

Abstract
Glia contribute to synapse elimination through phagocytosis in the central nervous system. Despite the important roles of this process in development and neurological disorders, the identity and regulation of the "eat‐me" signal that initiates glia‐mediated phagocytosis of synapses has remained incompletely understood. Here, we generated conditional knockout mice with neuronal‐specific deletion of the flippase chaperone Cdc50a, to induce stable exposure of phosphatidylserine, a well‐known "eat‐me" signal for apoptotic cells, on the neuronal outer membrane. Surprisingly, acute Cdc50a deletion in mature neurons causes preferential phosphatidylserine exposure in neuronal somas and specific loss of inhibitory post‐synapses without effects on other synapses, resulting in abnormal excitability and seizures. Ablation of microglia or the deletion of microglial phagocytic receptor Mertk prevents the loss of inhibitory post‐synapses and the seizure phenotype, indicating that microglial phagocytosis is responsible for inhibitory post‐synapse elimination. Moreover, we found that phosphatidylserine is used for microglia‐mediated pruning of inhibitory post‐synapses in normal brains, suggesting that phosphatidylserine serves as a general "eat‐me" signal for inhibitory post‐synapse elimination.
Keywords: “eat‐me” signal, Glia‐dependent synapse elimination, inhibitory synapse elimination, MERTK, phosphatidylserine
Subject Categories: Neuroscience
Mouse models with increased neuron‐specific exposure of an apoptotic cell‐defining phospholipid provide insight into the nature of the "eat‐me" signal and its recognition during synapse elimination.
Introduction
Synapse elimination, the process by which unnecessary synapses are selectively removed, occurs in the central nervous system during development and adult stages as well as in various neurological disorders (Chung & Barres, 2012). Recent studies have shown that microglia and astrocytes contribute to synapse elimination through complement and MERTK/MEGF10 phagocytic pathways, respectively (Stevens et al, 2007; Schafer et al, 2012; Chung et al, 2013; Lee et al, 2020). Interestingly, microglia have been shown to use the complement cascade, and eliminate either excitatory or inhibitory synapses during development and under disease conditions (Stevens et al, 2007; Schafer et al, 2012; Hong et al, 2016; Lui et al, 2016; Sekar et al, 2016; Vasek et al, 2016). However, the identity of the “eat‐me” signal that allows these glial phagocytic receptors to recognize specific synapses for subsequent elimination is still unclear. Moreover, whether microglia use other molecular mechanism than the complement cascade for eliminating synapses and whether excitatory and inhibitory synapses utilize the same or distinct mechanisms for presenting “eat‐me” signals are unknown.
Phosphatidylserine (PS) is a phospholipid that predominantly resides on the inner plasma membrane under normal physiological conditions. When cells undergo stress or receive apoptotic stimuli, PS flips to the outer plasma membrane, where it functions as an “eat‐me” signal for subsequent engulfment by neighboring phagocytes (Segawa & Nagata, 2015). Interestingly, recent studies have also shown that PS can be exposed transiently in restricted portion of the plasma membrane, such as neuronal processes (Sapar et al, 2018) or excitatory pre‐synapses (Li et al, 2020; Scott‐Hewitt et al, 2020), suggesting their presentation can be controlled locally within the cell. Among the upstream enzymes that control PS presentation in the inner or outer plasma membrane, the flippases, type IV P‐type adenosine triphosphatases (P4‐ATPases), were shown to use adenosine triphosphate (ATP) to translocate PS from the outer to the inner plasma membrane. Although many functionally redundant P4‐ATPases are expressed in various tissues, most P4‐ATPase family members require the activity of the flippase chaperone cell cycle control protein 50A (CDC50A) for proper localization on the plasma membrane (Segawa et al, 2014; Segawa & Nagata, 2015). Previous in vitro studies have shown that a Cdc50a null mutation causes constitutive PS exposure on the outer plasma membrane without inducing general apoptosis and that PS exposure is sufficient to initiate phagocytic engulfment by macrophages (Segawa et al, 2014). However, although various P4‐ATPases and CDC50A are highly expressed in the mammalian brain (Coleman & Molday, 2011; Zhang et al, 2017), their potential roles in synapse elimination have not yet been studied.
Here, by conditionally deleting Cdc50a in mature neurons, we reveal that CDC50A‐dependent PS exposure is required for eliminating inhibitory post‐synapses and generates excessive excitability with appearance of seizure. Surprisingly, CDC50A deletion induces PS exposure preferentially in neuronal soma, thereby inducing engulfment of inhibitory post‐synapses by microglial phagocytosis, without affecting excitatory synapses or inhibitory pre‐synapses. Importantly, ablating microglia or deleting microglial Mertk, a critical phagocytic receptor, rescues the loss of inhibitory post‐synapses and seizure phenotype in Cdc50a‐deleted mice. These data indicate that microglial MERTK phagocytic pathway can specifically phagocytose and eliminate PS‐exposed inhibitory post‐synapses without damaging other cellular membranes. Moreover, we find that inhibitory post‐synapses in normal brains also use PS for synapse pruning by microglia, suggesting PS may serve as a general “eat‐me” signal for inhibitory post‐synapse elimination.
Results
Audiogenic seizure phenotype of Cdc50a cKO mice
In the mammalian brains, various P4‐ATPases are expressed, potentially performing redundant functions (Andersen et al, 2016). Therefore, to understand the roles of PS in synapse elimination, we decided to acutely delete Cdc50a and block the activity of the P4‐ATPase family members in mature neurons. First, we investigated whether CDC50A is expressed in neurons in the mouse brain. In heterozygous Cdc50a knockout‐first allele mice, in which the lacZ gene is knocked in at the Cdc50a locus, we found that β‐galactosidase was highly localized in both CAMKII‐positive and GAD67‐positive neurons (Fig 1A), indicating that Cdc50a is expressed by both excitatory and inhibitory neurons. To generate loxP‐floxed Cdc50a (Cdc50afl /+) mice, we crossed mice with the Cdc50a knockout‐first allele with Act‐FLPe mice to remove FRT sites. Then, the mice with the Cdc50afl /+ allele were subsequently crossed with Thy1‐CreERT2, ‐EYFP mice to generate inducible neuron‐specific Cdc50a conditional KO (cdc50a cKO) mice (Fig EV1A). To delete Cdc50a specifically in mature neurons, we injected oil or 4‐hydroxytamoxifen (4‐OHT) into 1‐month‐old control and cdc50a cKO mice (Fig EV1B). Unlike control mice, Cdc50a cKO mice showed rapid lethality within 2 weeks after 4‐OHT injection (Fig 1B) without significant changes in body weight or brain morphology (Fig EV1C and D). Interestingly, there was no difference in the expression of cleaved caspase‐3 (cCas3), terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining or the number of NeuN‐positive neuronal cells between the brains of Cdc50a cKO and those of control mice (Figs 1C and EV1E–G). We also examined the axonal processes of Thy1‐CreERT2, ‐EYFP‐positive motor neurons in the diaphragm muscles and neuronal morphology of sparsely labeled Thy1‐CreERT2, ‐EYFP‐positive neurons with a floxed tdTomato (Rosa26‐CAG‐tdTomato) reporter, and found that they were unaltered in Cdc50a cKO mice (Fig EV1H and J). These data indicate that acute deletion of neuronal CDC50A induces rapid lethality in mice without inducing cell death.
Figure 1. Neuronal Cdc50a deletion induces rapid lethality and audiogenic seizures.

-
ARepresentative confocal z‐stack images of co‐immunostaining for β‐galactosidase (green) and CAMKII (red, upper) or GAD67 (magenta, lower) in neurons from the brains of Cdc50a knockout‐first allele (Cdc50a lacZ/+) mice. Scale bar, 20 μm.
-
BSurvival rates of control and Cdc50a cKO mice after oil or 4‐OHT injection (n = 5 for each group, ***P = 0.0003).
-
CSchematic diagram of the experiment. Mice of both genotypes were sacrificed 13 days after oil or 4‐OHT injection. Representative confocal z‐stack images of cleaved caspase‐3 (cCas3, red)‐ and Thy1‐CreERT2, ‐EYFP (green)‐expressing cells from control and Cdc50a cKO mice. Scale bar, 20 μm.
-
DSchematic diagram of behavioral tests for measuring audiogenic seizures (0: normal behavior, 1: wild running, 2: seizure, and 3: respiratory arrest and death) following auditory stimuli.
-
E, FBar graphs showing audiogenic seizure scores following auditory stimuli (5, 10, 15, 20, 40 kHz or white noise (WN) at 70, 80, 90, or 100 dB) in control (E) and Cdc50a cKO (F) mice (n = 8 for each group).
-
GRepresentative confocal z‐stack images showing cFos (magenta)‐positive neurons from control and Cdc50a cKO mice (CTX: cortex, HP: hippocampus, and IC: inferior colliculus). Scale bar, 20 μm.
-
HBar graphs showing the relative area of cFos‐positive cells in the brains of control and Cdc50a cKO mice (n = 5 for each group, **P = 0.0030, n.s., not significant).
Data information: The individual dots indicate experimental replicates (3 to 5 images were taken from 2 brain slices per an animal, H). The data are the mean ± SEM and were analyzed by the log‐rank (Mantel‐Cox) test (B) or two‐tailed unpaired t‐test (H).
Figure EV1. Acute neuronal Cdc50a deletion does not induce cell death.
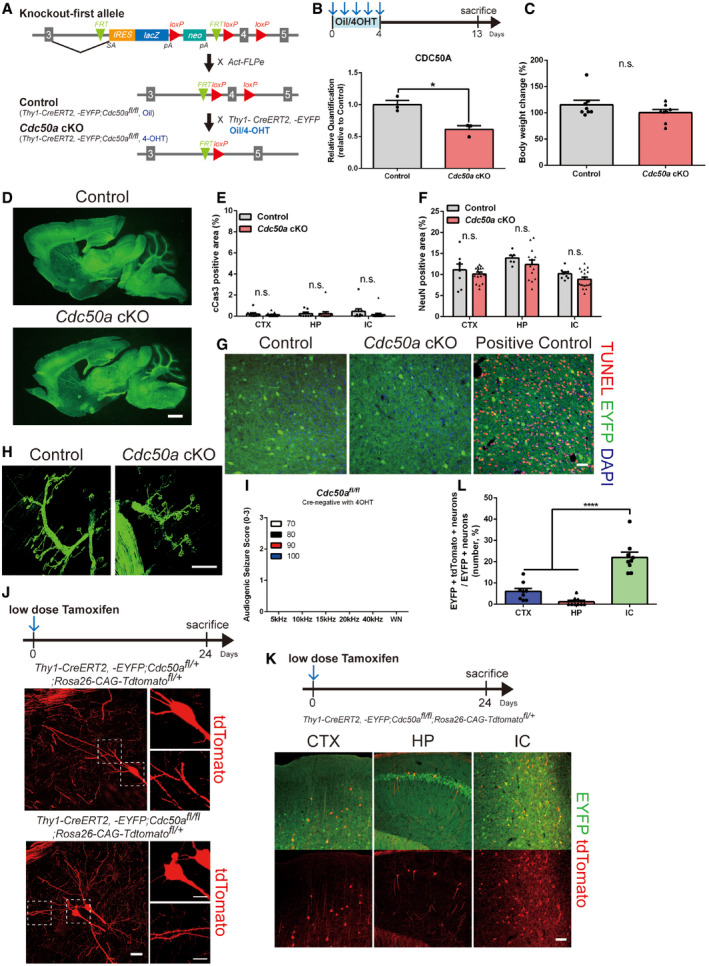
- Schematic diagram of the control and Cdc50a cKO mice. For control and Cdc50a cKO mice, Thy1‐creERT2, ‐EYFP;Cdc50afl / fl mice were injected with oil or 4‐OHT at 1 month of age, respectively.
- Schematic diagram of the experiment. Bar graphs showing relative protein expression of CDC50A in the IC (inferior colliculus) of control and Cdc50a cKO mice (n = 3 for each group, *P = 0.0127). Please see Fig EV1 source data.
- Bar graphs showing the body weight changes in control and Cdc50a cKO mice (n = 8 for each group, n.s., not significant).
- Representative images of Thy1‐creERT2, ‐EYFP (green)‐expressing neurons in sagittal brain sections from control and Cdc50a cKO mice. Scale bar, 100 μm.
- Bar graphs showing the area of cCas3‐positive cells in the CTX (cortex), HP (hippocampus), and IC of control and Cdc50a cKO mice (n = 5 for each group).
- Bar graphs showing the area of NeuN‐positive cells in the CTX, HP, and IC of control and Cdc50a cKO mice (n = 4 for each group).
