

Abstract
The structural elucidation of native macromolecular assemblies has been a subject of considerable interest in native mass spectrometry (MS), and more recently in tandem with ion mobility spectrometry (IMS-MS), for a better understanding of their biochemical and biophysical functions. In the present work, we describe a new generation trapped ion mobility spectrometer (TIMS), with extended mobility range (K0 = 0.185 – 1.84 cm2.V−1.s−1), capable of trapping high molecular weight (MW) macromolecular assemblies. This compact 4-cm long TIMS analyzer utilizes a convex electrode, quadrupolar geometry with increased pseudopotential penetration in the radial dimension, extending the mobility trapping to high MW species under native state (i.e., lower charge states). The TIMS capabilities to perform variable scan rate (Sr) mobility measurements over short time (100–500 ms), high mobility resolution, and ion-neutral collision cross section (CCSN2) measurements are presented. The trapping capabilities of the convex electrode TIMS geometry and ease of operation over a wide gas flow, rf range and electric field trapping range are illustrated for the first time using a comprehensive list of standards varying from CsI clusters (n=6–73), Tuning Mix oligomers (n=1–5), common proteins (e.g., ubiquitin, cytochrome C, lysozyme, concanavalin (n=1–4), carbonic anhydrase, β clamp (n=1–4), topoisomerase IB, bovine serum albumin (n=1–3), topoisomerase IA, alcohol dehydrogenase), IgG antibody (e.g., avastin), protein-DNA complexes, and macromolecular assemblies (e.g., GroEL and RNA Polymerase (n=1–2)) covering a wide mass (up to m/z 19,000) and CCS range (up to 22,000 Å2 with < 0.6% RSD).
Graphical Abstract
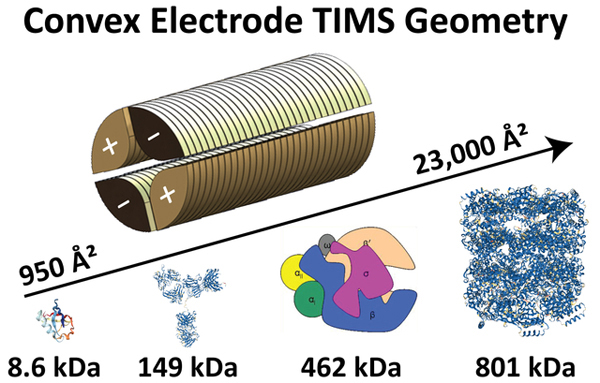
Native mass spectrometry (MS) has demonstrated significant advances for the investigation of protein and protein assemblies, with applications ranging from protein identification, ligand binding and dynamics of protein assembly.1–3 Recent advances in MS technology have enabled the characterization of protein assemblies of high molecular weights (MW),4–6 with new MS instruments approaching near one megadalton mass range with sensitivity down to the detection of single ions.6 Further characterization of protein and protein assemblies analysis relies on the measurement of their three-dimensional structures, as a way to better understand their biological function depending on the cell environment.7 The low abundance of protein and protein assemblies combined with the biological heterogeneity of many of the protein assemblies make their structural information not readily accessible using traditional structural biology tools, such as X-ray crystallography and nuclear magnetic resonance spectroscopy. Since 2005, several efforts have shown the advantages of coupling ion mobility with mass spectrometry (IM-MS) for the study of high MW proteins and protein assemblies during native conditions.8–13 In 2011, ambient pressure differential mobility analyzer measurements of high MW protein complexes using a TOF MS analyzer (DMA-MS) showed narrow mobility peaks (R ~ 50).14 Drift tube - ion mobility spectrometry (DT-IMS) platforms have been used to investigate proteins and protein complexes that reach relatively high resolving powers (R ~ 30–60).15–17 Recently, a periodic focusing DTIMS system reported R ~ 60 for MW species greater than ~200 kDa.17
Different from previous IMS concepts, we introduced the trapped IMS (TIMS) coupled to TOF MS as a field dispersive mobility separation in 2011.18,19 Over the years, it has been shown TIMS capabilities to perform variable scan rate (Sr) mobility analysis at reduced pressures leading to high mobility resolutions (R ~ 100–400) over a short analysis time (e.g., 20–500 ms).20,21 TIMS-MS studies from native solvent conditions have included mainly the molecular level characterization of small and middle size proteins,22–28 being the largest MW reported a ~ 150 kDa system (IgG, avastin).22 The challenges for high MW TIMS studies relies on, different from other transmission based IMS variants, that the TIMS mobility separation is based on holding the ions stationary using an axial electric field (E) against a moving buffer gas, while a radial field is applied to avoid collision with the electrodes.18,19 In the case of TIMS, the electrode geometry and electrodynamic trapping conditions define the number of ions and mobility range that can be effectively trapped.18,19
In the present work, we describe for the first time the use of a convex electrode TIMS quadrupolar cell geometry, capable of trapping over an extended mobility range. This new generation TIMS technology, implemented in a custom built TIMS-TOF MS instrument (Figure S1a), permits the study of high MW/low charge state (native) species. In the following discussion, a special emphasis is placed on the performance over a wide gas flow, rf range and electric field trapping range. This study comprises several years of instrument optimization and a comprehensive list of CCS values from CsI clusters (n=6–73), Tuning Mix oligomers (n=1–5), common proteins (e.g., ubiquitin, cytochrome C, lysozyme, concanavalin (n=1–4), carbonic anhydrase β clamp (n=1–4), topoisomerase IB, bovine serum albumin (n=1–3), topoisomerase IA, alcohol dehydrogenase), IgG antibody (e.g., avastin), protein-DNA complexes and macromolecular assemblies (e.g., GroEL and RNA Polymerase (n=1–2)). The term “native analysis” refers to the use of physiologically friendly solvent condition (e.g., 100 mM ammonium acetate) followed by gentle ionization/injection/analysis conditions to prevent potential collisional activation and structural/conformational rearrangements prior and during TIMS -MS analysis.
