

Abstract
Background:
Fetal akinesia and arthrogryposis are clinically and genetically heterogeneous and have traditionally been refractive to genetic diagnosis. The widespread availability of affordable genome-wide sequencing has facilitated accurate genetic diagnosis and gene discovery in these conditions.
Methods:
We performed next generation sequencing (NGS) in 190 probands with a diagnosis of arthrogryposis multiplex congenita, distal arthrogryposis, fetal akinesia deformation sequence or multiple pterygium syndrome. This sequencing was a combination of bespoke neurogenetic disease gene panels and whole exome sequencing. Only Class 4 and 5 variants were reported, except for two cases where the identified variants of unknown significance (VUS) are most likely to be causative for the observed phenotype. Co-segregation studies and confirmation of variants identified by NGS were performed where possible. Functional genomics was performed as required.
Results:
Of the 190 probands, 81 received an accurate genetic diagnosis. All except two of these cases harboured Class 4 and/or 5 variants based on ACMG guidelines. We identified phenotypic expansions associated with CACNA1S, CHRNB1, GMPPB and STAC3. We describe a total of 50 novel variants, including a novel missense variant in the recently identified gene for arthrogryposis with brain malformations – SMPD4.
Conclusions:
Comprehensive gene panels give a diagnosis for a substantial proportion (42%) of fetal akinesia and arthrogryposis cases, even in an unselected cohort. Recently identified genes account for a relatively large proportion, 32%, of the diagnoses. Diagnostic-research collaboration was critical to the diagnosis and variant interpretation in many cases, facilitated genotype-phenotype expansions and reclassified VUS through functional genomics.
Keywords: fetal akinesia, arthrogryposis, next generation sequencing, genotype-phenotype, functional genomics, SMPD4, STAC3, CHRNB1, GLDN, GMPPB
INTRODUCTION
Fetal akinesia or hypokinesia can result in a range of clinical presentations, including; fetal akinesia deformation sequence (FADS),1 arthrogryposis multiplex congenita (AMC), distal arthrogryposis (DA),2 lethal congenital contracture syndromes (LCCS), and multiple pterygium syndrome (MPS). Many of these conditions have overlapping features including: joint contractures, pterygia, fetal hydrops, lung hypoplasia and dysmorphic features. While fetal akinesia is frequently diagnosed in utero, following abnormal indications on routine ultrasounds and/or maternal reporting of reduced fetal movements, many cases are not detected until birth, when they present with contractures and associated complications.3 For the purposes of this manuscript, the term fetal akinesia is used as an umbrella description for all entities described above.
Fetal akinesia can arise from gene defects or maternal/external factors. In recent years the contribution of variants in genes that encode critical neuromuscular system proteins has become increasingly recognised as a cause of fetal akinesia. Variants in >70 of these genes are now known to cause fetal akinesia.4–7
As was foreshadowed for rare diseases,8 many novel phenotypes have been attributed to variants in known disease genes – this is also true for the fetal akinesias. In particular, bi-allelic null variants in genes previously associated with autosomal dominant neuromuscular disease have now been associated with fetal akinesia, e.g. recessive CACNA1S,9 DNM2,10 RYR1,11 SCN4A,12 TOR1A,13 and TTN14 disease.
In this study we present our findings from massively parallel sequencing in a cohort of 190 probands presenting with fetal akinesia. We achieved an accurate genetic diagnosis in 81 of these cases, identified two then novel disease genes,15,16 described 50 novel variants and extended the phenotypes associated with known neuromuscular disease genes.
METHODS
Cohort details
This cohort includes 190 probands with a clinical diagnosis of arthrogryposis multiplex congenita, distal arthrogryposis, fetal akinesia deformation sequence, multiple pterygium syndrome (Escobar-variant or lethal) or Schwartz Jampel Syndrome [n=2]. Additional features included CNS involvement (lissencephaly [n=3], polymicrogyria [n=5]), neuropathy (n=2), and congenital myopathy (n=5).
Recruitment into this project was via two arms: (1) cases submitted for diagnostic testing via the PathWest neurogenetic gene panels (some of which were then enrolled in research testing, if no genetic diagnosis was provided in the diagnostic setting), and (2) cases consented for research that underwent testing via the PathWest neurogenetic gene panels and/or whole exome sequencing (WES). Of the 190 patients, 146 underwent diagnostic testing, 16 were studied in both the diagnostic and research setting, and 28 were analysed in the research setting only. Of the 190 probands, all except 15 were from Australia/NZ. These 15 cases were part of the research recruitment arm of this study.
Testing was performed via the PathWest neurogenetic gene panel alone for 150 probands. Combined panel and WES was performed in 29 probands whilst in the remaining 11 probands only WES was performed. The number of probands sequenced on each version of the panel were: 33 (v1), 37 (v2) and 109 (v3).
NGS: Neurogenetic disease gene panel sequencing, mapping and analysis
Next generation sequencing was performed on custom-designed neurogenetic disease gene panels (versions 1–3). Details of versions 1 and 2 of the panel are outlined in Beecroft et al.17 For design of the third iteration of the panel, the genes were split into two panels: a muscle disease gene panel (v3 muscle) and a neurogenic disease gene panel (v3 neuro), with some overlap as appropriate. Sequencing for v3 was performed using Illumina Nextera Rapid Capture Custom Enrichment Kit and sequenced on an Illumina Benchtop Sequencer. Average coverage was 309 fold for versions 1 and 2 of the panel and 350-fold for v3. The minimum accepted coverage cutoffs were as follows; average coverage> 150-fold (v1–3), and 92% (v1–2) or 95% (v3) of te target regions covered to >20x. Details of the genes included in each iteration of the panel are included in Supplementary Table 1. During the iterative process, panels were spiked with probes to known low-coverage regions to facilitate deeper sequencing.17 Base calling, mapping, and variant calling were performed using Torrent Suite 3.6.2 or 4.2 with germline high stringency settings (GRCh37), with default values as outlined at: https://github.com/iontorrent/TS/blob/master/plugin/variantCaller/pluginMedia/parameter_sets/4_4/ampliseqexome_germline_highstringency_p1_parameters.json. For v3 of the panel all mapping and calling was done by the BWA Enrichment App v2.1.2 on the Illumina Basespace Sequence Hub using our custom bed files.
In the diagnostic setting, Cartagenia BenchLab/Alissa software (Agilent Technologies) was used for annotation and analysis. Bioinformatic filters were used as a first pass to restrict analysis to a sub-panel of genes associated with each clinical phenotype. If no pathogenic variant was identified, rare coding variants across all genes on the panel were analysed. Variants with a minor allele frequency of >2% were filtered from the analysis. The diagnostic laboratory adopted the American College of Medical Genetics (ACMG) guidelines to classify variants when they became available in 2015.18
The research team was able to interpret variants on a research-only basis in Cartagenia/Alissa software, without analysis restricted to sub-panels of genes. If a likely reportable variant was found (Class 4 or 5), interpretation was referred to PathWest for validation in the Australian National Association of Testing Authorities (NATA) accredited laboratory or, in the case of overseas samples, the research findings were reported to the consenting clinician for confirmation by their corresponding diagnostic genomics laboratory. Class 3 variants were followed up with appropriate literature searchs, consultation with international experts, and functional genomics where possible.
This project was approved by the Human Research Ethics Committee of the University of Western Australia (approval number RA/4/20/1008) and participants provided informed consent.
NGS - Whole exome sequencing, mapping and analysis
Ampliseq whole exome sequencing was performed by the Lotterywest State Biomedical Facility Genomics (LSBFG) as previously described.5,16 Illumina whole exome sequencing (WES) was performed at either the Australian Genomics Research Facility (AGRF) or the Center for Mendelian Genomics (CMG; The Broad). For AGRF and LSBFG exomes, processing from FASTQ to VCF was performed as per the GATK best practice guidelines version 3.6 (https://software.broadinstitute.org/gatk). VCFs and pedigree information were compiled in a GEMINI database for variant annotation and querying.19 Variant annotation was performed as described previously20 with additional filtering: variants with a minor allele frequency of >1%, or which were seen in the homozygous state in ExAC and gnomAD were excluded from downstream analysis. Variants observed >10 times within the GEMINI database were also removed, which removed many sequencing artefacts. Scripts were used to extract variants compatible with the presumed modes of inheritance (autosomal recessive and de novo dominant).
CMG exomes were analysed in seqr (https://seqr.broadinstitute.org). CNV detection was performed using The Broad’s GATK gCNV calling pipeline, as outlined (https://github.com/broadinstitute/gatk-protected/blob/master/docs/CNVs/CNV-methods.pdf).
