

Abstract
The 593 kbp 16p11.2 copy number variation (CNV) affects the gene dosage of 29 protein coding genes, with heterozygous 16p11.2 microduplication or microdeletion implicated in about 1% of autism spectrum disorder (ASD) cases. The 16p11.2 CNV is frequently associated with macrocephaly or microcephaly indicating early defects of neurogenesis may contribute to subsequent ASD symptoms, but it is unknown which 16p11.2 transcripts are expressed in progenitors and whose levels are likely, therefore, to influence neurogenesis. Analysis of human fetal gene expression data revealed that KIF22, ALDOA, HIRIP3, PAGR1, and MAZ transcripts are expressed in neural progenitors with ALDOA and KIF22 significantly enriched compared to post-mitotic cells. To investigate the possible roles of ALDOA and KIF22 proteins in human cerebral cortex development we used immunohistochemical staining to describe their expression in late first and early second trimester human cerebral cortex. KIF22 protein is restricted to proliferating cells with its levels increasing during the cell cycle and peaking at mitosis. ALDOA protein is expressed in all cell types and does not vary with cell-cycle phase. Our expression analysis suggests the hypothesis that altered neurogenesis in the cerebral cortex contributes to ASD in 16p11.2 CNV patients.
Keywords: ALDOA, autism, cerebral cortex, CNV, KIF22
Introduction
Large, recurrent Copy Number Variations (CNVs) are implicated in many neuropsychiatric disorders including autism spectrum disorders (ASD), epilepsy, intellectual disability (ID), and schizophrenia (McCarthy et al. 2009; Girirajan and Eichler 2010; Levy et al. 2011; Sanders et al. 2011; Malhotra and Sebat 2012). The 16p11.2 CNV (OMIM 611913) encompasses a 593 kb DNA sequence in the p11.2 region of human chromosome 16 (BP4-BP5). This region harbors 29 protein coding genes and is strongly linked to neurodevelopmental disorders (NDDs) including ASD (Kumar et al. 2008; Bijlsma et al. 2009; Rosenfeld et al. 2010; Shinawi et al. 2010; Zufferey et al. 2012). This 16p11.2 region is flanked by two homologous 147kbp sequences that arose after the evolutionary divergence of humans from other primates, generating a hot-spot for mis-aligned recombination that explains the high frequency of the 16p11.2 CNV in the human population and also the high frequency of de novo 16p11.2 CNV (Nuttle et al. 2016). In humans, the 16p11.2 microdeletion is associated with transient infant brain overgrowth (macrocephaly) and focal thickening of the cerebral cortex, while the 16p11.2 microduplication is associated with reduced brain size (microcephaly) (Qureshi et al. 2014; Blackmon et al. 2018). The early manifestation of anatomical phenotype in newborns, along with the onset of ASD symptoms in infancy, suggests crucial roles for 16p11.2 genes during neural development. 16p11.2 is the most prevalent CNV associated with ASD, approximately 1% incidence, making this CNV particularly intriguing and providing motivation for investigating the role played by 16p11.2 genes in brain development and function (Weiss et al. 2008). Available lines of evidence from 16p11.2 rodent models, 16p11.2 patient derived lymphoblastoid cell lines, and human induced pluripotent stem cells genetically engineered to harbor the 16p11.2 CNV indicate that all 16p11.2 messenger RNAs’ (mRNAs’) levels reflect the altered gene dosage of 16p11.2 genes (50% in microdeletion and 150% in microduplication heterozygotes) (Horev et al. 2011; Blumenthal et al. 2014; Pucilowska et al. 2015; Tai et al. 2016). This indicates that multiple 16p11.2 transcript levels are affected by the 16p11.2 CNV and that the pathology of the 16p11.2 CNV could stem from altered dosage of one or more them.
Although none of the individual 16p11.2 genes have been identified as sole causative genes for the 16p11.2 phenotype, MAPK3, QPRT, KCTD13, ALDOA, TAOK2, and KIF22 have each been individually associated with a variety of neural phenotypes in non-human models. These include cell proliferation, neuronal morphology, axonal projection and spine morphogenesis, altered head size, and behavioral phenotypes (Blaker-Lee et al. 2012; de Anda et al. 2012; Golzio et al. 2012; Pucilowska et al. 2015, 2018) (Escamilla et al. 2017) (Richter et al. 2019) (Yadav et al. 2017) (Ultanir et al. 2014).
The cellular mechanisms by which the 16p11.2 CNV causes the patient phenotype are poorly understood. One plausible hypothesis is that disrupted neurogenesis causes changes in neuronal output, which produce a brain with abnormal cell number or composition and that this contributes to the 16p11.2 pathology. Consistent with this hypothesis, the 16p11.2 deletion mouse model exhibits proliferation defects in cortical progenitors during prenatal brain development and subsequently develops ASD-like symptoms (Horev et al. 2011; Pucilowska et al. 2015). However, it is unknown which of the proteins produced by 16p11.2 CNV genes are expressed by progenitor cells in the developing human cerebral cortex and are therefore candidates for regulating neurogenesis.
Excitatory (glutamatergic) neurons in the human cerebral cortex are derived from progenitors that reside in the ventricular and subventricular zones of the dorsal telencephalon while inhibitory (GABAergic) interneurons are derived from progenitors that reside in the ventral telencephalon and migrate into the cerebral cortex (Clowry et al. 2010) (Bystron et al. 2008) (Ma et al. 2013) Here we focused on the potential for the 16p11.2 CNV to affect neurogenesis of excitatory neurons in the developing human cerebral cortex by identifying 16p11.2 genes that are highly expressed in cerebral cortex progenitors in the ventricular zones and down-regulated as cells become post-mitotic. We analyzed previously published human fetal cortex single cell RNA sequencing (scRNA-seq) data (Pollen et al. 2015) (Zhong et al. 2018) to identify candidate genes and characterize their expression in sections of developing human fetal cerebral cortex from the late first and early second trimester.
Material and Methods
Human Tissue
Human embryos ranging in age from 12 to 16 post-conceptual weeks (PCW) were obtained from the Medical Research Council (MRC)/Wellcome-Trust funded Human Developmental Biology Resource at Newcastle University (HDBR, http://www.hdbr.org/) with appropriate maternal written consent and approval from the Newcastle and North Tyneside NHS Health Authority Joint Ethics Committee. HDBR is regulated by the UK Human Tissue Authority (HTA; www.hta.gov.uk) and operates in accordance with the relevant HTA Codes of Practice. For cryosections 12 PCW week brains were fixed in 4%PFA/PBS for 1 week then cryoprotected with 30% sucrose/PBS and then embedded in 50:50 30%sucrose: OCT, flash frozen and sectioned at 12 μm using a Leica Cryostat. Stages used for this study: 12 PCW (two brains), 14 PCW (one brain) and 16 PCW (one brain).
scRNA-seq Analysis
The publicly available scRNA-seq data sets (Pollen et al. 2015; Zhong et al. 2018) were used to identify candidate genes. Prior to dataset publication the reads were aligned, we normalized the RPKMs as log (x + 1). Analysis was performed using R studio. To determine genes with significant changes a Wilcox test by FindAllMarkers in Seurat package was used. Monocle2 R package was used to order cells in pseudotime. To identify cell-cycle phase specific transcripts we used function CellCycleScoring from Seurat R package.
Immunohistochemistry
Immunohistochemistry was carried out on paraffin sections obtained from HDBR. Antigen retrieval consisting of boiling sections in 10 mM sodium citrate pH 6 for 10 min was used for all stains. Primary antibodies were diluted in 20% blocking serum in pH 7.6 Tris buffered saline (TBS) and sections incubated overnight at 4°C. Primary antibodies used: KID 1/5000 DAB, 1/2000 fluorescent (Invitrogen PA5–29490), KI67 1/800 (Novus Biologicals NBP2-22112), ALDOA 1/100 (Sigma HPA004177).
For colourmetric stains, sections were incubated 1 h at room temperature with biotinylated secondary antibody (1/200) followed by incubation for 1 h with ABC (Vector Labs) and developed with diaminobenzidine solution (Vector Labs), washed, counterstained with nuclear fast red, dehydrated and then mounted using DPX.
For immunofluorescence sections were incubated with secondary antibodies 1/200 1 h room temperature, counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI;ThermoFisher) and mounted with Vectashield H1400 Hardset Mounting Medium (Vector Labs). Extensive TBS washes were carried out between each step.
In Situ Hybridisation
PCR primers used to clone in situ probes from human cDNA into pGEMTeasy for preparation of DIG labeled RNA were as follows: ALDOA, CTG TCA CTG GGA TCA CCT T, & GTG ATG GAC TTA GCA TTC AC; KIF22,: CGA GAG CGG ATG GTG CTA AT &: GAG ACC CAG GAT GTT TGC CT; PAGR1, ATG ATG AGC CAG TGA CAC CA & TCT GCC TCT CCC TTC AAG TG; HIRIP3, TGG TGC CCA TCG AAA CTA CA & TGG CCC AAA ATA CAG GAG GT; & MAZ, CAC GAG GAG AAA GTG CCA TG & GAG AGA AGA GGA CCG TCG AG.
