

Abstract
Rationale:
The heartbeat is organized by the cardiac conduction system (CCS), a specialized network of cardiomyocytes. Patterning of the CCS into atrial node versus ventricular conduction system (VCS) components with distinct physiology is essential for the normal heartbeat. Distinct node versus VCS physiology has been recognized for more than a century, but the molecular basis of this regional patterning is not well understood.
Objective:
To study the genetic and genomic mechanisms underlying node versus VCS distinction and investigate rhythm consequences of failed VCS patterning.
Methods and Results:
Using mouse genetics, we found that the balance between T-box transcriptional activator, Tbx5, and T-box transcriptional repressor, Tbx3, determined the molecular and functional output of VCS myocytes. Adult VCS-specific removal of Tbx5 or overexpression of Tbx3 re-patterned the fast VCS into slow, nodal-like cells based on molecular and functional criteria. In these cases, gene expression profiling showed diminished expression of genes required for VCS-specific fast conduction but maintenance of expression of genes required for nodal slow conduction physiology. Action potentials (APs) of Tbx5-deficient VCS myocytes adopted nodal-specific characteristics, including increased AP duration and cellular automaticity. Removal of Tbx5 in-vivo precipitated inappropriate depolarizations in the atrioventricular (His)-bundle associated with lethal ventricular arrhythmias. TBX5 bound and directly activated cis-regulatory elements at fast conduction channel genes required for fast physiological characteristics of the VCS action potential, defining the identity of the adult VCS.
Conclusions:
The CCS is patterned entirely as a slow, nodal ground state, with a T-box dependent, physiologically dominant, fast conduction network driven specifically in the VCS. Disruption of the fast VCS gene regulatory network (GRN) allowed nodal physiology to emerge, providing a plausible molecular mechanism for some lethal ventricular arrhythmias.
Keywords: Tbx5, Tbx3, T-box, Ryr2, Kcnk3, cardiac conduction system (CCS), ventricular conduction, arrhythmia, heart rhythm disorder, atrioventricular node (AVN), His bundle/atrioventricular bundle (AVB), animal model cardiovascular disease, gene expression/regulation, ventricular tachycardia
Subject Terms: Arrythmias, Basic Science, Cardiovascular Disease, Electrophysiology, Gene Expression and Regulation
Graphical Abstract
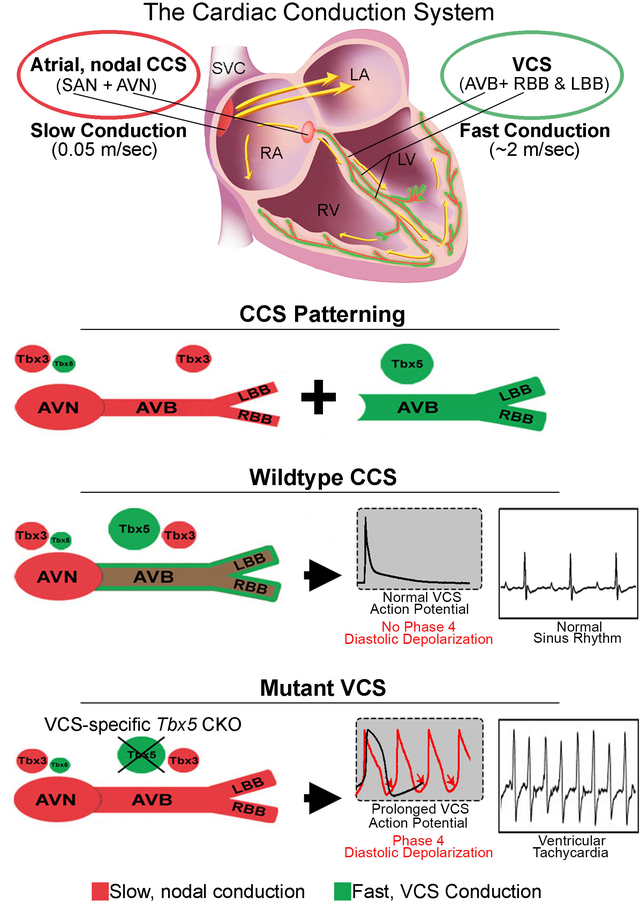
The cardiac conduction system (CCS) is a specialized network of cardiomyocytes that organizes the heartbeat. Patterning of the CCS into slow atrial (nodal) and fast VCS components has been recognized for more than a century, however its molecular basis is poorly described. We found that the balance between Tbx5 and Tbx3, two cardiogenic transcription factor genes, was responsible for distinguishing the VCS from the node. Normally, Tbx5 expression predominated in the VCS. Removal of Tbx5 or overexpression of Tbx3 in the VCS of the adult mouse heart converted the fast VCS into a slow, nodal-like region. VCS-specific gene expression for fast conduction physiology was lost but gene expression for slow nodal physiology was maintained, revealing a nodal-like ground state in the VCS. Moreover, Tbx5-deficient VCS myocytes autonomously depolarized. These depolarizations, normally suppressed in the VCS, initiated lethal ventricular arrhythmias in Tbx5-deficient mice. TBX5 bound and activated enhancers at fast conduction loci. This work revealed that the CCS is patterned entirely as a slow, nodal ground state, with a T-box dependent, physiologically dominant, fast conduction GRN driven by TBX5 specifically in the VCS. The underlying nodal potential of the VCS, here uncovered in the absence of Tbx5, may contribute to the mechanisms underlying VCS-based ventricular arrhythmias.
INTRODUCTION
The mammalian heartbeat is organized by the cardiac conduction system (CCS), a specialized network of cardiomyocytes that is patterned into functional regions with slow conduction or fast conduction that were described over 100 years ago (1–6). Slow conducting atrial nodes include the sinoatrial node (SAN) and atrioventricular node (AVN), while the fast conducting ventricular conduction system (VCS) is comprised of the AV (His) bundle (AVB), left and right bundle branches (BBs) and His-Purkinje network (7–9). Abnormalities in cardiac conduction leading to arrhythmias are a major source of morbidity and mortality worldwide (7, 9–12). For example, slowed ventricular conduction is a major morbidity risk factor and the VCS has been recognized as a substrate for multiple life-threatening ventricular arrhythmias (7, 12–15). We investigated the molecular mechanisms that establish and maintain regional functionality of mature CCS domains and that prevent central conduction system-based arrhythmias.
T-box transcription factors play important roles in the CCS. TBX5, a transcriptional activator, and TBX3, a transcriptional repressor, have both been associated with cardiac conduction speed and arrhythmias in humans with structurally normal hearts by Genome Wide Association Studies (GWAS) (reviewed in 7, 16). Dominant mutations in human TBX5 cause Holt-Oram syndrome (HOS, OMIM:142900), which includes ventricular conduction system slowing, and Tbx5 is expressed at high levels in the mature VCS (17–20). Dominant mutations in human TBX3 cause Ulnar-Mammary syndrome (OMIM:181450) (21, 22), including conduction system defects (23). Tbx3 is selectively expressed in the heart throughout the CCS (24, 25) and Tbx3 knockout mice display abnormal atrial nodal function (25) and lethal arrhythmias (26). Therefore, decreasedTbx5 dose primarily impacts VCS function whereas decreased Tbx3 dose primarily impacts atrial nodal function.
We hypothesized that relative TBX5/TBX3 dose determined the regional specification of the CCS. We examined the impact of altered Tbx5 and Tbx3 dose on the molecular and physiologic state of the VCS using mouse genetics. We found that the CCS is patterned entirely as a slow conduction system ground state with a T-box dependent, physiologically dominant fast conduction system network driven specifically in the VCS. The balance of TBX5 versus TBX3 determined the molecular and functional output of VCS myocytes. VCS-specific removal of TBX5 or overexpression of TBX3 re-patterned the fast VCS into a slow, nodal-like system. After removal of Tbx5, VCS myocytes demonstrated autonomous depolarizations and action potentials characteristic of nodal cells, and initiated lethal ventricular arrhythmias. The underlying nodal potential of the VCS, uncovered in the absence of Tbx5, provided a mechanism for VCS-based ventricular arrhythmias. TBX5 bound and directly activated cis-regulatory elements at the fast conduction loci, directly driving genes that contribute to the fast VCS physiology, which overrides nodal potential in the VCS.
METHODS
Data Availability
The authors declare that all supporting data and materials presented within this manuscript and its Online Supplemental Materials are available from the corresponding author upon reasonable request. TBX5 ChIP-seq data has been deposited to the Gene Expression Omnibus (GEO) database, accession number GSE139803.
A detailed description of the methods is provided in the Online Supplemental Materials.
Please see the Major Resources Table in the Online Supplemental Materials
RESULTS
We hypothesized that TBX5/TBX3 balance may determine regional identity within the mature central conduction system. Therefore, we investigated relative Tbx5 and Tbx3 expression levels in wild-type AVN and VCS cardiomyocytes isolated from adult mouse hearts (Figure 1A–C; Online Expanded Methods). TBX3 expression predominated in AVN cardiomyocytes with Tbx5/Tbx3 ratio of 0.4:1 by qRT-PCR and by western blot. In contrast, TBX5 expression predominated in VCS cardiomyocytes with Tbx5/Tbx3 ratio of 6.6:1 by qRT-PCR and 3.2:1 by western blot. (Figure 1A, B). These observations indicate that expression of Tbx3 predominated over Tbx5 in the slow AV-nodal cardiomyocytes and that Tbx5 predominated over Tbx3 in the fast VCS cardiomyocytes (Figure 1C).