Variant confirmation and interpretation
Bi-directional Sanger sequencing was used to confirm missense and indel variants and to determine co-segregation with disease where parental and sibling DNAs were available. CNVs were confirmed using multiplex ligation-dependent probe amplification (MLPA).
In the case of apparent de novo variants, samples from the parents and affected individual were sequenced across a panel of microsatellites to check that the samples were compatible with the provided family structure (i.e. to confirm paternity and rule out the possibility of sample mix-ups).
For missense variants, likely pathogenicity was explored using in silico tools including: CADD,21, MuPro,22 MutationAssessor, MutationTaster, PolyPhen-223 PROVEAN24 and SIFT.25
cDNA studies
For muscle cDNA studies, 10 µm frozen sections were cut on a Leica ultramicrotome and collected into a pre-chilled microfuge tube. RNA extraction was performed using the RNeasy (fibrous tissue) kit (QIAGEN). cDNA was generated using Superscript III reverse transcriptase (ThermoFisher Scientific). Sanger sequencing of regions of interest was performed using standard protocols. Primer details are available upon request.
Minigene assays were performed as previously described,26,27 with slight modifications. This assay relies on the use of a minigene vector which contains a fragment of the SERPING1 gene, with two exonic regions separated by an intron, cloned into the mammalian expression vector pcDNA 3.1(-), downstream of the cytomegalovirus (CMV) promoter. Following transient transfection into human cells, chimeric transcripts can be analysed by reverse transcriptase PCR (RT-PCR) and sequencing. In brief, the exon of interest and approximately 150 bp of flanking intronic sequence either side, were PCR amplified from patient and control DNA using 2X SuperFi (ThermoFisher Scientific) and cloned into the pCas2.1 minigene construct (a kind gift from Dr. Alexandra Martins) using BamHI/MluI (ThermoFisher Scientific) to generate pCas2.1-STAC3_e3 (https://benchling.com/s/seq-MpmgmQUFtsZtWsXCHHS9). Wild-type and variant-containing plasmids were purified using a QIAprep miniprep kit (QIAGEN) and sequences verified by Sanger sequencing. Expression constructs were then transfected into human HEK293FT cells using Lipofectamine 3000 (ThermoFisher Scientific), and RNA harvested after 24 hours using an RNeasy mini kit (QIAGEN). cDNA was generated using SuperScript III reverse transcriptase (ThermoFisher Scientific) and chimeric cDNA amplified by RT-PCR using 2X GoTaq G2 (Promega) using forward primer KO1F (5’-TGACGTCGCCGCCCATCAC) and reverse primer pCAS2R (5’-ATTGGTTGTTGAGTTGGTTGTC) and 30 cycles of amplification. RT-PCR products were analysed on a 2% agarose gel stained with ethidium bromide (0.5 µg/mL), and gel extracted and purified using a QIAquick gel extraction kit (QIAGEN) for Sanger sequencing. Splicing differences were assessed by comparing RT-PCR and sequencing results for the wild-type and variant (c.312T>G) constructs. Sequence alignments were performed in benchling (benchling.com) using the MAFFT algorithm.
GLDN cell assay
The plasmid expressing human GLDN (hGLDN) with a myc-tag epitope at the extracellular C-terminus was obtained from Dr Jerome Devaux.28 Site-directed mutagenesis was performed by Genscript to generate a construct containing the p.Leu20Pro substitution. HEK cells were grown in tissue culture plates as per standard conditions and transfected with either the WT or mutant hGLDN constructs using Lipofectamine 3000. After 48 hours the live cells were incubated for 2 hours with a c-myc (dylight 550) monoclonal antibody (Myc.A7, ThermoFisher Scientific) to label GLDN at the cell surface. Cells were washed 3 times for 5 minutes with PBS to remove unbound antibody and then fixed with 2% paraformaldehyde for 5 minutes. Cells were washed again 3x for 5 minutes in PBS and then blocked and permeabilised with 10% FCS and 0.1% saponin in PBS for 30 minutes. Cells were then incubated with a c-myc FITC-labeled monoclonal antibody (FITCR953–25, ThermoFisher Scientific) to label total GLDN and then washed in PBS containing Hoechst to label nuclei. Imaging was performed on a Nikon inverted microscope.
RESULTS AND DISCUSSION
Diagnostic yield
Through diagnostic and/or research testing (including bespoke targeted neurogenetic disease gene panels and whole exome sequencing) we investigated the genetics of a cohort of 190 Australasian cases with a primary diagnosis of arthrogryposis, fetal akinesia or multiple pterygium syndrome. A genetic diagnosis was obtained in 81 cases (42.6% diagnostic yield, Table 1, Supplementary Table 2). Fifty variants not previously associated with disease were identified (Supplementary Table 2).
Table 1:
Causative genes identified within the 81 genetically diagnosed probands
| Gene | Cases (n) | Gene | Cases (n) |
|---|---|---|---|
| TTN | 8 | CACNA1S | 1 |
| TNNI2 | 7 | CHANB1 | 1 |
| BICD2 | 6 | FBN2 | 1 |
| NEB | 5 | FLNC1 | 1 |
| CHRNG | 4 | GBE1 | 1 |
| COL6A1 | 4 | GLDN* | 1 |
| MYH3 | 4 | KLHL40 | 1 |
| ECEL1 | 3 | MYL1* | 1 |
| MAGEL2 | 3 | MYO18B | 1 |
| PIEZO2 | 3 | NUP88* | 1 |
| TPM2 | 3 | RAPSN | 1 |
| TRPV4 | 3 | RYR1 | 1 |
| ZC4H2 | 3 | SMPD4* | 1 |
| ACTA1 | 2 | STAC3 | 1 |
| DYNC1H1 | 2 | TNNT3 | 1 |
| GMPPB | 2 | ||
| NALCN | 2 | ||
| SCN4A | 2 |
Genes in bold denote genes that were added into v2 or v3 of the panel. Genes tagged with an “*” represent then novel human disease genes identified by WES (i.e. not on v1–3 of the panel)
TTN was the most frequently identified causative gene (n=8). Of the eight TTN cases, two showed striking amyoplasia – a feature which has previously been described in congenital titinopathy.14 Five patients harboured variants within the triplicate repeat region of TTN, which is frequently poorly sequenced on exome sequencing.29 The most frequently observed pathogenic variant within the cohort is the common 1 bp duplication in CHRNG (rs774279192)30 which was present on five alleles (allele frequency 0.0132). The allele frequency of this variant in gnomAD is 0.000343 (97 of 282,834 alleles).
Recently described genes represent a substantial proportion of diagnoses
Of the 81 cases, 26 harboured variants in genes which were added in v2 and v3 iterations of the targeted gene panel (BICD2, ECEL1, MAGEL2, MYO18B, NALCN, PIEZO2, STAC3, ZC4H2), or within genes not then or only recently associated with disease (GLDN, MYL1, NUP88, SMPD4), i.e. none were on v3 gene panel. Thus, within our cohort, the more recently identified genes for arthrogryposis and fetal akinesia made up a substantial proportion of the genetic diagnoses (32.1%, Table 1). To illustrate this point, the diagnostic yield has increased with each iteration of the comprehensive panel (v1 = 30.3%, v2 = 37.8% and v3 = 44.0%). The improved diagnostic yield in v3 of the panel is also in part due to improved coverage and uniformity of coverage thus facilitating calling of variants within the repetitive regions of NEB and TTN and the improved ability to detect CNVs due to greater and more even coverage. Three variants in NEB and four in TTN occurred within the repetitive regions which are typically refractory to mapping and sequencing with exome sequencing.
Genotype-phenotype expansions
BICD2:
Interestingly, six cases harboured de novo variants in BICD2. This includes three novel variants not previously reported in the literature or in ClinVar (c.628C>A, p.His201Asn; c.1559T>C, p.Leu520Pro; c.2113G>A, pGlu705Lys). One patient with a de novo p.His210Asn substitution presented with arthrogryposis multiplex congenita, as did cases with the recurrent de novo p.Arg694Cys substitution.31,32 A series of papers originally described heterozygous BICD2 variants in families with an SMA-LED phenotype;33–35 BICD2-opathies now account for cases with lethal arthrogryposis through to asymptomatic individuals with mild subclinical features (i.e. myopathic MRI).32 Over a similar period, the PathWest Diagnostic Genomics service has performed sequencing using v2 of the panel (which includes BICD2) on >1,100 neuromuscular disease patients. BICD2 variants were not identified by the PathWest Diagnostic Genomics service in any other neuromuscular disease groups. Thus, at least in the Australasian setting BICD2 variants seem to cause arthrogryposis more frequently than the SMA-LED phenotype for which BICD2 variants were first described.
