

Abstract
Efficient degradation of by‐products of protein biogenesis maintains cellular fitness. Strikingly, the major biosynthetic compartment in eukaryotic cells, the endoplasmic reticulum (ER), lacks degradative machineries. Misfolded proteins in the ER are translocated to the cytosol for proteasomal degradation via ER‐associated degradation (ERAD). Alternatively, they are segregated in ER subdomains that are shed from the biosynthetic compartment and are delivered to endolysosomes under control of ER‐phagy receptors for ER‐to‐lysosome‐associated degradation (ERLAD). Demannosylation of N‐linked oligosaccharides targets terminally misfolded proteins for ERAD. How misfolded proteins are eventually marked for ERLAD is not known. Here, we show for ATZ and mutant Pro‐collagen that cycles of de‐/re‐glucosylation of selected N‐glycans and persistent association with Calnexin (CNX) are required and sufficient to mark ERAD‐resistant misfolded proteins for FAM134B‐driven lysosomal delivery. In summary, we show that mannose and glucose processing of N‐glycans are triggering events that target misfolded proteins in the ER to proteasomal (ERAD) and lysosomal (ERLAD) clearance, respectively, regulating protein quality control in eukaryotic cells.
Keywords: ERAD, ERLAD, ER‐phagy, N‐glycan processing, Protein quality control
Subject Categories: Autophagy & Cell Death; Membrane & Intracellular Transport; Post-translational Modifications, Proteolysis & Proteomics
Cycles of N‐glycan deglucosylation/glucosylation and persistent binding to Calnexin target α1‐antitrypsin‐Z polymers and misfolded pro‐collagen to ERLAD in mammalian cells.
Introduction
Proteins synthesized in the ER are covalently modified with pre‐assembled oligosaccharides on asparagine residues in ‐N‐X‐S/T‐ sequons (Fig 1A, single letter code for asparagine (N), any amino acid but proline (X), serine (S), or threonine (T)) (Aebi et al, 2010). Rapid removal of glucose residues 1 and 2 by sequential action of the castanospermine (CST)‐sensitive ER α‐glucosidases I and II (GI and GII, Fig 1A) (Elbein, 1991) generates a mono‐glucosylated trimming intermediate that recruits Calnexin (CNX) (Hammond et al, 1994). CNX is an ER lectin chaperone that assists folding of newly synthesized polypeptides and protects them from unwanted selection for ERAD via the ubiquitin proteasome system (UPS) (Moore & Spiro, 1993; Hammond et al, 1994; Helenius, 1994; Kearse et al, 1994; Hebert et al, 1996; Liu et al, 1997, 1999; Ayalon‐Soffer et al, 1999; Molinari et al, 2002; Molinari et al, 2003; Oda et al, 2003; Helenius & Aebi, 2004; Molinari, 2007; Olivari & Molinari, 2007; Lederkremer, 2009; Aebi et al, 2010; Adams et al, 2019; Sun & Brodsky, 2019). Folding‐defective polypeptides are subject of extensive de‐mannosylation of their N‐linked oligosaccharides (i.e., removal of the light green mannose residues by members of the glycosyl hydrolase 47 family including the kifunensine (KIF)‐sensitive ER‐resident α‐mannosidase I (αMI) and EDEM proteins, Fig 1A) (Moremen & Molinari, 2006). This has two consequences: first, removal of mannose A (Fig 1A) eliminates the acceptor for the re‐glucosylation of the oligosaccharide’s branch A by the ER quality control factor UDP‐glucose:glycoprotein glucosyltransferase (UGGT1, Fig 1A). This prevents the generation of mono‐glucosylated N‐glycans and irreversibly extracts non‐native polypeptides from the CNX chaperone system (Olivari et al, 2006). Second, it engages ER lectins that bind N‐linked oligosaccharides displaying dark green terminal mannoses (Fig 1A). These mannose‐binding lectins (OS‐9 and XTP3‐B) shuttle misfolded polypeptides to supramolecular complexes built around E3 ubiquitin ligases. This promotes dislocation of terminally misfolded polypeptides across the ER membrane for degradation by cytosolic proteasomes (ERAD) (Bernasconi et al, 2008; Christianson et al, 2008; Hosokawa et al, 2009; Bernasconi et al, 2010; Sun & Brodsky, 2019).
Figure 1. N‐glycan processing and delivery of ATZNNN to endolysosomes.
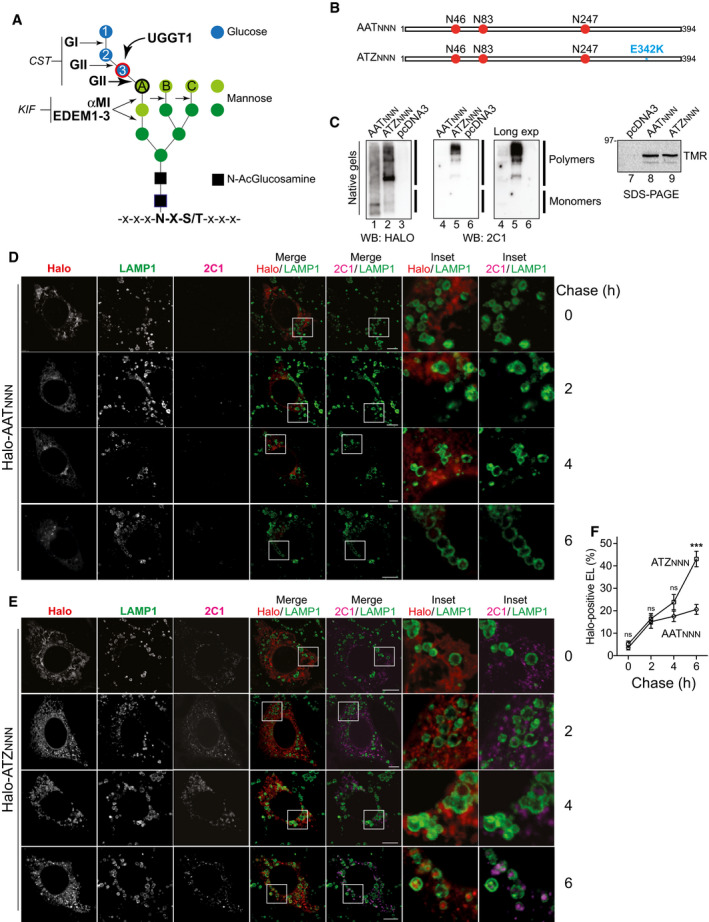
- N‐glycan. Glucoses, blue; mannoses, green; N‐acetylglucosamines, black. GI removes glucose 1. GII removes glucoses 2 and 3. UGGT1 adds‐back glucose 3. UGGT1 can only modify the terminal mannose on branch A (black circle). αMI and EDEM1, EDEM2, and EDEM3 remove light green, α1,2‐bonded, mannose residues. CST inhibits GI and GII. KIF inhibits α1,2mannosidases.
- AATNNN and ATZNNN. N‐glycans in red. The E342K mutation of ATZNNN is in blue.
- Control of polymerization propensity for the Halo‐tagged versions of AAT (lanes 1 and 4) and of ATZ (lanes 2 and 5). Polymers and monomers corresponding to (Miranda et al, 2010; Ronzoni et al, 2021) are seen with anti‐Halo (lanes 1‐3) or with the polymer‐specific 2C1 antibody (lanes 4‐6). Synthesis control in SDS‐polyacrylamide gel by visualization of tetramethylrhodamine (TMR) ligand‐labeled Halo‐AATNNN and Halo‐ATZNNN (lanes 7‐9).
- HaloTag pulse‐chase analysis in WT MEF monitoring fluorescent Halo‐AATNNN during up to 6 h of chase in the presence of BafA1. The fluorescent JF646‐Halo ligand and the polymer‐specific 2C1 antibody stains are shown.
- Same as (D) for fluorescent Halo‐ATZNNN.
- Quantification of accumulation of fluorescent Halo‐AATNNN and Halo‐ATZNNN within LAMP1‐positive endolysosomes. Mean ± SEM, n = 20, 11, 10, 13 for Halo‐AATNNN, n = 18, 12, 12, 13 for Halo‐ATZNNN. Two‐way ANOVA and Sidak’s multiple comparison test, ns P > 0.05, ***P < 0.001.
Data information: Scale bars 10 µm.
Source data are available online for this figure.
Misfolded proteins that cannot be dislocated across the ER membrane for ERAD are segregated in specialized ER subdomains displaying ER‐phagy receptors at the limiting membrane (Fregno et al, 2018; Schultz et al, 2018; Cui et al, 2019; De Leonibus et al, 2019; Forrester et al, 2019; Fregno & Molinari, 2019; Hubner & Dikic, 2020; Wilkinson, 2020; Molinari, 2021). ER‐phagy receptors including FAM134B drive, with contribution of yet elusive additional factors, the scission of these ER subdomains from the bulk ER (Bhaskara et al, 2019; Jiang et al, 2020) and recruit select cytosolic autophagy gene products. These control delivery to lysosomes and clearance of the ER‐derived vesicles containing misfolded proteins via client‐specific avenues including LC3‐dependent vesicular transport (Fregno et al, 2018), macro‐ (Cui et al, 2019; Forrester et al, 2019), or micro‐ER‐phagy (Omari et al, 2018). In this manuscript, we use the term ER‐to‐Lysosome‐Associated Degradation (ERLAD) to define the catabolic pathways that remove ERAD‐resistant misfolded proteins from the ER (Fregno et al, 2018; De Leonibus et al, 2019; Forrester et al, 2019; Fregno & Molinari, 2019; Sun & Brodsky, 2019; Wilkinson, 2019, 2020; Hubner & Dikic, 2020; Molinari, 2021).
The Z mutation of the SERPINA1 gene characterizes about 95% of the patients affected by alpha‐1 antitrypsin (AAT) deficiency, a lung and liver disease. The Z mutation results in a Glu342Lys substitution that encodes for alpha‐1 antitrypsin‐Z (ATZ) (Strnad et al, 2020). Misfolded ATZ is translocated across the ER membrane for proteasomal degradation. However, a significant fraction of the polypeptide undergoes polymerization and cannot be translocated across the ER membrane. Instead, ATZ polymers are delivered to the endolysosomal district in an ERLAD pathway that relies on ATG5 and ATG7 (Teckman & Perlmutter, 2000; Kamimoto et al, 2006; Kroeger et al, 2009; Hidvegi et al, 2010; Perlmutter, 2011; Pastore et al, 2013; Yamamura et al, 2014; Fregno et al, 2018; Cui et al, 2019; Strnad et al, 2020). ATG5 and ATG7 regulate the covalent association of the ubiquitin‐like protein LC3 with the membrane lipid phosphatidyl ethanolamine. Lipidated LC3 displayed at the limiting membrane of autophagosomes or endolysosomes is engaged by autophagy receptors in catabolic processes, where cells clear their own components (Bento et al, 2016).
We and others recently identified FAM134B (and Atg40p, the yeast paralogue of FAM134B) as the LC3‐binding ER‐phagy receptor that regulates removal of ER portions containing ERAD‐resistant misfolded proteins from mammalian or yeast cells (Fregno et al, 2018; Schultz et al, 2018; Cui et al, 2019; Forrester et al, 2019). Relevant for this study, ERAD‐resistant ATZ polymers (Fregno et al, 2018) are segregated in ER subdomains enriched in FAM134B. These vesiculate and ER‐derived vesicles directly release their content within LAMP1/RAB7‐positive endolysosomes under control of the SNAREs Syntaxin17 and VAMP8. Misfolded Pro‐collagen is also segregated in FAM134B‐positive ER‐derived vesicles, but these are first captured by autophagosomes that then fuse with endolysosomes (Forrester et al, 2019).
Puzzlingly, CNX, which we recall is an ER‐resident lectin that protects misfolded N‐glycosylated polypeptides from proteasomal degradation, is a major interactor of the ER‐phagy receptor FAM134B (Grumati et al, 2017; Fregno et al, 2018). CNX deletion significantly inhibits FAM134B‐driven ERLAD of ATZ polymers via LC3‐dependent vesicular transport (Fregno et al, 2018) and FAM134B‐driven ERLAD of misfolded Pro‐collagen via macro‐ER‐phagy (Forrester et al, 2019). This led us to hypothesize that the role of N‐glycans as quality control appendices, whose extensive de‐mannosylation determines selection of misfolded proteins for ERAD, could also extend to define, upon appropriate processing, a signal that marks aberrant polypeptides for ERLAD.
