

Abstract
Understanding how protein function has evolved and diversified is of great importance for human genetics and medicine. Here, we tackle the problem of describing the whole transcript variability observed in several species by generalizing the definition of splicing graph. We provide a practical solution to construct parsimonious evolutionary splicing graphs where each node is a minimal transcript building block defined across species. We show a clear link between the functional relevance, tissue regulation, and conservation of alternative transcripts on a set of 50 genes. By scaling up to the whole human protein-coding genome, we identify a few thousand genes where alternative splicing modulates the number and composition of pseudorepeats. We have implemented our approach in ThorAxe, an efficient, versatile, robust, and freely available computational tool.
Eukaryotes have evolved a transcriptional mechanism that can augment the protein repertoire without increasing genome size. A gene can be transcribed, spliced, and matured into several transcripts by choosing different initiation/termination sites or by selecting different exons (Graveley 2001). Alternative splicing, as well as alternative promoter usage or alternative polyadenylation (hereafter referred to as AS), concerns almost all multiexon genes in vertebrates (Wang et al. 2008), and many organ-specific splicing patterns have diverged rapidly during vertebrate evolution (Barbosa-Morais et al. 2012; Merkin et al. 2012). This mechanism can affect transcript maturation and post-transcriptional regulation or result in protein isoforms (“proteoforms”) with different shapes (Birzele et al. 2008), interaction partners (Yang et al. 2016), and functions (Kelemen et al. 2013; Baralle and Giudice 2017). Its misregulation is associated with the development of cancer, among other diseases (Wang and Cooper 2007; Ward and Cooper 2010; Lim et al. 2011; Scotti and Swanson 2016; Climente-González et al. 2017). Moreover, the influence of natural isoform variations between human populations on disease susceptibility is increasingly recognized (Park et al. 2018). Hence, understanding how AS contributes to protein function diversification is of utmost importance for human genetics and medicine.
In recent years, the advent of high-throughput sequencing technologies like RNA-seq has made possible in-depth surveys of transcriptome complexity (Sultan et al. 2008; Wang et al. 2008). However, evaluating how many of the detected transcripts are translated and functional in the cell remains challenging (Wang et al. 2018). This difficulty has stimulated the development of integrative approaches combining gene annotations, RNA-seq data, and also data generated by other high-throughput techniques (De La Grange et al. 2010; Gonzàlez-Porta et al. 2013; Rodriguez et al. 2013; Ezkurdia et al. 2015; Weatheritt et al. 2016; Tapial et al. 2017; Tranchevent et al. 2017; Denti et al. 2018; Sterne-Weiler et al. 2018; Agosto et al. 2019; Ait-hamlat et al. 2020; de la Fuente et al. 2020; Louadi et al. 2020; Marti-Solano et al. 2020) toward a better characterization of the AS landscape. Recent studies underscore AS functional impact and contribution to protein diversity (Agosto et al. 2019; Marti-Solano et al. 2020).
Evolutionary conservation can arguably serve as a reliable indicator of function. Indeed, we expect splice variants selected over millions of years of evolution to comply with physical and environmental constraints and thus to play a functional role. The classical approach for assessing AS evolutionary conservation first identifies orthologous exons between species and then compares their inclusion/exclusion rates across cell/tissue types. A common practice for orthology detection is the BLAST (Altschul et al. 1990) “all-against-all” methodology (Nichio et al. 2017). When dealing with exons, more specialized protocols based on pairwise genomic sequence alignments (Modrek and Lee 2003; Xing and Lee 2005; Barbosa-Morais et al. 2012; Abascal et al. 2015; Herrero et al. 2016; Mei et al. 2017) or multiple alignments of genomic or protein sequences (Christinat and Moret 2012; Merkin et al. 2012; Szalkowski 2012) have been proposed. Challenges associated with this task include correctly handling large indel events, finding plausible matches for highly divergent sequences, and resolving ambiguities arising from highly similar sequences (e.g., resulting from in-gene duplication) or very short sequences. The alternative usage of orthologous exons may then be investigated using compact representations of transcript variability, such as splicing graphs (Heber et al. 2002), where the nodes represent the exons, and the edges denote exon junctions. Hence, a way to assess AS conservation between two genes would be to compare the environment of their orthologous exons in the two corresponding splicing graphs. However, until now, there exists no method combining these two layers of information.
In this work, we addressed the problem of exon orthology detection in the context of AS. Our specific aims were to develop a novel and general method revisiting splicing graph representation to account for the whole transcript variability observed in a set of species and to apply this method at the protein-coding genome scale to provide, for the first time, granular estimates of AS evolutionary conservation and significantly improve our knowledge on the amount of variations that are functionally relevant.
Results
Evolution-informed model describes transcript variability
Our method maps all transcriptomic information coming from many species on an evolutionary splicing graph, where the nodes represent minimal transcript building blocks defined across species (Fig. 1). Classically, a splicing graph (SG) contains nodes representing exons, that is, genomic intervals, and edges indicating co-occurrences of contiguous exons in a set of transcripts observed for a gene. In the present work, we use a slightly different definition, in which the nodes are the genomic intervals supplemented by their reading frames (Supplemental Methods). In practice, we work with the corresponding translated amino acid sequences (Fig. 1A). Moreover, the nodes may actually represent subexons, because donor or acceptor sites can be located inside an exon (Fig. 1A, n1, and n2). We distinguish the edges induced by the subexon boundaries (Fig. 1A, n1 → n2) from the structural edges arising from intron boundaries (Fig. 1A, n1 → n3).
Figure 1.

Principle of the method. (A) Two transcripts are depicted, in which each gray box represents a genomic interval and contains the corresponding protein sequence. (Below) The minimal SG is shown, with the nodes (n1, n2, n3, n4) corresponding to subexons. The start and end nodes are added for convenience. Each structural edge in red corresponds to some intron, and each induced edge in green corresponds to a junction located inside the initial genomic interval (such as the donor site of exon AEIGV). (B) Close-up view of three SGs corresponding to three orthologous genes coming from human, gorilla, and cow, along with three examples of ESGs summarizing the same information. The nodes in the ESGs represent s-exons, or multiple sequence alignments (MSAs) of exonic regions. The details of the ESG scores, computed from Equation 1, are given in the inset table, with σmatch = 1, σmismatch = −0.5, and σgap = 0 for the MSA scores, and edge penalties σS = 0.5 and σI = 2. The best-scored ESG shows at the same time compactness (parsimony) and good-quality alignments. (C) Main steps of the ESG construction in ThorAxe. The input genes and transcripts are depicted on top, with exons displayed as boxes. ThorAxe first step consists in grouping similar exons together. Here, three clusters are identified, colored in red (1), cyan (2), and blue (3); note that cluster 2 groups to multiple exons in human and cow. Then, subexons are defined based on intra-species transcript variability. For instance, the first exon from gorilla is split into two subexons. The subexons would be the nodes in the species-specific minimal SGs, although the latter are not explicitly computed by ThorAxe. The next step consists in aligning the sequences belonging to each cluster (with some padding “X” between mutually exclusive subexons) and identifying the spliced exons (s-exons) as blocks in the alignment. We keep track of the cluster IDs in the s-exon IDs, to ease interpretability. Finally, ThorAxe builds an ESG in which the nodes are the s-exons. For the sake of clarity, multiedges are visualized as single edges.
