

Abstract
Type 2 diabetes (T2D) is highly prevalent and a strong contributor for cardiovascular disease (CVD). However, there is significant heterogeneity in disease pathogenesis and the risk of complications. Enormous progress has been made in our ability to catalog genetic variation associated with T2D-risk and variation in disease relevant quantitative traits. These discoveries hold the potential to shed light on tractable targets and pathways for safe and effective therapeutic development, but the promise of precision medicine has been slow to be realized. Recent studies have identified subgroups of individuals with differential risk for intermediate phenotypes (e.g., lipid levels, fasting insulin, BMI), which contribute to T2D risk, helping to account for the observed clinical heterogeneity. These “partitioned genetic risk scores” have not only the potential to identify patients at greatest risk of CVD and rapid disease progression, but could also aid patient stratification bridging the gap towards precision medicine for T2D.
Keywords: Type 2 diabetes, genetics, precision medicine
Condensed abstract
Type 2 diabetes (T2D) has significant heterogeneity in its pathogenesis and disease course/complications. Recent advances have identified subgroups within T2D, helping to account for the clinical heterogeneity observed. In particular, genetic approaches to clustering subgroups of T2D have the potential to identify patients at greater risk of downstream complications (e.g., heart disease) and disease progression (e.g., insulin requiring T2D). Genetic approaches have identified and validated new drug targets for T2D and may also allow more targeted therapeutic approaches. Together, clustering methods have the potential to bring precision medicine into the clinic for T2D prevention and treatment.
Introduction
Type 2 diabetes (T2D) is one of the most common and fastest growing diseases, with an estimated worldwide prevalence of nearly 700 million by 2045(1). Cardiovascular disease (CVD), including coronary artery disease and ischemic stroke, is the leading cause of mortality in T2D, while diabetic microvascular disease (chronic kidney disease, neuropathy and retinopathy) contributes significantly to morbidity(1). Despite its prevalence and high human cost, T2D is currently treated as a homogenous entity(2, 3) – diagnosed by an abnormally high blood glucose level and treated algorithmically(4) – without taking into account the varied mechanisms that can contribute to its pathogenesis. To address this, recent work from a number of groups has focused on identifying sub-groups within T2D through clustering analyses of either clinical/laboratory data(5–7) or through genetic data(8–10). Both methods identify numerous, clinically distinct subgroups within the broader diagnostic umbrella of T2D, with variable disease progression (e.g., need for insulin) and risk of micro- and macrovascular complications(6, 8). This review will focus on genetic methods to cluster patients into pathologic subgroups, as genes are fixed with respect to time and do not vary by environmental exposure(11, 12). In particular, we will focus on the potential for precision medicine through implementation of genetic clustering and partitioned polygenic risk scores (pPRS(11)) to identify and potentially prevent or treat the primary pathologic mechanisms leading to T2D in an individual patient. We also highlight recent studies that have helped to unravel the molecular mechanisms through which effector genes at genome wide association study (GWAS) loci affect T2D risk. Finally, we highlight recent methodologic advances in genetics that allow for drug target discovery and validation.
Defining subtypes of T2D using clustering approaches
Clinicians have long appreciated the significant heterogeneity among their patient’s risk of developing T2D and T2D-related sequelae. For instance, many patients with traditional T2D risk factors (e.g., obesity) do not develop T2D(8). Disease progression is also variable, with some patients rapidly developing insulin requiring T2D, whilst others can be stably treated for decades with metformin alone(12). Complications related to T2D-mediated hyperglycemia are similarly heterogeneous, with randomization control trial data consistently showing intensive glycemic control reduces the risk of microvascular disease(13, 14). In comparison, while early trials showed that T2D-mediated hyperglycemia was not associated with CVD(15), newer evidence suggests a nuanced view, whereby there is longitudinally-reduced CVD risk during periods of time with intensive glycemic control(16). This is consistent with Mendelian randomization (see below) data, which demonstrates a causal role for hyperglycemia and CVD risk(17). These data suggested that within the homogeneous diagnosis of T2D there are numerous subgroups of individuals who have a variable clinical course, prompting investigation into the identification of these patient clusters.
Clustering based on non-genetic, clinical data:
In 2015, Li et al performed one of the first, large attempts at clustering patients using clinical/laboratory data(18). Drawing on electronic medical records (EMRs) for 11,210 multi-ethnic participants, they used machine learning methodology and identified three subtypes of T2D patients. Building upon this work, Ahlqvist et al leveraged Bayesian machine learning methods in a cohort of 8,980 Swedish patients newly diagnosed with T2D(6). These authors used six pre-selected variables in their clustering analyses: glutamate decarboxylase (GAD) antibodies, age at T2D diagnosis, body mass index (BMI), hemoglobin A1c (HbA1c), and the homeostatic model assessments of pancreatic beta cell function and insulin resistance (HOMA2- β and HOMA2-IR, respectively, (6)). They stratified their cohort into 5 clusters with differential disease progression and risk of complications (Table 1). This method of clustering T2D patients by carefully selected clinical characteristics set off a flurry of replication studies in different populations including European(7), American/Chinese cohorts(19), and randomization control trials(5), showing the generalizability of the clinical covariate clustering approach.
Table 1.
Comparison of T2D subgroups identified by representative clinical covariate vs genetic clustering methods.
| Clinical & Biochemical Clustering Ahlqvist et al (6) | Genetic Clustering Udler et al (8) and Mahajan et al (37) | ||||||
|---|---|---|---|---|---|---|---|
| Cluster | Characteristics (6) | Outcomes | Cluster | Characteristics | Outcomes (8) | Example Genes captured in cluster | |
| β-cell | Autoimmune | Autoantibody + ↓ Insulin (plasma) | ↑ insulin dependence | β-cell | ↑ proinsulin, ↓ insulin |
↑ CAD, ↑ stroke |
HNF1A, SLC30A8 |
| Insulin deficiency | ↓ Insulin (plasma) | ↑ Retinopathy, ↑ insulin dependence |
Proinsulin | ↓ insulin, ↓ proinsulin |
- | KCNJ11 | |
| Mixed β-cell + insulin resistance (from (37)) | - | - | - | Mixed | ↓ insulin, ↓ proinsulin, ↑ HOMA2-β |
- | PAM, RREB1 |
| Insulin Resistance | Insulin resistance | ↑ HOMA2-IR, ↑ HOMA2-β |
↑ DKD | Lipodystrophy | ↓ BMI, ↑ insulin, ↑ TG | ↑ CAD, ↑ DKD, ↑ HTN |
KLF14, FAM13A |
| NAFLD/Lipid | ↓ TG | ↓ DKD | TM6SF2, GCKR | ||||
| Obesity | ↑ BMI; mild T2D | - | Obesity/Adiposity | ↑ BMI, ↑insulin | - | FTO/IRX3, MCR4 | |
| ? | Aging | ↑ Age, normal BMI, mild T2D | - | - | - | - | - |
Abbreviations: BMI – body mass index, CAD – coronary artery disease, DKD – diabetic kidney disease, HOMA-β - homeostatic model assessment of pancreatic beta cell function, HOMA2-IR - homeostatic model assessments of insulin resistance, HTN - hypertension, LADA – latent autoimmune diabetes in adults, NAFLD – nonalcoholic fatty liver disease, T1D – type 1 diabetes, TG - triglycerides
In the latest work in this space, Wagner et al applied clustering methods on a longitudinal cohort of non-diabetic patients with sophisticated clinical variables and identified 6 clusters(20). Three of these clusters corresponded to very-low, low, and obese-but-low risk groups. The remaining three clusters were defined by beta-cell failure, insulin-resistance/NAFLD, and visceral fat/nephropathy. The beta-cell and insulin-resistance/NAFLD groups were both at high-risk of imminent T2D, while the visceral fat/nephropathy cluster had moderate T2D risk but increased kidney disease and mortality risk(20). Interestingly, Wagner et al also applied the genetic risk score developed by Udler(8) and found that the two highest risk clusters (beta-cell and insulin-resistance/NAFLD) had concurrently increased genetic risk, highlighting the role of non-modifiable genetic risk in T2D development(20).