CACNA1S:
In a Caucasian family (Figure 1A) that presented with recurrent fetal akinesia, panel sequencing identified two missense VUS in the CACNA1S gene (c.665T>A, p.Met222Lys and c.2365C>T, p.Arg789Cys) in the proband (II:4). Sanger sequencing confirmed the variants and showed that the c.665T>A variant was paternally inherited and the c.2365C>T variant was maternally inherited. The affected sibling (II:1) was compound heterozygous for these variants, while both healthy siblings carried only the maternal variant. Both variants were predicted to be disease-causing by MutationAssessor, MutationTaster, PolyPhen-2, PROVEAN, and SIFT. MuPro predicted that both variants reduce protein stability. Both substitutions alter highly conserved amino acids (p.Met222 to D. rerio and p.Arg789 to C. elegans, Figure 1B) and return high CADD scores (p.Met222Lys = 27.1, p.Arg789Cys = 28.9). The p.Met222Lys and p.Arg789Cys substitutions occur within an ion transport domain and an intracellular loop domain of CACNA1S, respectively. The c.665T>A variant is absent from gnomAD and c.2365C>T is present on only 2 of 157,372 alleles. Heterozygous dominantly acting variants in the CACNA1S gene cause malignant hyperthermia, hypokalemic periodic paralysis and thyrotoxic periodic paralysis (OMIM 114208). More recently, Schartner et al. identified dominant and recessive CACNA1S variants associated with congenital myopathy.9 Ophthalmoplegia, ptosis and high-arched palate were common within this cohort. On muscle biopsy, central nuclei, cores and myofibre size variation were observed. Antenatal onset was detected in three of the seven families based on decreased fetal movements on ultrasound.9 Affected individuals in two of these families had bi-allelic null alleles, and the third had a single de novo missense variant.(9) Hunter et al. described an isolated case with bi-allelic CACNA1S variants and a congenital myopathy with ophthalmoplegia.36 The proband (II:4) in our family was a fetus in which polyhydramnios, scalp oedema, wrist contractures and talipes were detected on ultrasound. Fetal movements were reported to be reduced by the mother and the pregnancy was terminated at 26 weeks gestation (wg). Mild facial dysmorphic features were noted on autopsy, including low anterior hairline, mild hypertelorism, and moderate retrognathia. Muscle biopsy did not detect atrophy or myofibre disorganisation. A previously affected sibling (II:1) was delivered by emergency Caesarean section at week 32/40 due to placental abruption and died at 10 days of age. The pregnancy was complicated by unexplained polyhydramnios. In retrospect, reduced fetal movements (II:1) were noted compared subsequent pregnancies (II:2/II:3). Subjectively, this baby had ptosis (based on photographic review) and a broad nasal tip (II:1). Unfortunately, cell lines and/or muscle biopsy material was not available for either case for follow-up of the identified class 3 CACNA1S variants.
Figure 1: Bi-allelic class 3 variants in CACNA1S and SCN4A associated with fetal akinesia.
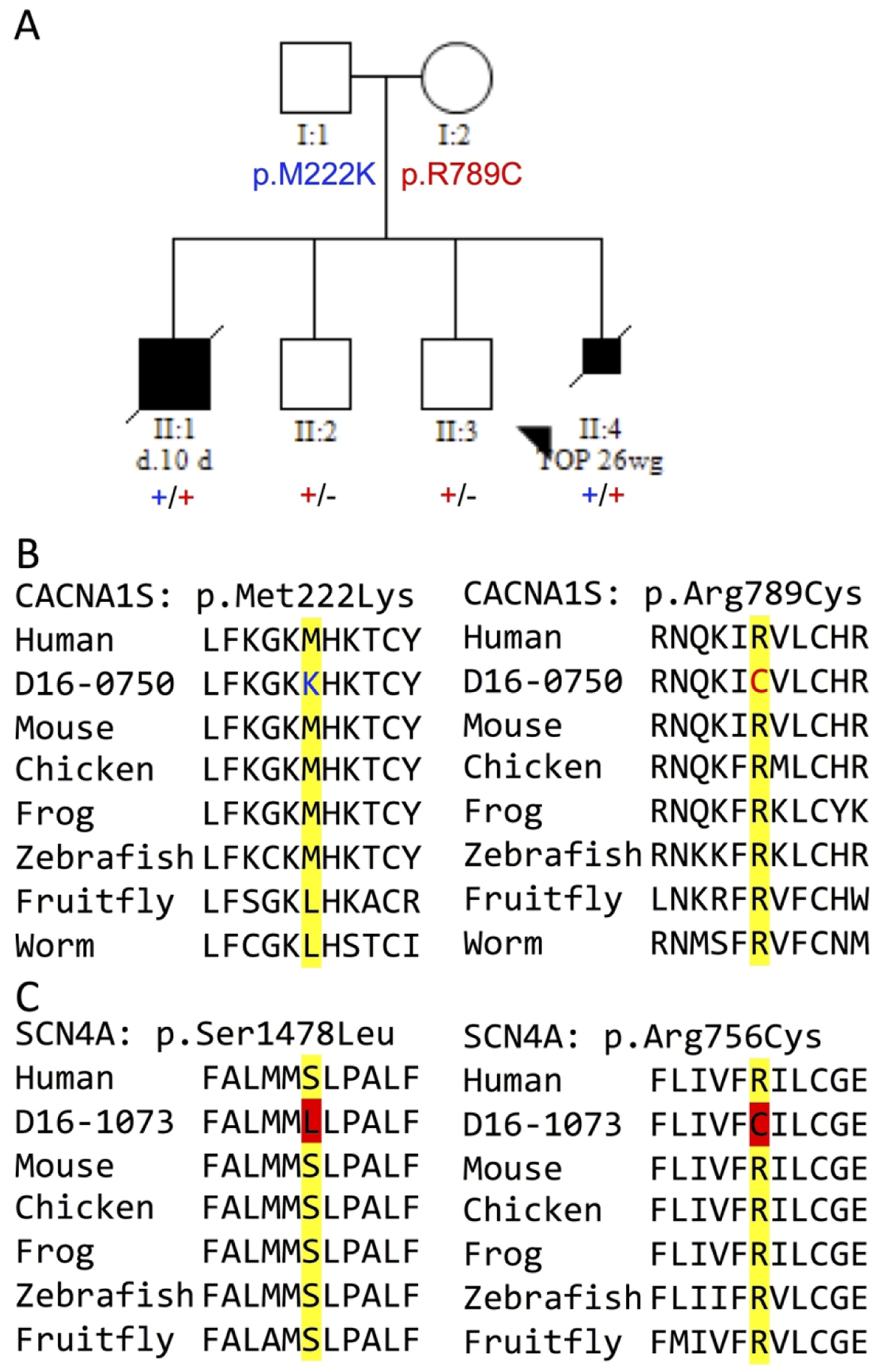
(A) A pedigree co-segregating bi-allelic missense variants in CACNA1S with fetal akinesia and (B) conservation across species at the observed substitutions: p.Met222 and p.Arg789. (C) Evolutionarily conservation at two SCN4A residues substituted, bi-allelically, in another case of fetal akinesia. Importantly, previous work substituting p.Ser1478 to cysteine showed enhanced inactivation of the Na+ channel. Abbreviations: d. 10 d = died at 10 days of age, TOP 26wg = termination of pregnancy at 26 weeks gestation.
CHRNB1:
In a non-consanguineous family of mixed European ancestry (Figure 2A) with recurrent lethal multiple pterygium syndrome, a novel homozygous deletion of CHRNB1 exon 8 was identified by WES and gCNV at the Broad Center for Mendelian Genomics (Figure 2B). Both parents were shown to be carriers. This deletion was also present in the NSES data; however it was not called due to variable coverage across neighbouring exons. The deletion was confirmed by MLPA. CHRNB1 variants are a rare cause of autosomal dominant congenital myasthenic syndrome (OMIM 616313) and have been identified to cause recessive CMS in one family (OMIM 616314). Affected individuals in this published recessive family harboured compound heterozygous variants: a 9bp in-frame deletion and skipping of exon 8 on the other allele.37 Skipping of exon 8 resulted in almost complete loss of pentameric AChR expression. The in-frame deletion resulted in ~70% reduced acetylcholine receptor (AChR) expression.37 In 2016, a Danish group published the identification of a homozygous 1 bp deletion in exon 1 of Chrnb1 in Red dairy cattle with arthrogryposis multiplex congenita.38 Three related stillborn calves showed severe generalised contractures of the joints of the spine and limbs. In patients with CHRNB1-related CMS there is some residual AChR expression, however in the calves, complete loss of CHRNB1 resulted in a lethal presentation. These data support the severe phenotype in our family with homozygous exon 8 deletion.