Figure 1. Tbx5/Tbx3 balance determines the regional functional output within the specialized mature CCS.
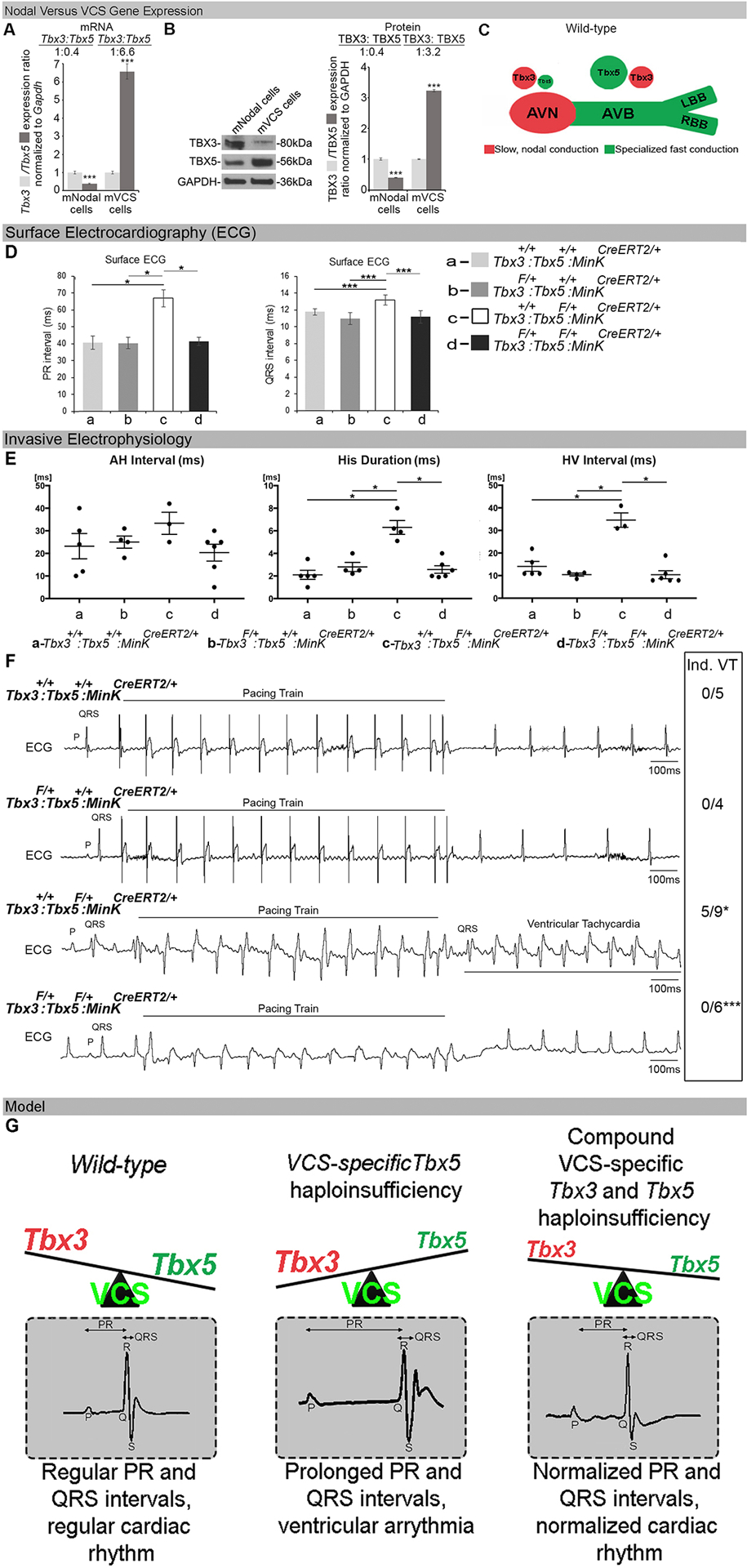
(A-C) Relative Tbx3/Tbx5 expression ratio in wild-type AVN and VCS cardiomyocytes isolated from adult mouse hearts. The qRT-PCR (A) and western blot (B) demonstrated that Tbx3 expression predominates over Tbx5 in the slow AV-nodal cardiomyocytes and that Tbx5 predominates over Tbx3 expression in the fast VCS cardiomyocytes. The AVN cardiomyocytes used in these studies were marked by EGFP expression and isolated from Tbx3BAC-Egfp mice (27) at 10 weeks of age. VCS cardiomyocytes, used in these studies were marked by EYFP expression using a VCS-specific tamoxifen (TM) inducible Cre transgenic mouse line (MinKCreERT2) (14) in combination with conditional EYFP expression from the ROSA locus (R26EYFP/+; MinKCreERT2/+) (7) and microdissected at 10 weeks of age. (C) Model of Tbx3/Tbx5 expression ratio in cardiac conduction system. (D-G) VCS-specific Tbx3 haploinsufficiency in Tbx3:Tbx5 compound heterozygous mice (Tbx3fl/+;Tbx5fl/+;R26EYFP/+;MinKCreERT2/+) rescues VCS defects caused by VCS-specific Tbx5 haploinsufficiency, in Tbx5 heterozygous mice (Tbx5fl/+;R26EYFP/+;MinKCreERT2/+). PR (D, left graph), QRS (D, right graph) and H-V (E, right graph) intervals prolongation, widening of the His duration (E, middle graph), as well as ventricular arrhythmias including reproducible pacing into ventricular tachycardia (VT) (F), observed in VCS-specific Tbx5 heterozygous mice (Tbx5fl/+;R26EYFP/+; MinKCreERT2/+), all got rescued in VCS-specific Tbx3:Tbx5 compound heterozygous mice (Tbx3fl/+;Tbx5fl/+;R26EYFP/+; MinKCreERT2/+). Control VCS-specific Tbx3 heterozygous mice (Tbx3fl/+;R26EYFP/+;MinKCreERT2/+) showed neither conduction nor electrophysiological defects (D-F). Note, ventricular pacing at 100ms S1 drive train followed by 50ms S2 single extrastimulus induced VT in 5/9 Tbx5fl/+;R26EYFP/+; MinKCreERT2/+mice (F, upper panel, representative image), but failed to induced VT in all Tbx3/Tbx5 compound Tbx3fl/+;Tbx5fl/+;R26EYFP/+; MinKCreERT2/+ mice (F, lower panel, representative image). (G) Summary of genetic interactions between Tbx5 and Tbx3 in the adult VCS. Data are presented as mean±SD. For (A), n=3 biological replicates/ analyzed tissue sample (AVN or VCS cardiomyocytes pooled from 30 mice per each biological replicate). (B), n=2 biological replicates/ analyzed tissue sample (AVN or VCS cardiomyocytes pooled from 50 mice per each biological replicate). For (D), n=4 animals/ genotype. For (E, F), n=4–9 animals/ genotype. For (A, B, D – QRP, F), Welch’s t-test; *P<0.05, ***P<0.01. For (D-PR, E), Mann-Whitney U test; *P<0.05. Abbreviations: PR, PR-interval duration; QRS, QRS complex, H-V, Hisio-Ventricular interval; Hd, His-duration.
We directly examined the importance of relative Tbx5/Tbx3 levels on VCS function by analyzing genetic interactions between Tbx5 and Tbx3 in the VCS (Figure 1D–G and Online Figure I and II). We interrogated VCS-specific Tbx5 heterozygous mice (Tbx5fl/+;R26EYFP/+;MinKCreERT2/+ and observed specific conduction and rhythm defects including PR and QRS interval prolongation (Figure 1D, Online Figure I), H-V interval prolongation, widening of the His duration Hd, (Figure 1E, Online Figure I), and ventricular arrhythmias including reproducible pacing into monomorphic ventricular tachycardia (VT) as well as spontaneous VT (Figure 1F, 4D, Online Figure IB, C). VCS-specific Tbx5 heterozygous mice demonstrated split-His potentials (Online Figure I), indicating significant slowing of conduction through the His-Purkinje system, and frequent spontaneous competing AV junctional rhythms (Online Figure I) indicating an increase in the automaticity in VCS structures. VCS-specific Tbx3 heterozygous mice (Tbx3fl/+;R26EYFP/+;MinKCreERT2/+) showed neither conduction nor electrophysiological defects (Figure 1D–G, Online Figure I). Remarkably, all of the observed conduction and rhythm defects caused by VCS-specific Tbx5 haploinsufficiency were rescued in VCS-specific compound Tbx3:Tbx5 double heterozygous (Tbx3fl/+;Tbx5fl/+;R26EYFP/+;MinKCreERT2/+) mice (Figure 1D–G and Online Figure I). Consistent with the use of a VCS-specific Cre (Mink-Cre), no changes in the refractory/recovery periods of atrium, ventricle, or nodes (AERP, VERP, AVERP, AVWB-PPS, or SNRT-SCL) were observed in any genotypic class (Online Figure II). The rescue of VCS defects observed in VCS-specific Tbx5 haploinsufficiency by VCS-specific Tbx3 haploinsufficiency was consistent with a deterministic role for Tbx5/Tbx3 balance in VCS phenotype.