Results
Formation and delivery of ATZ polymers to endolysosomes for clearance
The wild‐type AAT protein and the disease‐causing ATZ variant display 3 N‐glycans on asparagine residues at position N46, N83, and N247 (Fig 1B, AATNNN and ATZNNN, respectively). The Z mutation (blue in Fig 1B) enhances formation of ERAD‐resistant polymers, which are delivered to lysosomal compartments for clearance both in mammalian and in yeast cells (Teckman & Perlmutter, 2000; Kamimoto et al, 2006; Kroeger et al, 2009; Hidvegi et al, 2010; Perlmutter, 2011; Pastore et al, 2013; Yamamura et al, 2014; Fregno et al, 2018; Cui et al, 2019; Strnad et al, 2020). ATZNNN polymers are recognized by the monoclonal antibody 2C1 (Miranda et al, 2010; Ronzoni et al, 2021). They are transported from the ER to degradative endolysosomal compartments, where they accumulate upon inhibition of lysosomal hydrolases (Fregno et al, 2018). Separation of Halo‐tagged versions of AATNNN and ATZNNN in non‐denaturing gels confirms the higher polymerization propensity of the latter (Fig 1C, lane 2 vs. 1 and 5 vs. 4). The fate of AATNNN and ATZNNN has been monitored in mouse embryonic fibroblasts (MEF) by HaloTag pulse‐chase, which allows time‐resolved analyses of the intracellular trafficking of fluorescently labeled proteins in confocal laser scanning microscopy (Fregno et al, 2018). Briefly, cells expressing Halo‐AATNNN (Fig 1D) or Halo‐ATZNNN (Fig 1E) are shortly incubated with the “black ligand” 6‐Chlorohexanol to occupy the ligand binding pocket of the HaloTag. Subsequently, cells are incubated with the cell permeable HaloTag fluorescent ligand JF646 (fluorescent pulse of newly synthesized Halo‐AATNNN or Halo‐ATZNNN). After 15 min, JF646 is replaced by the “black ligand” 7‐Bromoheptanol to stop fluorescence incorporation and start a chase of 0, 2, 4, or 6 h (Fig 1D–F). Bafilomycin A1 (BafA1) is added to the cell culture media during the chase to ensure accumulation within the LAMP1‐positive compartments of the fluorescently labeled protein selected for lysosomal clearance (Klionsky et al, 2008; Fregno et al, 2018). The 15 min pulse generates a population of fluorescent Halo‐AATNNN (Fig 1D, Halo, 0 h chase) or Halo‐ATZNNN (Fig 1E, Halo, 0 h chase). Image analyses and unbiased quantification with LysoQuant, a deep learning approach for segmentation and classification of fluorescence images (Morone et al, 2020), show significant and progressive accumulation within LAMP1‐positive endolysosomes only for Halo‐ATZNNN (Fig 1E, 2C1/LAMP1 and Insets, quantification in Fig 1F, 0 to 6 h chase). This conforms to the fact that the secretory protein AATNNN is efficiently released from cells, whereas a fraction of ATZNNN enters polymers that are retained intracellularly and are eventually cleared by lysosomal enzymes (Fig 2B) (Teckman & Perlmutter, 1996, 2000; Kamimoto et al, 2006; Kroeger et al, 2009; Hidvegi et al, 2010; Perlmutter, 2011; Pastore et al, 2013; Yamamura et al, 2014; Fregno et al, 2018; Cui et al, 2019).
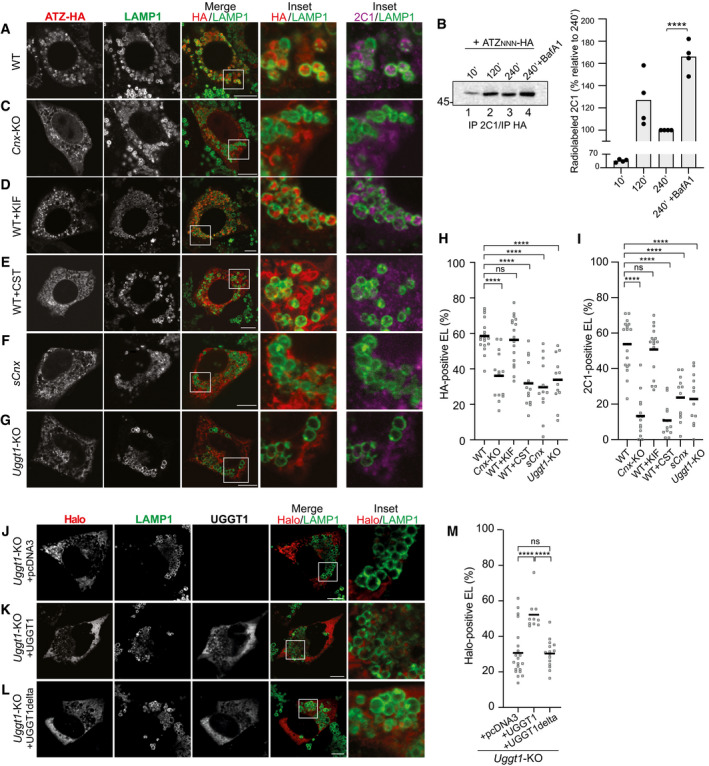
Figure 2. Role of CNX and of N‐glycan processing in delivery of ATZNNN to the endolysosomes.
-
AConfocal laser scanning microscopy analyses in cells treated with BafA1. Total ATZNNN‐HA (red) or ATZNNN polymers (magenta) in LAMP1‐positive endolysosome (green) in WT MEF.
-
BRadiolabeled ATZNNN enters in 2C1‐positive polymers that are immunoisolated at the end of the chase times. BafA1 inhibits polymers degradation. Quantification for n = 4. Unpaired two‐tailed t‐test, ****P < 0.0001.
-
CSame as (A) in Cnx‐KO MEF.
-
DSame as (A) in WT MEF exposed to kifunensine (KIF).
-
ESame as (A) in WT MEF exposed to Castanospermine (CST).
-
FSame as (A) in sCNX MEF.
-
GSame as (A) in Uggt1‐KO MEF.
-
H, I(H) Quantification of HA‐positive or (I) of 2C1‐positive endolysosomes (mean, n = 18, 14, 17, 13, 12, 12 cells for panels (A, C–G), respectively). One‐way ANOVA and Dunnett’s multiple comparison test, ns P > 0.05, ****P < 0.0001.
-
JHalo‐ATZNNN in Uggt1‐KO MEF mock‐transfected (empty pcDNA3 plasmid).
-
K, L(K) Same as (J), with a plasmid for expression of active or (L) inactive UGGT1.
-
MQuantification of Halo‐ATZNNN‐positive endolysosomes (mean, n = 22, 10, 15 cells for panels (J–L), respectively). One‐way ANOVA and Dunnett’s multiple comparison test, ns P > 0.05, ****P < 0.0001.
Data information: Scale bars 10 μm.
Source data are available online for this figure.
The ER‐phagy receptor FAM134B, and not the functional homologs SEC62 and TEX264, is involved in lysosomal delivery of ERAD‐resistant ATZNNN polymers
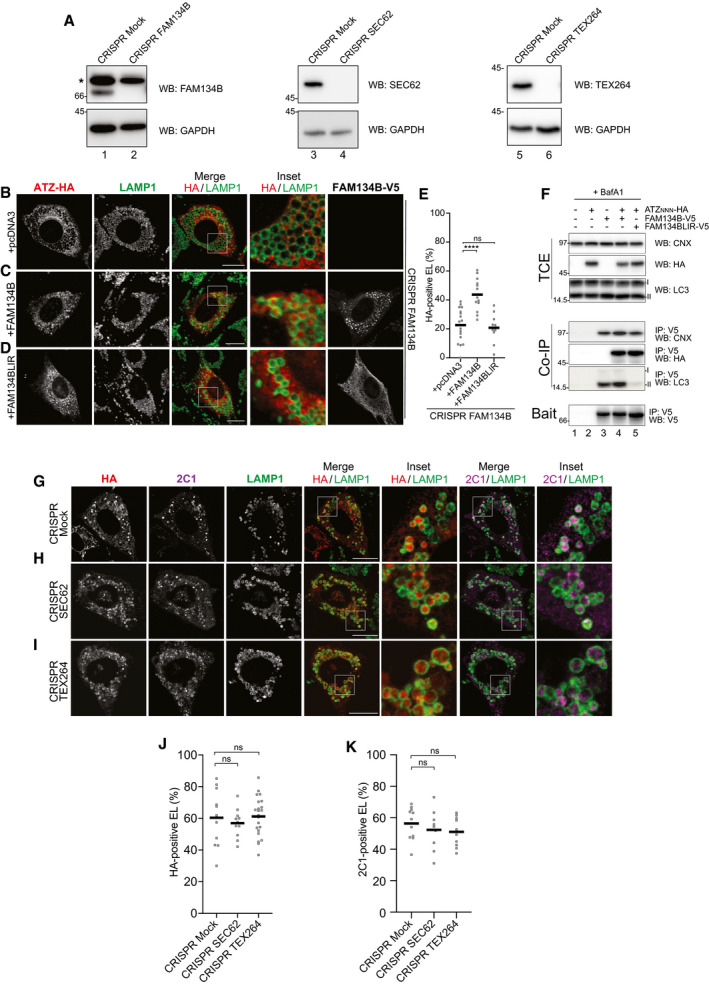
In wild‐type MEF, confocal laser scanning microscopy with anti‐HA (Fig 2A, HA/LAMP1, Fig 2H) and 2C1 antibodies (Fig 2A, 2C1/LAMP1, Fig 2I) reports on delivery of HA‐tagged ATZNNN polymers within LAMP1‐positive endolysosomes, where ATZNNN accumulates upon inactivation of hydrolytic enzymes with BafA1 (Fig 2A). Consistently, cell exposure to BafA1 substantially delays the clearance of 2C1‐positive polymers (Fig 2B) (and thoroughly analyzed in (Fregno et al, 2018)). In MEF lacking the ER‐phagy receptor FAM134B (Fig EV1A, lanes 1–2), delivery of ATZNNN polymers within the LAMP1‐positive degradative compartments is defective (Fig EV1B and E, cells expressing HA‐tagged ATZNNN and mock‐transfected with empty pcDNA3 plasmid). Delivery of ATZNNN polymers within endolysosomes is rescued upon back‐transfection of V5‐tagged FAM134B (Fig EV1C and E), but not upon back‐transfection of FAM134BLIR (Fig EV1D and E). This latter is a mutant form of FAM134B, where the LC3‐interacting region ‐453DDFELL458‐ has been mutated to ‐453AAAAAA458‐, to abolish LC3 binding and to render the ER‐phagy receptor inactive in starvation‐induced ER‐phagy and in ERLAD (Khaminets et al, 2015; Fregno et al, 2018). A co‐immunoprecipitation assay confirms that both FAM134B and FAM134BLIR do associate with CNX (Fig EV1F, Co‐IP, lanes 4 and 5, upper panel) (Fregno et al, 2018) and with the ERLAD client ATZNNN (Co‐IP, lanes 4 and 5, middle panel). However, only FAM134B engages LC3 (Co‐IP, lanes 4 and 5, lower panel), which is required for formation of ER‐derived vesicles containing polymers and their delivery to the degradative endolysosomal district (Fig EV1C–E) (Fregno et al, 2018). Deletion of SEC62 or of TEX264 (Fig EV1A, lanes 3‐4 and 5‐6, respectively), two ER‐phagy receptors that regulate ER clearance during recovery from ER stress (Fumagalli et al, 2016; Loi et al, 2019; Loi & Molinari, 2020) or upon nutrient deprivation (An et al, 2019; Chino et al, 2019; Liang et al, 2020), does not affect delivery of polymeric ATZNNN to the degradative LAMP1‐positive compartment (Fig EV1G–K). These results reveal that FAM134B is required and sufficient for lysosomal clearance of ATZNNN polymers generated in the ER (Fregno et al, 2018).
Figure EV1. ER‐phagy receptors involvement in delivery of ATZNNN polymers to endolysosomal compartments.
-
AControl of genetic Fam134B‐, Sec62‐, and Tex264‐KO, respectively. * shows a non‐specific cross‐reaction of the antibody.
- B
-
CSame as (B) in cells back‐transfected with active FAM134B.
-
DSame as (B) in cells back‐transfected with inactive FAM134B (FAM134BLIR), which cannot bind LC3.
-
EQuantification of HA‐positive endolysosomes (B‐D) (mean, n = 17, 13 and 10). One‐way ANOVA and Dunnett’s multiple comparison test, ns P > 0.05, ****P < 0.0001.
-
FHEK293 cells transfected with empty vector (lane 1), ATZ‐HA (2), FAM134B‐V5 (3), FAM134B‐V5 and ATZ‐HA (4), or FAM134BLIR‐V5 and ATZ‐HA (5), treated for 6 h with 100 nM BafA1 and then cross‐linked with DSP before lysis (see Materials and Methods). FAM134BLIR‐V5 interacts with CNX and ATZ‐HA, but not with LC3.
-
G–I(G) Same as (B) in control CRISPR cells and (H) in cells edited with CRISPR/Cas9 to delete Sec62 (Fumagalli et al, 2016) or (I) Tex264.
-
J, K(J) Quantification of (G–I) for HA‐positive endolysosomes, (mean, n = 11, 11, 22) and (K) for 2C1‐positive endolysosomes (mean, n = 11, 9, 11). One‐way ANOVA and Dunnett’s multiple comparison test, ns P > 0.05.
Data information: Scale bars 10 μm.
Source data are available online for this figure.
CNX and glucose processing enzymes regulate delivery of ERAD‐resistant ATZNNN polymers (and misfolded Pro‐Col2A1) to LAMP1‐positive endolysosomes
N‐glycosylation and processing of N‐linked oligosaccharides (i.e., deglucosylation by GI and GII, re‐glucosylation by the UGGT1, de‐mannosylation by αMI and EDEM proteins, Fig 1A) are crucial events in ER protein quality control. They enhance solubility of unfolded nascent chains in the crowded ER environment and determine their access and retention in the CNX chaperone system. CNX association promotes structural maturation and protects not‐yet‐folded polypeptides from unwanted selection for ERAD (Moore & Spiro, 1993; Hammond et al, 1994; Helenius, 1994; Kearse et al, 1994; Hebert et al, 1996; Ayalon‐Soffer et al, 1999; Liu et al, 1999; Molinari et al, 2002; Molinari et al, 2003; Oda et al, 2003; Helenius & Aebi, 2004; Molinari, 2007; Olivari & Molinari, 2007; Lederkremer, 2009; Aebi et al, 2010; Adams et al, 2019; Sun & Brodsky, 2019).