In situ hybridisation was performed on cryosections of 12 PCW brain as described previously (Radonjić et al. 2014). Briefly, 12 μm cryosections were dried at 37 °C for 3 h then incubated overnight at 70 °C in hybridization mix containing ×1 salts (200 mM NaCl, 10 mM Tris HCl (pH 7.5), 1 mM Tris Base, 5 mM NaH2PO42H2O, 5 mM Na2HPO4, 0.5 M EDTA: Sigma-Aldrich), 50% deionized formamide, 10% dextran sulfate, 1 mg/mL rRNA, ×1 Denhardt’s, and DIG-labeled RNA probe. Next day sections were washed three times at 70°C in wash buffer comprising ×1 SSC, 50% formamide, 0.1% Tween-20 and then three times at RT in ×1 MABT (20 mM Maleic acid, 30 mM NaCl, 0.5% Tween-20 and pH adjusted to 7.5 with 10 mM NaOH). Sections were incubated 1 h RT in ×1 MABT blocking solution (20% sheep serum, 2% blocking reagent) and then incubated overnight with anti-DIG antibody 1:1500 in blocking solution at 4°C followed by color reaction overnight at RT.
Microscopy and Imaging
DAB and in situ hybridisation images were taken using a Leica DMNB microscope with an attached Leica DFC480 Camera. Fluorescence images were obtained with a Leica DM5500B epifluorescence microscope with a DFC360FX camera. Confocal images were obtained using Nikon A1R FILM microscope and analyzed in ImageJ.
Image Analysis and Quantification
For DAB stains and in situ hybridisation the images were stitched in ImageJ using the stitching plugin (Preibisch et al. 2009).
For KIF22 analysis of DAB stains rectangular counting boxes (34x88μm) were overlaid across the section. Using ImageJ cell counting plugin cells in each box were counted and denoted KIF22+ (brown) or KIF22- (red). The distinction between the regions (VZ, SVZ, and IZ/CP) was determined anatomically by cell density. The count for each box was averaged with other boxes in the region to provide the final value.
For analysis of KIF22/KI67 double staining counting boxes (×20145 μm) were overlaid over the VZ and SVZ (determined based on cell density). For determining intensity cells were randomly selected on the DAPI channel, the nucleus outlined and intensity of KIF22 and KI67 recorded. 20 cells were selected per box and the counts from individual boxes combined to give final values.
For subcellular ALDOA analysis counting boxes (20 × 145 μm) were overlaid over the VZ and SVZ. Cells were randomly selected on the DAPI channel, far enough apart to ensure their cytoplasm would not overlap, the Z plane through the center of the cell was used and the nucleus outlined. The KI67 and ALDOA intensity was measured constituting the nuclear value. To obtain ALDOA cytoplasmic intensity the nuclear outline was duplicated and extended 4 pixels allowing a reading of just the cytoplasmic area to be obtained (see Fig. 6e). This was performed for 10 cells in each box and the counts from individual boxes combined to give final values.
Figure 6 .
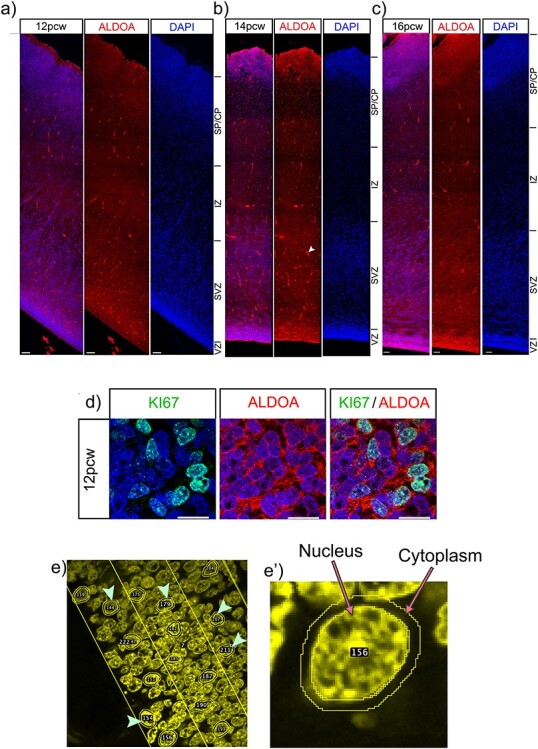
ALDOA protein expression in the cortex. ALDOA protein expression across the telencephalic wall at a) 12, b) 14 and c) 16 pcw. Scale bars = 100 μm. White arrow indicates non-specific binding to blood vessels. d) high magnification immunofluorescence of ALDOA and KI67 proteins, scale bar = 10 μm. e) low power image showing how cells were randomly selected for analysis using the DAPI channel. e’) high power image showing how the nucleus and cytoplasm were delineated for analysis
Data Analysis and Statistics
Where error bars are shown they are expressed as mean ± SEM. Boxplots show median and upper and lower quartiles. Statistical comparison between two groups was performed with a t test. Statistical comparison between more than two groups was performed with ANOVA followed by post hoc test. P < 0.05 was considered statistically significant. Analysis was performed using GraphPad Prism.
Results
Analysis of scRNA-seq Data Identifies KIF22 and ALDOA as Progenitor-Enriched 16p11.2 Transcripts in the Developing Human Fetal Cerebral Cortex
The 16p11.2 CNV involves microduplication or microdeletion of a 593 kb locus on human chromosome 16 containing 29 protein coding genes (Fig. 1a). The aim of the current study is to identify 16p11.2 genes that are potential candidates for being involved in neurogenesis in the developing human cerebral cortex (Fig. 1b,c) and whose altered dosage in 16p11.2 microdeletion or microduplication patients may disrupt neurogenesis and contribute to the CNV phenotype. We reasoned that 16p11.2 genes important for neurogenesis would be highly expressed in proliferating progenitor cells and downregulated as cells became postmitotic.
Figure 1 .
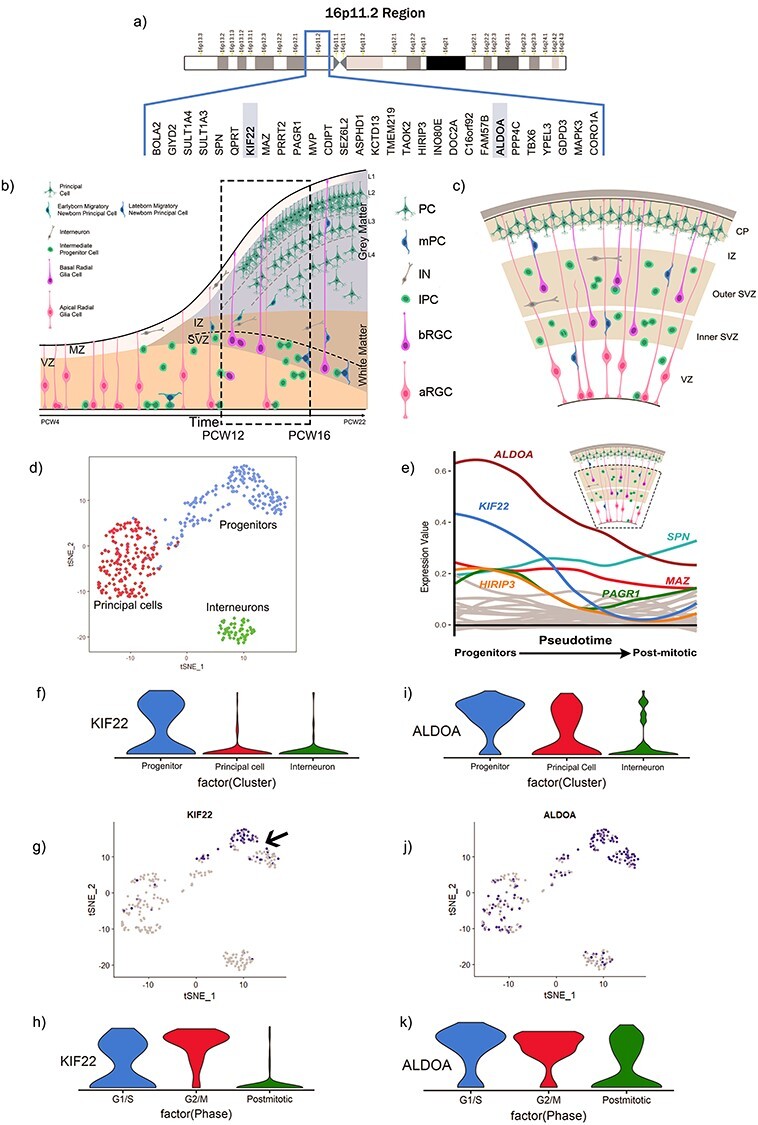
Bioinformatics analysis of scRNA-seq from the VZ and SVZ of the 16-18GW human fetal cortex. (a) 16p11.2 region and genes. (b) schematic adapted from Budday et al. 2015 shows the process of human fetal cortex development over time. Dotted box indicates approximately the time-period of interest for our study; 12-16PCW. (c) schematic of human cortical structure during development. (d) tSNE clustering of cell types. (e) changing mRNA expression levels of 16p11.2 genes as cells move from progenitors to neurons with KIF22 and ALDOA transcripts identified as changing significantly. Schematic of human cortex with dotted box indicates the compartments of the brain that were used to generate this dataset—the germinative VZ and SVZ. (f) Violin plots showing distribution of KIF22 in different cell types. (g) KIF22 gradient plot (arrow indicates progenitors with a subset expressing high levels of KIF22 mRNA). (h) Violin plots showing distribution of KIF22 at different cell cycle stages. (i) Violin plots showing distribution of ALDOA mRNA levels in different cell types. (j) ALDOA gradient plot. k) Violin plots showing distribution of ALDOA mRNA at different cell cycle stages.