Figure 4. Spontaneous ventricular tachycardia (VT) in VCS-specific Tbx5-deficient adult mice originates in the VCS and is caused by inappropriate automaticity within the VCS.
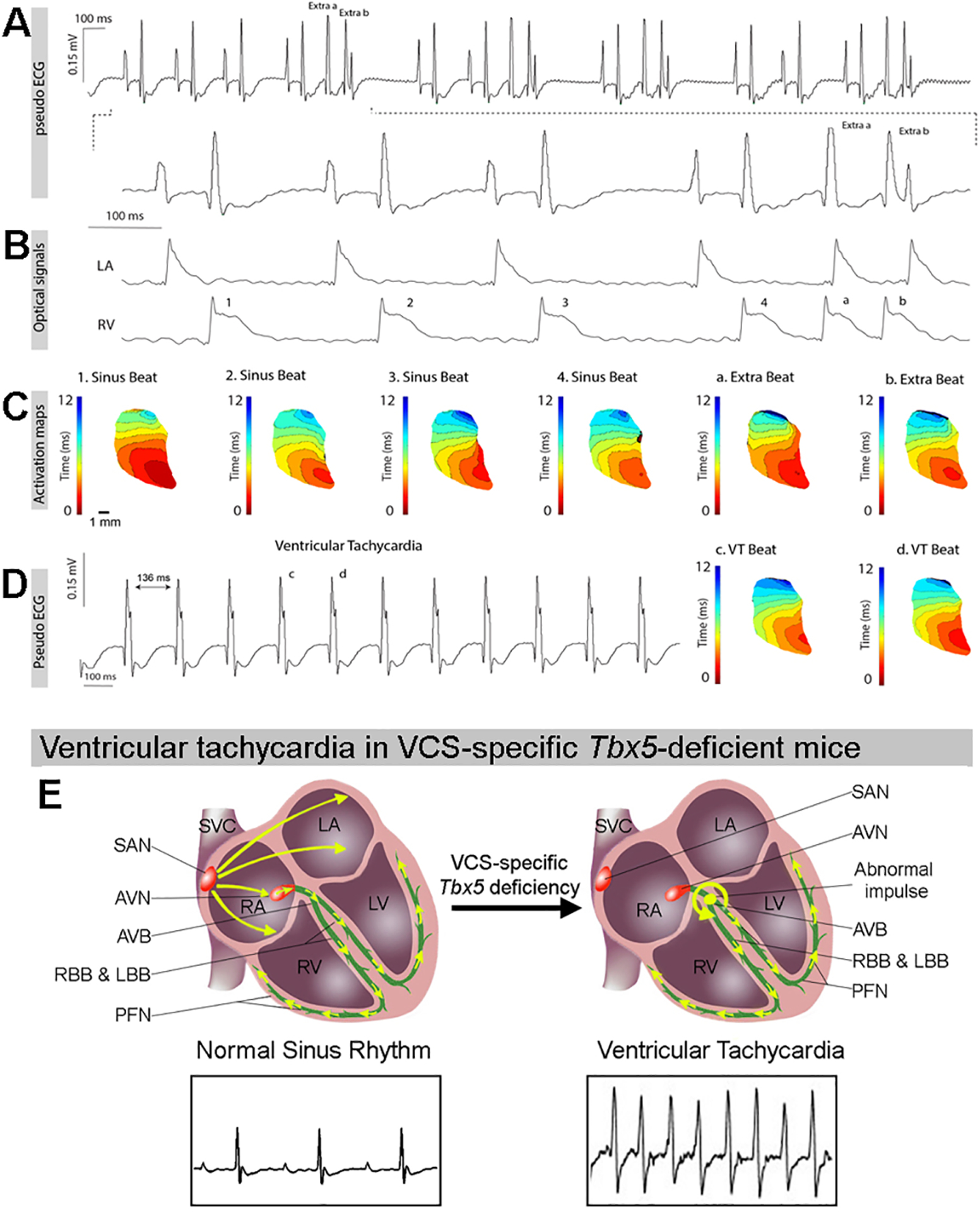
(A). Electrocardiograms recorded from adult VCS-specificTbx5-deficient mice during Langendorff-perfusion showing sinus rhythm and ectopy (extra a-b). (B) Simultaneously recorded optical action potentials showed that ectopic beat originated in the ventricle. (C) Representative voltage activation maps from VCS-specific Tbx5–deficient mice have been reconstructed from sinus beat and two consecutive beats during ventricular ectopy. (D) ECG trace of episode of spontaneous VT (right) and reconstructed activation maps during two consecutive beats (left). Note, spontaneous VT was only detected in Tbx5-deficient mice, not in control mice. The activation patterns during sinus rhythm (SR) and VT are similar. (E) Graphic model of inappropriate activation pattern observed within the VCS of VCS-specific Tbx5 mutant mice that results in spontaneous VT. For (A-D), n=4 or 6 animals/ genotype.
As Tbx5 expression predominated in the VCS, we hypothesized that VCS identity was Tbx5-dependent. Therefore, we investigated Tbx5-dependent expression of VCS genes. Based on the literature, we identified 2 cohorts of VCS expressed genes: genes expressed throughout the entire conduction system (Pan-CCS) and implicated in slow, nodal physiology, such as Hcn4, Cacna1g, Cacna1h, Cx30.2 and Cx45 (27–30), and genes expressed at higher levels in the VCS and implicated in fast conduction physiology, such as Cx40, Scn5a, Ryr2, Kcnk3, Kcnj2, Kcnj3, Kcnj4, and Kcnj12 (Online Figure III; 28, 30–33). Using adult VCS-specific knockout of Tbx5 (Tbx5fl/fl;R26EYFP/+;MinKCreERT2/+ (7)), we examined Tbx5-dependent gene expression for both cohorts of CCS genes in the VCS (Figure 2A, B). We observed that the Pan-CCS slow physiology genes did not demonstrate Tbx5-dependence in the VCS (Figure 2A). In contrast, VCS-high fast conduction physiology genes, including Cx40, Scn5a, Ryr2, Kcnk3, Kcnj2, Kcnj3, Kcnj4, and Kcnj12 (28, 30–33), all demonstrated Tbx5-dependent expression in the VCS (Figure 2B). Immunoblotting analysis confirmed Tbx5-dependent expression in the VCS (Figure 2C). Adult VCS-specific Tbx5 knockout mice therefore retain slow conduction physiology gene expression but lose fast conduction physiology gene expression, transforming the gene expression profile of the VCS into a slow conduction nodal-like pattern (Figure 2D). These observations suggested that the Tbx5-driven VCS-specific fast conduction network is normally superimposed on a Tbx5-independent Pan-CCS nodal gene regulatory network (GRN) in the VCS.
Figure 2. Proper Tbx3/Tbx5 balance is required to maintain expression of VCS-specific fast conduction genes.
