

Abstract
Human intestinal organoids (HIOs) are millimeter-scale models of the human intestinal epithelium and hold tremendous potential for advancing fundamental and applied biomedical research. HIOs resemble the native gut in that they consist of a fluid-filled lumen surrounded by a polarized epithelium and associated mesenchyme; however, their topologically-closed, spherical shape prevents flow through the interior luminal space, making the system less physiological and leading to the buildup of cellular and metabolic waste. These factors ultimately limit experimentation inside the HIOs. Here, we present a millifluidic device called the Gut Organoid Flow Chip (GOFlowChip), which we use to “port” HIOs and establish steady-state liquid flow through the lumen for multiple days. This long-term flow is enabled by the use of laser-cut silicone gaskets, which allow liquid in the device to be slightly pressurized, suppressing bubble formation. To demonstrate the utility of the device, we establish separate luminal and extraluminal flow and use luminal flow to remove accumulated waste. This represents the first demonstration of established liquid flow through the luminal space of a gastrointestinal organoid over physiologically relevant time scales. Flow cytometry results reveal that HIO cell viability is unaffected by long-term porting and luminal flow. We expect the real-time, long-term control over luminal and extraluminal contents provided by the GOFlowChip will enable a wide variety of studies including intestinal secretion, absorption, transport, and co-culture with intestinal microorganisms.
Introduction
Human intestinal organoids (HIOs) are millimeter-scale experimental models of the intestinal epithelium1–3. These tissues are grown in the lab through directed differentiation of human pluripotent stem cells (iPSC)1 and have become a standard for basic and applied biomedical research3–13. HIOs are spherical in shape and consist of an inner, liquid-filled space enclosed by a polarized epithelial shell that mimics the cellular complexity of the intestinal epithelium. The shell is comprised of several epithelial lineages, including stem cells, progenitors, and absorptive enterocytes, as well as secretory goblet, enteroendocrine, and Paneth cell precursors1, 14. These cells are bound to one another through tight junctions and thus provide a physical barrier between the lumen and the outside environment. As in the human gut, the HIO barrier is dynamic and both actively and passively mediates the transport of molecules and water15–17. Quite remarkably, HIOs exist as topologically closed, self-contained systems for human gut research.
HIOs are particularly useful for studying interactions between bacteria and human host tissue. For example, the natural microbial colonization of immature intestinal epithelium, such as that in newborn infants, has been modeled by co-culturing microorganisms inside HIOs3. HIOs also represent a new and unconventional model for understanding enteric dysfunction, which can be caused by pathogenic bacteria and viruses10–12. To study such interactions, microbes have been injected into the luminal space using a micropipette3, 10–12. After injection, the epithelial shell rapidly heals, and both HIO and microbes can be cultured together. The topologically closed surface of the epithelial shell is beneficial in that it acts to contain the microorganisms, thus allowing for short-term assays. However, the human gut is not a closed system and transport into and out of the intestine is critical to clear human and microbial cellular waste. The lack of liquid advection through the luminal space in HIOs and other gastrointestinal organoids leads to abuildup of waste and cellular debris that can eventually lead to “popping” events18. Thus, enclosed HIOs do not adequately mimic natural luminal flow through the human gut.
Short-term luminal flow has been established through human gastric organoids (HGOs) for tens of minutes19; however, to perform physiologically relevant experiments such as real-time monitoring and control of luminal contents, luminal flow must be established for multiple days, and extending flow time by more than two orders of magnitude presents significant engineering challenges. For example, the flow of biological media in millifluidic devices is plagued by the formation of bubbles, which disrupt the luminal space and interfere with organoid imaging20. Moreover, luminal waste that dislodges during long-term flow leads to device clogging. These issues preclude long-term imaging and require new engineering solutions. Without a fluidic system that better reflects in vivo conditions with controlled luminal flow over multiple days, new avenues of long-term experimentation involving gastrointestinal organoids will be precluded.
Here, we present a multilayer millifluidic device used to establish long-term internal liquid HIO flow in parallel with external flow around the outer surface of the HIO. We call this device the Gut Organoid Flow Chip (GOFlowChip). Internal liquid flow allows for the removal of accumulated waste from the lumen, while the extraluminal flow exchanges nutrients and waste to mimic collection by mesenteric arteries and portal vein transport. Luminal flow through the organoid is provided by tapered glass capillaries, which are used to puncture the HIO and establish a flow for periods as long as t = 65 h. This long-term flow is enabled by the use of laser-cut silicone gaskets, which allow liquid in the device to be slightly pressurized, suppressing bubble formation. Flow around the outside of the organoid is provided by an additional channel cut into the device. The seal which forms at each puncture site is sufficient to maintain separation between the inner and outer contents of the organoid under physiologically relevant flow conditions. This device can be used for a broad range of experimentation and imaging. For example, independent control of luminal and extra-luminal liquid flow allows for the introduction of molecules or colloidal-scale objects, such as bacteria, into the luminal space. Additionally, fine-scale control of HIO luminal flow will enable continuous sampling of the luminal contents.
Background
HIOs are routinely grown from stem cells into multi-lineage, millimeter-scale, enclosed spheres with an internal lumen (Fig. 1a)1, 2. The spheres are generated and propagated within a bio-compatible hydrogel21–23, and during growth are exposed to growth factors necessary for cellular differentiation and proliferation13. HIOs are considered fully differentiated once they have reached several millimeters in diameter, which requires six to eight weeks of growth. Functional and physiological assays conducted on HIOs at this stage reveal the presence of brush borders on enterocytes, production of mucin by goblet cells, peptide transport systems, and barrier-forming tight junctions13, 24. HIOs older than eight weeks become dense with accumulated waste and the epithelial shell can lose mechanical integrity. HIOs can be maintained as long-term cultures for periods longer than a year, but this requires periodically cutting open mature spheres into individual pieces25, which then reform into intact, closed spherical organoids15, 16. Thus, methods for establishing control over luminal and extraluminal transport are clearly needed.
Fig. 1. Time-lapse microscopy imaging of a human intestinal organoid (HIO) reveals waste accumulation.
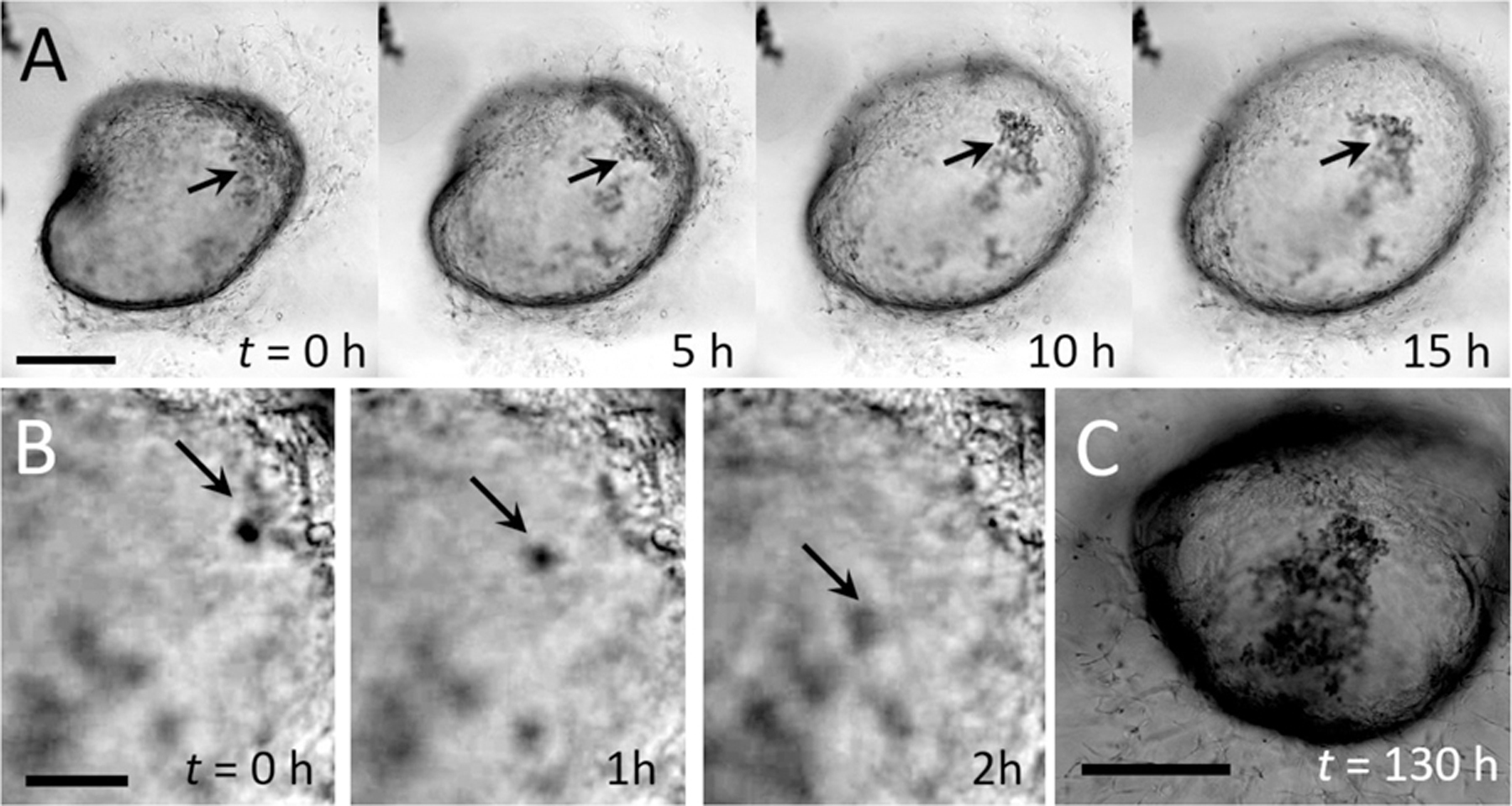
(A) Image series. Closed-shell structure formed by an HIO. The epithelial sheet acts as semipermeable membrane, limiting transport between the luminal and extraluminal space. In the early stages of organoid growth, HIOs are optically transparent, but cellular debris accumulates over time (black arrows). Scale bar represents 1 mm. (B) Image series. High magnification time-lapse images of small debris (black arrows) sloughing off the inner surface and settling to the bottom of the interior space. Images are separated by 1 h each. Scale bar represents 0.1 mm. (C) After 130 h, the HIO from (A) has darkened significantly and waste has continued to accumulate in the luminal space
A common approach for establishing well-defined flow control within a tissue culture is to integrate the tissue into a microfluidic or millifluidic device26–28. This typically involves directing microscale fluid flow together with engineered cell scaffolds to replicate the structure and function of a specific human tissue or organ. For example, human “organ-on-a-chip” systems that are designed to replicate the kidney29–32, heart33–38, lung39–45, intestine40, 46–52, liver44, 47, 51, 53–64, blood vessels42, 43, 65–67, bone68–70, marrow71, nerve72–77, muscle78 and cornea79 have been developed. The exquisite control of liquid flow provided by fluidic devices can be used to deliver minute quantities of chemical or biological material with spatial and temporal precision27, 28, allows for on-demand monitoring and analysis of nanoliter and picoliter liquid volumes26, and can be used to maintain chemostasis28.
