

Abstract
Alternate isoforms are important contributors to phenotypic diversity across eukaryotes. Although short-read RNA-sequencing has increased our understanding of isoform diversity, it is challenging to accurately detect full-length transcripts, preventing the identification of many alternate isoforms. Long-read sequencing technologies have made it possible to sequence full-length alternative transcripts, accurately characterizing alternative splicing events, alternate transcription start and end sites, and differences in UTR regions. Here, we use Pacific Biosciences (PacBio) long-read RNA-sequencing (Iso-Seq) to examine the transcriptomes of five organs in threespine stickleback fish (Gasterosteus aculeatus), a widely used genetic model species. The threespine stickleback fish has a refined genome assembly in which gene annotations are based on short-read RNA sequencing and predictions from coding sequence of other species. This suggests some of the existing annotations may be inaccurate or alternative transcripts may not be fully characterized. Using Iso-Seq we detected thousands of novel isoforms, indicating many isoforms are absent in the current Ensembl gene annotations. In addition, we refined many of the existing annotations within the genome. We noted many improperly positioned transcription start sites that were refined with long-read sequencing. The Iso-Seq-predicted transcription start sites were more accurate and verified through ATAC-seq. We also detected many alternative splicing events between sexes and across organs. We found a substantial number of genes in both somatic and gonadal samples that had sex-specific isoforms. Our study highlights the power of long-read sequencing to study the complexity of transcriptomes, greatly improving genomic resources for the threespine stickleback fish.
The ability to generate alternative isoforms from a finite number of genes is a widespread phenomenon across eukaryotes that has been hypothesized to play a key role in the evolution of phenotypic diversity (for reviews, see Graveley 2001; Keren et al. 2010; Baralle and Giudice 2017). Alternative isoforms can arise through multiple mechanisms. For one, the coding sequence can be altered by alternative splicing. This can be achieved through the retention of introns, the inclusion or exclusion of entire exons, or the usage of alternative splice sites within exons (for reviews, see Smith et al. 1989; Keren et al. 2010). Isoform diversity can also be increased through the inclusion of alternate transcription start sites (TSSs) or transcription termination sites (TTSs), leading to differences in the 5′ or 3′ untranslated regions (UTRs). These variants do not alter the underlying coding sequence but can alter transcriptional regulation and underlying stability of the mRNA transcript (Gupta et al. 2014; Wang et al. 2016b; Zhang et al. 2017). These positions are also important for some experimental applications such as scRNA-seq, which uses 3′ tags to select or enrich transcripts during library preparation (Hashimshony et al. 2012; Macosko et al. 2015).
Short-read RNA-seq has greatly expanded our ability to survey the complexity of transcriptomes (for review, see Costa et al. 2010), including the computational prediction of alternative splicing events (Trapnell et al. 2009; Anders et al. 2012; Kim et al. 2015, 2019). However, short-read RNA-seq cannot accurately detect all isoforms. To detect all splice junctions among alternative isoforms, there must be sufficient read depth at alternative exon–exon boundaries (Bryant et al. 2012; Steijger et al. 2013). For experiments with insufficient read coverage, lowly expressed isoforms are challenging to recover and predict (for review, see Conesa et al. 2016). Alternative isoform identification is further confounded as short reads map to multiple isoforms, causing reads to collapse into a single isoform. To properly differentiate alternative transcripts, full-length transcripts are needed (Steijger et al. 2013; Wang et al. 2016a).
Long-read sequencing technologies have made it possible to sequence a single full-length transcript with high accuracy (Wang et al. 2016a, 2019).With sufficient sequencing coverage, isoforms can be identified unambiguously, classifying the complete catalog of splice junctions and alternate TSSs and TTSs (Wang et al. 2019). This technology has been successfully applied to multiple species of plants and animals (Sharon et al. 2013; Abdel-Ghany et al. 2016; Wang et al. 2016a; Cheng et al. 2017; Kuo et al. 2017; Li et al. 2018; Nudelman et al. 2018; Deslattes Mays et al. 2019; Zhang et al. 2019). Long-read RNA sequencing has refined existing gene annotations as well as characterized pervasive alternative splicing among organs (Abdel-Ghany et al. 2016; Wang et al. 2016a; Kuo et al. 2017; Li et al. 2018; Zhang et al. 2019).
Although transcriptome complexity has been increasingly studied at the organ level, comparatively little is known about the sex specificity of isoforms. Sexual dimorphism in alternative splicing may be important in regulating many of the phenotypic differences observed between sexes. For instance, male and female somatic differentiation in Drosophila is controlled by alternatively spliced transcripts of the doublesex gene (Dmel\dsx) (Burtis and Baker 1989). In addition, alternative splicing can be a mechanism to resolve intralocus sexual antagonism, where the expression of a gene is beneficial to one sex yet harmful to the other (for reviews, see Ellegren and Parsch 2007; Stewart et al. 2010). Alternative splicing could allow antagonistic exons to be restricted to a single sex, or alternative TSSs and TTSs could create opportunities for sex-specific transcriptional regulation. At a genome level, there is growing evidence that alternative splicing is widespread between sexes (McIntyre et al. 2006; Blekhman et al. 2010; Brown et al. 2014; Gibilisco et al. 2016; Rogers et al. 2020). However, all surveys have used either short-read RNA-seq or microarray probes targeting known transcripts. This raises the possibility that the true amount of alternative splicing between sexes may be underestimated.
Here we use Pacific Biosciences (PacBio) long-read RNA sequencing (Iso-Seq) to explore the extent of alternative isoforms across organs and between sexes in threespine stickleback fish (Gasterosteus aculeatus). Threespine stickleback fish are an emerging genetic model system for evolutionary biology, ecology, behavior, physiology, and toxicology (Bell and Foster 1994; Barber and Nettlseship 2010; Hendry et al. 2013). Although the genome sequence has been curated well (Peichel et al. 2001, 2017; Jones et al. 2012a; Glazer et al. 2015; Nath et al. 2021), the gene annotations are based entirely on the Ensembl annotation pipeline (Yates et al. 2020), which incorporates expressed sequencing tags (ESTs), publicly available short-read RNA-seq, and known homology from other organisms (Yates et al. 2020). Our study uses Iso-Seq to sequence the transcriptomes of five organs to high coverage in both sexes (liver, brain, pronephros, testis, and ovary) to expand and refine the existing Ensembl annotations. In addition, we survey the extent of alternative isoforms among organs and sexes. These transcriptomes will be an important resource in exploring the overall contribution of alternative isoforms to sexual dimorphism as threespine stickleback fish show pronounced phenotypic differences between the sexes (Kitano et al. 2007; Leinonen et al. 2011; Kotrschal et al. 2012; McGee and Wainwright 2013).
Results
The Iso-Seq3 pipeline produces an accurate transcriptome
We extracted total RNA from the brain (including the optic bulb), liver, pronephros or head kidney, and meiotic gonads (ovary and testis) from both sexes. The entire organ was extracted, and both nuclear and cytoplasmic RNAs were collected for long-read RNA sequencing (Iso-Seq). On average, 618,000 circular consensus sequences (CCSs) per organ were produced from the raw subreads (Supplemental Table S1). These CCS reads were then filtered, clustered into full-length transcripts, and polished, producing an average of 41,000 high-quality consensus reads per sample (Supplemental Table S1). We then tested the effect of different aligners on the produced transcriptomes. minimap2 identified 26,432 isoforms, whereas deSALT found 48,345 isoforms. Using universal single-copy orthologs (BUSCO) (see Supplemental Methods: Optimizing Long-Read Transcriptome Pipeline), we found the minimap2 transcriptome was more complete, containing more complete single-copy orthologs than the deSALT transcriptome (Supplemental Fig. S1). Therefore, we used minimap2 for the final Iso-Seq transcriptome.
We explored whether the number of BUSCO genes we observed in the Iso-Seq transcriptome was affected by the stringent filtering implemented in the Iso-Seq3 pipeline. We identified isoforms using only the CCS reads, created before running Iso-Seq3, from the female brain as a representative sample (see Supplemental Methods: Optimizing Long-Read Transcriptome Pipeline). The CCS transcriptome produced more isoforms, but the full Iso-Seq transcriptome produced 10-fold more complete single-copy orthologs (Supplemental Fig. S2). This suggests the Iso-Seq3 pipeline may not be driving the difference in complete single-copy orthologs compared with the Ensembl annotations, although it is important to note we cannot fully rule out the effect of size selection bias in the sequencing protocol (see size bias described below).
PacBio long-read sequencing identifies several thousand isoforms
Using the Iso-Seq3 pipeline and SQANTI filtering, we recovered a final Iso-Seq transcriptome composed of 26,432 isoforms (13,703 genes; annotated genes: 7754; novel genes: 5949) (Supplemental Files S1–S4, Supplemental Table S2). In comparison, the Ensembl transcriptome (build 97) had 22,443 genes and 29,245 isoforms. Although we found a smaller number of genes in the Iso-Seq transcriptome, we identified a similar number of isoforms, suggesting that alternative splicing and/or alternative TSSs and TTSs may be more pervasive than predicted through the Ensembl annotations. Consistent with this, we identified 18,271 novel isoforms that did not match previously annotated Ensembl isoforms. Furthermore, within single genes we also observed a greater breadth of alternative isoforms. There was a higher percentage of genes that had two or more isoforms in the Iso-Seq transcriptome compared with the Ensembl transcriptome (Ensembl: 24%; Iso-Seq: 31%) (Supplemental Fig. S3).