We took advantage of a published single cell RNA-sequencing (scRNA-seq) data-set acquired from 393 cells of the ventricular zone (VZ) and subventricular zone (SVZ) of gestational week (GW) 16–18 human fetal cerebral cortex (equivalent to post conception week (PCW) 14–16) to perform an unbiased screen to identify 16p11.2 transcripts that matched this expression profile (Pollen et al. 2015). Dimensional reduction of the scRNA-seq data separated the cells into three clusters based on transcriptome similarity (Fig. 1d—each dot on the tSNE plot represents an individual cell) that were subsequently identified as the three cardinal cell classes of progenitors (blue), post-mitotic neurons/principal cells (red) and interneurons (green), by expression of cell-type specific transcripts. We next used the monocle2 R package to order the cells in pseudotime using the normalized expression levels of selected differentially expressed genes (DEGs) as input to order the cells (Trapnell et al. 2014; Qiu et al. 2017) (Fig. 1e) moving from the progenitor state (left) to post mitotic state (right) along the X-axis. We plotted the average expression of each 16p11.2 transcript at each pseudotime-point on the Y-axis. We found that two genes, KIF22 (blue line) and ALDOA (brown line), were notable for having high expression in progenitors that declined as cells became post-mitotic. A Wilcox test identified ALDOA and KIF22 as the only 16p11.2 transcripts that were significantly higher in progenitor than neuronal populations (P < < 0.05). Although not significantly enriched in progenitors HIRIP3 (orange line), MAZ (red line), and PAGR1 (green line) were expressed in progenitors at higher levels than the remaining 16p11.2 transcripts (shown as gray lines), many of which were barely expressed at all.
Violin plots of the numbers of cells expressing different levels of KIF22 mRNA in the different cardinal cell classes show that KIF22 is expressed predominantly in progenitors (Fig. 1f) and mapping the expression level of KIF22 onto the tSNE plot (Fig. 1g) revealed that KIF22 expression is highest in a subset of the progenitor cluster (arrow in Fig. 1g) with a substantial proportion of progenitor cells expressing relatively low levels of KIF22. Very few post-mitotic neurons, both interneurons and principal cells, express appreciable levels of KIF22 (Fig. 1f,g). The expression of KIF22 in a subset of progenitors prompted us to ask whether its expression was related to the cell-cycle phase. We used the expression of cell-cycle phase specific transcripts using function CellCycleScoring from Seurat R package to divide the cells into three classes (Macosko et al. 2015; Tirosh et al. 2016), G1/S, G2/M, and post-mitotic neurons, and compared KIF22 transcript levels between these three groups using a violin plot (Fig. 1h). We found that the majority of cells in G2/M phase expressed higher levels of KIF22 (red plot), cells in G1/S expressed lower levels (blue plot) while the vast majority of post-mitotic cells expressed low levels of KIF22 (green plot). Similar analysis for ALDOA show that while a greater proportion of cells expressing the highest levels of ALDOA are progenitors (blue plot) there are also a substantial number of principal cells (red plot) expressing similarly high levels of ALDOA transcripts although very few interneurons (green plot). Mapping ALDOA expression level onto the tSNE plot (Fig. 1j) shows cells expressing high levels of ALDOA are evenly distributed throughout the progenitor cluster with appreciable numbers of principal cells expressing high levels of ALDOA and a much lower proportion of interneurons. In contrast to KIF22, there is no clear difference in the partitioning of ALDOA expression level between different phases of the cell-cycle (Fig. 1k). We performed the same analysis on another developing human cortex scRNAseq data set that spanned a wider developmental interval (PCW10–28) and also included cells from all layers of the cerebral cortex (Zhong et al. 2018). This analysis produced similar results. KIF22 transcripts showing them to be enriched in progenitors (Supplementary Figure 1b, e) and in G2/M phase of the cell cycle (Supplementary Fig. 1g). Although ALDOA transcript levels were highest in progenitors they did not decline as much as KIF22 (Supplementary Fig. 1b,f) in postmitotic cells and showed no clear difference between different phases of the cell-cycle (Supplementary Fig. 1h).
We next used in situ hybridisation to visualize the expression of KIF22, ALDOA, HIRIP3, PAGR1, and MAZ transcripts in the different layers of the 12 PCW brain. We identified the VZ and SVZ based on cytoarchitecture, however, to validate our delineation we stained a section with PAX6, expressed by progenitor cells (Fig. 2a-a’*). This confirmed the absence of progenitor cells in the IZ and CP and allowed us a guidance for estimating cellular location in other sections.
Figure 2 .
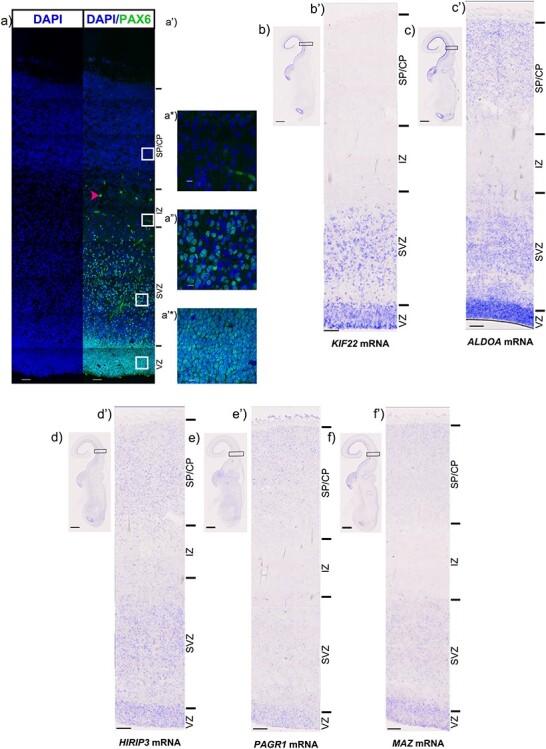
In situ hybridisation of candidate genes. (a) PAX6 protein (green) at 12pcw. a-a’*) show high magnification images of PAX6 protein expression in the (a’) SP/CP, a*) IZ, a”) SVZ and a’*) VZ. Low magnification scale bars = 50 μm, high magnification scale bars = 10 μm. b) Low magnification image of KIF22 mRNA in the 12pcw human fetal cortex, b’) High magnification showing KIF22 mRNA (blue) to be predominantly expressed in the germinative zones. c) Low magnification image of ALDOA mRNA in the 12pcw human fetal cortex, (b’) High magnification showing ALDOA mRNA (blue) to be predominantly expressed in the germinative zones but also some expression in the IZ and CP. (d) Low magnification image of HIRIP3 mRNA in the 12pcw human fetal cortex, (d’) High magnification showing HIRIP3 mRNA (blue) to be expressed throughout the telencephalic wall. e) Low magnification image of PAGR1 mRNA in the 12pcw human fetal cortex, e’) High magnification showing PAGR1 mRNA (blue) to be expressed throughout the telencephalic wall. (f) Low magnification image of MAZ mRNA in the 12pcw human fetal cortex, (f’) High magnification showing MAZ mRNA (blue) to be expressed throughout the telencephalic wall. For ISH, low magnification scale bars = 2 mm and high magnification scale bars = 100 μm.
Consistent with the scRNAseq data KIF22 and ALDOA transcripts are the most clearly differentially expressed between zones containing progenitors (VZ and SVZ) and the more superficial layers that are mainly composed of postmitotic cells (IZ and SP/CP). KIF22 mRNA expression is most prominent in the VZ and SVZ with a few expressing cells in the IZ and SP/CP (Fig. 2b,b′). Although ALDOA mRNA expression is most prominent in the proliferative VZ and SVZ there are substantial numbers of ALDOA expressing cells in the SP/CP (Fig. 2c,c′). HIRIP3 (Fig. 2d,d′), PAGR1 (Fig. 2e,e′), and MAZ (Fig. 2f,f′) mRNAs are expressed in the VZ/SVZ and also in cells of the SP/CP. In addition to being expressed in the proliferative zones of the cerebral cortex KIF22, ALDOA, HIRIP3, PAGR1, and MAZ are also expressed in the ventricular zone of the ganglionic eminences where interneuron progenitors reside suggesting the hypothesis that interneuron development may be affected by alterations in their dosage in the 16p11.2 CNV.