While powerful, there are caveats to using clustering approaches on clinical data. First, clinical data are not static and are strongly influenced by the environmental(21, 22): e.g., there will be patients who after being diagnosed with T2D will undergo diet and exercise changes to successfully lower their HbA1c(11, 12). However, these participants will be mis-classified moving forward as “T2D patients” despite having successfully implemented lifestyle interventions to treat their T2D. As well, there is the issue of which clinical variables are included in the model for cluster building(6). As discussed below, the variables chosen for cluster analyses have a significant impact on what subgroups are identified.
In comparison to clinical methods of clustering, genetic methods have several advantages: first, the included variants have been vetted by GWAS and have a significant association with T2D(9, 10, 23–37). Second, germline genetic data does not vary with time or environmental changes(21, 22) and therefore can be used at any point in life to understand underlying pathologic susceptibilities to a complex disease such as T2D(11, 12). As a result, genetic clustering of T2D patients has been an ongoing area of research for nearly a decade to help bring precision medicine into the clinical setting.
Historical approaches to genetic clustering analyses:
Hints at the varied genetic architecture of T2D first arose in 2010, when Voight et al found that variants associated with T2D-risk were more likely to be associated with measures of pancreatic beta cell function than of insulin sensitivity(27). In a separate study, Ingelsson et al performed a hypothesis-driven analyses and demonstrated that the GWAS loci for T2D identified at that time could broadly be categorized into those which influenced T2D-risk through defects in insulin processing/secretion or peripheral insulin sensitivity(38). These categories were later confirmed through the use of “hard” hierarchical clustering of T2D genes and clinical traits(39). Of note, with “hard” hierarchical clustering, each genetic variant can only belong to one clinical trait “cluster” or group.
Scott et al later performed an iteratively improved analysis with a larger sample size and the inclusion of BMI and lipid traits in their clustering analysis(32). Using hierarchical clustering methods, they identified three major pathologic groups for genes associated with T2D: insulin secretion/processing, disruption of insulin action, and a new cluster of dyslipidemia(32).Building upon the dyslipidemia genetic cluster(32) and the classical observation that lipodystrophy-related phenotypes were associated with insulin resistance and T2D in normal-to-underweight patients(40), Lotta et al created a genetic risk score that defined a lipodystrophy-like subset of T2D(41). They noted with increasing genetic risk, there was a concomitant decrease in body fat percentage and BMI, with a paradoxical increase in waist-to-hip ratio. To confirm their observations, they applied their genetic risk score to an independent cohort and confirmed that patients at the highest genetic risk had lower fat mass, but greater risk of incident T2D, suggesting that this lipodystrophy-like phenotype was itself predictive of T2D. These studies have stimulated follow-up work by several groups which has begun to tease out the underlying biology of the causal genes in these loci. For instance, we and others confirmed that FAM13A is the causal “lipodystrophy-like” gene in one locus and perturbations of this gene affect adipose biology and fat distribution both in vitro and in vivo(42).
“Soft” hierarchical clustering approaches to genetic clustering:
To account for clinical heterogeneity in pathogenesis for complex diseases (e.g., that a given person may have variants that alter their risk of both obesity and insulin secretion, both of which contribute to T2D risk), Udler et al developed a “soft” hierarchical clustering method(8). In this method each genetic variant can belong to more than one cluster of clinical phenotypes, thereby allowing for modeling of the pleiotropic effects of genes/loci(8).
Using 94 genetic variants and 47 T2D-related clinical traits, Udler et al identified and replicated 5 partially overlapping genetic clusters (Table 1 and Figure 1(8)). The first 2 clusters were strongly associated with insulin processing/secretion with the genes all associated with deficiency in circulating insulin. The remaining 3 clusters were all related to tissue-specific response to insulin. Cluster 3, the “obesity/adiposity” cluster, was defined by increased waist circumference, BMI, body fat, and fasting insulin, suggesting that this cluster represented classic obesity mediated T2D. Cluster 4, the “lipodystrophy” cluster, was characterized by increased insulin resistance, triglycerides, and waist-to-hip ratio (in women only), with an accompanying decrease in BMI, suggesting that this cluster represented the contribution of visceral fat to both insulin resistance and T2D risk. Cluster 5, the “liver/lipid” cluster, was associated with decreased serum triglycerides but increased NAFLD risk, suggesting a role of liver metabolism in T2D risk(8). Notably, these 5 genetic clusters were largely able to recapitulate the clinical clusters previously identified(6). Moreover, the identified genetic clusters showed remarkable similarity to subsequent work by Mahajan et al, who also used soft clustering methods, but identified an additional cluster corresponding to a “mixed” picture with aspects of both insulin secretion and insulin resistance (Table 1, (37)).
Figure 1. Genetically defined clusters and example genes.
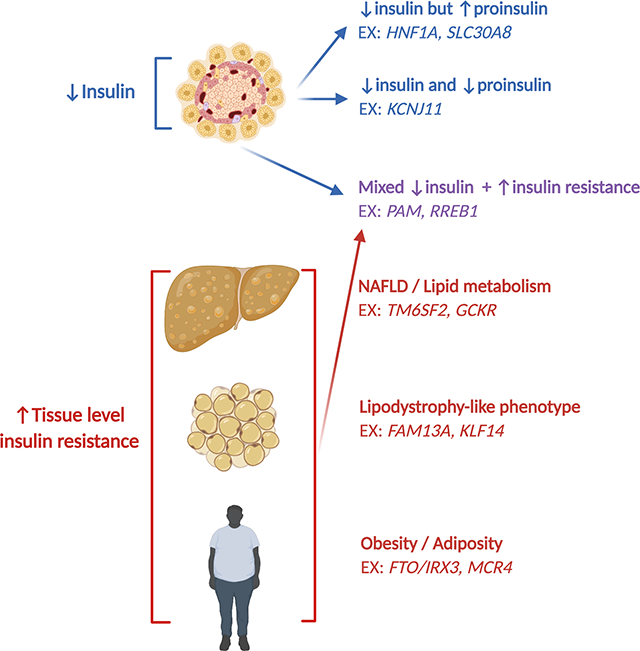
Schematic representation of the genetic clusters identified by Udler et al, with the additional “mixed” genetic cluster concurrently identified by Mahajan et al (see main text and Table 1), with example genes. Abbreviations: EX – for example, NAFLD – nonalcoholic fatty liver disease.
Application of this method to independent replication cohorts demonstrated that 30% of participants were in the top 10% of genetic risk for a single cluster(8). When comparing the mean characteristics of these individuals at the highest genetic risk, Udler et al found characteristic clinical profiles defining each genetic cluster: for example, those in the highest genetic risk for the obesity cluster had significantly higher BMI, body fat, and waist circumference. In contrast, those in the highest genetic risk for the lipodystrophy had significantly lower BMI, body fat, and HDL levels(8). In summary, these results suggested that each genetic cluster represented a specific pathophysiology through which T2D risk was affected through intermediate phenotypes.