- Representative confocal z‐stack images of TUNEL staining (red) and DAPI (blue) in Thy1‐CreERT2, ‐EYFP (green)‐expressing IC neurons. Scale bar, 40 μm.
- Representative confocal z‐stack images of Thy1‐creERT2, ‐EYFP (green)‐expressing axons and neuromuscular junctions of motor neurons in the diaphragms of control and Cdc50a cKO mice. Scale bar, 100 μm.
- Bar graphs showing audiogenic seizure scores following auditory stimuli (5, 10, 15, 20, 40 kHz, or white noise (WN) at 70, 80, 90, or 100 dB) in Cre‐negative Cdc50afl / fl mice injected with 4‐OHT (n = 5).
- Schematic diagram of the experiments and representative confocal z‐stack images of tdTomato (red)‐positive HP neurons in control and Cdc50a cKO mice after low‐dose tamoxifen injection. The dashed line represents where the high magnified insert was taken from. Scale bar, 10 μm.
- Representative confocal z‐stack images showing Cre‐mediated recombination in Thy1‐CreERT2, ‐EYFP;Cdc50afl / fl;Rosa26‐CAG‐tdTomatofl /+ mice after low‐dose tamoxifen injection. tdTomato (red) expression in Thy1‐CreERT2, ‐EYFP (green)‐expressing CTX, HP, and IC (CTX: cortex, HP: hippocampus, IC: inferior colliculus) neurons. Scale bar, 100 μm.
- Bar graphs showing the percentage of EYFP and tdTomato double‐positive neurons relative to EYFP‐positive neurons in the CTX, HP, and IC (n = 3 for each group, P**** < 0.0001).
Data information: The individual dots indicate experimental replicates (3–5 images were taken from 2 brain slices per an animal, E, F, and L). The data are the mean ± SEM and were analyzed by two‐tailed unpaired t‐test (B, C, E, and F) or one‐way ANOVA with Tukey’s test (L).
Source data are available online for this figure.
Unexpectedly, while monitoring the behaviors of Cdc50a cKO mice, we found they showed convulsive seizures upon exposure to a loud noise (Movie EV1). To test whether Cdc50a cKO mice develop seizures in response to sounds, we provided auditory stimuli and scored seizure behaviors (0: normal behavior, 1: wild running, 2: seizure, and 3: tonic–clonic seizures with loss of righting reflex, Movie EV2). Interestingly, we found that unlike control mice, Cdc50a cKO mice showed severe audiogenic seizures (wild running and seizures) at a high frequency between 10 and 20 kHz (Figs 1D–F and EV1I). In agreement with the audiogenic seizure phenotype, neuronal activity as measured by cFos protein expression, was significantly upregulated in the inferior colliculus (IC) (Fig 1G and H), the principal midbrain nucleus of the auditory pathway (Lee & Sherman, 2010) and primary brain region for audiogenic seizure (Faingold et al, 1992; Snyder‐Keller & Pierson, 1992; Yang et al, 2001; Faingold, 2002), in Cdc50a cKO mice compared with control mice 13 days after 4‐OHT injection.
The observed audiogenic seizure phenotype and cFos upregulation in the IC are likely due to strong Thy1 promoter activity in IC neurons. When we sparsely labeled Thy1‐CreERT2, ‐EYFP‐positive neurons with a floxed tdTomato (Rosa26‐CAG‐tdTomato) reporter by injecting low doses of tamoxifen, we found more frequent and robust genetic recombination in the IC than in other brain regions, such as the cortex and hippocampus (Fig EV1K and L).
Inhibitory post‐synapse loss in Cdc50a cKO brains
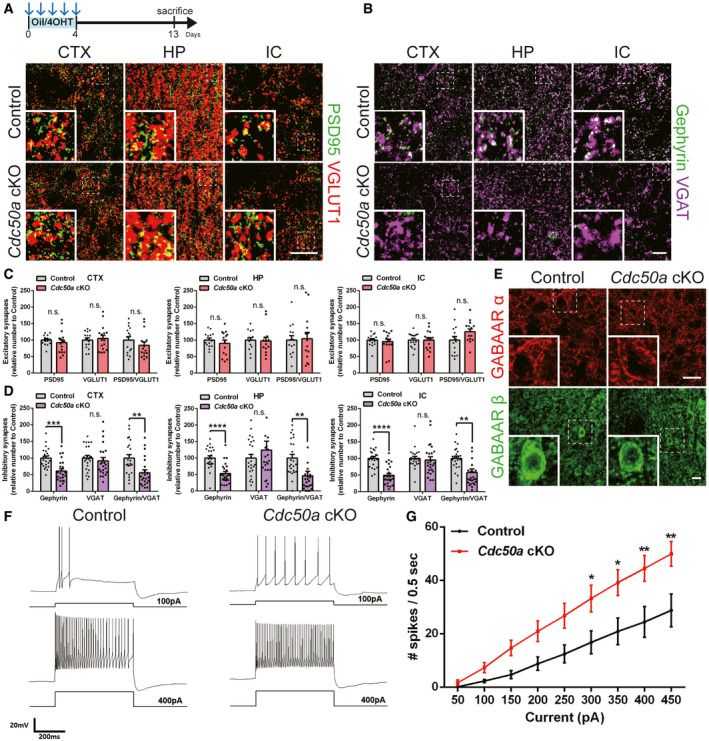
Next, to test whether audiogenic seizures in Cdc50a cKO mice are due to synaptic imbalance (Fritschy, 2008), we measured the number of excitatory and inhibitory synapses in various brain regions. Interestingly, we found that the number of VGLUT1/2‐ and PSD95‐positive excitatory synapses was not changed in Cdc50a KO mice compared with control mice (Figs 2A and C and EV2A and B). Moreover, there was no significant difference in the number of VGAT‐positive inhibitory pre‐synapses (Figs 2B and D). However, surprisingly, we found that the number of Gephyrin‐positive inhibitory post‐synapses was significantly lower in Cdc50a cKO mice than in control mice in the cortex, hippocampus, and IC (Figs 2B and D and EV2C and D). Due to the reduced number of Gephyrin‐positive inhibitory post‐synapses, the total number of inhibitory synapses containing both Gephyrin and VGAT was significantly reduced (Fig 2D).
Figure 2. Neuronal Cdc50a deletion induces loss of inhibitory post‐synapses.
-
A, BSchematic diagram of the experiment. Representative confocal z‐stack images of PSD95 (green)‐ and VGLUT1 (red)‐positive excitatory (A) and Gephyrin (green)‐ and VGAT (magenta)‐positive inhibitory (B) synapses in the CTX, HP, and IC (CTX: cortex, HP: hippocampus, and IC: inferior colliculus) of control and Cdc50a cKO mice. The dashed line represents where the high magnified insert was taken from. Scale bar, 20 μm.
-
C, DBar graphs showing the numbers of excitatory (C, PSD95 and VGLUT1) and inhibitory (D, Gephyrin and VGAT) synapses in the CTX, HP, and IC of control and Cdc50a cKO mice (n = 5 for each group, for excitatory synapses: n.s., not significant, for inhibitory synapses: CTX, ***P = 0.0001, **P = 0.0023, HP, **P = 0.0038, IC, **P = 0.0023, ****P < 0.0001).
-
ERepresentative confocal z‐stack images of GABAA receptor α (red) and β (green) staining in the IC in control and Cdc50a cKO mice. The dashed line represents where the high magnified insert was taken from. Scale bar, 20 μm.
-
FRepresentative current clamp traces from the IC of control and Cdc50a cKO mice; +100 pA (upper) and +400 pA (lower) currents were injected into control mice (left) and Cdc50a cKO mice (right) for 500 ms.
-
GFrequency–current (F‐I) curve of the IC of control and Cdc50a cKO mice. The number of spikes per 500 ms was measured and averaged for each current (each, n = 9 and 13 cells, 300 pA: *P = 0.0203, 350 pA: *P = 0.0133, 400 pA **P = 0.0098, 450 pA: **P = 0.0072).
Data information: The individual dots indicate experimental replicates (3–5 images were taken from 2 brain slices per an animal, C and D). The data are the mean ± SEM and were analyzed by two‐tailed unpaired t‐test (C and D) or two‐way ANOVA (G).
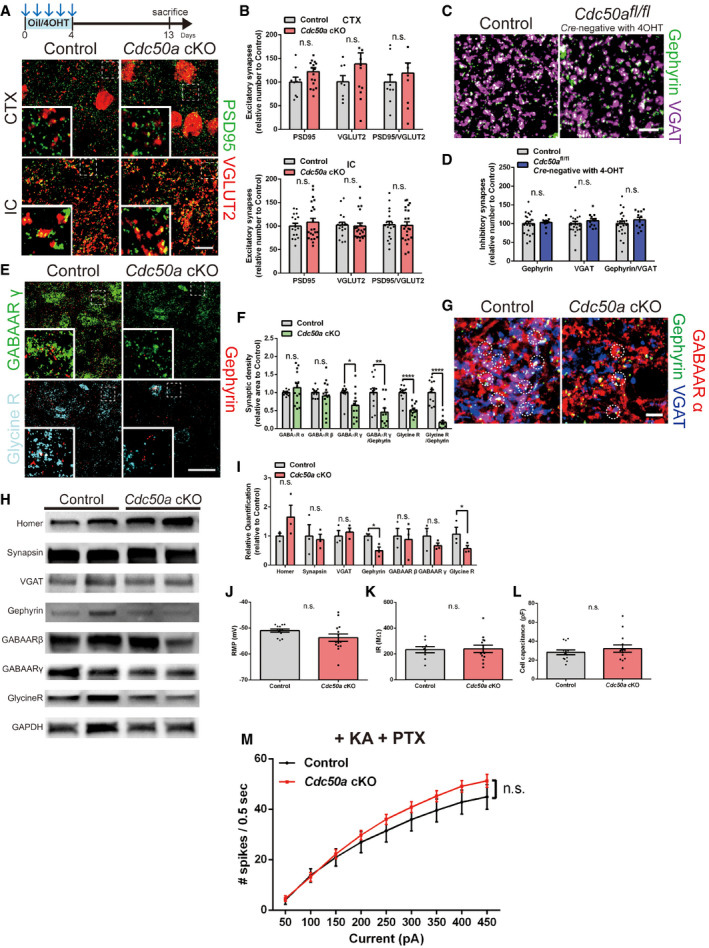
Figure EV2. Neuronal Cdc50a deletion reduces the number of inhibitory synapses without affecting excitatory synapses.
-
ASchematic diagram of the experiment. Representative confocal z‐stack images of PSD95 (green)‐ and VGLUT2 (red)‐positive excitatory synapses in the CTX and IC (CTX: cortex and IC: inferior colliculus) of control and Cdc50a cKO mice. The dashed line represents where the high magnified insert was taken from. Scale bar, 10 μm.
-
BBar graphs showing the number of excitatory (PSD95‐ and VGLUT2‐positive) synapses in the CTX, HP (hippocampus), and IC of control and Cdc50a cKO mice (n = 5 for each group).
-
CRepresentative confocal z‐stack images of Gephyrin (green)‐ and VGAT (magenta)‐positive inhibitory synapses in control and Cre‐negative Cdc50afl / fl mice injected with 4‐OHT. Scale bar, 10 μm.
-
DBar graphs showing the number of inhibitory (Gephyrin‐ and VGAT‐positive) synapses in the IC of control and Cre‐negative Cdc50afl / fl mice injected with 4‐OHT (n = 3 to 5 for each group, n.s., not significant).
-
ERepresentative confocal z‐stack images of GABAA receptor γ (green), glycine receptor (cyan), and gephyrin (red) in the IC of control and Cdc50a cKO mice. The dashed line represents where the high magnified insert was taken from. Scale bar, 20 μm.
-
FBar graphs showing the relative inhibitory synaptic density in the IC of control and Cdc50a cKO mice (n = 4 for each group, *P = 0.0123, **P = 0.0042, ****P < 0.0001).
-
GRepresentative confocal z‐stack images of GABAA receptor α (red), Gephyrin (green), and VGAT (magenta) in the IC of control and Cdc50a cKO mice. The white circles indicate colocalization of GABAA receptor α with Gephyrin and VGAT. Scale bar, 20 μm.
-
HRepresentative Western blot images of synaptic proteins (Homer, Synapsin, VGAT, Gephyrin, GABAAR β, GABAAR γ, and Glycine R) and GAPDH in the IC of control and Cdc50a cKO mice. Please see Fig EV2 source data.