EXPERIMENTAL SECTION
Materials and Reagents.
Low-concentration Tuning Mix calibration standard (G24221A) was purchased from Agilent Technologies (Santa Clara, CA). Cesium iodide (CsI) was purchased from Sigma-Aldrich (Saint-Louis, MO) and was prepared at 2 mg/mL in water. An immunoglobulin G (IgG, avastin, 149 kDa) was obtained from Genentech Inc. (San Francisco, CA). Ubiquitin (8.6 kDa), cytochrome C (12 kDa), lysozyme (14.3 kDa), carbonic anhydrase (29 kDa), bovine serum albumin (66 kDa), concanavalin A (103 kDa), alcohol dehydrogenase (147 kDa) and GroEL (801 kDa) were purchased from Sigma-Aldrich. The E. coli topoisomerase IA (97.5 kDa) was expressed and purified as described elsewhere.29 The variola virus topoisomerase IB (38.5 kDa) was expressed and purified as described previously.30 The E. coli RNA polymerase holoenzyme (462 kDa) was purified by the method of Hager et al.31 The dnaN gene encoding the β clamp was expressed and purified as previously described.32 All protein assembly solutions were analyzed at a concentration of 5 μM in 100 mM aqueous ammonium acetate (NH4Ac).
TIMS-MS Instrumentation.
Ion mobility experiments were performed on a custom built nanoESI-TIMS coupled to an Impact Q-ToF mass spectrometer (Bruker Daltonics Inc., Billerica, MA, Figure S1a).18,19 NanoESI emitters were pulled in-house from quartz capillaries (O.D. = 1.0 mm and I.D. = 0.70 mm) using a Sutter Instrument Co. P2000 laser puller. A custom built XYZ positioner holds a laser pulled-tip capillary <5 mm from the orifice of the atmospheric pressure-vacuum interface.33 Sample solution is biased at ~1kV relative to interface entrance. Small differences in the charge state distribution (shift of one charge state on average toward higher m/z) were observed as compared to recent literature reports15,16 that could be related to the use of in-house laser pulled glass capillary tips in addition to the low capillary voltage applied, which may affect the charge state distribution upon ionization. The general fundamentals of TIMS as well as the calibration procedure have been described in the literature.34–39 Briefly, the ion mobility separation in a TIMS device depends on the gas flow velocity (vg), elution voltage (Ve), ramp voltage (Vramp), ramp time (tramp) and base voltage (Vout). The reduced mobility, K0, is defined by:
| (1) |
where, A is a constant related to vg define by P1 and P2 and the TIMS geometry. Ve is experimentally determined by varying the tramp for a constant Vramp. The constant A is determined using calibration standards (e.g., Tuning Mix) of known reduced mobilities.34 The measured mobilities are converted into collision cross section (CCS, Ω in Å2) using the Mason-Schamp equation:
| (2) |
where, z is the charge of the ion, kB is the Boltzmann constant, N* is the number density of the bath gas and mi and mb refer to the masses of the ion and bath gas, respectively.40
In the presented design, the TIMS analyzer section is composed of 27 printed circuit (PC) boards including the base plates at entrance and exit, electrically insulated, with each board containing two pairs of opposite electrodes at the same voltage and radiofrequency phase. The shape of the electrode is in a convex geometry with an internal diameter of 6 mm (Figure 1a). The TIMS unit is controlled by an in-house software in LabView (National Instruments) and synchronized with the MS platform controls.19
Figure 1.

(a) Designs of the TIMS analyzer in a concave/convex electrode geometry. (b) Equipotential lines using SIMION are illustrated for the concave and convex electrode geometries at a given applied voltage (equipotential lines shown over 20 – 200 V/mm in 20 V steps), showing that the proposed convex geometry produces a stronger pseudopotential.
TIMS separation was carried out using nitrogen (N2) at ambient temperature (T) with vg set by the pressure difference between the funnel entrance (P1 = 1.7 and 2.6 mbar) and exit (P2 = 0.8 mbar, Figure S1a). An rf voltage of 175–250 Vpp at 450, 800 and 2040 kHz was applied to all electrodes. In particular, an rf voltage of 220 Vpp at 450 kHz with P1 = 2.6 mbar was used for all investigated proteins. Ions were softly transferred and injected into the TIMS analyzer to avoid potential activation, by keeping a low ΔV (ΔV = 20–50 V) between the deflector (Vdef) and the funnel entrance (Vfun) as well as between the funnel entrance and the TIMS analyzer (Vramp) in order to generate native-like mobility distributions (Figure S1a). Changes in the mobility profiles were not observed for the systems reported under native conditions over 100–500 ms time range after desolvation. All resolving power (R) values reported herein were determined as R = CCS/ΔCCS, where ΔCCS is the full peak width at half maximum (FWHM) of the IMS signal. A gaussian peak fitting algorithm with non-linear least squares functions (Levenberg–Marquardt algorithm) using OriginPro 2016 was used to evaluate the FWHM of each IMS band.