Most organ-on-chip systems use traditional cell cultures. By contrast, the integration of organoid cultures into fluidic systems has been limited. In one approach, cells from disrupted human gastrointestinal organoids have been templated with microfluidic channels80, 81: a promising method that controls organoid structure and provides access to the luminal space. In another approach, preformed organoids including liver, cardiac, and vascular organoids were combined into a single, circulatory microfluidic system to create a “body-on-a-chip” platform83, 84. The use of intact, preformed organoids precludes the use of artificial scaffolding and allows the tissue culture to form in an environment more representative of in vitro conditions before integration into the chip; however, for gastrointestinal organoids, access to the luminal space remains a challenge. Short-term luminal flow has been established through human gastric organoids (HGOs)19, but the time scale of flow has been limited, and the impact of luminal flow on organoid viability is not clear. A fluidic device capable of establishing long-term flow through gastrointestinal organoids is still needed.
Results and Discussion
To demonstrate the transport limitations caused by the closed epithelial shell, we follow the accumulation of waste inside the lumen of an HIO over several days using time-lapse light microscopy. Our imaging reveals that colloidal-scale cell debris sloughs off from the inner surface of the HIO and settles to the bottom of the lumen where it remains for multiple days (image series, Fig. 1a, b). As waste builds, HIOs eventually darken and become optically opaque, as shown by the image in Fig. 1c. While the images in Fig. 1 provide a visual depiction of waste accumulation, dissolved molecular-scale waste, and metabolites which are not visible also likely accumulate due to the semipermeable nature of the epithelial shell13, 15–17, 24. Accumulation of waste impacts organoid viability and physiology and limits the time period over which organoids remain viable for experimentation13.
To establish real-time control over luminal contents, we develop a chip-based fluidic device for establishing luminal liquid flow. Mature HIOs are millimeters in diameter; so, a fabrication method capable of generating a device with millimeter-scale features and channels is needed. To achieve device features at this scale, we use a laser to cut thin acrylic sheets into precise shapes. Our device consists of three layers: a middle layer containing the organoid and a channel for extraluminal flow, and an upper and lower layer which enclose the middle layer (Fig. 2a). These layers are then sandwiched together to form a three-dimensional millifluidic device (Fig. 2b). Thin, laser-cut silicone rubber sheets are also included between acrylic layers to seal the device and prevent leaking. Holes in the upper acrylic layer allow for the introduction of liquid into and out of the upper (extraluminal) flow channel, and the bottom layer forms the floor of the device. Laser-cut cylindrical side channels with long axes normal to the side walls of the device and perpendicular to the layer plane (Fig. 2c) allow for tapered glass capillaries to be inserted into the HIO. Luminal flow is established in one of two ways: using a single double-lumen capillary (Fig. 2d, bottom) or two single-lumen capillaries (Fig. 2d, top). Laser-cut silicone rubber gaskets are used on the sides as compression seals to prevent leakage. The modular design of the device allows each layer to be designed independently and the device to be disassembled and reassembled for sterilization and repeated use. The transparency of the acrylic allows for optical imaging of a ported HIO.
Fig. 2. Multilayer millifluidic chip for establishing distinct luminal and extraluminal flow.
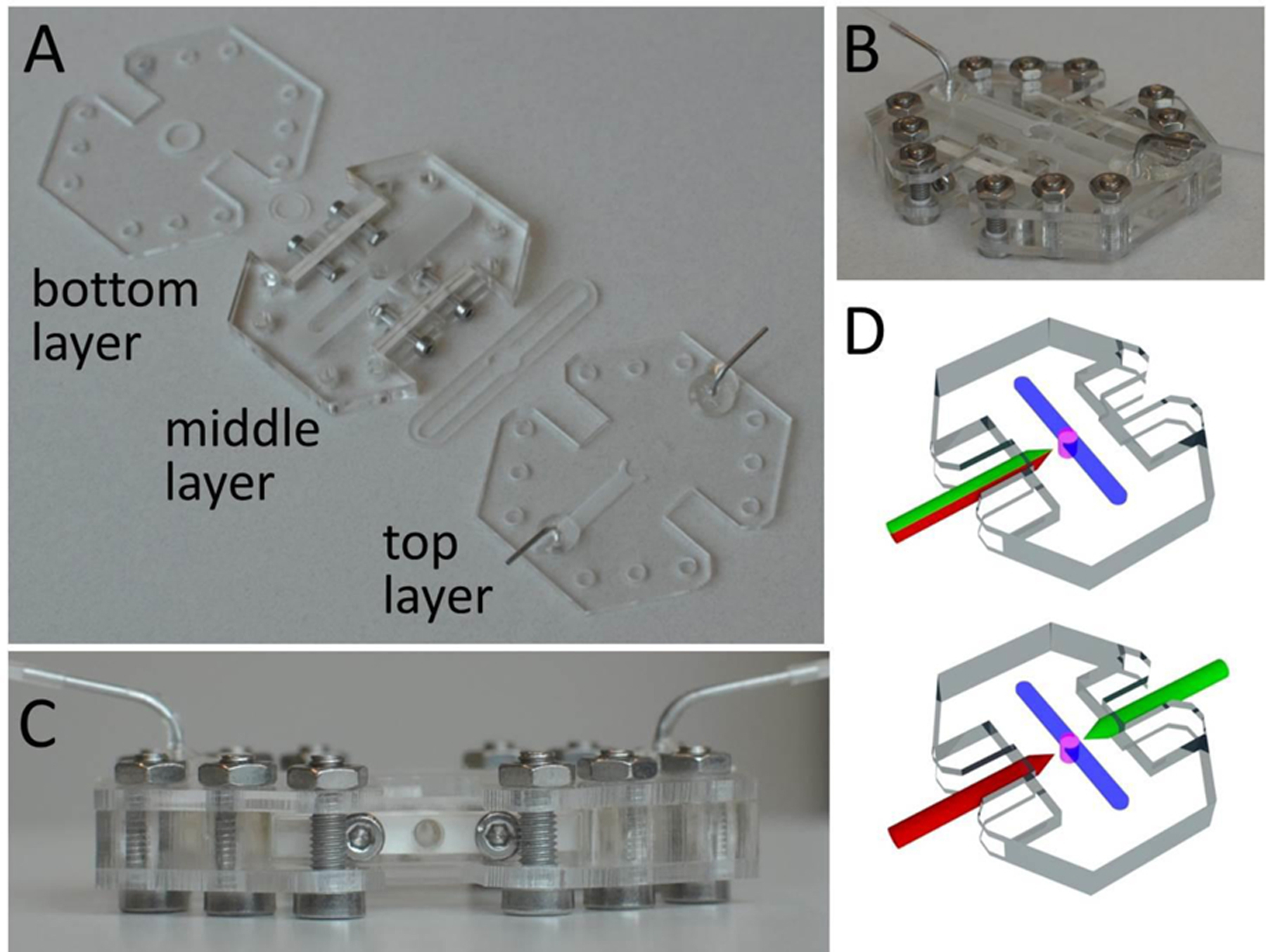
(A) The device is composed of three layers. The HIO is contained in a central well in the middle layer. Extraluminal flow is guided by an engraved channel in the top layer just above the organoid. The bottom layer encloses the bottom of the device. Arrows indicate laser-cut silicone gaskets. (B) Orthogonal view of the assembled device with fluidics assembled for extraluminal flow but no capillaries yet inserted for HIO porting. (C) Side view of the assembled device with the side port gasket (white) clearly visible. (D) Illustrations highlighting two different configurations for HIO porting. Upper: Porting with a single, double-lumen capillary (green-red). Lower: Porting with two, single-lumen capillaries (red and green).