One possibility why we did not capture more single-copy orthologs was that our libraries may not have been sequenced to an adequate depth. To test this, we created subsamples of the CCS reads (Workman et al. 2018). We recovered at least 90% of the predicted isoforms with only 35%–85% of the original CCS reads (Supplemental Fig. S4). This indicates that each library was nearly saturated with reads and that including additional sequencing of these organs would not greatly increase the transcriptome completeness.
Long-read sequencing technologies are biased toward sequencing short isoforms (≤2 kb) (Byrne et al. 2019; Amarasinghe et al. 2020). To examine if our Iso-Seq transcriptome was enriched for short isoforms, we compared the isoform lengths between the Iso-Seq transcriptome and the Ensembl isoforms missing from our data set. The Iso-Seq transcriptome was missing 23,794 isoforms that were annotated in the Ensembl transcriptome. Among these isoforms, 18,283 isoforms (76.7%) were <2 kb in length. This was enriched compared with the percentage of isoforms <2 kb in length in the remaining Iso-Seq transcriptome (15,091 isoforms; 57.1%; chi-squared test; X-squared: 2189.7; P < 0.001). These results suggest many of the missing transcripts in our assembly may be owing to a size bias against short transcripts in the sequencing protocol.
We used short-read RNA sequencing to verify the accuracy of alternative splicing among the Iso-Seq isoforms. We sequenced each sample to high coverage in order to target a complete set of alternative splice junctions (approximately 166 million reads per sample were produced) (Supplemental Table S3; Supplemental File S5). We searched for the presence of uniquely mapping and multimapping short reads that spanned the splice junctions of isoforms with two or more exons (17,853 isoforms). The short-read sequencing showed that the Iso-Seq was highly accurate at detecting alternative isoforms. A majority of the isoforms (16,826 isoforms, 94% of isoforms with two or more exons) had all splice junctions confirmed by short-read sequencing through both uniquely mapping reads and multimapping reads. Only 321 isoforms (2%) had no short reads supporting the splice junctions. The remaining 706 isoforms (4%) had one or more splice junctions that were not confirmed by the short-read data.
Full-length isoform sequencing refines many of the previously predicted TSSs and transcription end sites
SQANTI characterizes isoforms into nine categories based on the splice junctions between exons (Tardaguila et al. 2018). We condensed these categories into three broad classes: Ensembl isoform matches, novel isoforms, and novel genes. Ensembl isoform matches corresponded to known Ensembl gene and transcript annotations, whereas novel isoforms matched a known Ensembl gene but did not match any of the annotated transcripts. Novel genes did not match any annotated gene in Ensembl. We also aligned the Iso-Seq transcriptome to the most recent genome assembly (Supplemental Table S4; Nath et al. 2021). Of the 26,432 isoforms, only 6410 exactly matched the Ensembl predicted splicing (FSM; 25%) (Fig. 1; Table 1). The remainder of the isoforms overlapping Ensembl annotations did not match the existing splicing annotations fully and likely represent alternative splicing events that exclude some internal exons (ISM; 1551 isoforms) (Fig. 1; Table 1). We found that a majority of isoforms that fully matched internal Ensembl splice junctions had differences in TSSs and TTSs (FSM isoforms that did not match annotated TSSs: 99%; FSM isoforms that did not match annotated TTSs: 98%). On average, the TSSs were 99 bp upstream of the annotated TSS (Fig. 2A). This pattern was more pronounced in the TTSs where the average distance from the Ensembl TTS was 500 bp upstream (Fig. 2B).
Figure 1.

In-depth characterization of isoforms by SQANTI for Iso-Seq transcriptome. Isoforms are classified into nine different splice categories by SQANTI: full-splice match (FSM), incomplete splice match (ISM), novel in catalog (NIC), novel not in catalog (NNC), genic, antisense, fusion, intergenic, and intronic. Each splice category is divided into predicted protein-coding isoforms (gold) and predicted non-protein-coding isoforms (gray).
Table 1.
Characterization of Iso-Seq transcripts
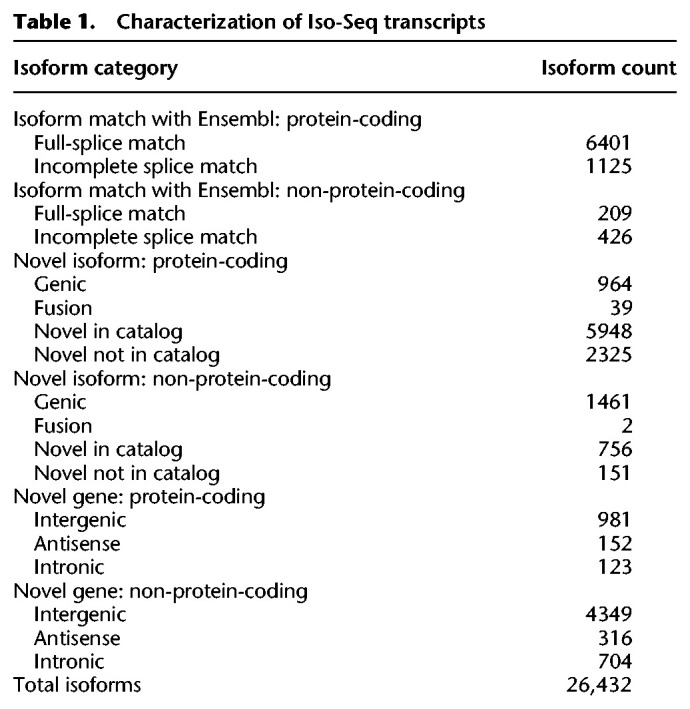
Figure 2.

Iso-Seq full-splice matches have different transcription start sites (TSSs) and transcription termination sites (TTSs) compared with that of Ensembl annotations. Full-splice matches are isoforms that have the same exon boundaries as Ensembl transcripts (6610 isoforms). (A) On average, full-splice match isoform TSSs are located 99 bp upstream of the annotated TSS. (B) Full-splice match isoform TTSs are located on average 500 bp upstream of the annotated TTS.
We confirmed the new TSSs by using liver ATAC-seq chromatin accessibility data from a different population of fish. ATAC-seq reads are expected to be enriched around the nucleosome-free region of TSSs (Mavrich et al. 2008; Buenrostro et al. 2013; Meers et al. 2018). We compared the read coverage in the 4 kb surrounding Ensembl-annotated TSSs and in the 4 kb surrounding the new Iso-Seq TSSs. We found an increased enrichment of ATAC-seq reads narrowed exactly at TSSs identified by Iso-Seq (Fig. 3; Supplemental Fig. S5). This enrichment was much weaker around Ensembl TSSs, indicating greater inaccuracy in placement. There may be some variation in enrichment of ATAC-seq reads around TSSs owing to population-specific differences in liver transcript expression (the ATAC-seq and Iso-Seq data were derived from different populations of fish). However, the strong enrichment we observed around Iso-Seq TSSs indicates many of the transcripts must be common across populations.
Figure 3.

Accessible chromatin is localized in narrow peaks around the Iso-Seq TSSs. We compared ATAC-seq read coverage at all Ensembl TSSs and Iso-Seq TSSs across the autosomes in the male liver (A) and female liver (B). ATAC-seq reads show an enrichment at the Iso-Seq TSS compared with the Ensembl TSS. This indicates a more accurate positioning of the TSS using Iso-Seq. A second male and female replicate is shown in Supplemental Figure S5.
Work in other species has shown chromatin accessibility at TSSs is a good predictor of transcriptional activity, but it does not accurately predict overall expression level of genes (Connelly et al. 2014). We examined whether the depth of ATAC-seq reads at the newly defined TSSs were positively correlated with RNA-seq expression levels. We found weak positive correlations in both male and female liver samples (male liver: Spearman's rank correlation, average rho = 0.049, P > 0.05; female liver: Spearman's rank correlation, average rho = 0.082, P < 0.05) (Supplemental Fig. S6), indicating the degree of chromatin accessibility in the sample is not a predictor of gene expression level in threespine stickleback fish.