To conclude, of all the 29 16p11.2 transcripts, five, KIF22, ALDOA, HIRIP3, PAGR1, and MAZ, are expressed in the ventricular and subventricular zones at higher levels than in post-mitotic cells. Of these only two, KIF22 and ALDOA, are significantly enriched in progenitors compared to post-mitotic cells making them candidates for having specific roles in neurogenesis in the developing human fetal cerebral cortex. Although both are enriched in progenitors, KIF22 and ALDOA transcript expression shows notable differences: KIF22 transcripts are more restricted to progenitors and their levels vary as the cell-cycle progresses. We next describe the expression of KIF22 and ALDOA protein over a range of developmental stages.
KIF22 Protein is Expressed in Germinal Zones of 12, 14, and 16 PCW Cortex
Here we characterize KIF22 protein expression during human corticogenesis. Coronal cortex sections spaced along the rostral-caudal axis were immunostained for KIF22 protein and counterstained with Nuclear Fast Red (NFR) to show cytoarchitecture. KIF22+ (brown) and KIF22− (red) cells were counted for each region in the telencephalic wall (VE, VZ, SVZ, IZ, and CP) (see methods for details of sampling) and lamination was identified by cell density (Bayer and Altman 2002, 2005). These data are shown for three developmental stages, 12 PCW (Fig. 3a–d), 14 PCW (Fig. 3a′–d′), and 16 PCW (Fig. 3a*–d*). At all stages and rostro-caudal positions KIF22 expressing cells appear most abundant in the VE followed by the VZ and SVZ with the IZ and CP presenting a very low to complete absence of KIF22 (Fig. 3c,c′,c* with higher magnification of boxed regions from each zone shown in d, d′, and d* respectively, green arrows indicate examples of individual KIF22+ cells).
Figure 3 .
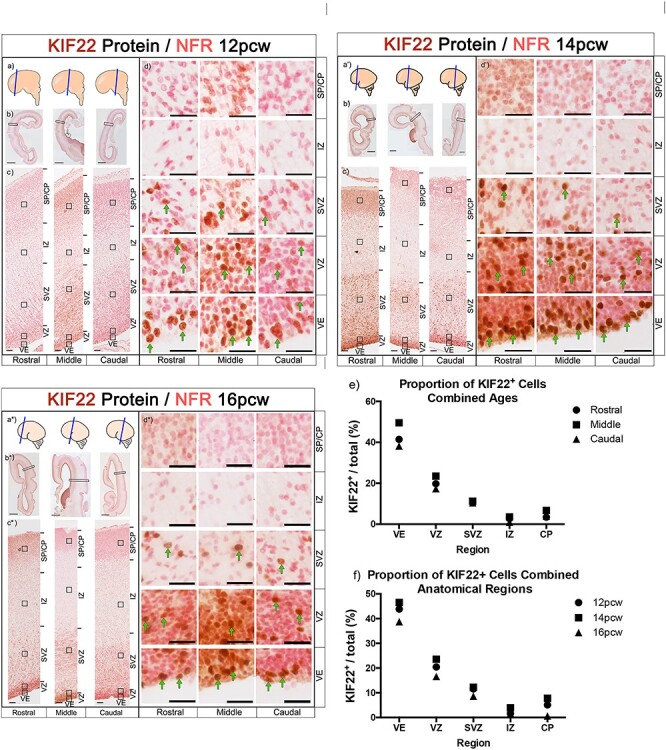
KIF22 protein expression levels in the cerebral cortex at 12, 14, and 16 PCW (a, a’, a*) schematic showing brain regions sectioned. (b, b’, b*) images of whole brain section, scale bars =2 mm. c, c’, c*) sections spanning the rostral-caudal axis showing KIF22 expression in the telencephalic wall, scale bars = 100 μm. (d, d’, d*) high magnification images of different cortical zones rostral-caudal. KIF22+ cells in brown and examples indicated by green arrows, KIF22− cells in pink, scale bars =25 μm. (e) Quantification of KIF22 expressing cells with all three ages combined. (f) Quantification of KIF22 expressing cells with rostral, middle, caudal values combined.
We next pooled KIF22+ cell count data in two ways to compare between all ages (Fig. 3e) and anatomical regions (Fig. 3f) and found that the percentage of KIF22+ cells in the VE (40–50%) was consistently higher than other regions, followed by the VZ (20–30%) and SVZ (10%), with even fewer cells (<10%) in the IZ, and CP (Fig. 3e,f). This result describes KIF22 protein as predominantly restricted to a subset of cells in the germinal zones of the developing cortex at all stages studied.
KIF22 Protein Expression is Restricted to Proliferating Cells
KIF22 protein expression is almost exclusively restricted to a subset of cells in the proliferative regions. From the scRNA-seq data, we expect these to be progenitor cells (Fig. 1). To identify these KIF22 positive cells we performed double immunofluorescence for KIF22 and KI67 (a protein expressed in all proliferating cells (Scholzen and Gerdes 2000; Miller et al. 2018)). KIF22+ cells were predominantly located in the VE, VZ, and SVZ (Fig. 3), therefore these regions were examined for analysis. Low magnification of KI67/KIF22 staining is shown in 12 PCW (Fig. 4a) and 14 PCW (Fig. 4b) with higher magnification showing individual cells in Fig. 4c. Cell counts for KIF22+/KI67+-labeled cells show that the majority (80–90%) of KI67+ cells also express KIF22 both at 12 (Fig. 4d) and 14 PCW (Fig. 4e) or across the rostral-caudal axis. Combining the data for anatomical locations and ages revealed significantly more KIF22+/KI67+ cells than KIF22+/KI67− and KIF22−/KI67+ cells (Fig. 4f).
Figure 4 .
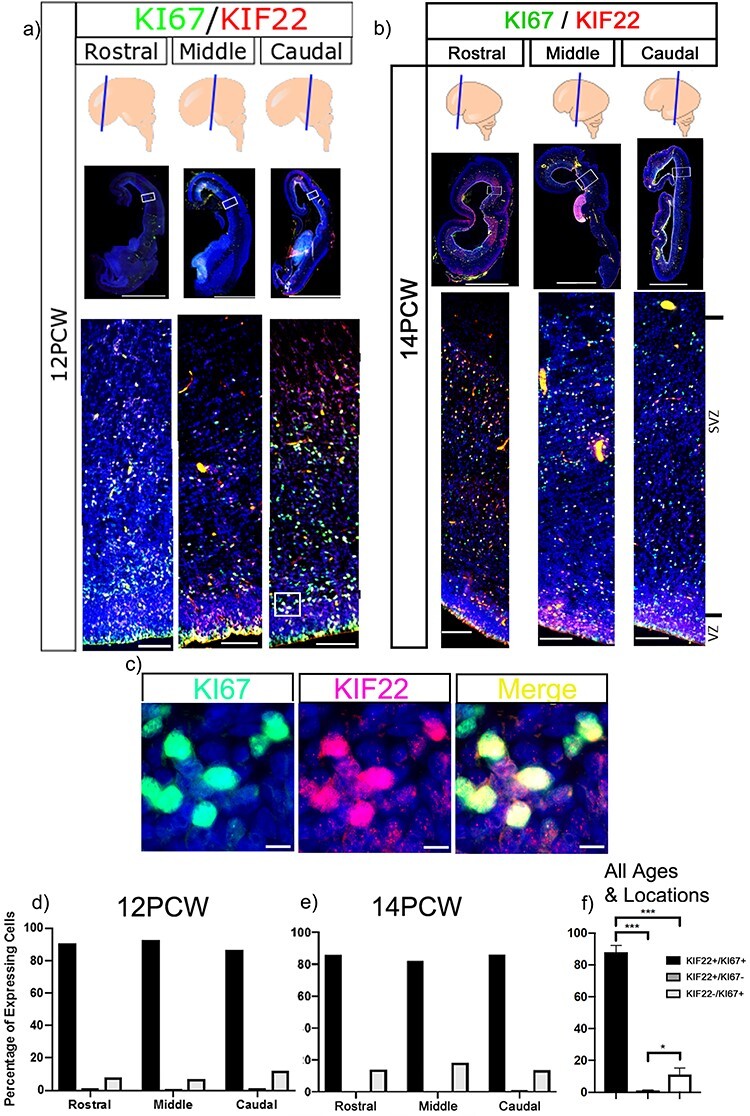
Immunofluorescence of KIF22 and KI67 proteins in the cortex. (a) KIF22 and KI67 at 12 PCW, low magnification scale bars = 4 mm, high magnification scale bars = 100 μm. (b) KIF22 and KI67 at 14 PCW, low magnification scale bars = 4 mm, high magnification scale bars = 100 μm. c) high magnification of KI67/KIF22 expressing cells. Scale bars = 10 μm. (d) Percentage of cells expressing KIF22, KI67, or both at 12 PCW. (e) Percentage of cells expressing KIF22, KI67, or both at 14 PCW. (f) Combined data of percentage of cells expressing KIF22, KI67, or both.