To establish flow through an organoid, the lower and middle layers (Fig. 2a) are assembled under sterile conditions. An HIO embedded in Matrigel is then placed in the circular well formed by the two layers, and the organoid is punctured on one or two sides by manipulating tapered capillaries using independent three-axis micromanipulators. An HIO before and after puncturing is depicted in Fig. 3a and Fig. 3b, respectively. Most organoids, when punctured, deflate slightly but do not immediately collapse. This is due to the fact that the outer surface of the organoid attaches to the surrounding Matrigel through cellular adhesions. Matrigel is viscoelastic with a characteristic relaxation time on the order of tens of minutes, which provides sufficient time to puncture both sides and establish flow. We find that HIOs with diameters between 2 mm and 3 mm are ideal for porting in our device; HIOs with diameters larger than 3 mm typically have a large amount of accumulated waste, which leads to clogging of the outlet capillary, and organoids with diameters smaller than 1 mm are difficult to manipulate with our current setup. While this lower size limit precludes the use of mouse organoids and human organoids derived from primary tissues, which are an order of magnitude smaller in diameter than HIOs derived from iPSCs 18, 25, a smaller version of a GOFlowChip imaged with appropriate optics could be used to port organoids and spheroids with sub-mm scale sizes. After the HIO is punctured, the two upper layers are added to the assembly, and the device is sealed by compression. To drive luminal liquid flow through the HIO, the outer ends of the glass capillaries are attached through microfluidic tubing to computer-controlled syringe pumps, two of which provide independent control over the infusion and withdrawal of liquid from the organoid. To drive liquid flow through the extraluminal flow channel, ferrules inserted into the top layer of the chip (Fig. 2a-c) are connected through microfluidic tubing to other computer-controlled syringe pumps. A detailed description of the loading and assembly protocol, including the most commonly encountered problems, is included in Supplementary Information along with the design files needed to fabricate the device.
Fig. 3. HIO Porting Process.
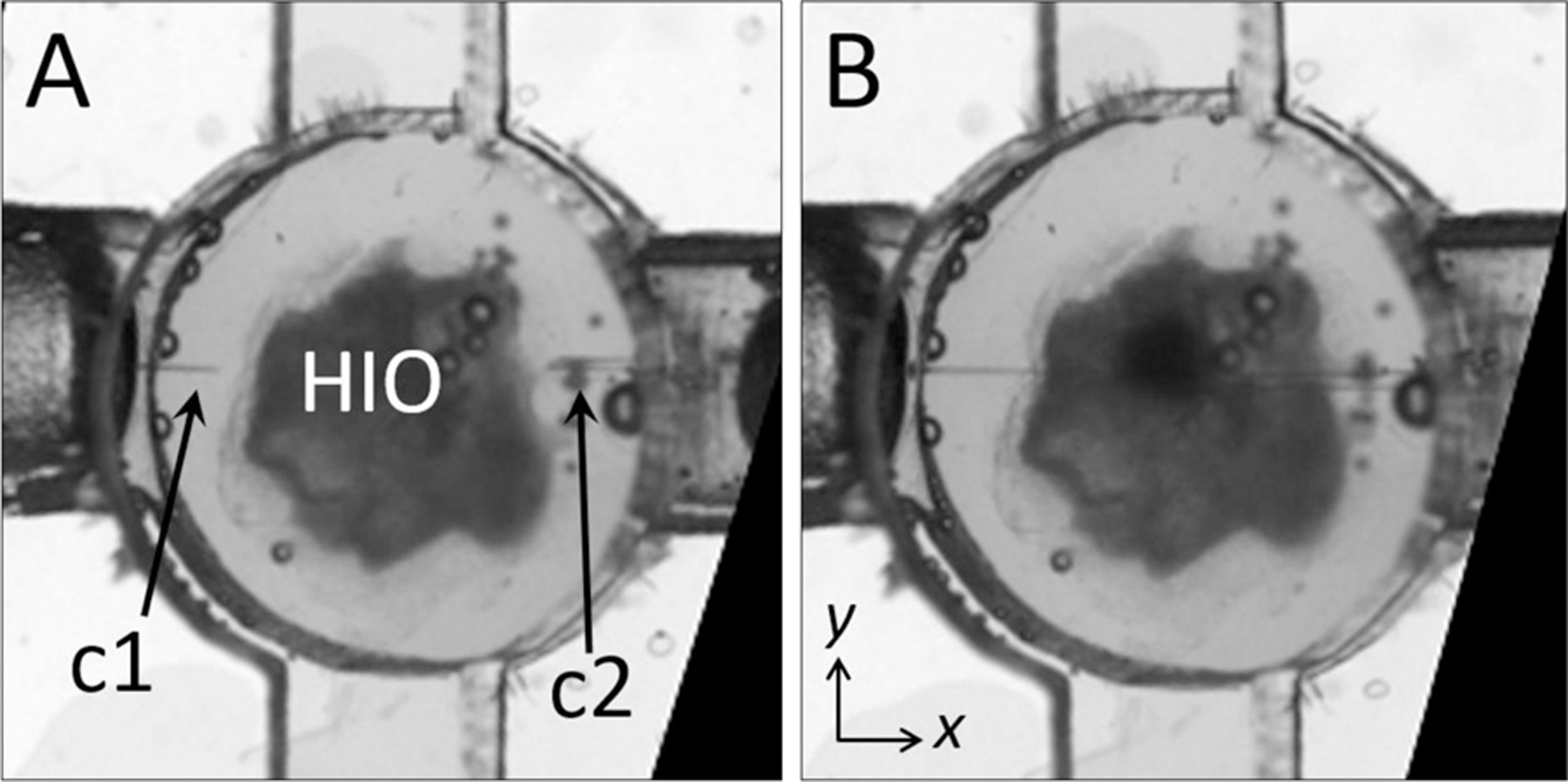
(A) Microscopy image of HIO in an assembled device before puncturing with capillaries (c1, c2). (B) Image of the same HIO after puncturing on either side. The diameter of the circular well in (A) and (B) is 4 mm.
A ported HIO will ideally possess the same barrier integrity found in the native gastrointestinal tract. In an unported HIO, barrier integrity is maintained by the epithelial shell via intact cellular tight junctions; however, when the shell is punctured, integrity is contingent on the formation of a seal at the puncture site. To test the seal of a fully ported HIO, we pressurize the HIO by flowing liquid through the left-side capillary, c1 (qin = 5 μL/h) while suppressing liquid outflow through the right-side capillary, c2 (qout = 0). Thus, a seal forms at the puncture site which allows for dramatic inflation without any leakage of liquid even over several hours, as shown by the series of images in Fig. 4a. High-resolution imaging suggests that Matrigel plays a role in maintaining this seal by closing around the capillary until the ruptured epithelium regrows and adheres to the capillary. This is not surprising, given that gut organoids are commonly punctured, injected with material, and the capillary removed without observable deflation or ejection of luminal contents12. We note that the temporary barrier provided by Matrigel differs from that provided by intact epithelium; while Matrigel suppresses liquid flow and the diffusion of colloidal-scale objects85, molecules smaller than the mesh size of the gel (ξ ≈ 10 nm) diffuse through the gel86. Thus, for experiments where barrier integrity immediately following HIO puncture is critical, the nature of the seal and time needed to ensure complete epithelium healing should be investigated further. To establish luminal flow and verify the integrity of the ported seal, we design a flow sequence that should result in organoid inflation and deflation. The inflation condition is achieved by infusing liquid media through c1, while preventing flow through c2, resulting in a positive net flow of media into the organoid, Δq. Here, Δq = qin- qout, where qin is the total flow rate into the organoid and qout is the total flow rate out of the organoid. The deflation condition is achieved by withdrawing liquid from c2, while preventing flow through c1, resulting in a negative net flow of liquid (qin < qout). We initiate this flow sequence, alternating between the two flow conditions, and observe that the organoid undergoes striking volume changes in response to flow, as depicted by the series of images in Fig. 4b and Fig. 4c. To quantify this volume change, we plot the net imposed flow rate as a function of time (Fig. 3d, top plot) along with the maximum diameter of the organoid measured along the y-axis as a function of time (Fig. 3d, bottom plot), and observe that the diameter changes consistently in response to the imposed flow. This represents, to our knowledge, the first demonstration of luminal flow through a topologically closed gastrointestinal organoid.
Fig. 4. Demonstration of puncture seal and luminal flow.