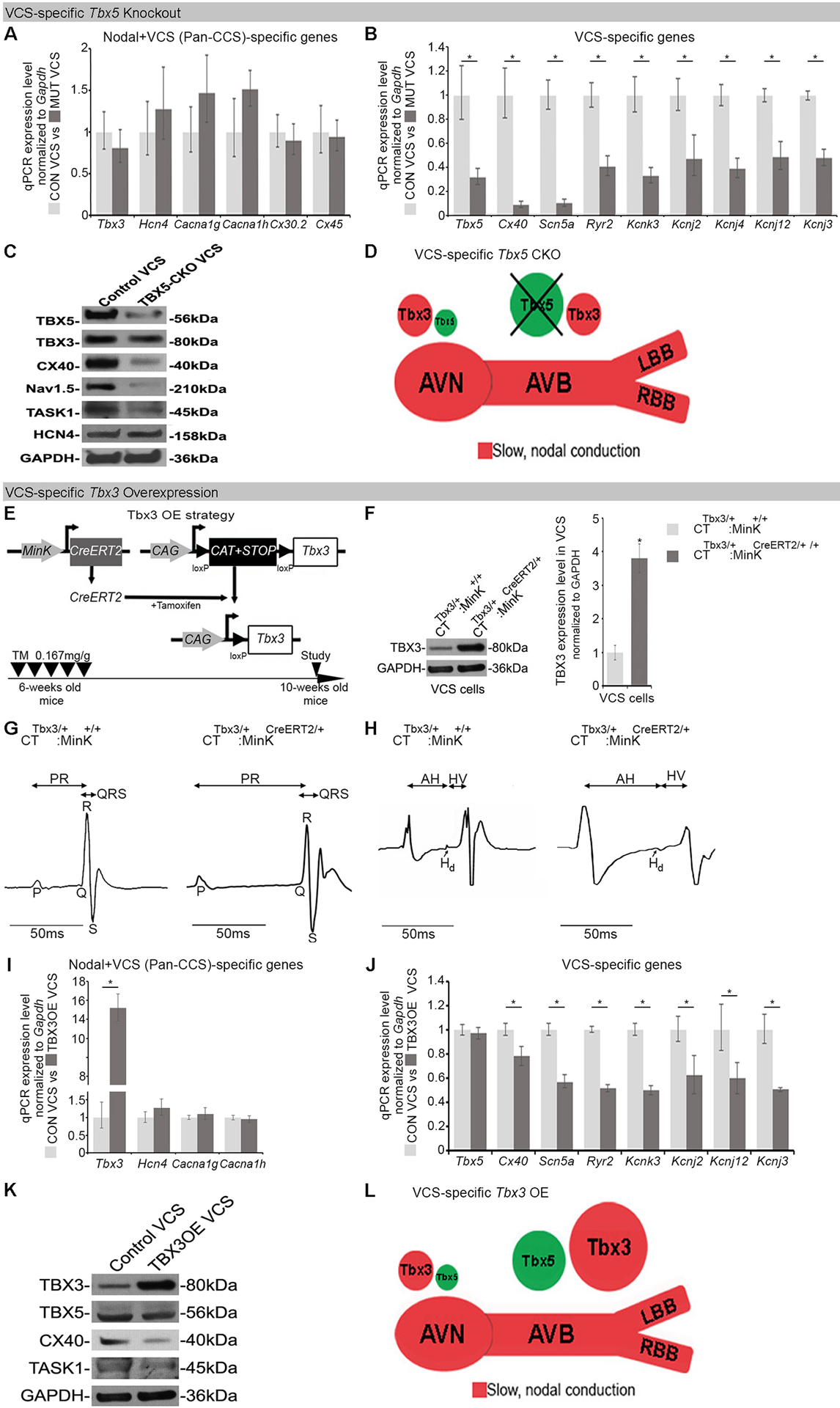
(A-D) Characterization of Tbx5-dependent gene expression in the adult mouse VCS. Tbx5 was specifically removed from the mature VCS in Tbx5fl/fl;R26EYFP/+;MinKCreERT2/+ mice (7), TM treated at 6-weeks of age and analyzed by qRT-PCR with two sets of CCS-specific markers at 10 weeks of age. (A) One set of markers comprised genes normally expressed in both the node and VCS (Pan-CCS) and important for the slow conducting nodal phenotype. None of these genes demonstrated Tbx5-dependent VCS expression by qRT-PCR. (B) The second set comprised genes normally expressed specifically in the VCS (excluded from the nodes) and essential for VCS function. Expression of all these genes was profoundly Tbx5-dependent by qRT-PCR. (C) Immunoblotting analysis confirmed Tbx5-dependent expression in the VCS. (D) Graphical summary of transcriptional changes observed in VCS of VCS-specific Tbx5-deficient mice. Genetic deletion of Tbx5 from the mature VCS uncovers its slow conduction, nodal molecular phenotype. (E-L) VCS-specific TBX3 overexpression in adult CTTbx3/+;MinKCreERT2/+ mice. (E) Strategy to generate VCS-specific TBX3 overexpression in adult CTTbx3/+;MinKCreERT2/+ mice. CTTbx3 transgenic mice carrying a Cre-inducible TBX3 expression construct (CAG–CAT–Tbx3) (24) were crossed to VCS-specific tamoxifen inducible Cre transgenic mouse line (MinKCreERT2) (14) to generate CTTbx3/+;MinKCreERT2/+ mice. The control (CTTbx3/+:MinK+/+) and mutant (CTTbx3/+:MinKCreERT2/+) mice were tamoxifen administrated at 6 week of age. The effects of VCS-specific TBX3 overexpression were assessed 4 weeks after tamoxifen administration (at 10 weeks of age). (F) Western blotting analysis demonstrates 3.8 fold induced overexpression of TBX3 in the mutant VCS cardiomyocytes compared to their control littermates. (G,H) VCS-specific TBX3 overexpression causes significant VCS conduction slowing in adult CTTbx3/+;MinKCreERT2/+ mice. (G) Ambulatory telemetry ECG analysis showed increased PR and QRS intervals in CTTbx3/+:MinKCreERT2/+ mice compared to littermate CTTbx3/+;MinK+/+ control mice. Moreover, intracardiac electrophysiology detected increased A-H, H-V, and His duration (Hd) intervals (H), all indicative of VCS conduction slowing, only in CTTbx3/+:MinKCreERT2/+ mutants never in CTTbx3/+;MinK+/+ controls. (I,J) qRT-PCR analysis of molecular changes driven by Tbx3 overexpression in VCS of adult mice. VCS-specific Tbx3 overexpression did not affect expression of Pan-CCS-specific slow conduction genes (I), but caused a significant reduction in expression of VCS-specific fast conducting genes (J) in CTTbx3/+;MinKCreERT2/+ mutants compared to littermate CTTbx3/+;MinK+/+ controls. (K) Immunoblotting analysis confirmed a significant reduction in expression of VCS-specific fast conducting genes in CTTbx3/+;MinKCreERT2/+ mutants compared to littermate CTTbx3/+;MinK+/+ controls (L) Graphical summary of transcriptional changes observed in mice with induced VCS-specific Tbx3 overexpression. Data are presented as mean±SD. For (A, B, I, J), n=3 biological replicates/ genotype (VCS cardiomyocytes pooled from 30 mice per each biological replicate). For (C, F, K), n=2 biological replicates/ genotype (VCS cardiomyocytes pooled from 50 mice per each biological replicate). For (G), n=3 or 5 animals/ genotype. For (H), n=3 or 4 animals/ genotype. For (A, B, I, J), Welch’s t-test or Mann-Whitney U test, multiple testing correction using Benjamini & Hochberg procedure; *: FDR≤0.15. For (F-H), Welch’s t-test; *P<0.05. Abbreviations: OE, overexpression; A-H, Atrio-Hisian interval; H-V, Hisio-Ventricular interval; Hd, His-duration.
We reasoned that if the TBX5/TBX3 balance determined regional conduction system function, then VCS-specific Tbx3 overexpression should phenotypically resemble the VSC-specific Tbx5 knockout. TBX3 was overexpressed in the mature VCS (3.8-fold), but not in the atria or ventricles, using a Cre-inducible Tbx3 allele (CTTbx3; CAG–CAT–Tbx3) (24) with VCS-specific MinKCreERT2 (Figure 2E, F; Online Figure IV; Online Expanded Methods). VCS-specific overexpression of TBX3 in adult CTTbx3/+;MinKCreERT2/+ mutant mice resulted in significant VCS conduction slowing, including increased PR and QRS intervals by ambulatory telemetry compared to their littermate CTTbx3/+;MinK+/+ controls (Figure 2G). Moreover, intracardiac electrophysiology showed increased A-H, H-V, and His duration (Hd) intervals (Figure 2H), all indicative of VCS conduction slowing. VCS-specific Tbx3 overexpression caused significantly diminished expression of VCS-high fast conduction genes (Figure 2J, K), but did not alter Pan-CCS (Node + VCS) slow conduction gene expression (Figure 2I). Thus, either decreased Tbx5 or increased Tbx3 expression transforms the VCS from a fast to a slow conduction phenotype (Figure 2D and 2L).
We hypothesized that the functional and molecular transformation of the Tbx5-deficient VCS to a nodal-like phenotype reflected cell autonomous defects of VCS cardiomyocyte electrophysiology. We investigated this prediction by isolating VCS cardiomyocytes and performing cellular electrophysiology. Using whole-cell patch-clamp, we observed that the action potential (AP) of VCS-specific, Tbx5-mutant cardiomyocytes demonstrated a slower upstroke (phase 0), prolonged plateau (phase 2), slower repolarization (phase 3) and enhanced phase 4 depolarization, all nodal-like characteristics, compared to the VCS-control cardiomyocytes (Figure 3A, B). The AP from control non-conduction ventricular myocytes isolated from VCS-specific Tbx5-mutant mice showed a rapid phase 0 upstroke, minimal phase 2, rapid phase 3 and no phase 4 depolarization using current clamp recordings in the whole-cell patch-clamp configuration (Figure 3C, D).
Figure 3. Electrophysiological characterization of mice with VCS-specific TBX5-deficiency.