Figure 2: Homozygous deletion within CHRNB1 causes lethal multiple pterygia syndrome.

(A) A nonconsanguineous family with recurrent lethal multiple pterygia syndrome; IUFD: in utero fetal demise and TOP: termination of pregnancy . (B) Visualisation of the CHRNB1 exon 8 deletion in the exome sequencing data from the proband (blue trace) and both parents (purple traces). The grey lines represent other samples in the same batch of CNV calling and show the average amount of noise across the region. The y-axis denotes the copy number as inferred by gCNV, and the x-axis shows the position along the gene in kilobases. The dots on the plot represent a probe for exon capture, which roughly represent exons. The copy number estimation is expected to hover around 2.0 for autosomes. For the X chromosome, the copy number estimation should hover around 2.0 for females and 1.0 for males.
FLNC:
A proband (Figure 3) presented at birth with hip dislocation, clenched hands, adducted thumbs, small mouth and high palate, and posteriorly rotated ears. On examination, she had mild arthrogryposis, reduced shoulder movement, elbow dimples and scoliosis. She remained undiagnosed after testing on a commercial arthrogryposis gene panel (143 genes). Trio whole exome sequencing identified a missense variant in FLNC (c.3557C>T, p. Ala1186Val). FLNC variants are associated with myofibrillar myopathy and distal myopathy39 and increasingly with cardiomyopathies. Recently, Kiselev et al. described a series of four cases with early-onset restrictive cardiomyopathy (RCM) and congenital myopathy.40 Three of these cases harboured the same de novo variant identified in our family and two of these also presented with arthrogryposis at birth. The RCM presented between 6 months and 15 years of age with the median onset of ~2 years of age.40 Follow-up echocardiogram at 5 years of age in our proband showed a mildly dilatated left atrium, with otherwise normal heart structure and function. Cardiac surveillance in these cases is important and this work highlights the importance of an accurate genetic diagnosis for optimal clinical care.
Figure 3: Clinical presentation in a case with mild distal arthrogryposis due to a de novo missense variant in FLNC.

Images A and B were taken at age 8 months and image C was taken at age 2 months. The images demonstrate reduced elbow extension with dimples, excessive ankle hypermobility, and subtle facial findings including plagiocephaly and micrognathia.
GMPPB:
In a case from the NICU, that was born after a history of reduced fetal movements, known biallelic GMPPB variants were identified (c.220C>T, p.Arg74* and c.1081G>A, p.Asp361Asn). In another family, 18 week gestation ultrasound detected hydrops, pterygia and talipes; the pregnancy was terminated at 20 wg. Post mortem reported facial dysmorphism with wide-set eyes, bilaterally low-set ears, retrognathia and a wide mouth. There was a cleft of the soft and hard palate. Multiple pterygia were noted, including the elbows, shoulders, knees and hips; there was also severe bilateral talipes. Palmar and finger creases were absent. There was thin muscle in all limbs and the psoas appeared similarly. CNS examination showed an overall small brain with a small hypoplastic cerebellum and suggestion of delayed sulcation. There was ambiguous genitalia. Babygram was normal. CGH array revealed a normal female karyotype. Bi-allelic missense variants were identified in GMPPB (c.95C>T, p.Pro32Leu and c.1069G>A, p.Val357Ile). Recessive variants in GMPPB were initially described in patients with congenital and limb-girdle muscular dystrophies.41 Bi-allelic GMPPB variants have since been described in patients with diverse phenotypes including congenital myasthenic syndrome and isolated episodic rhabdomyolysis.20,42 Our cases represent a substantial phenotypic expansion of GMPPB disease.
SCN4A:
Two families within this cohort harboured bi-allelic variants in SCN4A. We previously described one of these families, that presented with recurrent lethal amyoplasia, in an autosomal recessive SCN4A cohort paper.12 In the second family, bi-allelic novel missense variants were identified (c.2266C>T, p.Arg756Cys and c.4433C>T, p.Ser1478Leu) in a fetus with distal arthrogryposis. This case had contractures of all limbs and polyhydramnios. The pregnancy was terminated at 22 wg. Both substitutions alter highly conserved amino acids (conserved to D. melanogaster, Figure 1C) and are predicted to be deleterious by SIFT and probably-damaging by PolyPhen-2; CADD scores of 32 (p.R756C) and 27.4 (p.S1478L). The p.Ser1478 residue lies within the S4–5 loop of the fourth domain which is involved in channel inactivation. Previous work showed that substitution of p.Ser1478 to cysteine enhanced channel in-activation, i.e. partial loss of function.43 It would be anticipated that the p.Ser1478Leu substitution would also generate a channel with partial loss-of-function. Based on this, these variants are highly suspicious but remain Class 3 variants under ACMG guidelines; functional validation of these variants is required to substantiate causality. More recently, SCN4A variants were postulated to cause sudden infant death syndrome.44
STAC3:
In a consanguineous family (Figure 4A) with a history of recurrent fetal akinesia and limb contractures detected on first-trimester ultrasound, we identified a homozygous missense variant in STAC3 (exon 3, c.312T>G, p.Asp104Glu) in the proband (II:3). Both parents were found to be carriers, and sequencing of DNA from formalin-fixed paraffin-embedded tissue of the first affected fetus (II:1) showed this case was also homozygous for the STAC3 variant. This variant is absent from gnomAD and affects a highly conserved amino acid. Splicing predictors in Alamut suggested the variant introduced a cryptic donor splice-site. To investigate this possibility, we generated minigene constructs containing the normal exon 3 or the variant exon 3. Studies of RNA (produced in HEK cells) showed that the variant resulted in skipping of the last 22 nucleotides of exon 3 from the mRNA (Figure 4B-C). The consequence of this variant is then (c.313_334del, p.Asp104Glufs*73) which is likely to lead to a loss of function. A homozygous missense variant (p.Trp284Ser) in STAC3 was originally identified as the cause of Native American myopathy.45 This variant has since been identified in other populations. Zaharieva et al. identified 18 patients from 12 families presenting with a congenital myopathy with dysmorphic features and susceptibility to malignant hyperthermia.46 Seventeen cases were homozygous for the p.Trp284Ser variant, and another proband was compound heterozygous for this variant and an essential splice site variant (c.997–1G>T). Functional analysis has shown reduced sarcoplasmic reticulum Ca2+ release in response to KCl depolarisation in patient myotubes.(46) Affected cases from our family are likely STAC3 nulls and thus the very early and severe presentation in our cases fit with the genotype. In support of this, Stac3 null (Stac3-/-) mice are born at near Mendelian ratios but are all found dead at birth, and show curved bodies and dropped forelimbs.47 When dissected from the uterus, Stac3-/- fetuses did not move or respond to touch but did have a heartbeat. Histologically, muscle from Stac3-/- mice showed central nuclei, reduced myofibril number and size and deranged sarcomeres.47 STAC3 appears to be critical to normal muscle development and function. Thus, bi-allelic loss-of-function variants in genes critical to excitation-contraction coupling (CACNA1S, SCN4A, STAC3, RYR1)5,9,11,12,48 cause lethal early-onset disease.
Figure 4: Homozygous variant in STAC3.

(A) Pedigree showing segregation of a homozygous variant in STAC3. (B) 2% agarose gel of RT-PCR products from the STAC3 minigene assay. WT – normal exon 3 of STAC3, MUT – exon 3 containing the c.312T>G variant. +P or -P indicates the cells were grown in the presence (+) or absence (-) of puromycin (to inhibit potential nonsense-mediated decay). NTC = no template control. The red arrowhead indicates the smaller product in the samples containing the variant. Sanger sequencing of the RT-PCR product shows that this smaller product corresponds to loss of the 22 nucleotides following the variant in the cDNA (C).
Identification of variants in novel human disease genes
Four cases harboured variants in disease genes that had not been described at the time of whole exome sequencing (GLDN,28 MYL1,16 NUP8815 and SMPD449).