Formation of 2C1‐immunoreactive ATZNNN polymers does not require the lectin chaperone CNX (Fig 2C, panel 2C1, Fig EV2A, Cnx‐KO, Fig EV2B, lane 4) (Fregno et al, 2018). However, delivery of ATZNNN polymers within the degradative LAMP1‐positive compartment for clearance is substantially impaired in Cnx‐KO MEF (Fig 2C, 2C1/LAMP1, Fig 2H and I). Characterization of another folding‐defective polypeptide, the disease‐causing Arg989Cys (R989C) variant of Pro‐Col2A1 (Fig EV3A) (Hintze et al, 2008; Zhang et al, 2011), another emblematic ERLAD client (Fig EV3B–F) (Forrester et al, 2019), confirms defective polypeptides delivery from the ER to degradative endolysosomes in cells lacking CNX (Figs EV3G and 3H and M). These data confirm that delivery of misfolded polypeptides from the ER to the endolysosomal district (i.e., ERLAD) requires CNX association (Fregno et al, 2018; Forrester et al, 2019). Interestingly, dislocation across the ER membrane and proteasomal degradation of misfolded polypeptides (i.e., ERAD) is in contrast prevented by CNX association (Moore & Spiro, 1993; Hammond et al, 1994; Helenius, 1994; Kearse et al, 1994; Hebert et al, 1996; Liu et al, 1997, 1999; Ayalon‐Soffer et al, 1999; Molinari et al, 2002; Molinari et al, 2003; Oda et al, 2003; Helenius & Aebi, 2004; Molinari, 2007; Olivari & Molinari, 2007; Lederkremer, 2009; Aebi et al, 2010; Adams et al, 2019; Sun & Brodsky, 2019).
Figure EV2. Control of polymerization propensity.
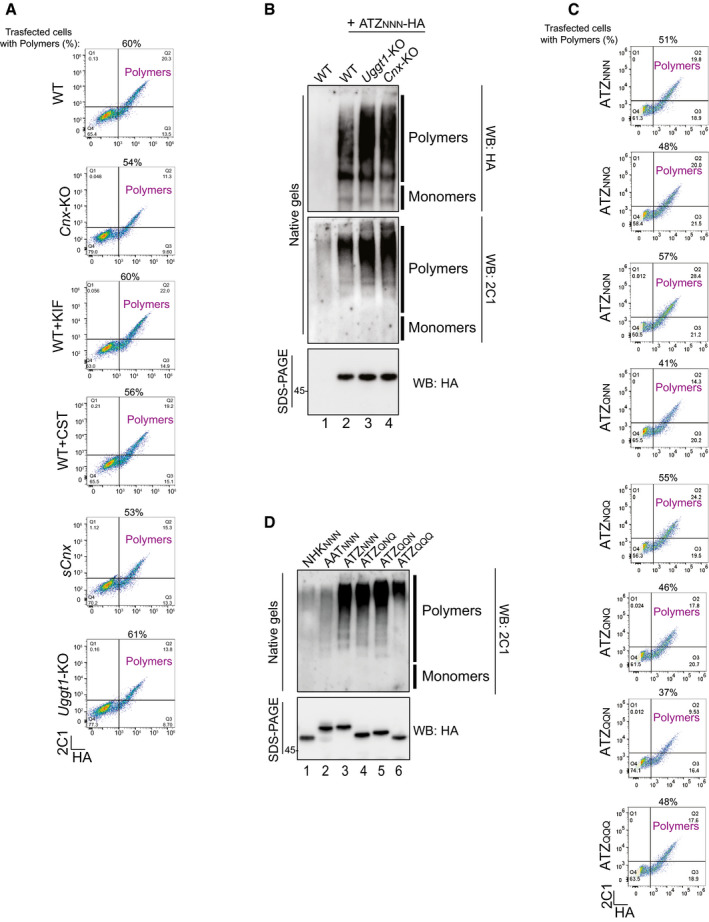
- Flow cytometric analyses of intracellular ATZNNN detected with anti‐HA (X‐axis) and anti‐2C1 antibody (Y‐axis). Cells containing ATZNNN polymers are in the upper right quadrant being immunoreactive to both anti‐HA and 2C1 antibody. Analyses have been performed for the conditions and cell lines shown in Fig 2A–G.
- Control of polymerization propensity for the ATZNNN in WT (lanes 1, 2), Uggt1‐KO (lane 3), and Cnx‐KO MEF (lane 4). Polymers and monomers are seen with anti‐HA (upper panel) or with the polymer‐specific 2C1 antibody (middle panel). Total protein in SDS‐polyacrylamide gels in lower panel.
- Same as (A) for cells expressing glycosylation mutants as in Fig 3B–I.
- Same as (B) for the 4 glycosylation mutants shown in Fig 4, for AAT and for the non‐polymerogenic, disease‐causing NHK variant of AAT expressed in HEK293 cells.
Source data are available online for this figure.
Figure EV3. Requirements for endolysosomal delivery of Pro‐collagens.
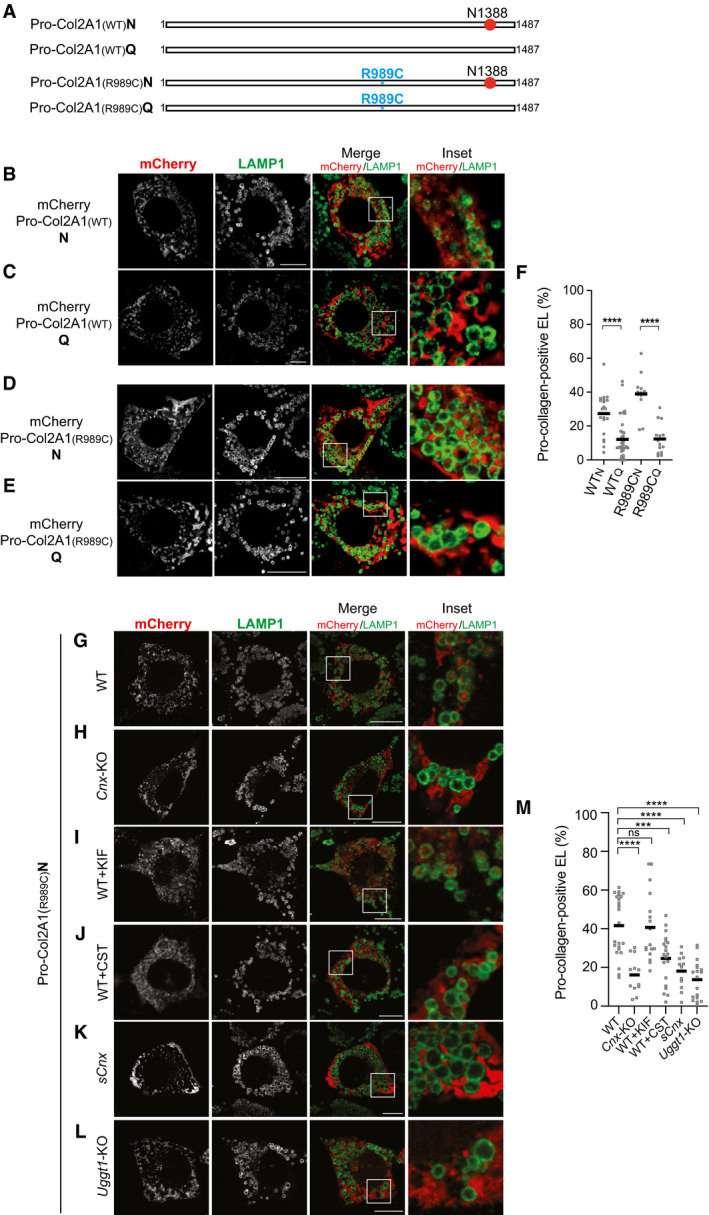
-
ASchematic representation of glycosylated Pro‐Col2A1(WT)N and Pro‐Col2A1(R989C)N and of non‐glycosylated Pro‐Col2A1(WT)Q and Pro‐Col2A1(R989C)Q (N‐glycan in red, mutation in blue).
-
B–EConfocal laser scanning microscopy analyses as in Fig 2, for Pro‐Col2A1(WT)N/Q and Pro‐Col2A1(R989C)N/Q.
-
FQuantification of Pro‐collagen‐positive endolysosomes (B–E) (mean, n = 21, 35, 13 and 15 cells). One‐way ANOVA and Dunnett’s multiple comparison test, ****P < 0.0001.
-
GSame as (D).
-
HSame as (G) in Cnx‐KO cells.
-
ISame as (G) in kifunensine (KIF)‐treated cells.
-
JSame as (G) in castanospermine (CST)‐treated cells.
-
KSame as (G) in sCnx cells.
-
LSame as (G) in Uggt1‐KO cells.
-
MQuantification of (G–L) (mean, n = 23, 12, 17, 20, 12 and 18 cells). One‐way ANOVA and Dunnett’s multiple comparison test, ns P > 0.05, ***P < 0.001, ****P < 0.0001.
Data information: Scale bars 10 μm.
Figure 3. N‐glycosylation at position 83 is required and sufficient for ATZ delivery to endolysosomes.
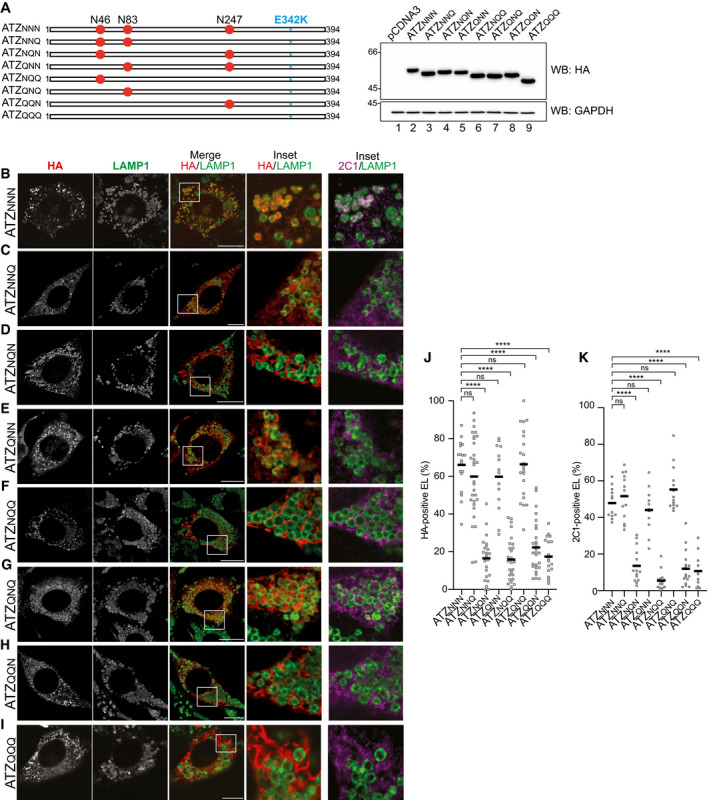
-
AATZ glycosylation mutants and their electrophoretic mobility. N is the acceptor site asparagine; Q is for the mutation to glutamine that prevents glycosylation. The E342K mutation of ATZNNN is in blue.
-
B–IConfocal laser scanning microscopy analyses (as in Fig 2) in WT MEF treated with BafA1.
-
J, K(J) Quantification of ATZxxx‐positive endolysosomes and (K) of ATZxxx polymers‐positive endolysosomes (mean, n = 18, 30, 24, 14, 27, 22, 27, 18 cells for HA stain and n = 12, 14, 14, 11, 14, 14, 17, 11 cells for 2C1 stain of panels B‐I, respectively). One‐way ANOVA and Dunnett’s multiple comparison test, ns P > 0.05, ****P < 0.0001.
Data information: Scale bars 10 μm.
Source data are available online for this figure.
Further experiments reveal that pharmacologic modulation of N‐glycan processing affects ERLAD and ERAD pathways differently. For instance, lysosomal delivery of ATZNNN polymers is unaffected in cells exposed to KIF (Figs 2D and H and I and EV3I and M for Pro‐Col2A1R989C). Thus, mannose removal from N‐glycans, which is a potent trigger of ERAD (Aebi et al, 2010; Adams et al, 2019; Sun & Brodsky, 2019), is dispensable for ERLAD. On the same line, CST, which abolishes glycoproteins association with CNX and substantially accelerates ERAD (Aebi et al, 2010; Adams et al, 2019; Sun & Brodsky, 2019), drastically reduces the selection of ERAD‐resistant ATZNNN polymers for lysosomal delivery (Figs 2E and H and I and EV3J and M for Pro‐Col2A1R989C).
FAM134B is inserted in the ER membrane but it does not protrude in the ER lumen (Khaminets et al, 2015). Thus, client association is mediated by its interacting partner CNX (Fregno et al, 2018; Forrester et al, 2019). Notably, lysosomal delivery of ATZNNN is defective in cells that express a short (s)Cnx variant (Figs 2F and H and I, and EV3K and M for Pro‐Col2A1R989C). sCNX conserves the transmembrane and cytosolic domains that ensure the formation of the functional complex with FAM134B. However, it is characterized by splicing between bases 307‐725, corresponding to a deletion from residues 104 to 242 in the polypeptide chain. The deletion removes D118, C161, Y165, K167, Y186, M189, C195, E217, which are essential for the lectin function of CNX (Denzel et al, 2002; Pieren et al, 2005). Consistently, like CNX (Fig EV1F, Co‐IP, upper panel, lanes 3‐5, Fig EV4A, Co‐IP, lower panel, lane 1), sCnx binds FAM134B (Fig EV4A, Co‐IP, lower panel, lane 2). However, lacking the lectin site, sCnx does not bind clients (Denzel et al, 2002; Pieren et al, 2005) and, consistently, it does not bind ATZNNN (Fig EV4A, Co‐IP, upper panel, lane 2). This prevents the formation of a complex that contains functional FAM134B:lipidated LC3‐II:CNX:ATZ, which is required for productive ERLAD (Fig EV1F, Co‐IP, lanes 3‐5 and Fig EV4A, Co‐IP, lanes 1‐4) (Fregno et al, 2018).
Figure EV4. FAM134B:CNX:ATZNNN complexes and intracellular localization of ATZ polymers.
