.S. The human serum metabolome. PloS One. 2011;6 doi: 10.1371/journal.pone.0016957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja T.N., Hu T.H., Kadir K.A., Mohamad D.S.A., Rosli N., Wong L.L., Hii K.C., Simon Divis P.C., Singh B. Naturally acquired human Plasmodium cynomolgi and P. knowlesi infections, Malaysian borneo. Emerg. Infect. Dis. 2020;26:1801–1809. doi: 10.3201/eid2608.200343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel G.W., Clark M.A., Kanjee U., Lim C., Shaw-Saliba K., Menezes M.J., Mascarenhas A., Chery L., Gomes E., Rathod P.K., Ferreira M.U., Duraisingh M.T. Enhanced ex vivo Plasmodium vivax intraerythrocytic enrichment and maturation for rapid and sensitive parasite growth assays. Antimicrob. Agents Chemother. 2018;62:e02519–17. doi: 10.1128/AAC.02519-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottmann M., McNamara C., Yeung B.K., Lee M.C., Zou B., Russell B., Seitz P., Plouffe D.M., Dharia N.V., Tan J., Cohen S.B., Spencer K.R., Gonzalez-Paez G.E., Lakshminarayana S.B., Goh A., Suwanarusk R., Jegla T., Schmitt E.K., Beck H.P., Brun R., Nosten F., Renia L., Dartois V., Keller T.H., Fidock D.A., Winzeler E.A., Diagana T.T. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell B., Suwanarusk R., Malleret B., Costa F.T., Snounou G., Kevin Baird J., Nosten F., Renia L. Human ex vivo studies on asexual Plasmodium vivax: the best way forward. Int. J. Parasitol. 2012;42:1063–1070. doi: 10.1016/j.ijpara.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Russell B.M., Udomsangpetch R., Rieckmann K.H., Kotecka B.M., Coleman R.E., Sattabongkot J. Simple in vitro assay for determining the sensitivity of Plasmodium vivax isolates from fresh human blood to antimalarials in areas where P. vivax is endemic. Antimicrob. Agents Chemother. 2003;47:170–173. doi: 10.1128/AAC.47.1.170-173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge G.G., Bohme U., Sanders M., Reid A.J., Cotton J.A., Maiga-Ascofare O., Djimde A.A., Apinjoh T.O., Amenga-Etego L., Manske M., Barnwell J.W., Renaud F., Ollomo B., Prugnolle F., Anstey N.M., Auburn S., Price R.N., McCarthy J.S., Kwiatkowski D.P., Newbold C.I., Berriman M., Otto T.D. Plasmodium malariae and P. ovale genomes provide insights into malaria parasite evolution. Nature. 2017;542:101–104. doi: 10.1038/nature21038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharrock W.W., Suwanarusk R., Lek-Uthai U., Edstein M.D., Kosaisavee V., Travers T., Jaidee A., Sriprawat K., Price R.N., Nosten F., Russell B. Plasmodium vivax trophozoites insensitive to chloroquine. Malar. J. 2008;7:94. doi: 10.1186/1475-2875-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B., Kim Sung L., Matusop A., Radhakrishnan A., Shamsul S.S., Cox-Singh J., Thomas A., Conway D.J. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004;363:1017–1024. doi: 10.1016/S0140-6736(04)15836-4. [DOI] [PubMed] [Google Scholar]
- Sutherland C.J., Haustein T., Gadalla N., Armstrong M., Doherty J.F., Chiodini P.L. Chloroquine-resistant Plasmodium falciparum infections among UK travellers returning with malaria after chloroquine prophylaxis. J. Antimicrob. Chemother. 2007;59:1197–1199. doi: 10.1093/jac/dkm104. [DOI] [PubMed] [Google Scholar]
- Sutherland C.J., Tanomsing N., Nolder D., Oguike M., Jennison C., Pukrittayakamee S., Dolecek C., Hien T.T., do Rosario V.E., Arez A.P., Pinto J., Michon P., Escalante A.A., Nosten F., Burke M., Lee R., Blaze M., Otto T.D., Barnwell J.W., Pain A., Williams J., White N.J., Day N.P., Snounou G., Lockhart P.J., Chiodini P.L., Imwong M., Polley S.D. Two nonrecombining sympatric forms of the human malaria parasite Plasmodium ovale occur globally. J. Infect. Dis. 2010;201:1544–1550. doi: 10.1086/652240. [DOI] [PubMed] [Google Scholar]
- Ta T.H., Hisam S., Lanza M., Jiram A.I., Ismail N., Rubio J.M. First case of a naturally acquired human infection with Plasmodium cynomolgi. Malar. J. 2014;13:68. doi: 10.1186/1475-2875-13-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W., Jensen J.B. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- van Schalkwyk D.A., Blasco B., Davina Nunez R., Liew J.W.K., Amir A., Lau Y.L., Leroy D., Moon R.W., Sutherland C.J. Plasmodium knowlesi exhibits distinct in vitro drug susceptibility profiles from those of Plasmodium falciparum. Int J Parasitol Drugs Drug Resist. 2019;9:93–99. doi: 10.1016/j.ijpddr.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schalkwyk D.A., Burrow R., Henriques G., Gadalla N.B., Beshir K.B., Hasford C., Wright S.G., Ding X.C., Chiodini P.L., Sutherland C.J. Culture-adapted Plasmodium falciparum isolates from UK travellers: in vitro drug sensitivity, clonality and drug resistance markers. Malar. J. 2013;12:320. doi: 10.1186/1475-2875-12-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schalkwyk D.A., Moon R.W., Blasco B., Sutherland C.J. Comparison of the susceptibility of Plasmodium knowlesi and Plasmodium falciparum to antimalarial agents. J. Antimicrob. Chemother. 2017;72:3051–3058. doi: 10.1093/jac/dkx279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schalkwyk D.A., Riscoe M.K., Pou S., Winter R.W., Nilsen A., Duffey M., Moon R.W., Sutherland C.J. Novel endochin-like quinolones exhibit potent in vitro activity against Plasmodium knowlesi but do not synergize with proguanil. Antimicrob. Agents Chemother. 2020;64 doi: 10.1128/AAC.02549-19. e02549-02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . World Health Organization; Geneva: 2020. World Malaria Report 2020: 20 Years of Global Progress and Challenges. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.