KIF22 Levels Vary with Cell-Cycle Phase
From the scRNA-seq analysis, and the variable KIF22 protein levels in KI67+ cells, we hypothesized that KIF22 protein levels change throughout the cell cycle. To test this, we quantified nuclear immunofluorescence intensity of KIF22 and KI67 in two 12 PCW brains (see methods for details of sampling procedure). KI67 protein levels vary during the cell cycle: lowest in G1 phase, increasing through S and G2 to peak in mitosis (Fig. 5o) (Scholzen and Gerdes 2000; Miller et al. 2018). We found a strong correlation between KIF22 and KI67 intensity (Brain 1 rostral R2 = 0.8095, middle R2 = 0.8139, and caudal R2 = 0.6691. Brain 2 rostral R2 = 0.7489, middle R2 = 0.7447, caudal R2 = 0.7763) (Fig. 5d–f). To ensure the correlation observed was not a result of nucleus size changing with cell cycle, we confirmed that KIF22 protein levels did not correlate with nuclear size by DAPI staining (Brain 1 rostral KIF22 R2 = 0.104, middle KIF22 R2 = 0.0874, caudal KIF22 R2 = 0.2969. Brain 2 rostral KIF22 R2 = 0.1183, middle KIF22 R2 = 0.0512, and caudal KIF22 R2 = 0.2287). A strong positive correlation was also observed at 14 PCW (Brain 1 rostral R2 = 0.7465, middle R2 = 0.6668, caudal R2 = 0.634, Fig. 5j–l). Again, we confirmed that KIF22 protein levels did not correlate with nuclear size (rostral KIF22 R2 = 0.1239, middle KIF22 R2 = 0.0599, caudal KIF22 R2 = 0.0229). This demonstrates that the correlation between KI67 and KIF22 is consistent between ages and rostral-caudal location. Combining all values of KI67/KIF22 nuclear intensity values showed that KIF22 was expressed at significantly higher levels in KI67+ cells (Fig. 5m) with a strong correlation (R2 = 0.7236) between KIF22 and KI67 levels (Fig. 5n). Although KIF22 expressing cells were scattered throughout the VE, VZ, and SVZ there was a general trend for the cells expressing the highest levels of KIF22 to be closest to the apical surface (yellow colored dots on scatterplots Fig. 5d–f,j–l,n) with lower expressing cells tending to be further from the apical surface (blue colored dots on scatterplots Fig. 5d–g, j–l,n). During interkinetic nuclear movement radial glial cell nuclei move to the apical surface to perform mitosis so this spatial distribution suggests KIF22 is expressed at high levels by radial glial cells undergoing mitosis at the apical surface of the VZ. Cerebral cortex progenitor that do not undergo interkinetic movement (intermediate progenitors and outer sub-ventricular zone progenitors) cannot be assigned to cell cycle phase using their position as we were able to do for radial glial cells which do undergo interkinetic movement. As KIF22 transcript levels were at highest in all progenitors in M-phase (Fig. 1h) we suspect that cells expressing high levels of KIF22 and KI67 further away from the apical surface are cortical progenitors in M-phase not undergoing interkinetic movement but further experiments would be needed to show this.
Figure 5 .
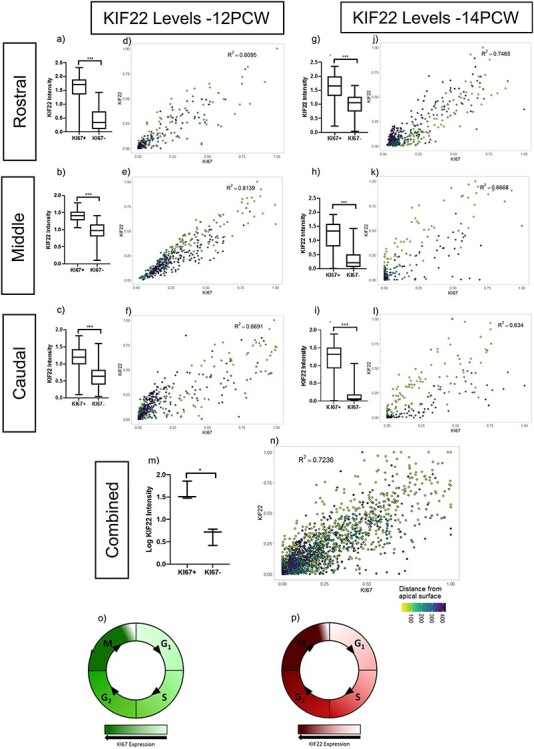
Quantification of KIF22 protein levels. (a, b, c) 12 PCW quantification of KIF22 fluorescence intensity in KI67+/KI67- cells (raw data transformation = +1(log), unpaired t-test with Welch’s correction,P = < 0.001). (d, e, f) intensity correlations of KIF22 and KI67 nuclear fluorescence intensity at 12 PCW. (g, h, i) 14 PCW quantification of KIF22 fluorescence intensity in KI67+/KI67- cells (raw data transformation = +1(log), unpaired t-test with Welch’s correction, P = < 0.001). j, k, l) intensity correlations of KIF22 and KI67 nuclear fluorescence intensity at 14 PCW. m) quantification of KIF22 fluorescence intensity in KI67+/KI67-cells 12 and 14 weeks combined (raw data transformation = (log), paired t-test, P = 0.0122). n) intensity correlations of KIF22 and KI67 nuclear fluorescence intensity for rostral-caudal points at 12 and 14 PCW with distance from apical surface indicated by dot color. o) diagram of KI67 protein levels throughout the cell cycle. p) model based on our results of KIF22 protein levels throughout the cell cycle.
From these data we show that, for radial glial cells, KIF22 protein levels change throughout the cell cycle in positive correlation with KI67: KIF22 is present in G1 and increases through S and G2 phase to peak in mitosis (Fig. 5p).
ALDOA Protein is Highest in the Germinal Zones of the Cortex
Bioinformatics analysis and in situ hybridisation show ALDOA mRNA levels decrease as progenitor cells move towards a neuronal fate (Fig. 1). Here we used immunofluorescence to characterize ALDOA protein expression across the telencephalic wall at 3 developmental time points; at 12, 14, and 16 PCW, ALDOA immunofluorescence is most intense in the VZ and SVZ before decreasing in the cortical plate (Fig. 6a–c). Double immunofluorescence for KI67 and ALDOA viewed at high magnification shows that ALDOA protein is primarily localized outside DAPI+ nuclei in the cytoplasm and that ALDOA is expressed by KI67+ progenitor cells and also by cells that do not express KI67 (Fig. 6d). The schematic (Fig. 6e,e′) illustrates the areas used for quantification of nuclear and whole cell ALDOA fluorescence presented in Fig. 6.
ALDOA Protein Levels Do Not Correlate with Proliferation
Although examination of ALDOA mRNA expression indicated it was enriched in progenitors we were unable to find a significant difference in ALDOA protein levels between KI67+ and KI67− cells at 12 (Fig. 7a), 14 (Fig. 7b), and 16 PCW (Fig. 7c) in the human cortex. To look for any fluctuation in ALDOA levels with the cell cycle we quantified immunofluorescence for KI67 and cell body ALDOA (nucleus and adjacent cell body) using the same analysis as that described above for KIF22, and found no correlation or discernible pattern at 12 (Fig. 7e) (R2 = 0.018), 14 (Fig. 7f) (R2 = 2e-4) or 16 PCW (Fig. 7g) (R2 = 0.00992). These data show that in human cortex development, cellular ALDOA protein levels do not correlate with proliferation or fluctuate with cell cycle.
Figure 7 .