Using Genetics to Predict Risk for T2D
In recent years a number of groups have proposed the concept of partitioned polygenic risk scores (pPRS) to identify patients at high risk of T2D through a specific, intermediate pathway (e.g., insulin deficiency) for early intervention(11). However, pPRS builds upon prior work over the past two decades to build polygenic risk scores based on candidate genes for complex diseases(43–47) and more recent work creating global polygenic risk scores (gPRS) for CVD(48) and T2D(9, 10, 48).
Early attempts at combining risk alleles:
Using 3 well-replicated variants in candidate genes (KCNJ11, PPARG, and TCF7L2), Weedon et al created the first restricted polygenic risk score (rPRS; see Figure 2) for T2D by calculating the area under the receiver-operator curve (AUROC) to determine the benefit conferred by this rPRS in predicting T2D. From this, they reported that the AUROC using their rPRS was 0.58 – improved from what would be expected from chance alone (0.5) – but falling well short of the threshold typically required for clinical implementation(43).
Figure 2. Visualization of the different polygenic risk score types.

From GWAS results (visualized as a Manhattan plot), investigators can choose only the significantly associated SNVs and create a restricted polygenic risk score (rPRS). Alternatively, they can create a genome-wide polygenic risk score (gPRS), which accounts for nearly all the variation in the genome. Finally, as proposed by Udler et al, investigators can create specific, partitioned polygenic risk scores (pPRS) for intermediate phenotypes that confer risk of the disease in question. These polygenic risk scores often underscore pathologic mechanisms through which the end-phenotype of disease can be achieved. Abbreviations: GWAS – genome wide association study, NAFLD – nonalcoholic fatty liver disease.
With the release of GWAS data for T2D and the identification of dozens of novel loci(24–26), several independent groups attempted to create new sets of rPRS for prediction of T2D(44–46). In all studies, the AUROC was improved; however, the added discriminatory benefit of including SNVs for T2D was only 1–2% in all 3 studies compared to a model using clinical covariates alone (e.g., improvement from an AUROC of 0.74 → 0.75 (44)).
Global polygenic risk scores provide risk estimates on par with monogenic diseases:
Acknowledging the limited prognostic value of rPRS for complex disease, Khera et al recently took a different approach of using variants across the genome to construct a “global” polygenic risk score (gPRS, see Figure 2(48)). Using a bioinformatic machine-learning tool(49), they leveraged genetic inheritance patterns (“linkage disequilibrium” or “LD”) in European ancestry populations to create a gPRS that accounted for nearly all the common variation in a genome, rather than a subset of several dozen variants (as used in prior rPRS(44–46)). Participants in the top 5% of gPRS distribution for T2D risk had a 2.75x increase in odds of T2D, as compared to the remaining 95% of participants, a risk similar to that seen in some monogenic diseases, which could lead to actionable precision medicine in the clinic (48).
Using an updated GWAS for T2D performed in close to one million individuals, Mahajan et al created a similar gPRS and applied it to data from the entirety of the UK Biobank(9). In their updated gPRS, those in the top 2.5% of risk for T2D had an increase in odds of 3.4x as compared to those with median (50%) T2D gPRS risk. Unfortunately, direct comparison to the estimates of Khera et al(48) was not possible, as they used different reference populations for calculation of their odds ratios (i.e., bottom 95% versus median 50%) despite applying their different gPRS to the same UK Biobank population(9). Follow-up work by Vujkovic et al(10) used the gPRS developed by Mahajan et al(9) in the multi-ancestry Million Veteran Program (MVP) and found that those in the highest risk group for T2D gPRS had significantly increased odds of T2D microvascular complications (DKD, neuropathy, retinopathy(10)).
Partitioned polygenic risk scores apply to individual pathways/intermediate phenotypes leading to disease:
In addition to rPRS and gPRS, a number of groups have proposed pPRS (Figure 2 shows a schematic differentiating rPRS, gPRS, and pPRS) for identification of genetic risk specific to intermediate phenotypes that represent possible pathologic mechanisms to disease (e.g., lipodystrophy-like genetic cluster leading to T2D(11)). These pPRSs integrate with the palette model proposed by McCarthy(50). In the palette model of complex disease, the focus is not on the end phenotype of T2D, but rather on the various intermediate phenotypes representing different pathogenesis to T2D (e.g., obesity/adiposity, lipodystrophy, insulin deficiency, and NAFLD/lipid metabolism). Each of these intermediate phenotypes are themselves complex genetic traits, requiring use of pPRS to define level of variance for an individual patient. Conceptually, each intermediate phenotype can be visualized as a primary color. Hence, by calculating pPRS for each intermediate phenotype, one can define germline genetic risk for T2D via known genetic clusters/pathologic pathways. These colors can then be combined or “mixed” and an individual’s risk of T2D aggregated, with the various contributions of individual pathways visualized (Figure 3). Then, for those patients at highest risk for a specific intermediate phenotype/pathologic pathway, clinicians can attempt targeted prevention/treatment(11, 50).
Figure 3. Potential clinical use of partitioned polygenic risk scores.

Each row in the “pPRS genotypes” represents presence/absence of risk alleles for a specific pPRS pathway. Combination of risk from different pPRS pathways (each represented by a different color) leads to an overall “palette” of color. In this figure, a primary color/pPRS pathway is predominant for each specific patient. We do note that this figure is likely an oversimplification – most patients will not have a specific dominant process driving their T2D pathogenesis. In the original work by Udler et al, only 30% of participants segregated into the top decile of pPRS genetic risk for a given genetic cluster. Abbreviations: BMI – body mass index, GLP-1Ra – glucagon-like peptide-1 receptor agonist, HLD - hyperlipidemia, HTN - hypertension, M – man, NAFLD – nonalcoholic fatty liver disease, SGLT2i – sodium-glucose cotransporter 2 inhibitor, T2D – type 2 diabetes, Tx – treatment, W – woman, yo – years old.
Use of polygenic risk scores for T2D:
To date, the clinical use of various polygenic risk scores in T2D has been limited. Using a 65 variant rPRS for T2D, Li et al reported that those with higher genetic risk had a greater reduction in HbA1c at 1-year follow-up in response to therapy with sulfonylureas(51).Separately, Jiang et al demonstrated that each increase in standard deviation for their 123 variant T2D rPRS increased risk of developing an insulin-dependence by 7%, with replication in an independent cohort(52). Using a gPRS for T2D, Wagner et al reported that increased pancreatic steatosis interacted with decreased insulin secretion(53). Interestingly, Wagner et al performed sensitivity analyses across the 5 clusters of T2D defined by Udler et al(8) and found that the interaction was driven by a pPRS defining the liver metabolism/NAFLD gene cluster(53).Finally, recent work using a pPRS for lipodystrophy found that despite having lower BMI/waist circumference, those with a higher lipodystrophy pPRS had significantly higher insulin resistance and low-density lipoprotein, portending possible increased CVD risk in this specific subpopulation(54). Further work will be needed to elucidate the relationship between pPRS and risk prediction/stratification in the clinical context.
Genetics and the potential for tailored therapy in T2D:
For illustration of the concept of pPRS and the palette model for tailoring of treatments, we can consider the concrete example from monogenic diabetes. Monogenic diabetes can be partitioned by the specific genetic mutations, each of which has different treatment strategies available(55). Heterozygous inactivating mutations in the key glycolytic enzyme glucokinase (GCK) cause stable mild fasting hyperglycemia from birth which is refractory to pharmacological intervention and usually successfully managed by diet alone(56–58). In contrast, mutations in the transcription factor hepatocyte nuclear 1 alpha (HNF1A) cause a more severe progressive phenotype which has a risk of complications similar to T2D(59, 60). Identifying patients with HNF1A-monogenic diabetes is important because they are optimally treated with low dose sulfonylureas(61, 62). Similarly, patients with heterozygous activating mutations in either of the genes encoding the components of the pancreatic beta cell KATP channel (KCNJ11 or ABCC8) can be treated with high dose sulfonylureas rather than insulin(63–65). Providing the optimal medication based on the genetic etiology improves glycemic control (55).