-
IBar graphs showing the quantification of the Western blot data of synaptic proteins (Homer, Synapsin, VGAT, Gephyrin, GABAAR β, GABAAR γ, and Glycine R) for the IC of control and Cdc50a cKO mice (n = 3 for each group, Gephyrin, *P = 0.0240 and Glycine receptor, *P = 0.0412).
-
J–LResting membrane potential (RMP, J), input resistance (IR, K), and cell capacitance (L) of the IC of control and Cdc50a cKO mice (each, n = 13 cells).
-
MFrequency‐current (F‐I) curve of the IC of control and Cdc50a cKO mice with kainate (KA) and picrotoxin (PTX). The number of spikes per 500 ms was measured and averaged for each current (each, n = 10 and 11 cells).
Data information: The individual dots indicate experimental replicates (3–5 images were taken from 2 brain slices per an animal, B, D, and F). The data are the mean ± SEM and were analyzed by two‐tailed unpaired t‐test.
Source data are available online for this figure.
Gephyrin is a critical inhibitory post‐synaptic element that binds and stabilizes both glycine and γ‐aminobutyric acid type A (GABAA) receptors for inhibitory synaptic transmission (Brickley & Mody, 2012). We found significantly fewer glycinergic and GABAergic post‐synapses, as detected by glycine receptor and GABAA receptor γ subunit antibodies, respectively, in Cdc50a cKO mice than in control mice (Fig EV2E–I). However, GABAA receptor α and β subunits, which are also localized in the extrasynaptic membrane as well (Choii & Ko, 2015), appeared to be intact in Cdc50a cKO mice (Figs 2E and EV2G–I), suggesting that Cdc50a deletion in neurons specifically reduces the number of inhibitory post‐synapses without disrupting neuronal membranes.
Importantly, when we evaluated the basic electrophysiological properties of IC neurons from control and Cdc50a cKO mice, we found that there were no significant changes in resting membrane potential (RMP), input resistance (IR), and cell capacitance (Fig EV2J–L), showing that membrane integrity was unaffected in Cdc50a cKO neurons. However, when we measured the frequency–current relationship (F‐I curve) by holding the membrane potential at −60 mV and injecting the current from +50 pA to +450 pA for 500 ms each, there were significantly more spikes in Cdc50a cKO neurons than in control neurons (Fig 2F and G), suggesting that neuronal excitability was increased by Cdc50a deletion. Interestingly, this difference in excitability was significantly diminished by applying both kainate (KA) and picrotoxin (PTX), AMPA/kainate agonist and GABAA receptor antagonist, respectively (Fig EV2M). Thus, our data suggest that increased excitability of Cdc50a cKO IC neurons to sound‐evoked stimuli is due to synaptic imbalance and may underlie audiogenic seizure behaviors in Cdc50a cKO mice.
PS exposure in Cdc50a cKO neuronal somas
Why are only inhibitory post‐synapses affected when Cdc50a is deleted from neurons? When we expressed Cdc50a‐mCherry in neurons in vitro and in vivo, we found that CDC50A‐mCherry remained in somas of neurons instead of spreading to their processes (Fig 3A and B). Nissl staining of rough endoplasmic reticulum (rER) revealed that CDC50A‐mCherry was localized to the rER, as previously suggested (Byrne & Roberts, 2009). Since rER resides mostly within the cell soma without extending to the distal processes of neurons (Ramirez & Couve, 2011), we hypothesized that Cdc50a cKO neurons might expose PS preferentially in the neuronal soma. To test our hypothesis, we cultured cortical neurons from Cdc50afl / fl mice in vitro and transfected them with hSyn‐mCherry or hSyn‐Cre‐p2a‐dTomato to generate control and Cdc50a cKO neurons, respectively (Fig 3C). After deleting Cdc50a, we monitored the extent of PS exposure on the outer plasma membranes of neurons by utilizing polarity‐sensitive indicator of viability (pSIVA), a probe that reversibly binds PS and emits green fluorescence (Kim et al, 2010). Surprisingly, after hSyn‐Cre‐p2a‐dTomato transfection, we found that PS exposure preferentially occurred in the soma first in Cdc50a cKO neurons (Fig 3D). Next, to detect this preferential PS exposure in the brains of Cdc50a cKO mice, we adopted the secreted Annexin V (secA5) system (van Ham et al, 2010), in which neighboring cells, in our case astrocytes, express and secrete Annexin V fused with mCherry (Fig 3E). AAV9‐GFAP‐secA5‐mCherry was injected to allow secA5‐mCherry to be expressed by and secreted from astrocytes, enabling the detection of PS‐exposed material in the brain (Fig 3E). Consistent with our in vitro data, we found that secA5 puncta were mostly localized in the neuronal soma rather than in processes in the brains of Cdc50a cKO mice (Fig 3F), suggesting that the neuronal soma is the major site of PS exposure when Cdc50a is deleted. Importantly, we also found that the number of Gephyrin‐positive inhibitory post‐synapses was significantly lower in the somas and proximal dendrites, but not in the distal dendrites, in the hippocampal CA1 region in Cdc50a cKO mice (Fig 3G and H). Thus, since neuronal somas harbor many more inhibitory post‐synapses than other types of synapses (Keith & El‐Husseini, 2008; Kubota et al, 2016), our data suggest that preferential PS exposure in the somas of Cdc50a‐deleted neurons is responsible for the specific loss of inhibitory post‐synapses.
Figure 3. Neuronal Cdc50a deletion induces PS exposure preferentially in the cell soma.
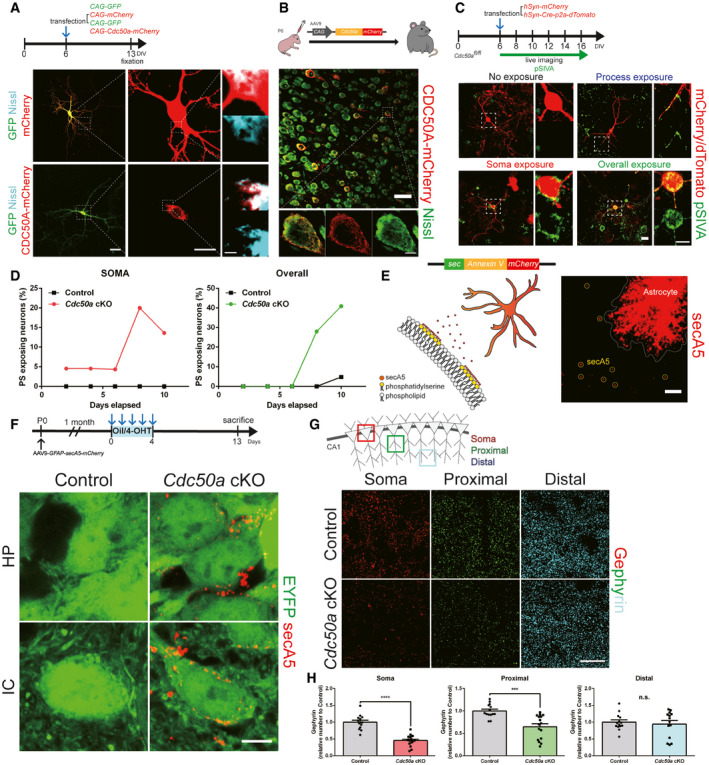
- Schematic diagram of the in vitro CDC50A‐mCherry expression experiment. After 6 days in vitro (DIV), cortical neurons were transfected with CAG‐GFP and CAG‐mCherry or CAG‐GFP and CAG‐Cdc50a‐mCherry. Representative confocal z‐stack images of the rER (Nissl staining, cyan) with GFP (green) and mCherry/CDC50A‐mCherry (red). The dashed line represents where the high magnified insert was taken from. Scale bars, 50 (left), 20 (middle), and 5 μm (right).
- Schematic diagram of in vivo CDC50A‐mCherry expression experiment. Neonatal (P0) wild‐type mice were injected with AAV9‐CAG‐Cdc50a‐mCherry. Representative confocal z‐stack images of the rER (Nissl staining, green) and CDC50A‐mCherry (red) in the CTX (cortex). The dashed line represents where the high magnified insert was taken from. Scale bars, 20 μm (upper) and 5 μm (lower).
- Schematic diagram of the experiment in which PS exposure was detected in control and Cdc50a cKO neurons in vitro. After 6 DIV, cortical Cdc50afl / fl neurons were transfected with either hSyn‐mCherry or hSyn‐Cre‐p2a‐dTomato to generate control and Cdc50a cKO neurons, respectively. Representative live confocal z‐stack images showing PS exposure (pSIVA, green) in mCherry/dTomato (red)‐expressing neurons. Scale bar, 20 μm.
- The bar graphs show the relative percentages of neurons showing PS exposure only in the soma (left) and overall PS exposure (right) (n = 30–40 cells for each group) in control and Cdc50a cKO mice.
- Schematic diagram of the method in which PS exposure was detected in vivo using secreted Annexin V (secA5) fused with mCherry (red). Representative confocal z‐stack images of secA5‐mCherry (red) expression in the brains of wild‐type mice using AAV9‐GFAP‐secA5‐mCherry injection. The white circles indicate PS exposure detected by secA5. Scale bar, 10 μm.
- Schematic diagram of the method in which PS exposure was detected in the brains of control and Cdc50a cKO mice in vivo after AAV9‐GFAP‐secA5‐mCherry was injected in the neonatal stage (P0). Representative confocal single‐plane images showing PS exposure (secA5‐mCherry, red) in the somas of Thy1‐CreERT2, ‐EYFP‐expressing HP (hippocampus) CA1 neurons or IC (inferior colliculus) neurons (green) from control and Cdc50a cKO mice. Scale bar, 5 μm.
- Schematic diagram of imaging of the neuronal somas and proximal and distal dendrites of neurons in the CA1 region of the HP. Representative confocal z‐stack images showing Gephyrin‐positive inhibitory post‐synapses in the somas (red) and proximal (green) and distal dendrites (cyan) of neurons in the CA1 region of the HP in control and Cdc50a cKO mice. Scale bar, 20 μm.
- Bar graphs showing the number of Gephyrin‐positive inhibitory post‐synapses in the somas and proximal and distal dendrites of neurons in the CA1 of the HP in control and Cdc50a cKO mice (n = 4 for each group, ***P = 0.0002, ****P < 0.0001).
Data information: The individual dots indicate experimental replicates (3–5 images were taken from 2 brain slices per an animal, H). The data are the mean ± SEM and were analyzed by two‐tailed unpaired t‐test.
Inhibitory post‐synapse elimination by microglial MERTK
Then, how does Cdc50a deletion and subsequent PS exposure induce the loss of inhibitory post‐synapses? We hypothesized that microglia and/or astrocytes might phagocytose inhibitory post‐synapses by recognizing outer membrane‐exposed PS due to Cdc50a deletion. To test our hypothesis, we first measured glial activation by detecting IBA1 and GFAP, and found that both microglia and astrocytes were significantly activated in the IC of Cdc50a cKO mice (Fig EV3A–C). In addition, the area of Cathepsin D‐positive lysosomes and the area of CD68‐positive microglial lysosomes were both dramatically upregulated in Cdc50a cKO mice compared with control mice (Figs 4A and EV3D–F). Interestingly, we found that Gephyrin‐positive puncta, as detected by staining with a Gephyrin antibody (Fig 4B and C) and the expression of mCherry‐Gephyrin fusion protein (Fig EV3I and J), were highly localized inside of CD68‐positive microglial lysosomes in Cdc50a cKO mice. These Gephyrin‐positive puncta inside microglia expressed secA5 as well (Fig EV3K and L), indicating that microglia phagocytose PS‐exposed inhibitory post‐synapses in Cdc50a cKO mice. Since astrocytes also play critical roles in synapse elimination (Chung et al, 2013; Lee et al, 2020), we also examined whether astrocytes also phagocytose Gephyrin in Cdc50a cKO mice. Although we found that the number of LAMP2‐positive lysosomes in astrocytes was higher in the cortex, hippocampus, and IC in Cdc50a cKO mice than in control mice (Fig EV3G and H), mCherry‐Gephyrin was mostly localized in microglia rather than astrocytes (Fig EV3I and J), indicating that microglia are responsible for phagocytosing inhibitory post‐synapses in Cdc50a cKO mice.
Figure EV3. Neuronal Cdc50a deletion increases the number of glial lysosomes.

-
ASchematic diagram of the experiment. Representative confocal z‐stack images of activated astrocyte (GFAP, red) and microglia (IBA1, green) in the IC (inferior colliculus) of control and Cdc50a cKO mice. Scale bar, 20 μm.