RESULTS AND DISCUSSION
Convex electrode TIMS geometry.
In TIMS operation, an rf is applied to the electrodes of the TIMS analyzer to generate a radially confining pseudopotential, while an axial electric field gradient is produced across the electrodes to counteract the drag force exerted by the gas flow, effectively leading to the trapping of the ions. Ions are then eluted from the TIMS analyzer region by decreasing the axial electric field (Figure S1a). The pursuit for new electrode TIMS geometries focuses on the need to apply higher penetration radial fields in order to extend the mobility range. In this new TIMS design, the shape of the electrode was varied from concave to convex (Figure 1a) geometries while keeping a quadrupolar form. This change leads to higher penetration pseudopotentials. Ion dynamic simulation using an elastic hard sphere model using the SIMION (v 8.0) package, as previously described,34 showed similar ion behavior despite the electrode shape (Figure S1b). The convex electrode geometry requires smaller rf Vpp amplitudes than the concave electrode geometry to trap ions (Figure 1b). This is a key feature since the rf Vpp amplitude can be a limiting factor during IMS operation at reduced pressures due to potential electric discharges at high values.
Extended mobility range in a convex TIMS geometry.
The performance evaluation of the convex electrode TIMS geometry, using a Tuning Mix calibration standard (Figure S2a), resulted in the trapping of a wide m/z range of species (up to m/z 13,650, Figure 2a) at P1 = 1.7 mbar. Closer inspection showed the trapping of single charge species of Tuning Mix oligomers (n = 1–5) over a large mobility range (K0 = 0.18 – 0.72 cm2.V−1.s−1, Figure 2a and Table S1). This is the first report of mobility trapping Tuning Mix monomers (Figure S2b), dimers (Figure S2c), trimers (Figure S2d), tetramers (Figure S2e) and pentamers (Figure S2f) over a mobility range of 0.45 – 0.72, 0.30 – 0.40, 0.24 – 0.27, 0.20 – 0.23, 0.18 – 0.20 cm2.V−1.s−1, respectively. Note that the observed Tuning Mix oligomers are not necessarily homo-oligomers, but can also be composed of a hetero-combination of monomeric units (Figure S2). In addition, the protonated species of the Tuning Mix oligomers were only observed for the monomers while only one ammonium, sodium and potassium adduct was observed for all the Tuning Mix oligomers, for which the ammonium adducts were the most abundant ions as a general trend (Figure S2). The Tuning Mix ions were trapped over the P1 = 2.6–1.7 mbar range, where the mobility range observed is directly proportional to the axial electric field range for single charge ions.
Figure 2.

TIMS-MS analysis of Tuning Mix showing (a) K0 vs m/z map and (b) a plot of the trapping as a function of the rf frequency, where the green, pink, orange, blue and brown regions illustrate the monomers (1M), dimers (2M), trimers (3M), tetramers (4M) and pentamers (5M), respectively. (c) MS and TIMS profiles for the multiply protonated species of ubiquitin as a function of the rf frequency, where the blue and black dashed traces were obtained with a 800 and 450 kHz rf, respectively.
For comparison purposes, we also evaluated the convex electrode TIMS geometry using a cesium iodide solution (Figure S3).41 A known caveat of using a concentrated CsI solution is the contamination of the instrument; the CsI cluster distribution is proportional to the starting concentration. From the nESI of a 2 mg/mL solution, we observed the trapping of CsI clusters (n=6–73) with charges 1+−3+, covering an m/z up to 8000 at P1 = 2.6 mbar (Figure S3a and Table S2). Note that we did not pursue the observation of higher CsI cluster since they will require more concentrated solutions leading to further instrument contamination. Closer inspection exhibited the trapping of multiply charged species of CsI clusters, mainly including the singly (Figure S3b), doubly (Figure S3c) and triply (Figure S3d) charged species over a mobility (K0) range of 0.99 – 0.40, 0.99 – 0.55 and 0.96 – 0.65 cm2.V−1.s−1, respectively (Table S2). Notice that the increase in charge state with the size of the CsI cluster resulted in a smaller mobility range than that observed for the single charged Tuning Mix oligomers.
Influence of rf frequency and Vpp amplitude in a convex TIMS geometry.