A majority of the Iso-Seq transcriptome was previously unannotated
Nearly 70% of isoforms detected in the Iso-Seq transcriptome (18,271 isoforms) were classified as novel isoforms or novel genes. Novel isoforms represent a completely new isoform of a previously annotated gene in the Ensembl transcriptome, whereas novel genes represent a completely novel gene not previously annotated. A majority of the novel isoforms (64%; 11,646) overlap currently annotated genes, indicating long-read sequencing captures alternative splicing events not readily annotated by current pipelines or identifies errors in current exon/intron boundaries (Table 1). The remaining isoforms represented completely novel genes (36%; 6625), either located entirely within an Ensembl intergenic region, within an Ensembl-annotated intron, or on the antisense strand (Table 1). To examine the functions of the novel protein-coding isoforms (11,646), we searched for Gene Ontology (GO) matches and similarities to known protein domains. We found 10,107 novel isoforms that shared sequence identity with at least one known protein domain (Supplemental File S6). Many GO terms were enriched for general components of the cell (e.g., membrane components or organelle components) and general molecular functions such as catalytic activity and biogenesis (Supplemental Fig. S7; Supplemental Table S5). There was also enrichment in various biological processes such as biological regulation, response to stimulus, reproduction, and immune processes (Supplemental Fig. S7; Supplemental Table S5). Over 50% of the novel isoforms appear to be unique to threespine stickleback fish (Supplemental Fig. S8).
Non-protein-coding isoforms are enriched for long noncoding RNAs
We identified 8374 non-protein-coding isoforms (32% of the 26,432 total isoforms). This fraction was much larger than the proportion of non-protein-coding isoforms currently annotated in the Ensembl transcriptome (10%; 2767 non-protein-coding isoforms of 29,245 total isoforms), indicating long-read sequencing may have captured a broader sampling of regulatory noncoding RNAs (ncRNAs). We first examined whether any of our isoforms overlapped with currently annotated regulatory ncRNAs. We recovered 29% of the previously annotated Ensembl ncRNAs in the Iso-Seq transcriptome. This low percentage of previously annotated ncRNAs recovered in our data set is likely owing to the limited number of organs we sequenced. Of the 2767 annotated Ensembl ncRNAs, we found only 209 exactly matched the Iso-Seq annotations (from the Ensembl isoform match: non-protein-coding category) (Table 1), and 582 partially overlapped an existing ncRNA annotation (distributed among the Ensembl isoform match: non-protein-coding category and the novel isoform: non-protein-coding category) (Table 1).
We characterized the remaining novel non-protein-coding isoforms and genes (7583 total distributed among the novel gene: non-protein-coding and novel isoform: non-protein-coding categories) (Table 1) by overall length and genomic location. Short ncRNAs are under 200 bp in length and include miRNAs, endo-siRNAs, and piRNAs (for reviews, see Farazi et al. 2008; Pauli et al. 2011). Long ncRNAs (lncRNAs) are >200 bp in length (Mercer et al. 2009). A majority of the novel short and long ncRNAs we identified were classified as intergenic (short ncRNAs: 306 isoforms, 69%; lncRNAs: 4135 isoforms, 58%). Far fewer ncRNAs were intronic (short ncRNAs: 44 isoforms, 10%; lncRNAs: 584 isoforms, 8%) or antisense (short ncRNAs: 27 isoforms; 6%; lncRNAs: 498, 7%). There were 69 (16%) short ncRNAs and 1920 (27%) lncRNAs that did not fall into these three regions. These uncategorized ncRNAs did not overlap with known ncRNAs but did overlap with other annotations.
Many isoforms have sex-specific alternative splicing
Alternative splicing plays a key role in increasing protein diversity using a limited number of genes. Isoforms of the same gene can regulate specific developmental and physiological processes (Graveley 2001; Baralle and Giudice 2017). Alternative splicing is important for sex determination in Drosophila (Burtis and Baker 1989) and for the development of sex-specific organs in other species (Telonis-Scott et al. 2009; Gibilisco et al. 2016; Planells et al. 2019). However, the overall importance of sex-specific alternative splicing remains largely underexplored (McIntyre et al. 2006) because detecting full-length isoforms from short-read RNA-seq is difficult. We searched for evidence of sex-specific alternative splicing among the somatic and gonadal samples in our data set. Using the female and male transcriptomes (Supplemental Table S1), we found that a substantial number of isoforms were specific to one sex. Of genes expressed in both sexes (4842 total genes), we found 1590 (33%) had female-specific isoforms and 2103 (43%) had male-specific isoforms (Supplemental File S7). In total, there were 2363 female-specific isoforms and 3664 male-specific isoforms. Of these isoforms, nearly half showed alternative splicing in only one sex (female specific: 1146 isoforms, 49%; male specific: 1531 isoforms, 42%). The remainder of isoforms had alternate TSSs/TTSs (female specific: 425 isoforms, 18%; male specific: 968 isoforms, 26%) or showed both alternative splicing and alternate TSSs/TTSs (female specific: 792 isoforms, 34%; male specific: 1165 isoforms, 32%).
We explored whether the alternative sex-specific isoforms we identified were driven by the inclusion of the gonads by analyzing the male and female somatic transcriptomes separately (brain, liver, and pronephros combined) (Supplemental Table S1). After removal of the gonads, we recovered a similar number of alternative isoforms in females (2218, 94% of the total sex-specific isoforms recovered from all samples combined) but a reduced number from males (2579, 70% of the total sex-specific isoforms recovered from all samples combined). This suggests that the ovary transcriptome does not contribute greatly to sex-specific alternative isoforms. The testis transcriptome, on the other hand, contains many genes with isoforms unique to males, suggesting this organ has a much greater transcriptional complexity.
We verified that the sex specificity we found among the isoforms was not simply owing to variation in expression among the organs. If an isoform was only expressed in a single organ, there would be a greater chance to falsely categorize the isoform as sex specific if this organ was not sequenced to sufficient coverage in the other sex. Our saturation analysis suggested variability among organs was not driven by an inadequate sequencing depth, but we also explored this by quantifying the expression level of all sex-specific alternatively spliced isoforms in the female and male organs. Across female-specific isoforms, 88.3% of the isoforms were expressed in all four female samples (Supplemental Fig. S9). In males, 89.1% of the isoforms were expressed across all four male samples (Supplemental Fig. S9). This indicates that most sex-specific isoforms are expressed across multiple organs and are likely robust to sampling artifacts.
Sex-biased genes are often enriched on sex chromosomes (for reviews, see Ellegren and Parsch 2007; Dean and Mank 2014). Gene expression has revealed that X Chromosomes can become feminized over time (Leder et al. 2010; White et al. 2015). However, feminization in the context of alternative isoforms has not been explored. We found female-specific isoforms were highly enriched on the X Chromosome compared with the autosomes (X Chromosome: 160; average autosomes: 98.2; Fisher's exact test; P < 0.001) (Supplemental Table S6). Male-specific isoforms, on the other hand, were under-enriched (X Chromosome: 111; average autosomes: 159.9; Fisher's exact test; P < 0.001) (Supplemental Table S6). Our results highlight feminization of the X Chromosome also involves the evolution of female-specific isoforms.
The testis has the greatest number of alternative isoforms
More than 25% of all genes in the male pronephros, female pronephros, and testis had two or more isoforms per gene (Fig. 4A). For the remaining organs, only 5%–15% of all genes had more than two isoforms. The pronephros samples from both sexes had the largest percentage of novel isoforms (female pronephros: 57%; male pronephros: 55%) (Fig. 4B). Most of the isoforms were predicted to be protein-coding (Supplemental Fig. S10).
Figure 4.

Novel isoforms are found across all samples and sexes. (A) More than 25% of genes in the testis and pronephros had more than one isoform. For the remaining organs, <15% of the genes had more than one isoform. (B) Over half of the isoforms identified in the pronephros samples are novel isoforms. The testis has the next largest count of novel isoforms.
To determine if an organ produced more alternative transcripts relative to the other organs, we limited the comparison to genes that were expressed in all organs. Among these genes, both the testis and ovary had the largest number of alternative transcripts (testis: 787 isoforms, 47%; ovary: 145 isoforms, 28%) (Supplemental Table S7; Supplemental File S8). A majority of these transcripts were alternatively spliced (brain: 74%; liver: 68%; pronephros: 66%; testis: 44%; ovary: 53%) (Supplemental Table S7). The testis had the largest proportion of alternate TSSs/TTSs or both alternative splicing and alternate TSSs/TTSs (Supplemental Table S7). These results suggest that although the pronephros has the largest percentage of organ-specific isoforms, this was largely driven by organ-specific gene expression.
We explored whether the stickleback testis showed any expression patterns in common with the mammalian testis. In mammals, the testis is the most transcriptionally complex, expressing more protein-coding genes than any other organ (Ramsköld et al. 2009). Using the organ-specific alternatively spliced isoforms, we observed a similar pattern in threespine stickleback fish, where the testis had more protein-coding genes compared with all organs except for the pronephros (Supplemental Table S8). The mammalian testis also has a disproportionately large number of lncRNAs (Soumillon et al. 2013). However, we found that the threespine stickleback testis had the smallest number of lncRNAs (Supplemental Table S8). Intron retention is the most common type of splicing within the mammalian testis (Soumillon et al. 2013). From the SQANTI categories, full or partial intron retention can be observed in NIC isoforms as well as genic isoforms. We found that the threespine stickleback testis had the lowest percentage of intron retention relative to other organs (testis: 16.5%; ovary: 27.4%; pronephros: 50.7%; liver: 54.7%, brain: 43.6%). We also expanded our analysis to identify isoforms overlapping Ensembl annotations with known functions in spermatogenesis, using the following search terms: meiosis, spindle, sperm, male germ line, and recombination. We found that 36 isoforms (6.2%) contained at least one of these terms (Supplemental Table S9).