-
B, CBar graphs showing the relative area of GFAP (B) and IBA1 (C) in the CTX (cortex), HP (hippocampus), and IC of control and Cdc50a cKO mice (n = 7 for each group, ****P < 0.0001 and n.s., not significant).
-
DRepresentative confocal z‐stack images of Cathepsin D (magenta)‐positive lysosomes in the IC of control and Cdc50a cKO mice. Scale bar, 40 μm.
-
EBar graphs showing the relative area of Cathepsin D in the CTX, HP, and IC of control and Cdc50a cKO mice (n = 4 for each group, **P = 0.0058, n.s., not significant).
-
FBar graphs showing the relative CD68 area in the CTX, HP, and IC of control and Cdc50a cKO mice (n = 4 for each group and ****P < 0.0001).
-
GRepresentative confocal z‐stack images of LAMP2 (red) and S100β (green) in the IC of control and Cdc50a cKO mice. Scale bar, 20 μm.
-
HBar graphs showing the relative colocalization area of LAMP2 and S100β in the CTX, HP, and IC of control and Cdc50a cKO mice (n = 4 for each group, CTX, **P = 0.0036, HP, *P = 0.0377, IC, *P = 0.0197).
-
ISchematic diagram of the experiments in which glial phagocytosis of inhibitory post‐synapses was measured in control and Cdc50a cKO mice by injecting AAV9‐hSyn‐mCherry‐Gephyrin in the neonatal stage (P0). Oil or 4‐OHT was injected into 1‐month‐old mice for 5 consecutive days. Representative confocal z‐stack and colocalization images of mCherry‐Gephyrin (green), S100β (red), and IBA1 (magenta) in the CTX of control and Cdc50a cKO mice. The yellow arrows indicate colocalization of mCherry‐Gephyrin with S100β and IBA1. Scale bar, 10 μm.
-
JBar graphs showing the percentages of engulfed mCherry‐Gephyrin area by glial cells in the CTX of control and Cdc50a cKO mice (n = 3 for each group, control S100β versus Cdc50a cKO IBA1, **P = 0.0035, Cdc50a cKO S100β versus Cdc50a cKO IBA1, **P = 0.0091, control IBA1 versus Cdc50a cKO IBA1, *P = 0.0126).
-
KSchematic diagram of the experiment in which microglial phagocytosis of PS‐exposed inhibitory post‐synapses was measured in control and Cdc50a cKO mice by injecting AAV9‐GFAP‐secA5‐mCherry in the neonatal stage (P0). Oil or 4‐OHT was injected into 1‐month‐old mice for 5 consecutive days. Representative confocal z‐stack and colocalization images of Gephyrin (green), secA5 (red), and IBA1 (blue) in the IC of control and Cdc50a cKO mice. The yellow arrows indicate colocalization of secA5 with Gephyrin in IBA1‐positive microglia. Scale bar, 20 μm.
-
LBar graphs showing the relative colocalization area of Gephyrin and secA5 in microglia in the IC of control and Cdc50a cKO mice (n = 3 for each group, **P = 0.018).
Data information: The individual dots indicate experimental replicates (3–5 images were taken from 2 brain slices per an animal, B, C, E, F, H, J, and L). The data are the mean ± SEM and were analyzed by two‐tailed unpaired t‐test (B, C, E, F, H, and L) or one‐way ANOVA with Tukey’s test (J).
Figure 4. Neuronal Cdc50a deletion increases microglial phagocytosis of inhibitory post‐synapses and prevents the loss of inhibitory post‐synapses by microglial ablation.

- Representative confocal z‐stack images of microglial lysosomes (CD68, green) in the IC (inferior colliculus) of control and Cdc50a cKO mice. Scale bar, 20 μm.
- Representative confocal z‐stack and colocalization images of Gephyrin (green), IBA1 (blue), and CD68 (red) in the IC of control and Cdc50a cKO mice. Scale bar, 20 μm.
- Bar graphs showing the relative colocalization area of Gephyrin and CD68 in the IC of control and Cdc50a cKO mice (n = 3 for each group, **P = 0.0024).
- Schematic diagram of microglial ablation in control and Cdc50a cKO mice using DMSO‐ or PLX3397‐containing chow. DMSO‐ or PLX33997‐containing chow was administered for 2 weeks before oil or 4‐OHT injection. Bar graphs showing the audiogenic seizure scores of Cdc50a cKO mice treated with DMSO or PLX3397 after a 20 kHz/90 dB auditory stimulus (n = 7 for Cdc50a cKO;DMSO and 3 for Cdc50a cKO;PLX3397).
- Representative confocal z‐stack images of microglia (IBA1, green, upper) and inhibitory synapses (Gephyrin: green, VGAT: red, lower) in the IC of control;DMSO, control;PLX3397, Cdc50a cKO;DMSO and Cdc50a cKO;PLX3397 mice. The dashed line represents where the high magnified insert was taken from. Scale bar, 20 μm.
- Bar graphs showing the relative area of IBA1‐positive cells in the IC of control;DMSO, control;PLX3397, Cdc50a cKO;DMSO and Cdc50a cKO;PLX3397 mice (n = 3 for each group, control;DMSO versus control;PLX3397, *P = 0.0428, control;DMSO versus Cdc50a cKO;DMSO, ***P = 0.0002, Cdc50a cKO;DMSO versus Cdc50a cKO;PLX3397, ***P = 0.0005).
- Bar graphs showing the number of inhibitory (Gephyrin‐ and VGAT‐positive) synapses in the IC of control;DMSO, control;PLX3397, Cdc50a cKO;DMSO and Cdc50a cKO;PLX3397 mice (n = 3 for each group, ****P < 0.0001).
Data information: The individual dots indicate experimental replicates (3–5 images were taken from 2 brain slices per an animal, C, F and G). The data are the mean ± SEM and were analyzed by two‐tailed unpaired t‐test (C and D) or one‐way ANOVA with Tukey’s test (F and G).
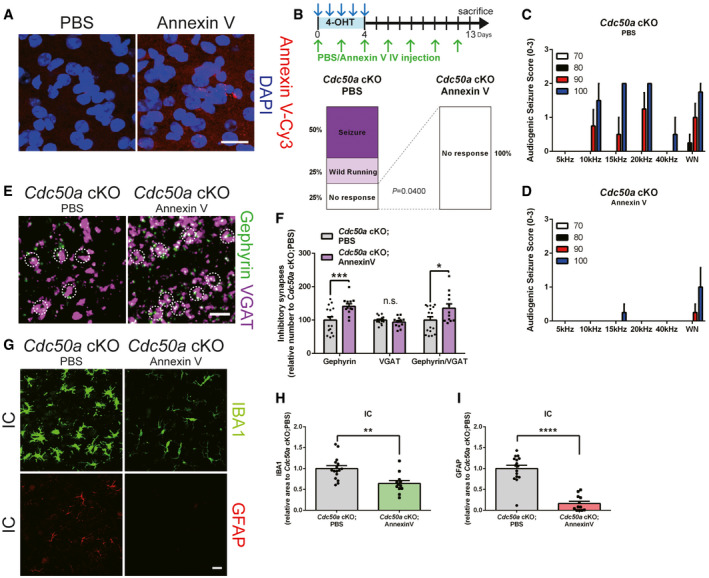
Next, to test whether PS exposure is necessary for glial activation and inhibitory synapse loss, we intravenously injected an excessive amount of Annexin V to quench and block outer membrane‐exposed PS from binding with its binding partners, such as bridging molecules and phagocytic receptors (Derecki et al, 2012). Surprisingly, we found that consecutive intravenous Annexin V injection was sufficient to rescue audiogenic seizure and block glial activation as well as inhibitory post‐synapse loss in Cdc50a KO mice (Fig EV4A–I), indicating that PS exposure is necessary for inducing such phenotypes in Cdc50a‐deleted neurons.
Figure EV4. PS exposure is necessary for glial activation and inhibitory post‐synapse loss in Cdc50a cKO mice.
-
ARepresentative confocal z‐stack images for Annexin V (red) and DAPI (magenta) in the IC (inferior colliculus) of PBS‐ or Annexin V‐Cy3‐injected wild‐type mice. Scale bar, 20 μm.
-
BSchematic diagram of the experiment in which Annexin V was intravenously injected (every 2 days) to block PS in Cdc50a cKO mice. Bar graphs showing the audiogenic seizure scores of in PBS‐ or Annexin V‐injected Cdc50a cKO mice after a 20 kHz/90 dB auditory stimulus (n = 5 for each group).
-
C, DBar graphs showing audiogenic seizure scores following auditory stimuli (5, 10, 15, 20, 40 kHz or white noise (WN) at 70, 80, 90, or 100 dB) in PBS‐ (C) or Annexin V‐ (D) injected Cdc50a cKO mice (n = 5 for each group).
-
ERepresentative confocal z‐stack images for Gephyrin (green)‐ and VGAT (magenta)‐positive inhibitory synapses in the IC of PBS‐ or Annexin V‐injected Cdc50a cKO mice. The white circles indicate inhibitory synapses. Scale bar, 10 μm.
-
FBar graphs showing the number of inhibitory (Gephyrin‐ and VGAT‐positive) synapses in the IC of PBS‐ or Annexin V‐injected Cdc50a cKO mice (n = 3 for each group, P*** = 0.0004, P* = 0.0347, n.s., not significant).
-
GRepresentative confocal z‐stack images showing reactive microglia (IBA1, green) and astrocytes (GFAP, red) in the IC of PBS‐ or Annexin V‐injected Cdc50a cKO mice. Scale bar, 20 μm.
-
H, IBar graphs showing the relative area of IBA1‐ (H) and GFAP‐ (I) positive cells in the IC (inferior colliculus) of PBS‐ or Annexin V‐injected Cdc50a cKO mice (n = 3 for each group, **P = 0.0014, ****P < 0.0001).
Data information: The individual dots indicate experimental replicates (3–5 images were taken from 2 brain slices per an animal, F, H, and I). The data are the mean ± SEM and were analyzed by two‐tailed unpaired t‐test.
To directly address the roles of microglia in the loss of inhibitory post‐synapses in Cdc50a cKO mice, we adopted two different approaches. First, we depleted microglia in the brains of Cdc50a cKO using PLX3397, a well‐characterized colony‐stimulating factor 1 receptor (CSF1R) kinase inhibitor that can efficiently deplete microglia from the brain (Elmore et al, 2014). Cdc50a cKO and control mice were fed PLX3397‐ or DMSO‐containing chow for 14 days before being injected with 4‐OHT or oil and for an additional 13 days before analysis (Fig 4D). PLX3397‐treated Cdc50a cKO mice showed variations in the extent of microglia depletion (Appendix Fig S1C and D). Surprisingly, we found that the audiogenic seizure phenotype was only fully rescued in PLX3397‐treated Cdc50a cKO mice when the vast majority of microglia were depleted (Fig 4E and F, Appendix Fig S1A and B). In these PLX3397‐treated Cdc50a cKO mice that exhibited behavioral rescue, the number of Gephyrin‐positive inhibitory post‐synapses was also restored to the level observed in DMSO‐treated control mice (Fig 4E and G). Interestingly, some PLX3397‐treated Cdc50a cKO mice failed to show behavioral rescue (no behavioral rescue group). When we analyzed the brains of these mice, we found that a significant number of microglia remained (Appendix Fig S1E) and that there was a significant correlation between the number of remaining microglia in the IC and audiogenic seizure score (Appendix Fig S1B). Moreover, in this no behavioral rescue group in which microglia were unsuccessfully depleted, the number of Gephyrin‐positive inhibitory post‐synapses was not restored to the control level (Appendix Fig S1C and E), indicating that microglial depletion is necessary to prevent the audiogenic seizure and inhibitory synapse loss phenotype in Cdc50a cKO mice.
As a second approach, we deleted critical phagocytic receptors from microglia in Cdc50a cKO mice. Previous studies have shown that microglia phagocytose excitatory and inhibitory synapses through the classical complement pathway during development and neurological diseases (Stevens et al, 2007; Schafer et al, 2012; Hong et al, 2016; Lui et al, 2016; Sekar et al, 2016; Vasek et al, 2016). Thus, we crossed complement 3 (C3) KO mice with Cdc50a cKO mice to address whether microglia utilize the complement pathway to phagocytose inhibitory post‐synapses. Interestingly, although the general expression of C1q, an initiator of the complement pathway, was significantly enhanced in the brains of Cdc50a cKO mice compared with control mice (Appendix Fig S2A and B), there was no increase in C1q‐tagged Gephyrin in Cdc50a cKO compared with control mice (Appendix Fig S2C and D). Moreover, we found that C3 KO failed to rescue audiogenic seizures, loss of inhibitory synapses, and reactive gliosis in Cdc50a cKO mice (Fig 5A–C and Appendix Fig S2E–G). These data indicate that the complement pathway is not directly associated with regulating the number of inhibitory synapses in Cdc50a cKO mice.