Different from drift tube IMS devices and analogous to any rf driven ion guide/trap, the trapping in the TIMS analyzers can be limited by the rf frequency and amplitude (see theoretical high-end m/z ~21,300 estimate cutoff in Figure 2b for the new convex quadrupolar geometry based on a linear quadrupolar ion trap and delhmoltz pseudopential42). Empirical evaluations showed no apparent experimental high-end cutoff using a 450 kHz rf for the case of Tuning Mix oligomers (Figure 2b blue trace). However, a low m/z trapping cutoff was observed below m/z 922. Using an 800 kHz rf (Figure 2b, red trace), a high-end m/z cut off was observed at m/z 8200 (Tuning Mix trimers), with a low m/z trapping cutoff at m/z 322. Using a 2040 kHz rf (Figure 2b, black trace), no low m/z cut off was observed for the Tuning Mix ions, but a high-end m/z cut-off at m/z 1522 was observed. Low and high m/z cut-offs are expected depending on the rf frequency for all rf ion guides and trapping devices.42 Depending of the analytical challenge, the rf frequency can be chosen for effective trapping using the convex electrode TIMS geometry. Compare to the concave electrode TIMS geometry, the most remarkable feature is the substantial extension of the high-end cutoff at 45o kHz and use of lower Vpp for ion confinement.
Preservation of native-like protein conformations in a convex TIMS geometry.
The ability of an IMS device to study biological relevant issues relies on its capability to preserve native-like structures present in the solution.43 This was initially assessed using a well-known and characterized protein – ubiquitin, which is known to display differences in the IMS profiles due to ion heating.25,27 Previous studies demonstrated that the concave electrode TIMS geometry is able to retain native-like structures of ubiquitin; moreover, the use of elevated dc/rf-electric field can induce unfolding noticeable in the IMS profiles.24,25
The nESI-TIMS-MS analysis of ubiquitin under native starting solution condition (e.g. 100 mM aqueous NH4Ac) exhibited a narrow charge state distribution, ranging from [M + 4H]4+ to [M + 6H]6+ molecular ion species (Figure 2c). The TIMS settings were optimized for “soft” injection and analysis conditions: a low ΔV (20 V) between Vdef and Vfun as well as between Vfun and Vramp (Figure S1a). Inspection of the [M + 4H]4+, [M + 5H]5+ and [M + 6H]6+ ions showed a single IMS band, centered at ~1124, ~1146 and ~1204 Å2, respectively. The measured CCS were found consistent with previously reported CCS values for native-like ubiquitin using a drift tube IMS (Table S3)44–47 and concave electrode TIMS geometry.24,27 These observations demonstrate that native-like mobility distributions can be obtained with the convex electrode TIMS geometry. Note that the single wide CCS distribution of the [M + 6H]6+ ions suggests the existence of several conformations. No differences were observed between the 800 kHz (blue traces) and 450 kHz (black dashed lines) rf frequencies (Figure 2c). During native TIMS analysis, the relative simplicity of the sample facilitates the operation of the quadrupolar cell TIMS design without the observation of space charge effects despite the ion confinement being stronger in the convex electrode geometry. As control experiments, non-native ubiquitin distribution were achieved (evidenced by an additional IMS band with larger CCS value ~ 1370 Å2 for the [M + 6H]6+) when the rf amplitudes exceeds a 250 Vpp threshold (Figure S4a–b). In addition, TIMS experiments as a function of the trapping time (100–500 ms) did not evidence any unfolding events for the [M + 6H]6+ ions of ubiquitin but only a small narrowing of the IMS band (Figure S4c and Table S3). This means that the effective temperature in the TIMS analyzer is not high enough to induce unfolding which deviates from the observations previously reported for the case of proteins48 and are more in adequation with the observations reported by Bleiholder et al.49
Native macromolecular assemblies in a convex TIMS geometry.
The analysis of native concanavalin A (ConA), a homo-tetramer of 103 kDa, resulted in the observation of the native IMS and MS profiles consisting of a single IMS band per charge state over the [M + 17H]17+ - [M + 21H]21+ range (Figure 3a). Inspection of the TIMS profiles were consistent with recently reported CCS values for the native-like conformational states of ConA (Table S3).15,16,46,50 An apparent mobility resolution of R ~ 55 was observed using fast Sr (1.11 V/ms, Figure 3a), whereas recent data obtained from a drift tube reported apparent mobility resolution estimated at R ~ 40.15
Figure 3.

MS and TIMS profiles are shown for (a) concanavalin A, (b) avastin, (c) E. coli RNA polymerase holoenzyme and (d) GroEL. The scan rate (Sr = ΔVramp / ttrap) and time values are given.
The nESI-TIMS-MS analysis of the native IgG (avastin, 149 kDa) antibody resulted in a narrow charge state distribution ([M + 19H]19+ − [M + 25H]25) with TIMS and MS profiles consistent with recent data obtained from a drift tube IMS (Table S3).51,52 Closer inspection to the TIMS profiles showed heterogeneous distributions, with an apparent mobility resolution of R ~ 60 using a relatively slow Sr of 0.25 V/ms (Figures 3b and S5). Note that the Gaussian fit for avastin IMS profiles can be found in Figure S5. The use of native conditions resulted in the trapping of lower charge state (19+ to 25+) when compared to previous concave electrode TIMS experiments (25+ to 27+) under similar trapping conditions.22
The nESI-TIMS-MS study of native E. coli RNA polymerase holoenzyme (RNAP) and a bacterial chaperonin GroEL resulted in the observation of a narrow charge state distribution [M + 33H]33+ - [M + 41H]41+ and [M + 59H]59+ - [M + 66H]66+, respectively (Figures 3c–d). The E. coli RNA polymerase holoenzyme (462 kDa) consists of 2 identical and 4 non-identical subunits, designated αI/αII (36.5 kDa), β (150.6 kDa), β´ (155.2 kDa), ω (10.2 kDa) and σ70 (72.4 kDa).53,54 The bacterial chaperonin GroEL is a homo-oligomeric complex of 801 kDa composed of 14 identical subunits of 57.2 kDa.55 All the measured CCS were found consistent with previously reported CCS values obtained from a drift tube IMS for the native-like conformational states of GroEL (Table S3).46 The TIMS profiles of the RNAP exhibited a single, wide band distribution (apparent mobility resolution R ~ 40 with a Sr = 1.48 V/ms, Figure 3c), as compared to GroEL, for which single, narrower IMS bands were observed (apparent mobility resolution R ~ 80 using a Sr = 0.74 V/ms, Figure 3d). The narrow IMS bands observed for the GroEL complex evidences the high ordering level expected from this complex as evidenced by NMR and X-ray crystallography.55 This suggests that the RNAP presents a high degree of structural heterogeneity, probably arising from the large number of possible movements and/or interactions between the subunits.