Gonad-specific isoforms are enriched on the Y Chromosome but not the X Chromosome
We investigated whether the Y Chromosome (Peichel et al. 2020) had accumulated male-specific isoforms. Using all male organs, we identified 961 Y-specific isoforms (Supplemental Table S4). Y Chromosomes tend to accumulate genes important for spermatogenesis (Skaletsky et al. 2003; Murphy et al. 2006; Hughes et al. 2010, 2020; Paria et al. 2011; Soh et al. 2014; Janečka et al. 2018). We therefore explored whether testis-specific isoforms were enriched on the Y Chromosome of threespine stickleback fish. Among the 146 testis-specific genes detected genome-wide, there was a strong enrichment on the Y Chromosome (44 of the 146; 30.1%; Fisher's exact test; P < 0.001). These 44 genes had 72 isoforms (Supplemental Tables S10, S11).
We also examined the distribution of ovary-specific genes to look for enrichment on the X Chromosome (Supplemental Table S10). There were 140 ovary-specific genes with alternatively spliced isoforms. Unlike testis-specific genes on the Y Chromosome, ovary-specific genes were not enriched on the X Chromosome (seven of 140; 5%; Fisher's exact test; P = 0.591) In addition, there were only eight ovary-specific isoforms on the X Chromosome from the following genes: chtf8 (two isoforms), arf5, kti12, imo2, slc25a44a, ppcdc, and hmg20a.
Discussion
PacBio Iso-Seq greatly improves the gene annotations in threespine stickleback
Using long-read sequencing of several organ transcriptomes, we refined the existing isoform annotations across the threespine stickleback genome, adding previously undocumented isoforms, modifying existing splice junctions, and correcting previous estimates of TSSs and TTSs. The modified splice junctions were highly accurate, verified through deep RNA-seq. TSSs and TTSs may still have some error in their exact location as the clustering algorithm used by Iso-Seq3 allows for 100 bp of variability at the 5′ end and 30 bp of variability at the 3′ end of the transcript. Transcripts with start or end positions within this range are collapsed into a single isoform, creating a small window of possible TSS and TTS locations (see Supplemental Methods: Long-Read RNA Alignment and Isoform Identification). Despite this potential variability, we show many of the Iso-Seq TSSs were accurate, confirmed by patterns of accessible chromatin from ATAC-seq. Correct TSSs/TTSs are particularly important for future work in threespine stickleback fish in understanding gene regulation. Additional work mapping accessible chromatin with ATAC-seq in multiple organs will be useful to further refine annotations.
We detected many new ncRNAs, similar to patterns seen in other systems using long-read technologies (Kuo et al. 2017). These ncRNAs were found across all samples, where the brain contained the highest percentage. ncRNAs are known to perform a variety of functions in the cell, including housekeeping and regulatory functions, and contain ribosomal RNA and transfer RNAs (for reviews, see Jacquier 2009; Pauli et al. 2011). lncRNAs were previously reported to be important for the evolution of the human brain and are associated with specific regions of the brain in mice (Mercer et al. 2008). We also found lncRNAs were prevalent in pronephros samples. The teleost fish pronephros is an integral component of immune response, containing cytokine-producing lymphoid cells (for review, see Geven and Klaren 2017). In mammals, lncRNAs are important in the development of immune cell lineages (Atianand et al. 2017; for review, see Ahmad et al. 2020). Functional characterization will be necessary to determine if these lncRNAs have a similar role in the threespine stickleback pronephros.
Although we captured >50% of complete Metazoan BUSCO orthologs, our Iso-Seq transcriptome did not approach the completeness within the Ensembl transcriptome. This is not surprising as we only sequenced five organs, whereas the Ensembl transcriptome compiles data across a wider representation of organs and also incorporates protein homology from other species to form gene predictions. Similar patterns of reduced completeness have been reported in other species in which only a few organs were examined (Workman et al. 2018; Minio et al. 2019). We showed that increasing sequencing depth at an individual sample would not increase the total number of genes and isoforms detected in our data set. However, we did see an underrepresentation of short isoforms in our Iso-Seq transcriptome relative to the Ensembl transcriptome (<2 kb in length). Long-read sequencing is biased against short transcripts (Byrne et al. 2019; Amarasinghe et al. 2020), suggesting part of the incompleteness may be a technical artifact. To survey transcriptome diversity at a greater number of genes, future work focused on additional organs will be necessary. Additionally, measures should be taken during library preparation to prevent bias against short transcripts through enrichment for longer transcripts.
Sex-specific alternative transcripts are ubiquitous across organs in threespine stickleback
We found >30% of alternative transcripts annotated in the Iso-Seq transcriptome were present in only males or females, regardless of organ. Alternative transcripts may therefore be a common mechanism to achieve sex-specific functions across organs in addition to sex-biased gene expression. Sex-specific alternative transcripts have been documented in Drosophila, albeit among a smaller proportion of genes than we observed in threespine stickleback fish (McIntyre et al. 2006; Telonis-Scott et al. 2009; Chang et al. 2011; Gibilisco et al. 2016). These surveys used exon-specific microarrays or short-read RNA-seq, raising the possibility that the degree of alternative splicing was underestimated owing to limitations in the sequencing technologies. This could also be a species-specific phenomenon, in which alternative transcripts are more widespread among genes in threespine stickleback fish, including those that are processed in a sex-specific manner. Some surveys have suggested that there is a greater number of genes with alternative transcripts in vertebrates compared with invertebrates (Kim et al. 2007). Additional long-read sequencing of transcriptomes will help clarify how extensive alternative transcripts are among taxa.
Sex chromosomes evolve sex-biased gene content owing to different selection pressures in males and females (Rice 1984). The X Chromosome is transmitted two-thirds of the time through females, leading to a favorable environment for the accumulation of female-beneficial mutations and loss of mutations detrimental to males. This has led to the feminization of the X Chromosome in many species (Reinke et al. 2000; Parisi et al. 2003; Gurbich and Bachtrog 2008; Reinius et al. 2012). Although this has been extensively explored in the context of differential gene expression, a detailed characterization of sex-biased alternative isoforms has not been conducted on sex chromosomes. We found that female-specific isoforms were enriched on the X Chromosome but not male isoforms. This suggests that similar selection pressures may be acting to feminize transcript processing on the X Chromosome. Additional characterization will be necessary to determine the function of these isoforms in female development.
Overall transcriptome complexity varies among threespine stickleback organs
Transcriptome complexity can be defined as the number of expressed genes, by transcript diversity, and through gene expression differences (Ramsköld et al. 2009). In mammals, the brain is one of the most transcriptionally complex organs, with the largest number of isoforms and organ-specific alternative splicing events (Xu et al. 2002; Kan et al. 2005; Barbosa-Morais et al. 2012; Mele et al. 2015). Unlike mammals, we found the threespine stickleback brain has relatively low complexity compared with that of other organs. This difference may be even more striking considering the mammalian brain transcriptome was surveyed using short-read RNA-seq, which can underestimate the total number of isoforms (Bryant et al. 2012; Steijger et al. 2013; Conesa et al. 2016; Wang et al. 2016a). This large difference in overall brain transcriptome complexity is likely owing to the increased number of neuronal cell classes that has accompanied mammalian evolution (for review, see Northcutt 2002). Indeed, distinct patterns of alternative splicing have been found for many of the neuronal cell types in the mammalian brain (Zhang et al. 2014).
The testis showed a greater degree of transcriptome complexity in the threespine stickleback, relative to the brain. Across mammals, the testis consistently shows one of the highest transcriptome complexities, outside of the brain (Xu et al. 2002; Kan et al. 2005; Ramsköld et al. 2009; Barbosa-Morais et al. 2012; Schmid et al. 2013; Soumillon et al. 2013). This pattern may be explained by the different cell types present during continuous spermatogenesis (Schulz et al. 2010). In addition to support cells, adult testes contain uninterrupted waves of spermatogenesis, with spermatogonia, spermatocytes, spermatids, and spermatozoa present at any given time. Mammalian testes have more protein-coding genes compared with other organs (Ramsköld et al. 2009) with the greatest number of expressed protein-coding genes at the early stages of spermatogenesis (spermatogonia), as well as within the supporting Sertoli cells (Soumillon et al. 2013). During the later stages of mammalian spermatogenesis (spermatids and spermatozoa), cells are enriched for lncRNAs as well as splice variants with retained introns (Soumillon et al. 2013). Threespine stickleback fish offer an interesting comparison to these patterns as they undergo synchronous spermatogenesis rather than continuous. In this form of spermatogenesis, a majority of the cells in the testes are at the same stage (Craig-Bennett 1931; Borg and Van Veen 1982). The juvenile testes we sequenced contained cells actively undergoing meiosis (spermatocytes) as well as spermatogonia and support cells. Consistent with the mammalian patterns, we found protein-coding genes were highly expressed in these early cell types. However, we did not see an enrichment of lncRNAs and isoforms with intron retention. In mice, alternative splicing has an important role in meiosis, affecting meiotic progression (Schmid et al. 2013). Key proteins involved in early meiosis and spermatogenesis also have multiple isoforms such as spo11, meig1, and mns1 (Bellani et al. 2010; Kauppi et al. 2011). We found at least 36 isoforms with predicted functions in meiosis. This characterization is likely an underestimate as we were limited to the existing annotations in the Ensembl database, and we did not query the large number of novel genes identified by SQANTI. These isoforms will require further functional characterization to clarify the extent alternative isoforms are involved in regulating meiosis in teleost fish.