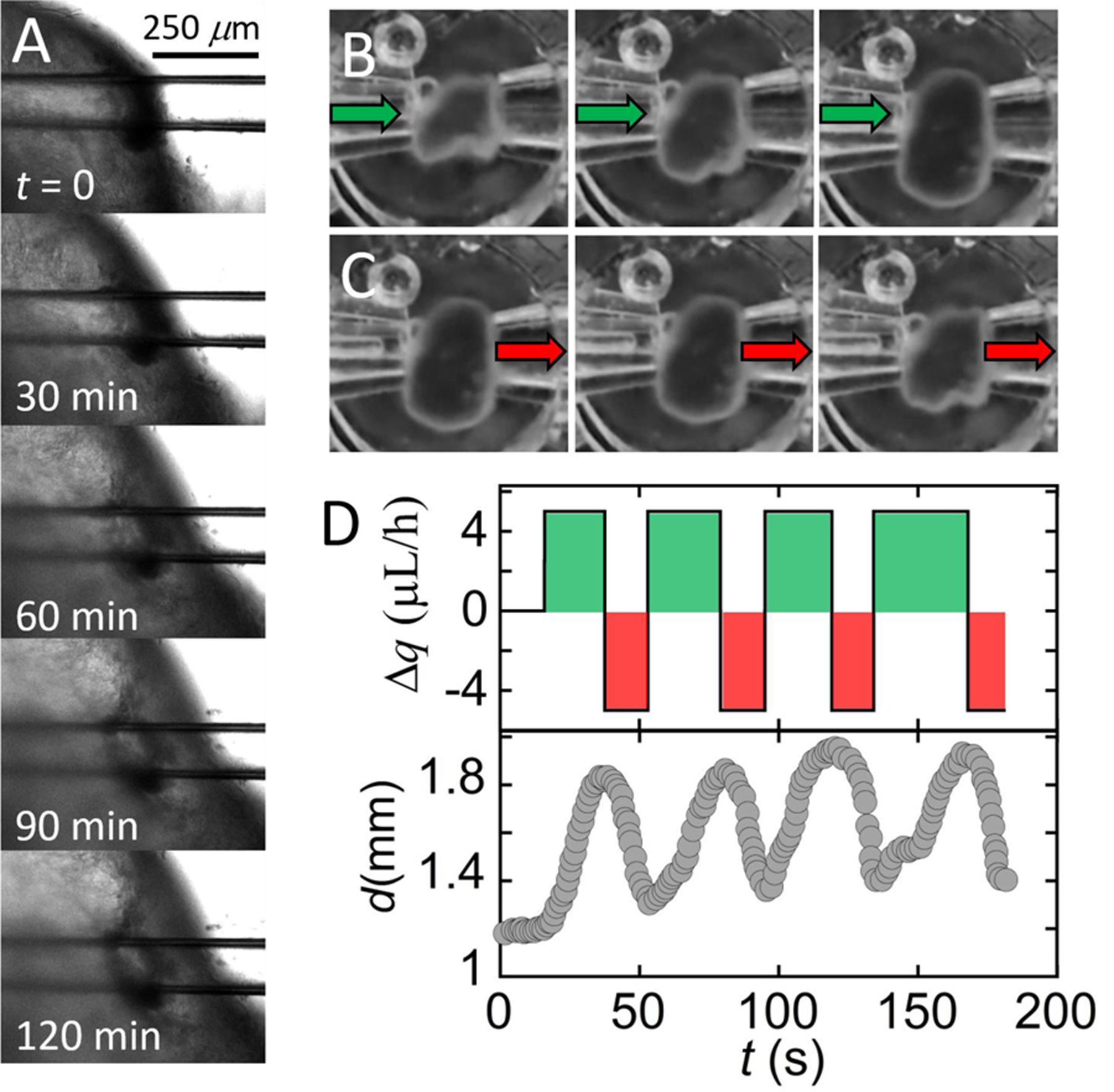
(A) Microscopy image series. An HIO punctured on both sides and subjected to a net influx of liquid (qin = 5 μL/h; qout = 0 μL/h) swells, but no leaking of liquid at the puncture site is detected. (B) Microscopy image series. An HIO punctured on both sides swells as aqueous media is infused from the left capillary (qin = 5 μL/h). No liquid is removed from the HIO through the right capillary (qout = 0 μL/h). Time between images is 20 s. (C) Microscopy image series. The HIO shrinks as media is withdrawn through the right capillary (qout = 5 μL/h; qin = 0 μL/h). Images in each series (B, C) are separated by Δt = 20s. (D) Upper plot. Net flow of media, Δq = qin-qout, into or out of the HIO is plotted as a function of time. Lower plot. Corresponding change in HIO diameter, d (y-axis) as a function of time in response to infusion and withdrawal.
For long-term experimentation, the chip must be capable of maintaining luminal flow through an HIO for hours and even days. In preliminary attempts, we find that establishing flow for this length of time is limited mainly by clogging of the exit capillary. Clogging presents a problem because syringe pumps are flow rate-controlled rather than pressure-controlled and thus insensitive to pressure buildup in the HIO; if blockage of the exit capillary occurs, liquid is driven into the HIO until it ruptures. To mitigate this, we explore a range of exit tip diameters (20 μm ≤ d ≤ 120 μm) and find that for fully differentiated organoids containing a significant amount of waste, an exit capillary with a tip diameter d ≈ 80 μm and a flow rate qout ≤ 5 μL/h is ideal; capillaries with smaller tip diameters tend to clog, and capillaries with larger tip diameters are difficult to puncture HIOs. We also find that during long-term experiments bubbles form in the overflow liquid, which negatively impacts image quality and HIO barrier integrity. Thus, to suppress bubble formation, we pressurize the liquid slightly by constricting the outlet of the overflow liquid (see Methods). After significant optimization we regularly establish steady-state flow through HIOs for t ≥ 65 h using both the single, double-lumen capillary and double, single-lumen capillary porting methods; longer flow experiments could be achieved but we were limited by microscope access in our shared facility. Luminal flow on the order of days will enable a wide variety of future experiments.
A continuous luminal flow could be used to introduce materials to the lumen or remove and sample luminal contents. To demonstrate the value of luminal flow, we port an HIO containing significant accumulated waste and use flow to remove waste. The HIO is ported using capillaries with tip diameters c1 = 40 μm and c2 = 80 μm, steady state flow is established by setting flow into and out of organoid equal (qin = qout = 5 μL/h), and the organoid is imaged for 20 h. During this time, the organoid undergoes significant fluctuations, moving in and out of our objective focus; however, no leaking, signs of cell death, or loss of barrier integrity are observed. At the 20 h mark, when the microscope is refocused, it is apparent that the HIO has clarified, and that waste is being removed by liquid flow through the exit capillary (Fig 5a). The organoid continues to clarify over the next 7 hours as depicted by the series of microscopy images in Fig 5b-d. Waste is observed exiting the organoid through c2. Images of the flow profile along c2 reveal the movement of large objects moving from left to right as they are carried by liquid flow, as shown by the series of microscopy images in Fig 5e. To determine the velocity of waste exiting the HIO, we measure the intensity profile along the center of the capillary as a function of time and plot the data as a kymograph in Fig. 5f. In this format, the y-axis represents the grayscale pixel intensity along a horizontal line bisecting the images in Fig. 5e, and the x-axis represents time. Thus, the lines moving from the bottom left to the upper right represent the movement of objects from left to right within c2, and the slope of these lines provides their velocity. The dark line represents the large piece of waste depicted by the image series in Fig. 5e with a velocity, v = 27 μm/s. The green crosses in Fig. 5e represent the positions associated with the pixels marked by the green crosses in Fig. 5f. This result demonstrates qualitatively that luminal flow can be used to perform a useful function: the removal of accumulated waste.
Fig. 5. Clearing waste with luminal flow.

(A) Microscopy image series. Steady state luminal flow is established in an HIO with significant waste accumulation by setting qin = qout = 5 μL/h. Flow is from left to right. After 20 h, the HIO is still viable, and no blebbing or leaking is observed. (B-D) Over time, the HIO becomes more transparent as waste is carried by liquid flow through c2. Images correspond to region in (A), red dashed box. The clarified region of the lumen is labeled in (D). (E) Series of images of c2 depict movement of the waste exiting the organoid from left to right. Images correspond to region in (D), blue dashed box. (F) Kymograph representing the intensity profile along a line in the center of the channel in c2 (E) is plotted (y-axis) as a function of time (x-axis). The dark line indicated by the green crosses represents a large piece of waste moving from left to right along the channel. The scale of the y-axis represents 150 μm and the scale of the x-axis represents 150 minutes. The slope of the line represents the velocity, v = 27 μm/s.