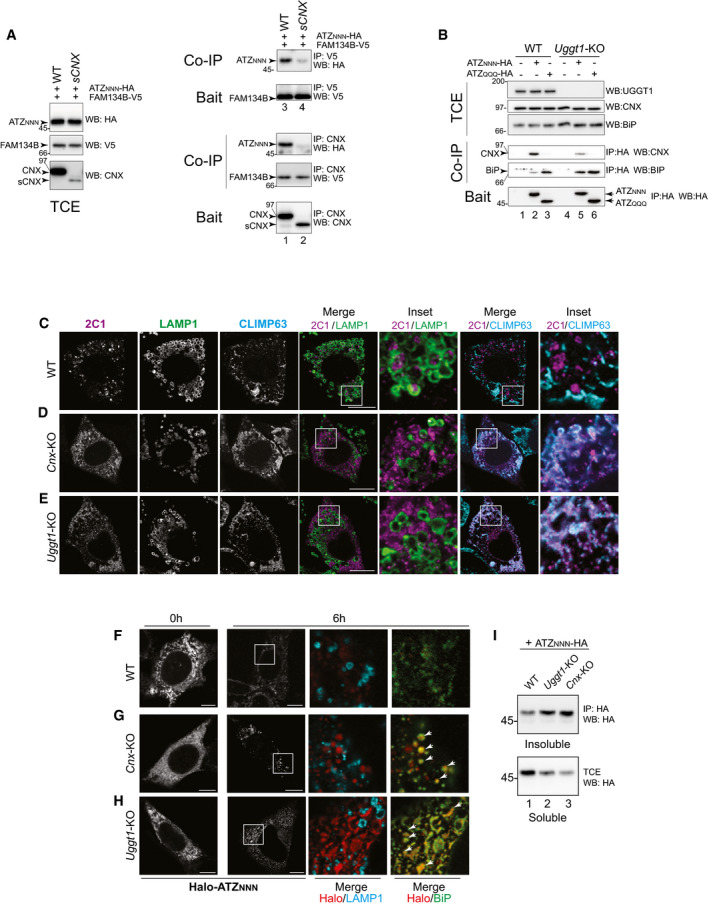
-
ASame as EV1F in WT and sCNX MEF, without DSP.
-
BSame as (A) in WT and Uggt1‐KO MEF.
-
CSame as Fig 2A with the endolysosomal marker LAMP1 and the ER marker CLIMP63.
-
DSame as (C) for Cnx‐KO.
-
ESame as (C) for Uggt1‐KO.
-
F–HHaloTag pulse‐chase analysis in WT, Cnx‐KO, and Uggt1‐KO MEF monitoring fluorescent Halo‐ATZNNN generated during a 45 min pulse with JF646, at 6 h of chase in the absence of BafA1. Arrowheads show select clusters of JF646‐labeled Halo‐ATZNNN co‐localizing with BiP.
-
IAnalyses of soluble and insoluble ATZNNN in WT, Cnx‐KO, and Uggt1‐KO MEF.
Data information: Scale bars 10 µm.
Source data are available online for this figure.
Thus, the role of the CNX:FAM134B complex in client delivery to endolysosomes for clearance requires the LIR function of FAM134B (Fig EV1B–E) and the glucose‐binding function of CNX (for ATZNNN, Figs 2A and C and E and F and H and I, and EV4A; for Pro‐Col2A1R989C, Fig EV3G and H, and J, and K, and M).
The quality control factor UGGT1 selects ERAD‐resistant ATZNNN polymers (and misfolded Pro‐Col2A1) for ERLAD
Newly synthesized proteins engage CNX via mono‐glucosylated oligosaccharides generated by the sequential removal of the two outermost glucose residues by GI and GII (Fig 1A, thin arrows). Execution of the folding program is followed by removal of glucose 3 by GII (Fig 1A, thick arrow), release from CNX, and transport of the native polypeptide at the intra‐ or extra‐cellular site of activity (Adams et al, 2019). Release of non‐native proteins such as ATZNNN from CNX is delayed by repeated cycles of GII‐mediated de‐glucosylation (Fig 1A, thick arrow) followed by immediate re‐glucosylation by the ER quality control factor UGGT1 (Fig 1A, curved arrow) (Adams et al, 2019). This can be disabled upon deletion of UGGT1, with an overall reduction of CNX engagement (Molinari et al, 2005). For ATZNNN, hampered CNX engagement is shown by the reduction in CNX that co‐precipitates with ATZNNN in cells lacking UGGT1 compared to WT cells (Fig EV4B, Co‐IP, top panel, lanes 2 vs. 5). In Uggt1‐KO MEF, ATZNNN abundantly associates with the Hsp70 family member BiP (Fig EV4B, Co‐IP, lower panel, lanes 2 vs. 5) that prevents secretion of misfolded proteins eventually released by CNX (Molinari et al, 2005). In Uggt1‐KO MEF, ATZNNN polymers are not delivered to the endolysosomal compartment (Fig 2G–J and M). Lysosomal delivery is rescued on back‐transfection of catalytically active UGGT1 (Fig 2K and M), but not when cells are complemented with a mutant form of UGGT1 (Fig 2L and M) that cannot re‐glycosylate misfolded proteins because it lacks the 1452DQDLPN1457 residues of the active site (Arnold et al, 2000). These experiments show that the catalytic activity of UGGT1 is required for lysosomal delivery of ATZNNN polymers and imply that multiple de‐glucosylation/re‐glucosylation cycles and persistent retention by CNX are required for ERLAD selection.
All in all, ATZNNN polymers are delivered within LAMP1‐positive endolysosomes for clearance (Fig EV5C, 2C1/LAMP1) (Teckman & Perlmutter, 2000; Kamimoto et al, 2006; Kroeger et al, 2009; Hidvegi et al, 2010; Perlmutter, 2011; Pastore et al, 2013; Yamamura et al, 2014; Fregno et al, 2018; Strnad et al, 2020). The fraction of polymeric ATZNNN that accumulates within endolysosomes upon cell exposure to BafA1 does not co‐localize with the ER marker CLIMP63 (Fig EV5C, 2C1/CLIMP63). In cells lacking CNX or UGGT1, ATZNNN polymers are not delivered within LAMP1‐positive endolysosomes (Fig EV5D and E, 2C1/LAMP1). Rather, they remain in the ER (or in a ER‐derived compartment) as shown by their co‐localization with the ER marker CLIMP63 (Fig EV5D and E, 2C1/CLIMP63).
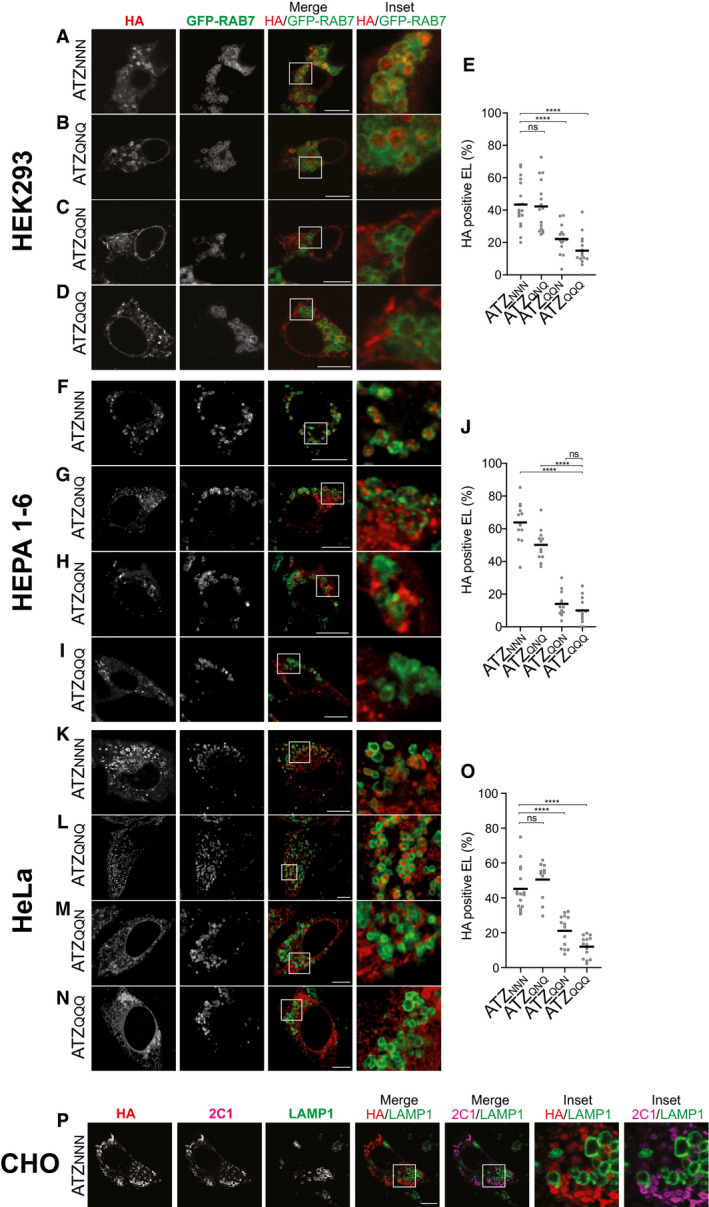
Figure EV5. Delivery of ATZXXX within endolysosomes in human, mouse, and hamster cell lines.
-
A–DSame as Fig 3 in HEK293 cells. Endolysosomes are labeled with GFP‐RAB7.
-
EQuantification of HA‐positive endolysosomes (A‐D) (mean, n = 18, 16, 16, 15 cells).
-
F–ISame as Fig 3 in HEPA1‐6 cells.
-
JQuantification of (F–I) (mean, n = 13, 12, 12, 13 cells).
-
K–NSame as Fig 3 in HeLa cells.
-
O(J) Quantification of (K–N) (mean, n = 16, 11, 14, 14 cells).
-
PSame as Fig 3 in CHO cells. The polymer‐specific 2C1 antibody stain is shown.
Data information: One‐way ANOVA and Dunnett’s multiple comparison test, ns P > 0.05, ****P < 0.0001. Scale bars 10 µm.
Time course, confocal light scanning microscopy analyses of the defective delivery of ATZNNN polymers to the endolysosomal district in Cnx‐ and in Uggt1‐KO MEF
HaloTag pulse‐chase reveals that in WT MEF exposed to BafA1, fluorescently labeled, polymeric, Halo‐ATZNNN progressively accumulates within LAMP1‐positive, inactive endolysosomes (Fig 1E, Insets, 6 h chase, Fig 1F). If the same experiment is performed in the absence of BafA1, fluorescent Halo‐tagged polymers of ATZNNN are degraded as soon as they are delivered within the LAMP1‐positive degradative compartment rather than accumulating in their lumen (Fig EV4F). In these cells, monomeric Halo‐ATZNNN is degraded by ERAD and polymeric Halo‐ATZNNN by ERLAD (Perlmutter, 2011), with overall disappearance of the labeled protein from cells (Fig EV4F, 0 vs. 6 h chase).
In cells lacking CNX (Fig EV4G) or UGGT1 (Fig EV4H), the fraction of Halo‐tagged ATZNNN labeled during the 45 min fluorescent pulse (red in Halo/LAMP1) remains in the ER (or in a ER‐derived compartment), where it extensively co‐localizes with BiP (Halo/BiP). ATZNNN expressed in Cnx‐ or in Uggt1‐KO cells with inactive ERLAD is more prone to enter in a detergent‐insoluble fraction, from which it is extracted upon dissolution in 8 M urea (Fig EV4I, lanes 2 and 3). This is a phenotype that we will again observe and discuss below, where analysis of ERLAD‐incompetent glycosylation variants of ATZ reveals their proneness to enter in a detergent‐insoluble fraction.
Context‐specific N‐oligosaccharides select ERAD‐resistant ATZNNN polymers (and misfolded Pro‐Col2A1) for lysosomal delivery
To definitely assess the role of N‐linked oligosaccharides in client selection for ERLAD, we monitored the intracellular fate of the full series of ATZNNN glycosylation mutants generated by individual and combined mutations of the N‐glycans acceptor asparagine residues (N) at positions 46, 83, and 247 to glutamine (Q) (Fig 3A). All 8 glycosylation variants do form 2C1‐immunoreactive polymers as shown in confocal laser scanning microscopy analyses (Fig 3B–I, 2C1), in flow cytometry and native gels (Fig EV2C–D) and by immunoisolation (Fig 4A and B). However, only ATZ polymers displaying the N‐glycan at position 83 are efficiently delivered within LAMP1‐positive, degradative compartments (i.e., ATZN N N (Fig 3B and J, and K), ATZN N Q (Fig 3C and J, and K), ATZQ N N (Fig 3E and J, and K), ATZQ N Q (Fig 3G and J, and K)). The variants lacking this oligosaccharide (i.e., ATZN Q N (Fig 3D and J, and K), ATZN Q Q (Fig 3F and J, and K), ATZQ Q N (Fig 3H and J, and K), and ATZQ Q Q (Fig 3I–K)) are very inefficiently, if at all, delivered within the lysosomal compartments. Thus, the oligosaccharide at position 83 is the N‐glycan whose modification marks ATZ for lysosomal delivery. These data were confirmed in a variety of cell lines transiently or stably expressing the model proteins, including human embryonic kidney cells (HEK293, Fig EV5A–E), mouse hepatocytes (HEPA 1‐6, Fig EV5F–J), human HeLa cells (Fig EV5K–O), and mouse 3T3 cells (see Fig 4G–J). The only notable exception is Chinese Hamster Ovary (CHO) cells, where ATZNNN is not delivered within LAMP1‐positive endolysosomes (Fig EV5P). Consistently, a recent work shows that BafA1 has no effect on clearance of soluble ATZ polymers in CHO cells and reports on the substantial deposition of this species in detergent insoluble pellet (Ronzoni et al, 2021). We have no explanation as for the reasons of defective transport of ATZ polymers from the ER to endolysosomes in CHO cells, but we consider this finding worth‐reporting because CHO cells are widely used in cell biology labs, including those investigating ATZ biology.
Figure 4. Intracellular fate of ERLAD‐competent vs. ERLAD‐incompetent ATZXXX glycosylation variants.