Our main contribution is to extend the definition of a splicing graph to a set of orthologous genes G. We describe the whole transcript variability of G with an evolutionary splicing graph (ESG) (Fig. 1B), where each node is a spliced-exon (s-exon) and represents a multiple sequence alignment (MSA) of subexons or subexon parts coming from different species. The edge set comprises the ensemble of edges linking the nodes in the individual SGs, possibly augmented by some edges induced by the definition of the s-exons (for more formal definitions, see Supplemental Methods). As a consequence, two nodes in may be linked by several edges (at most one per species), and we designate this set as a multiedge. There are many possible ways of grouping the exonic sequences coming from the different genes, and hence of defining the s-exons (Fig. 1B). For instance, one may define each s-exon in the ESG by taking at most one subexon from each SG (Fig. 1B, solutions 1 and 3). In that case, the edge set is exactly the set of edges coming from the individual SGs. Alternatively, it may be advantageous to split a subexon into two or more subsequences and assign them to different s-exons (Fig. 1B, solution 2, in which AEI and GV come from the human subexon AEIGV). This strategy can lead to better MSAs, but it increases the complexity of the graph. Ideally, one would like to find a representation as compact as possible and at the same time, conveying meaningful evolutionary information. To estimate both properties, we define the score of the ESG as
| (1) |
where σ(v) is the score of the MSA associated to the node v. σ can be, for instance, a consensus score or a sum-of-pairs score and may additionally penalize very short MSAs (fewer than three columns). (respectively, ) are the numbers of induced (respectively, structural) edges in the multiedge e with associated fixed penalty σI (respectively, σS). In practice, we set σI ≫ σS to avoid small s-exons induced by ambiguous alignment columns in the MSAs. As an example, with a simple sum-of-pair scoring for σ and an induced edge penalty σI = 4 · σS, the best-scored ESG in Figure 1B (solution 1) comprises the smallest numbers of s-exons, induced edges, and gaps. In general, determining the best-scored ESG is a NP-hard problem (Methods).
Here, we provide a practical solution to construct a meaningful parsimonious ESG given a set of input transcripts (Fig. 1C; Supplemental Fig. S1). Our heuristic procedure first preclusters exons using pairwise alignments. Then, within each cluster, it concatenates the sequences coming from each species in the order of their genomic coordinates and aligns the obtained sequences using ProGraphMSA (Supplemental Fig. S2; Szalkowski 2012). The latter allows better handling of AS-induced deletions and insertions than classical progressive alignment methods. Moreover, using the genomic coordinates as ordering constraints helps to disentangle orthology from paralogy relationships between similar sequences (Supplemental Fig. S2C). Finally, we locally solve the problem exposed in Equation 1 by realigning some sequences and by maximizing the agreement between subexon boundaries across different species (Supplemental Fig. S3). Controlling the creation of (penalizing) induced edges allows us to implicitly evaluate the ESG score. We implemented the heuristic in the fully automated tool ThorAxe.
In the following, we show that ThorAxe allows obtaining simple and meaningful representations for evolution in the context of AS. We primarily rely on gene annotations from Ensembl (Yates et al. 2016), and we complement the computed ESG with RNA-seq data from the NCBI Sequence Read Archive (SRA) (Leinonen et al. 2011), together with the tissue annotations compiled from Bgee (Supplemental Methods; Komljenovic et al. 2016). We focus on one-to-one orthologous genes across 12 species, namely three primates, two rodents, four other mammals, one amphibian, one fish, and nematode. Our motivation for this choice was to span different evolutionary distances and to ensure that enough RNA-seq data would be available. We take human as reference for selecting the genes, but ThorAxe ESG construction is reference-free.
ThorAxe recapitulates known functional AS events
We tested ThorAxe on a curated set of 50 genes representing 16 families (Supplemental Table S1; Supplemental Methods), in which several splice variants have been associated with diverse protein functions. ThorAxe detected 448 alternative splicing, initiation, and termination events. RNA-seq splice junctions mapping onto the ESGs provided additional support for about one-quarter of them and uncovered 101 more events. Detailed information is available on the accompanying website (http://www.lcqb.upmc.fr/ThorAxe) (Supplemental Fig. S4). We report here the results for a set of 30 documented events influencing partner binding affinity, selectivity, or specificity (Supplemental Tables S2, S3). We observed tissue-regulation patterns well-conserved across mammals for most of them; in amphibians, for seven of them (Fig. 2A; see also Supplemental Table S3). Although the gene annotations and the RNA-seq data show a good overall agreement, many subpaths are contributed solely by RNA-seq in platypus, cow, and zebrafish (Fig. 2B).
Figure 2.

Conservation and tissue regulation of a set of documented AS events. (A) Each event is designated by the name of the gene where it occurs and its rank in ThorAxe output, the latter reflecting its relative conservation level. In the ESG, an event corresponds to a pair of subpaths, one being canonical and the other alternative. Within each species, either none of the paths are supported by the data (gray), or only one path is supported (light orange), or both paths are supported (orange and dark orange). As data, we consider the gene annotations from Ensembl and the RNA-seq evidence compiled from public databases. When both paths are supported, we highlight the cases in which they are differentially expressed in at least one tissue in dark orange. The white cells indicate species in which a one-to-one ortholog of the human query gene could not be found. (B) For each species, the percentages of events supported by both gene annotations and RNA-seq (in green), by only RNA-seq (in yellow), by only gene annotations (in blue), or unsupported (in gray) are reported. An event is considered to be supported only if both its canonical and alternative subpaths are detected.
The ESG computed for CAMK2B linker region provides an illustrative example for which, despite a very high AS-generated complexity, ThorAxe results are interpretable, meaningful, and consistent with what has been reported in the literature (Fig. 3A). For instance, one can readily see that the shortest isoform lacking the linker (Fig. 3B, “7”) has low evolutionary support. This is in line with recent findings emphasizing the importance of the linker for regulating the protein activity (Bhattacharyya et al. 2020). Moreover, all the s-exons defined by ThorAxe are conserved at least as far as amphibians. The smallest s-exon (25_1) contains only one column of alanines and corresponds to a well-documented internal splice site (Sloutsky and Stratton 2020). Finally, the two documented functional AS events are clearly identifiable on the ESG (Fig. 3A, gray areas). This observation still holds true when removing the two best-annotated species, namely, human and mouse (Supplemental Fig. S5A), and when scaling up to about 100 species (Supplemental Fig. S5B). Furthermore, RNA-seq mapping revealed evolutionarily conserved tissue regulation for both events (Fig. 3C). For instance, the alternatively spliced F-actin binding region comprised of the s-exons 15_0 and 15_1 is specifically expressed in the brain and muscles of primates and rodents (Fig. 3C, on the left).
Figure 3.

Transcript variability in the CAMK2B linker. (A) Evolutionary splicing subgraph computed by ThorAxe starting from 63 transcripts annotated in 10 species. It corresponds to the region linking the kinase and hub domains of CAMK2B. The colors of the nodes and the edges indicate conservation levels, from yellow (low) to dark purple (high). Conservation is measured as the species fraction for the nodes (proportion of species where the s-exon is present) and as the averaged transcript fraction for the edges (averaged transcript inclusion rate of the s-exon junction). For ease of visualization, we filtered out the s-exons present in only one species. The events documented in the literature are located in the gray areas. (B) On top, genomic structure of the human gene. Each gray box corresponds to a genomic exon (nomenclature taken from Sloutsky and Stratton 2020). (Below) List of human transcripts. All of them have been described in the literature, referred to as β (Bulleit et al. 1988), βM (Bayer et al. 1998), βe (Brocke et al. 1995), β′e (Brocke et al. 1995), βe− (Cook et al. 2018), α (Bulleit et al. 1988), 7 (Wang et al. 2000), and 6 (Wang et al. 2000). The functional roles of some exons (Bayer et al. 1998; Khan et al. 2019) are given. (C) Percent-spliced in (PSI) computed from RNA-seq splice junctions for the two documented AS events. The two pairs of alternative subpaths depicted on top are also highlighted on A.