Ideally, flow through a ported HIO will mimic flow through the human gut. The topology of a dual-ported HIO, with an inlet and outlet on opposing sides, is identical to that of the gut, but the dimensions and aspect ratio differ significantly. A dual-ported HIO is a short tube with equal diameter and length (d ≈ 3–5 mm); by comparison, the lumen of the human intestine is an order of magnitude wider in diameter (d ≈ 2–3 cm), and the length of the human intestine is three orders of magnitude longer (ℓ ≈ 2–3 m) than an HIO. Because of these differences in size and aspect ratio, matching the volumetric flow rate would result in flow conditions that are unrealistically fast, which could result in the removal of not just waste, but also key molecules that are critical to epithelial health and function. Instead of flow rate, liquid velocity, v appears to be the relevant parameter as it controls the rate at which materials are transported to and from the inner wall of the epithelium, as well as determining the stress exerted by the luminal contents on the inner lining of the gut, which is critical for gut physiology. The average velocity through the human gut is reported to be on the order of 0.4 mm/s87, 88. For comparison, the average velocity in our pulsatile experiment (Fig. 3), where q = 25 μL/min, d = 2r ≈ 1.5 mm, is v = q/πr2 ≈ 0.24 mm/s. Thus, the luminal flow velocity through our HIOs is comparable to that in the human gut. We note that the shear stress exerted by luminal contents on the inner wall of the gut is also governed by the topography of the gut lining and the rheological properties of the material in the lumen, and the impact of these parameters on HIO physiology warrant further investigation.
Similarly, flow outside a ported HIO should ideally mimic flow around the outside of the human gut. To determine the relevant range for extraluminal flow in our HIO chip, we begin by considering the frequency of media exchange needed to maintain HIOs under standard culture conditions. Cultured HIOs require media replacements of 50 μl per HIO every 48 h, which is approximately equal to 1 μl/h of continuous flow in our chip for a single HIO. In the native human gut, the transport of blood through gut tissue is on the order of 10 μl/h per mg of tissue, which corresponds to continuous flow on the order of 10s of μl/h in our chip for a single HIO (see Methods). For practical purposes, in the experiments described here we use flow rates in the range 50 μl/h ≤ q ≤ 300 μl/h, but to explore the impact of extraluminal flow on HIO physiology, q could be significantly reduced. In the future, additional changes could be made to mimic physiologically relevant conditions. For example, media composition could be altered such that oxygen-poor, nutrient-rich liquid is delivered to the lumen and oxygen-rich, nutrient-poor liquid is delivered to the basolateral surface. Additionally, the structure of the material around the HIO could be engineered to mimic the complex, layered tissues around the native gut, which govern liquid flow and transport and contain vasculature.
To determine if puncturing the epithelium and subjecting HIOs to long-term luminal and extraluminal flow adversely affects HIO viability, we perform measurements of cell viability using flow cytometry. Ported HIOs subjected to flow for t = 65h are removed from the device, individually dispersed as single cell suspensions, stained with a fluorescent indicator of membrane integrity (intracellular/extracellular amines strained with a Live/Dead cell stain, ThermoFisher Inc.) that differentially labels viable and nonviable cells, and assayed using flow cytometry (see Methods). A histogram of stain fluorescence intensity reveals a bimodal distribution (Fig. 6a), representing the fraction of live and dead cells from each HIO. When the data are compared (Fig. 6c), we find that the fraction of dead cells in ported HIOs (mean ± SD: 0.1296 ± 0.063, n = 3) was not statistically different from our two controls: unported HIOs assayed individually (0.1555 ± 0.1115, n = 4) and unported HIOs combined and assayed together (0.177 ± 0.0512, n = 3)(ANOVA: F2, 7 0.64, P = 0.55). This critical experiment and positive result support our microscopy observations that HIO viability is not adversely affected by porting and luminal flow. In the future, the impact of flow velocity, luminal content rheology, and nutrient concentration on gene expression, cellular differentiation, and cellular proliferation should be investigated to determine how these parameters impact the distribution of cell types and behaviors in an HIO.
Fig. 6. Flow cytometry results show that HIO viability is not adversely affected by porting and luminal flow.
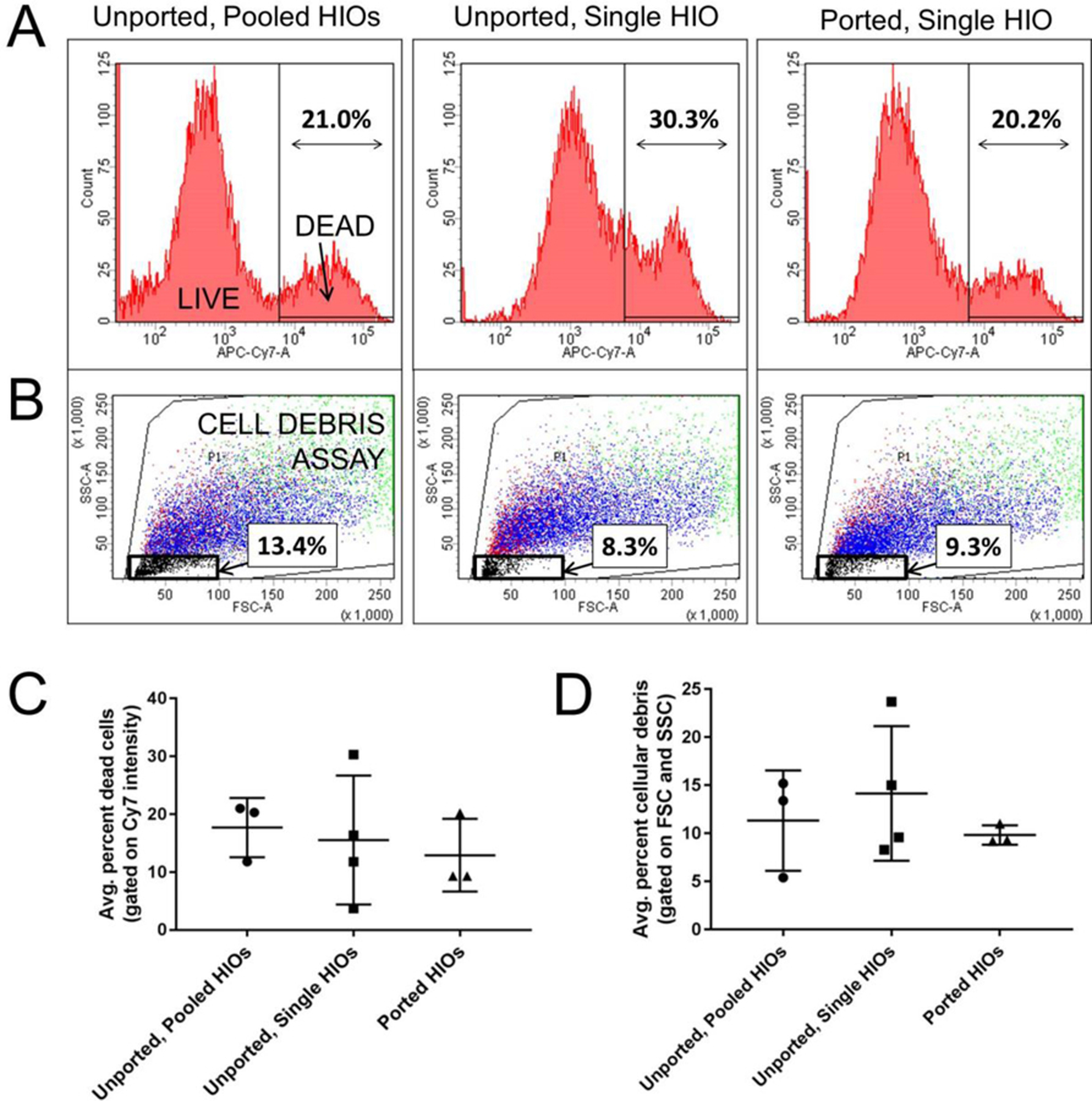
(A) Representative flow cytometry histograms of near-IR fluorescence intensity (APC-Cy7-A) from homogenized HIOs reveal two populations of cells: live (low intensity) and dead (high intensity). (B) Forward (FSC) and side (SSC) scattering at low intensities provide a measure of cellular debris as indicated by the black box and percentage values. (C) The average percent of dead cells present in HIOs that were either unported or ported, as determined by the LIVE/DEAD cell staining data represented in (A). (D) The average percent of cell debris present in HIOs that were either unported or ported, as determined by the scattering data represented in (A).
Flow cytometry can also provide a measure of insoluble cellular debris; particles with low intensity forward and side scattering are characteristic of suspended particles with sizes smaller than a cell (Fig. 6b). When these data are compared (Fig. 6d), we also find that the fraction of scattering events in ported HIOs corresponding to cellular debris (mean ± SD: 0.098 ± 0.01, n = 3) was not statistically different from controls: unported HIOs assayed individually (0.1415 ± 0.07, n = 4) and unported HIOs assayed collectively (0.1133 ± 0.0522, n = 3)(ANOVA: F2, 7 = 1.42, P = 0.30). While this is somewhat surprising given the dramatic removal of luminal waste depicted in Fig. 5, it could be that luminal HIO waste is solubilized during preparation for flow cytometry and no longer scatters light. It is also possible that even though more debris is being removed, the epithelium is producing more waste because it is more active in the ported state. Regardless, it is clearly apparent from microscopy that luminal material is being removed by the porting and flow process in manner which is physiologically relevant. In the future, more specific chemical or biochemical assays for quantifying the luminal concentrations of specific metabolic byproducts and waste as a function of luminal flow should be explored.