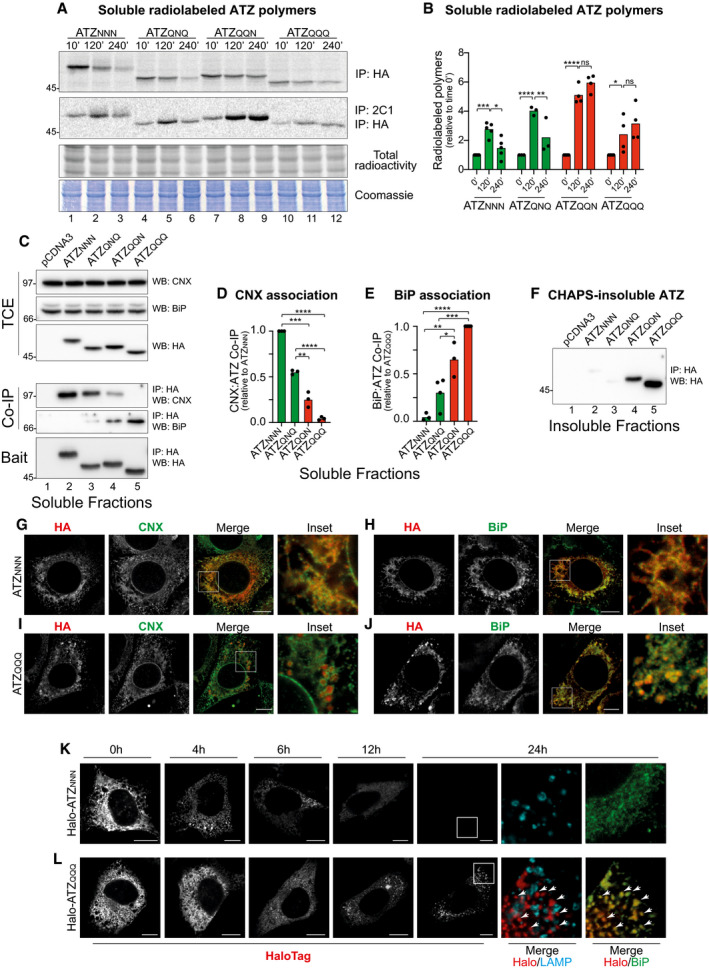
- Radiolabeled ATZXXX enter in 2C1‐positive polymers that are immunoisolated at the end of the given chase times. Total ATZNNN is immunoisolated with HA. Protein load and 35S‐methionine/cysteine incorporated in nascent polypeptides are shown.
- Quantification of radiolabeled polymers. Green columns are for the ERLAD‐competent ATZNNN and ATZQNQ variants (see Fig 3B and G and, J and K), red columns are for the ERLAD‐incompetent ATZQQN and ATZQQQ (see Fig 3H–K) (mean, n = 5 for ATZNNN, n = 3 for ATZQNQ, n = 4 ATZQQN and n = 4 for ATZQQQ). Two‐way ANOVA and Sidak’s multiple comparison test, ns P > 0.05, *P < 0.005, **P < 0.01, ***P < 0.001, ****P < 0.0001.
- Co‐immunoprecipitation of ATZXXX (Bait, lower panel) with CNX (upper panel) and BiP (middle panel). Protein content in the total cell extracts (TCE) is shown.
- Quantification of (C) for CNX association (n = 3). Unpaired two‐tailed t‐test, **P < 0.01***P < 0.001, ****P < 0.0001.
- Quantification of (C) for BiP association (n = 3 for ATZNNN and ATZQQN; n = 4 for ATZQNQ and ATZQQQ). Unpaired two‐tailed t‐test, *P < 0.005, **P < 0.01, ***P < 0.001, ****P < 0.0001.
- Detergent‐insoluble fraction of (C).
- Confocal laser scanning microscopy in Flp‐In 3T3 cells stably expressing ATZNNN in the absence of BafA1 (to allow clearance of misfolded model proteins delivered within endolysosomes). Co‐localization with Calnexin (CNX).
- Same as (G) for BiP co‐localization.
- Same as (G) for ATZQQQ.
- Same as (H) for ATZQQQ.
- HaloTag pulse‐chase analysis in MEF monitoring fluorescent Halo‐ATZNNN during up to 24 h.
- Same as (K) for fluorescent Halo‐ATZQQQ. Arrowheads show select clusters of JF646‐labeled Halo‐ATZQQQ. Arrowheads show co‐localization Halo‐ATZQQQ:BiP.
Data information: Scale bars 10 μm.
Source data are available online for this figure.
The relevance of N‐glycans as a tag for lysosomal delivery of ERAD‐resistant polypeptides is reinforced by data showing that for both the WT and the R989C form of Pro‐Col2A1, the elimination of the protein‐bound oligosaccharide at position 1388 (Fig EV3A) substantially impairs delivery within LAMP1 degradative compartments for clearance (Fig EV3B–F).
Chaperone engagement and formation of insoluble aggregates by ERLAD‐competent versus ERLAD‐incompetent ATZ glycosylation variants
Next, we selected 2 ERLAD‐competent (ATZNNN and ATZQNQ) and 2 ERLAD‐resistant variants of ATZ (ATZQQN and ATZQQQ) to monitor polymer’s fate and chaperone association (Fig 4, note that all experiments shown in this figure are performed in the absence of BafA1). Cells expressing the individual model proteins were metabolically labeled with 35S methionine and cysteine and chased for 10, 120, and 240 min. At the end of each chase time, the residual population of radiolabeled ATZXXX and ATZXXX polymers was immunoisolated from detergent‐lysates with anti‐HA (Fig 4A, IP:HA) or with the polymer‐specific 2C1 antibody (IP:2C1), respectively. For the ERLAD‐competent variants, radiolabeled polymers peak 120 min after synthesis (ATZNNN and ATZQNQ, Fig 4A, IP:2C1, lanes 1–3 and 4–6, respectively, Fig 4B). Their disappearance at longer chase times (Fig 4A, lanes 3 and 6, Fig 4B) is ascribed to their lysosomal delivery and clearance (Fig 3B and G, and J, and K) (Teckman & Perlmutter, 2000; Kamimoto et al, 2006; Kroeger et al, 2009; Hidvegi et al, 2010; Perlmutter, 2011; Pastore et al, 2013; Yamamura et al, 2014; Fregno et al, 2018). In contrast, the ERLAD‐incompetent ATZQQN and ATZQQQ polymers, which are not delivered to the endolysosomal district for clearance (Fig 3H–K), accumulate during the chase time (Fig 4A, lanes 7–9 and 10–12, Fig 4B).
Further analyses performed by immunoisolation of ATZ with the associated molecular chaperones (see methods section) reveal that the two ERLAD substrates ATZNNN and ATZQNQ abundantly associate with CNX as expected by the involvement of the lectin chaperone in selection for their lysosomal delivery (Fig 4C, Co‐IP, upper panel, lanes 2, 3, Fig 4D). In contrast, the two ERLAD‐resistant variants ATZQQN and ATZQQQ only poorly do so, if at all (Fig 4C, Co‐IP, upper panel, lanes 4, 5 and Fig 4D). Rather, ATZQQN and ATZQQQ associate more abundantly with BiP (Fig 4C, Co‐IP, lower panel, lanes 4, 5, Fig 4E).
To sum up, in cells with active ERLAD (WT MEF, Fig EV4B, lanes 1–3), the ERLAD client ATZNNN abundantly associates with CNX (Fig EV4B, Co‐IP, upper panel, lane 2). The ERLAD‐resistant ATZQQQ abundantly associates with BiP (Co‐IP, lower panel, lane 3). In cells with inactive ERLAD (Uggt1‐KO MEF, Fig EV4B, lanes 4‐6), the association of ATZNNN with CNX is substantially reduced (compare Co‐IP, upper panel, lane 5 vs. lane 2) and the association with BiP is enhanced (Co‐IP, lower panel, lane 5 vs. lane 2). These data further support the notion that association with CNX plays a crucial role to channel ERAD‐resistant polypeptides in the ERLAD pathway and may reveal a role of BiP for ER retention of those misfolded polypeptides that fail to access the ERLAD pathway.
Analyses of the detergent‐insoluble fractions show the propensity of the ERLAD‐incompetent ATZQQN and ATZQQQ to enter in detergent‐insoluble fractions, from which they are extracted upon dissolution in 8 M urea (Fig 4F, lanes 4, 5).
Stationary and time course, confocal light scanning microscopy analyses of lysosomal delivery of ATZ glycosylation variants
The different intracellular fate of ERLAD‐competent versus ERLAD‐incompetent ATZ glycosylation variants was monitored by confocal laser scanning microscopy in the absence of BafA1. Under these experimental conditions, ATZNNN polymers are degraded as soon as they are delivered within the LAMP1‐positive endolysosomes (see above and Fig EV4F). Consequently, at steady state, ATZNNN is mostly in the ER, where it is synthesized and co‐localizes with the ER chaperones CNX (Fig 4G) and BiP (Fig 4H). In contrast, ATZQQQ is seen in the biosynthetic compartment, but it also accumulates in large clusters that exclude CNX (Fig 4I) and co‐localize with BiP (Fig 4J). Altogether, the results are consistent with a model, where ERLAD clients associates with CNX and are efficiently delivered within endolysosomes for clearance, whereas terminally misfolded proteins that inefficiently associate with CNX (or that are in cells characterized by defective function of the CNX chaperone system) are inefficiently delivered within endolysosomes, are captured by BiP and eventually enter insoluble aggregates.
This was further confirmed by the same HaloTag pulse‐chase approach shown in Fig EV4F–H (i.e., performed in the absence of BafA1). Briefly, cells expressing Halo‐ATZNNN or Halo‐ATZQQQ (Fig 4K and L, respectively) are incubated with the cell permeable HaloTag fluorescent ligand JF646. After 45 min, JF646 is replaced by the “black ligand” 7‐bromoheptanol to stop fluorescence incorporation. Fluorescent Halo‐ATZNNN gradually disappears from cells within 24 h, consistent with its efficient lysosomal clearance (Fig 4K, gray scale, and see also Fig EV4F). Fluorescent Halo‐ATZQQQ clusters in intracellular spots during the 24 h of chase (Fig 4L, gray scale). Characterization of the ATZQQQ clusters (red, Fig 4L) shows that they form outside the LAMP1‐positive endolysosomes (Fig 4L, panel Merge, Halo/LAMP) and that they co‐localize with BiP (Fig 4L, arrowheads). Thus, ERLAD clients expressed in ERLAD‐incompetent cells that lack functional CNX or functional glucosidases and glucosyltransferase (Figs 2C and E–G, and J, and EV1B and Figs EV3H and J–L, and EV4D and E, and G, and H) or ERLAD clients depleted of the N‐glycan required for ERLAD selection (Figs 3D and F, and H, and I, and 4I and J, and L, and EV3C and E, and EV5C and D, and H, and I, and M, and N) fail to be delivered to the lysosomal compartments for clearance and are more prone to form insoluble intracellular deposits.
Discussion
The ER is site of protein synthesis. Folding‐incompetent and terminally misfolded polypeptides with highly divergent structure, size, modifications, physicochemical properties engage a variety of client‐, cell‐, tissue‐specific and general pathways to be eventually removed from the biosynthetic compartment and degraded. A large fraction of them is delivered, via specialized translocation machineries built around membrane‐embedded E3 ubiquitin ligases, across the ER membrane, for degradation by the cytosolic ubiquitin/proteasome system in catabolic processes collectively defined as ER‐associated degradation (ERAD, upper part in Fig 5) (Sun & Brodsky, 2019).
Figure 5. Selection of misfolded polypeptides for proteasomal (ERAD) or endolysosomal degradation (ERLAD).
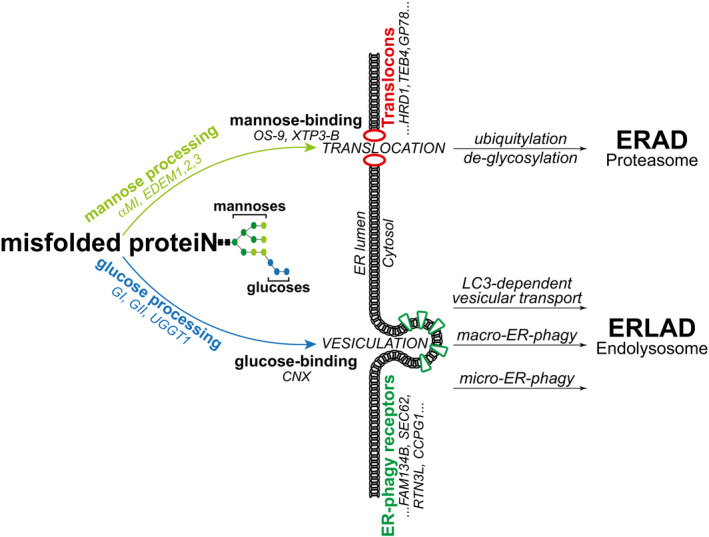
De‐mannosylation (i.e., removal of light green mannoses by ER α‐mannosidase I (αMI) and EDEM proteins) generates ligand for mannose‐binding lectins (OS‐9 and XTP‐3B). These shuttle misfolded proteins to translocation sites. Translocons are built around ER membrane‐embedded E3 ligases (e.g., HRD1, TEB4, GP78) and include co‐factors that confer client‐specificity and promote cytosolic delivery. Misfolded proteins are de‐glycosylated, poly‐ubiquitylated, and eventually degraded by 26S‐proteasomes (ERAD, upper part) (Sun & Brodsky, 2019). De/re‐glucosylation cycles and persistent association with Calnexin (CNX) select ERAD‐resistant misfolded proteins for segregation in ER subdomains. ER vesiculation is directly or indirectly promoted by ER‐phagy receptors (e.g., FAM134B, SEC62, RTN3L, CCPG1). ER‐derived vesicles containing misfolded polypeptides are delivered to the endolysosomal district by LC3‐dependent transport, macro‐ or micro‐ ER‐phagy (ERLAD, lower part) (Molinari, 2021).