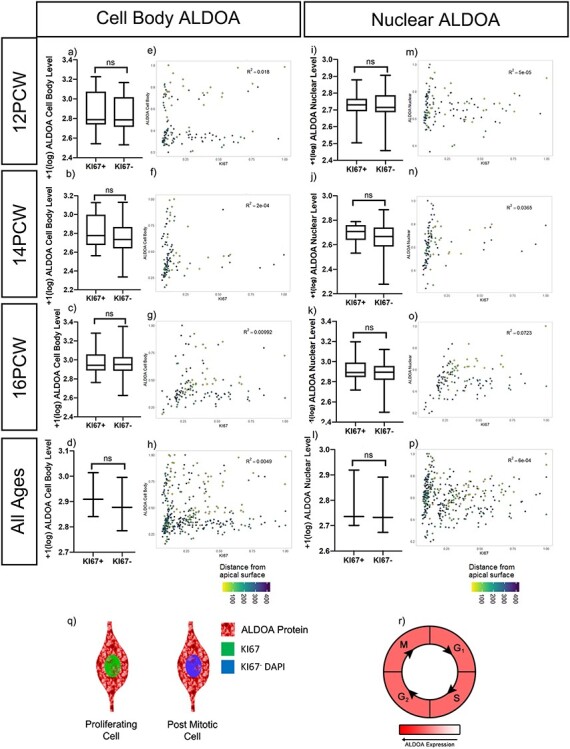
ALDOA protein quantification. (a–d) Cell body ALDOA protein fluorescent intensity in KI67+ and KI67− cells at (a) 12 PCW (raw data transformation = +1(log), bimodal distribution, Mann–Whitney test, p = 0.3702), (b) 14 PCW (raw data transformation = +1(log), normal distribution, unpaired t-test with Welch’s correction, P = 0.2032), (c) 16 PCW (raw data transformation = +1(log), normal distribution, unpaired t-test with Welch’s correction, P = 0.3523). (d) ALDOA cell body protein fluorescent intensity in KI67+ and KI67- cells, 12, 14, and 16 PCW individual datasets averaged, (raw data transformation = +1(log)), paired t-test, P = 0.0836. (e–h) ALDOA cellular protein intensity levels ls correlated to nuclear KI67 protein intensity at (e)12, (f)14 and (g)16 PCW with distance from ventricular edge indicated. (h) ALDOA whole cell protein intensity levels correlated to nuclear KI67 protein intensity pooled 12, 14, 16 PCW. (i–l) Nuclear ALDOA protein fluorescent intensity in KI67+ and KI67− cells at (i) 12 PCW (raw data transformation = +1(log), normal distribution, unpaired t-test with Welch’s correction, P = 0.7543), (j) 14 PCW (raw data transformation = +1(log), normal distribution, unpaired t-test with Welch’s correction, P = 0.0694), (k) 16 PCW (raw data transformation = +1(log), normal distribution, unpaired t-test with Welch’s correction, P = 0.0772). (l) ALDOA nuclear protein fluorescent intensity in KI67+ and KI67− cells, 12, 14, and 16 PCW individual datasets averaged, (raw data transformation = +1(log)), paired t-test, P = 0.1330. (m–p) ALDOA nuclear protein intensity levels is correlated to nuclear KI67 protein intensity at (m)12, (n)14, and o)16 PCW with distance from ventricular edge indicated. (p) ALDOA nuclear protein intensity levels correlated to nuclear KI67 protein intensity pooled 12, 14, and 16 PCW. (q) schematic demonstrating ALDOA protein is predominantly in the cytoplasm and lower in the nucleus in both KI67+ proliferating cells and KI67- post mitotic cells. (r) model showing ALDOA levels do not change with the cell cycle.
Previous work in different models demonstrated nuclear ALDOA level is greater in proliferating cells (Mamczur et al. 2010; Mamczur et al. 2013). To see if this was the case in human cortex development, we quantified nuclear ALDOA and KI67 (Fig. 6e) but found no significant difference in nuclear ALDOA fluorescence between KI67+ and KI67− cells at 12 (Fig. 7i), 14 (Fig. 7j), or 16 PCW (Fig. 7k). We next tested if nuclear ALDOA levels in proliferating cells varied with cell cycle. Analysis of ALDOA and KI67 nuclear intensity established no correlation or pattern at 12 (Fig. 7m) (R2 = 5e-05), 14 (Fig. 7n) (R2 = 0.0365), or 16 PCW (Fig. 7o) (R2 = 0.0723). This shows nuclear ALDOA levels do not increase with proliferation, nor fluctuate with cell cycle. We combined results across the 12, 14, and 16 PCW. There was no significant difference between KI67+ and KI67− cells when examining ALDOA protein intensity in the whole cell (Fig. 7d) or the nucleus (Fig. 7i). Using the pooled data, there was no correlation or discernible pattern when nuclear ALDOA intensity was graphed against nuclear KI67 level for the whole cell (Fig. 7h) (R2 = 0.0049) or the nucleus (Fig. 7p) (R2 = 6e-04).
Discussion
16p11.2 Transcript Expression during Human Neurogenesis
The 16p11.2 CNV is a polygenic mutation that causes NDDs and the current study identified a number of the 29 16p11.2 transcripts expressed in progenitor cells of the cerebral cortex. In addition to ALDOA and KIF22 that are significantly enriched in progenitors, several other transcripts (e.g., HIRIP3, PAGR1, and MAZ) are also expressed in progenitors albeit at lower levels and are not significantly down-regulated as cells become postmitotic. The simultaneous expression of multiple 16p11.2 genes in cells undergoing neurogenesis suggests that these cells may be particularly vulnerable to simultaneous alteration in their dosage as a consequence of the 16p11.2 microdeletion or microduplication. This lends support to the hypothesis that neurogenesis is disrupted in 16p11.2 CNV patients and that this contributes to subsequent development of NDDs.
In this study we focused on the expression of 16p11.2 genes in progenitors of the developing cerebral cortex that will give rise to excitatory neurons. However, KIF22, ALDOA, HIRIP3, PAGR1, and MAZ transcripts are also expressed in the ventricular zone of the ganglionic eminences where the progenitors of the inhibitory interneurons that subsequently migrate into the cerebral cortex reside. Simultaneous increase or decrease of these transcripts in interneuron progenitors resulting from the 16p11.2 CNV may therefore have an impact on the neurogenesis or differentiation of interneurons.
KIF22
KIF22 is a multifunctional protein that can regulate cell proliferation through at least two distinct mechanisms. First, KIF22 is a kinesin-like microtubule-based motor that binds microtubules and chromosomes during mitosis and regulates mitotic spindle microtubule stability and symmetric/asymmetric cell division (Tokai et al. 1996; Tokai-Nishizumi et al. 2005; Sun and Hevner 2014). Second, KIF22 regulates the expression of the cell-cycle regulator CDC25C. During cell division, CDC25C dephosphorylates CDK1, thus activating the CDK1-cyclinB complex while the CDK1-cyclin B complex phosphorylates CDC25C, causing an amplification loop to drive cells to mitosis (Nilsson and Hoffmann 2000). KIF22 directly transcriptionally represses CDC25C and inhibits mitosis; this transcriptional repression of CDC25C is dependent on KIF22 being phosphorylated on Thr463 (Ohsugi et al. 2003; Yu et al. 2014). KIF22 depletion in a tumor cell line accelerates the G2/M transition and slows M/G1 transition (Yu et al. 2014).
Overall, it therefore appears that KIF22 can act at several different points in the cell cycle making it difficult to predict how increased or decreased dosage of KIF22 in the 16p11.2 microduplication or microdeletion respectively would impact cell cycle in the specific context of cerebral cortex neural progenitors especially in light of the concomitant altered dosage of other 16p11.2 genes co-expressed with KIF22 in progenitors. Our observation that KIF22 mRNA and KIF22 protein levels both increase during the cell cycle to achieve the highest levels in G2/M phase that drop as cells enter G1 phase implies that KIF22 protein does not persist for long after it is translated and is degraded at the end of M-phase suggesting that both transcriptional and post-transcriptional mechanisms regulate its levels. A clear outcome of our study is that KIF22 levels positively correlate with KI67 in neural progenitors and steadily rise as the cell progresses through G1 > S > G2 > M phases culminating in the maximum level during M-phase. One possibility is that KIF22 is required to reach a threshold for mitosis to occur, after which its levels must decrease sufficiently to allow mitotic exit. Whether cells undertake proliferative or neurogenic divisions is a process heavily controlled by cell cycle length (Borrell and Calegari 2014). Perturbing KIF22 gene dosage as a consequence of the 16p11.2 CNV might affect the timing of KIF22 protein reaching this threshold in neural progenitors and therefore affect cell-cycle kinetics and perturb neurogenesis and neuronal output. Our results suggest the hypothesis that KIF22 regulates neurogenesis in the human developing cortex through cell-cycle regulation.
ALDOA
The process of brain development requires a vast and consistent supply of energy. Glucose is the predominant energy substrate for the fetal brain (Gustafsson 2009), therefore efficient and controlled glycolysis is essential for normal brain development. ALDOA is required for the fourth step of glycolysis, conversion of fructose 1,6-biphospate to dihyroxyacetone phosphate and gluteraldehyde 3-phosphate. The metabolic role of cytoplasmic ALDOA is well established, and ALDOA also has other non-glycolytic ‘moonlighting’ roles such as regulating mitochondrial function and cytoskeleton stability (Orosz et al. 1988; Pagliaro and Taylor 1992; Kao et al. 1999; Jewett and Sibley 2003; Buscaglia et al. 2006). In addition to its cytoplasmic roles, ALDOA has been observed in the nucleus (Mamczur and Dzugaj 2008; Mamczur et al. 2010; Mamczur et al. 2013) where it has been suggested to impact cell cycle by positively regulating cyclin D1 expression to mediate G1/S progression (Ritterson Lew and Tolan 2012; Fu et al. 2018). Cell-culture studies show ALDOA sub-cellular localization depends on the availability of energetic substrates, with addition of glucose driving ALDOA protein to the cytoplasm (Mamczur et al. 2013). Therefore, it is likely the primary role for ALDOA is metabolic when cells require, and have available to them, large amounts of energy. The majority of ALDOA studies have used highly abnormal cancer tissue, or artificial cell culture systems in which glycolytic enzymes have been shown to be increased (Ritterson Lew and Tolan 2012; Mamczur et al. 2013; Fu et al. 2018; Pollen et al. 2019). How these observations of ALDOA in a variety of systems relate to its role in human cerebral cortex development is unclear.