We expect that the results presented here will enable a wide variety of experiments. Luminal flow provides a means of introducing materials to the luminal space as well as extracting materials from this space; so, the device will be ideal for experiments exploring the establishment and stability of the microbiome, including the introduction of microbes, monitoring microbial dynamics with fluorescence microscopy, and detecting the presence of detached microbes and dissolved waste products and metabolites in the luminal effluent. In addition, maintained barrier integrity and independent control of luminal and extraluminal liquid streams will allow researchers to explore the transport of substances across the epithelial shell. For example, the absorption of nutrients and pharmaceutical compounds from the lumen through the apical surface of the epithelium could be explored. Inversely, the excretion of materials such as mucus and fluid into the lumen could also be studied. The functionality of the millifluidic chip presented here could be enhanced through the integration of a variety of soft, PDMS-based microfluidic modules such as flow-focusing drop makers and detection and sorting capabilities89, 90. While the design presented here could be parallelized to port small numbers of HIOs, the delicate porting process is not amenable to high-throughput testing. Truly massive parallelization would require the development of an automated porting method as well as improvements in HIO culture techniques to generate large numbers of HIOs with monodisperse sizes.
Conclusions
In conclusion, the Gut Organoid Flow Chip (GOFlowChip), presented here represents the first device engineered to establish liquid flow through the lumen of a gastrointestinal organoid for multiple days. This is achieved by pressurizing the device to suppress bubble formation and optimizing device design to prevent clogging. While the limits of HIO culturing and experimentation have not been fully explored, this prototype provides a significant advancement by mimicking a critically important physiologic parameter of the human gut: long-term luminal flow. Moreover, the chip holds several other advantages over previous gut organoid chip designs: the multilayer design allows for straightforward assembly, disassembly, sterilization, and reuse; a seal which forms at each epithelial puncture site allows for independent control of luminal and extraluminal liquid flow conditions, and biological assays of cell viability confirm the long-term viability of HIOs in the device. Thus, the GOFlowChip opens the field for broader application of HIO models in biomedical research.
Experimental
Device Fabrication.
Multilayer devices composed of three distinct layers and silicone rubber gaskets were cut from clear cast acrylic plastic sheets (McMaster Carr; dimensions: 12” x 12”) and silicone rubber sheets (McMaster Carr, Durometer 40A, White; dimensions: 12” x 12”, 1/16” thickness) using an automated laser cutter (Universal). Top and bottom layers were cut from sheets with thicknesses of h = 2.0 mm and h = 1.5 mm, respectively. The middle layer was cut from h = 4.5 mm thick sheets. Layers and gaskets were designed using AutoCAD software and design files are included in the supplementary section. Layers were sealed through gaskets between each layer and compressing the assembled layers using nuts and bolts (McMaster Carr; 316 stainless steel, M3 × 0.3 mm thread, 10 mm length). For the single-lumen, two capillary setup; tapered glass capillaries for puncturing the HIOs were created by pulling thin-wall borosilicate glass capillaries (World Precision Instruments TW150–6) using a micropipette puller (Sutter Instruments, P-97). Capillaries with tip diameters between 40 μm and 80 μm and taper lengths of 4 mm and 3.5 cm were used. For dual-lumen, single capillary setup; septum theta borosilicate glass capillaries (World Precision Instruments TST150–6) were pulled in the same way to obtain a 100 μm tip with 3 mm taper length. Capillaries were mounted to three-axis translational micromanipulators (Quater Research; XYZ 300 ML) with capillary holders designed in Fusion 360 and 3D printed using SLA 3D printer (see design files) for precision control during organoid puncturing.
Liquid Flow.
Luminal and extraluminal liquids were introduced to the device by connecting liquid-filled syringes (Hamilton 500μL and BD 10 mL, respectively) fitted with blunt-tip stainless steel dispensing needles (McMaster Carr; luminal flow: 26 gauge and 17; extraluminal flow: 16 gauge) to medical grade polyethylene micro-tubing (Scientific Commodities Inc., PE/9, ID = 1.40 mm, OD = 1.91 mm). For luminal flow, tubing was connected to the non-tapered ends of the glass capillaries. For extraluminal flow, tubing was connected to blunt-tip stainless steel dispensing needles (McMaster Carr, 90° angle, 20 gauge) inserted into holes in the upper layer of the device. Both luminal and extraluminal liquid flow was driven by programmable, computer-controlled New Era NE-1000 syringe pumps for precise delivery and withdrawal of small volumes of liquid. For short-term periodic flow experiments, phosphate-buffered saline (PBS) was used for both luminal and extraluminal flow. For long-term, steady-state flow experiments, HIO growth media was used for both luminal and extraluminal flow. To establish a baseline for extraluminal flow, we estimate the transport of blood through gut tissue in the native human gut. We estimate cardiac output to be 5L/min of which 20% is shared between the spleen, liver, stomach, small intestine and large intestine which are approximately 5 kg in total91–94. This corresponds to approximately 10 μl/h per mg of tissue. The HIOs used in our experiments contain tissue mass on the order of 2–5 mg, so we estimate the baseline for extraluminal flow to be 20–50 μl/h.
Sterilization.
The glass transition of the cast acrylic sheets is below our autoclave temperature range (T ≈ 121–132 C), so sterilizing the millifluidic device using heat is not feasible. Instead, the device was sterilized by disassembling individual layers and soaking them in pure ethanol for five minutes, followed by rinsing in autoclaved distilled water. After assembly, the device and associated tubing and connectors were again flushed with pure ethanol followed by autoclaved distilled water.
Bubble suppression.
The pressure required to drive liquid flow through a microfluidic device is usually sufficient to suppress air bubble formation in the device; however, this is not the case for millifluidic devices. To suppress bubble formation, we pressurized the liquid by attaching a tapered capillary at the outlet of the overflow channel. We observed that flow rates of 100–150 μl/hr and exit tip diameters of 40–60 μm corresponding to pressure drops of 70–140 Pa were sufficient to suppress bubble formation during multiday flow experiments.
Imaging.
Time-lapse video microscopy measurements were performed using a laser scanning confocal microscope (Leica SP5 II) equipped with an environmental control chamber (Life Imaging Services) maintained at 37°C. Fluorescence and brightfield images were collected with 10X air objective (Leica 506505, HC PL Fluotar 10X/0.03) and 1.25 X air objective (Leica 506215, HCX PL Fluotar 1.25X/0.04). Time-lapse measurements were also collected using a stereomicroscope (Leica, M205 FA) equipped with color CMOS video camera (Leica, DFC3000 G). After collection, images were processed and analyzed using IMARIS, MetaMorph and ImageJ image analysis software.
Organoid Culture.
Derivation and maintenance of HIOs followed published protocols1, 25. Briefly, HIOs were embedded in Matrigel (BD Biosciences) and overlaid with Advanced DMEM-F12 medium (Invitrogen, Carlsbad, CA) containing 1X B27 supplement (Invitrogen), 1X GlutaMAX (Life Technologies, Carlsbad, CA), 10 µM Hepes, 10% pen/strep, 100 ng/mL rhNoggin (R&D Systems), 100 ng/mL epidermal growth factor (R&D Systems), and approximately 500 ng/mL R-Spondin1 (RSPO1). RSPO1 was obtained from conditioned media collected from a HEK293 cell line that was stably transfected and zeocin-selected for the RSPO1 expression vector. Media was changed every two to four days, and HIOs were transferred to fresh Matrigel once a week until they reached approximately 2 to 3 mm in diameter for experiments. This size was reached on average 48 days after initial spheroid formation.
Cell Viability and Cellular Debris Assays:
Cell viability and cellular debris assays were determined using a LIVE/DEAD Fixable Dead Cell Stain Kit (ThermoFisher) and flow cytometry. Ported HIOs were collected after being subjected to luminal flow for t ≥ 65 h and preparation of the HIOs for the LIVE/DEAD stain occurred within 1 hour of collection. To disperse HIOs as single-cell suspensions, individual HIOs were washed with PBS, incubated in 0.25% trypsin-EDTA, and subjected to mechanical shear by passing the HIO through a P1000 pipette tip or 21 gauge needle. Cells were then washed in PBS, incubated with fluorescent dye, and fixed with formaldehyde following the manufacturer’s instructions. Cell viability, as determined by near-IR fluorescence intensity, was quantified using a LSRFortessa flow cytometer, fluorescence-activated cell sorting (FACS), and FACSDiva Software (BD Biosciences). Gating single cells was based on forward and side-scatter profiles using an isotype control made from a pooled sample of four unported HIOs that were maintained under static cell culture conditions. The percentages of live and dead cells were determined by using the manufacturer’s recommended settings and guidelines. After removing doublets and cell clumps from analysis, infrared staining was analyzed to determine the best fit of separation between live cells and dead cells, which are represented by low and high APC-Cy7-A emission intensity, respectively. As an additional control, unported HIOs similar in size and age to the ported HIOs were collected and analyzed individually following the protocol above.
Statistical Analysis:
A one-way ANOVA with multiple comparisons was performed to test statistical differences between the means of three groups: unported, pooled HIOs; unported, single HIOs; and ported HIOs.