ThorAxe summarizes within- and across-species variations at the human proteome scale
We further assessed ThorAxe on the whole human proteome (18,226 genes) (Supplemental Methods). ThorAxe analysis across 12 species completed in less than 20 h with 15 cores. The genes are well-represented in all mammals (Supplemental Fig. S6), except in platypus, which covers only 40% of the human protein-coding genome. Frog, zebrafish, and nematode cover about 65%, 50%, and 14% of the genes, respectively (Supplemental Fig. S6). ThorAxe produced ESGs with 26 s-exons, on average, and at most 354 (Supplemental Table S4). They are either very lowly or very highly conserved, as measured by the species fraction (SF) that is the proportion of species where a s-exon is found (Supplemental Figs. S7, S8).
We distinguish the species-specific s-exons detected in only one species and thus containing only one sequence in their MSA, from the s-exons conserved in at least two species. The proportion of species-specific s-exons goes from <10% in mammals and zebrafish to 23% in frog and 72% in nematode (Fig. 4A; Supplemental Table S5), emphasizing the high sequence divergence of this organism. These s-exons are often located at the transcript extremities (Fig. 5A,B, nodes in yellow) and tend to be smaller than the conserved ones (Supplemental Fig. S9). The vast majority of the latter are well-conserved from primates to amphibians, and their species representativity strongly correlates with the evolutionary distance they span (Fig. 4A). For instance, almost all the conserved s-exons present in frog also comprise sequences coming from primates, non-primate eutherians, and noneutherian mammals (Fig. 4A, pink curve). This correlation is even more evident in the subset of 13,558 genes with one-to-one orthologs in more than seven species (Supplemental Figs. S10, S11). Overall, ∼40% of the conserved s-exons present in human span an evolutionary time of more than 400 million years, up to zebrafish (Fig. 4A). The proportion drops down to <10% outside of vertebrates. Nevertheless, we identified 295 genes for which ThorAxe assigned most of the exonic sequences contributed by nematode to well-conserved s-exons. This set is enriched in genes coding for proteins involved in the transcription or the translation (RNA polymerases, ribosome, spliceosome, chaperones) or in protein degradation (proteasome).
Figure 4.

S-exon evolutionary profiling over the whole protein-coding human genome. (A) Percentages of s-exons conserved at different evolutionary distances from human (represented by dashed vertical lines). Each curve is centered on its corresponding species. The values at the origin are the percentages of conserved (i.e., not species-specific) s-exons. Conservation is then assessed at each evolutionary distance according to the s-exons possessing at least one representative in each phylogroup. For instance, we report 73%–76% of the s-exons of frog (pink curve) as conserved among eutherians (second dashed line) in the sense that they are also conserved in at least one primate (among human, gorilla, macaque) and at least one nonprimate eutherian (among rat, mouse, boar, cow). Likely, conservation up to mammals (68%–72% for frog) would imply at least one primate, one nonprimate eutherian, and one noneutherian mammal. See also Supplemental Figure S10 for a version of this plot focusing on genes with one-to-one orthologs in more than seven species. (B) Cumulative distributions of s-exon species fraction. On the y-axis we report the percentage of s-exons with a species fraction greater than the x-axis value. The different curves correspond to all s-exons (All), only those involved in at least an event (Any event), or only those involved in a specific type of event. (Alter-S) alternative start; (Alter-I) alternative (internal); (Alter-E) alternative end; (Del) deletion; (Insert) insertion. (C) Heatmap of the s-exon phastCons median scores versus the s-exon species fractions. Only the s-exons longer than 10 residues and belonging to genes with one-to-one othologs in more than seven species are shown. (D) Proportions of conserved s-exons displaying very poor (negative score) to very good (score close to one) alignment quality. The MSA score of a s-exon is computed as a normalized sum of pairs. A score of 1 indicates 100% sequence identity without any gap. The proportions are given for different s-exon selections (same labels as in B).
Figure 5.

Examples of evolutionarily conserved events with in-gene paralogy. (A,B) ESGs computed by ThorAxe (left) and the best 3D templates found by HHblits (right); PDB codes 2w49:abuv (Wu et al. 2010) and 2dfs:H (Liu et al. 2006) for TPM1 and MYO1B. On the ESGs, the colors indicate conservation levels, species fraction for the nodes and averaged transcript fraction for the edges (Supplemental Methods). The nodes in yellow are species-specific, whereas those in dark purple are present in all species. The 3D structures show complexes between the query proteins (black) and several copies of their partners (light gray). The s-exons involved in conserved events are highlighted with colored spheres. (C) S-exon consensus sequence alignments within a gene family (TPM on top, MAPK in the middle) or a gene (MYO1B, at the bottom). Each letter reported is the amino acid conserved in all sequences of the corresponding MSA (allowing one substitution). The color scheme is that of Clustal X (Thompson et al. 1997). The subgraphs show the events in which the s-exons are involved. The symbols α and β on the right indicate groups of s-exons defined across paralogous genes based on sequence similarity (Supplemental Methods). The symbols at the bottom denote highly conserved positions across the gene family: (dot) fully conserved position; (square) position conserved only within each s-exon group; (upward triangle) position conserved in the α group only; (downward triangle) position conserved in the β group only. For MYO1B, the start and canonical sequence of the CALM1-binding IQ motif are indicated. The motifs resulting from different combinations of the depicted s-exons are numbered 4, 4/5, and 4/6 in the literature (Greenberg and Ostap 2013).
As for the s-exon usage, almost a third is involved in some event (Fig. 4B). The most (respectively, least) conserved ones, are involved in deletions (respectively, insertions) (green and pink curves). This observation can be explained by the fact that ThorAxe detects events as variations from a reference canonical transcript chosen for its high conservation and length (Supplemental Methods). The alternatively expressed s-exons located at the beginning or in the middle of the protein (gold and light blue curves) tend to be more conserved than at the end of the protein (dark blue curve).
The s-exons accurately measure sequence conservation
To evaluate ThorAxe ability to correctly match exonic sequences between more or less distant species, we compared the s-exon species fractions with estimates of evolutionary conservation deduced from whole-genome alignments between human and 99 other vertebrates, available as phastCons scores through the UCSC Genome Browser (Supplemental Methods; Siepel and Haussler 2005; Siepel et al. 2005). Overall, the two measures agree very well (Fig. 4C). For instance, most of ThorAxe species-specific or very lowly conserved s-exons (SF<0.3) are not expected to be evolutionarily conserved based on genomic alignments (Fig. 4C, left column). Nevertheless, ThorAxe seems to underestimate the conservation level of 1508 s-exons (Fig. 4C, top left corner). We investigated whether these s-exons could share significant sequence similarity with some other s-exons defined across distinct species, and we found that only 24 of them may be considered as “false negatives” (Supplemental Table S6). Hence, the low conservation estimated by ThorAxe likely reflects the lack of annotated transcripts in certain species rather than errors in the heuristic. Reciprocally, ThorAxe seems to detect more conservation signal than whole-genome alignments for more than 7000 s-exons (SF>0.3) (Fig. 4C, bottom row), without any particular trend in their alternative usage (Supplemental Fig. S12). They display high sequence identity (Supplemental Fig. S12C,D), suggesting that they are indeed conserved across many species and not “false positives.” The collagen type XVIII alpha 1 chain (COL18A1) on its own contributes 12 such s-exons, conserved from primates to amphibians according to ThorAxe (SF>0.8) but with very low phastCons scores (<0.1). The COL18A1 protein is highly enriched in glycines and prolines and the 12 s-exons fall within regions of low sequence complexity (Supplemental Fig. S13). We can hypothesize that this low-complexity context confounds the whole-genome alignments but not ThorAxe heuristic. To get a better view on the s-exon sequence divergence, we computed the sum-of-pair scores of the associated MSAs (Supplemental Methods). Overall, almost half of the conserved s-exons have very high-quality MSAs with very few mismatches and gaps (Fig. 4D, score > 0.75). This proportion increases up to about 70% on the 50-gene set (Supplemental Fig. S14). A very small proportion (about 1%) of s-exons have very poor quality MSAs (Fig. 4D, in black), and those are typically short (Supplemental Fig. S15). Moreover, the inserted and, to a lesser extent, alternatively expressed s-exons display lower-quality MSAs (Fig. 4D, Insert and Alter-I). Finally, we checked the relationship between structural order/disorder and s-exon sequence divergence. Structurally disordered s-exons (∼40% of the ensemble) tend to be less conserved and to have lower-quality MSAs than well-folded ones (Supplemental Fig. S16). However, the differences between the two groups are rather small.