The ER also generates misfolded polypeptides that fail to engage the ERAD machinery. Large size or aggregation/polymerization propensity generally prevent translocation across the ER membrane and activate alternative catabolic pathways that engage components of the ER‐phagy machinery. ER‐phagy programs are activated by pleiotropic or ER‐centric signals that lead cells to eat their own ER to gain nutrients, to control ER size and function or, as in the case of misfolded proteins, to remove ER portions that contain them (Molinari, 2021). Under the control of a family of ER‐phagy receptors, portions of the ER vesiculate and are delivered to the endolysosomal compartments for clearance. In the case of ER‐to‐lysosomes‐associated degradation (ERLAD), the ER portions detaching from the biosynthetic compartment contain ERAD‐resistant misfolded polypeptides, which are delivered for lysosomal degradation via LC3‐dependent vesicular transport, macro‐ and micro‐ER‐phagy (ERLAD, lower part in Fig 5) (Fregno & Molinari, 2019).
How proteins are selected for ERAD is well understood. Strong experimental evidences support a mannose timer model, first proposed by Ari Helenius in the mid ‘90s (Helenius, 1994). In this model, aberrant gene products generated in the ER are marked for proteasomal, ER‐associated degradation (ERAD) upon removal of α1,2‐bonded mannose residues (light green in Fig 1A). De‐mannosylation is operated by members of the glycosyl hydrolase 47 family including the ER‐resident α‐mannosidase I and EDEM proteins. It interrupts CNX binding and generates N‐glycan species engaging the mannose‐binding ERAD lectins OS‐9 and XTP3‐B, which deliver terminally misfolded proteins to retro‐translocation sites built around membrane‐embedded E3 ubiquitin ligases (upper part in Fig 5) (Aebi et al, 2010; Adams et al, 2019; Sun & Brodsky, 2019). Mannose processing to select terminally misfolded proteins for ERAD and intervention of mannose‐binding lectins to deliver terminally misfolded proteins to retro‐translocation sites is conserved in yeast (Buschhorn et al, 2004; Bhamidipati et al, 2005; Kim et al, 2005; Szathmary et al, 2005; Gauss et al, 2006; Kanehara et al, 2007; Clerc et al, 2009; Gauss et al, 2011).
The study of how ERAD‐resistant proteins are selected for ERLAD is still in its infancy. Our analyses of the fate of ERAD‐resistant ATZ polymers and Pro‐Col2A1 variants in 5 mouse and human cell lines reveal, for ERLAD as well, the crucial role of N‐linked glycans, their position within the polypeptide chain, and their processing. Our data reveal that selection of a misfolded polypeptide for ERLAD relies on GI/GII/UGGT1‐driven cycles of CNX association/release.
An interesting result emerging from our study is that one of the three ATZNNN oligosaccharides is required and sufficient for selection of ATZNNN polymers for ERLAD. The functional prevalence of one oligosaccharide in the context of multi‐glycosylated proteins has previously been reported for N‐glycan‐regulated protein folding and for N‐glycan selection for ERAD. For protein folding, Dan Hebert et al, showed that of the 7 N‐linked oligosaccharides of influenza virus hemagglutinin, only the oligosaccharide at position 81 is absolutely required for maturation (Hebert et al, 1997). For ERAD, by studying the mechanistic details of clearance of the tetra‐glycosylated CPY* and of the di‐glycosylated PrA* proteins from the yeast ER, Spear and Ng revealed that only the oligosaccharide at position 479 of CPY* and the one at position 107 of PrA* are required to channel these two folding‐defective polypeptides into the ERAD pathway (Spear & Ng, 2005). Here, by determining the fate of the 2C1‐immunoreactive polymers formed by the 8 glycosylation variants of ATZ, we show that only the 4 variants displaying the oligosaccharide at position 83 enter the FAM134B:CNX‐driven ERLAD pathway. Thus, for protein folding (Hebert et al, 1997), for selection of misfolded proteins for proteasomal (ERAD, (Spear & Ng, 2005)), and for lysosomal degradation (ERLAD, this work) one of the oligosaccharides displayed on multi‐glycosylated polypeptides plays a pivotal role as signaling appendix.
Surprisingly, our tests in a variety of cell lines revealed the lack of ATZNNN polymers delivery to endolysosomes in CHO cells. This confirms indirect evidences of defective ERLAD in CHO cells published by others (e.g., (Ronzoni et al, 2021)) and is in contrast to the efficient lysosomal delivery and clearance of ER portions containing ATZNNN polymers in human and mouse cell lines, primary hepatocytes, in a mouse model of the disease and even in yeast cells (Teckman & Perlmutter, 2000; Kamimoto et al, 2006; Kroeger et al, 2009; Hidvegi et al, 2010; Perlmutter, 2011; Pastore et al, 2013; Yamamura et al, 2014; Fregno et al, 2018; Cui et al, 2019; Strnad et al, 2020 and this work). We did not further investigate this issue, but we think that this merits a note in our discussion.
Multiple client‐specific ERAD pathways ensure rapid removal by the ubiquitin proteasome system of the large array of misfolded polypeptides generated within the eukaryotic cells ER (Leto et al, 2019; Sun & Brodsky, 2019; Molinari, 2021). It is therefore fair to predict that multiple client‐specific pathways also exist to ensure intraluminal segregation and lysosomal removal of the vast array of misfolded polypeptides that do not enter ERAD pathways. These client‐specific ERLAD pathways may rely on selective engagement of one of the many ER‐phagy receptors expressed by nucleated cells (Molinari, 2021). ER‐phagy receptors play major role in ERLAD. This prediction is supported by evidences including (i) the defective lysosomal clearance of ATZ polymers ((Fregno et al, 2018) and this work), misfolded Pro‐collagen (Forrester et al, 2019), or NPC1 variants (Schultz et al, 2018) in cells lacking the ER‐phagy receptor FAM134B (or of its yeast ortholog Atg40p (Cui et al, 2019)); (ii) the ER expansion upon accumulation of insoluble acinar secretory enzymes in mice expressing a non‐functional form of the ER‐phagy receptor CCPG1 (Smith et al, 2018); (iii) the accumulation of aberrant secretory proteins in the hepatocyte’s ER of mice lacking the liver‐specific FAM134B‐2 splice variant (Kohno et al, 2019) and reviewed in (Molinari, 2021). As it happened for the identification of the large variety of ERAD pathways, further studies involving analysis of other model, ERAD‐resistant polypeptides will certainly reveal the involvement of other ER‐phagy receptors in ERLAD. On this line, ER‐phagy receptors TEX264, SEC62, ATL3, and FAM134B are expressed ubiquitously, with the latter highly expressed in peripheral neurons and in the brain; its splice variant FAM134B‐2 in kidney, liver, white adipose tissue, and spleen; CCPG1 in pancreas, liver, kidney, and stomach (Smith et al, 2018; Chino et al, 2019; Kohno et al, 2019; Keles et al, 2020). Moreover, select ER‐phagy receptors have specific distribution within the ER, with TEX264 in three‐way ER junctions (An et al, 2019; Chino et al, 2019), FAM134 variants in ER sheets (Khaminets et al, 2015), ATL3 and RTN3L in ER tubuli (Grumati et al, 2017; Chen et al, 2019). We predict that misfolded polypeptides expressed in tissue‐specific or in sub‐organellar‐restricted manner will engage specific ERLAD pathways driven by the relevant ER‐phagy receptor.
Why it is important to investigate ERLAD in molecular details? In about 10% of patients carrying the Z mutation of the SERPINA1 gene, clearance of ATZ polymers from the ER is defective. This results in clinically significant hepatotoxicity, which is the major inherited cause of pediatric liver disease and transplantation (Sharp et al, 1969; Eriksson et al, 1986; Wu et al, 1994; Hidvegi et al, 2010; Perlmutter, 2011; Roussel et al, 2011; Marciniak et al, 2016). Notably, cell lines derived from patients with liver disease (susceptible cells) and without liver disease (protected cells) degrade a model ERAD substrate with equal efficiency. However, susceptible cells degrade ATZ and other polymerogenic serpins less efficiently (Teckman & Perlmutter, 1996). Altogether, we speculate that cells of susceptible hosts carrying the PiZZ mutation leading to expression of ATZ have additional mutations leading to a loss‐of‐function in the ERLAD pathway. This would enhance accumulation of mutant ATZ within the ER of liver cells and make the patients more susceptible to liver injury.
Finally, our report underscores the role of N‐glycans as tag for protein quality control. Their context‐specific localization determines their processing by ER‐resident glycanases and the signal they will encode to eventually release the protein into the secretory, the ERAD or the ERLAD pathways. Specialized lectins must decode the signal to ensure the appropriate assistance to folded (mature, ready‐for‐secretion), folding (immature, to be retained in the folding environment), or terminally misfolded polypeptide (to be cleared by proteasomes or lysosomes). ER‐targeted therapeutic approaches are sought to intervene in diseases caused by protein misfolding, to interfere with pathogens that hijack these machineries to invade host cells and generate their progeny, and to enhance production of proteins‐of‐interest. Understanding how N‐glycosylation and processing of N‐linked glycans determine polypeptide’s fate allows pharmacologic/genetic intervention to enhance production of functional proteins or to optimize proteasomal (via ERAD) or lysosomal (via ERLAD) removal of defective ones.
Materials and Methods
Expression plasmids and antibodies
HA‐tagged ATZ, FAM134B‐V5, and FAM134BLIR‐V5 are subcloned in pcDNA3.1, mCherry‐Pro‐collagen 2A1 and the R989C disease‐linked variant and UGGT1 expression plasmid were described in (Arnold et al, 2000; Fregno et al, 2018; Forrester et al, 2019). Halo‐ATZNNN‐HA, Halo‐AATNNN‐HA Halo‐ATZQQQ‐HA are subcloned in pCDNA5 vector. The ATZ glycosylation variants were prepared by replacing asparagine 46, 83 and/or 247 of the glycosylation sequons with glutamine residues (fwd N46Q 5′‐GAAGAAGATATTGGTGCTATTGGACTGGTGTGCCAGCTG‐3′, rev N46Q 5′‐CAGCTGGCACACCAGTCCAATAGCACCAATATCTTCTTC‐3′; fwd N83Q 5′‐CCGGAATCTCCGTGAGATTGAAATTCAGGCCCTCCA‐3′, rev N83Q 5′‐TGGAGGGCCTGAATTTCAATCTCACGGAGATTCCGG‐3′; fwd N247Q 5′‐GAAGAAGATGGCGGTGGCATTGCCCAGGTATTTCATCAG‐3′, rev N247Q 5′‐CTGATGAAATACCTGGGCAATGCCACCGCCATCTTCTTC‐3′). The mCherry‐Pro‐collagen glycosylation variant was prepared by replacing asparagine 1388 of the glycosylation sequon with a glutamine residue (fwd 5′‐GCAGTGGTAGGTGATCTGCTGGGAGCCTTCCGT‐3′; rev 5′‐ACGGAAGGCTCCCAGCAGATCACCTACCACTGC‐3′). Antibodies used in this study are the monoclonal 2C1 anti‐polymeric ATZ (Hycult Biotech) (Miranda et al, 2010), polyclonal anti‐HA (Sigma), monoclonal anti‐HA probe F7 (Santa Cruz Biotech.), anti‐LAMP1 (Hybridoma Bank, 1D4B deposited by J.T. August), anti‐GAPDH (Merck), anti‐V5 (Invitrogen), anti‐CLIMP63 (Proteintech), anti‐BiP (Stressgen), anti‐UGGT1 (Sigma), anti‐CNX (kind gift A. Helenius), anti‐FAM134B (kind gift I. Kurth), anti‐SEC62 (kind gift R. Zimmermann), anti‐HaloTag® monoclonal (Promega), anti‐HA probe (F7, Santa Cruz), anti‐LC3B (Sigma), and anti‐TEX264 (Novus). Alexa‐conjugated secondary antibodies were purchased from Thermo Fisher Scientific and Jackson ImmunoResearch; HRP‐conjugated from SouthernBiotech, Invitrogen and Bio‐Rad; Protein A and Protein G beads were from Sigma and Thermo Fisher, respectively.
Cell culture, transient transfection, and inhibitors
MEF, HEK293, HeLa, CHO, and HEPA 1‐6 cells were grown in DMEM supplemented with 10% FBS at 37°C and 5% CO2. Cnx‐KO and sCnx (Denzel et al, 2002; Pieren et al, 2005; Kraus et al, 2010) and Uggt1‐KO MEF (Molinari et al, 2005; Soldà et al, 2007) have been previously described. Flp‐In™‐3T3 cells (Thermo Fisher) stably expressing Halo‐ATZNNN‐HA or Halo‐ATZQQQ‐HA were generated following manufacturer instructions and cultured in DMEM supplemented with 10% FCS and 150 mg/ml Hygromycin. Transient transfections were performed using JetPrime transfection reagent (PolyPlus) following manufacturer’s protocol. BafA1 (Calbiochem) was used at 50 nM for 12 h or 100 nM for 6 h. CST (Sigma) and KIF (Toronto Research Chemicals) were used at 1 mM, and 200 µM, respectively.
CRISPR
Cas9 engineered MEF cell lines SEC62‐deficient MEF cells (CRISPR SEC62) and FAM134B‐deficient MEF cells (CRISPR FAM134B) were engineered using CRISPR/Cas9 system as previously described (Fumagalli et al, 2016; Fregno et al, 2018). For generation of TEX264 KO cells: the guideRNA‐Cas9 plasmid, lenti‐CRISPRv2‐puro system (Addgene52961) was purchased from Addgene (http://www.addgene.org). Guide sequences were obtained from the Cas9 target design tools (crispr.mit.edu:8079 and/or www.addgene.org/pooled‐library) and synthesized by Microsynth. Two annealed oligonucleotides (5′‐CACCG GCTGTGTGCTCACCCTCGCC‐3′, 5′‐AAACGGCGAGGGTGAGCACACAGCC‐3′ for murine TEX264) were inserted into the lenti‐CRISPRv2‐puro vector using the BsmBI restriction site. After transfection of MEF cells with JetPrime (Polyplus), cells were cultured in DMEM supplemented with 10% FBS. Two days after transfection, the medium was supplemented with 2 µg/ml puromycin. Puromycin‐resistant clones were picked after 10 days, and gene KO was verified by WB analysis.