Supplementary Material
Funding
This work was supported in part by the Bill & Melinda Gates Foundation, Grant OPP1108199 (S.T.W. and J.R.S.), the National Science Foundation, DMR-1455247 (J.N.W.) and the National Institutes of Health, R01GM131408–01 (J.N.W.).
Bibliography
- 1.Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn AM, Shroyer NF and Wells JM, Nature, 2011, 470, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sato T, Stange DE, Ferrante M, Vries RGJ, van Es JH, van den Brink S, van Houdt WJ, Pronk A, van Gorp J, Siersema PD and Clevers H., Gastroenterology, 2011, 141, 1762–1772. [DOI] [PubMed] [Google Scholar]
- 3.Hill DR, Huang S, Nagy MS, Yadagiri VK, Fields C, Mukherjee D, Bons B, Dedhia PH, Chin AM, Tsai YH, Thodla S, Schmidt TM, Walk S, Young VB and Spence JR, Elife, 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finkbeiner SR, Zeng XL, Utama B, Atmar RL, Shroyer NF and Estes MK, Mbio, 2012, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G, Ivanov JA, Fu YY, Takashima S, Hua GQ, Martin ML, O’Rourke KP, Lo YH, Mokry M, Romera-Hernandez M, Cupedo T, Dow LE, Nieuwenhuis EE, Shroyer NF, Liu C, Kolesnick R, van den Brink MRM and Hanash AM, Nature, 2015, 528, 560–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nozaki K, Mochizuki W, Matsumoto Y, Matsumoto T, Fukuda M, Mizutani T, Watanabe M and Nakamura T., J Gastroenterol, 2016, 51, 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wells JM and Spence JR, Development, 2014, 141, 752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aurora M and Spence JR, Dev Biol, 2016, 420, 230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dedhia PH, Bertaux-Skeirik N, Zavros Y and Spence JR, Gastroenterology, 2016, 150, 1098–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forbester JL, Goulding D, Vallier L, Hannan N, Hale C, Pickard D, Mukhopadhyay S and Dougan G., Infect Immun, 2015, 83, 2926–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leslie JL, Huang S, Opp JS, Nagy MS, Kobayashi M, Young VB and Spence JR, Infect Immun, 2015, 83, 138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engevik MA, Yacyshyn MB, Engevik KA, Wang J, Darien B, Hassett DJ, Yacyshyn BR and Worrell RT, Am J Physiol-Gastr L, 2015, 308, G510–G524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sinagoga KL and Wells JM, Embo J, 2015, 34, 1149–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finkbeiner Stacy R., Hill David R., Altheim Christopher H., Dedhia Priya H., Taylor Matthew J., Tsai Y-H, Chin Alana M., Mahe Maxime M., Watson Carey L., Freeman Jennifer J., Nattiv R, Thomson M, Klein Ophir D., Shroyer Noah F., Helmrath Michael A., Teitelbaum Daniel H., Dempsey Peter J. and Spence Jason R., Stem Cell Reports, 2015, 4, 1140–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buckley A and Turner JR, Csh Perspect Biol, 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hill DR, Huang S, Tsai YH, Spence JR and Young VB, J Vis Exp, 2017, DOI: 10.3791/56960. [DOI] [PMC free article] [PubMed]
- 17.Gray H and Standring S., Gray’s anatomy: the anatomical basis of clinical practice, Churchill Livingstone, 2008. [Google Scholar]
- 18.Sebrell TA, Sidar B, Bruns R, Wilkinson RA, Wiedenheft B, Taylor PJ, Perrino BA, Samuelson LC, Wilking JN and Bimczok D., Cell and Tissue Research, 2018, 371, 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee KK, McCauley HA, Broda TR, Kofron MJ, Wells JM and Hong CI, Lab Chip, 2018, 18, 3079–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin XL, Mead BE, Safaee H, Langer R, Karp JM and Levy O., Cell Stem Cell, 2016, 18, 25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cruz-Acuña R, Quirós M, Farkas AE, Dedhia PH, Huang S, Siuda D, García-Hernández V, Miller AJ, Spence JR, Nusrat A and García AJ, Nature Cell Biology, 2017, 19, 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cruz-Acuña R, Quirós M, Huang S, Siuda D, Spence JR, Nusrat A and García AJ, Nature protocols, 2018, 13, 2102–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capeling MM, Czerwinski M, Huang S, Tsai Y-H, Wu A, Nagy MS, Juliar B, Sundaram N, Song Y, Han WM, Takayama S, Alsberg E, Garcia AJ, Helmrath M, Putnam AJ and Spence JR, Stem Cell Reports, 2019, 12, 381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zachos NC, Kovbasnjuk O, Foulke-Abel J, In J, Blutt SE, de Jonge HR, Estes MK and Donowitz M., J Biol Chem, 2016, 291, 3759–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCracken KW, Howell JC, Wells JM and Spence JR, Nature protocols, 2011, 6, 1920–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.deMello AJ, Nature, 2006, 442, 394–402. [DOI] [PubMed] [Google Scholar]
- 27.El-Ali J, Sorger PK and Jensen KF, Nature, 2006, 442, 403–411. [DOI] [PubMed] [Google Scholar]
- 28.Maschmeyer I, Lorenz AK, Schimek K, Hasenberg T, Ramme AP, Hubner J, Lindner M, Drewell C, Bauer S, Thomas A, Sambo NS, Sonntag F, Lauster R and Marx U., Lab Chip, 2015, 15, 2688–2699. [DOI] [PubMed] [Google Scholar]
- 29.Baudoin R, Griscom L, Monge M, Legallais C and Leclerc E., Biotechnol Progr, 2007, 23, 1245–1253. [DOI] [PubMed] [Google Scholar]
- 30.Jang KJ, Mehr AP, Hamilton GA, McPartlin LA, Chung SY, Suh KY and Ingber DE, Integr Biol-Uk, 2013, 5, 1119–1129. [DOI] [PubMed] [Google Scholar]
- 31.Jang KJ and Suh KY, Lab Chip, 2010, 10, 36–42. [DOI] [PubMed] [Google Scholar]
- 32.Snouber LC, Letourneur F, Chafey P, Broussard C, Monge M, Legallais C and Leclerc E., Biotechnol Progr, 2012, 28, 474–484. [DOI] [PubMed] [Google Scholar]
- 33.Agarwal A, Goss JA, Cho A, McCain ML and Parker KK, Lab Chip, 2013, 13, 3599–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng W, Klauke N, Sedgwick H, Smith GL and Cooper JM, Lab Chip, 2006, 6, 1424–1431. [DOI] [PubMed] [Google Scholar]
- 35.Giridharan GA, Nguyen MD, Estrada R, Parichehreh V, Hamid T, Ismahil MA, Prabhu SD and Sethu P., Anal Chem, 2010, 82, 7581–7587. [DOI] [PubMed] [Google Scholar]
- 36.Grosberg A, Alford PW, McCain ML and Parker KK, Lab Chip, 2011, 11, 4165–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khanal G, Chung K, Solis-Wever X, Johnson B and Pappas D., Analyst, 2011, 136, 3519–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen MD, Tinney JP, Ye F, Elnakib AA, Yuan FP, El-Baz A, Sethu P, Keller BB and Giridharan GA, Anal Chem, 2015, 87, 2107–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fritsche CS, Simsch O, Weinberg EJ, Orrick B, Stamm C, Kaazempur-Mofrad MR, Borenstein JT, Hetzer R and Vacanti JP, Int J Artif Organs, 2009, 32, 701–710. [DOI] [PubMed] [Google Scholar]
- 40.Henry OYF, Villenave R, Cronce MJ, Leineweber WD, Benz MA and Ingber DE, Lab Chip, 2017, 17, 2264–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huh D, Fujioka H, Tung YC, Futai N, Paine R, Grotberg JB and Takayama S., P Natl Acad Sci USA, 2007, 104, 18886–18891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huh D, Leslie DC, Matthews BD, Fraser JP, Jurek S, Hamilton GA, Thorneloe KS, McAlexander MA and Ingber DE, Sci Transl Med, 2012, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY and Ingber DE, Science, 2010, 328, 1662–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sin A, Chin KC, Jamil MF, Kostov Y, Rao G and Shuler ML, Biotechnol Progr, 2004, 20, 338–345. [DOI] [PubMed] [Google Scholar]
- 45.Tavana H, Zamankhan P, Christensen PJ, Grotberg JB and Takayama S., Biomed Microdevices, 2011, 13, 731–742. [DOI] [PubMed] [Google Scholar]