The comparison of similar s-exons unveils functionally relevant signatures
ThorAxe allows exploring how function diversification may arise through the alternative usage of similar sequences within and across genes. We illustrate the power of the approach on three gene families (Fig. 5; see also Supplemental Tables S2, S3), focusing on a set of events involving two or more highly conserved s-exons with similar consensus sequences. The origin of the events can be traced back to the ancestor common to mammals, amphibians, and fishes. The first example is given by the tropomyosin family (Fig. 5A,C), whose protein members (TPM1,2,3,4) serve as integral components of the actin filaments forming the cell cytoskeleton. Several conserved events detected in the ESGs have direct implications for actin binding (Fig. 5A; Wu et al. 2010; Pathan-Chhatbar et al. 2018). Among them, the internal mutually exclusive pair displays high sequence similarity and strong sequence conservation across species and between paralogous genes (Fig. 5C, on top, α and β groups). Fourteen specificity-determining sites (SDS) can be identified (Fig. 5C). SDS are key positions with specific conservation patterns, and they play a role in diversifying protein function in evolution (Chakraborty and Chakrabarti 2015). Given two groups, here α and β, type I SDS are conserved in one group and variable in the other one, indicating different functional constraints between the groups. For instance, position 24 is occupied by a glutamate in all the s-exons from the β group, whereas it is variable in the a group. Type II SDS are conserved in both groups, but each group displays a different amino acid. This is the case of positions 14 and 15, where the Thr-Asn couple of the α group is replaced by Asp-Gln in the β group. These SDS may be responsible for the differences observed in actin filaments formation, mobility, and myosin recruitment ability between the isoforms (Pathan-Chhatbar et al. 2018). The Mitogen-activated protein kinase (MAPK) family (MAPK8,9,10) gives another example with even higher sequence identities (Fig. 5C, in the middle). Among the eight identified SDSs, three positively charged residues—His, Lys, and Arg in positions 16, 17, and 23—are specifically conserved in the α group, whereas the β group is characterized by Lys, Gly, and Thr in positions 15, 16, and 23. These observations are in line with our previous study highlighting differences in the dynamical behavior of these residues (Ait-hamlat et al. 2020) and their potential implication for substrate selectivity (Waetzig and Herdegen 2005). As a third example, myosin IB comprises a set of consecutive similar s-exons overlapping with calmodulin(CALM1)-binding so-called IQ motifs (Fig. 5B). The alternative inclusion of two s-exons, which share almost 50% identity (Fig. 5C, bottom), results in different binding motifs. Compared to the motif's canonical form (IQXXXRGXXXR) (Houdusse et al. 1996; Bähler and Rhoads 2002), they all lack the glutamine in the IQ residue pair and the arginine in the RG pair. These differences could explain their lower affinity compared to the constitutive s-exons (Greenberg and Ostap 2013).
Alternative usage of similar sequences is not a rare phenomenon
At the human genome scale, we identified 2190 genes (12% of the protein-coding genome) with evidence of evolutionarily conserved alternative usage of similar exonic sequences (Fig. 6). The corresponding proteins tend to be involved in cell organization and muscle contraction (cytoskeleton, collagen, fibers, etc.), and in intercellular communication (Supplemental Fig. S17). Our strategy here was to look for similar s-exon pairs involved in some event (Supplemental Methods). We found a total of 31,031 pairs, among which 446 are mutually exclusive (Fig. 6A,B, MEX). This case scenario highlights the exclusive usage of one or the other version of a protein region. The 232 concerned proteins are enriched in transporters and channels (Supplemental Fig. S17). Another 438 pairs (coming from 134 genes) are alternatively used without mutual exclusivity (Fig. 6A,B, ALT). In about half of the MEX or ALT s-exon pairs, one of the s-exons is conserved in all studied species, and the other one in more than half of them (Fig. 6C). In 3813 pairs, one s-exon is included in the canonical or alternative subpath of an event, whereas the other one serves as a “canonical anchor” for the event (Fig. 6A,B, REL). This highlights the AS-induced modulation of the number of nonidentical consecutive copies of a protein region. The remaining 26,334 pairs correspond to cases in which one of the s-exons participates in an event (on the canonical or alternative subpath), whereas the other one is located outside the event in the canonical transcript (UNREL). The full lists of s-exons are given in Supplemental Tables S7–S9. This resource overlaps well with a previously reported manually curated set of 97 human genes with mutually exclusive homologous exon pairs (Abascal et al. 2015). It extends it by one order of magnitude and represents a more diverse range of AS-mediated relationships between similar protein regions. Although most of the identified genes contain only one or a few similar s-exons pair(s), almost 50 genes have several hundreds or thousands of pairs (Fig. 6D). Nebulin gives the most extreme example, with 2380 detected pairs. This giant skeletal muscle protein has evolved through several duplications of nebulin domains, and a definition of pertinent nebulin evolutionary units was proposed (Björklund et al. 2010). These units correspond to parts of exons, in line with ThorAxe s-exons MSAs.
Figure 6.
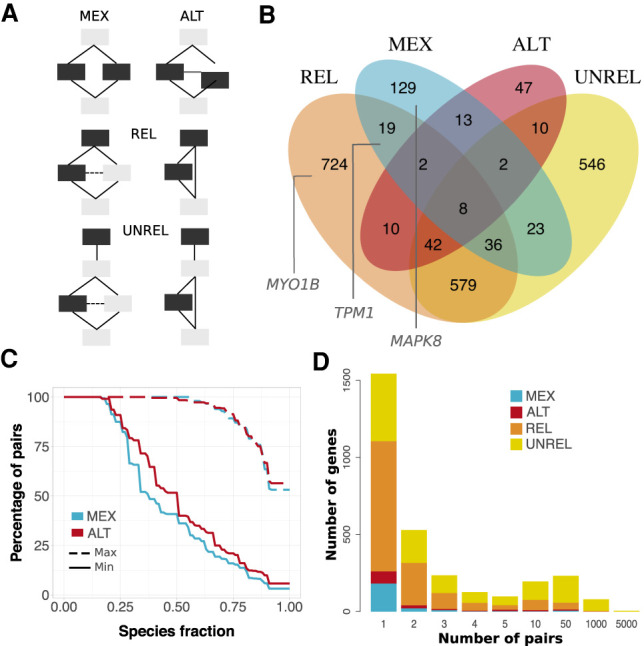
Alternative usage of similar s-exons. (A) Evolutionary splicing subgraphs depicting different alternative usage scenarios. The detected s-exon pairs are colored in black. (MEX) mutual exclusivity; (ALT) alternative (non-mutually exclusive) usage; (REL) one s-exon is in the canonical or alternative subpath of an event (of any type), whereas the other one serves as a “canonical anchor” for the event; (UNREL) one s-exon is in the canonical or alternative subpath of an event (of any type), whereas the other one is located outside the event in the canonical transcript. Each detected pair is assigned to only one category with the following priority rule: MEX > ALT > REL > UNREL. (B) Venn diagram of the genes containing similar pairs of s-exons. The genes shown in Figure 5 are highlighted in the corresponding subsets. (C) Cumulative distributions of s-exon conservation. On the y-axis we report the percentage of s-exon pairs with species fraction greater than the x-axis value. The solid (respectively, dashed) curve corresponds to the highest (respectively, lowest) species fraction among the two s-exons in the pair. We report values only for the MEX (blue) and ALT (red) categories. (D) Distribution of per-gene s-exon pair number within each of the four categories. For instance, the yellow rectangle at x = 50 gives the number of genes with more than 10 and up to 50 UNREL s-exon pairs.