Protein cross‐linking with DSP
Cells were washed with PBS followed by addition of 1 mM DSP in PBS. After 30 min incubation at room temperature, the reaction was stopped with 1 M Tris, pH 7.8 to a final concentration of 20 mM and incubated for 15 min at room temperature. Cells were washed with PBS containing 20 mM NEM and lysed with RIPA buffer (1% Triton X‐100, 0.1% SDS, 0.5% sodium deoxycholate in HBS, pH 7.4) for 20 min on ice.
Cell lysis, immunoprecipitation, and Western blot
After treatments, cells were washed with ice‐cold PBS containing 20 mM NEM and lysed with 2% CHAPS (Anatrace) in HBS pH 6.8 supplemented with protease inhibitors and Apyrase (Sigma) to prevent client proteins release from BiP. Post‐nuclear supernatants (PNS) were collected after centrifugation at 10,600 g for 10 min. Detergent‐insoluble material was solubilized in 8 M Urea and dissolved in 0.1 M ammonium bicarbonate for further analyses. For immunoprecipitations, PNSs were diluted with lysis buffer and incubated with Protein A or Protein G beads (1:10 w/v, swollen in PBS) and select antibodies at 4°C. After three washes of the immunoprecipitates with 0.5% CHAPS in HBS pH 7.4, beads were denatured for 5 min at 95°C and subjected to SDS–PAGE. Proteins were transferred to PVDF membranes using the Trans‐Blot Turbo Transfer System (Bio‐Rad). Membranes were blocked with 10% (w/v) non‐fat dry milk (Bio‐Rad) in TBS‐T and stained with primary antibodies diluted in TBS‐T followed by HRP‐conjugated secondary antibodies or Protein A diluted in TBS‐T. Membranes were developed using Western Bright ECL or Quantum (Advansta) and signals captured on Fusion FX (Vilber). Images were quantified with the Evolution Capture Edge (Vilber). Membrane stripping for probing additional antigens was done using Re‐Blot Plus Strong Solution (Millipore) following manufacturer’s instructions.
Native gel electrophoresis
After CHAPS lysis, PNSs were incubated for 15 min at RT in Native Sample Buffer (Bio‐Rad) and run on 7.5% native acrylamide gel in Tris/Glycine Buffer (Bio‐Rad). Proteins were then transferred onto PVDF membrane in Tris/Glycine Buffer (Bio‐Rad). Immunoblot analysis was performed as described above.
Metabolic labeling
Seventeen hours after transient transfections, cells were pulsed with 0.05 mCi [35S]methionine/cysteine mix and chased for the indicated time points with DMEM supplemented with 5mM cold methionine and cysteine. Cells were detergent‐solubilized and radiolabeled proteins were revealed with Typhoon FLA 9500, version 1.0 (GE Healthcare). Radioactive signals were quantified using ImageQuant software (Molecular Dynamics, GE Healthcare).
Confocal laser scanning microscopy
Cells were plated on Alcian Blue‐treated glass coverslips (for HEK293 cells, glass coverslips were pre‐treated with Poly‐Lys). After two washes with PBS cells were fixed at room temperature for 20 min in 3.7% formaldehyde diluted in PBS. Cells were permeabilized for 15 min with permeabilization solution (PS) (0.05% saponin, 10% goat serum, 10mM HEPES, 15mM glycine). Primary antibodies were diluted 1:100 in PS for 90 min, washed for 15 min in PS, and then incubated with Alexa Fluor‐conjugated secondary antibodies diluted 1:300 in PS for 45 min. Cells were rinsed with PS and water and mounted with Vectashield (Vector Laboratories) supplemented with or without 4’,6‐diamidino‐2‐phenylindole (DAPI). Confocal pictures were acquired on a Leica TCS SP5 microscope with a Leica HCX PL APO lambda blue 63.0X1.40 OIL UV objective. Image analyses and quantification were performed with LysoQuant as described in (Morone et al, 2020). Image processing was also performed with Photoshop (Adobe).
HaloTag pulse‐chase
HaloTag covalently and irreversibly binds synthetic ligands, including cell permeable ligands that are coupled to fluorescent probes (England et al, 2015). When placed at the N‐terminus of ATZ, the HaloTag does not affect polymerization and lysosomal clearance of ATZ (Dickens et al, 2016; Fregno et al, 2018). The HaloTag pulse‐chase protocol is described in (Fregno et al, 2018). Briefly, cells expressing Halo‐ATZ are incubated for 30 min with a cell permeable “black ligand” (i.e., 6‐chlorohexanol, Sigma) to quench fluorescent detection of cellular Halo‐ATZ. The “black ligand” is replaced by the cell permeable fluorescent, far red, HaloTag ligands JF646 or tetramethylrhodamine (TMR) (Promega) to label newly synthesized Halo‐ATZ for 15 or 45 min (fluorescent pulse). The fluorescent ligand is replaced with the “black ligand” 7‐Bromoheptanol (Sigma) to rapidly interrupt fluorescent labeling of Halo‐ATZ (chase). Cells were then fixed at the indicated time points for 20 min in 3.7% formaldehyde diluted in PBS or detergent‐solubilized. The intracellular fate of the ATZ population synthesized during the fluorescent pulse is monitored by confocal laser scanning microscopy.
Flow cytometry
Cells were transfected as described above and, after 24 h, detached from Petri dishes, washed with PBS, fixed with 3.7% PFA in PBS for 20 min at RT, washed 2 times with PBS, and permeabilized with saponin solution (10% goat serum, 15 mM glycine, 0.05% saponin, 10 mM HEPES in PBS). Permeabilized cells were exposed for 90 min at RT to anti‐HA and 2C1 antibodies diluted in saponin solution. Fluorophore‐conjugated secondary antibodies diluted in saponin solution were added after 2 washes in PBS for 60 min at RT. Finally, cells were washed 2 times in saponin solution, resuspended in MACS buffer (PBS, 2% FCS, 2 mM EDTA), and run on a FACSCanto I flow cytometer (BD Biosciences). Data were analyzed and annotated using FlowJo software (FlowJo LLC).
Statistical analysis
Plots and statistical analyses were performed using GraphPad Prism 8 (GraphPad Software Inc.). In this study, one‐way ANOVA with Dunnett’s multiple comparisons test, two‐way ANOVA and Sidak’s multiple comparison test, and unpaired two‐tailed t‐test were used to assess statistical significance. An adjusted P‐value < 0.05 was considered as statistically significant.
Author contributions
Conceptualization: IF, EF, TS, CG, and MM; Methodology: IF, EF, TS, CG, and MM; Investigation: MM, IF, EF, TS, CG, and MM; Writing original draft: MM; Writing, review, editing: IF, EF, TS, CG, and MM; Supervision: MM.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank Marika Kucinska for experiments with the ER‐phagy receptor TEX264, Diego Morone for assistance with microscopy, Carmine Settembre and the members of Molinari’s lab for discussions. M.M. is supported by AlphaONE Foundation (Application ID: 681136), Foundation for Research on Neurodegenerative Diseases, Swiss National Science Foundation (310030_184827), Comel, Mizutani (No.190010), and Gelu Foundations.
The EMBO Journal (2021) 40: e107240.
Data availability
No primary datasets produced in the study.
References
- Adams BM, Oster ME, Hebert DN (2019) Protein quality control in the endoplasmic reticulum. Protein J 38: 317–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebi M, Bernasconi R, Clerc S, Molinari M (2010) N‐glycan structures: recognition and processing in the ER. Trends Biochem Sci 35: 74–82 [DOI] [PubMed] [Google Scholar]
- An H, Ordureau A, Paulo JA, Shoemaker CJ, Denic V, Harper JW (2019) Tex264 is an endoplasmic reticulum‐resident ATG8‐interacting protein critical for ER remodeling during nutrient stress. Mol Cell 74: 891–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SM, Fessler LI, Fessler JH, Kaufman RJ (2000) Two homologues encoding human UDP‐glucose:glycoprotein glucosyltransferase differ in mRNA expression and enzymatic activity. Biochemistry 39: 2149–2163 [DOI] [PubMed] [Google Scholar]
- Ayalon‐Soffer M, Shenkman M, Lederkremer GZ (1999) Differential role of mannose and glucose trimming in the ER degradation of asialoglycoprotein receptor subunits. J Cell Sci 112: 3309–3318 [DOI] [PubMed] [Google Scholar]
- Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, Menzies FM, Rubinsztein DC (2016) Mammalian autophagy: how does it work? Annu Rev Biochem 85: 685–713 [DOI] [PubMed] [Google Scholar]
- Bernasconi R, Galli C, Calanca V, Nakajima T, Molinari M (2010) Stringent requirement for HRD1, SEL1L, and OS‐9/XTP3‐B for disposal of ERAD‐LS substrates. J Cell Biol 188: 223–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernasconi R, Pertel T, Luban J, Molinari M (2008) A dual task for the Xbp1‐responsive OS‐9 variants in the mammalian endoplasmic reticulum: inhibiting secretion of misfolded protein conformers and enhancing their disposal. J Biol Chem 283: 16446–16454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhamidipati A, Denic V, Quan EM, Weissman JS (2005) Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol Cell 19: 741–751 [DOI] [PubMed] [Google Scholar]
- Bhaskara RM, Grumati P, Garcia‐Pardo J, Kalayil S, Covarrubias‐Pinto A, Chen W, Kudryashev M, Dikic I, Hummer G (2019) Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER‐phagy. Nat Commun 10: 2370–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschhorn BA, Kostova Z, Medicherla B, Wolf DH (2004) A genome‐wide screen identifies Yos9p as essential for ER‐associated degradation of glycoproteins. FEBS Lett 577: 422–426 [DOI] [PubMed] [Google Scholar]
- Chen QZ, Xiao Y, Chai PY, Zheng PL, Teng JL, Chen JG (2019) ATL3 is a tubular ER‐phagy receptor for GABARAP‐mediated selective autophagy. Curr Biol 29: 846–855 [DOI] [PubMed] [Google Scholar]
- Chino H, Hatta T, Natsume T, Mizushima N (2019) Intrinsically disordered protein TEX264 mediates ER‐phagy. Mol Cell 74: 909–921 [DOI] [PubMed] [Google Scholar]
- Christianson JC, Shaler TA, Tyler RE, Kopito RR (2008) OS‐9 and GRP94 deliver mutant alpha1‐antitrypsin to the Hrd1/SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol 10: 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc S, Hirsch C, Oggier DM, Deprez P, Jakob C, Sommer T, Aebi M (2009) Htm1 protein generates the N‐glycan signal for glycoprotein degradation in the endoplasmic reticulum. J Cell Biol 184: 159–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Parashar S, Zahoor M, Needham PG, Mari M, Zhu M, Chen S, Ho H‐C, Reggiori F, Farhan H et al (2019) A COPII subunit acts with an autophagy receptor to target endoplasmic reticulum for degradation. Science 365: 53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leonibus C, Cinque L, Settembre C (2019) Emerging lysosomal pathways for quality control at the endoplasmic reticulum. FEBS Lett 593: 2319–2329 [DOI] [PubMed] [Google Scholar]
- Denzel A, Molinari M, Trigueros C, Martin JE, Velmurgan S, Brown S, Stamp G, Owen MJ (2002) Early postnatal death and motor disorders in mice congenitally deficient in calnexin expression. Mol Cell Biol 22: 7398–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickens JA, Ordonez A, Chambers JE, Beckett AJ, Patel V, Malzer E, Dominicus CS, Bradley J, Peden AA, Prior IA et al (2016) The endoplasmic reticulum remains functionally connected by vesicular transport after its fragmentation in cells expressing Z‐alpha(1)‐antitrypsin. FASEB J 30: 4083–4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbein AD (1991) Glycosidase inhibitors: inhibitors of N‐linked oligosaccharide processing. FASEB J 5: 3055–3063 [DOI] [PubMed] [Google Scholar]
- England CG, Luo HM, Cai WB (2015) HaloTag technology: a versatile platform for biomedical applications. Bioconjug Chem 26: 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson S, Carlson J, Velez R (1986) Risk of cirrhosis and primary liver‐cancer in alpha‐1‐antitrypsin deficiency. N Engl J Med 314: 736–739 [DOI] [PubMed] [Google Scholar]
- Forrester A, De Leonibus C, Grumati P, Fasana E, Piemontese M, Staiano L, Fregno I, Raimondi A, Marazza A, Bruno G et al (2019) A selective ER‐phagy exerts procollagen quality control via a Calnexin‐FAM134B complex. EMBO J 38: e99847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregno I, Fasana E, Bergmann TJ, Raimondi A, Loi M, Solda T, Galli C, D'Antuono R, Morone D, Danieli A et al (2018) ER‐to‐lysosome‐associated degradation of proteasome‐resistant ATZ polymers occurs via receptor‐mediated vesicular transport. EMBO J 37: e99259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregno I, Molinari M (2019) Proteasomal and lysosomal clearance of faulty secretory proteins: ER‐associated degradation (ERAD) and ER‐to‐lysosome‐associated degradation (ERLAD) pathways. Crit Rev Biochem Mol Biol 54: 153–163 [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Noack J, Bergmann T, Cebollero E, Pisoni G, Fasana E, Fregno I, Galli C, Loi M, Soldà T et al (2016) Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol 18: 1173–1184 [DOI] [PubMed] [Google Scholar]
- Gauss R, Jarosch E, Sommer T, Hirsch C (2006) A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat Cell Biol 8: 849–854 [DOI] [PubMed] [Google Scholar]
- Gauss R, Kanehara K, Carvalho P, Ng DT, Aebi M (2011) A complex of Pdi1p and the mannosidase Htm1p initiates clearance of unfolded glycoproteins from the endoplasmic reticulum. Mol Cell 42: 782–793 [DOI] [PubMed] [Google Scholar]