In addition to these systems, other common proteins (e.g., cytochrome C, lysozyme, carbonic anhydrase, bovine serum albumin and alcohol dehydrogenase) were studied using native conditions nESI-TIMS-MS and summarized in Table S3. In some cases, non-specific oligomers for common proteins were detected as a consequence of the “soft” injection and TIMS analysis conditions. The TIMS CCS data is in good agreement with recently reported CCS values from “soft” drift tube IMS studies (Table S3).15,16,45 In general, a slightly higher apparent mobility resolution was observed using the convex TIMS geometry relative to the reported soft drift tube IMS studies: for example, ubiquitin 4+ (R = 36–40, Sr = 0.85–0.22 V/ms vs. R ~2015,45), cytochrome C 6+ (R = 29, Sr = 1.23 V/ms vs. R ~1915,45), carbonic anhydrase 9+ (R = 31–40, Sr = 1.85–0.74 V/ms vs. R ~2845), bovine serum albumin 15+ (R = 30–44, Sr = 0.77–0.20 V/ms vs. R ~1715), concanavalin A 19+ (R = 44–67, Sr = 1.85–0.39 V/ms vs. R ~4115) and alcohol dehydrogenase 24+ (R = 35–54, Sr = 2.50–0.89 V/ms vs. R ~3115).
CCS/Mobility range and apparent mobility resolution in a convex TIMS geometry.
Most of the proteins and protein complexes illustrated in Table S3 were commercially available and covered a wide mass and mobility range (Figures 4a and 4b). Details on the analysis of other native protein and protein-DNA systems can be found elsewhere (e.g., topoisomerases in complex with DNA,56 β clamp,57 and bovine serum albumin58). Briefly, the “soft” analysis condition of the convex quadrupolar TIMS geometry allowed the investigation of the microheterogeneity of topoisomerase IA/IB and their DNA-bound states,56 the dynamics of the E. coli β clamp dimer interface and its influence on DNA loading,57 and the structural changes by thermal denaturation of bovine serum albumin.58 The RNAP dimers (924 kDa) are currently the largest native complex evaluated using the convex electrode nESI-TIMS-TOF MS technology with CCS values up to 23,000 Å2 (Figure 4a) and narrow charge state and mobility distributions (52+−60+ over 0.524–0.534 cm2.V−1.s−1, Figure 4b). All mobility experiments were conducted on different days from at least triplicate measurements. The intraday and interday analysis showed good reproducibility with less than 0.6% variation in the CCS measurements (Figure S6). A relatively large database of K0 and TIMSCCSN2 values was collected using standard calibrants (Tuning Mix and CsI clusters) and native-like proteins and protein complexes including 22 protein species under the convex electrode TIMS geometry (Figures 4a and 4b). The comparison of literature CCS values with those determined from this work showed excellent agreement (see linearity in Figure 4c with R2 of 0.9989).15,16,45,46,52 This agreement provides further confidence on the use of TIMS-MS technology for structural biology studies.
Figure 4.

Plots illustrating the (a) CCS as a function of MW, (b) 1/K0 as a function of m/z for all proteins investigated and (c) comparison of CCSN2 values obtained in this work with literature values obtained from a drift tube using comparable solution conditions (Table S3). The blue linear fit line (R2 = 0.9989) highlights the good agreement between drift tube and convex electrode TIMS geometry. Plots illustrating the resolving power as a function of 1/K0 for (d) calibrants (P1 = 1.7 mbar), (e) calibrants (P1 = 2.6 mbar), (f) protein (P1 = 2.6 mbar) and as a function of Sr for selected species at (g) P1 = 2.6 mbar and (h) P1 = 1.7 mbar.