Altering ALDOA dosage in the developing brain will likely impact energy metabolism by altering the flow of metabolites through the glycolytic pathway and impacting subsequent pathways which feed on outputs of glycolysis. Disruption to energy metabolism during development has previously been linked to ASD and ADHD (Rash et al. 2018). The offspring of hyperglycemic mice presented microcephaly, a phenocopy of the microcephaly observed in 16p11.2 microduplication patients (Rash et al. 2018) and disruptions to energy metabolism may contribute to the microcephaly seen in the offspring of Zika infected mothers (Gilbert-Jaramillo et al. 2019). No homozygous null ALDOA patients have been identified suggesting it is essential for life, but patients with changes to ALDOA levels have been identified; one patient with reduced ALDOA activity presented microcephaly (Kreuder et al. 1996) and another presented intellectual disability (Beutler et al. 1973). Of particular interest is the finding of schizophrenia patients with upregulated cortical ALDOA levels (Beasley et al. 2006) and 16p11.2 microduplication is strongly associated with risk of schizophrenia. This information, coupled with our results that ALDOA is expressed in all cell types, make it clear that any changes to ALDOA dose will perturb energy metabolism at many stages in the brain, impacting its development.
ALDOA is much more abundant in the cytoplasm and we also found no clear relationship between cell body ALDOA levels and cell proliferation status. Nuclear ALDOA has been linked to cell proliferation (Mamczur et al. 2010; Mamczur et al. 2013; Fu et al. 2018) but we found no clear relationship between nuclear ALDOA protein levels and cell proliferation status. Although ALDOA mRNA levels are higher in proliferating cells compared to non-proliferating cells, quantitative analysis of ALDOA protein revealed that ALDOA protein persists once cells exit mitosis. Therefore, while ALDOA protein is abundant in progenitor cells of the developing human cerebral cortex and so may play a role in neurogenesis phenotypes, the persistent expression of ALDOA protein as cells become post-mitotic argues against a specific role in neurogenesis and raises the additional possibility that ALDOA also plays roles in differentiated neurons.
Conclusion
Our study of 16p11.2 gene expression in developing human fetal cerebral cortex indicates that altered dosage of KIF22, ALDOA, HIRIP3, PAGR1, and MAZ caused by the 16p11.2 microduplication or microdeletion may impact on neurogenesis in the developing human cortex and we identified KIF22 being a strong candidate for having a specific role in neurogenesis. Further studies are required to unpick the mechanisms involved, but given the nature of the tissue, the scope for studying this in vivo is currently limited. However, growth of new model systems in which 16p11.2 gene expression can be manipulated such as human cerebral organoids will provide the opportunity address these questions.
Supplementary Material
Contributor Information
Sarah Morson, Simons Initiative for the Developing Brain, Hugh Robson Building, Edinburgh Medical School Biomedical Sciences, The University of Edinburgh, Edinburgh EH8 9XD, UK; Centre for Discovery Brain Sciences, Hugh Robson Building, Edinburgh Medical School Biomedical Sciences, The University of Edinburgh, Edinburgh EH8 9XD, UK.
Yifei Yang, Simons Initiative for the Developing Brain, Hugh Robson Building, Edinburgh Medical School Biomedical Sciences, The University of Edinburgh, Edinburgh EH8 9XD, UK; Centre for Discovery Brain Sciences, Hugh Robson Building, Edinburgh Medical School Biomedical Sciences, The University of Edinburgh, Edinburgh EH8 9XD, UK.
David J Price, Simons Initiative for the Developing Brain, Hugh Robson Building, Edinburgh Medical School Biomedical Sciences, The University of Edinburgh, Edinburgh EH8 9XD, UK; Centre for Discovery Brain Sciences, Hugh Robson Building, Edinburgh Medical School Biomedical Sciences, The University of Edinburgh, Edinburgh EH8 9XD, UK.
Thomas Pratt, Simons Initiative for the Developing Brain, Hugh Robson Building, Edinburgh Medical School Biomedical Sciences, The University of Edinburgh, Edinburgh EH8 9XD, UK; Centre for Discovery Brain Sciences, Hugh Robson Building, Edinburgh Medical School Biomedical Sciences, The University of Edinburgh, Edinburgh EH8 9XD, UK.
Notes
We thank Katherine Howe for her assistance with designing in situ probes and undergraduate student Emma Fowler (EF funded by a WR Henderson Scholarship) for her assistance with quantification.
Funding
The Biotechnology and Biological Sciences Research Council (BBSRC) grant (BB/M00693X/1); the Simons Initiative for the Developing Brain (SFARI - 529085); a BBSRC EASTBIO PhD studentship (to S.M.); Joint Medical Research Council (MRC)/Wellcome (MR/R006237/1 for the human embryonic and fetal material); Human Developmental Biology Resource (www.hdbr.org).
References
- Bayer SA, Altman J. 2002. The Human Brain During the Late First Trimester. England: Taylor & Francis Group (Head Office) United Kingdom. [Google Scholar]
- Bayer SA, Altman J. 2005. The Human Brain During The Second Trimester. England: Taylor & Francis Group (Head Office) United Kingdom. [Google Scholar]
- Beasley CL, Pennington K, Behan A, Wait R, Dunn MJ, Cotter D. 2006. Proteomic analysis of the anterior cingulate cortex in the major psychiatric disorders: evidence for disease-associated changes. Proteomics. 6:3414–3425. [DOI] [PubMed] [Google Scholar]
- Beutler E, Scott S, Bishop A, Margolis N, Matsumoto F, Kuhl W. 1973. Red cell aldolase deficiency and hemolytic anemia: a new syndrome. Trans Assoc Am Physicians. 86:154–166. [PubMed] [Google Scholar]
- Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, Haeringen A, Fransen van de Putte DE, Anderlid BM, Lundin J, Lapunzina P, Pérez Jurado LA, Delle Chiaie B, et al. 2009. Extending the phenotype of recurrent rearrangements of 16p11.2: deletions in mentally retarded patients without autism and in normal individuals. Eur J Med Genet. 52:77–87. [DOI] [PubMed] [Google Scholar]
- Blackmon K, Thesen T, Green S, Ben-Avi E, Wang X, Fuchs B, Kuzniecky R, Devinsky O. 2018. Focal cortical anomalies and language impairment in 16p11.2 deletion and duplication syndrome. Cereb Cortex. 28:2422–2430. [DOI] [PubMed] [Google Scholar]
- Blaker-Lee A, Gupta S, McCammon JM, De Rienzo G, Sive H. 2012. Zebrafish homologs of genes within 16p11.2, a genomic region associated with brain disorders, are active during brain development, and include two deletion dosage sensor genes. Dis Model Mech. 5:834–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal I, Ragavendran A, Erdin S, Klei L, Sugathan A, Guide JR, Manavalan P, Zhou JQ, Wheeler VC, Levin JZ, et al. 2014. Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. Am J Hum Genet. 94:870–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrell V, Calegari F. 2014. Mechanisms of brain evolution: regulation of neural progenitor cell diversity and cell cycle length. Neurosci Res. 86:14–24. [DOI] [PubMed] [Google Scholar]
- Budday S, Steinmann P, Ellen K. 2015. Physical biology of human brain development. Front Cell Neurosci. 9:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscaglia CA, Penesetti D, Tao M, Nussenzweig V. 2006. Characterization of an aldolase-binding site in the Wiskott-Aldrich syndrome protein. J Biol Chem. 281:1324–1331. [DOI] [PubMed] [Google Scholar]
- Bystron I, Blakemore C, Rakic P. 2008. Development of the human cerebral cortex: boulder committee revisited. Nat Rev Neurosci. 9:110–122. [DOI] [PubMed] [Google Scholar]
- Clowry G, Molnar Z, Rakic P. 2010. Renewed focus on the developing human neocortex. J Anat. 217:276–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anda FC, Rosario AL, Durak O, Tran T, Graff J, Meletis K, Rei D, Soda T, Madabhushi R, Ginty DD, et al. 2012. Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat Neurosci. 15:1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escamilla CO, Filonova I, Walker AK, Xuan ZX, Holehonnur R, Espinosa F, Liu S, Thyme SB, Lopez-Garcia IA, Mendoza DB, et al. 2017. Kctd 13 deletion reduces synaptic transmission via increased rho a. Nature. 551:227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Gao H, Qi X, Zhao L, Wu D, Bai Y, Li H, Liu X, Hu J, Shao S. 2018. Aldolase a promotes proliferation and G1/S transition via the EGFR/MAPK pathway in non-small cell lung cancer. Cancer Commun. 38:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert-Jaramillo J, Garcez P, James W, Molnár Z, Clarke K. 2019. The potential contribution of impaired brain glucose metabolism to congenital Zika syndrome. J Anat. 235:468–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Eichler EE. 2010. Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet. 19:R176–R187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, et al. 2012. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 485:363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson J. 2009. Neonatal energy substrate production. Indian J Med Res. 130:618–623. [PubMed] [Google Scholar]
- Horev G, Ellegood J, Lerch JP, Son YE, Muthuswamy L, Vogel H, Krieger AM, Buja A, Henkelman RM, Wigler M, et al. 2011. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc Natl Acad Sci U S A. 108:17076–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett TJ, Sibley LD. 2003. Aldolase forms a bridge between cell surface adhesins and the actin cytoskeleton in apicomplexan parasites. Mol Cell. 11:885–894. [DOI] [PubMed] [Google Scholar]