Harnessing physiology to guide the molecular characterization of T2D GWAS variants
The translation of genetic signals into biological and clinical insights has not kept a pace with their discovery. This is in part due to the fact that unlike monogenic forms of diabetes, which are largely due to variants that alter the protein sequence, most GWAS signals are in non-coding regions of the genome with a presumed regulatory function, making it challenging to be certain which is the “effector” gene. Gene regulation is highly context dependent, with the degree of gene expression (and resulting magnitude of association with a trait/outcome) conditional on numerous factors, including: specific cell type, developmental time point and/or chromatin accessibility, and environmental conditions. All of these context-cues are critically important for interpreting the effect of a T2D-associated regulatory variant. Large scale efforts to characterize tissue-specific regulatory landscapes such as Genotype-Tissue Expression consortium (GTEx) have been critical in this regard(66). Investigations into the physiological processes that are influenced by T2D-associated variants have been enormously helpful in providing information on which tissue and/or cell type should be studied to understand the disease mechanism. Below we provide examples from the literature, which exemplify different physiological/molecular mechanisms and showcase how researchers move from GWAS signal to a molecular mechanism. We first highlight colocalization: a method through which investigators can fine-map hundreds-to-thousands of GWAS signals in a genetic region to the likely causal variant.
Colocalization can identify causal variants/genes, prioritizing translation:
A powerful way to identify effector transcripts at GWAS loci is to identify colocalization of association signals for disease risk and gene expression. Colocalization of genetic signals is generally performed using summary-level statistics from GWAS for disease (e.g., T2D) and GWAS for expression quantitative trait loci (eQTL) data(67), though any other type of genome-wide study data could also be used in colocalization methods (i.e., DNA methylation data, histone methylation data, etc). eQTL data reflects genome wide variant associations with individual gene expression levels within a specific tissue or cell type (e.g., pancreatic islet). eQTLs can be detected in multiple tissues; however, working with disease relevant tissues is the preferred approach, as eQTLs are frequently tissue-specific. Many genes are highly dependent on context-specificity, with their expression levels dependent on a combination of cell-type, environmental perturbation (e.g., starvation vs hyperglycemia), and/or epigenome structure. In particular, epigenetics has become a focus in the last decade with mounting evidence that aging and disease alter the physical structure of DNA, potentially limiting gene expression even in cases where the context-specificity is otherwise met(68). In this context, methods such as chromatin segregation to highlight the active and repressed regions of DNA represent promising avenues through which to integrate many data sources and identify effector transcripts(69).
Identifying colocalization of a T2D-GWAS signal and an eQTL in a diabetes relevant tissue, such as pancreatic islets, offers evidence for a potential causal link (and thereby identifies the effector transcript). In this causal relationship, the genetic variant influences pancreatic islet gene expression, which in turn affects T2D risk (Figure 4). In addition to the GTEx database(66), recent publications of eQTL data for skeletal muscle(70), adipose(71), and pancreatic islet tissue(72), have contributed to deciphering possible mechanistic links for T2D GWAS variants.
Figure 4. Colocalization of genetic signals offers evidence of causality.

(A) Example of an eQTL study, where the outcome is gene expression and variant genotypes are tested across the genome to find specific loci that are associated with differential gene expression (eQTL levels). (B) Colocalization analysis of GWAS and eQTL studies, with colocalization of signals offering mechanistic evidence for a potential causal variant. In the case of non-colocalization, the two variants are in linkage disequilibrium (LD) meaning they are often inherited together as a haplotype. Abbreviations: eQTL – expression quantitative trait loci, GWAS – genome-wide association study, LD – linkage disequilibrium.
Examples of translation of T2D GWAS signals into understanding of molecular mechanism/pathogenesis:
The early identification of two independent coding variant signals in the PAM gene for T2D risk provided a molecular “signpost” to the effector transcript (i.e., strongly implicating PAM as the causal gene), accelerating efforts to understand the molecular mechanisms of PAM(73). Further work highlighted the association of PAM with both T2D and beta-cell function(73). Thomsen et al reported that both coding alleles in the PAM protein result in a loss of function, reducing the ability of this amidating enzyme to increase the biological potency of glycine-extended neuropeptides(74). Loss of PAM in a human beta-cell model resulted in reduced insulin content and secretion an effect also observed in isolated primary islets from carriers of the PAM T2D-risk allele (74).
Even when the effector transcript and the tissue of effect are known, there can still be challenges. The SLC30A8 locus provides an example of the complexity. Early GWAS for T2D(25) identified a nonsynonymous coding allele in SLC30A8 associated with T2D-risk; however, whether T2D risk was associated with loss vs. gain-of-function in the gene was unclear from early functional and rodent studies(75–77) but human genetic evidence provided compelling support that loss-of-function in SLC30A8 was associated with diabetes protection(78). A recent study, combining detailed physiological characterization in human carriers of the SLC30A8 loss-of-function variants coupled with cellular studies in a suite of human beta-cell models, demonstrated that the SLC30A8 T2D-protective alleles cause enhanced insulin secretion (79).
TM6SF2 was first identified as a missense variant (hence identifying TM6SF2 as the likely effector transcript) causing low low-density lipoprotein cholesterol(80) (LDL-C), but paradoxically increased NAFLD risk(81). Several groups found that TM6SF2 was primarily expressed by the liver(82) and congruent work by Smagris et al and Mahdessian et al found that the likely mechanism of action for TM6SF2 was through impaired very-low density lipoprotein (VLDL) release from the liver, leading to NAFLD via fat accumulation but also lower LDL-C in serum(82, 83). Subsequent work supports this proposed mechanism and has found that TM6SF2 is associated with incident T2D(84), a finding that is supported by large-scale GWAS that later identified TM6SF2 as a T2D risk locus(31).
A locus containing FTO has long been associated with obesity and Fto knockout in mice reduces BMI(85). However, recent evidence suggests that FTO is not the causal gene at this locus. The T2D risk allele in the locus increased both IRX3 and IRX5 gene expression(86). Adding further weight to these convincing human studies, Irx3 knockout mice recapitulated the prior Fto knockout phenotype(85): substantially reduced body weight and increased brown adipose tissue, suggesting that IRX3, rather than FTO, is the causal gene causing obesity and increased T2D/CVD risk(86). The example of FTO/IRX3 illustrates the complexity in untangling the molecular mechanism of T2D GWAS variants that are regulatory in nature, and highlights the importance of context (correct tissue, at the right developmental time point, and with appropriate environmental conditions).
Genetic methodologies informing and/or validating drug discovery in T2D
Given the saturation of genetic data in the scientific literature (>400 signals at >350 loci associated with T2D(9, 36)), deciding which specific gene to target for functional validation and drug development has been difficult. Prior mechanistic studies on candidate genes were largely driven by biologic plausibility. Novel methods offer can prioritize genetic loci for translational research and also have improved drug discovery pipelines.