Comparison with other studies
We evaluated the ability of ThorAxe to detect the events reported in two reference studies dealing with the evolution of AS (Barbosa-Morais et al. 2012; Merkin et al. 2012). We could map 40 exons from Barbosa-Morais et al. (2012) and 323 exons from Merkin et al. (2012), all displaying conserved tissue-specific splicing patterns, onto our ESGs (Supplemental Methods). For >75% of the 40 exons, we found events conserved across mammals and ranked first or second in the ESG (Supplemental Fig. S18A, dark green). The conservation signal extends to amphibians for 18 events and to teleosts for five. For the 323-exon set, we detected 277 events, 90% of which are in the top three most conserved of ThorAxe ESGs. About 70% are well-conserved in mammals, 82 in amphibians, and 19 in teleosts (Supplemental Fig. S18B). In particular, we can mention exon 3 from the eukaryotic translation elongation factor 1 delta (EEF1D-ex3) and exon 20 from the tight junction protein 1 (TJP1-ex20) highlighted in Figures 2 and 4 from Merkin et al. (2012). In both cases, the deletion of the matching s-exon(s) is the most conserved event of the ESG and is observed in human, mouse, and pig. The deletion of EEF1D-ex3 (s-exons 1_1 and 1_2) is also conserved in gorilla and cow, whereas that of TJP1-ex20 (s-exon 6_0) is also conserved in macaque. We can also pinpoint the six exons intersecting with our curated set (Supplemental Fig. S19). ThorAxe detected events well-conserved across mammals for all of them (and in amphibians, for two of them), with RNA-seq evidence of conserved tissue-specific AS patterns for all but one. One of the matching s-exons, 11_1 from MYO1B, is part of a couple of alternatively spliced pseudorepeats (Fig. 5B). Although we found conserved tissue-regulation patterns for 11_3, the other s-exon in the couple, it was not reported in Merkin et al. (2012). This example highlights the difficulty of assessing the tissue-specific expression of several instances of (pseudo-)repeated sequences, and showcases ThorAxe's ability to deal with such complexity.
Comparison with other methods
We assessed the pertinence of the ThorAxe heuristic by performing an ablation study and by comparing it with two popular exon orthology detection methods. For the ablation study, one strategy was to skip the exon clustering step (Methods, step b), and the other was to rely solely on global multiple sequence alignment, which means both the exon clustering step and the s-exon refinement step are skipped (Methods, steps b and e). Compared to these two strategies, ThorAxe produces longer and higher-quality s-exon MSAs (Supplemental Fig. S18C,D). Specifically, the clustering step helps to improve the s-exon sequence identities (Supplemental Fig. S18D, cf. blue and orange boxes) by reducing the space of sequences to align. The final local optimization step increases the lengths of the s-exons (Supplemental Fig. S18C, cf. blue and red boxes) by minimizing subexon boundaries violations. As popular exon orthology detection methods, we chose the Reciprocal Best Blast Hit (RBBH) method and Ensembl Compara (Herrero et al. 2016). The RBBH method consists in finding the best matching subexon pairs across any two species using BLAST. One of the drawbacks of this method is that many subexons remain without any match in other species (Supplemental Fig. S18E, orange box). By allowing for one-to-many subexon matching between species, ThorAxe covers a much higher proportion of subexons (Supplemental Fig. S18E, blue box). ThorAxe strategy is justified by the fact that exons may undergo truncation or elongation in the course of evolution, and thus we do not expect a one-to-one relationship between them across a pair of species. Moreover, ThorAxe increased subexon coverage is not at the expense of sequence identity (Supplemental Fig. S20). Another drawback of RBBH is that defining s-exons from a set of pairwise alignments of subexons is a difficult task. Finally, Ensembl Compara relies on whole-genome alignments. However, it does not include all the species for which annotated transcripts are available in Ensembl. Moreover, one can expect that working with DNA sequences produces lower-quality alignments compared to working with protein sequences, as is done by ThorAxe.
Assessment of the default parameter choices
All parameters in ThorAxe are customizable by the user, enabling a rapid adaptation of the method to specific contexts and questions. We investigated the pertinence of some of the default values. For instance, by default, ThorAxe filters out the transcripts flagged in Ensembl as lowly supported (Transcript Support Level [TSL]<3). This restriction only concerns human and mouse, because the other species do not have any TSL annotations. By varying the TSL value between 1 and 5, we observed that, as expected, the less stringent the TSL criterion, the higher the number of transcripts and of events (Supplemental Fig. S21A,B). However, very little change is observed in the definition of the canonical transcript and in the conservation levels of the s-exons, suggesting that the results and their interpretation are robust to this parameter (Supplemental Fig. S21C–E). Globally, the biggest changes are observed either when only top-quality transcripts are retained (TSL ≤ 1) or when transcripts are not filtered at all (TSL ≤ 5). We thus recommend using intermediate TSL values (2–4).
The first step of the ThorAxe algorithm consists in grouping similar exonic sequences together, with the aim of reducing the complexity of the subsequent construction of the MSAs. By default, ThorAxe applies a sequence identity cutoff of 30% to define the clusters. To assess the suitability of this cutoff value for handling divergent sequences, we looked at the MSA quality with respect to the species fraction (Supplemental Fig. S22). Although the two measures are correlated, a significant portion of s-exons display high species fractions (>0.8) but low MSA scores (≤0.5). This observation suggests that ThorAxe is able to cluster together divergent sequences that are difficult to align. The cutoff may be adapted by the user depending on the level of sequence divergence expected in the input data.
To ease interpretability of the results and ensure that the s-exons represent groups of one-to-one orthologous exonic regions, ThorAxe default mode considers only one-to-one orthologous genes, as annotated in Ensembl. This means that organisms with additional round(s) of whole-genome duplications and/or separated by long evolutionary distances will likely be excluded from the analysis. We found that taking into account many-to-many gene orthology relationships leads to a better detection of the documented events (Supplemental Fig. S23). The improvement is particularly visible for zebrafish, in which we now have eight events with both the canonical and alternative subpaths supported by Ensembl annotations (Supplemental Fig. S23, cf. A and B). The detection in rat, cow, and platypus is also improved. Finally, we tested the impact of excluding the two best-annotated species, namely, human and mouse. As a result of this exclusion, three documented events are lost. Nevertheless, the conservation profiles of the other events remain almost identical (Supplemental Fig. S23, cf. A and C).
Discussion
We have presented a novel method to describe transcript variability in evolution. Our approach provides a double generalization, by extending the definition of SG to the case of multiple species and by providing a way to combine MSAs over structures with a partial order. The heuristic is general enough to deal with very different genes (in terms of length, structure, degree of conservation, number of transcripts, etc.). Its identification of transcript minimal building blocks (the s-exons) is the first and necessary step for inferring evolutionary scenarios explaining AS-induced protein function diversification (Ait-hamlat et al. 2020). Our data structure is reminiscent of current developments on pangenome graphs. However, pangenome approaches keep track of variations across a population, whereas we highlight conservation across species in the context of AS. To the best of our knowledge, we are the first to do it. Effectively, we consider a pan-transcriptome across multiple species. As a consequence, we do not need to rely on a central species and project the transcripts on it. In the analysis conducted here, human was taken as a reference only to find orthologous genes in other species.
To illustrate the potential of the method, we assessed the evolutionary conservation and tissue regulation of a set of documented AS events we compiled from the literature. This set could serve as a reference for future studies. We then scaled up to the human protein-coding genome, and found that AS is conserved across a wide range of evolutionary distances, is not limited to ancient events, and does not generate conserved alternative isoforms in all of the proteins. We have shown that the alternative usage of repeats in protein is not a rare phenomenon in the human proteome and that it is of ancient evolutionary origin. Although we focused on one-to-one orthologs, thereby limiting the contribution of nematode, our analysis can be readily extended to one-to-many orthology relationships to better compare vertebrates with other organisms.