- Grumati P, Morozzi G, Holper S, Mari M, Harwardt MI, Yan R, Muller S, Reggiori F, Heilemann M, Dikic I (2017) Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 6: e25555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Braakman I, Helenius A (1994) Role of N‐linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc Natl Acad Sci U S A 91: 913–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert DN, Foellmer B, Helenius A (1996) Calnexin and calreticulin promote folding, delay oligomerization and suppress degradation of influenza hemagglutinin in microsomes. EMBO J 15: 2961–2968 [PMC free article] [PubMed] [Google Scholar]
- Hebert DN, Zhang JX, Chen W, Foellmer B, Helenius A (1997) The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J Cell Biol 139: 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A (1994) How N‐linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol Biol Cell 5: 253–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A, Aebi M (2004) Roles of N‐linked glycans in the endoplasmic reticulum. Annu Rev Biochem 73: 1019–1049 [DOI] [PubMed] [Google Scholar]
- Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S et al (2010) An Autophagy‐enhancing drug promotes degradation of mutant alpha 1‐antitrypsin Z and reduces hepatic fibrosis. Science 329: 229–232 [DOI] [PubMed] [Google Scholar]
- Hintze V, Steplewski A, Ito H, Jensen DA, Rodeck U, Fertala A (2008) Cells expressing partially unfolded R789C/p.R989C type II procollagen mutant associated with spondyloepiphyseal dysplasia undergo apoptosis. Hum Mutat 29: 841–851 [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Kamiya Y, Kamiya D, Kato K, Nagata K (2009) Human OS‐9, a lectin required for glycoprotein endoplasmic reticulum‐associated degradation, recognizes mannose‐trimmed N‐glycans. J Biol Chem 284: 17061–17068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner CA, Dikic I (2020) ER‐phagy and human diseases. Cell Death Differ 27: 833–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Wang X, Ding X, Du M, Li B, Weng X, Zhang J, Li L, Tian R, Zhu Q et al (2020) FAM134B oligomerization drives endoplasmic reticulum membrane scission for ER‐phagy. EMBO J 39: e102608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, Yoshimori T (2006) Intracellular inclusions containing mutant alpha1‐antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem 281: 4467–4476 [DOI] [PubMed] [Google Scholar]
- Kanehara K, Kawaguchi S, Ng DT (2007) The EDEM and Yos9p families of lectin‐like ERAD factors. Semin Cell Dev Biol 18: 743–750 [DOI] [PubMed] [Google Scholar]
- Kearse KP, Williams DB, Singer A (1994) Persistence of glucose residues on core oligosaccharides prevents association of TCR alpha and TCR beta proteins with calnexin and results specifically in accelerated degradation of nascent TCR alpha proteins within the endoplasmic reticulum. EMBO J 13: 3678–3686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keles U, Iscan E, Yilmaz HE, Karakulah G, Suner A, Bal E, Tasdemir N, Cavga AD, Ekin U, Mutlu Z et al (2020) Differential expression of full‐length and NH2 terminally truncated FAM134B isoforms in normal physiology and cancer. Am J Physiol Gastrointest Liver Physiol 319: G733–G747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N et al (2015) Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522: 354–358 [DOI] [PubMed] [Google Scholar]
- Kim W, Spear ED, Ng DT (2005) Yos9p detects and targets misfolded glycoproteins for ER‐associated degradation. Mol Cell 19: 753–764 [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC (2008) Does bafilomycin A(1) block the fusion of autophagosomes with lysosomes? Autophagy 4: 849–850 [DOI] [PubMed] [Google Scholar]
- Kohno S, Shiozaki Y, Keenan AL, Miyazaki‐Anzai S, Miyazaki M (2019) An N‐terminal‐truncated isoform of FAM134B (FAM134B‐2) regulates starvation‐induced hepatic selective ER‐phagy. Life Sci Alliance 2: e201900340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus A, Groenendyk J, Bedard K, Baldwin TA, Krause K‐H, Dubois‐Dauphin M, Dyck J, Rosenbaum EE, Korngut L, Colley NJ et al (2010) Calnexin deficiency leads to dysmyelination. J Biol Chem 285: 18928–18938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeger H, Miranda E, MacLeod I, Perez J, Crowther DC, Marciniak SJ, Lomas DA (2009) Endoplasmic Reticulum‐associated Degradation (ERAD) and autophagy cooperate to degrade polymerogenic mutant serpins. J Biol Chem 284: 22793–22802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederkremer GZ (2009) Glycoprotein folding, quality control and ER‐associated degradation. Curr Opin Struct Biol 19: 515–523 [DOI] [PubMed] [Google Scholar]
- Leto DE, Morgens DW, Zhang LC, Walczak CP, Elias JE, Bassik MC, Kopito RR (2019) Genome‐wide CRISPR analysis identifies substrate‐specific conjugation modules in ER‐associated degradation. Mol Cell 73: 377–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JR, Lingeman E, Luong T, Ahmed S, Muhar M, Nguyen T, Olzmann JA, Corn JE (2020) A Genome‐wide ER‐phagy screen highlights key roles of mitochondrial metabolism and ER‐resident UFMylation. Cell 180: 1160–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Choudhury P, Cabral CM, Sifers RN (1997) Intracellular disposal of incompletely folded human alpha1‐antitrypsin involves release from calnexin and post‐translational trimming of asparagine‐linked oligosaccharides. J Biol Chem 272: 7946–7951 [DOI] [PubMed] [Google Scholar]
- Liu Y, Choudhury P, Cabral CM, Sifers RN (1999) Oligosaccharide modification in the early secretory pathway directs the selection of a misfolded glycoprotein for degradation by the proteasome. J Biol Chem 274: 5861–5867 [DOI] [PubMed] [Google Scholar]
- Loi M, Molinari M (2020) Mechanistic insights in recov‐ER‐phagy: micro‐ER‐phagy to recover from stress. Autophagy 16: 385–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi M, Raimondi A, Morone D, Molinari M (2019) ESCRT‐III‐driven piecemeal micro‐ER‐phagy remodels the ER during recovery from ER stress. Nat Commun 10: 5058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Ordonez A, Dickens JA, Chambers JE, Patel V, Dominicus CS, Malzer E (2016) New concepts in alpha‐1 antitrypsin deficiency disease mechanisms. Ann Am Thorac Soc 13(Suppl 4): 289–296 [DOI] [PubMed] [Google Scholar]
- Miranda E, Perez J, Ekeowa UI, Hadzic N, Kalsheker N, Gooptu B, Portmann B, Belorgey D, Hill M, Chambers S et al (2010) A novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with alpha(1)‐antitrypsin deficiency. Hepatology 52: 1078–1088 [DOI] [PubMed] [Google Scholar]
- Molinari M (2007) N‐glycan structure dictates extension of protein folding or onset of disposal. Nat Chem Biol 3: 313–320 [DOI] [PubMed] [Google Scholar]
- Molinari M (2021) ER‐phagy responses in yeast, plants, and mammalian cells and their crosstalk with UPR and ERAD. Dev Cell 56: 949–966 [DOI] [PubMed] [Google Scholar]
- Molinari M, Calanca V, Galli C, Lucca P, Paganetti P (2003) Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299: 1397–1400 [DOI] [PubMed] [Google Scholar]
- Molinari M, Galli C, Piccaluga V, Pieren M, Paganetti P (2002) Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J Cell Biol 158: 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari M, Galli C, Vanoni O, Arnold SM, Kaufman RJ (2005) Persistent glycoprotein misfolding activates the glucosidase II/UGT1‐driven calnexin cycle to delay aggregation and loss of folding competence. Mol Cell 20: 503–512 [DOI] [PubMed] [Google Scholar]
- Moore SE, Spiro RG (1993) Inhibition of glucose trimming by castanospermine results in rapid degradation of unassembled major histocompatibility complex class I molecules. J Biol Chem 268: 3809–3812 [PubMed] [Google Scholar]
- Moremen KW, Molinari M (2006) N‐linked glycan recognition and processing: the molecular basis of endoplasmic reticulum quality control. Curr Opin Struct Biol 16: 592–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morone D, Marazza A, Bergmann TJ, Molinari M (2020) Deep learning approach for quantification of organelles and misfolded polypeptide delivery within degradative compartments. Mol Biol Cell 31: 1512–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y, Hosokawa N, Wada I, Nagata K (2003) EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299: 1394–1397 [DOI] [PubMed] [Google Scholar]
- Olivari S, Cali T, Salo KE, Paganetti P, Ruddock LW, Molinari M (2006) EDEM1 regulates ER‐associated degradation by accelerating de‐mannosylation of folding‐defective polypeptides and by inhibiting their covalent aggregation. Biochem Biophys Res Commun 349: 1278–1284 [DOI] [PubMed] [Google Scholar]
- Olivari S, Molinari M (2007) Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding‐defective glycoproteins. FEBS Lett 581: 3658–3664 [DOI] [PubMed] [Google Scholar]
- Omari S, Makareeva E, Roberts‐Pilgrim A, Mirigian L, Jarnik M, Ott C, Lippincott‐Schwartz J, Leikin S (2018) Noncanonical autophagy at ER exit sites regulates procollagen turnover. Proc Natl Acad Sci U S A 115: E10099–E10108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore N, Blomenkamp K, Annunziata F, Piccolo P, Mithbaokar P, Maria Sepe R, Vetrini F, Palmer D, Ng P, Polishchuk E et al (2013) Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha‐1‐anti‐trypsin deficiency. EMBO Mol Med 5: 397–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter DH (2011) Alpha‐1‐antitrypsin deficiency: importance of proteasomal and autophagic degradative pathways in disposal of liver disease‐associated protein aggregates. Annu Rev Med 62: 333–345 [DOI] [PubMed] [Google Scholar]
- Pieren M, Galli C, Denzel A, Molinari M (2005) The use of calnexin and calreticulin by cellular and viral glycoproteins. J Biol Chem 280: 28265–28271 [DOI] [PubMed] [Google Scholar]
- Ronzoni R, Heyer‐Chauhan N, Fra A, Pearce AC, Rudiger M, Miranda E, Irving JA, Lomas DA (2021) The molecular species responsible for alpha1 ‐antitrypsin deficiency are suppressed by a small molecule chaperone. FEBS J 288: 2222–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel BD, Irving JA, Ekeowa UI, Belorgey D, Haq I, Ordóñez A, Kruppa AJ, Duvoix A, Rashid ST, Crowther DC et al (2011) Unravelling the twists and turns of the serpinopathies. FEBS J 278: 3859–3867 [DOI] [PubMed] [Google Scholar]
- Schultz ML, Krus KL, Kaushik S, Dang D, Chopra R, Qi L, Shakkottai VG, Cuervo AM, Lieberman AP (2018) Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6‐dependent ERAD. Nat Commun 9: 3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp HL, Bridges RA, Krivit W, Freier EF (1969) Cirrhosis associated with alpha‐1‐antitrypsin deficiency ‐ a previously unrecognized inherited disorder. J Lab Clin Med 73: 934–939 [PubMed] [Google Scholar]
- Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A , Behrends C, Wilkinson S (2018) CCPG1 Is a non‐canonical autophagy cargo receptor essential for ER‐phagy and pancreatic ER proteostasis. Dev Cell 44: 217–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldà T, Galli C, Kaufman RJ, Molinari M (2007) Substrate‐specific requirements for UGT1‐dependent release from calnexin. Mol Cell 27: 238–249 [DOI] [PubMed] [Google Scholar]
- Spear ED, Ng DT (2005) Single, context‐specific glycans can target misfolded glycoproteins for ER‐associated degradation. J Cell Biol 169: 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strnad P, McElvaney NG, Lomas DA (2020) Alpha1‐antitrypsin deficiency. N Engl J Med 382: 1443–1455 [DOI] [PubMed] [Google Scholar]
- Sun Z, Brodsky JL (2019) Protein quality control in the secretory pathway. J Cell Biol 218: 3171–3187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szathmary R, Bielmann R, Nita‐Lazar M, Burda P, Jakob CA (2005) Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol Cell 19: 765–775 [DOI] [PubMed] [Google Scholar]
- Teckman JH, Perlmutter DH (1996) The endoplasmic reticulum degradation pathway for mutant secretory proteins alpha1‐antitrypsin Z and S is distinct from that for an unassembled membrane protein. J Biol Chem 271: 13215–13220 [DOI] [PubMed] [Google Scholar]
- Teckman JH, Perlmutter DH (2000) Retention of mutant alpha(1)‐antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol 279: G961–G974 [DOI] [PubMed] [Google Scholar]
- Wilkinson S (2019) ER‐phagy: shaping up and destressing the endoplasmic reticulum. FEBS J 286: 2645–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S (2020) Emerging principles of selective ER autophagy. J Mol Biol 432: 185–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Whitman I, Molmenti E, Moore K, Hippenmeyer P, Perlmutter DH (1994) A lag in intracellular degradation of mutant alpha 1‐antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1‐antitrypsin deficiency. Proc Natl Acad Sci U S A 91: 9014–9018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura T, Ohsaki Y, Suzuki M, Shinohara Y, Tatematsu T, Cheng J, Okada M, Ohmiya N, Hirooka Y, Goto H et al (2014) Inhibition of Niemann‐Pick‐type C1‐like1 by ezetimibe activates autophagy in human hepatocytes and reduces mutant alpha1‐antitrypsin Z deposition. Hepatology 59: 1591–1599 [DOI] [PubMed] [Google Scholar]
- Zhang Z, He JW, Fu WZ, Zhang CQ, Zhang ZL (2011) Identification of three novel mutations in the COL2A1 gene in four unrelated Chinese families with spondyloepiphyseal dysplasia congenita. Biochem Biophys Res Commun 413: 504–508 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Data Availability Statement
No primary datasets produced in the study.