Mendelian Randomization (MR) for validation and identification of potential drug targets:
Mendelian Randomization (MR) is a powerful technique that leverages the random assortment of alleles during meiosis to assess potential causality between SNV genotype and outcomes (Figure 5 and (87) for a topical review on interpretation of MR studies in cardiometabolic disease). MR has already been used to demonstrate a causal link between hyperglycemia and CVD risk(17). A further example of the utility of MR comes from PCSK9 inhibition. In 2005, Cohen et al identified two nonsense mutations in PCSK9 that resulted in drastically lower LDL-C, spurring drug development of PCSK9 inhibitors(88). A subsequent MR study demonstrated that loss-of-function variants in PCSK9 were associated with decreased LDL-C and CVD, suggesting a causal pathway(89). Concurrent to this MR study, PCSK9 inhibition was found to reduce both LDL-C and CVD events(90).
Figure 5. Mendelian randomization identifies causal associations between an allele/outcome.

For evidence of the causal association between lower LDL-C and T2D, please refer to (89)and (94). * denotes that meiosis during gamete production acts as a randomization event for inheritance of a given genetic variant allele. Abbreviations: LDL-C – low-density lipoprotein cholesterol; T2D – type 2 diabetes.
MR can also be done at large-scale, allowing for rapid screening of multiple candidates for potential drug development. For example, Folkerson et al recently reported on genetic variants influencing 90 serum protein levels (a pQTL study) and performed a MR study that implicated a causal role for RAGE protein in T2D(91), whereby AGER (the gene encoding RAGE) SNVs caused higher RAGE protein levels, and subsequent lower T2D risk. Of note, their MR analysis did not find evidence of an association with Alzheimer’s disease, for which a RAGE inhibitor had previously been studied (NCT00566397). This study, in conjunction with prior work on PCSK9 inhibition, demonstrates the utility of MR in identifying novel drug targets and validating drugs under development.
Although classically described for SNV alleles, MR can also be used to assess if a given pPRS is causally associated with an outcome via an intermediate phenotype. For example, Sanna et al used a pPRS for proprionate, a fecal short-chain fatty acid, and found that the pPRS was associated with both increased proprionate levels and increased T2D risk(92). These results suggested that genetically controlled proprionate levels were causally linked with differential T2D risk (see (93) for in-depth review on the gut microbiome and T2D risk). Similarly, Sinnott- Armstrong et al leveraged deep phenotyping with protein and urine biomarkers to demonstrate causal relationships across many traits and diseases including highlighting an emerging causal role for decreased LDL-cholesterol and increased T2D risk ((94), Figure 5).
Phenome-wide association studies (PheWAS) to identify potential non-label use for existing drugs:
A final genetic tool that has broad relevance in drug repurposing and identification of potential adverse reactions is the phenome-wide association study (PheWAS). A PheWAS inverts the study design of a GWAS: a single SNV is tested for association with multiple phenotypes; mostly commonly through use of ICD-10 codes in EMR data (see Figure 6). Through such large-scale SNV-disease associations, investigators can identify off-target effects (OTEs) that may represent targets for drug repurposing and also potential adverse drug reactions (ADRs). However, after identification of an OTE, further investigation must be undertaken to ensure that the direction of effect for the SNV-disease association is consistent with beneficence. For example, although TM6SF2 is associated with decreased LDL-C and CVD(80), it is concomitantly associated with increased NAFLD(81).
Figure 6. PheWAS identifies potential off target effects and adverse drug reactions.

From a genome-wide association study (GWAS) “Manhattan plot”, the top gene variant associated with an outcome can be tested against multiple phenotypes (e.g., International Classification of Disease-10 (ICD-10) codes) to identify pleiotropic associations with other diseases and adverse reactions. In this example, Gene X encodes protein X, for which an inhibitor X is currently on the market for treatment of rheumatoid arthritis (RA). PheWAS of the top variant in Gene X finds OTEs for type 2 diabetes (T2D) and cardiovascular disease (CVD) – suggesting that inhibitor X may be repurposed for treatment of these diseases. However, an association alone does not suggest that an OTE may exist – examining the direct of effect is important in secondary analyses (see main text) – hence, the potential OTE for T2D is nullified in this example. An ADR is also identified via an association with bacterial sepsis, warranting caution in prescribing inhibitor X.
As an example of PheWAS, Cai et al recently analyzed an IL6R SNV and found a significant association with abdominal aortic aneurysm, representing a possible OTE(95). Tocilizumab, an IL-6R antagonist, has since been approved for use in giant cell arteritis, of which a significant complication is aortic aneurysm. Moreover, Cai et al found that decreased IL-6R levels was associated with increased risk of ADRs eczema, conjunctivitis, and pleuritis, consistent with trial data for tocilizumab. These results offer a glimpse into the power of PheWAS for drug discovery and repurposing in T2D, as well as in identifying the likely side effect profile of a given agent.
Future directions for research into the genetics of T2D
Precision Medicine for T2D using gPRS/pPRS:
gPRS can be used to risk-stratify patients, enabling earlier interventions and prevention of T2D pathogenesis and complications(48). We envision that gPRS can be used to identify asymptomatic patients at high risk of developing T2D based on their non-modifiable genetic risk. Clinicians can then intervene with intensive lifestyle modification, medications (e.g., metformin), and/or counseling to prevent onset and progression of T2D(11, 12). gPRS-based risk stratification can also be implemented in conjunction with clinical covariates, as recently demonstrated by Wagner et al(20), as simple clinical covariates are excellent predictors of T2D-related complications(5).
As we move toward more precise measurements and targeting of pathways, pPRS for intermediate phenotypes has the potential to further refine our diagnostics and therapeutics with regard to complex diseases like T2D. Already, pPRS methods have highlighted unknown or unappreciated pathways (such as NAFLD or lipodystrophy) leading to T2D (11, 12). Although in the seminal work by Udler et al only 30% of patients segregated to the highest decile of genetic risk for a given pPRS(8), we suspect that this proportion will increase with larger/improved GWAS (as demonstrated in follow-up work by Mahajan(37)), particularly in non-European populations (see below). More clinically relevant is the question of how pPRS will change management of T2D: although we infer that T2D caused by lipodystrophy-like vs beta-cell failure processes can likely be optimally managed with different combinations of medications/lifestyle interventions (Figure 3), there is yet a paucity of evidence to support these hypotheses. Hence, moving forward with gPRS/pPRS in clinical settings, we strongly advocate for randomized studies to evaluate the effect of these genetic risk tools in clinical decision making and outcomes, simultaneous to its implementation(96). With such an evidence base available, we are hopeful that the treatment of T2D can mirror that of monogenic diabetes (see above), whereby specific therapies are tailored to genotype (55).
Limited studies in non-European ancestry populations:
Perhaps the greatest limitation of research into the genomics of T2D and CVD is that the vast majority of studies have been performed in European-ancestry populations. Recently, several high-profile GWAS and other genetic studies for T2D have been performed for Asian-ancestry(36, 52) and African-ancestry individuals(10, 97, 98). Further GWAS/genetic studies in non-European-ancestry populations are of crucial importance, as they serve as the input data for gPRS/pPRS, which in turn have the potential to identify patients at high-risk for chronic diseases, thereby enabling clinicians to implement strategies for aggressive prevention/treatment. However, algorithms are strongly influenced by their input data and have already been shown to be less predictive in African-ancestry individuals(99). Already, early application of rPRS has found that efficacy is reduced when applied to African-ancestry subjects for T2D(47). Similar application of rPRS/gPRS for CVD found attenuated effects in African-ancestry populations(100). Factoring in multi-ethnic GWAS data to create gPRS/pPRS while accounting for complex population genetics will likely complicate matters further(101), as there is evidence of latent population structure even within the relatively racially-homogenous UK Biobank(102). Moreover, the largest studies currently are from European-ancestry populations, and gPRS/pPRS derived from these data perform poorly when extrapolated to other populations, hence exacerbating existing health disparities in non-White populations(103). In line with the recent International Common Disease Alliance (ICDA) white paper(104), we believe that greater efforts must be made to ensure that all genetic ancestry groups are represented in biobanks linking genetic data and EMRs, and the resulting discovery genetic work.