On the one hand, a limitation of the approach is that it mainly relies on gene annotations, which may be partial, incomplete, or erroneous (Salzberg 2019). To avoid errors, we chose to select only high-quality transcripts, with the risk of biasing the results toward species with more fully annotated alternative splicing landscapes. Another strategy could be to use APPRIS annotations, but we expect a reduction in the overall input transcript variability, therefore limiting ThorAxe potential to discover AS events. Moreover, APPRIS annotations are derived from an analysis accounting for transcript sequence conservation, which would be somewhat redundant with ThorAxe's own analysis of AS conservation. On the other hand, an important advantage of ThorAxe is its robustness with respect to the presence of highly divergent sequences and the creation of species-specific s-exons. Indeed, the latter simply contribute single nodes to the ESG without preventing the detection of conserved AS events. In a way, detecting too many species-specific s-exons would not be a problem because this would only slightly diminish ThorAxe conservation estimates without hampering the interpretation of the ESGs. Traditional methods may recover genomic conservation at lower levels of sequence similarity, but by disregarding the whole transcript structure, they may not properly evaluate AS conservation.
Future work could benefit from the development of more accurate approximations of the general problem stated here. Another direction is to expand the application field to transcriptomes coming from patients or human populations. In the coming years, we expect a tremendous increase in available transcriptomic data, including transcriptome annotations generated by long-read sequencing technologies (Byrne et al. 2019). Methods addressing the complexity of these data will become instrumental in understanding the evolution of a disease, for example, cancer, and the phenotypic variability among human populations and individuals (Lonsdale et al. 2013; Park et al. 2018). ThorAxe could be easily adapted to deal with these data, and, along this line, we have already implemented the possibility to give additional “user-defined” transcripts as input.
Methods
Complexity of the problem: determining a minimal ESG is NP-hard
To illustrate the complexity of determining a minimal ESG, consider a case example with n input transcripts observed in n species (i.e., one transcript per species). Moreover, because the problem is theoretically independent of the penalties σI and σS (Equation 1), an algorithm that would solve it in the general case would also be valid for σI = σS = +∞. In this scenario, a minimal ESG has no edge and maximizes the sum-of-pair-score σ. Thus, the problem of building a minimal ESG is equivalent to solving the problem of multiple sequence alignment with sum-of-pair-score σ on the n input transcripts. Because the n input transcripts can be any string (over the amino acid alphabet), and finding a MSA of any string with sum-of-pair-score is NP-hard (Wang and Jiang 1994), it follows that finding a minimal ESG is NP-hard.
Description of ThorAxe algorithm and parameters
Given a gene name and a list of species as input, ThorAxe extracts and exploits gene annotations from Ensembl (and, optionally, input transcripts provided by the user) to build an ESG maximizing the sequence similarity within each node (or s-exon) and minimizing the number of induced edges, which indirectly implies that the number of nodes is minimized. The heuristic approximates the best-scored solution of Equation 1 by controlling the creation of induced edges, without explicitly computing ESG scores. It unfolds in six main steps (Fig. 1C; Supplemental Fig. S1).
Data acquisition and preprocessing. ThorAxe downloads the gene tree, the transcripts annotated as protein coding and their exons (genomic coordinates, sequences, and phases) for the query gene and its (by default one-to-one) orthologs in the selected species from Ensembl. ThorAxe then removes incomplete or lowly supported (TSL < 3, adjustable by the user) transcripts, and translates the retained transcripts’ DNA sequences into amino acid sequences using the exon phases. Transcripts or exonic regions leading to the same protein sequence are merged, but the same genomic region may lead to the generation of several protein sequences if it is associated with more than one frame (Supplemental Methods). ThorAxe can additionally take as input user-defined transcripts (from any species). The format is similar to the one in Ensembl and includes exon coordinates, their rank, frame, and nucleotide sequence.
Pairwise-alignment-based exon preclustering. ThorAxe clusters the input exons based on their sequence similarity (Fig. 1C, three clusters colored in red, cyan, and blue). This step provides a coarse-grained partitioning of the sequence space that reduces the complexity of step d (see below). We perform pairwise local alignments using a modified version of the Hobohm I algorithm (Supplemental Methods). We use a relatively low default sequence identity threshold of 30% to ensure homology detection across many species. As illustrated in Figure 1C by cluster 2, pairs of duplicated mutually exclusive exons coming from the same species will likely be grouped in the same cluster (see also Supplemental Fig. S1).
Redundancy reduction. ThorAxe defines a set of subexons for each species. This implies systematically detecting overlapping exons and replacing them by nonredundant distinct subexons. In the example shown in Figure 1C, one exon from gorilla leads to the definition of two subexons (in red; for their sequences, see also Supplemental Fig. S2A). This step relies only on the genomic coordinates of the exons and does not require aligning the exonic sequences. It is performed after exon clustering, because dealing with subexons at this early stage would add some unnecessary complexity by augmenting the number of comparisons and the ambiguity associated with small sequences.
MSA-based s-exon identification. ThorAxe defines a set of s-exons across all species, by aligning the exonic sequences belonging to each cluster defined in step b and identifying blocks in the constructed MSAs (Fig. 1C, see the three MSAs corresponding to the three clusters). The aim of this step is to determine a mapping of exonic sequences between different species (for details, see Supplemental Methods). To identify the s-exons from each MSA, ThorAxe scans the MSA from left to right and creates a new s-exon whenever there is a change of subexon in at least one sequence/species (Algorithm 1 in Supplemental Methods). This ensures that the identified s-exons can be used as building blocks to reconstruct any transcript in any species from the input data.
S-exon refinement. ThorAxe refines the s-exons’ definition by locally optimizing the MSAs built in step d. The aim is twofold, namely, to improve the quality of the MSAs associated to the s-exons and to reduce the number of s-exons, and hence the number of induced edges in the corresponding ESG. This step then represents a means to increase the ESG score expressed in Equation 1 without explicitly computing it. Specifically, we systematically detect lowly scored subexons and migrate them from one MSA to another, and we minimize the number of very small s-exons, comprising only one or two columns (Supplemental Methods).
ESG construction and annotation, and event detection. Once the s-exons have been identified, building the corresponding ESG is straightforward (Fig. 1C). ThorAxe annotates the nodes and the edges of the output graph with evolutionary information and summary statistics (Supplemental Fig. S2D; Supplemental Methods). Finally, it defines a canonical transcript and detects a set of events as variations between this reference transcript and each input transcript. Ideally, the canonical transcript should be well-represented across species; thus, to choose it, we rely on a combination of conservation measures computed over the ESG edges (Supplemental Methods; Supplemental Fig. S24). By default, the events are detected on a reduced version of the ESG, where the edges supported by only one transcript have been removed (Algorithm 2 in Supplemental Methods). We visualized the ESGs with Cytoscape V.3.7.2 (Shannon et al. 2003).
An additional output of ThorAxe is the list of input transcripts described as collections of s-exons (where each s-exon is designated by a symbol) and the gene tree representative of the selected species. These data can directly serve as input for PhyloSofS (Ait-hamlat et al. 2020), toward the reconstruction of transcripts’ phylogenetic forests. ThorAxe may also be easily interfaced with other tools requiring the same type of input.
Analysis of ThorAxe results
We give details about the calculation of the MSA scores, the detection of similar pairs of s-exons, the complementation of the ESGs with RNA-seq splice junctions, the characterization of isoform 3D structures and disorder content, the functional analysis of some genes, the comparison with phastCons scores, other studies, and other methods in the Supplemental Methods, Supplemental Figs. S25–S27, and Supplemental Table S10.