The identification of a proband within this cohort with a homozygous MYL1 missense variant, and another with compound heterozygous variants in NUP88, were recently described in separate publications, which also combined additional cases from other research centres with bi-allelic variants in these genes.15,16
GLDN:
In a family from Hong Kong (Figure 5A) with a history of recurrent fetal akinesia, we identified two VUS in the gliomedin gene (GLDN). The first affected baby was born at 36/40 and died at 1 day of age. She had hypoplastic heart and lungs and a high-arched palate. In the second pregnancy, the fetus presented at 18/40 with decreased fetal movements and the pregnancy was terminated at 22 wg. In the third pregnancy, reduced fetal movements were reported at 20/40 and the pregnancy was terminated at 22 wg. This case had fetal hydrops, multiple joint contractures, pterygia and increased muscle bulk. All three affected cases harboured bi-allelic GLDN variants: a maternally inherited missense variant, c.59C>T [p.Leu20Pro], and a paternally-inherited essential splice site (ESS) change, c.363+1G>A (Figure 5A). Analysis of cDNA derived from muscle from the second case indicated that the missense variant was homozygous at the transcript level, suggesting loss of expression from the allele harbouring the ESS change (Figure 5B). Using the myc-GLDN reporter assay defined in Maluenda et al.28 we showed that, unlike WT human GLDN (hGLDN), gliomedin containing the p.Leu20Pro substitution fails to localise at the surface membrane of HEK cells (Figure 5C). In comparison, Maluenda et al. found that gliomedin mutants failed to localise at the cell surface and also did not bind its axonal partner neurofascin-186. Thus, the GLDN variants identified in our family are likely functional nulls.
Figure 5: Novel GLDN variants identified in a family with recurrent fetal akinesia.

(A) Pedigree showing segregation of bi-allelic VUS in GLDN. (B) Sanger sequencing showed that the heterozygous c.59T>C on gDNA (in A) appeared homozygous in muscle cDNA, suggesting loss of expression from the allele containing the essential-splice site change. (C) Detection of hGLDN and mutant hGLDN fused to an extracellular myc-tag in HEK cells in culture (red) and post-fixation and permeabilization (green). Nuclei are stained with Hoechst.
SMPD4:
A consanguineous family, from Melbourne, presented with recurrent arthrogryposis multiplex congenita and complex brain malformations (Figure 6A). All cases presented with AMC, were small for gestation age and displayed hypoplasia of the corpus callosum (Figure 6B). Additional features present in two of the three cases included: congenital encephalopathy and microcephaly, cerebellar malformation and hypoplasia (Figure 6B) and hypomyelination (Figure 6C). We performed panel sequencing on the proband (II:3) with the v3 muscle and v3 neuro panels but did not identify any causative variants. We subsequently performed trio whole exome sequencing and identified a novel homozygous missense variant in the SMPD4 gene (c.575C>T, p.Pro192Leu) in the proband. Each parent was a carrier. This variant was confirmed by bi-directional Sanger sequencing and we also showed that an affected sibling (II:1) was homozygous for this variant. The SMPD4 variant was also homozygous in a clinical exome (performed elsewhere) on the most recently affected baby (II:4). The variant is predicted to be damaging by MutationTaster, SIFT and PolyPhen-2 and has a CADD score of 22.8. The variant is present on three alleles in gnomAD and alters a highly conserved amino acid (up to D. rerio, Figure 6D). SMPD4 was recently described as a likely disease gene for a syndrome presenting as a skeletal dysplasia with cortical malformations and epilepsy.50 In a follow-up study, 12 families with bi-allelic SMPD4 variants and a phenotype encompassing microcephaly, hypomyelination, cerebellar atrophy, congenital arthrogryposis and fetal/postnatal demise were described.49 SMPD4 encodes the neutral sphingomyelinase-3; sphingomyelinases are important for the properties of cell membranes and the regulation of transmembrane and peripheral membrane proteins, they are also highly enriched in the nervous system. Magini et al.49 showed via over-expression studies that SMPD4 localises to the ER and nuclear envelope. Immunoprecipitation assays revealed that SMPD4 interacts with several nuclear pore proteins; highlighting a role for the nuclear pore in the disease pathogenesis. In support of this, the role of the nuclear pore in human disease is becoming increasingly recognised.51,52
Figure 6: A family with recurrent arthrogryposis and central involvement due to a homozygous missense variant in SMPD4.

(A) Pedigree, (B) T1 midline sagittal image (Individual II:2, neonatal MRI brain scan) showing absence of the genu and rostrum, thinning and elongation of the callosal body. Microcephaly, hypoplasia of the inferior cerebellar vermis and prominent venous sinuses are also evident. (C) T2 axial image (Individual II:2) showing simplified gyration, compensatory ventriculomegaly and absent myelination. (D) Alignment showing evolutionary conservation of the p.Pro192 residue.
Conclusions
The utility of next generation sequencing in arthrogryposis and fetal akinesia is undeniable, yielding a genetic diagnosis in 42% of cases. In many instances, couples have gone on to have IVF and preimplantation genetic diagnosis or prenatal genetic diagnosis of subsequent pregnancies.
Of note is the substantial contribution of relatively recently identified disease genes and the large muscle genes (NEB, RYR1 and TTN) to disease burden. Previously, most cases did not receive a genetic diagnosis. The clinical and genetic heterogeneity, along with the sporadic nature of the disease or small families, meant that genetic testing was difficult or unattainable beyond a handful of known genes or gene hotspots. The number of genes known to cause these diseases and the phenotypic expansions associated with known genes has been quite remarkable. In this report we show that homozygous null variants in CHRNB1 or STAC3 and known GMPPB variants cause lethal fetal akinesia, expanding the phenotypes associated with each of these genes.
Moving forward, it is critically important that interdisciplinary teams discuss interpretation of VUS in candidate genes and perform functional genomics to reclassify VUS as likely pathogenic or benign. Alternative splicing appears to be a particularly important mechanism underlying protein regulation in skeletal muscle.53 Thus, splicing defects represent a substantial contribution to muscle disease burden.29,54
Submission of variants into well-curated gene databases (e.g. the LOVD) is also critical to the reclassification of VUS and mapping of known phenotype-genotype associations. Identification of the same rare VUS in two unrelated patients with a similar clinical presentation will facilitate reclassification as likely-pathogenic. By way of illustration, we identified in the research laboratory a then novel, de novo missense variant in BICD2 in a patient with arthrogryposis multiplex congenita. The diagnostic laboratory within PathWest had a similar case with the same BICD2 variant. Follow-up revealed that the variant in this diagnostic case had also arisen de novo.31 In the words of Johan den Dunnen: many VUS may be “variants of under sharing” (International Congress of the World Muscle Society 2019, Copenhagen).
As the affordability of and availability of massively parallel sequencing has improved, variant interpretation has become the new bottleneck in accurate genetic diagnosis. Scalable, relatively robust and affordable assays (e.g. the cell-surface localisation assay for gliomedin) that can be utilised to assay VUS in multiple genes should be an ideal that the rare diseases community work towards. Saturation mutagenesis together with appropriate functional read-outs55 have already been performed for some common disease genes including BRCA1 and PTEN.56,57 A historical mutagenesis study of a small region of the SCN4A protein to cysteines informed our interpretation of the likely pathogenicity of a Class 3 variant in this gene,43 and highlights the clinical utility of such experimental work.
Based on studies in model organisms,58,59 it is likely that many more fetal akinesia and arthrogryposis genes await discovery. Exome and genome sequencing, along with RNAseq in genetically unresolved cases, will likely identify additional novel disease genes.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the patients and their families for participating in this study. This work was supported by the Australian National Health and Medical Research Council (NHMRC) Fellowships APP1122952 and APP1117510 to GR and NGL, NHMRC project grant APP1080587 to GR and NGL, the Association Francaise contre les Myopathies (18724) to GR. Sequencing and analysis were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 and in part by National Human Genome Research Institute grant R01 HG009141.
Footnotes
ADDITIONAL RESOURCES
Benchling: https://www.benchling.com/
CADD: https://bio.tools/CADD_Phredh
GEMINI: https://github.com/arq5x/gemini
gnomAD: https://gnomad.broadinstitute.org/
MutationAssessor: http://mutationassessor.org/r3/
MuPro: https://omictools.com/mupro-tool
MutationTaster: http://mutationtaster.org/
PolyPhen-2: http://genetics.bwh.harvard.edu/pph2/
PROVEAN: http://provean.jcvi.org/index.php
COMPETING INTERESTS
The authors do not have any competing interests to declare.