One of the major advantages of a TIMS device is the capacity to achieve variable scan rate (Sr) mobility separation in short time scales (e.g., 50–500 ms) with good trapping and transmission efficiency. The mobility dependence for Tuning Mix and CsI cluster ions with R and Sr is summarized in Figures 4d–e, 5a–f and S7. A change of Sr from 1.25 to 0.05 V/ms resulted in a mobility resolution increase of ~2.5x. In addition to the scan rate, the gas velocity plays an important role in the mobility resolution. In fact, higher mobility resolution values were obtained at P1 = 2.6 mbar as compared to P1 = 1.7 mbar with an apparent increase of ~2x. For example, an apparent mobility resolution of 245 with Sr = 0.43 V/ms was observed for the Tuning Mix [M2721 + H]+ ions at P1 = 2.6 mbar, while R of 114 (red trace in Figure 5a) was observed at P1 = 1.7 mbar (Figures 4d–e). These observations are consistent with the initial TIMS reports.18,19 Note that higher apparent mobility resolution can be reached at P1 = 1.7 mbar (R up to 425, green trace in Figure 5d) as compared to P1 = 2.6 mbar (R up to 325), due to the ability of the convex electrode geometry to trap ions with low mobility at lower pressure which was never reported using the concave electrode geometry. In the case of CsI clusters, the influence of the scan rate is shown for [Cs12I11]+ (Figure 5e) and [Cs20I19]+ (Figure 5f) ions and summarized in (Table S2).
Figure 5.

TIMS-MS analysis of (a) Tuning Mix [M2721 + H]+, (b) Tuning Mix [2M2721 + NH4]+, (c) Tuning Mix [3M2721 + NH4]+, (d) Tuning Mix [4M2721 + NH4]+, (e) CsI cluster [Cs12I11]+, (f) CsI cluster [Cs20I19]+, (g) ConA [M + 19H]19+ and (h) GroEL [M + 64H]64+ as a function of the scan rates (Sr). The resolving power (R) and trapping conditions (E/P) values are given. Note that the mobility profiles were fitted using a Gaussian peak fitting algorithm for illustrative purposes.
Different from the calibrant ions (e.g., Tuning Mix and CsI cluster ions), which could contained a small structural diversity, the apparent mobility resolution observed in the case of biomolecules is largely defined by their structural diversity (Figures 4f–h). Over the same mobility range, there is a large difference in the mobility resolving power observed between the calibrant systems (R up to 325, Figure 4e) and the biomolecules (R up to 90, Figure 4f) considered in this study. A new and interesting observation is that the TIMS apparent mobility resolution increases at a different rate when the Sr decrease for native macromolecular assemblies (1.5x) relative to the calibrant ions and small peptides (2.5x). For example, in the case of [M + 19H]19+ ConA (Figure 5g) and [M + 64H]64+ GroEL (Figure 5h), a mobility resolution 1.5x increase was observed at Sr = 1.85 − 0.39 (R ~ 44 − 67) and 1.85 − 0.74 V/ms (R ~ 57 − 85), respectively. This effect is not the focus of the current study and requires further development of the theoretical models.
This data shows that the TIMS mobility resolution scales with the reduction of the scan rate; in good agreement with original observations in 201118,19 and proposed theoretical models for TIMS operation 36,37,59,60. In addition, the data also shows that the TIMS mobility resolution scales with the velocity of the gas, also in good agreement with early observation in 2011.18,19 However, there is a practical operational gas velocity limit for effective trapping defined by the strength of the radial confinement (Vpp amplitude without inducing and electric discharge) and the pumping capacity of the instrument. The gas velocity increase effectively translates in the trapping at higher E/p values within the low field limit; that is, species with higher 1/Ko should be observed at higher mobility resolutions. This trend as shown, is molecular species dependent, since the structural diversity defines the mobility resolution experimentally observed.
CONCLUSION
A convex quadrupolar TIMS electrode geometry design is introduced and implemented in an custom built nESI-TIMS-TOF MS instrument with extended mobility range The extended mobility range using soft trapping (axial and radial trapping) conditions is illustrated with a series of calibrants, known protein standards and macromolecular complexes. This new TIMS geometry enables the characterization of native macromolecular assemblies with wide applications in structural studies of intrinsically disordered proteins, functional proteomics (e.g., PTMs, mutagenesis, etc.), protein-protein and protein ligand interaction networks, among others. With a small footprint, mobility measurements with high mobility resolving power, direct CCS measurement, and short analysis time can be performed for native macromolecular assemblies. While the convex TIMS developments and results utilized a q-TOF MS analyzer, the technology can be easily implemented in ultra-high resolution, mass analyzers (e.g., FT-ICR MS) as well as with other analytical workflows (e.g., LC-TIMS-MS/MS).
Supplementary Material
ACKNOWLEDGEMENTS
The authors at FIU acknowledge the financial support from the National Science Foundation Division of Chemistry, under CAREER award CHE-1654274, with co-funding from the Division of Molecular and Cellular Biosciences to FFL and funding from National Institutes of General Medicine (R01GM134247 to FFL and R01GM054226 to YT) and National Institute of Allergy and Infectious Disease (1R21AI125973 to FL).
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at http://pubs.acs.org. TIMS-MS instrument showing the TIMS cell schematic and TIMS operation, TIMS-MS analysis of Tuning Mix oligomers, CsI clusters and avastin as well as of the [M+6H]6+ ions of ubiquitin as a function of the radial confinement in a 450 and 800 kHz rf frequency and trapping time, TIMS-MS metrics showing the CCS deviations, Plots showing the resolving power as a function of 1/K0 for Tuning Mix and CsI clusters and Tables of the measured mobility and CCS for Tuning Mix oligomers and CsI clusters as well as CCS for the investigated protein species.