The high transcriptome complexity in the pronephros is intriguing as the pronephros is present across vertebrates but only persists into adulthood in amphibians and fish (Smyth et al. 2017). Therefore, very little is known about this organ. In fish, the nephritic tissue degenerates over time, and the pronephros functions as part of the immune system (for review, see Geven and Klaren 2017). In mammals, transcriptome complexity of the immune system is high (for review, see Schaub and Glasmacher 2017) but often is below levels observed in testes and brain (Kan et al. 2005; Brawand et al. 2011; Soumillon et al. 2013; Mele et al. 2015). The high transcriptome complexity we observed in the pronephros may be a unique feature of this organ and could indicate the presence of a more heterogeneous cell population or a more diverse set of isoforms among fewer cell types. More work is necessary to fully understand the function of the pronephros and why the transcriptome of this organ is so diverse.
Methods
Ethics statement
All procedures using threespine stickleback fish were approved by the University of Georgia Animal Care and Use Committee (protocol A2018 10-003-A8).
Total RNA extraction and short-read and long-read sequencing
All samples were obtained from laboratory-reared threespine stickleback fish, originally collected from the Japanese Pacific Ocean population (Akkeshi, Japan). The fish were reared under a 16-h light/8-h dark light cycle mimicking the light cycle during the breeding season of wild threespine stickleback fish. For all samples, the entire organ was collected. Brain (including the olfactory bulb and excluding the pituitary) and liver samples were dissected from one adult male (1 yr old, 6.2 cm in standard length) and one adult female fish (1 yr old, 6.3 cm in standard length). The pronephros or head kidney samples were dissected from a separate adult male (1 yr old, 6.1 cm in standard length) and female fish (1 yr old, 6.1 cm in standard length). Gonads were dissected from a juvenile male (6 mo old, 4.6 cm in standard length) and a juvenile female (6 mo old, 4.8 cm in standard length). We selected juvenile stages to capture gonads that were actively undergoing meiosis (Craig-Bennett 1931; Borg and Van Veen 1982). Total RNA was extracted from all samples using TRIzol:chloroform RNA extraction, following the manufacturer's recommended protocols (Invitrogen). RNA from all samples was used for both the Iso-Seq library preparation and the Illumina strand-specific RNA library preparation. Iso-Seq library preparation and sequencing was completed at the Georgia Genomics and Bioinformatics Core (University of Georgia). Briefly, the Iso-Seq library preparation was completed using the SMRTbell template prep kit 1.0 (100-259-100), sequel binding kit 3.0 (101-613-900), and sequel sequencing plate 3.0 (101-613-700). Transcripts were selected using a standard bead concentration (1.6×) with the center of the transcript length distribution falling ∼2 kb. All samples were sequenced using a PacBio Sequel 1 machine for 26 h, using two SMRT cells per sample with 8 pM loading concentration. Illumina strand-specific RNA library preparation and sequencing was completed by GENEWIZ. Strand-specific libraries were sequenced on an Illumina HiSeq (2 × 150 bp).
Nuclei isolation and ATAC-seq library preparation
Liver samples were collected from two juvenile males (∼4.4 cm in standard length) and two juvenile females (∼4.3 cm in standard length), originally collected from Lake Washington. ATAC-seq library preparation was performed using previously established protocols (Lu et al. 2017) and primers (Supplemental Table S12; Supplemental Methods). ATAC-seq libraries were sequenced on Illumina NextSeq (2 × 150 bp; Georgia Genomics and Bioinformatics Core).
Long-read RNA alignment and isoform identification
The eight samples produced an average of 40.3 million raw subreads per sample (583 gigabytes) (Supplemental Table S1). We analyzed the raw subreads following the Iso-Seq3 pipeline (v3.1; https://github.com/PacificBiosciences/IsoSeq). CCSs were created from raw subreads, and the cDNA primers were removed using lima (Supplemental Methods). Nearly 70% of the CCS reads passed Lima default filters (Supplemental Table S1). Full-length reads were then filtered, clustered, and polished (Iso-Seq3, v3.1) (Supplemental Methods). The polished high-quality reads were aligned to the threespine stickleback genome (Ensembl build 97) (Jones et al. 2012b; Aken et al. 2016) using minimap2 (v2.13) with the following parameters: -ax splice -uf –secondary = no -C5 (Li 2018). We also aligned the high-quality reads using deSALT (v1.5.6) with the following parameters: -x ccs -T (Liu et al. 2019). Redundant isoforms were removed before running SQANTI isoform characterization. Alignments from both deSALT and minimap2 were characterized by SQANTI.
An in-depth characterization of isoforms and removal of artifacts was completed using SQANTI (Tardaguila et al. 2018). SQANTI classified isoforms into nine different descriptors (see Supplemental Methods). These nine categories were cataloged into three broad classes for the purposes of this study. If an isoform did not match a known Ensembl gene annotation, this isoform was classified as a novel gene. If an isoform matched a known Ensembl gene but represented a new isoform, it was classified as a novel isoform. Lastly, if an isoform matched both an Ensembl gene and an Ensembl isoform annotation, it was classified as an Ensembl isoform match. Isoform characterization was completed using sqanti_qc.py, and filtering was completed using sqanti_filter.py. The filtered data set was rerun through sqanti_qc.py for the final characterization. The SQANTI filtered isoforms were used for the remaining analyses.
Benchmarking universal single-copy orthologs
To assess transcriptome completeness, we used benchmarking universal single-copy orthologs (BUSCO, v4.0.6) (Simão et al. 2015; Seppey et al. 2019). BUSCO examines predicted genome annotations for completeness by using single-copy orthologs shared among Metazoans (see Supplemental Methods). All BUSCO analyses were run with the same parameters.
Assessing the completeness of each transcriptome
To further assess whether our samples were sequenced to an adequate depth, we used a subsampling approach (Workman et al. 2018). CCS reads were subsampled and then compared with the nucleotide sequences from the full-organ transcriptome using BLAST (v2.2.6, BLASTN, default parameters) (Altschul et al. 1990, 1997; Camacho et al. 2009). The BLAST results for each sample were filtered using custom Python scripts. All BLAST alignments that covered at least 50% of the subsampled CCS read and at least 50% of an isoform from the full sample transcriptome were retained. The total proportion of isoforms detected in each subsample compared with the full-sample transcriptome was calculated.
Comparisons between transcriptomes
We assembled several different transcriptomes using SMRTlink (v. 6). To examine the differences between the Ensembl transcriptome and the transcriptome produced using Iso-Seq, we used all eight samples (hereafter referred to as the Iso-Seq transcriptome). To examine sex specificity, all five organs (brain, liver, pronephros, testis, and ovary) were combined for each sex (hereafter referred to as the female transcriptome and the male transcriptome). We also examined sex specificity in somatic organs, combining only the brain, liver, and pronephros of each sex (hereafter referred to as the somatic female transcriptome and the somatic male transcriptome). Lastly, we compared each sample's transcriptome. All transcriptomes were subject to the same Iso-Seq3 pipeline beginning with the removal of cDNA primers with Lima.
We assigned universal isoform identifications for the full Iso-Seq transcriptome. BLAST was used to compare isoforms among individual organ transcriptomes (v2.2.6, BLASTN, default parameters) (Altschul et al. 1990, 1997; Camacho et al. 2009). Duplicate isoforms within the full Iso-Seq transcriptome were first removed by identifying any isoforms that aligned exactly to another isoform (i.e., BLAST alignments were identical between the isoforms). This removed 1139 isoforms from the full Iso-Seq transcriptome. The other transcriptomes (i.e., individual samples, female transcriptome, male transcriptome, somatic female transcriptome, or somatic male transcriptome) were compared with the full transcriptome with BLAST. The BLAST results were filtered using custom Python scripts. A positive alignment was identified if at least 50% of the query sequence matched at least 60% of the subject sequence. Query isoforms that matched more than one subject isoform were collapsed to a single isoform, keeping the longest alignment. Any isoforms that did not meet these criteria were discarded.