Figure 5. Mertk deficiency prevents the loss of inhibitory post‐synapses and audiogenic seizures in Cdc50a cKO mice.
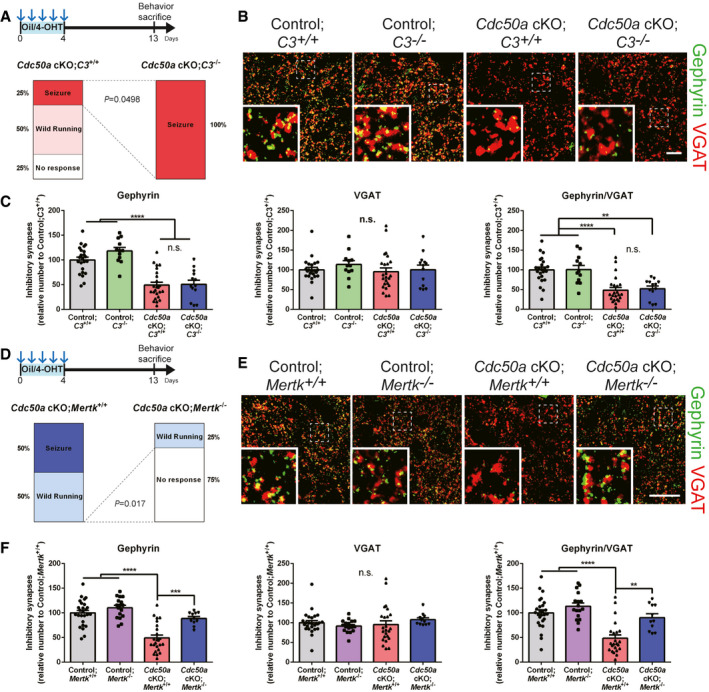
- Schematic diagram of the experiment. Bar graphs showing the audiogenic seizure scores of Cdc50a cKO;C3 +/+ and Cdc50a cKO;C3 −/− mice after a 20 kHz/90 dB auditory stimulus (n = 4 for each group).
- Representative confocal z‐stack images of Gephyrin (green)‐ and VGAT (red)‐positive inhibitory synapses in the IC (inferior colliculus) of control;C3 +/+, control;C3 −/−, Cdc50a cKO;C3 +/+ and Cdc50a cKO;C3 −/− mice. The dashed line represents where the high magnified insert was taken from. Scale bar, 10 μm.
- Bar graphs showing the number of inhibitory (Gephyrin‐ and VGAT‐positive) synapses in the IC of control;C3 +/+, control;C3 −/−, Cdc50a cKO;C3 +/+, and Cdc50a cKO;C3 −/− mice (n = 4 for each group, **P = 0.0022, ****P < 0.0001, n.s., not significant).
- Schematic diagram of the experiment. Bar graphs showing the audiogenic seizure scores of Cdc50a cKO;Mertk +/+ and Cdc50a cKO;Mertk −/− mice after a 20 kHz/90 dB auditory stimulus (n = 4 for each group).
- Representative confocal z‐stack images of Gephyrin (green)‐ and VGAT (red)‐positive inhibitory synapses in the IC of control;Mertk +/+, control;Mertk −/−, Cdc50a cKO;Mertk +/+, and Cdc50a cKO;Mertk −/− mice. The dashed line represents where the high magnified insert was taken from. Scale bar, 20 μm.
- Bar graphs showing the number of inhibitory (Gephyrin‐ and VGAT‐positive) synapses in the IC of control;Mertk +/+, control;Mertk −/−, Cdc50a cKO;Mertk +/+, and Cdc50a cKO;Mertk −/− mice (n = 4 for each group, ****P < 0.0001, ***P = 0.0002, **P = 0.0021).
Data information: The individual dots indicate experimental replicates (3–5 images were taken from 2 brain slices per an animal, C and F). The data are the mean ± SEM and were analyzed by two‐tailed unpaired t‐test (A and D) or one‐way ANOVA with Tukey’s test (C and F).
In addition to the complement pathway, microglia express the phagocytic receptor MERTK (Fourgeaud et al, 2016), a member of the TAM (Tyro3, Axl, and Mertk) tyrosine kinase receptor family, which can recognize PS as an “eat‐me” signal through bridging molecules, such as Gas6 and protein S, and initiate phagocytosis (Lemke, 2017). Although the expression of Gas6 was not changed in Cdc50a cKO mice (Appendix Fig S2H), we found that the expression of microglial MERTK and AXL was highly upregulated in Cdc50a cKO mice compared with control mice (Appendix Fig S2I–K). Moreover, to our surprise, introducing a Mertk KO in the Cdc50a cKO mice prevented audiogenic seizures as well as the loss of inhibitory post‐synapses (Fig 5D–F and Appendix Fig S2L). However, reactive gliosis occurred in the brains of Cdc50a cKO; Mertk KO mice (Appendix Fig S2M and N), suggesting that other phagocytic receptors than Mertk might also participate in glial activation in Cdc50a cKO mice. Taken together, our data strongly suggest that Cdc50a cKO mediates the loss of inhibitory synapses through MERTK‐dependent microglial phagocytosis.
PS as an “eat‐me” signal during normal inhibitory synaptic pruning
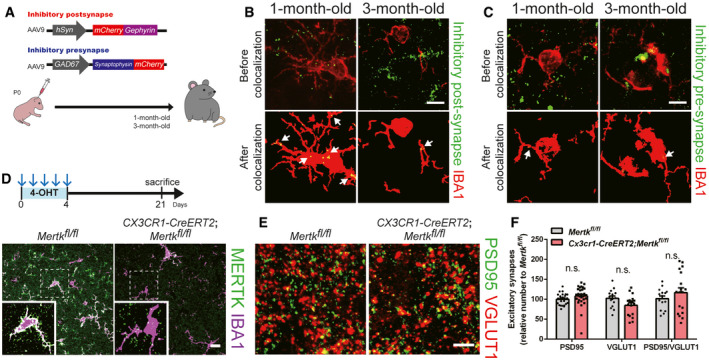
Then, does PS exposure‐mediated inhibitory post‐synapse elimination occur naturally during the normal synaptic pruning process? By utilizing our in vivo PS reporter system with secA5‐mCherry, we found that Gephyrin‐positive inhibitory post‐synapses showed a significantly higher chance of PS exposure than inhibitory pre‐synapses in the brains of 1‐month‐old mice (Fig 6A and B). Interestingly, we also found that this preferential PS exposure in inhibitory post‐synapses was not observed in the brains of 3‐month‐old mice (Fig 6A and B), indicating that this process is developmentally regulated. We injected AAV9‐hSyn‐mCherry‐Gephyrin and AAV9‐GAD67‐Synaptophysin‐mCherry to label inhibitory post‐synapses and pre‐synapses, respectively, and measure their engulfment (Fig EV5A). Consistent with our PS exposure data, we found that inhibitory post‐synapses, but not pre‐synapses were frequently found inside Iba1‐positive microglia in the brains of 1‐month‐old mice (Figs 6B and EV5B and C). In agreement with the observed age‐dependent changes in PS exposure, the preferential engulfment of inhibitory post‐synapses by microglia was not observed in the brains of 3‐month‐old mice (Figs 6C and EV5B and C). Importantly, when we intravenously injected Annexin V into 1‐month‐old wild‐type mice, we found that Annexin V injection was sufficient to increase the number of Gephyrin‐positive inhibitory post‐synapses without changing the number of inhibitory pre‐synapses (Fig 6D and E). Furthermore, we found that deleting Mertk specifically in microglia by crossing Mertkfl / fl mice with CX3CR1‐CreERT2 mice and injecting 4‐OHT at 1 month of age (Fig EV5D) significantly increased the number of Gephyrin‐positive inhibitory post‐synapses without changing the number of inhibitory pre‐synapses (Fig 6F and G). Excitatory synapses were not affected by deletion of microglial Mertk (Fig EV5E and F). Finally, the number of secA5 co‐localized inhibitory synapses was significantly higher in Mertk cKO mice than in control mice, as microglia failed to eliminate these inhibitory synapses (Fig 6H and I). Taken together, our data show that PS exposure is necessary for pruning inhibitory post‐synapses through MERTK‐dependent microglial phagocytosis, suggesting that PS serves as a general “eat‐me” signal for inhibitory post‐synapse elimination in the normal brain.
Figure 6. Microglial Mertk is required for normal pruning of PS‐exposed inhibitory post‐synapses.
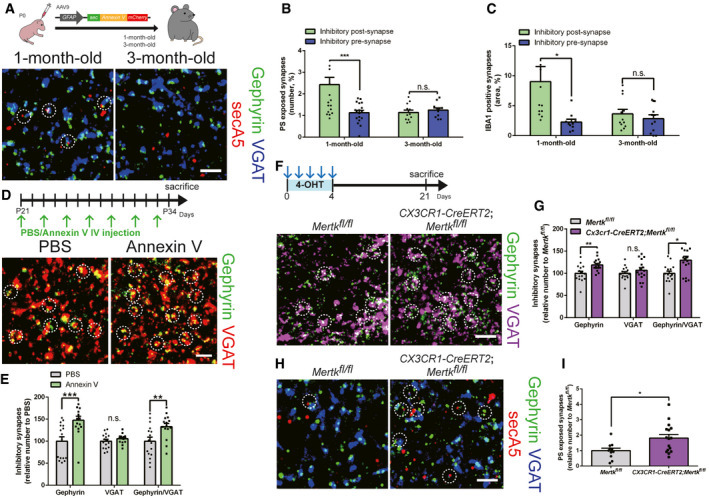
-
ASchematic diagram of the experiment in which PS exposure was measured in the brains of 1‐month‐ and 3‐month‐old wild‐type mice by injecting AAV9‐GFAP‐secA5‐mCherry in the neonatal stage (P0). Representative confocal z‐stack colocalization images of Gephyrin (green), VGAT (blue), and secA5 (red) in the HP (hippocampus) of 1‐month‐ and 3‐month‐old wild‐type mice. The white circles indicate secA5‐positive inhibitory synapses. Scale bar, 10 μm.
-
B, CBar graphs showing the expression of secA5 (B) and IBA1 (C) in inhibitory post‐synapses (Gephyrin‐positive, green) and pre‐synapses (VGAT‐positive, blue) in the HP of 1‐month‐ and 3‐month‐old wild‐type mice (n = 4 for each group, ***P = 0.0009, *P = 0.0248, n.s., not significant).
-
DSchematic diagram of the experiment in which Annexin V was injected intravenously (every 2 days) to block PS in the brains of 1‐month‐old wild‐type mice. Representative confocal z‐stack images of Gephyrin (green)‐ and VGAT (red)‐positive inhibitory synapses in the HP of 1‐month‐old wild‐type mice after PBS or Annexin V injection. The white circles indicate inhibitory synapses. Scale bar, 10 μm.
-
EBar graphs showing the number of inhibitory (Gephyrin‐ and VGAT‐positive) synapses in the HP of 1‐month‐old mice after PBS or Annexin V injection (n = 4 for each group, ***P = 0.0013, **P = 0.0090).
-
FSchematic diagram of the experiment in which microglial Mertk KO (CX3CR1‐CreERT2;Mertkfl / fl) and control (Mertkfl / fl) mice were generated. Both 1‐month‐old microglial Mertk KO mice and 1‐month‐old control mice were injected with 4‐OHT for 5 consecutive days. Representative confocal z‐stack images of Gephyrin (green)‐ and VGAT (magenta)‐positive inhibitory synapses in the HP of Mertkfl / fl and CX3CR1‐CreERT2;Mertkfl / fl mice. The white circles indicate inhibitory synapses. Scale bar, 10 μm.
-
GBar graphs showing the number of inhibitory (Gephyrin‐ and VGAT‐positive) synapses in the HP of Mertkfl / fl and CX3CR1‐CreERT2;Mertkfl / fl mice (n = 4 for each group, **P = 0.0027, *P = 0.0062).
-
HRepresentative confocal z‐stack colocalization images of Gephyrin (green), VGAT (blue) and secA5 (red) in the HP of Mertkfl / fl and CX3CR1‐CreERT2;Mertkfl / fl mice. The white circles indicate secA5‐positive inhibitory synapses. Scale bar, 10 μm.
-
IBar graphs showing the relative expression of secA5 in inhibitory synapses in the HP of Mertkfl / fl and CX3CR1‐CreERT2;Mertkfl / fl mice (n = 4 for each group, *P = 0.0015).