- 46.Shah P, Fritz JV, Glaab E, Desai MS, Greenhalgh K, Frachet A, Niegowska M, Estes M, Jäger C, Seguin-Devaux C, Zenhausern F and Wilmes P., Nature Communications, 2016, 7, 11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen WLK, Edington C, Suter E, Yu JJ, Velazquez JJ, Velazquez JG, Shockley M, Large EM, Venkataramanan R, Hughes DJ, Stokes CL, Trumper DL, Carrier RL, Cirit M, Griffith LG and Lauffenburger DA, Biotechnol Bioeng, 2017, 114, 2648–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Esch MB, Sung JH, Yang J, Yu CH, Yu JJ, March JC and Shuler ML, Biomed Microdevices, 2012, 14, 895–906. [DOI] [PubMed] [Google Scholar]
- 49.Kim HJ, Huh D, Hamilton G and Ingber DE, Lab Chip, 2012, 12, 2165–2174. [DOI] [PubMed] [Google Scholar]
- 50.Kim HJ and Ingber DE, Integr Biol-Uk, 2013, 5, 1130–1140. [DOI] [PubMed] [Google Scholar]
- 51.Lee DW, Ha SK, Choi I and Sung JH, Biomed Microdevices, 2017, 19. [DOI] [PubMed] [Google Scholar]
- 52.Mahler GJ, Esch MB, Glahn RP and Shuler ML, Biotechnol Bioeng, 2009, 104, 193–205. [DOI] [PubMed] [Google Scholar]
- 53.Allen JW and Bhatia SN, Biotechnol Bioeng, 2003, 82, 253–262. [DOI] [PubMed] [Google Scholar]
- 54.Carraro A, Hsu WM, Kulig KM, Cheung WS, Miller ML, Weinberg EJ, Swart EF, Kaazempur-Mofrad M, Borenstein JT, Vacanti JP and Neville C., Biomed Microdevices, 2008, 10, 795–805. [DOI] [PubMed] [Google Scholar]
- 55.Chao P, Maguire T, Novik E, Cheng KC and Yarmush ML, Biochem Pharmacol, 2009, 78, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng S, Prot JM, Leclerc E and Bois FY, Bmc Genomics, 2012, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kane BJ, Zinner MJ, Yarmush ML and Toner M., Anal Chem, 2006, 78, 4291–4298. [DOI] [PubMed] [Google Scholar]
- 58.Lee PJ, Hung PJ and Lee LP, Biotechnol Bioeng, 2007, 97, 1340–1346. [DOI] [PubMed] [Google Scholar]
- 59.Legendre A, Baudoin R, Alberto G, Paullier P, Naudot M, Bricks T, Brocheton J, Jacques S, Cotton J and Leclerc E., J Pharm Sci-Us, 2013, 102, 3264–3276. [DOI] [PubMed] [Google Scholar]
- 60.Novik E, Maguire TJ, Chao PY, Cheng KC and Yarmush ML, Biochem Pharmacol, 2010, 79, 1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sivaraman A, Leach JK, Townsend S, Iida T, Hogan BJ, Stolz DB, Fry R, Samson LD, Tannenbaum SR and Griffith LG, Curr Drug Metab, 2005, 6, 569–591. [DOI] [PubMed] [Google Scholar]
- 62.Toh YC, Lim TC, Tai D, Xiao GF, van Noort D and Yu HR, Lab Chip, 2009, 9, 2026–2035. [DOI] [PubMed] [Google Scholar]
- 63.Viravaidya K and Shuler ML, Biotechnol Progr, 2004, 20, 590–597. [DOI] [PubMed] [Google Scholar]
- 64.Esch MB, Prot JM, Wang YI, Miller P, Llamas-Vidales JR, Naughton BA, Applegate DR and Shuler ML, Lab Chip, 2015, 15, 2269–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Booth R and Kim H., Lab Chip, 2012, 12, 1784–1792. [DOI] [PubMed] [Google Scholar]
- 66.Liu MC, Shih HC, Wu JG, Weng TW, Wu CY, Lu JC and Tung YC, Lab Chip, 2013, 13, 1743–1753. [DOI] [PubMed] [Google Scholar]
- 67.Shin M, Matsuda K, Ishii O, Terai H, Kaazempur-Mofrad M, Borenstein J, Detmar M and Vacanti JP, Biomed Microdevices, 2004, 6, 269–278. [DOI] [PubMed] [Google Scholar]
- 68.Park SH, Sim WY, Min BH, Yang SS, Khademhosseini A and Kaplan DL, Plos One, 2012, 7. [DOI] [PMC free article] [PubMed]
- 69.Zhang WT, Lee WY, Siegel DS, Tolias P and Zilberberg J., Tissue Eng Part C-Me, 2014, 20, 663–670. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y, Gazit Z, Pelled G, Gazit D and Vunjak-Novakovic G., Integr Biol-Uk, 2011, 3, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Torisawa YS, Spina CS, Mammoto T, Mammoto A, Weaver JC, Tat T, Collins JJ and Ingber DE, Nat Methods, 2014, 11, 663–+. [DOI] [PubMed] [Google Scholar]
- 72.Park HS, Liu S, McDonald J, Thakor N and Yang IH, Ieee Eng Med Bio, 2013, 2833–2835. [DOI] [PubMed]
- 73.Shi MJ, Majumdar D, Gao YD, Brewer BM, Goodwin CR, McLean JA, Lib D and Webb DJ, Lab Chip, 2013, 13, 3008–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsantoulas C, Farmer C, Machado P, Baba K, McMahon SB and Raouf R., Plos One, 2013, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van de Wijdeven R, Ramstad OH, Bauer US, Halaas O, Sandvig A and Sandvig I., Biomed Microdevices, 2018, 20. [DOI] [PubMed] [Google Scholar]
- 76.Xiao RR, Zeng WJ, Li YT, Zou W, Wang L, Pei XF, Xie M and Huang WH, Anal Chem, 2013, 85, 7842–7850. [DOI] [PubMed] [Google Scholar]
- 77.Ziegler L, Grigoryan S, Yang IH, Thakor NV and Goldstein RS, Stem Cell Rev Rep, 2011, 7, 394–403. [DOI] [PubMed] [Google Scholar]
- 78.Grosberg A, Nesmith AP, Goss JA, Brigham MD, McCain ML and Parker KK, J Pharmacol Tox Met, 2012, 65, 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Puleo CM, Ambrose WM, Takezawa T, Elisseeff J and Wang TH, Lab Chip, 2009, 9, 3221–3227. [DOI] [PubMed] [Google Scholar]
- 80.Schweinlin M, Wilhelm S, Schwedhelm I, Hansmann J, Rietscher R, Jurowich C, Walles H and Metzger M., Tissue Eng Part C-Me, 2016, 22, 873–883. [DOI] [PubMed] [Google Scholar]
- 81.Workman MJ, Gleeson JP, Troisi EJ, Estrada HQ, Kerns SJ, Hinojosa CD, Hamilton GA, Targan SR, Svendsen CN and Barrett RJ, Cellular and Molecular Gastroenterology and Hepatology, 2018, 5, 669–677.e662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang YQ, Wang L, Guo YQ, Zhu YJ and Qin JH, Rsc Adv, 2018, 8, 1677–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Skardal A, Murphy SV, Devarasetty M, Mead I, Kang HW, Seol YJ, Zhang YS, Shin SR, Zhao L, Aleman J, Hall AR, Shupe TD, Kleensang A, Dokmeci MR, Lee SJ, Jackson JD, Yoo JJ, Hartung T, Khademhosseini A, Soker S, Bishop CE and Atala A., Sci Rep-Uk, 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang YS, Aleman J, Shin SR, Kilic T, Kim D, Shaegh SAM, Massa S, Riahi R, Chae S, Hu N, Avci H, Zhang W, Silvestri A, Nezhad AS, Manbohi A, De Ferrari F, Polini A, Calzone G, Shaikh N, Alerasool P, Budina E, Kang J, Bhise N, Ribas J, Pourmand A, Skardal A, Shupe T, Bishop CE, Dokmeci MR, Atala A and Khademhosseini A., P Natl Acad Sci USA, 2017, 114, E2293–E2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marshall LE, Koomullil R, Frost AR and Berry JL, Ann Biomed Eng, 2017, 45, 1027–1038. [DOI] [PubMed] [Google Scholar]
- 86.Miura T and Tanaka R., Math Model Nat Pheno, 2009, 4, 118–130. [Google Scholar]
- 87.Wang YT, Mohammed SD, Farmer AD, Wang D, Zarate N, Hobson AR, Hellstrom PM, Semler JR, Kuo B, Rao SS, Hasler WL, Camilleri M and Scott SM, Aliment Pharm Ther, 2015, 42, 761–772. [DOI] [PubMed] [Google Scholar]
- 88.Stevens CE and Hume ID, Comparative physiology of the vertebrate digestive system, Cambridge University Press, Cambridge; New York, 2nd edn., 1995. [Google Scholar]
- 89.Meng ZJ, Wang W, Liang X, Zheng WC, Deng NN, Xie R, Ju XJ, Liu Z and Chu LY, Lab Chip, 2015, 15, 1869–1878. [DOI] [PubMed] [Google Scholar]
- 90.Girabawe C and Fraden S., Sensor Actuat B-Chem, 2017, 238, 532–539. [Google Scholar]
- 91.Lowrey LG, The American Journal of Anatomy, 1911, 12, 131. [Google Scholar]
- 92.Chou CC, Fed Proc, 1983, 42, 1656–1657. [PubMed] [Google Scholar]
- 93.Baraki YM, Traverso P, Elariny HA and Fang Y., Surg Technol Int, 2010, 20, 167–171. [PubMed] [Google Scholar]
- 94.John W DJL Calvert, in Muscle, ed. Joseph A ENO. Hill, Academic Press, 2012, vol. 1, ch. Chapter 6, pp. 57–66. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.