Aligning to the Y Chromosome
All male organs were aligned to the threespine stickleback reference Y Chromosome assembly (Peichel et al. 2020) separately to identify Y-specific isoforms. The same Iso-Seq pipeline was run as for the rest of the genome. The individual male organs were also aligned to the Y assembly to identify testis-specific Y Chromosome transcripts.
ATAC-seq genome coverage at TSSs
Residual adapter sequences from the Nextera primers were trimmed using Trimmomatic (v0.36) (Bolger et al. 2014). Trimmed reads were aligned to the revised threespine stickleback genome (Nath et al. 2021) using Bowtie 2 (v2.3.5) (Langmead and Salzberg 2012). The read coverage per base pair was calculated using BEDTools (v2.26, genomecov -d) (Quinlan and Hall 2010). Custom Python scripts were used to average the read coverage across a 4-kb window surrounding Ensembl and Iso-Seq TSSs.
Characterizing ncRNAs by size and genomic location
ncRNAs were characterized by size and genomic location using custom Python scripts (Supplemental Methods). All ncRNAs did not have detectable protein-coding potential. ncRNAs are generally classified based on overall length: short ncRNAs are <200 bp, and lncRNAs are >200 bp (Jacquier 2009; Pauli et al. 2011). We then separated ncRNAs into these two length categories as well as three main classes: intergenic, intronic, or antisense. Any remaining ncRNAs were added to an unknown category.
Novel gene protein domain search through InterProScan
We used InterProScan (v.5.32) (Jones et al. 2014) to identify protein domains of novel proteins. The amino acid sequences from all novel protein-coding genes from the full Iso-Seq transcriptome were used. All available databases in IntroProScan were used. InterProScan was run with default parameters and GO terms, and pathway information was recorded.
GO analysis
GO enrichment analysis was completed using custom Python scripts (see Supplemental Methods). P-values were adjusted for multiple testing using a Bonferroni correction based on the total number of observed GO terms in each set. Enriched GO terms were visualized using Web Gene Ontology Annotation Plot (WEGO) (Ye et al. 2006, 2018).
Data access
The Iso-Seq and short-read RNA-seq data, as well as the liver ATAC-seq data, generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA633846 and PRJNA667175, respectively. The Iso-Seq transcriptome has also been submitted to the TSA repository (https://www.ncbi.nlm.nih.gov/genbank/tsa/) under accession GJAP00000000 (V GJAP01000000). All custom scripts are available as Supplemental Code and at GitHub (https://github.com/ASNaftaly/IsoSeq3_Stickleback).
Supplementary Material
Acknowledgments
This research was funded by the National Science Foundation IOS 1645170 (M.A.W.), the National Science Foundation MCB 1943283 (M.A.W.), the Office of the Vice President of Research at the University of Georgia (UGA; M.A.W.), the Jan and Kirby Alton Fellowship (Department of Genetics, UGA, to A.S.N.), and the Rosemary Grant Award (Society for the Study of Evolution to A.S.N.). We also thank Robert Schmitz and his laboratory at UGA for assistance and reagents for the ATAC-seq protocol. PacBio Sequencing and library preparations were conducted at the Georgia Genomics and Bioinformatics Core at UGA.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.274282.120.
Competing interest statement
The authors declare no competing interests.
References
- Abdel-Ghany SE, Hamilton M, Jacobi JL, Ngam P, Devitt N, Schilkey F, Ben-Hur A, Reddy AS. 2016. A survey of the sorghum transcriptome using single-molecule long reads. Nat Commun 7: 11706. 10.1038/ncomms11706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad I, Valverde A, Ahmad F, Naqvi AR. 2020. Long noncoding RNA in myeloid and lymphoid cell differentiation, polarization and function. Cells 9: 269. 10.3390/cells9020269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, Garcia Giron C, Hourlier T, et al. 2016. The Ensembl gene annotation system. Database (Oxford) 2016: baw093. 10.1093/database/baw093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarasinghe SL, Su S, Dong X, Zappia L, Ritchie ME, Gouil Q. 2020. Opportunities and challenges in long-read sequencing data analysis. Genome Biol 21: 30. 10.1186/s13059-020-1935-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Reyes A, Huber W. 2012. Detecting differential usage of exons from RNA-seq data. Genome Res 22: 2008–2017. 10.1101/gr.133744.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atianand MK, Caffrey DR, Fitzgerald KA. 2017. Immunobiology of long noncoding RNAs. Annu Rev Immunol 35: 177–198. 10.1146/annurev-immunol-041015-055459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baralle FE, Giudice J. 2017. Alternative splicing as a regulator of development and tissue identity. Nat Rev Mol Cell Biol 18: 437–451. 10.1038/nrm.2017.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber I, Nettlseship S. 2010. From “trash fish” to supermodel: the rise and rise of the threespine stickleback in evolution and ecology. Biologist 57: 15–21. [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. 2012. The evolutionary landscape of alternative splicing in vertebrate species. Science 338: 1587–1593. 10.1126/science.1230612 [DOI] [PubMed] [Google Scholar]
- Bell MA, Foster SA. 1994. The evolutionary biology of the threespine stickleback. Oxford University Press, Oxford. [Google Scholar]
- Bellani MA, Boateng KA, McLeod D, Camerini-Otero RD. 2010. The expression profile of the major mouse SPO11 isoforms indicates that SPO11β introduces double strand breaks and suggests that SPO11α has an additional role in prophase in both spermatocytes and oocytes. Mol Cell Biol 30: 4391–4403. 10.1128/MCB.00002-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blekhman R, Marioni JC, Zumbo P, Stephens M, Gilad Y. 2010. Sex-specific and lineage-specific alternative splicing in primates. Genome Res 20: 180–189. 10.1101/gr.099226.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg B, Van Veen T. 1982. Seasonal effects of photoperiod and temperature on the ovary of the three-spined stickleback, Gasterosteus aculeatus L. Can J Zool 60: 3387–3393. 10.1139/z82-428 [DOI] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478: 343–348. 10.1038/nature10532 [DOI] [PubMed] [Google Scholar]
- Brown JB, Boley N, Eisman R, May GE, Stoiber MH, Duff MO, Booth BW, Wen J, Park S, Suzuki AM, et al. 2014. Diversity and dynamics of the Drosophila transcriptome. Nature 512: 393–399. 10.1038/nature12962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DW, Priest HD, Mockler TC. 2012. Detection and quantification of alternative splicing variants using RNA-seq. In RNA abundance analysis: methods and protocols (ed. Jin H, Gassmann W). Springer, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burtis KC, Baker BS. 1989. Drosophila doublesex gene controls somatic sexual differentiation by producing alternatively spliced mRNAs encoding related sex-specific polypeptides. Cell 56: 997–1010. 10.1016/0092-8674(89)90633-8 [DOI] [PubMed] [Google Scholar]
- Byrne A, Cole C, Volden R, Vollmers C. 2019. Realizing the potential of full-length transcriptome sequencing. Philos Trans R Soc Lond B Biol Sci 374: 20190097. 10.1098/rstb.2019.0097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10: 421. 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PL, Dunham JP, Nuzhdin SV, Arbeitman MN. 2011. Somatic sex-specific transcriptome differences in Drosophila revealed by whole transcriptome sequencing. BMC Genomics 12: 364. 10.1186/1471-2164-12-364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Furtado A, Henry RJ. 2017. Long-read sequencing of the coffee bean transcriptome reveals the diversity of full-length transcripts. Gigascience 6: gix086. 10.1093/gigascience/gix086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szczesniak MW, Gaffney DJ, Elo LL, Zhang X, et al. 2016. A survey of best practices for RNA-seq data analysis. Genome Biol 17: 13. 10.1186/s13059-016-0881-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly CF, Wakefield J, Akey JM. 2014. Evolution and genetic architecture of chromatin accessibility and function in yeast. PLoS Genet 10: e1004427. 10.1371/journal.pgen.1004427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V, Angelini C, De Feis I, Ciccodicola A. 2010. Uncovering the complexity of transcriptomes with RNA-Seq. J Biomed Biotechnol 2010: 853916. 10.1155/2010/853916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig-Bennett A. 1931. The reproductive cycle of the three-spined stickleback, Gasterosteus aculeatus, Linn, Vol. 219. Harrison and Sons, Ltd., London. [Google Scholar]
- Dean R, Mank JE. 2014. The role of sex chromosomes in sexual dimorphism: discordance between molecular and phenotypic data. J Evol Biol 27: 1443–1453. 10.1111/jeb.12345 [DOI] [PubMed] [Google Scholar]
- Deslattes Mays A, Schmidt M, Graham G, Tseng E, Baybayan P, Sebra R, Sanda M, Mazarati JB, Riegel A, Wellstein A. 2019. Single-molecule real-time (SMRT) full-length RNA-sequencing reveals novel and distinct mRNA isoforms in human bone marrow cell subpopulations. Genes (Basel) 10: 253. 10.3390/genes10040253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. 2007. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet 8: 689–698. 10.1038/nrg2167 [DOI] [PubMed] [Google Scholar]