Data information: The individual dots indicate experimental replicates (3–5 images were taken from 2 brain slices per an animal, B, C, E, G, and I). The data are the mean ± SEM and were analyzed by two‐tailed unpaired t‐test.
Figure EV5. Microglial Mertk is required for eliminating inhibitory post‐synapse, but not excitatory synapses.
-
ASchematic diagram of the experiment in which microglial phagocytosis of inhibitory post‐ and pre‐synapses was measured in the brains of 1‐month‐ and 3‐month‐old wild‐type mice by injecting AAV9‐hSyn‐mCherry‐Gephyrin and AAV9‐GAD67‐Synaptophysin‐mCherry in the neonatal stage (P0).
-
B, CRepresentative confocal z‐stack and colocalization images showing IBA1 (red) expression in inhibitory post‐synapses (B, mCherry‐Gephyrin, green) and pre‐synapses (C, Synaptophysin‐mCherry, green) in the HP (hippocampus) of 1‐month‐ and 3‐month‐old wild‐type mice. The white arrows indicate expression of IBA1 in inhibitory synapses. Scale bar, 10 μm.
-
DSchematic diagram of the experiment. Representative confocal z‐stack images of MERTK (green) and IBA1 (magenta) in the HP of Mertkfl/fl and CX3CR1‐CreERT2;Mertkfl/fl mice. The dashed line represents where the high magnified insert was taken from. Scale bar, 20 μm.
-
ERepresentative confocal z‐stack images of PSD95 (green)‐ and VGLUT1 (red)‐positive excitatory synapses in the HP of Mertkfl/fl and CX3CR1‐CreERT2;Mertkfl/fl mice. Scale bar, 10 μm.
-
FBar graphs showing the number of excitatory (PSD95‐ and VGLUT1‐positive) synapses in the HP of Mertkfl/fl and CX3CR1‐CreERT2;Mertkfl/fl mice (n = 4 for each group, n.s., not significant).
Data information: The individual dots indicate experimental replicates (3–5 images were taken from 2 brain slices per an animal, F). The data are the mean ± SEM and were analyzed by two‐tailed unpaired t‐test.
Discussion
Since several phagocytic receptors and their bridging molecules expressed by glial cells recognize PS, it has been speculated that PS may function as an “eat‐me” signal for glia‐mediated synapse elimination (Mukherjee & Williams, 2017; Lemke, 2019; Nonaka & Nakanishi, 2019; Li et al, 2020; Scott‐Hewitt et al, 2020). Interestingly, recent two studies have demonstrated that exposed PS can be detected at excitatory pre‐synapses in developing retinogeniculate systems and hippocampus using fluorescent probe (Li et al, 2020; Scott‐Hewitt et al, 2020). However, in vivo genetic evidence addressing roles of PS in synapse elimination has been lacking. Herein, by deleting an upstream modulator of PS in mature neurons, we reveal that CDC50A‐dependent PS exposure is specifically required for elimination of inhibitory post‐synapses by microglia through MERTK pathway. Inhibiting microglial phagocytosis by (i) ablating microglia, (ii) deleting a Mertk phagocytic receptor specifically in microglia, or (iii) masking the outer membrane‐exposed PS by Annexin V IV‐injection was sufficient to prevent the loss of inhibitory post‐synapses in Cdc50a cKO mice.
Importantly, we found that microglia recognize and eliminate PS‐exposed inhibitory post‐synapses not by the complement pathway, but by the Mertk phagocytic receptor. Previously, we have reported that astrocytes mediate pruning of excitatory synapses by phagocytosing them through Megf10 and Mertk pathways (Chung et al, 2013; Lee et al, 2020). However, in this study, we found that it is microglia that use Mertk for phagocytosing inhibitory post‐synapses in Cdc50a cKO mice. Interestingly, recent studies have shown that microglia seem to preferentially monitor and respond to changes in neuronal somas, while astrocytes seem to be specialized in monitoring neuronal processes (Cserep et al, 2020; Damisah et al, 2020). In addition, previous studies have identified that PS‐exposed rod outer segment can be engulfed by retinal pigment epithelial cells (RPEs) through MERTK pathway (Nandrot et al, 2012; Ruggiero et al, 2012). Therefore, it is plausible that restricted PS exposure in neuronal somas by Cdc50a deletion is recognized by microglial Mertk rather than astrocytes, inducing specific elimination of inhibitory post‐synapses, which are heavily located in neuronal somas compared with other types of synapses. Moreover, microglia appear to specifically engulf PS‐exposed inhibitory post‐synapses without damaging cellular membranes, revealed by the intact localization of GABAA receptor α and β subunits in the extrasynaptic membrane (Fig 2E). Since neuronal soma and proximal dendrites are surrounded by heavy extracellular matrix and perineuronal nets (Kwok et al, 2011; Fawcett et al, 2019), it is also possible that inhibitory post‐synaptic area can be selectively vulnerable to glial intervention compared with other cellular membranes.
Our unexpected findings raise many intriguing questions. First, are other upstream modulators of PS, such as scramblases, involved in synapse elimination as well? Unlike flippases, scramblases are the enzyme that non‐specifically and bidirectionally translocate PS between the inner and outer membranes without ATP usage (Segawa & Nagata, 2015). A previous study has demonstrated that the constitutively active mutant form of scramblase is sufficient to expose PS in the outer membrane (Segawa et al, 2011). Moreover, in the Drosophila nervous system, it has been shown that overexpression of a calcium dependent scramblase causes neurite degeneration (Sapar et al, 2018). These studies suggest that scramblases may be responsible for presenting PS in the neuronal processes and initiate synapse elimination by glial phagocytosis. Manipulating specific scramblases in mature neurons would be required in addressing such hypothesis. Second, what is the relationship between neuronal activity and PS exposure at synapses? Importantly, it has been shown that either caspase activation or calcium elevation can induce PS exposure by inactivating flippases and activating scramblases (Segawa & Nagata, 2015). Since caspase‐3 activity as well as calcium response are critical in mediating synaptic long‐term depression (D'Amelio et al, 2012; Grienberger & Konnerth, 2012), neuronal activity may regulate activation states of flippases and scrambases at synapses and initiate glia‐mediated synapse elimination.
An excitatory–inhibitory (E‐I) imbalance has been reported in many neurological disorders, such as autism spectrum disorder, schizophrenia, frontotemporal dementia, and several forms of seizures (Gao & Penzes, 2015; Lui et al, 2016). Even though previous studies have suggested the association between activated microglia and E‐I imbalance in these disorders (Chen et al, 2014; Zhao et al, 2018; Andoh et al, 2019), molecular mechanisms were unclear. Our findings strongly suggest that such an E‐I imbalance can originate from abnormal PS exposure at inhibitory post‐synapses or hyperactivated MERTK‐dependent microglial phagocytosis. Therefore, modulating inhibitory synapse elimination by microglia may serve as a novel strategy for treating various brain disorders.
Materials and Methods
Mice
Cdc50a tm1a(KOMP)Wtsi mice were obtained from Knockout Mouse Project (KOMP) Repository and crossed with B6;SJL‐Tg(ACT‐FLPe)9205Dym/J mice gifted from Dr Jeong Ho Lee (KAIST) to remove the FRT sites. To generate loxp floxed Cdc50a (Cdc50a fl/+) mice, FRT‐deleted mice were bred with Tg(Thy1‐CreERT2,‐EYFP)HGfng/PyngJ mice (Jackson Laboratory). Then, Thy1‐CreERT2,‐EYFP;Cdc50afl / fl mice were used to generate control and Cdc50a cKO mice by intraperitoneally injecting oil and 4‐OHT (Sigma, 75 μg per body weight (g)), respectively. In general, 1‐month‐old mice were injected with oil or 4‐OHT once a day for 5 consecutive days and sacrificed after 13 days. To sparsely label Cdc50a KO cells, Thy1‐CreERT2‐EYFP;Cdc50afl / fl mice were crossed with B6. Cg‐Gt(ROSA)26Sortm3(CAG‐EYFP)Hze /J mice (Jackson Laboratory). Thy1‐CreERT2‐EYFP;Cdc50afl / fl ;B6. Cg‐Gt(ROSA)26Sortm3(CAG‐EYFP)Hze /J mice were intraperitoneally injected with tamoxifen once (Sigma, 7.5 μg per body weight (g)) at 1 month of age and sacrificed after 24 days. To delete phagocytic receptors in Cdc50a cKO mice, B6;129S4‐C3tm1Crr /J or B6;129‐Mertktm1Grl/J mice (Jackson Laboratory) were crossed with Thy1‐CreERT2,‐EYFP;Cdc50afl / fl mice. Both C3 and Mertk KO mice were backcrossed to the C57BL/6J background at least 10 generations. For microglial‐specific Mertk deletion, Mertkfl / fl mice (see below) were crossed with B6.129P2(Cg)‐Cx3cr1tm2.1(cre / ERT2)Litt /WganJ mice gifted from Dr Seyun Kim (KAIST). Mertkfl / fl and B6.129P2(Cg)‐Cx3cr1tm2.1(cre / ERT2)Litt /WganJ; Mertkfl / fl mice were intraperitoneally injected with 4‐OHT (Sigma, 75 μg per body weight (g)) to generate control and Mertk cKO mice, respectively. Both sexes were included in all experiments. Wild‐type C57BL/6N mice and wild‐type Sprague Dawley (SD) rats were purchased from DBL and Samtaco. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Korea Advanced Institute of Science and Technology.
Generation of loxp floxed Mertk mice
A loxp floxed Mertk (Mertkfl /+) mouse line was generated using clustered regularly interspaced short palindromic repeats (CRISPR) technology (Applied StemCell). Briefly, a mixture of two sets of active guide RNA molecules (gRNAs), two single‐stranded oligodeoxynucleotides (ssODNs), and appropriate Cas9 mRNA were injected into the cytoplasm of C57BL/6 embryos, and 2 LoxP cassettes were inserted into intron 1 and intron 2 to flank exon 2 of the Mertk locus. The following primers were used for genotyping: forward: 5′‐ CTTCATCATGCTCACCTCAAACC‐3′, reverse: 5′‐ GTGCAGAATATTCACCT GACTGC‐3′.
Immunohistochemistry and image analysis
Animals were anesthetized with isoflurane (Piramal) or avertin (2, 2, 2‐tribomoethanol, Sigma) and transcardially perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA, WAKO). The brains and diaphragms were removed, postfixed overnight in 4% PFA at 4°C, and then cryoprotected in 30% sucrose (Sigma)/PBS for 24 h at 4°C. After the brain samples were embedded and frozen in OCT compound (Leica), serial brain sections (30 μm) were collected in 24‐well plates filled with PBS. Then, each section was treated with blocking solution (4% bovine serum albumin, 0.3% Triton X‐100 in PBS) for 1 h at room temperature (RT) and incubated with the following primary antibodies in blocking solution: anti‐CAMKII (rabbit monoclonal from Abcam), anti‐β‐galactosidase (chicken polyclonal from Aves Labs), anti‐GAD67 (mouse monoclonal from Merck), anti‐cleaved Caspase‐3 (rabbit polyclonal from Cell Signaling Technology), anti‐NeuN (mouse monoclonal from Merck), anti‐cFos (rabbit polyclonal from Cell Signaling Technology), anti‐mCherry (rat monoclonal from Invitrogen), anti‐KDEL (rabbit monoclonal from Abcam), anti‐PSD95 (rabbit polyclonal from Invitrogen), anti‐VGLUT1 (guinea pig polyclonal from Merck), anti‐VGLUT2 (guinea pig polyclonal from Merck), anti‐Gephyrin (mouse monoclonal from SYSY and rabbit chimeric monoclonal from SYSY), anti‐VGAT (rabbit polyclonal from SYSY and guinea pig polyclonal from SYSY), anti‐GABAA receptor α1 (rabbit polyclonal from Alomone labs), anti‐GABAA receptor β 2,3 (mouse monoclonal from Merck), anti‐GABAA receptor γ (guinea pig polyclonal from SYSY), anti‐glycine receptor (mouse monoclonal from SYSY), anti‐IBA1 (rabbit polyclonal from Wako and goat polyclonal from Novus Biologicals), anti‐GFAP (mouse monoclonal from Merck), CD68 (rat monoclonal from Abcam), anti‐Cathepsin D (goat polyclonal from R&D systems), anti‐LAMP2 (rat monoclonal from Abcam), anti‐S100β (rabbit monoclonal from Abcam), anti‐MERTK (rat monoclonal from Invitrogen), and anti‐C1q (rabbit monoclonal from Abcam). The sections were rinsed five times with PBST (0.1% Tween 20 in PBS, Sigma) and incubated with Alexa Fluor‐conjugated secondary antibodies (Invitrogen) overnight at 4°C. After multiple rinses in PBST, the sections were coverslipped with antifade mounting medium with DAPI or without DAPI (Vectorshield). For anti‐GABAA receptor γ and glycine receptor antibody staining, antigen retrieval with citrate acid and incubation with methanol/acetic acid were performed before blocking. For the TUNEL assay, brain sections were treated with the In Situ Cell Death Detection Kit (Sigma) according to the manufacturer’s instructions. DNase I (Sigma) was administered to positive control sections for the TUNEL assay. For Nissl staining, brain sections were treated with NeuroTrace (Invitrogen) according to the manufacturer’s instructions.