REFERENCES
- 1.Hall JG (2009) Pena-Shokeir phenotype (fetal akinesia deformation sequence) revisited. Birth Defects Res A Clin Mol Teratol 85, 677–694 [DOI] [PubMed] [Google Scholar]
- 2.Hall JG (2014) Arthrogryposis (multiple congenital contractures): diagnostic approach to etiology, classification, genetics, and general principles. Eur J Med Genet 57, 464–472 [DOI] [PubMed] [Google Scholar]
- 3.Filges I, and Hall JG (2013) Failure to identify antenatal multiple congenital contractures and fetal akinesia--proposal of guidelines to improve diagnosis. Prenat Diagn 33, 61–74 [DOI] [PubMed] [Google Scholar]
- 4.Ravenscroft G, Sollis E, Charles AK, North KN, Baynam G, and Laing NG (2011) Fetal akinesia: review of the genetics of the neuromuscular causes. J Med Genet 48, 793–801 [DOI] [PubMed] [Google Scholar]
- 5.Todd EJ, Yau KS, Ong R, Slee J, McGillivray G, Barnett CP, Haliloglu G, Talim B, Akcoren Z, Kariminejad A, Cairns A, Clarke NF, Freckmann ML, Romero NB, Williams D, Sewry CA, Colley A, Ryan MM, Kiraly-Borri C, Sivadorai P, Allcock RJ, Beeson D, Maxwell S, Davis MR, Laing NG, and Ravenscroft G (2015) Next generation sequencing in a large cohort of patients presenting with neuromuscular disease before or at birth. Orphanet J Rare Dis 10, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beecroft SJ, Lombard M, Mowat D, McLean C, Cairns A, Davis M, Laing NG, and Ravenscroft G (2018) Genetics of neuromuscular fetal akinesia in the genomics era. J Med Genet 55, 505–514 [DOI] [PubMed] [Google Scholar]
- 7.Pehlivan D, Bayram Y, Gunes N, Coban Akdemir Z, Shukla A, Bierhals T, Tabakci B, Sahin Y, Gezdirici A, Fatih JM, Gulec EY, Yesil G, Punetha J, Ocak Z, Grochowski CM, Karaca E, Albayrak HM, Radhakrishnan P, Erdem HB, Sahin I, Yildirim T, Bayhan IA, Bursali A, Elmas M, Yuksel Z, Ozdemir O, Silan F, Yildiz O, Yesilbas O, Isikay S, Balta B, Gu S, Jhangiani SN, Doddapaneni H, Hu J, Muzny DM, Baylor-Hopkins Center for Mendelian, G., Boerwinkle E, Gibbs RA, Tsiakas K, Hempel M, Girisha KM, Gul D, Posey JE, Elcioglu NH, Tuysuz B, and Lupski JR (2019) The Genomics of Arthrogryposis, a Complex Trait: Candidate Genes and Further Evidence for Oligogenic Inheritance. Am J Hum Genet 105, 132–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boycott KM, Vanstone MR, Bulman DE, and MacKenzie AE (2013) Raredisease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet 14, 681–691 [DOI] [PubMed] [Google Scholar]
- 9.Schartner V, Romero NB, Donkervoort S, Treves S, Munot P, Pierson TM, Dabaj I, Malfatti E, Zaharieva IT, Zorzato F, Abath Neto O, Brochier G, Lornage X, Eymard B, Taratuto AL, Bohm J, Gonorazky H, Ramos-Platt L, Feng L, Phadke R, Bharucha-Goebel DX, Sumner CJ, Bui MT, Lacene E, Beuvin M, Labasse C, Dondaine N, Schneider R, Thompson J, Boland A, Deleuze JF, Matthews E, Pakleza AN, Sewry CA, Biancalana V, Quijano-Roy S, Muntoni F, Fardeau M, Bonnemann CG, and Laporte J (2017) Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol 133, 517–533 [DOI] [PubMed] [Google Scholar]
- 10.Koutsopoulos OS, Kretz C, Weller CM, Roux A, Mojzisova H, Bohm J, Koch C, Toussaint A, Heckel E, Stemkens D, Ter Horst SA, Thibault C, Koch M, Mehdi SQ, Bijlsma EK, Mandel JL, Vermot J, and Laporte J (2013) Dynamin 2 homozygous mutation in humans with a lethal congenital syndrome. Eur J Hum Genet 21, 637–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bharucha-Goebel DX, Santi M, Medne L, Zukosky K, Dastgir J, Shieh PB, Winder T, Tennekoon G, Finkel RS, Dowling JJ, Monnier N, and Bonnemann CG (2013) Severe congenital RYR1-associated myopathy: the expanding clinicopathologic and genetic spectrum. Neurology 80, 1584–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaharieva IT, Thor MG, Oates EC, van Karnebeek C, Hendson G, Blom E, Witting N, Rasmussen M, Gabbett MT, Ravenscroft G, Sframeli M, Suetterlin K, Sarkozy A, D’Argenzio L, Hartley L, Matthews E, Pitt M, Vissing J, Ballegaard M, Krarup C, Slordahl A, Halvorsen H, Ye XC, Zhang LH, Lokken N, Werlauff U, Abdelsayed M, Davis MR, Feng L, Phadke R, Sewry CA, Morgan JE, Laing NG, Vallance H, Ruben P, Hanna MG, Lewis S, Kamsteeg EJ, Mannikko R, and Muntoni F (2016) Loss-of-function mutations in SCN4A cause severe foetal hypokinesia or ‘classical’ congenital myopathy. Brain 139, 674–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kariminejad A, Dahl-Halvarsson M, Ravenscroft G, Afroozan F, Keshavarz E, Goullee H, Davis MR, Faraji Zonooz M, Najmabadi H, Laing NG, and Tajsharghi H (2017) TOR1A variants cause a severe arthrogryposis with developmental delay, strabismus and tremor. Brain 140, 2851–2859 [DOI] [PubMed] [Google Scholar]
- 14.Oates EC, Jones KJ, Donkervoort S, Charlton A, Brammah S, Smith JE 3rd, Ware JS, Yau KS, Swanson LC, Whiffin N, Peduto AJ, Bournazos A, Waddell LB, Farrar MA, Sampaio HA, Teoh HL, Lamont PJ, Mowat D, Fitzsimons RB, Corbett AJ, Ryan MM, O’Grady GL, Sandaradura SA, Ghaoui R, Joshi H, Marshall JL, Nolan MA, Kaur S, Punetha J, Topf A, Harris E, Bakshi M, Genetti CA, Marttila M, Werlauff U, Streichenberger N, Pestronk A, Mazanti I, Pinner JR, Vuillerot C, Grosmann C, Camacho A, Mohassel P, Leach ME, Foley AR, Bharucha-Goebel D, Collins J, Connolly AM, Gilbreath HR, Iannaccone ST, Castro D, Cummings BB, Webster RI, Lazaro L, Vissing J, Coppens S, Deconinck N, Luk HM, Thomas NH, Foulds NC, Illingworth MA, Ellard S, McLean CA, Phadke R, Ravenscroft G, Witting N, Hackman P, Richard I, Cooper ST, Kamsteeg EJ, Hoffman EP, Bushby K, Straub V, Udd B, Ferreiro A, North KN, Clarke NF, Lek M, Beggs AH, Bonnemann CG, MacArthur DG, Granzier H, Davis MR, and Laing NG (2018) Congenital Titinopathy: Comprehensive characterization and pathogenic insights. Ann Neurol 83, 1105–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonnin E, Cabochette P, Filosa A, Juhlen R, Komatsuzaki S, Hezwani M, Dickmanns A, Martinelli V, Vermeersch M, Supply L, Martins N, Pirenne L, Ravenscroft G, Lombard M, Port S, Spillner C, Janssens S, Roets E, Van Dorpe J, Lammens M, Kehlenbach RH, Ficner R, Laing NG, Hoffmann K, Vanhollebeke B, and Fahrenkrog B (2018) Biallelic mutations in nucleoporin NUP88 cause lethal fetal akinesia deformation sequence. PLoS Genet 14, e1007845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ravenscroft G, Zaharieva IT, Bortolotti CA, Lambrughi M, Pignataro M, Borsari M, Sewry CA, Phadke R, Haliloglu G, Ong R, Goullee H, Whyte T, Consortium UK, Manzur A, Talim B, Kaya U, Osborn DPS, Forrest ARR, Laing NG, and Muntoni F (2018) Bi-allelic mutations in MYL1 cause a severe congenital myopathy. Hum Mol Genet 27, 4263–4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beecroft SJ, Yau KS, Allcock RJN, Mina K, Gooding R, Faiz F, Atkinson VJ, Wise C, Sivadorai P, Trajanoski D, Kresoje N, Ong R, Duff RM, Cabrera-Serrano M, Nowak KJ, Pachter N, Ravenscroft G, Lamont PJ, Davis MR, and Laing NG (2020) Targeted gene panel use in 2249 neuromuscular patients: the Australasian referral center experience. Ann Clin Transl Neurol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, and Rehm HL (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paila U, Chapman BA, Kirchner R, and Quinlan AR (2013) GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol 9, e1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabrera-Serrano M, Ghaoui R, Ravenscroft G, Johnsen RD, Davis MR, Corbett A, Reddel S, Sue CM, Liang C, Waddell LB, Kaur S, Lek M, North KN, MacArthur DG, Lamont PJ, Clarke NF, and Laing NG (2015) Expanding the phenotype of GMPPB mutations. Brain 138, 836–844 [DOI] [PubMed] [Google Scholar]