- Farazi TA, Juranek SA, Tuschl T. 2008. The growing catalog of small RNAs and their association with distinct Argonaute/Piwi family members. Development 135: 1201–1214. 10.1242/dev.005629 [DOI] [PubMed] [Google Scholar]
- Geven EJW, Klaren PHM. 2017. The teleost head kidney: integrating thyroid and immune signalling. Dev Comp Immunol 66: 73–83. 10.1016/j.dci.2016.06.025 [DOI] [PubMed] [Google Scholar]
- Gibilisco L, Zhou Q, Mahajan S, Bachtrog D. 2016. Alternative splicing within and between drosophila species, sexes, tissues, and developmental stages. PLoS Genet 12: e1006464. 10.1371/journal.pgen.1006464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazer AM, Killingbeck EE, Mitros T, Rokhsar DS, Miller CT. 2015. Genome assembly improvement and mapping convergently evolved skeletal traits in sticklebacks with genotyping-by-sequencing. G3 (Bethesda) 5: 1463–1472. 10.1534/g3.115.017905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR. 2001. Alternative splicing: increasing diversity in the proteomic world. Trends Genet 17: 100–107. 10.1016/S0168-9525(00)02176-4 [DOI] [PubMed] [Google Scholar]
- Gupta I, Clauder-Münster S, Klaus B, Järvelin AI, Aiyar RS, Benes V, Wilkening S, Huber W, Pelechano V, Steinmetz LM. 2014. Alternative polyadenylation diversifies post-transcriptional regulation by selective RNA-protein interactions. Mol Syst Biol 10: 719. 10.1002/msb.135068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurbich TA, Bachtrog D. 2008. Gene content evolution on the X chromosome. Curr Opin Genet Dev 18: 493–498. 10.1016/j.gde.2008.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T, Wagner F, Sher N, Yanai I. 2012. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep 2: 666–673. 10.1016/j.celrep.2012.08.003 [DOI] [PubMed] [Google Scholar]
- Hendry AP, Peichel CL, Matthews B, Boughman JW, Nosil P. 2013. Stickleback research: the now and the next. Evol Ecol Res 15: 111–141. [Google Scholar]
- Hughes JF, Skaletsky H, Pyntikova T, Graves TA, van Daalen SK, Minx PJ, Fulton RS, McGrath SD, Locke DP, Friedman C, et al. 2010. Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature 463: 536–539. 10.1038/nature08700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Pyntikova T, Koutseva N, Raudsepp T, Brown LG, Bellott DW, Cho TJ, Dugan-Rocha S, Khan Z, et al. 2020. Sequence analysis in Bos taurus reveals pervasiveness of X–Y arms races in mammalian lineages. Genome Res 30: 1716–1726. 10.1101/gr.269902.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquier A. 2009. The complex eukaryotic transcriptome: unexpected pervasive transcription and novel small RNAs. Nat Rev Genet 10: 833–844. 10.1038/nrg2683 [DOI] [PubMed] [Google Scholar]
- Janečka JE, Davis BW, Ghosh S, Paria N, Das PJ, Orlando L, Schubert M, Nielsen MK, Stout TAE, Brashear W, et al. 2018. Horse Y chromosome assembly displays unique evolutionary features and putative stallion fertility genes. Nat Commun 9: 2945. 10.1038/s41467-018-05290-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones FC, Chan YF, Schmutz J, Grimwood J, Brady SD, Southwick AM, Absher DM, Myers RM, Reimchen TE, Deagle BE, et al. 2012a. A genome-wide SNP genotyping array reveals patterns of global and repeated species-pair divergence in sticklebacks. Curr Biol 22: 83–90. 10.1016/j.cub.2011.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, Swofford R, Pirun M, Zody MC, White S, et al. 2012b. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484: 55–61. 10.1038/nature10944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30: 1236–1240. 10.1093/bioinformatics/btu031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z, Garrett-Engele PW, Johnson JM, Castle JC. 2005. Evolutionarily conserved and diverged alternative splicing events show different expression and functional profiles. Nucleic Acids Res 33: 5659–5666. 10.1093/nar/gki834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi L, Barchi M, Baudat F, Romanienko PJ, Keeney S, Jasin M. 2011. Distinct properties of the XY pseudoautosomal region crucial for male meiosis. Science 331: 916–920. 10.1126/science.1195774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H, Lev-Maor G, Ast G. 2010. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 11: 345–355. 10.1038/nrg2776 [DOI] [PubMed] [Google Scholar]
- Kim E, Magen A, Ast G. 2007. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res 35: 125–131. 10.1093/nar/gkl924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37: 907–915. 10.1038/s41587-019-0201-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano J, Mori S, Peichel CL. 2007. Sexual dimorphism in the external morphology of the threespine stickleback (Gasterosteus aculeatus). Copeia 2007: 336–349. 10.1643/0045-8511(2007)7[336:SDITEM]2.0.CO;2 [DOI] [Google Scholar]
- Kotrschal A, Räsänen K, Kristjánsson BK, Senn M, Kolm N. 2012. Extreme sexual brain size dimorphism in sticklebacks: a consequence of the cognitive challenges of sex and parenting? PLoS One 7: e30055. 10.1371/journal.pone.0030055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo RI, Tseng E, Eory L, Paton IR, Archibald AL, Burt DW. 2017. Normalized long read RNA sequencing in chicken reveals transcriptome complexity similar to human. BMC Genomics 18: 323. 10.1186/s12864-017-3691-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder EH, Cano JM, Leinonen T, O'Hara RB, Nikinmaa M, Primmer CR, Merilä J. 2010. Female-biased expression on the X chromosome as a key step in sex chromosome evolution in threespine sticklebacks. Mol Biol Evol 27: 1495–1503. 10.1093/molbev/msq031 [DOI] [PubMed] [Google Scholar]
- Leinonen T, Cano JM, Merilä J. 2011. Genetic basis of sexual dimorphism in the threespine stickleback Gasterosteus aculeatus. Heredity (Edinb) 106: 218–227. 10.1038/hdy.2010.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Fang C, Fu Y, Hu A, Li C, Zou C, Li X, Zhao S, Zhang C, Li C. 2018. A survey of transcriptome complexity in Sus scrofa using single-molecule long-read sequencing. DNA Res 25: 421–437. 10.1093/dnares/dsy014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Liu Y, Li J, Guo H, Zang T, Wang Y. 2019. deSALT: fast and accurate long transcriptomic read alignment with de Bruijn graph-based index. Genome Biol 20: 274. 10.1186/s13059-019-1895-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Hofmeister BT, Vollmers C, DuBois RM, Schmitz RJ. 2017. Combining ATAC-seq with nuclei sorting for discovery of cis-regulatory regions in plant genomes. Nucleic Acids Res 45: e41. 10.1093/nar/gkw1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. 2015. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161: 1202–1214. 10.1016/j.cell.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF. 2008. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 18: 1073–1083. 10.1101/gr.078261.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee MD, Wainwright PC. 2013. Sexual dimorphism in the feeding mechanism of threespine stickleback. J Exp Biol 216: 835–840. 10.1242/jeb.074948 [DOI] [PubMed] [Google Scholar]
- McIntyre LM, Bono LM, Genissel A, Westerman R, Junk D, Telonis-Scott M, Harshman L, Wayne ML, Kopp A, Nuzhdin SV. 2006. Sex-specific expression of alternative transcripts in Drosophila. Genome Biol 7: R79. 10.1186/gb-2006-7-8-r79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meers MP, Adelman K, Duronio RJ, Strahl BD, McKay DJ, Matera AG. 2018. Transcription start site profiling uncovers divergent transcription and enhancer-associated RNAs in Drosophila melanogaster. BMC Genomics 19: 157. 10.1186/s12864-018-4510-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, et al. 2015. The human transcriptome across tissues and individuals. Science 348: 660–665. 10.1126/science.aaa0355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. 2008. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci 105: 716–721. 10.1073/pnas.0706729105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Mattick JS. 2009. Long non-coding RNAs: insight into functions. Nat Rev Genet 10: 155–159. 10.1038/nrg2521 [DOI] [PubMed] [Google Scholar]
- Minio A, Massonnet M, Figueroa-Balderas R, Vondras AM, Blanco-Ulate B, Cantu D. 2019. Iso-Seq allows genome-independent transcriptome profiling of grape berry development. G3 (Bethesda) 9: 755–767. 10.1534/g3.118.201008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy WJ, Pearks Wilkerson AJ, Raudsepp T, Agarwala R, Schaffer AA, Stanyon R, Chowdhary BP. 2006. Novel gene acquisition on carnivore Y chromosomes. PLoS Genet 2: e43. 10.1371/journal.pgen.0020043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath S, Shaw DE, White MA. 2021. Improved contiguity of the threespine stickleback genome using long-read sequencing. G3 (Bethesda) 11: jkab007. 10.1093/g3journal/jkab007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcutt RG. 2002. Understanding vertebrate brain evolution. Integ Comp Biol 42: 743–756. 10.1093/icb/42.4.743 [DOI] [PubMed] [Google Scholar]