All images were acquired with an Airyscan LSM880 confocal microscope (Carl Zeiss). All images were processed using ImageJ (Fiji), and 3D reconstruction was performed with Imaris software. For colocalization between secA5 and synapses, images were acquired near secA5 expressing astrocytes. SecA5‐expressing neurons by GFAP promoter were excluded in imaging analysis to clearly distinguish PS‐exposed synapses from endogenous neuronal expression of SecA5. For colocalization and synapse number analysis, each plane of z‐stack images was processed with the ImageJ Distance Analysis (DiAna) plugin (Gilles et al, 2017), and the z‐stack planes were merged to generate representative images.
Plasmid and virus preparation
To generate AAV‐CAG‐Cdc50a‐mCherry, Mus musculus Cdc50a cDNA and mCherry (Addgene) were amplified by overlapping PCR and subcloned into the AAV‐CAG‐GFP vector (Addgene) at BamHI and EcoRI sites. To obtain Cdc50a cDNA, total RNA was extracted from the brains of C57BL/6N mice using the RNeasy Plus Kit (Qiagen), and cDNA synthesis was performed using reverse transcriptase (Promega). The primers for Cdc50a cDNA were as follows: forward: 5′‐ATGGCGATGAACTATAGCGCGAA‐3′, reverse: 5′‐AATGGTGATGTCAGCAGTGT‐3′.
To generate AAV‐hSyn‐mCherry‐Gephyrin, Gephyrin was amplified by PCR from the pmCherryC2‐Gephyrin P1 vector (Addgene) and subcloned into the pAAV‐hSyn‐mCherry vector (Addgene) at the BsrGI site. AAV‐GAD67‐synaptophysin‐mCherry‐GFP was obtained from our previous study (Lee et al, 2020).
To generate AAV‐CAG‐secA5‐YFP, secA5‐YFP was amplified by PCR from the pBH‐UAS‐secA5‐YFP vector (Addgene) and subcloned into the pAAV‐CAG‐GFP vector (Addgene) at EcoRI and SalI sites. To produce AAV‐GFAP‐secA5‐mCherry, secA5 was amplified by PCR from the pBH‐UAS‐secA5‐YFP vector (Addgene), and mCherry was amplified by PCR from the pAAV‐hSyn‐mCherry vector (Addgene). secA5‐mCherry was amplified by overlapping PCR from secA5 and mCherry and subcloned into the pAAV‐GFAP‐EGFP vector (Addgene) at SalI and EcoRI sites.
For the in vitro studies, pAAV‐hSyn‐mCherry (Addgene) and hSyn‐Cre‐p2a‐dTomato (Addgene) plasmids were used.
AAV preparation was performed as previously described with minor modifications (Guo et al, 2012). Briefly, HEK293 cell lines were transfected with transfer plasmid (ITR‐containing plasmid), helper plasmid (pAdDeltaF6, Addgene), and capsid plasmid (pAAV9, Addgene) with polyethylenimine (PEI, Sigma). After 72 h, the transfected cells were harvested by cell scraping and centrifugation and lysed by freezing/thawing in hypotonic buffer containing Dulbecco’s modified Eagle’s medium (DMEM, Welgene) and deoxyribonuclease I (DNase I, Worthington). After the cell debris was removed by centrifugation, chloroform (Sigma) was applied to the supernatant, and the top aqueous phase was collected by centrifugation. PEG8000 (Sigma) and ammonium sulfate (Sigma) solutions were added to the aqueous phase, and the bottom clear phase was collected by centrifugation. Amicon Ultra (Merck) was used for concentration.
Virus injection
Wild‐type C57BL/6N pups (P0) were anesthetized by hypothermia (for no more than 4 min). Under anesthesia, virus with Trypan Blue (Gibco, 1 µl) was injected into the lateral ventricle using a glass pipette (WPI). After viral injections, the pups were allowed to recover on a heating pad and returned to their home cages. For AAV9‐CAG‐Cdc50a‐mCherry injection, the mice were sacrificed after 1 month. For AAV9‐hSyn‐mCherry‐Gephyrin, AAV9‐GAD67‐synaptophysin‐mCherry and AAV9‐GFAP‐secA5‐mCherry injection, the mice were sacrificed after 1 or 3 months for analysis. For control and Cdc50a cKO experiments, mouse pups (P0) were injected with AAV9‐hSyn‐mCherry‐Gephyrin or AAV9‐GFAP‐secA5‐mCherry and sacrificed 13 days after oil or 4‐OHT injection.
Primary neuron cultures, immunocytochemistry, and pSIVA live imaging
Primary neuron cultures were prepared from Cdc50afl / fl mouse pups (P0) or wild‐type SD rat pups (P0). Briefly, the cerebral cortex was removed, placed in Hank’s buffered salt solution (HBSS, Gibco), and digested with trypsin‐EDTA (Gibco). The digested cells were further dissociated by gentle trituration with minimum essential media (MEM, Gibco) containing 10% fetal bovine serum (FBS, Gibco), 25 mM glucose (Sigma), 2 mM glutamine (Gibco), and 1% penicillin/streptomycin (Gibco). The suspended cells were plated on poly‐D‐lysine‐ and laminin‐precoated glass coverslips (Corning). Two hours after cell plating, the medium was replaced with Neurobasal A (Gibco) containing 2 mM glutamine (Gibco), B27 (Gibco), and 1% penicillin/streptomycin (Gibco). The cells were incubated at 37°C in a humidified incubator with 5% CO2, and the medium was changed twice a week.
Lipofectamine 2000 (Thermo Fisher) was used to transfect the cells with CAG‐GFP and CAG‐mCherry as a control or CAG‐GFP and CAG‐Cdc50a‐mCherry to express CDC50A‐mCherry at 6 days in vitro (DIV), and the cells were fixed 7 days after transfection with 4% PFA (Wako) at RT for 10 min. Then, the cells were treated with blocking solution (4% bovine serum albumin and 0.3% Triton X‐100 in PBS) at RT for 1 h and incubated with anti‐mCherry (rat monoclonal from Invitrogen) at 4°C overnight. The cells were rinsed five times with PBST (0.1% Tween 20 in PBS, Sigma) and incubated with Alexa Fluor‐conjugated secondary antibodies (Invitrogen) and Neurotrace (Invitrogen) at RT for 1 h. After rinsing with PBST several times, the cells were coverslipped with antifade mounting medium without DAPI (Vectorshield).
To detect PS exposure in vitro, Lipofectamine 2000 (Thermo Fisher) was used to transfect cells from Cdc50afl / fl mouse brains with hSyn‐mCherry as a control or hSyn‐Cre‐p2a‐dTomato for Cdc50a cKO at 6 days in vitro (DIV), and the cells were imaged on DIV8, DIV10, DIV12, DIV14, and DIV16. Then, pSIVA‐IANBD (Abcam) was administered to transfected cells for 10 min before live‐cell imaging. All live images were acquired with an LSM880 confocal microscope (Carl Zeiss).
PLX3397 treatment
PLX3397 (pexidartinib) was purchased from SelleckChem and dissolved in 50 mM dimethyl sulfoxide (DMSO). PLX3397 was added to standard rodent chow (290 mg/kg). DMSO was also added to rodent standard chow as a control. Control and Cdc50a cKO mice were fed DMSO‐containing chow or PLX3397‐containing chow for 4 weeks.
Annexin V treatment
Annexin V was purchased from Biovision and dissolved in PBS. Annexin V (1 mg/kg) or PBS was intravenously injected into Cdc50a cKO and wild‐type mice once every 2 days. For the Cdc50a cKO rescue experiment, 4‐OHT and Annexin V/PBS were injected into 1‐month‐old Cdc50a cKO mice. For the wild‐type experiment, Annexin V/PBS was injected into 3‐week‐old wild‐type mice. Both Cdc50a cKO and wild‐type mice were sacrificed 13 days after PBS or Annexin V injection. For Annexin V‐Cy3 experiment, Annexin V‐CF594 (Biotium) or PBS was intravenously injected into 3‐week‐old wild‐type mice. Mice were sacrificed 1 day after injection.
Audiogenic seizure behavior
Auditory stimuli (5, 10, 15, 20, 40 kHz and white noise (WN); 60 s duration) were generated with sinusoidal ramps at the onset (50 ms) and the offset (50 ms) using a custom‐made MATLAB code. The sound pressure level (SPL) of the stimuli on the speaker (Avisoft‐Bioacoustics, Ultrasonic Dynamic Speaker Vifa) was calibrated using a calibrated polarized condenser microphone (Avisoft‐Bioacoustics, CM16/CMPA) placed at the center of an acrylic chamber. The auditory stimuli were presented as a cycle of each stimulus from low to high dB (70, 80, 90, and 100 dB). The mice were placed in an acrylic chamber (32*31*21.5 cm) covered with a transparent acrylic lid. All behaviors were recorded with a video camera. The behavior of each mouse was scored (0: normal response, 1: wild running, 2: seizure, and 3: tonic–clonic seizures with loss of righting reflex) during auditory stimulation.
Electrophysiology
Brain slices containing the IC were subjected to electrophysiological analysis. Control and Cdc50ac cKO mice were treated with oil or 4‐OHT at the age of 2 weeks and used for patch‐clamp experiments after 13 days. The mice were anesthetized with isoflurane and perfused with cutting solution, which was modified ice‐cold artificial cerebrospinal fluid (ACSF) containing sucrose rather than NaCl (220 mM sucrose, 26 mM NaHCO3, 2.5 mM KCl, 1 mM NaH2PO4, 5 mM MgCl2, 1 mM CaCl2, 10 mM glucose; pH 7.3–7.35). Then, the mice were decapitated and the brains were immediately submerged in cutting solution. Coronal sections (250 µm) of the IC were cut with a vibratome (Leica VT 1200S, Leica) and incubated in oxygenated storage solution (123 mM NaCl, 26 mM NaHCO3, 2.8 mM KCl, 1.25 mM NaH2PO4, 1.2 mM, MgSO4, 2.5 mM CaCl2, 10 mM glucose; pH 7.3–7.35) in a 34°C water bath for at least 30 min before recording.
The brain slices were transferred to the recording chamber and bathed in recording solution (126 mM NaCl, 26 mM NaHCO3, 2.8 mM KCl, 1.25 mM NaH2PO4, 1.2 mM MgSO4, 2.5 mM CaCl2, 5 mM glucose; pH 7.3–7.35). Epifluorescence was used to target fluorescent cells, after which infrared differential interference contrast imaging was performed to obtain whole‐cell recordings (Nikon Eclipse FN‐S2N equipped with a fixed stage and a QImaging optiMOS sCMOS camera). Electrophysiological signals were recorded using an Axopatch 700B amplifier (Molecular Devices), low‐pass filtered at 1 kHz and sampled at 20 kHz. The electrophysiological data were analyzed offline on a PC with Clampfit software (Molecular Devices).
Current‐clamp experiments were performed to measure the resting membrane potential and input resistance of and generate a frequency–current curve (F‐I curve) for control and Cdc50a cKO mice. The recording electrodes had a resistance of 3.5 ˜ 5 MΩ when filled with K+‐gluconate internal solution (Alexa Fluor 594: 120 mM potassium gluconate, 10 mM KCl, 10 mM HEPES, 1 mM CaCl2, 1 mM MgCl2, 5 mM EGTA, 2 mM Mg‐ATP; pH 7.29). The resting membrane potential was recorded by measuring the membrane potential for 10 s before the recording current step. The input resistance was recorded by measuring the voltage deflection during hyperpolarizing current pulse steps (from −50 pA to 0 pA, 500 ms each). After the current step, the membrane potential was fixed at −60 mV by injecting a stable current into the electrode. Then, depolarizing currents were injected (+50 pA to +450 pA, 500 ms each). The number of spikes for each current was calculated and analyzed according to the F‐I curve. For KA and PTX treatment, 1 mM kynurenic acid and 50μM picrotoxin in recording solution was treated at a flow rate of ˜2 ml/min. During drug treatment, depolarizing currents were injected (+50 pA to +450 pA, 500 ms each), and each spikes was calculated and analyzed.