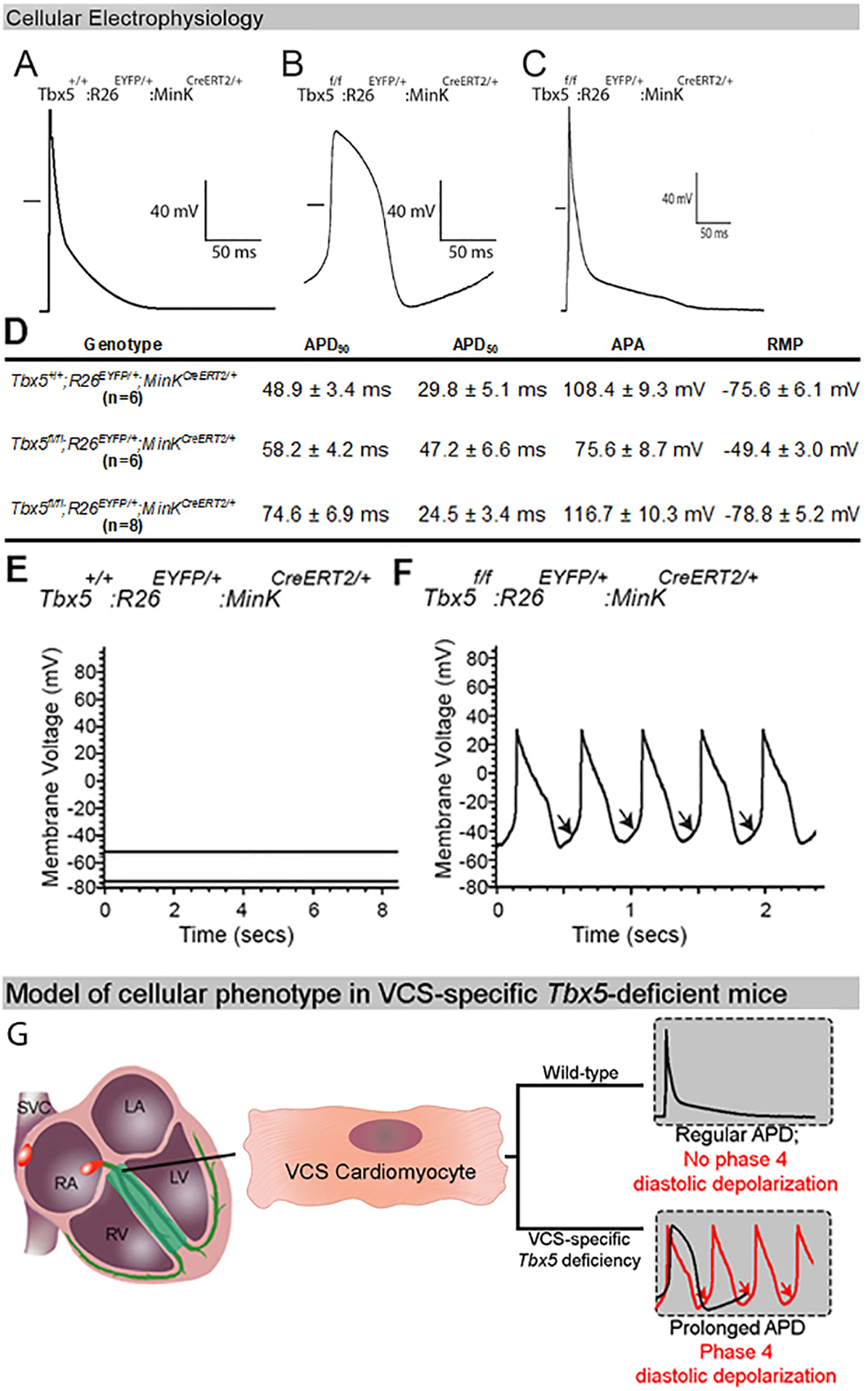
(A-C) Action potentials (APs) from a control VCS (A), Tbx5-deficient VCS (B) and ventricular (C) cardiomyocyte by whole-cell patch–clamp. (A-B) AP of the Tbx5-deficient VCS cardiomyocytes demonstrate a slower upstroke (phase 0), longer plateau (phase 2), delayed repolarization (phase 3) and enhanced phase 4 depolarization compared to the control VCS cardiomyocyte. (C) AP of a ventricular cardiomyocyte from a VCS-specific, Tbx5-deficient mouse shows a rapid upstroke (phase 0), minimal plateau (phase 2), rapid repolarization (phase 3) and no phase 4 depolarization. The short horizontal bar in each panel represents the zero potential level. (D) Table showing detailed AP parameters recorded from mutant VCS, control VCS and ventricular cardiomyocytes. (E, F) VCS-specific removal of Tbx5 induces cellular automaticity in adult VCS cardiomyocytes. (E) No evidence of autonomous electrical activity, including phase 4 depolarization or autonomous beating, were observed in VCS cardiomyocytes isolated from control mice. (F) Phase 4 depolarization (arrows) and autonomous beating were observed in all VCS cardiomyocytes isolated from Tbx5-deficient mice. (G) Summary of cell autonomous defect observed in adult, Tbx5-deficient VCS cardiomyocytes. For (A-F), n=6 or 8 biological replicates/ genotype; Kruskal-Wallis H test; *P<0.05. Abbreviations: APD50, 90, AP duration at 50 and 90% of repolarization; APA, AP amplitude, RMP, resting membrane potential.
We hypothesized that a cell autonomous defect in Tbx5-deficient VCS cardiomyocytes caused inappropriate automaticity. A defining characteristic of the nodal CCS cardiomyocyte is spontaneous beating, electrophysiologically defined as electrical automaticity with diastolic depolarization during phase 4 of the action potential (AP) (34). Using whole-cell patch-clamp, we found that VCS-specific Tbx5 deletion induced cellular automaticity in isolated VCS cardiomyocytes (Figure 3E–G). Autonomous beating and phase 4 depolarization (arrows), an electrophysiologic hallmark of automaticity, were observed in all VCS cardiomyocytes isolated from TM-treated Tbx5fl/fl;R26EYFP/+;MinKCreERT2/+ mice (Figure 3F), no evidence of autonomous electrical activity, neither phase 4 depolarizations nor autonomous beating, were observed in control VCS cardiomyocytes isolated from TM-treated Tbx5+/+;R26EYFP/+;MinKCreERT2/+ mice (Figure 3E).These observations indicated that VCS cardiomyocytes were physiologically transformed to acquire nodal-like APs and automaticity in the absence of Tbx5.
We hypothesized that the ventricular arrhythmias including ventricular tachycardia and sudden death observed in VCS-specific Tbx5 mutant mice (7) may be caused by the inappropriate acquisition of automaticity within VCS cardiomyocytes. To investigate the mechanism underlying ventricular arrhythmias, we analyzed the ventricular electrical activation pattern of adult VCS-specific Tbx5 mutant mice using optical mapping. Spontaneous ventricular tachycardia (VT) was only detected in VCS-specific Tbx5 mutant mice, not in control mice (4/6 Tbx5fl/fl;R26EYFP/+;MinKCreERT2/+ vs. 0/4 Tbx5+/+;R26EYFP/+;MinKCreERT2/+, p=0.04) (Figure 4C). During optical mapping, the ECG of spontaneous ectopic beats demonstrated QRS morphology similar to that observed during sinus rhythm (Figure 4A). Simultaneously recorded optical action potentials showed that the spontaneous complexes originated from the ventricle and caused retrograde activation of the atrium (Figure 4B). Reconstruction of right ventricular activation patterns indicated that spontaneous activation arose high in the ventricular conduction system, because the ventricular activation sequence during the spontaneous complex was similar to that observed during sinus rhythm (Figure 4D). The right ventricular activation patterns observed during spontaneous complexes of ventricular tachycardia and during sinus rhythm appeared identical. Together, these observations suggested that arrhythmia initiation occurred within a proximal anatomic component of the VCS (35) (Figure 4E).
We predicted that TBX5 may directly determine VCS fate by activating cis-regulatory elements (CREs) at fast conduction VCS-high expressed genes (Figures 2A, 3A and 5). To comprehensively identify CREs directly targeted by TBX5, we performed ChIP-seq for TBX5 from embryonic mouse hearts at E14.5, when the VCS has differentiated from non-VCS cardiomyocytes (9). TBX5 was found to localize to 12,436 sites genome-wide (Online Supplementary Data I), of which 3055 were classified as promoters (peak summit <2kb from nearest transcriptional start site or TSS) and 9381 sites as non-promoters (Figure 5A). Motif analysis of TBX5-bound regions showed a strong central enrichment for the published T-box family motif (Figure 5B). We investigated the overlap between TBX5-bound sites with chromatin marks of cis-regulatory function from mouse embryonic heart ENCODE data (36, 37) to categorize TBX5 sites. TBX5-bound sites were predominately defined as active promoters (2905/3055; defined by presence of H3K4me3, H3K27ac, or H3K9ac and absence of H3K9me3 or H3K27me3) or active enhancers (6237/9381; defined by presence of H3K27ac and absence of H3K27me3). TBX5 bound very few repressed promoter (54/3055) or non-promoter (87/9381) sites (Figure 5A). The most enriched biological process and cellular component GO terms for active promoters and non-promoters were all terms related to cardiac function (Online Figure VA).
Figure 5. TBX5 binds and directly activates cis-regulatory elements at VCS conduction loci, establishing a T-box-dependent gene regulatory network (GRN).