Metabolomics/Proteomics and T2D:
One growing area of research is the role of the metabolome/proteome in T2D pathogenesis(105). Although the metabolome/proteome is vast, the majority of research has focused on the strong link between branched-chain amino acids (BCAA) and T2D risk(105). Cross-sectional analyses first found that BCAA levels were higher in T2D patients as compared to healthy controls and positively correlated with insulin resistance(106). This finding was later confirmed in longitudinal data that showed a 2–3.5x increased risk of T2D with higher BCAA levels in 12-years of follow-up(107). The association between BCAA levels and T2D risk were later confirmed using MR(108, 109), demonstrating a likely causal relationship. Given the predictive value of BCAA levels and the wide-spread use of metabolomic assays for newborn diagnoses (e.g., phenylketonuria), one promising avenue of investigation is the use of BCAA levels for precision medicine in identifying patients at high-risk for T2D and implementing intensive preventative measures(105).
Conclusions and Perspectives
In summary, clustering methods allow for more precise sub-phenotyping of T2D and increased understanding of the pathophysiology leading to complex disease. In particular, genetic methodologies offer a wealth of information for a specific patient from a single peripheral blood draw. Use of germline genetics for patients in combination with pPRS has the potential to identify patients at high risk for specific intermediate phenotypes for T2D (e.g., NAFLD vs lipodystrophy), leading to earlier implementation of interventions tailored to the patient’s specific risk for various pathologic processes leading to T2D. In addition, development of novel methodologies has allowed for increased drug discovery and validation. Further work will be required to elucidate and demonstrate the clinical utility of germline genotyping and pPRS in T2D, particularly in non-European ancestry populations.
Central Figure. Disentangling T2D pathogenic mechanisms, leading to precision medicine.

Partitioned polygenic risk scores (pPRS) can identify the intermediate phenotypes that are driving T2D pathogenesis in a specific patient. With further research (preferably with randomization concurrent with clinical implementation), we aim to develop an evidence base similar to that with monogenic diabetes that tailors prevention/treatment strategies based upon a specific patient’s genetics and pPRS-identified pathologic pathways. Abbreviations: B-cell – pancreatic beta cell, NAFLD – nonalcoholic fatty liver disease, T2D – type 2 diabetes.
Highlights.
The association of type 2 diabetes (T2D) with cardiovascular disease accounts for substantial morbidity and mortality.
Genetic associations delineate mechanistic pathways influencing T2D phenotypes with implications for pathogenesis and treatment.
Further work is needed to characterize these genetic links across diverse ancestries.
Acknowledgements:
D.S.K. thanks Maja Ivanovic, Srikanth Palanisamy, Bryan Huebner, and Kevin Li for their excellent clinical care, allowing for the writing of this article. Illustrations created with Biorender.com.
Funding:
D.S.K. was formerly supported by grants from the National Institutes of Health (SNPG9895 and F31 MH101905) and the American Heart Association (AHA 16POST27250048).
A.L.G. is a Wellcome Senior Fellow in Basic Biomedical Science. A.L.G. is funded by the Wellcome (095101, 200837) and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (U01-DK105535; U01-DK085545, UM1DK126185), the Stanford Diabetes Research Center (NIDDK award P30DK116074), and a Pilot and Feasibility Grant.
J.W.K. is supported by grants from the NIDDK (1R01DK107437, 1R01 DK116750 and 1R01DK106236), the American Diabetes Association (1-19-JDF-108), the American Heart Association, the Stanford Diabetes Research Center (NIDDK award P30DK116074) and a Pilot and Feasibility Grant.
Abbreviations:
- BMI
body mass index
- CVD
cardiovascular disease
- eQTL
expression quantitative trait locus
- GWAS
genome-wide association study (can be plural or singular)
- HbA1c
hemoglobin A1c
- MR
Mendelian randomization
- NAFLD
non-alcoholic fatty liver disease
- PheWAS
phenome-wide association study
- PRS
polygenic risk score
- T2D
type 2 diabetes
Footnotes
Conflicts of Interest: A.L.G. declares that her spouse is an employee of Genentech and hold shares in Roche. D.S.K. and J.W.K. declare no financial conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cho NH, Shaw JE, Karuranga S, et al. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pr 2018;138:271–281. [DOI] [PubMed] [Google Scholar]
- 2.Dennis JM. Precision Medicine in Type 2 Diabetes: Using Individualized Prediction Models to Optimize Selection of Treatment. Diabetes 2020;69:2075–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung WK, Erion K, Florez JC, et al. Precision Medicine in Diabetes: A Consensus Report From the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2020;43:1617–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buse JB, Wexler DJ, Tsapas A, et al. 2019 Update to: Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2019;43:487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennis JM, Shields BM, Henley WE, Jones AG, Hattersley AT. Disease progression and treatment response in data-driven subgroups of type 2 diabetes compared with models based on simple clinical features: an analysis using clinical trial data. Lancet Diabetes Endocrinol 2019;7:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahlqvist E, Storm P, Käräjämäki A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol 2018;6:361–369. [DOI] [PubMed] [Google Scholar]
- 7.Zaharia OP, Strassburger K, Strom A, et al. Risk of diabetes-associated diseases in subgroups of patients with recent-onset diabetes: a 5-year follow-up study. Lancet Diabetes Endocrinol 2019;7:684–694. [DOI] [PubMed] [Google Scholar]
- 8.Udler MS, Kim J, von Grotthuss M, et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: A soft clustering analysis. Plos Med 2018;15:e1002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahajan A, Taliun D, Thurner M, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 2018;50:1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vujkovic M, Keaton JM, Lynch JA, et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet 2020;52:680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Udler MS, McCarthy MI, Florez JC, Mahajan A. Genetic Risk Scores for Diabetes Diagnosis and Precision Medicine. Endocr Rev 2019;40:1500–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Udler MS. Type 2 Diabetes: Multiple Genes, Multiple Diseases. Curr Diabetes Rep 2019;19:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Group UPDS (UKPDS). Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998;352:837–853. [PubMed] [Google Scholar]
- 14.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HAW. 10-Year Follow-up of Intensive Glucose Control in Type 2 Diabetes. New Engl J Medicine 2008;359:1577–1589. [DOI] [PubMed] [Google Scholar]
- 15.Boussageon R, Bejan-Angoulvant T, Saadatian-Elahi M, et al. Effect of intensive glucose lowering treatment on all cause mortality, cardiovascular death, and microvascular events in type 2 diabetes: meta-analysis of randomised controlled trials. Bmj 2011;343:d4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reaven PD, Emanuele NV, Wiitala WL, et al. Intensive Glucose Control in Patients with Type 2 Diabetes — 15-Year Follow-up. New Engl J Med 2019;380:2215–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leong A, Chen J, Wheeler E, et al. Mendelian Randomization Analysis of Hemoglobin A 1c as a Risk Factor for Coronary Artery Disease. Diabetes Care 2019;42:1202–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L, Cheng W-Y, Glicksberg BS, et al. Identification of type 2 diabetes subgroups through topological analysis of patient similarity. Sci Transl Med 2015;7:311ra174–311ra174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou X, Zhou X, Zhu Z, Ji L. Novel subgroups of patients with adult-onset diabetes in Chinese and US populations. Lancet Diabetes Endocrinol 2019;7:9–11. [DOI] [PubMed] [Google Scholar]