Software availability
ThorAxe is freely available at GitHub (https://github.com/PhyloSofS-Team/thoraxe) and as a stand-alone package and Python module as Supplemental Code. All data supporting the findings of this study are available via a supplementary webserver (http://www.lcqb.upmc.fr/ThorAxe) and as Supplemental Material.
Supplementary Material
Acknowledgments
A grant of the French National Research Agency (MASSIV project, ANR-17-CE12-0009) provided a salary to D.J.Z. and funded the work of S.L. We thank P. Charpentier and J. Cortes for their help in the systematic detection of the disordered regions and S. Grudinin for insightful comments. We thank F. Oteri and H. Ripoche for the technical support. We thank the reviewers for their comments, which greatly improved the manuscript.
Author contributions: D.J.Z., H.R., and E.L. designed the research. D.J.Z. and S.L. performed the implementation. D.J.Z., S.L., H.R., and E.L. produced and analyzed the results. A.B. contributed the proof of NP-hard complexity. E.L. wrote the manuscript with support and feedback from all authors. H.R. and E.L. supervised the project.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.274696.120.
Competing interest statement
The authors declare no competing interests.
References
- Abascal F, Tress ML, Valencia A. 2015. The evolutionary fate of alternatively spliced homologous exons after gene duplication. Genome Biol Evol 7: 1392–1403. 10.1093/gbe/evv076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agosto LM, Gazzara MR, Radens CM, Sidoli S, Baeza J, Garcia BA, Lynch KW. 2019. Deep profiling and custom databases improve detection of proteoforms generated by alternative splicing. Genome Res 29: 2046–2055. 10.1101/gr.248435.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait-hamlat A, Zea DJ, Labeeuw A, Polit L, Richard H, Laine E. 2020. Transcripts’ evolutionary history and structural dynamics give mechanistic insights into the functional diversity of the JNK family. J Mol Biol 432: 2121.– . 10.1016/j.jmb.2020.01.032 [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Bähler M, Rhoads A. 2002. Calmodulin signaling via the IQ motif. FEBS Lett 513: 107–113. 10.1016/S0014-5793(01)03239-2 [DOI] [PubMed] [Google Scholar]
- Baralle FE, Giudice J. 2017. Alternative splicing as a regulator of development and tissue identity. Nat Rev Mol Cell Biol 18: 437–451. 10.1038/nrm.2017.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. 2012. The evolutionary landscape of alternative splicing in vertebrate species. Science 338: 1587–1593. 10.1126/science.1230612 [DOI] [PubMed] [Google Scholar]
- Bayer KU, Harbers K, Schulman H. 1998. αKAP is an anchoring protein for a novel CaM kinase II isoform in skeletal muscle. EMBO J 17: 5598–5605. 10.1093/emboj/17.19.5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya M, Lee YK, Muratcioglu S, Qiu B, Nyayapati P, Schulman H, Groves JT, Kuriyan J. 2020. Flexible linkers in CaMKII control the balance between activating and inhibitory autophosphorylation. eLife 9: e53670. 10.7554/eLife.53670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birzele F, Csaba G, Zimmer R. 2008. Alternative splicing and protein structure evolution. Nucleic Acids Res 36: 550–558. 10.1093/nar/gkm1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björklund AK, Light S, Sagit R, Elofsson A. 2010. Nebulin: a study of protein repeat evolution. J Mol Biol 402: 38–51. 10.1016/j.jmb.2010.07.011 [DOI] [PubMed] [Google Scholar]
- Brocke L, Srinivasan M, Schulman H. 1995. Developmental and regional expression of multifunctional Ca2+/calmodulin-dependent protein kinase isoforms in rat brain. J Neurosci 15: 6797–6808. 10.1523/JNEUROSCI.15-10-06797.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulleit RF, Bennett MK, Molloy SS, Hurley JB, Kennedy MB. 1988. Conserved and variable regions in the subunits of brain type II Ca2+/calmodulin-dependent protein kinase. Neuron 1: 63–72. 10.1016/0896-6273(88)90210-3 [DOI] [PubMed] [Google Scholar]
- Byrne A, Cole C, Volden R, Vollmers C. 2019. Realizing the potential of full-length transcriptome sequencing. Philos Trans R Soc Lond B Biol Sci 374: 20190097. 10.1098/rstb.2019.0097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Chakrabarti S. 2015. A survey on prediction of specificity-determining sites in proteins. Brief Bioinform 16: 71–88. 10.1093/bib/bbt092 [DOI] [PubMed] [Google Scholar]
- Christinat Y, Moret BM. 2012. Inferring transcript phylogenies. BMC Bioinformatics 13 (Suppl 9): S1. 10.1186/1471-2105-13-s9-s1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Climente-González H, Porta-Pardo E, Godzik A, Eyras E. 2017. The functional impact of alternative splicing in cancer. Cell Rep 20: 2215–2226. 10.1016/j.celrep.2017.08.012 [DOI] [PubMed] [Google Scholar]
- Cook SG, Bourke AM, O'Leary H, Zaegel V, Lasda E, Mize-Berge J, Quillinan N, Tucker CL, Coultrap SJ, Herson PS, et al. 2018. Analysis of the CaMKIIα and β splice-variant distribution among brain regions reveals isoform-specific differences in holoenzyme formation. Sci Rep 8: 5448. 10.1038/s41598-018-23779-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente L, Arzalluz-Luque A, Tardáguila M, del Risco H, Martí C, Tarazona S, Salguero P, Scott R, Lerma A, Alastrue-Agudo A, et al. 2020. tappAS: a comprehensive computational framework for the analysis of the functional impact of differential splicing. Genome Biol 21: 199. 10.1186/s13059-020-02028-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Grange P, Gratadou L, Delord M, Dutertre M, Auboeuf D. 2010. Splicing factor and exon profiling across human tissues. Nucleic Acids Res 38: 2825–2838. 10.1093/nar/gkq008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denti L, Rizzi R, Beretta S, Della Vedova G, Previtali M, Bonizzoni P. 2018. ASGAL: aligning RNA-Seq data to a splicing graph to detect novel alternative splicing events. BMC Bioinformatics 19: 444. 10.1186/s12859-018-2436-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezkurdia I, Rodriguez JM, Carrillo-de Santa Pau E, Vázquez J, Valencia A, Tress ML. 2015. Most highly expressed protein-coding genes have a single dominant isoform. J. Proteome Res 14: 1880–1887. 10.1021/pr501286b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzàlez-Porta M, Frankish A, Rung J, Harrow J, Brazma A. 2013. Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome Biol 14: R70. 10.1186/gb-2013-14-7-r70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR. 2001. Alternative splicing: increasing diversity in the proteomic world. Trends Genet 17: 100–107. 10.1016/S0168-9525(00)02176-4 [DOI] [PubMed] [Google Scholar]
- Greenberg MJ, Ostap EM. 2013. Regulation and control of myosin-I by the motor and light chain-binding domains. Trends Cell Biol 23: 81–89. 10.1016/j.tcb.2012.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heber S, Alekseyev M, Sze SH, Tang H, Pevzner PA. 2002. Splicing graphs and EST assembly problem. Bioinformatics 18: S181–S188. 10.1093/bioinformatics/18.suppl_1.S181 [DOI] [PubMed] [Google Scholar]
- Herrero J, Muffato M, Beal K, Fitzgerald S, Gordon L, Pignatelli M, Vilella AJ, Searle SM, Amode R, Brent S, et al. 2016. Ensembl comparative genomics resources. Database (Oxford) 2016: baw053. 10.1093/database/baw053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houdusse A, Silver M, Cohen C. 1996. A model of Ca2+-free calmodulin binding to unconventional myosins reveals how calmodulin acts as a regulatory switch. Structure 4: 1475–1490. 10.1016/S0969-2126(96)00154-2 [DOI] [PubMed] [Google Scholar]