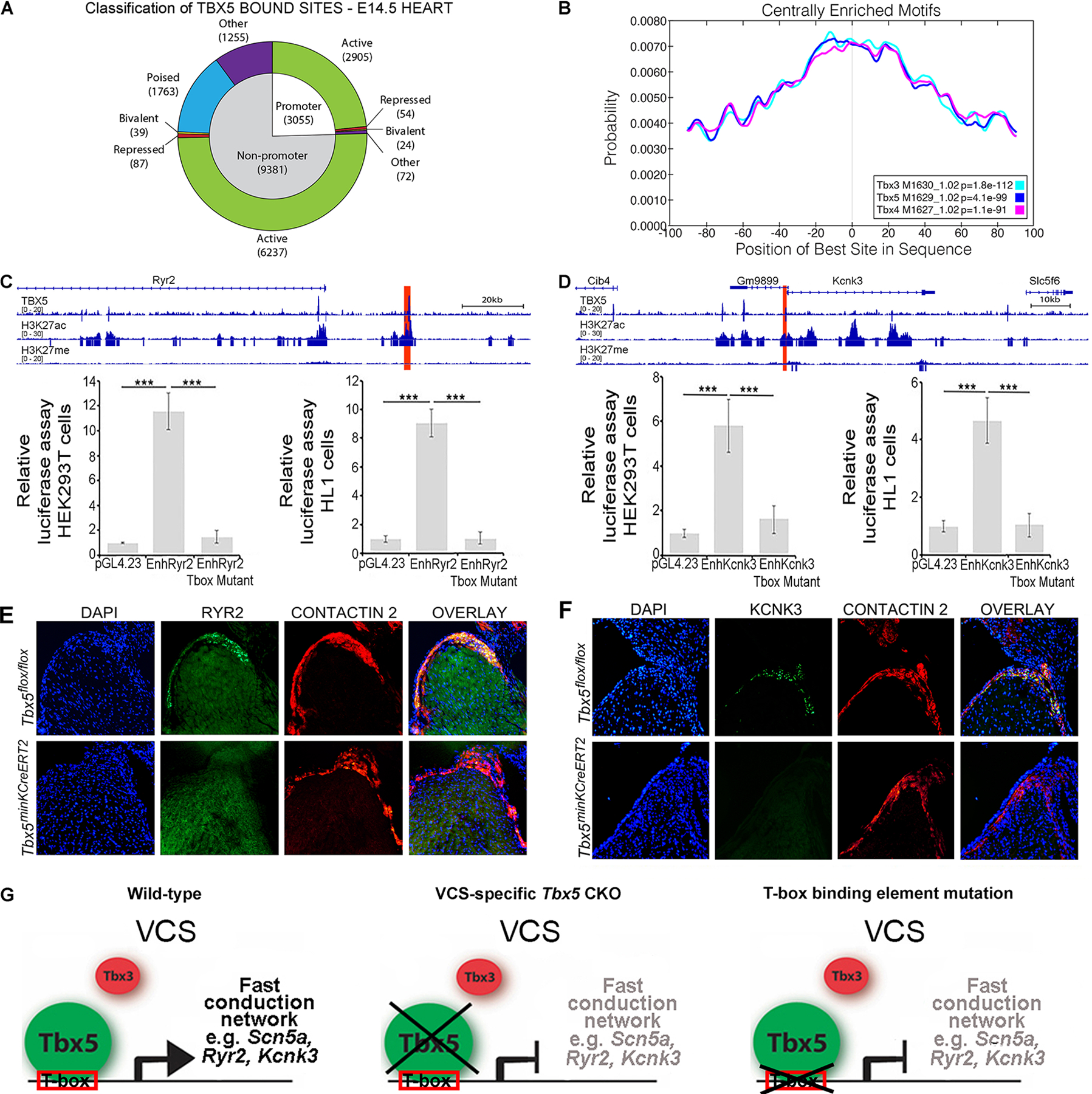
(A) Classification of the 12,436 peaks identified by TBX5 ChIP-seq from E14.5 whole heart based on distance to nearest transcription start site (TSS) and relevant histone data. (B) Central enrichment plots for the TBX5 ChIP-seq peaks. (C,D) Tbx5 directly regulates enhancer downstream of Ryr2 and upstream of Kcnk3. Genome browser views for the Ryr2 (C, upper panel) and Kcnk3 (D, upper panel) loci showing fold-enrichment tracks for TBX5, H3K27ac, and H3K27me3 ChIP-seq. Candidate cis-regulatory elements (CREs, highlighted red) were cloned into pGL4.23 vector to generate CRE-luciferase constructs. Both candidate enhancers demonstrated transcriptional activation in response to Tbx5 expression in dual luciferase reporter assays, in HEK-293T cells (lower panels of C and D, respectively), and in the HL-1 cardiomyocyte cell line with endogenous Tbx5 expression (lower panels of C and D, respectively). Enhancer activation was T-box dependent, as T-box binding element mutation neutralized enhancer activity in each case (lover panels of C and D, respectively). (E,F) Decreased RYR2 and KCNK3/TASK1 expression in the VCS following VCS-specific removal of Tbx5 in Tbx5f/f:R26EYFP/+;MinKCreERT2/+ mutant mice. Tbx5+/+:R26EYFP/+;MinKCreERT2/+ control and Tbx5f/f:R26EYFP/+;MinKCreERT2/+ mutant mice were administered tamoxifen at 6 weeks of age. RYR2 (E) and KCNK3/TASK1 (F) protein expression was evaluated by immunohistochemistry (green signal) 4–5 weeks later. VCS components (AV bundle and bundle branches) were identified by positive contactin-2 expression on serial sections from control and Tbx5 mutant hearts (E and F, red signal). The contactin-2 positive AV bundle expressed high levels of RYR2 (E, green signal) and KCNK3 (F, green signal) in Tbx5+/+:R26EYFP/+;MinKCreERT2/+ control hearts whereas RYR2 (E) and KCNK3/TASK1 (F) expression got drastically extinguished in the contactin-2 positive AV bundle of Tbx5f/f:R26EYFP/+;MinKCreERT2/+ mutant hearts. Nuclei were stained with DAPI. (G) TBX5 binds and directly activates cis-regulatory elements at fast conduction VCS-specific expressed genes, defining the identity of the adult VCS. In the absence of Tbx5 or mutation of T-box binding elements, the expression of the VCS fast conduction channel genes is suppressed. Data are presented as mean±SD normalized to blank vector control, Welch’s t-test; ***P≤0.01, original magnification, x20. For (C-F), n=3 biological replicates/ genotype.
We considered the possibility that TBX5 and TBX3 modulate gene expression through direct regulation of the same CREs. We intersected our TBX5 ChIP-seq with previously published TBX3 ChIP-seq (38) (Online Figure VB and C). Surprisingly, we found that only 27% of all TBX5 ChIP-seq peaks overlapped with TBX3 peaks. However, TBX3/5 overlap was highly significantly enriched at active cardiac promoters (46% overlap, p<2.2e–16) and active cardiac enhancers (28% overlap, p=0.012) (Online Figure VB). TBX3/5 overlap was significantly depleted at non-active promoters (6% overlap, p=6.8e–5), repressed non-promoters (13% overlap, p=0.0036), poised non-promoters (10% overlap, p<2.2e–16), and non-active non-promoters (3% overlap, p<2.2e–16) (Online Figure VB). These results were consistent with co-regulatory action of TBX5 and TBX3 at enhancers driving expression of genes central to cardiac function.
We hypothesized that TBX5 driven enhancers for fast conduction genes would be associated with active enhancer sites. We identified TBX5-bound sites at active enhancers at the loci of most fast conduction genes (8/9 genes, Figure 2C and E) that demonstrated Tbx5-dependent expression in the VCS (Figure 5C, D; Online Figures VD, VI and VII). We interrogated enhancers at genes required for the rapid VCS action potential, including the fast voltage gated sodium channel gene Scn5a, the calcium transporter channel gene Ryr2, and the potassium channel gene Kcnk3 (Figure 5C, D; Online Figure VD, VI and VII) (32, 39–44). TBX5 binding by ChIP was observed at two Scn5a enhancers (Online Figure V) that we previously showed are necessary and sufficient for Scn5a expression (45) (Online Figure VD). TBX5 binding by ChIP was identified at both Ryr2 (mm10 chr13:12132469–12133948) and Kcnk3 (mm10 chr5:30586101–30587731) in concert with epigenetic H3K27Ac from ENCODE derived from E14.5 mouse whole heart (37, 46) (Figure 5C, D). Both candidate enhancers demonstrated transcriptional activation in response to Tbx5 expression in dual luciferase reporter assays, in HEK-293T cells (Figure 5C and D, respectively), and in the HL-1 cardiomyocyte cell line with endogenous Tbx5 expression (Figure 5C and D, respectively). Enhancer activation was T-box dependent, as T-box binding element mutation neutralized enhancer activity in both cases (Figure 5C, D). Additionally, immunohistochemistry demonstrated extinguished expression for both of these channel genes specifically in the VCS of VCS-specific Tbx5 mutant mice (Figure 5E, F). These results indicated that TBX5 directly drives a fast conduction GRN composed of Na+, K+, and Ca++ channels responsible for cardiomyocyte membrane currents that generate the rapid VCS cellular phenotype.