- 20.Wagner R, Heni M, Tabák AG, et al. Pathophysiology-based subphenotyping of individuals at elevated risk for type 2 diabetes. Nat Med 2021:1–9. [DOI] [PubMed] [Google Scholar]
- 21.Lakhani CM, Tierney BT, Manrai AK, Yang J, Visscher PM, Patel CJ. Repurposing large health insurance claims data to estimate genetic and environmental contributions in 560 phenotypes. Nat Genet 2019;51:327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang K, Gaitsch H, Poon H, Cox NJ, Rzhetsky A. Classification of common human diseases derived from shared genetic and environmental determinants. Nat Genet 2017;49:1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 2003;33:177–182. [DOI] [PubMed] [Google Scholar]
- 24.Scott LJ, Mohlke KL, Bonnycastle LL, et al. A Genome-Wide Association Study of Type 2 Diabetes in Finns Detects Multiple Susceptibility Variants. Science 2007;316:1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sladek R, Rocheleau G, Rung J, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007;445:881–885. [DOI] [PubMed] [Google Scholar]
- 26.Saxena R, Voight BF, Lyssenko V, et al. Genome-Wide Association Analysis Identifies Loci for Type 2 Diabetes and Triglyceride Levels. Science 2007;316:1331–1336. [DOI] [PubMed] [Google Scholar]
- 27.Voight BF, Scott LJ, Steinthorsdottir V, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 2010;42:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strawbridge RJ, Dupuis J, Prokopenko I, et al. Genome-Wide Association Identifies Nine Common Variants Associated With Fasting Proinsulin Levels and Provides New Insights Into the Pathophysiology of Type 2 Diabetes. Diabetes 2011;60:2624–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012;44:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaulton KJ, Ferreira T, Lee Y, et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat Genet 2015;47:1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuchsberger C, Flannick J, Teslovich TM, et al. The genetic architecture of type 2 diabetes. Nature 2016;536:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott RA, Scott LJ, Mägi R, et al. An Expanded Genome-Wide Association Study of Type 2 Diabetes in Europeans. Diabetes 2017;66:2888–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Consortium E, Consortium M, Consortium G, et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat Genet 2018;50:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonàs-Guarch S, Guindo-Martinez M, Miguel-Escalada I, et al. Re-analysis of public genetic data reveals a rare X-chromosomal variant associated with type 2 diabetes. Nat Commun 2018;9:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Platform BG, Collaboration D, CHARGE, et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 2019;570:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spracklen CN, Horikoshi M, Kim YJ, et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature 2020;582:240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahajan A, Wessel J, Willems SM, et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat Genet 2018;50:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ingelsson E, Langenberg C, Hivert M-F, et al. Detailed Physiologic Characterization Reveals Diverse Mechanisms for Novel Genetic Loci Regulating Glucose and Insulin Metabolism in Humans. Diabetes 2010;59:1266–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dimas AS, Lagou V, Barker A, et al. Impact of Type 2 Diabetes Susceptibility Variants on Quantitative Glycemic Traits Reveals Mechanistic Heterogeneity. Diabetes 2013;63:2158–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawrence RD. LIPODYSTROPHY AND HEPATOMEGALY WITH DIABETES, LIPÆMIA, AND OTHER METABOLIC DISTURBANCES A CASE THROWING NEW LIGHT ON THE ACTION OF INSULIN. Lancet 1946;247:773–775. [DOI] [PubMed] [Google Scholar]
- 41.Lotta LA, Gulati P, Day FR, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet 2017;49:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fathzadeh M, Li J, Rao A, et al. FAM13A affects body fat distribution and adipocyte function. Nat Commun 2020;11:1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weedon MN, McCarthy MI, Hitman G, et al. Combining Information from Common Type 2 Diabetes Risk Polymorphisms Improves Disease Prediction. Plos Med 2006;3:e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lyssenko V, Jonsson A, Almgren P, et al. Clinical Risk Factors, DNA Variants, and the Development of Type 2 Diabetes. New Engl J Medicine 2008;359:2220–2232. [DOI] [PubMed] [Google Scholar]
- 45.Meigs JB, Shrader P, Sullivan LM, et al. Genotype Score in Addition to Common Risk Factors for Prediction of Type 2 Diabetes. New Engl J Medicine 2008;359:2208–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lango H, Consortium UT 2 DG, Palmer CNA, et al. Assessing the Combined Impact of 18 Common Genetic Variants of Modest Effect Sizes on Type 2 Diabetes Risk. Diabetes 2008;57:3129–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vassy JL, Hivert M-F, Porneala B, et al. Polygenic Type 2 Diabetes Prediction at the Limit of Common Variant Detection. Diabetes 2014;63:2172–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vilhjálmsson BJ, Yang J, Finucane HK, et al. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am J Hum Genetics 2015;97:576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCarthy MI. Painting a new picture of personalised medicine for diabetes. Diabetologia 2017;60:793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li JH, Szczerbinski L, Dawed AY, et al. A Polygenic Score for Type 2 Diabetes Risk is Associated with Both the Acute and Sustained Response to Sulfonylureas. Diabetes 2020:db200530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang G, Luk AO, Tam CHT, et al. Obesity, clinical, and genetic predictors for glycemic progression in Chinese patients with type 2 diabetes: A cohort study using the Hong Kong Diabetes Register and Hong Kong Diabetes Biobank. Plos Med 2020;17:e1003209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagner R, Jaghutriz BA, Gerst F, et al. Pancreatic steatosis associates with impaired insulin secretion in genetically predisposed individuals. J Clin Endocrinol Metabolism 2020;105:dgaa435–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Srinivasan S, Jablonski KA, Knowler WC, et al. A polygenic lipodystrophy genetic risk score characterizes risk independent of BMI in the Diabetes Prevention Program. J Endocr Soc 2019;3:1663–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic β-cell diabetes. Nat Clin Pract Endoc 2008;4:200–213. [DOI] [PubMed] [Google Scholar]
- 56.Gloyn AL, Odili S, Zelent D, et al. Insights into the Structure and Regulation of Glucokinase from a Novel Mutation (V62M), Which Causes Maturity-onset Diabetes of the Young. J Biol Chem 2005;280:14105–14113. [DOI] [PubMed] [Google Scholar]
- 57.Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S, Hattersley AT. Prevalence of Vascular Complications Among Patients With Glucokinase Mutations and Prolonged, Mild Hyperglycemia. Jama 2014;311:279–286. [DOI] [PubMed] [Google Scholar]
- 58.Chakera AJ, Steele AM, Gloyn AL, et al. Recognition and Management of Individuals With Hyperglycemia Because of a Heterozygous Glucokinase Mutation. Diabetes Care 2015;38:1383–1392. [DOI] [PubMed] [Google Scholar]
- 59.Yamagata K, Oda N, Kaisaki PJ, et al. Mutations in the hepatocyte nuclear factor-1α gene in maturity-onset diabetes of the young (MODY3). Nature 1996;384:455–458. [DOI] [PubMed] [Google Scholar]
- 60.Surmely JF, Guenat E, Philippe J, et al. Glucose utilization and production in patients with maturity-onset diabetes of the young caused by a mutation of the hepatocyte nuclear factor-1alpha gene. Diabetes 1998;47:1459–1463. [DOI] [PubMed] [Google Scholar]
- 61.Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 2003;362:1275–1281. [DOI] [PubMed] [Google Scholar]
- 62.Shepherd M, Pearson ER, Houghton J, Salt G, Ellard S, Hattersley AT. No Deterioration in Glycemic Control in HNF-1 Maturity-Onset Diabetes of the Young Following Transfer From Long-Term Insulin to Sulphonylureas. Diabetes Care 2003;26:3191–3192. [DOI] [PubMed] [Google Scholar]