- Kao AW, Noda Y, Johnson JH, Pessin JE, Saltiel AR. 1999. Aldolase mediates the association of F-actin with the insulin-responsive glucose transporter GLUT4. J Biol Chem. 274:17742–17747. [DOI] [PubMed] [Google Scholar]
- Kreuder J, Borkhardt A, Repp R, Pekrun A, Göttsche B, Gottschalk U, Reichmann H, Schachenmayr W, Schlegel K, Lampert F. 1996. Brief report: inherited metabolic myopathy and hemolysis due to a mutation in aldolase a. N Engl J Med. 334:1100–1104. [DOI] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH Jr, Dobyns WB, et al. 2008. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 17:628–638. [DOI] [PubMed] [Google Scholar]
- Levy D, Ronemus M, Yamrom B, Lee YH, Leotta A, Kendall J, Marks S, Lakshmi B, Pai D, Ye K, et al. 2011. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 70:886–897. [DOI] [PubMed] [Google Scholar]
- Ma T, Wang C, Wang L, Zhou X, Tian M, Zhang Q, Zhang Y, Li J, Liu Z, Cai Y, et al. 2013. Subcortical origins of human and monkey neocortical interneurons. Nat Neurosci. 16:1588–1597. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. 2015. Highly parallel genome-wide expression profiling of individual cells using Nanoliter droplets. Cell. 161:1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Sebat J. 2012. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 148:1223–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamczur P, Dzugaj A. 2008. Aldolase a is present in smooth muscle cell nuclei. Acta Biochim Pol. 55:799–805. [PubMed] [Google Scholar]
- Mamczur P, Gamian A, Kolodziej J, Dziegiel P, Rakus D. 2013. Nuclear localization of aldolase a correlates with cell proliferation. Biochim Biophys Acta. 1833:2812–2822. [DOI] [PubMed] [Google Scholar]
- Mamczur P, Mazurek J, Rakus D. 2010. Ubiquitous presence of gluconeogenic regulatory enzyme, fructose-1,6-bisphosphatase, within layers of rat retina. Cell Tissue Res. 341:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium, Wellcome Trust Case Control Consortium, McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, et al. 2009. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 41:1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller I, Min M, Yang C, Tian C, Gookin S, Carter D, Spencer SL. 2018. Ki67 is a graded rather than a binary marker of proliferation versus quiescence. Cell Rep. 24:1105–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson I, Hoffmann I. 2000. Cell cycle regulation by the Cdc25 phosphatase family. Prog Cell Cycle Res. 4:107–114. [DOI] [PubMed] [Google Scholar]
- Nuttle X, Giannuzzi G, Duyzend MH, Schraiber JG, Narvaiza I, Sudmant PH, Penn O, Chiatante G, Malig M, Huddleston J, et al. 2016. Emergence of a Homo sapiens-specific gene family and chromosome 16p11.2 CNV susceptibility. Nature. 536:205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsugi M, Tokai-Nishizumi N, Shiroguchi K, Toyoshima YY, Inoue J, Yamamoto T. 2003. Cdc2-mediated phosphorylation of kid controls its distribution to spindle and chromosomes. EMBO J. 22:2091–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orosz F, Christova TY, Ovádi J. 1988. Modulation of phosphofructokinase action by macromolecular interactions. Quantitative analysis of the phosphofructokinase-aldolase-calmodulin system. Biochim Biophys Acta. 957:293–300. [DOI] [PubMed] [Google Scholar]
- Pagliaro L, Taylor DL. 1992. 2-Deoxyglucose and cytochalasin D modulate aldolase mobility in living 3T3 cells. J Cell Biol. 118:859–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollen AA, Bhaduri A, Andrews MG, Nowakowski TJ, Meyerson OS, Mostajo-Radji MA, Di Lullo E, Alvarado B, Bedolli M, Dougherty ML, et al. 2019. Establishing cerebral organoids as models of human-specific brain evolution. Cell. 176:743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, Shuga J, Liu SJ, Oldham MC, Diaz A, et al. 2015. Molecular identity of human outer radial glia during cortical development. Cell. 163:55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, Tomancak P. 2009. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 25:1463–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucilowska J, Vithayathil J, Pagani M, Kelly C, Karlo JC, Robol C, Morella I, Gozzi A, Brambilla R, Landreth GE. 2018. Pharmacological inhibition of ERK Signaling rescues pathophysiology and Behavioral phenotype associated with 16p11.2 chromosomal deletion in mice. J Neurosci. 38:6640–6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucilowska J, Vithayathil J, Tavares EJ, Kelly C, Karlo JC, Landreth GE. 2015. The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J Neurosci. 35:3190–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma Y-A, Trapnell C. 2017. Single-cell mRNA quantification and differential analysis with census. Nat Methods. 14:309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi AY, Mueller S, Snyder AZ, Mukherjee P, Berman JI, Roberts TP, Nagarajan SS, Spiro JE, Chung WK, Sherr EH, et al. 2014. Opposing brain differences in 16p11.2 deletion and duplication carriers. J Neurosci. 34:11199–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radonjić NV, Ortega JA, Memi F, Dionne K, Jakovcevski I, Zecevic N. 2014. The complexity of the calretinin-expressing progenitors in the human cerebral cortex. Front Neuroanat. 8:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash BG, Micali N, Huttner AJ, Morozov YM, Horvath TL, Rakic P. 2018. Metabolic regulation and glucose sensitivity of cortical radial glial cells. Proc Natl Acad Sci U S A. 115:10142–10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M, Murtaza N, Scharrenberg R, White SH, Johanns O, Walker S, Yuen RKC, Schwanke B, Bedürftig B, Henis M, et al. 2019. Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol Psychiatry. 24:1329–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritterson Lew C, Tolan DR. 2012. Targeting of several glycolytic enzymes using RNA interference reveals aldolase affects cancer cell proliferation through a non-glycolytic mechanism. J Biol Chem. 287:42554–42563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Coppinger J, Bejjani BA, Girirajan S, Eichler EE, Shaffer LG, Ballif BC. 2010. Speech delays and behavioral problems are the predominant features in individuals with developmental delays and 16p11.2 microdeletions and microduplications. J Neurodev Disord. 2:26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. 2011. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 70:863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholzen T, Gerdes J. 2000. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 182:311–322. [DOI] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, et al. 2010. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 47:332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Hevner RF. 2014. Growth and folding of the mammalian cerebral cortex: from molecules to malformations. Nat Rev Neurosci. 15:217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai DJC, Ragavendran A, Manavalan P, Stortchevoi A, Seabra CM, Erdin S, Collins RL, Blumenthal I, Chen X, Shen Y, et al. 2016. Engineering microdeletions and microduplications by targeting segmental duplications with CRISPR. Nat Neurosci. 19:517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. 2016. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352:189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokai N, Fujimoto-Nishiyama A, Toyoshima Y, Yonemura S, Tsukita S, Inoue J, Yamamota T. 1996. Kid, a novel kinesin-like DNA binding protein, is localized to chromosomes and the mitotic spindle. EMBO J. 15:457–467. [PMC free article] [PubMed] [Google Scholar]
- Tokai-Nishizumi N, Ohsugi M, Suzuki E, Yamamoto T. 2005. The chromokinesin kid is required for maintenance of proper metaphase spindle size. Mol Biol Cell. 16:5455–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. 2014. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 32:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ultanir SK, Yadav S, Hertz NT, Oses-Prieto JA, Claxton S, Burlingame AL, Shokat KM, Jan LY, Jan YN. 2014. MST3 kinase phosphorylates TAO1/2 to enable myosin Va function in promoting spine synapse development. Neuron. 84:968–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, et al. 2008. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 358:667–675. [DOI] [PubMed] [Google Scholar]
- Yadav S, Oses-Prieto JA, Peters CJ, Zhou J, Pleasure SJ, Burlingame AL, Jan LY, Jan YN. 2017. TAOK2 kinase mediates PSD95 stability and dendritic spine maturation through Septin 7 phosphorylation. Neuron. 93:379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wang XY, Sun L, Wang YL, Wan YF, Li XQ, Feng YM. 2014. Inhibition of KIF22 suppresses cancer cell proliferation by delaying mitotic exit through upregulating CDC25C expression. Carcinogenesis. 35:1416–1425. [DOI] [PubMed] [Google Scholar]
- Zhong S, Zhang S, Fan X, Wu Q, Yan L, Dong J, Zhang H, Li L, Sun L, Pan N, et al. 2018. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature. 555:524–528. [DOI] [PubMed] [Google Scholar]
- Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, Mace A, Ferrari C, Kutalik Z, Andrieux J, et al. 2012. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet. 49:Jb 660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.