- 21.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, and Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng J, Randall A, and Baldi P (2006) Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 62, 1125–1132 [DOI] [PubMed] [Google Scholar]
- 23.Grimm DG, Azencott CA, Aicheler F, Gieraths U, MacArthur DG, Samocha KE, Cooper DN, Stenson PD, Daly MJ, Smoller JW, Duncan LE, and Borgwardt KM (2015) The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum Mutat 36, 513–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi Y, and Chan AP (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng PC, and Henikoff S (2001) Predicting deleterious amino acid substitutions. Genome Res 11, 863–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaildrat P, Killian A, Martins A, Tournier I, Frebourg T, and Tosi M (2010) Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods Mol Biol 653, 249–257 [DOI] [PubMed] [Google Scholar]
- 27.Gaildrat P, Krieger S, Di Giacomo D, Abdat J, Revillion F, Caputo S, Vaur D, Jamard E, Bohers E, Ledemeney D, Peyrat JP, Houdayer C, Rouleau E, Lidereau R, Frebourg T, Hardouin A, Tosi M, and Martins A (2012) Multiple sequence variants of BRCA2 exon 7 alter splicing regulation. J Med Genet 49, 609–617 [DOI] [PubMed] [Google Scholar]
- 28.Maluenda J, Manso C, Quevarec L, Vivanti A, Marguet F, Gonzales M, Guimiot F, Petit F, Toutain A, Whalen S, Grigorescu R, Coeslier AD, Gut M, Gut I, Laquerriere A, Devaux J, and Melki J (2016) Mutations in GLDN, Encoding Gliomedin, a Critical Component of the Nodes of Ranvier, Are Responsible for Lethal Arthrogryposis. Am J Hum Genet 99, 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, Bolduc V, Waddell LB, Sandaradura SA, O’Grady GL, Estrella E, Reddy HM, Zhao F, Weisburd B, Karczewski KJ, O’Donnell-Luria AH, Birnbaum D, Sarkozy A, Hu Y, Gonorazky H, Claeys K, Joshi H, Bournazos A, Oates EC, Ghaoui R, Davis MR, Laing NG, Topf A, Genotype-Tissue Expression C, Kang PB, Beggs AH, North KN, Straub V, Dowling JJ, Muntoni F, Clarke NF, Cooper ST, Bonnemann CG, and MacArthur DG (2017) Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall JG (2013) Pretibial linear vertical creases or indentations (shin dimples) associated with arthrogryposis. Am J Med Genet A 161A, 737–744 [DOI] [PubMed] [Google Scholar]
- 31.Ravenscroft G, Di Donato N, Hahn G, Davis MR, Craven PD, Poke G, Neas KR, Neuhann TM, Dobyns WB, and Laing NG (2016) Recurrent de novo BICD2 mutation associated with arthrogryposis multiplex congenita and bilateral perisylvian polymicrogyria. Neuromuscul Disord 26, 744–748 [DOI] [PubMed] [Google Scholar]
- 32.Storbeck M, Horsberg Eriksen B, Unger A, Holker I, Aukrust I, Martinez-Carrera LA, Linke WA, Ferbert A, Heller R, Vorgerd M, Houge G, and Wirth B (2017) Phenotypic extremes of BICD2-opathies: from lethal, congenital muscular atrophy with arthrogryposis to asymptomatic with subclinical features. Eur J Hum Genet 25, 1040–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neveling K, Martinez-Carrera LA, Holker I, Heister A, Verrips A, Hosseini-Barkooie SM, Gilissen C, Vermeer S, Pennings M, Meijer R, te Riele M, Frijns CJ, Suchowersky O, MacLaren L, Rudnik-Schoneborn S, Sinke RJ, Zerres K, Lowry RB, Lemmink HH, Garbes L, Veltman JA, Schelhaas HJ, Scheffer H, and Wirth B (2013) Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am J Hum Genet 92, 946–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oates EC, Rossor AM, Hafezparast M, Gonzalez M, Speziani F, MacArthur DG, Lek M, Cottenie E, Scoto M, Foley AR, Hurles M, Houlden H, Greensmith L, Auer-Grumbach M, Pieber TR, Strom TM, Schule R, Herrmann DN, Sowden JE, Acsadi G, Menezes MP, Clarke NF, Zuchner S, Muntoni F, North KN, and Reilly MM (2013) Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am J Hum Genet 92, 965–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peeters K, Litvinenko I, Asselbergh B, Almeida-Souza L, Chamova T, Geuens T, Ydens E, Zimon M, Irobi J, De Vriendt E, De Winter V, Ooms T, Timmerman V, Tournev I, and Jordanova A (2013) Molecular defects in the motor adaptor BICD2 cause proximal spinal muscular atrophy with autosomal-dominant inheritance. Am J Hum Genet 92, 955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hunter JM, Ahearn ME, Balak CD, Liang WS, Kurdoglu A, Corneveaux JJ, Russell M, Huentelman MJ, Craig DW, Carpten J, Coons SW, DeMello DE, Hall JG, Bernes SM, and Baumbach-Reardon L (2015) Novel pathogenic variants and genes for myopathies identified by whole exome sequencing. Mol Genet Genomic Med 3, 283–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quiram PA, Ohno K, Milone M, Patterson MC, Pruitt NJ, Brengman JM, Sine SM, and Engel AG (1999) Mutation causing congenital myasthenia reveals acetylcholine receptor beta/delta subunit interaction essential for assembly. J Clin Invest 104, 1403–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agerholm JS, McEvoy FJ, Menzi F, Jagannathan V, and Drogemuller C (2016) A CHRNB1 frameshift mutation is associated with familial arthrogryposis multiplex congenita in Red dairy cattle. BMC Genomics 17, 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duff RM, Tay V, Hackman P, Ravenscroft G, McLean C, Kennedy P, Steinbach A, Schoffler W, van der Ven PF, Furst DO, Song J, Djinovic-Carugo K, Penttila S, Raheem O, Reardon K, Malandrini A, Gambelli S, Villanova M, Nowak KJ, Williams DR, Landers JE, Brown RH Jr., Udd B, and Laing NG (2011) Mutations in the N-terminal actin-binding domain of filamin C cause a distal myopathy. Am J Hum Genet 88, 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kiselev A, Vaz R, Knyazeva A, Khudiakov A, Tarnovskaya S, Liu J, Sergushichev A, Kazakov S, Frishman D, Smolina N, Pervunina T, Jorholt J, Sjoberg G, Vershinina T, Rudenko D, Arner A, Sejersen T, Lindstrand A, and Kostareva A (2018) De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy. Hum Mutat 39, 1161–1172 [DOI] [PubMed] [Google Scholar]