DISCUSSION
The transcriptional architecture of CCS regional specification provides a molecular understanding of regionalized conduction system function and a homeostatic mechanism for the fidelity of the cardiac contraction cycle. Our observations support a model in which Tbx5/Tbx3 balance determines regionalized functional and molecular output within the specialized mature CCS (Figure 1C, 2D, L, and 6). The AVN endogenously possesses a low Tbx5/Tbx3 expression ratio, whereas the VCS is characterized by a high Tbx5/Tbx3 expression ratio (Figure 1A–C and 6) (29, 47). We posit that Tbx3, expressed in a Pan-CCS pattern and required for normal nodal function, generates a nodal ground state throughout the entire specialized CCS, including the VCS (Figure 6A) (17, 24, 25, 27, 48, 49). Tbx5, expressed at high levels specifically in the VCS (Figure 1A–C and 6A), directly drives a fast conduction network to establish fast VCS identity and physiology (Figure 6C) (7, 45). Tbx5-dependent gene expression contributing to fast physiology apparently overrides nodal identity in the mature VCS (Figure 6). Removal of Tbx5 from the VCS eliminates Tbx5-dependent expression of the fast physiology network to uncover underlying nodal potential (Figure 6) (17, 24, 25, 27, 48, 49). This study details the organization of the entire conduction system as having nodal potential, with a Tbx5-driven functionally dominant fast conduction gene regulatory network expressed in the VCS.
Figure 6. Model of transcriptional architecture in specialized mature CCS.
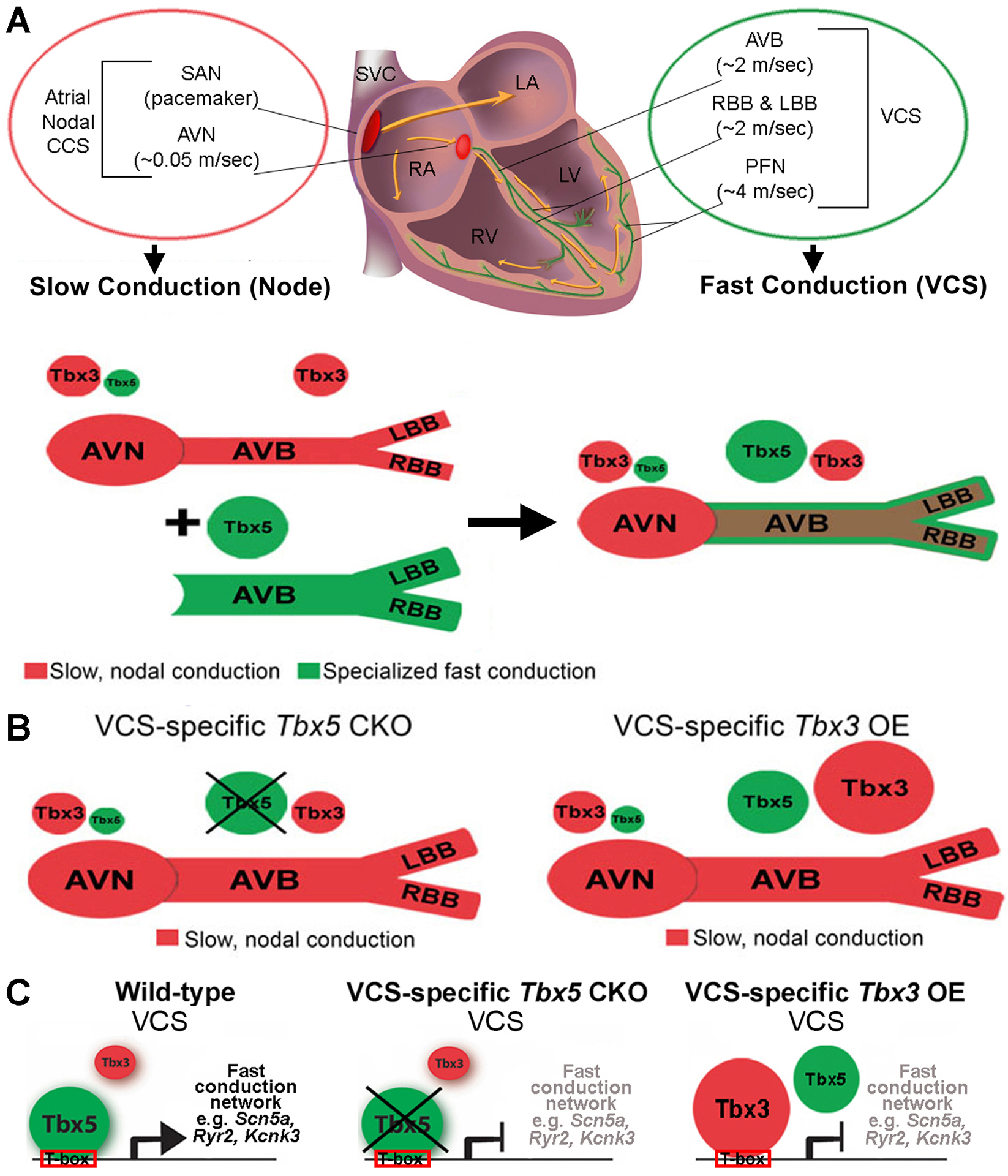
(A) CCS regional specialization is driven by local expression of Tbx5-dependent fast conduction network in the VCS, which overlaps underlying Pan-CCS expression of nodal, slow conduction network. (B) VCS-specific removal of Tbx5 or overexpression of Tbx3 re-patterned the fast VCS into a slow, nodal-like system. (C) The balance of Tbx5, a T-box transcriptional activator, versus Tbx3, a T-box transcriptional repressor, determined the molecular and functional output of VCS myocytes. TBX5 binds and directly activates cis-regulatory elements at fast conduction loci, defining the identity of the adult VCS. In the absence of TBX5 or overexpression of TBX3, the expression of the VCS fast conduction channel genes is suppressed.
This study indicates that changing of the Tbx5/Tbx3 dose in adult mouse VCS in favor of Tbx3, through VCS-specific Tbx5 removal or Tbx3 overexpression, transforms the VCS into a nodal-like conduction system. We have tested this model in the VCS genetically by interaction studies, in which the VCS conduction slowing and arrhythmia propensity cause by Tbx5 haploinsufficiency can be rescued by Tbx3 haploinsufficiency (Figure 1D–G and Online Figure I), and molecularly, in which removal of Tbx5 or overexpression of Tbx3 each effectively removed VCS-specific expression of fast conduction genes to repattern the normally fast VCS into a slow nodal phenotype (Figure 2, Figure 3, Online Figure IV). We uncovered a gene regulatory network with T-box-dependent enhancers driving sodium, potassium and calcium handling genes for fast conduction in the VCS, suggesting that TBX5 functions as a fast conduction master regulator (Figure 5C–G, Online Figure VD). This work provides evidence that the regional specialization of mature CCS is patterned by a direct TBX5-driven fast conduction GRN in the VCS, overlaying Pan-CCS expression of a nodal, slow conduction network (Figure 6). The molecular basis underlying the VCS-specific expression of Tbx5 at high levels to effect this patterning is the basis for future studies.
The clinical consequences of VCS disorders are serious. The VCS has been recognized as a substrate for life-threatening ventricular arrhythmias, including bundle branch reentry tachycardia, idiopathic fascicular tachycardia, short-coupled torsade de pointes, and ventricular fibrillation and slowed VCS conduction is associated with increased morbidity and mortality (7, 12–15). The genetic basis and the molecular mechanisms underlying VCS-based arrhythmias or slowed conduction are not well-known. We observe that the emergence of slow, nodal-like functional potential in the VCS following Tbx5 removal causes ventricular tachycardia, which appears to be initiated in the VCS and mimics well-recognized VCS-based human arrhythmias (Figure 2A–D, 3, and 4D) (17, 24, 25, 27, 48, 49). Our work provides a plausible explanation for VCS- initiated arrhythmias. This work suggests that ventricular arrhythmias are a possible consequence of the inappropriate transformation from fast to slow conduction phenotype in the VCS, here observed following removal of Tbx5 or upregulation of Tbx3 (Figure 4). The node-like physiology ground state of the VCS, uncovered in the absence of Tbx5, includes spontaneous depolarizations. Such depolarizations, normal in nodal tissue, may contribute to the genesis of VCS-based arrhythmias, especially in the context of slowed VCS-conduction (Figure 3F, 4D and 6). We hypothesize that the emergence of underlying nodal potential in the VCS (Figure 3B, F) enables autonomous depolarizations as a plausible arrhythmia trigger (Figure 4D). The observed ventricular arrhythmias are unlikely to result from generalized ventricular dysfunction in our model. Our previous studies of VCS-specific Tbx5-deficient mice confirmed normal cardiac function during sinus rhythm and the immediate recovery of normal cardiac function after episodes of spontaneous VT (7). Additionally, whole adult murine heart histology demonstrated no histological differences between Tbx5 mutant and control mice (7). These results indicated that the VT and cellular electrophysiology alterations observed in VCS-specific Tbx5-deficient mice were unlikely to be a secondary consequence of myocardial dysfunction, but were instead associated with the molecular transformation of the Tbx5-deficient VCS to a nodal-like phenotype. The transformation from VCS to nodal physiology therefore provides a possible mechanism for the genesis of human VCS-based ventricular arrhythmias. Future work to uncover the genetic basis of VCS-based ventricular arrhythmias will be essential to test this model.