- Kelemen O, Convertini P, Zhang Z, Wen Y, Shen M, Falaleeva M, Stamm S. 2013. Function of alternative splicing. Gene 514: 1–30. 10.1016/j.gene.2012.07.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Downing KH, Molloy JE. 2019. Architectural dynamics of CaMKII-actin networks. Biophys J 116: 104–119. 10.1016/j.bpj.2018.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. 2014. A draft map of the human proteome. Nature 509: 575–581. 10.1038/nature13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komljenovic A, Roux J, Wollbrett J, Robinson-Rechavi M, Basian FB. 2016. BgeeDB, an R package for retrieval of curated expression datasets and for gene list expression localization enrichment tests. F1000Res 5: 2748. 10.12688/f1000research.9973.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R, Sugawara H, Shumway M, International Nucleotide Sequence Database Collaboration. 2011. The sequence read archive. Nucleic Acids Res 39: D19–D21. 10.1093/nar/gkq1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KH, Ferraris L, Filloux ME, Raphael BJ, Fairbrother WG. 2011. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc Natl Acad Sci USA 108: 11093–11098. 10.1073/pnas.1101135108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Taylor DW, Krementsova EB, Trybus KM, Taylor KA. 2006. Three-dimensional structure of the myosin V inhibited state by cryoelectron tomography. Nature 442: 208–211. 10.1038/nature04719 [DOI] [PubMed] [Google Scholar]
- Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, et al. 2013. The genotype-tissue expression (GTEx) project. Nat Genet 45: 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louadi Z, Yuan K, Gress A, Tsoy O, Kalinina OV, Baumbach J, Kacprowski T, List M. 2020. Digger: exploring the functional role of alternative splicing in protein interactions. Nucleic Acids Res 49(D1): D309.– . 10.1093/nar/gkaa768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Solano M, Crilly SE, Malinverni D, Munk C, Harris M, Pearce A, Quon T, Mackenzie AE, Wang X, Peng J, et al. 2020. Combinatorial expression of GPCR isoforms affects signalling and drug responses. Nature 588: E24. 10.1038/s41586-020-2999-9 [DOI] [PubMed] [Google Scholar]
- Mei W, Boatwright L, Feng G, Schnable JC, Barbazuk WB. 2017. Evolutionarily conserved alternative splicing across monocots. Genetics 207: 465–480. 10.1534/genetics.117.300189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, Burge CB. 2012. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 338: 1593–1599. 10.1126/science.1228186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek B, Lee CJ. 2003. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat Genet 34: 177–180. 10.1038/ng1159 [DOI] [PubMed] [Google Scholar]
- Nichio BT, Marchaukoski JN, Raittz RT. 2017. New tools in orthology analysis: a brief review of promising perspectives. Front Genet 8: 165. 10.3389/fgene.2017.00165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Pan Z, Zhang Z, Lin L, Xing Y. 2018. The expanding landscape of alternative splicing variation in human populations. Am J Hum Genet 102: 11–26. 10.1016/j.ajhg.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathan-Chhatbar S, Taft MH, Reindl T, Hundt N, Latham SL, Manstein DJ. 2018. Three mammalian tropomyosin isoforms have different regulatory effects on nonmuscle myosin-2B and filamentous β-actin in vitro. J Biol Chem 293: 863–875. 10.1074/jbc.M117.806521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JM, Maietta P, Ezkurdia I, Pietrelli A, Wesselink JJ, Lopez G, Valencia A, Tress ML. 2013. APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res 41(Database issue): D110–D117. 10.1093/nar/gks1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL. 2019. Next-generation genome annotation: we still struggle to get it right. Genome Biol 16: 92. 10.1186/s13059-019-1715-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti MM, Swanson MS. 2016. RNA mis-splicing in disease. Nat Rev Genet 17: 19–32. 10.1038/nrg.2015.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Haussler D. 2005. Phylogenetic hidden Markov models. In Statistical methods in molecular evolution (ed. Nielsen R), pp. 325–351. Springer, New York. [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050. 10.1101/gr.3715005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloutsky R, Stratton MM. 2020. Functional implications of CaMKII alternative splicing. Eur J Neurosci 10.1111/ejn.14761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterne-Weiler T, Weatheritt RJ, Best AJ, Ha KC, Blencowe BJ. 2018. Efficient and accurate quantitative profiling of alternative splicing patterns of any complexity on a laptop. Mol Cell 72: 187–200.e6. 10.1016/j.molcel.2018.08.018 [DOI] [PubMed] [Google Scholar]
- Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, et al. 2008. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321: 956–960. 10.1126/science.1160342 [DOI] [PubMed] [Google Scholar]
- Szalkowski AM. 2012. Fast and robust multiple sequence alignment with phylogeny-aware gap placement. BMC Bioinformatics 13: 129. 10.1186/1471-2105-13-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapial J, Ha KCH, Sterne-Weiler T, Gohr A, Braunschweig U, Hermoso-Pulido A, Quesnel-Vallières M, Permanyer J, Sodaei R, Marquez Y, et al. 2017. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res 27: 1759–1768. 10.1101/gr.220962.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25: 4876–4882. 10.1093/nar/25.24.4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranchevent LC, Aubé F, Dulaurier L, Benoit-Pilven C, Rey A, Poret A, Chautard E, Mortada H, Desmet FO, Chakrama FZ, et al. 2017. Identification of protein features encoded by alternative exons using exon ontology. Genome Res 27: 1087–1097. 10.1101/gr.212696.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waetzig V, Herdegen T. 2005. Context-specific inhibition of JNKs: overcoming the dilemma of protection and damage. Trends Pharmacol Sci 26: 455–461. 10.1016/j.tips.2005.07.006 [DOI] [PubMed] [Google Scholar]
- Wang GS, Cooper TA. 2007. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 8: 749–761. 10.1038/nrg2164 [DOI] [PubMed] [Google Scholar]
- Wang L, Jiang T. 1994. On the complexity of multiple sequence alignment. J Comput Biol 1: 337–348. 10.1089/cmb.1994.1.337 [DOI] [PubMed] [Google Scholar]
- Wang P, Wu YL, Zhou TH, Sun Y, Pei G. 2000. Identification of alternative splicing variants of the β subunit of human Ca2+/calmodulin-dependent protein kinase II with different activities. FEBS Lett 475: 107–110. 10.1016/S0014-5793(00)01634-3 [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476. 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Codreanu SG, Wen B, Li K, Chambers MC, Liebler DC, Zhang B. 2018. Detection of proteome diversity resulted from alternative splicing is limited by trypsin cleavage specificity. Mol Cell Proteomics 17: 422–430. 10.1074/mcp.RA117.000155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AJ, Cooper TA. 2010. The pathobiology of splicing. J Pathol 220: 152–163. 10.1002/path.2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatheritt RJ, Sterne-Weiler T, Blencowe BJ. 2016. The ribosome-engaged landscape of alternative splicing. Nat Struct Mol Biol 23: 1117–1123. 10.1038/nsmb.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Liu J, Reedy MC, Tregear RT, Winkler H, Franzini-Armstrong C, Sasaki H, Lucaveche C, Goldman YE, Reedy MK, et al. 2010. Electron tomography of cryofixed, isometrically contracting insect flight muscle reveals novel actin-myosin interactions. PLoS One 5: e12643. 10.1371/journal.pone.0012643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Lee C. 2005. Assessing the application of Ka/Ks ratio test to alternatively spliced exons. Bioinformatics 21: 3701–3703. 10.1093/bioinformatics/bti613 [DOI] [PubMed] [Google Scholar]
- Yang X, Coulombe-Huntington J, Kang S, Sheynkman GM, Hao T, Richardson A, Sun S, Yang F, Shen YA, Murray RR, et al. 2016. Widespread expansion of protein interaction capabilities by alternative splicing. Cell 164: 805–817. 10.1016/j.cell.2016.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, et al. 2016. Ensembl 2016. Nucleic Acids Res 44: D710–D716. 10.1093/nar/gkv1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.