REFERENCES
- (1).Bergendahl LT; Marsh JA Sci. Rep 2017, 7, 4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Heck AJ; Van Den Heuvel RH Mass Spectrom. Rev 2004, 23, 368–389. [DOI] [PubMed] [Google Scholar]
- (3).Sharon M; Robinson CV Annu. Rev. Biochem 2007, 76, 167–193. [DOI] [PubMed] [Google Scholar]
- (4).Sobott F; Hernández H; McCammon MG; Tito MA; Robinson CV Anal. Chem 2002, 74, 1402–1407. [DOI] [PubMed] [Google Scholar]
- (5).van den Heuvel RH; van Duijn E; Mazon H; Synowsky SA; Lorenzen K; Versluis C; Brouns SJ; Langridge D; van der Oost J; Hoyes J; Heck AJ Anal. Chem 2006, 78, 7473–7483. [DOI] [PubMed] [Google Scholar]
- (6).Rose RJ; Damoc E; Denisov E; Makarov A; Heck AJ Nat. Methods 2012, 9, 1084–1086. [DOI] [PubMed] [Google Scholar]
- (7).Sali A; Glaeser R; Earnest T; Baumeister W Nature 2003, 422, 216–225. [DOI] [PubMed] [Google Scholar]
- (8).Ruotolo BT; Giles K; Campuzano I; Sandercock AM; Bateman RH; Robinson CV Science 2005, 310, 1658–1661. [DOI] [PubMed] [Google Scholar]
- (9).Ruotolo BT; Benesch JLP; Sandercock AM; Hyung S-J; Robinson CV Nat. Protoc. 2008, 3, 1139–1152. [DOI] [PubMed] [Google Scholar]
- (10).Duijn E. v.; Barendregt A; Synowsky S; Versluis C; Heck AJR J. Am. Chem. Soc 2009, 131, 1452–1459. [DOI] [PubMed] [Google Scholar]
- (11).Zhong Y; Hyung SJ; Ruotolo BT Expert Rev. Proteomics 2012, 9, 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Giles K; Williams JP; Campuzano I Rapid Commun. Mass Spectrom 2011, 25, 1559–1566. [DOI] [PubMed] [Google Scholar]
- (13).Lanucara F; Holman SW; Gray CJ; Eyers CE Nat. Chem 2014, 6, 281–294. [DOI] [PubMed] [Google Scholar]
- (14).Hogan CJ; Ruotolo BT; Robinson CV; Fernandez de la Mora J J. Phys. Chem. B 2011, 115, 3614–3621. [DOI] [PubMed] [Google Scholar]
- (15).France AP; Migas LG; Sinclair E; Bellina B; Barran PE Anal. Chem 2020, 92, 4340–4348. [DOI] [PubMed] [Google Scholar]
- (16).Stiving AQ; Jones BJ; Ujma J; Giles K; Wysocki VH Anal. Chem 2020, 92, 4475–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).McCabe JW; Mallis CS; Kocurek KI; Poltash ML; Shirzadeh M; Hebert MJ; Fan L; Walker TE; Zheng X; Jiang T; Dong S; Lin CW; Laganowsky A; Russell DH Anal. Chem 2020, 92, 11155–11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fernandez-Lima FA; Kaplan DA; Suetering J; Park MA Int. J. Ion Mobil. Spectrom 2011, 14, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Fernandez-Lima FA; Kaplan DA; Park MA Rev. Sci. Instrum 2011, 82, 126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Adams KJ; Montero D; Aga D; Fernandez-Lima F Int. J. Ion Mobil. Spectrom 2016, 19, 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jeanne Dit Fouque K; Ramirez CE; Lewis RL; Koelmel JP; Garrett TJ; Yost RA; Fernandez-Lima F Anal. Chem 2019, 91, 5021–5027. [DOI] [PubMed] [Google Scholar]
- (22).Benigni P; Marin R; Molano-Arevalo JC; Garabedian A; Wolff JJ; Ridgeway ME; Park MA; Fernandez-Lima F Int. J. Ion Mobil. Spectrom 2016, 19, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Molano-Arevalo JC; Jeanne Dit Fouque K; Pham K; Miksovska J; Ridgeway ME; Park MA; Fernandez-Lima F Anal. Chem 2017, 89, 8757–8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Liu FC; Ridgeway ME; Park MA; Bleiholder C Analyst 2018, 143, 2249–2258. [DOI] [PubMed] [Google Scholar]
- (25).Liu FC; Kirk SR; Bleiholder C Analyst 2016, 141, 3722–3730. [DOI] [PubMed] [Google Scholar]
- (26).Butcher D; Bernad S; Derrien V; Sebban P; Miksovska J; Fernandez-Lima F Int. J. Mass Spectrom 2018, 430, 37–43. [Google Scholar]
- (27).Ridgeway ME; Silveira JA; Meier JE; Park MA Analyst 2015, 140, 6964–6972. [DOI] [PubMed] [Google Scholar]
- (28).Liu FC; Cropley TC; Ridgeway ME; Park MA; Bleiholder C Anal. Chem 2020, 92, 4459–4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Xu X; Leng F Protein Expr. Purif 2011, 77, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ahmad M; Xue Y; Lee SK; Martindale JL; Shen W; Li W; Zou S; Ciaramella M; Debat H; Nadal M; Leng F; Zhang H; Wang Q; Siaw GE; Niu H; Pommier Y; Gorospe M; Hsieh TS; Tse-Dinh YC; Xu D; Wang W Nucleic Acids Res. 2016, 44, 6335–6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hager DA; Jin DJ; Burgess RR Biochemistry 2002, 29, 7890–7894. [DOI] [PubMed] [Google Scholar]