- 63.Gloyn AL, Pearson ER, Antcliff JF, et al. Activating Mutations in the Gene Encoding the ATP-Sensitive Potassium-Channel Subunit Kir6.2 and Permanent Neonatal Diabetes. New Engl J Medicine 2004;350:1838–1849. [DOI] [PubMed] [Google Scholar]
- 64.Pearson ER, Flechtner I, Njølstad PR, et al. Switching from Insulin to Oral Sulfonylureas in Patients with Diabetes Due to Kir6.2 Mutations. New Engl J Medicine 2006;355:467–477. [DOI] [PubMed] [Google Scholar]
- 65.Babenko AP, Polak M, Cavé H, et al. Activating Mutations in the ABCC8 Gene in Neonatal Diabetes Mellitus. New Engl J Medicine 2006;355:456–466. [DOI] [PubMed] [Google Scholar]
- 66.Consortium TGte. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020;369:1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. Plos Genet 2014;10:e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Horvath S DNA methylation age of human tissues and cell types. Genome Biol 2013;14:3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods 2012;9:473–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taylor DL, Jackson AU, Narisu N, et al. Integrative analysis of gene expression, DNA methylation, physiological traits, and genetic variation in human skeletal muscle. Proc National Acad Sci 2019;116:201814263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raulerson CK, Ko A, Kidd JC, et al. Adipose Tissue Gene Expression Associations Reveal Hundreds of Candidate Genes for Cardiometabolic Traits. Am J Hum Genetics 2019;105:773–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Viñuela A, Varshney A, van de Bunt M, et al. Genetic variant effects on gene expression in human pancreatic islets and their implications for T2D. Nat Commun 2020;11:4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huyghe JR, Jackson AU, Fogarty MP, et al. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat Genet 2013;45:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thomsen SK, Raimondo A, Hastoy B, et al. Type 2 diabetes risk alleles in PAM impact insulin release from human pancreatic β-cells. Nat Genet 2018;50:1122–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pound LD, Sarkar SA, Benninger RKP, et al. Deletion of the mouse Slc30a8 gene encoding zinc transporter-8 results in impaired insulin secretion. Biochem J 2009;421:371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wijesekara N, Dai FF, Hardy AB, et al. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia 2010;53:1656–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nicolson TJ, Bellomo EA, Wijesekara N, et al. Insulin Storage and Glucose Homeostasis in Mice Null for the Granule Zinc Transporter ZnT8 and Studies of the Type 2 Diabetes–Associated Variants. Diabetes 2009;58:2070–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Flannick J, Thorleifsson G, Beer NL, et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet 2014;46:357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dwivedi OP, Lehtovirta M, Hastoy B, et al. Loss of ZnT8 function protects against diabetes by enhanced insulin secretion. Nat Genet 2019;51:1596–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Holmen OL, Zhang H, Fan Y, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet 2014;46:345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kozlitina J, Smagris E, Stender S, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mahdessian H, Taxiarchis A, Popov S, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc National Acad Sci 2014;111:8913–8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J Biol Chem 2016;291:10659–10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim DS, Jackson AU, Li YK, et al. Novel association of TM6SF2 rs58542926 genotype with increased serum tyrosine levels and decreased apoB-100 particles in Finns. J Lipid Res 2017;58:1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fischer J, Koch L, Emmerling C, et al. Inactivation of the Fto gene protects from obesity. Nature 2009;458:894–898. [DOI] [PubMed] [Google Scholar]
- 86.Smemo S, Tena JJ, Kim K-H, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014;507:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Holmes MV, Ala-Korpela M, Smith GD. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat Rev Cardiol 2017;14:577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet 2005;37:161–165. [DOI] [PubMed] [Google Scholar]
- 89.Ference BA, Robinson JG, Brook RD, et al. Variation in PCSK9 and HMGCR and Risk of Cardiovascular Disease and Diabetes. New Engl J Medicine 2016;375:2144–2153. [DOI] [PubMed] [Google Scholar]
- 90.Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. New Engl J Medicine 2017;376:1713–1722. [DOI] [PubMed] [Google Scholar]
- 91.Folkersen L, Gustafsson S, Wang Q, et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat Metabolism 2020;2:1135–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sanna S, van Zuydam NR, Mahajan A, et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat Genet 2019;51:600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol 2021;19:55–71. [DOI] [PubMed] [Google Scholar]
- 94.FinnGen Sinnott-Armstrong N, Tanigawa Y, et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat Genet 2021:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cai T, Zhang Y, Ho Y-L, et al. Association of Interleukin 6 Receptor Variant With Cardiovascular Disease Effects of Interleukin 6 Receptor Blocking Therapy: A Phenome-Wide Association Study. Jama Cardiol 2018;3:849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li R, Chen Y, Ritchie MD, Moore JH. Electronic health records and polygenic risk scores for predicting disease risk. Nat Rev Genet 2020;21:493–502. [DOI] [PubMed] [Google Scholar]
- 97.Adeyemo AA, Zaghloul NA, Chen G, et al. ZRANB3 is an African-specific type 2 diabetes locus associated with beta-cell mass and insulin response. Nat Commun 2019;10:3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen J, Sun M, Adeyemo A, et al. Genome-wide association study of type 2 diabetes in Africa. Diabetologia 2019;62:1204–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Obermeyer Z, Powers B, Vogeli C, Mullainathan S. Dissecting racial bias in an algorithm used to manage the health of populations. Science 2019;366:447–453. [DOI] [PubMed] [Google Scholar]
- 100.Dikilitas O, Schaid DJ, Kosel ML, et al. Predictive Utility of Polygenic Risk Scores for Coronary Heart Disease in Three Major Racial and Ethnic Groups. Am J Hum Genetics 2020;106:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gurdasani D, Barroso I, Zeggini E, Sandhu MS. Genomics of disease risk in globally diverse populations. Nat Rev Genet 2019;20:520–535. [DOI] [PubMed] [Google Scholar]
- 102.Haworth S, Mitchell R, Corbin L, et al. Apparent latent structure within the UK Biobank sample has implications for epidemiological analysis. Nat Commun 2019;10:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet 2019;51:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Alliance ICD. International Common Disease Alliance: Recommendations and White Paper. undefined 2020. Available at: https://www.icda.bio/.
- 105.Chen Z-Z, Gerszten RE. Metabolomics and Proteomics in Type 2 Diabetes. Circ Res 2020;126:1613–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Newgard CB, An J, Bain JR, et al. A Branched-Chain Amino Acid-Related Metabolic Signature that Differentiates Obese and Lean Humans and Contributes to Insulin Resistance. Cell Metab 2009;9:311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang TJ, Larson MG, Vasan RS, et al. Metabolite profiles and the risk of developing diabetes. Nat Med 2011;17:448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mahendran Y, Jonsson A, Have CT, et al. Genetic evidence of a causal effect of insulin resistance on branched-chain amino acid levels. Diabetologia 2017;60:873–878. [DOI] [PubMed] [Google Scholar]
- 109.Wang Q, Holmes MV, Smith GD, Ala-Korpela M. Genetic Support for a Causal Role of Insulin Resistance on Circulating Branched-Chain Amino Acids and Inflammation. Diabetes Care 2017;40:1779–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]