- 41.Carss KJ, Stevens E, Foley AR, Cirak S, Riemersma M, Torelli S, Hoischen A, Willer T, van Scherpenzeel M, Moore SA, Messina S, Bertini E, Bonnemann CG, Abdenur JE, Grosmann CM, Kesari A, Punetha J, Quinlivan R, Waddell LB, Young HK, Wraige E, Yau S, Brodd L, Feng L, Sewry C, MacArthur DG, North KN, Hoffman E, Stemple DL, Hurles ME, van Bokhoven H, Campbell KP, Lefeber DJ, Lin YY, and Muntoni F (2013) Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of alpha-dystroglycan. Am J Hum Genet 93, 29–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belaya K, Rodriguez Cruz PM, Liu WW, Maxwell S, McGowan S, Farrugia ME, Petty R, Walls TJ, Sedghi M, Basiri K, Yue WW, Sarkozy A, Bertoli M, Pitt M, Kennett R, Schaefer A, Bushby K, Parton M, Lochmuller H, Palace J, Muntoni F, and Beeson D (2015) Mutations in GMPPB cause congenital myasthenic syndrome and bridge myasthenic disorders with dystroglycanopathies. Brain 138, 2493–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lerche H, Peter W, Fleischhauer R, Pika-Hartlaub U, Malina T, Mitrovic N, and Lehmann-Horn F (1997) Role in fast inactivation of the IV/S4-S5 loop of the human muscle Na+ channel probed by cysteine mutagenesis. J Physiol 505 ( Pt 2), 345–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mannikko R, Wong L, Tester DJ, Thor MG, Sud R, Kullmann DM, Sweeney MG, Leu C, Sisodiya SM, FitzPatrick DR, Evans MJ, Jeffrey IJM, Tfelt-Hansen J, Cohen MC, Fleming PJ, Jaye A, Simpson MA, Ackerman MJ, Hanna MG, Behr ER, and Matthews E (2018) Dysfunction of NaV1.4, a skeletal muscle voltage-gated sodium channel, in sudden infant death syndrome: a case-control study. Lancet 391, 1483–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horstick EJ, Linsley JW, Dowling JJ, Hauser MA, McDonald KK, Ashley-Koch A, Saint-Amant L, Satish A, Cui WW, Zhou W, Sprague SM, Stamm DS, Powell CM, Speer MC, Franzini-Armstrong C, Hirata H, and Kuwada JY (2013) Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nat Commun 4, 1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaharieva IT, Sarkozy A, Munot P, Manzur A, O’Grady G, Rendu J, Malfatti E, Amthor H, Servais L, Urtizberea JA, Neto OA, Zanoteli E, Donkervoort S, Taylor J, Dixon J, Poke G, Foley AR, Holmes C, Williams G, Holder M, Yum S, Medne L, Quijano-Roy S, Romero NB, Faure J, Feng L, Bastaki L, Davis MR, Phadke R, Sewry CA, Bonnemann CG, Jungbluth H, Bachmann C, Treves S, and Muntoni F (2018) STAC3 variants cause a congenital myopathy with distinctive dysmorphic features and malignant hyperthermia susceptibility. Hum Mutat 39, 1980–1994 [DOI] [PubMed] [Google Scholar]
- 47.Reinholt BM, Ge X, Cong X, Gerrard DE, and Jiang H (2013) Stac3 is a novel regulator of skeletal muscle development in mice. PLoS One 8, e62760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amburgey K, Bailey A, Hwang JH, Tarnopolsky MA, Bonnemann CG, Medne L, Mathews KD, Collins J, Daube JR, Wellman GP, Callaghan B, Clarke NF, and Dowling JJ (2013) Genotype-phenotype correlations in recessive RYR1-related myopathies. Orphanet J Rare Dis 8, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Magini P, Smits DJ, Vandervore L, Schot R, Columbaro M, Kasteleijn E, van der Ent M, Palombo F, Lequin MH, Dremmen M, de Wit MCY, Severino M, Divizia MT, Striano P, Ordonez-Herrera N, Alhashem A, Al Fares A, Al Ghamdi M, Rolfs A, Bauer P, Demmers J, Verheijen FW, Wilke M, van Slegtenhorst M, van der Spek PJ, Seri M, Jansen AC, Stottmann RW, Hufnagel RB, Hopkin RJ, Aljeaid D, Wiszniewski W, Gawlinski P, Laure-Kamionowska M, Alkuraya FS, Akleh H, Stanley V, Musaev D, Gleeson JG, Zaki MS, Brunetti-Pierri N, Cappuccio G, Davidov B, Basel-Salmon L, Bazak L, Shahar NR, Bertoli-Avella A, Mirzaa GM, Dobyns WB, Pippucci T, Fornerod M, and Mancini GMS (2019) Loss of SMPD4 Causes a Developmental Disorder Characterized by Microcephaly and Congenital Arthrogryposis. Am J Hum Genet 105, 689–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, Alowain M, Alzaidan H, Alsayed M, Subhani S, Cupler E, Faden M, Alhashem A, Qari A, Chedrawi A, Aldhalaan H, Kurdi W, Khan S, Rahbeeni Z, Alotaibi M, Goljan E, Elbardisy H, ElKalioby M, Shah Z, Alruwaili H, Jaafar A, Albar R, Akilan A, Tayeb H, Tahir A, Fawzy M, Nasr M, Makki S, Alfaifi A, Akleh H, Yamani S, Bubshait D, Mahnashi M, Basha T, Alsagheir A, Abu Khaled M, Alsaleem K, Almugbel M, Badawi M, Bashiri F, Bohlega S, Sulaiman R, Tous E, Ahmed S, Algoufi T, Al-Mousa H, Alaki E, Alhumaidi S, Alghamdi H, Alghamdi M, Sahly A, Nahrir S, Al-Ahmari A, Alkuraya H, Almehaidib A, Abanemai M, Alsohaibaini F, Alsaud B, Arnaout R, Abdel-Salam GMH, Aldhekri H, AlKhater S, Alqadi K, Alsabban E, Alshareef T, Awartani K, Banjar H, Alsahan N, Abosoudah I, Alashwal A, Aldekhail W, Alhajjar S, Al-Mayouf S, Alsemari A, Alshuaibi W, Altala S, Altalhi A, Baz S, Hamad M, Abalkhail T, Alenazi B, Alkaff A, Almohareb F, Al Mutairi F, Alsaleh M, Alsonbul A, Alzelaye S, Bahzad S, Manee AB, Jarrad O, Meriki N, Albeirouti B, Alqasmi A, AlBalwi M, Makhseed N, Hassan S, Salih I, Salih MA, Shaheen M, Sermin S, Shahrukh S, Hashmi S, Shawli A, Tajuddin A, Tamim A, Alnahari A, Ghemlas I, Hussein M, Wali S, Murad H, Meyer BF, and Alkuraya FS (2019) Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population. Am J Hum Genet 104, 1182–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Juhlen R, and Fahrenkrog B (2018) Moonlighting nuclear pore proteins: tissue-specific nucleoporin function in health and disease. Histochem Cell Biol 150, 593–605 [DOI] [PubMed] [Google Scholar]
- 52.Fichtman B, Harel T, Biran N, Zagairy F, Applegate CD, Salzberg Y, Gilboa T, Salah S, Shaag A, Simanovsky N, Ayoubieh H, Sobreira N, Punzi G, Pierri CL, Hamosh A, Elpeleg O, Harel A, and Edvardson S (2019) Pathogenic Variants in NUP214 Cause “Plugged” Nuclear Pore Channels and Acute Febrile Encephalopathy. Am J Hum Genet 105, 48–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakka K, Ghigna C, Gabellini D, and Dilworth FJ (2018) Diversification of the muscle proteome through alternative splicing. Skelet Muscle 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gonorazky HD, Naumenko S, Ramani AK, Nelakuditi V, Mashouri P, Wang P, Kao D, Ohri K, Viththiyapaskaran S, Tarnopolsky MA, Mathews KD, Moore SA, Osorio AN, Villanova D, Kemaladewi DU, Cohn RD, Brudno M, and Dowling JJ (2019) Expanding the Boundaries of RNA Sequencing as a Diagnostic Tool for Rare Mendelian Disease. Am J Hum Genet 104, 466–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gasperini M, Starita L, and Shendure J (2016) The power of multiplexed functional analysis of genetic variants. Nat Protoc 11, 1782–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mighell TL, Evans-Dutson S, and O’Roak BJ (2018) A Saturation Mutagenesis Approach to Understanding PTEN Lipid Phosphatase Activity and Genotype-Phenotype Relationships. Am J Hum Genet 102, 943–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, and Shendure J (2018) Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, Meehan TF, Weninger WJ, Westerberg H, Adissu H, Baker CN, Bower L, Brown JM, Caddle LB, Chiani F, Clary D, Cleak J, Daly MJ, Denegre JM, Doe B, Dolan ME, Edie SM, Fuchs H, Gailus-Durner V, Galli A, Gambadoro A, Gallegos J, Guo S, Horner NR, Hsu CW, Johnson SJ, Kalaga S, Keith LC, Lanoue L, Lawson TN, Lek M, Mark M, Marschall S, Mason J, McElwee ML, Newbigging S, Nutter LM, Peterson KA, Ramirez-Solis R, Rowland DJ, Ryder E, Samocha KE, Seavitt JR, Selloum M, Szoke-Kovacs Z, Tamura M, Trainor AG, Tudose I, Wakana S, Warren J, Wendling O, West DB, Wong L, Yoshiki A, International Mouse Phenotyping, C., Jackson L, Infrastructure Nationale Phenomin, I. C. d. l. S., Charles River L, Harwell MRC, Toronto Centre for, P., Wellcome Trust Sanger, I., Center, R. B., MacArthur DG, Tocchini-Valentini GP, Gao X, Flicek P, Bradley A, Skarnes WC, Justice MJ, Parkinson HE, Moore M, Wells S, Braun RE, Svenson KL, de Angelis MH, Herault Y, Mohun T, Mallon AM, Henkelman RM, Brown SD, Adams DJ, Lloyd KC, McKerlie C, Beaudet AL, Bucan M, and Murray SA (2016) High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dawes R, Lek M, and Cooper ST (2019) Gene discovery informatics toolkit defines candidate genes for unexplained infertility and prenatal or infantile mortality. NPJ Genom Med 4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.