- Nudelman G, Frasca A, Kent B, Sadler KC, Sealfon SC, Walsh MJ, Zaslavsky E. 2018. High resolution annotation of zebrafish transcriptome using long-read sequencing. Genome Res 28: 1415–1425. 10.1101/gr.223586.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paria N, Raudsepp T, Pearks Wilkerson AJ, O'Brien PC, Ferguson-Smith MA, Love CC, Arnold C, Rakestraw P, Murphy WJ, Chowdhary BP. 2011. A gene catalogue of the euchromatic male-specific region of the horse Y chromosome: comparison with human and other mammals. PLoS One 6: e21374. 10.1371/journal.pone.0021374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Nuttall R, Naiman D, Bouffard G, Malley J, Andrews J, Eastman S, Oliver B. 2003. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science 299: 697–700. 10.1126/science.1079190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A, Rinn JL, Schier AF. 2011. Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet 12: 136–149. 10.1038/nrg2904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peichel CL, Nereng K, Ohgi KA, Cole BL, Colosimo PF, Buerkle CA, Schluter D, Kingsley DM. 2001. The genetic architecture of divergence between threespine stickleback species. Nature 414: 901–905. 10.1038/414901a [DOI] [PubMed] [Google Scholar]
- Peichel CL, Sullivan ST, Liachko I, White MA. 2017. Improvement of the threespine stickleback genome using a Hi-C-based proximity-guided assembly. J Hered 108: 693–700. 10.1093/jhered/esx058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peichel CL, McCann SR, Ross JA, Naftaly AFS, Urton JR, Cech JN, Grimwood J, Schmutz J, Myers RM, Kingsley DM, et al. 2020. Assembly of the threespine stickleback Y chromosome reveals convergent signatures of sex chromosome evolution. Genome Biol 21: 177. 10.1186/s13059-020-02097-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planells B, Gómez-Redondo I, Pericuesta E, Lonergan P, Gutiérrez-Adán A. 2019. Differential isoform expression and alternative splicing in sex determination in mice. BMC Genomics 20: 202. 10.1186/s12864-019-5572-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsköld D, Wang ET, Burge CB, Sandberg R. 2009. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol 5: e1000598. 10.1371/journal.pcbi.1000598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius B, Johansson MM, Radomska KJ, Morrow EH, Pandey GK, Kanduri C, Sandberg R, Williams RW, Jazin E. 2012. Abundance of female-biased and paucity of male-biased somatically expressed genes on the mouse X-chromosome. BMC Genomics 13: 607. 10.1186/1471-2164-13-607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke V, Smith HE, Nance J, Wang J, Van Doren C, Begley R, Jones SJM, Davis EB, Scherer S, Ward S, et al. 2000. A global profile of germline gene expression in C. elegans. Mol Cell 6: 605–616. 10.1016/S1097-2765(00)00059-9 [DOI] [PubMed] [Google Scholar]
- Rice WR. 1984. Sex chromosomes and the evolution of sexual dimorphism. Evolution (N Y) 38: 735–742. 10.1111/j.1558-5646.1984.tb00346.x [DOI] [PubMed] [Google Scholar]
- Rogers TF, Palmer DH, Wright AE. 2020. Sex-specific selection drives the evolution of alternative splicing in birds. Mol Biol Evol 38: 519–530. 10.1093/molbev/msaa242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub A, Glasmacher E. 2017. Splicing in immune cells-mechanistic insights and emerging topics. Int Immunol 29: 173–181. 10.1093/intimm/dxx026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid R, Grellscheid SN, Ehrmann I, Dalgliesh C, Danilenko M, Paronetto MP, Pedrotti S, Grellscheid D, Dixon RJ, Sette C, et al. 2013. The splicing landscape is globally reprogrammed during male meiosis. Nucleic Acids Res 41: 10170–10184. 10.1093/nar/gkt811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz RW, de Franca LR, Lareyre J-J, Le Gac F, Chiarini-Garcia H, Nobrega RH, Miura T. 2010. Spermatogenesis in fish. Gen Comp Endocr 165: 390–411. 10.1016/j.ygcen.2009.02.013 [DOI] [PubMed] [Google Scholar]
- Seppey M, Manni M, Zdobnov EM. 2019. BUSCO: assessing genome assembly and annotation completeness. In Methods in molecular biology, Vol. 1962 (ed. Kollmar M). Humana, New York. [DOI] [PubMed] [Google Scholar]
- Sharon D, Tilgner H, Grubert F, Snyder M. 2013. A single-molecule long-read survey of the human transcriptome. Nat Biotechnol 31: 1009–1014. 10.1038/nbt.2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, Pyntikova T, Ali J, Bieri T, et al. 2003. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423: 825–837. 10.1038/nature01722 [DOI] [PubMed] [Google Scholar]
- Smith CWJ, Patton JG, Nadal-Ginard B. 1989. Alternative splicing in the control of gene expression. Annu Rev Genet 23: 527–577. 10.1146/annurev.ge.23.120189.002523 [DOI] [PubMed] [Google Scholar]
- Smyth IM, Cullen-McEwen LA, Caruana G, Black MJ, Bertram JF. 2017. Development of the kidney: morphology and mechanisms. In Fetal and neonatal physiology (ed. Polin RA, et al. ), Vol. 2, pp. 953–964. Elsevier, Philadelphia. [Google Scholar]
- Soh YQ, Alföldi J, Pyntikova T, Brown LG, Graves T, Minx PJ, Fulton RS, Kremitzki C, Koutseva N, Mueller JL, et al. 2014. Sequencing the mouse Y chromosome reveals convergent gene acquisition and amplification on both sex chromosomes. Cell 159: 800–813. 10.1016/j.cell.2014.09.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H, Barthès P, Kokkinaki M, Nef S, Gnirke A, et al. 2013. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep 3: 2179–2190. 10.1016/j.celrep.2013.05.031 [DOI] [PubMed] [Google Scholar]
- Steijger T, Abril JF, Engström PG, Kokocinski F, Consortium R, Hubbard TJ, Guigó R, Harrow J, Bertone P. 2013. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods 10: 1177–1184. 10.1038/nmeth.2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AD, Pischedda A, Rice WR. 2010. Resolving intralocus sexual conflict: genetic mechanisms and time frame. J Hered 101: S94–S99. 10.1093/jhered/esq011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardaguila M, de la Fuente L, Marti C, Pereira C, Pardo-Palacios FJ, Del Risco H, Ferrell M, Mellado M, Macchietto M, Verheggen K, et al. 2018. SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res 28: 396–411. 10.1101/gr.222976.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telonis-Scott M, Kopp A, Wayne ML, Nuzhdin SV, McIntyre LM. 2009. Sex-specific splicing in Drosophila: widespread occurrence, tissue specificity and evolutionary conservation. Genetics 181: 421–434. 10.1534/genetics.108.096743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Tseng E, Regulski M, Clark TA, Hon T, Jiao Y, Lu Z, Olson A, Stein JC, Ware D. 2016a. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat Commun 7: 11708. 10.1038/ncomms11708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hou J, Quedenau C, Chen W. 2016b. Pervasive isoform-specific translational regulation via alternative transcription start sites in mammals. Mol Syst Biol 12: 875. 10.15252/msb.20166941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Kumar V, Olson A, Ware D. 2019. Reviving the transcriptome studies: an insight into the emergence of single-molecule transcriptome sequencing. Front Genet 10: 384. 10.3389/fgene.2019.00384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Kitano J, Peichel CL. 2015. Purifying selection maintains dosage-sensitive genes during degeneration of the threespine stickleback Y chromosome. Mol Biol Evol 32: 1981–1995. 10.1093/molbev/msv078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman RE, Myrka AM, Wong GW, Tseng E, Welch KC Jr, Timp W. 2018. Single-molecule, full-length transcript sequencing provides insight into the extreme metabolism of the ruby-throated hummingbird Archilochus colubris. Gigascience 7: giy009. 10.1093/gigascience/giy009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Modrek B, Lee C. 2002. Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res 30: 3754–3766. 10.1093/nar/gkf492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, et al. 2020. Ensembl 2020. Nucleic Acids Res 48: D682–D688. 10.1093/nar/gkz966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, et al. 2006. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34: W293–W297. 10.1093/nar/gkl031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Zhang Y, Cui H, Liu J, Wu Y, Cheng Y, Xu H, Huang X, Li S, Zhou A, et al. 2018. WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res 46: W71–W75. 10.1093/nar/gky400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. 2014. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34: 11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Dimont E, Ha T, Swanson DJ, Consortium F, Hide W, Goldowitz D. 2017. Relatively frequent switching of transcription start sites during cerebellar development. BMC Genomics 18: 461. 10.1186/s12864-017-3834-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nyong AT, Shi T, Yang P. 2019. The complexity of alternative splicing and landscape of tissue-specific expression in lotus (Nelumbo nucifera) unveiled by Illumina- and single-molecule real-time-based RNA-sequencing. DNA Res 26: 301–311. 10.1093/dnares/dsz010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.