Western blot
The brains of control and Cdc50a cKO mice were removed and lysed in EzRIPA lysis buffer (Atto) containing protease inhibitors (Atto) and phosphatase inhibitor (Atto). After the protein concentration was quantified by the Bradford method (Bio‐Rad), the lysates were mixed with 5× SDS sample buffer (250 mM Tris–HCl pH 6.8, 5% 2‐mercaptoethanol, 10% sodium dodecyl sulfate, 0.5% bromophenol blue, and 50% glycerol). The samples were loaded on SDS–PAGE gels (Bio‐Rad) and transferred to polyvinylidene fluoride (PVDF, Atto) membranes. The membranes were blocked in blocking solution (10% skim milk in Tris‐buffered saline, TBS) at RT for 1 h and incubated with the following primary antibodies in blocking solution (5% skim milk in TBS) at 4°C: anti‐CDC50A (mouse monoclonal gifted from Dr. Robert Molday), anti‐Homer (mouse monoclonal from SYSY), anti‐Synapsin (rabbit monoclonal from Cell Signaling Technology), anti‐VGAT (guinea pig polyclonal from SYSY), anti‐Gephyrin (mouse monoclonal from SYSY), anti‐GABAA receptor β 2, 3 (mouse monoclonal from Merck), anti‐GABAA receptor γ (guinea pig polyclonal from SYSY), anti‐glycine receptor (mouse monoclonal from SYSY), anti‐Gas6 (goat polyclonal from R&D Systems), and anti‐GAPDH (rabbit monoclonal from Santa Cruz). The membranes were rinsed three times in TBST (0.1% Tween 20 in TBS) and incubated with HRP‐conjugated secondary antibodies (Santa Cruz and Invitrogen) at RT for 1 h. After washing with TBST three times, the membranes were developed by enhanced chemiluminescence (ECL, GE Healthcare) and visualized with Chemidoc XRS+ (Bio‐Rad). The data were analyzed with ImageJ software (Fiji).
Quantification and statistical analysis
GraphPad Prism 6 software was used for all statistical analyses. Data from two samples were compared using unpaired t‐test or multiple t‐tests with 95% confidence. To compare multiple samples, one‐way or two‐way analysis of variance (ANOVA) with Tukey’s test was used. For normalization to the control, baseline correction was applied. For survival analysis, survival curves were analyzed by the log‐rank (Mantel‐Cox) test with 95% confidence. For the correlation test, Pearson’s correlation coefficients were used. All data are presented as the means and standard error of the mean (SEM).
Author contributions
Project design: W‐SC and JP; All the experiments including plasmid cloning, virus production, cell culture, immunohistochemistry, confocal imaging, Western blot, and behavior analysis: JP Slice electrophysiology: YC and J‐WS; Software with auditory stimulus: EJ and S‐HL; Manuscript writing: W‐SC and JP.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Expanded View and Appendix
Acknowledgements
We thank all members of the Chung laboratory for helpful discussion. This work was supported by grants from the Samsung Science & Technology Foundation (SSTF‐BA1701‐18, W.‐S. C.). This work was also supported from the National Research Foundation of Korea (NRF) (2020M3E5D9079912 to S.‐H.L. and 2019R1A2C2005161 to J.‐W.S.).
The EMBO Journal (2021) 40: e107121.
Data availability
This study includes no data deposited in external repositories.
References
- Andersen JP, Vestergaard AL, Mikkelsen SA, Mogensen LS, Chalat M, Molday RS (2016) P4‐ATPases as phospholipid flippases‐structure, function, and enigmas. Front Physiol 7: 275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoh M, Ikegaya Y, Koyama R (2019) Synaptic pruning by microglia in epilepsy. J Clin Med 8: 2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Mody I (2012) Extrasynaptic GABA(A) receptors: their function in the CNS and implications for disease. Neuron 73: 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne JH, Roberts JL (2009) From molecules to networks: an introduction to cellular and molecular neuroscience. Amsterdam, Boston: Academic Press/Elsevier; [Google Scholar]
- Chen Z, Jalabi W, Hu W, Park H‐J, Gale JT, Kidd GJ, Bernatowicz R, Gossman ZC, Chen JT, Dutta R et al (2014) Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat Commun 5: 4486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choii G, Ko J (2015) Gephyrin: a central GABAergic synapse organizer. Exp Mol Med 47: e158 [DOI] [PubMed] [Google Scholar]
- Chung WS, Barres BA (2012) The role of glial cells in synapse elimination. Curr Opin Neurobiol 22: 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W‐S, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C et al (2013) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504: 394–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JA, Molday RS (2011) Critical role of the beta‐subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the P4‐ATPase ATP8A2. J Biol Chem 286: 17205–17216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserép C, Pósfai B, Lénárt N, Fekete R, László ZI, Lele Z, Orsolits B, Molnár G, Heindl S, Schwarcz AD et al (2020) Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 367: 528–537 [DOI] [PubMed] [Google Scholar]
- D'Amelio M, Sheng M, Cecconi F (2012) Caspase‐3 in the central nervous system: beyond apoptosis. Trends Neurosci 35: 700–709 [DOI] [PubMed] [Google Scholar]
- Damisah EC, Hill RA, Rai A, Chen F, Rothlin CV, Ghosh S, Grutzendler J (2020) Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci Adv 6: eaba3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, Kipnis J (2012) Wild‐type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484: 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore M, Najafi A, Koike M, Dagher N, Spangenberg E, Rice R, Kitazawa M, Matusow B, Nguyen H, West B et al (2014) Colony‐stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82: 380–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faingold CL (2002) Role of GABA abnormalities in the inferior colliculus pathophysiology ‐ audiogenic seizures. Hear Res 168: 223–237 [DOI] [PubMed] [Google Scholar]
- Faingold CL, Naritoku DK, Copley CA, Randall ME, Riaz A, Anderson CA, Arneric SP (1992) Glutamate in the inferior colliculus plays a critical role in audiogenic seizure initiation. Epilepsy Res 13: 95–105 [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Oohashi T, Pizzorusso T (2019) The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nat Rev Neurosci 20: 451–465 [DOI] [PubMed] [Google Scholar]
- Fourgeaud L, Través PG, Tufail Y, Leal‐Bailey H, Lew ED, Burrola PG, Callaway P, Zagórska A, Rothlin CV, Nimmerjahn A et al (2016) TAM receptors regulate multiple features of microglial physiology. Nature 532: 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy JM (2008) Epilepsy, E/I balance and GABA(A) receptor plasticity. Front Mol Neurosci 1: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Penzes P (2015) Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr Mol Med 15: 146–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles JF, Dos Santos M, Boudier T, Bolte S, Heck N (2017) DiAna, an ImageJ tool for object‐based 3D co‐localization and distance analysis. Methods 115: 55–64 [DOI] [PubMed] [Google Scholar]
- Grienberger C, Konnerth A (2012) Imaging calcium in neurons. Neuron 73: 862–885 [DOI] [PubMed] [Google Scholar]
- Guo P, El‐Gohary Y, Prasadan K, Shiota C, Xiao X, Wiersch J, Paredes J, Tulachan S, Gittes GK (2012) Rapid and simplified purification of recombinant adeno‐associated virus. J Virol Methods 183: 139–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham TJ, Mapes J, Kokel D, Peterson RT (2010) Live imaging of apoptotic cells in zebrafish. FASEB J 24: 4336–4342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja‐Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres Ba et al (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352: 712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith D, El‐Husseini A (2008) Excitation control: balancing PSD‐95 function at the synapse. Front Mol Neurosci 1: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Chen J, Chan JR, Langen R (2010) Engineering a polarity‐sensitive biosensor for time‐lapse imaging of apoptotic processes and degeneration. Nat Methods 7: 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Karube F, Nomura M, Kawaguchi Y (2016) The diversity of cortical inhibitory synapses. Front Neural Circuits 10: 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok JC, Dick G, Wang D, Fawcett JW (2011) Extracellular matrix and perineuronal nets in CNS repair. Dev Neurobiol 71: 1073–1089 [DOI] [PubMed] [Google Scholar]
- Lee CC, Sherman SM (2010) Drivers and modulators in the central auditory pathways. Front Neurosci 4: 79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Kim JY, Noh S, Lee H, Lee SY, Mun JY, Park H, Chung WS (2020) Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 590: 612–617 [DOI] [PubMed] [Google Scholar]
- Lemke G (2017) Phosphatidylserine is the signal for TAM receptors and their ligands. Trends Biochem Sci 42: 738–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke G (2019) How macrophages deal with death. Nat Rev Immunol 19: 539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chiou B, Gilman CK, Luo R, Koshi T, Yu D, Oak HC, Giera S, Johnson‐Venkatesh E, Muthukumar AK et al (2020) A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. EMBO J 39: e104136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui H, Zhang J, Makinson S, Cahill M, Kelley K, Huang H‐Y, Shang Y, Oldham M, Martens L, Gao F et al (2016) Progranulin deficiency promotes circuit‐specific synaptic pruning by microglia via complement activation. Cell 165: 921–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Williams DW (2017) More alive than dead: non‐apoptotic roles for caspases in neuronal development, plasticity and disease. Cell Death Differ 24: 1411–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandrot EF, Silva KE, Scelfo C, Finnemann SC (2012) Retinal pigment epithelial cells use a MerTK‐dependent mechanism to limit the phagocytic particle binding activity of alphavbeta5 integrin. Biol Cell 104: 326–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka S, Nakanishi H (2019) Microglial clearance of focal apoptotic synapses. Neurosci Lett 707: 134317 [DOI] [PubMed] [Google Scholar]
- Ramirez OA, Couve A (2011) The endoplasmic reticulum and protein trafficking in dendrites and axons. Trends Cell Biol 21: 219–227 [DOI] [PubMed] [Google Scholar]
- Ruggiero L, Connor MP, Chen J, Langen R, Finnemann SC (2012) Diurnal, localized exposure of phosphatidylserine by rod outer segment tips in wild‐type but not Itgb5−/− or Mfge8−/− mouse retina. Proc Natl Acad Sci USA 109: 8145–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapar ML, Ji H, Wang B, Poe AR, Dubey K, Ren X, Ni JQ, Han C (2018) Phosphatidylserine externalization results from and causes neurite degeneration in Drosophila . Cell Rep 24: 2273–2286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer D, Lehrman E, Kautzman A, Koyama R, Mardinly A, Yamasaki R, Ransohoff R, Greenberg M, Barres B, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron 74: 691–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott‐Hewitt N, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, Carey A, Faggiani E, Schuetz LT, Mason S et al (2020) Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J 39: e105380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S (2014) Caspase‐mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344: 1164–1168 [DOI] [PubMed] [Google Scholar]
- Segawa K, Nagata S (2015) An apoptotic 'eat me' signal: phosphatidylserine exposure. Trends Cell Biol 25: 639–650 [DOI] [PubMed] [Google Scholar]
- Segawa K, Suzuki J, Nagata S (2011) Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci USA 108: 19246–19251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V et al (2016) Schizophrenia risk from complex variation of complement component 4. Nature 530: 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder‐Keller AM, Pierson MG (1992) Audiogenic seizures induce c‐fos in a model of developmental epilepsy. Neurosci Lett 135: 108–112 [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B et al (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131: 1164–1178 [DOI] [PubMed] [Google Scholar]
- Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Yu J, Perez‐Torres C, Frouin A, Wilton DK et al (2016) A complement‐microglial axis drives synapse loss during virus‐induced memory impairment. Nature 534: 538–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Long C, Faingold CL (2001) Audiogenic seizure susceptibility is induced by termination of continuous infusion of gamma‐aminobutyric acid or an N‐methyl‐D‐aspartic acid antagonist into the inferior colliculus. Exp Neurol 171: 147–152 [DOI] [PubMed] [Google Scholar]
- Zhang L, Yang Y, Li S, Zhang S, Zhu X, Tai Z, Yang MU, Liu Y, Guo X, Chen Bo et al (2017) Loss of Tmem30a leads to photoreceptor degeneration. Sci Rep 7: 9296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Liao Y, Morgan S, Mathur R, Feustel P, Mazurkiewicz J, Qian J, Chang J, Mathern GW, Adamo MA et al (2018) Noninflammatory changes of microglia are sufficient to cause Epilepsy. Cell Rep 22: 2080–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Expanded View and Appendix
Data Availability Statement
This study includes no data deposited in external repositories.