- (32).Fang J; Engen JR; Beuning PJ Biochemistry 2011, 50, 5958–5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Garabedian A; Butcher D; Lippens JL; Miksovska J; Chapagain PP; Fabris D; Ridgeway ME; Park MA; Fernandez-Lima F Phys. Chem. Chem. Phys 2016, 18, 26691–26702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hernandez DR; Debord JD; Ridgeway ME; Kaplan DA; Park MA; Fernandez-Lima F Analyst 2014, 139, 1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ridgeway ME; Lubeck M; Jordens J; Mann M; Park MA Int. J. Mass Spectrom 2018, 425, 22–35. [Google Scholar]
- (36).Michelmann K; Silveira JA; Ridgeway ME; Park MA J. Am. Soc. Mass Spectrom 2015, 26, 14–24. [DOI] [PubMed] [Google Scholar]
- (37).Silveira JA; Michelmann K; Ridgeway ME; Park MA J. Am. Soc. Mass Spectrom 2016, 27, 585–595. [DOI] [PubMed] [Google Scholar]
- (38).Chai M; Young MN; Liu FC; Bleiholder C Anal. Chem 2018, 90, 9040–9047. [DOI] [PubMed] [Google Scholar]
- (39).Silveira JA; Ridgeway ME; Park MA Anal. Chem 2014, 86, 5624–5627. [DOI] [PubMed] [Google Scholar]
- (40).Revercomb HE; Mason EA Anal. Chem 1975, 47, 970–983. [Google Scholar]
- (41).Campuzano I; Giles K Nanoproteomics: Methods and Protocols 2011, 790, 57–70. [DOI] [PubMed] [Google Scholar]
- (42).Blain MG; Riter LS; Cruz D; Austin DE; Wu G; Plass WR; Cooks RG Int. J. Mass Spectrom 2004, 236, 91–104. [Google Scholar]
- (43).Bleiholder C; Bowers MT Annu. Rev. Anal. Chem 2017, 10, 365–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).May JC; Jurneczko E; Stow SM; Kratochvil I; Kalkhof S; McLean JA Int. J. Mass Spectrom 2018, 427, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Harrison JA; Kelso C; Pukala TL; Beck JL J. Am. Soc. Mass Spectrom 2019, 30, 256–267. [DOI] [PubMed] [Google Scholar]
- (46).Bush MF; Hall Z; Giles K; Hoyes J; Robinson CV; Ruotolo BT Anal. Chem 2010, 82, 9557–9565. [DOI] [PubMed] [Google Scholar]
- (47).Bleiholder C; Johnson NR; Contreras S; Wyttenbach T; Bowers MT Anal. Chem 2015, 87, 7196–7203. [DOI] [PubMed] [Google Scholar]
- (48).Morsa D; Hanozin E; Eppe G; Quinton L; Gabelica V; Pauw E Anal. Chem 2020, 92, 4573–4582. [DOI] [PubMed] [Google Scholar]
- (49).Bleiholder C; Liu FC; Chai M Anal. Chem 2020, 92, 16329–16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).El-Baba TJ; Clemmer DE Int. J. Mass Spectrom 2019, 443, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Pacholarz KJ; Porrini M; Garlish RA; Burnley RJ; Taylor RJ; Henry AJ; Barran PE Angew. Chem. Int. Ed 2014, 53, 7765–7769. [DOI] [PubMed] [Google Scholar]
- (52).Campuzano IDG; Larriba C; Bagal D; Schnier PD Ion Mobility and Mass Spectrometry Measurements of the Humanized IgGk NIST Monoclonal Antibody, 2015; Vol. 1202, p 75–112. [Google Scholar]
- (53).Finn RD; Orlova EV; Gowen B; Buck M; van Heel M EMBO J. 2000, 19, 6833–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Ilag LL; Westblade LF; Deshayes C; Kolb A; Busby SJ; Robinson CV Structure 2004, 12, 269–275. [DOI] [PubMed] [Google Scholar]
- (55).Krishna K; Rao G; Rao K Curr. Protein Pept. Sci 2007, 8, 418–425. [DOI] [PubMed] [Google Scholar]
- (56).Jeanne Dit Fouque K; Garabedian A; Leng F; Tse-Dinh YC; Fernandez-Lima F ACS Omega 2019, 4, 3619–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Koleva BN; Gokcan H; Rizzo AA; Lim S; Jeanne Dit Fouque K; Choy A; Liriano ML; Fernandez-Lima F; Korzhnev DM; Cisneros GA; Beuning PJ Biophys. J 2019, 117, 587–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Jeanne Dit Fouque K; Fernandez-Lima FJ Phys. Chem. B 2020, 124, 6257–6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Bleiholder C Int. J. Mass Spectrom 2016, 399–400, 1–9. [Google Scholar]
- (60).Larriba-Andaluz C; Chen X; Nahin M; Wu T; Fukushima N Anal. Chem 2019, 91, 919–927. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.