We provide evidence that a fate transformation from one cardiac conduction region into another can be caused in-vivo by the removal of a single master regulator. This observation imports concepts from developmental biology to explain adult organ homeostasis, and thus provides a novel paradigm for the understanding how the breakdown of adult organ homeostasis can cause adult disease (50–53). Given the limitations of current therapies for arrhythmias and their consequences, which include sudden cardiac death, understanding the molecular etiology of cardiac rhythm homeostasis and arrhythmias is of high significance. This work may thereby contribute to the blueprint for the generation of CCS sub-lineages ex-vivo, and thus have implications for regenerative and cell-based therapeutic approaches. Further studies are necessary to investigate the genomic establishment of the slow versus fast gene regulatory networks to explain how two states, “slow” vs “fast”, are observed in the conduction system with no intermediate phenotypes. Incomplete removal of the fast network, by decreased but not eliminated expression of Tbx5 or its targets, are associated with arrhythmia risk in mice and men. The degree to which these perturbations afford the uncovering of underlying nodal potential as an arrhythmia risk factor remains to be determined. The patterning model for the CCS provides a model for the homeostatic control of the cardiac contraction cycle.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
The mammalian heartbeat is organized by the cardiac conduction system (CCS), a specialized network of cardiomyocytes that is specified into distinct regions: physiologically slow atrial nodes and physiologically fast ventricular conduction system (VCS).
Slowed ventricular conduction is a major morbidity risk factor and the VCS has been recognized as a substrate for multiple life-threatening ventricular arrhythmias.
Developmentally important T-box transcription factors, Tbx5, a transcriptional activator, and Tbx3, a transcriptional repressor, have both been associated with cardiac conduction speed and arrhythmias in humans and mice with structurally normal hearts.
What New Information Does This Article Contribute?
High Tbx5/Tbx3 balance determined the normal phenotype of the mature VCS.
Decreased Tbx5 or increased Tbx3 expression caused loss of fast-VCS-specific gene expression and uncovered a nodal-like ground state.
Transformation of VCS to node-like phenotype after removal of Tbx5 caused autonomous depolarizations in the VCS that provided a mechanism for lethal ventricular arrhythmias.
TBX5 bound and directly activated enhancers at the fast conduction loci required for the fast VCS action potential.
This work supports a model in which the entire CCS is patterned as a slow, nodal ground state, with a physiologically dominant Tbx5-driven fast conduction gene regulatory network (GRN) in the VCS.
ACKNOWLEDGEMENTS
We thank Dr. Vincent Christoffels (Academic Medical Center, University of Amsterdam, Amsterdam, the Netherlands) for generously providing the Tbx3BAC-Egfp and Tbx3CT (CAG-CAT-TBX3) transgenic mice. Histone ChIP-seq data was produced by the laboratory of Bing Ren and made publicly available through the ENCODE Consortium.
SOURCES OF FUNDING
This work was supported by National Institutes of Health (R01 HL126509, R01 HL148719, and R01 HL147571 to IPM, R33 HL123857 to IPM and MAN, T32 GM007183 and HL007381 to JDS, and 5T32HL007381-37 to RDN), National Institute on Aging (1R21AG054770-01A1 to KI and IPM), Foundation Leducq Transatlantic Networks of Excellence (to IPM), American Heart Association (7CSA33610126 to IPM and 13POST17290028 to OBT) and University of Chicago (MSTP 5T32GM007281-41 to RDN).
Nonstandard Abbreviations and Acronyms:
- ACUP
animal care and use protocol
- AERP
atrial effective refractory period
- A-H
atrio-hisian interval
- APA
AP amplitude
- APD50, 90
AP duration at 50 and 90% of repolarization
- AP
action potential
- AVB
atrioventricular bundle
- AVERP
atrioventricular nodal effective refractory period
- AVN
atrioventricular node
- BAC
bacterial artificial chromosome
- BBs
bundle branches
- Cacna1g
calcium voltage-gated channel subunit alpha1 G
- Cacna1h
calcium voltage-gated channel subunit alpha1 H
- CCS
cardiac conduction system
- CREs
cis-regulatory elements
- Cx30.2/Gjd3
gap junction protein gamma 3
- Cx40/Gja5
gap junction protein alpha 5
- Cx45/Gjc1
gap junction protein gamma 1
- Egfp
enhanced green fluorescent protein
- Eyfp
enhanced yellow fluorescent protein
- FACS
fluorescent-activated cell sorting
- FDR
false discovery rate
- GEO
gene expression omnibus database
- GO
gene ontology
- GRN
gene regulatory network
- GWAS
genome wide association studies
- Hcn4
hyperpolarization activated cyclic nucleotide gated potassium channel 4
- Hd
his-duration
- HOS
Holt-Oram syndrome
- H-V
hisio-ventricular interval
- IACUC
institutional animal care and use committee
- Kcnj2
potassium inwardly rectifying channel subfamily J member 2
- Kcnj3
potassium inwardly rectifying channel subfamily J member 3
- Kcnj4
potassium inwardly rectifying channel subfamily J member 4
- Kcnj12
potassium inwardly rectifying channel subfamily J member 12
- Kcnk3
potassium two pore domain channel subfamily K member 3
- OE
overexpression
- OMIM
online mendelian inheritance in man
- QRS
QRS complex
- PR
PR-interval duration
- RMP
resting membrane potential
- Ryr2
ryanodine receptor 2
- SAN
sinoatrial node
- Scn5a
sodium voltage-gated channel alpha subunit 5
- SNRT-SCL
corrected sinus node recovery time
- Tbx3
T-box transcriptional factor 3
- Tbx5
T-box transcriptional factor 5
- TM
tamoxifen
- TSS
transcriptional start site
- VCS
ventricular conduction system
- VERP
ventricular effective refractory period
- VT
ventricular tachycardia
Footnotes
REFERENCES
- 1.Silverman ME, Grove D, Upshaw CB. Why does the heart beat? The discovery of the electrical system of the heart. Circulation. 2006;113:2775–2781. [DOI] [PubMed] [Google Scholar]
- 2.Tawara S. Das Reizleitungssystem des Säugetierherzens. Gustav Fischer; Jena, Germany, 1906:135–149. [Google Scholar]
- 3.Keith A, Flack MW. The auriculo-ventricular bundle of the human heart. Lancet. 1906;2:359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kent AFS. Researches on the structure and function of the mammalian hearts. J Physiol. 1893;14:233–254. [PMC free article] [PubMed] [Google Scholar]
- 5.His W Jr. Die Thätigkeit des embryonalen Herezens und deren Bedeutung für die Lehre von der Herzbewegung bein Erwachsenen. Arb Med Klinik Leipzig. 1893;1:14–49. [Google Scholar]
- 6.Purkinje JE. Mikrosckopisch neurologische Beobachtungen. Arch Anat Physiol Wiss Med. 1845;12:281–295. [Google Scholar]
- 7.Arnolds DE, Liu F, Fahrenbach JP, Kim GH, et al. TBX5 drives Scn5a expression to regulate cardiac conduction system function. J Clin Invest. 2012;122:2509–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park DS, Fishmann GI. The cardiac conduction system. Circulation. 2011;123:904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moskowitz IPG, Kim JB, Moore ML, et al. A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system. Development Cell. 2007;129:1365–1376. [DOI] [PubMed] [Google Scholar]
- 10.Munshi NV. Gene regulatory networks in cardiac conduction system development. Circ Res. 2012;110:1525–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005;115:2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345:1473–1482. [DOI] [PubMed] [Google Scholar]
- 13.van Duijvenboden K, Ruijter JM, Christoffels VM. Gene regulatory elements of the cardiac conduction system. Brief Funct Genomics. 2014;13:28–38. [DOI] [PubMed] [Google Scholar]
- 14.Arnolds DE, Moskowitz IP. Inducible recombination in the cardiac conduction system of minK:CreERT2 BAC transgenic mice. Genesis. 2011;49:878–884. [DOI] [PubMed] [Google Scholar]
- 15.Scheinman MM. Role of the His-Purkinje system in the genesis of cardiac arrhythmia. Heart Rhythm. 2009;6:1050–10158. [DOI] [PubMed] [Google Scholar]
- 16.Arnolds DE, Chu A, McNally EM, Nobrega MA, Moskowitz IP. The emerging genetic landscape underlying cardiac conduction system function. Birth Defects Res A Clin Mol Teratol. 2011;91:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoogaars WM, Barnett P, Moorman AF, Christoffels VM. T-box factors determine cardiac design. Cell Mol Life Sci. 2007;64:646–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDermott DA, Bressan MC, He J, et al. TBX5 genetic testing validates strict clinical criteria for Holt-Oram syndrome. Pediatr Res. 2005;58:981–986. [DOI] [PubMed] [Google Scholar]
- 19.Li QY, Newbury-Ecob RA, Terrett JA, et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet. 1997;15:21–29. [DOI] [PubMed] [Google Scholar]
- 20.Basson CT, Cowley GS, Solomon SD, Weissman B, Poznanski AK, Traill TA, Seidman JG, Seidman CE. The clinical and genetic spectrum of the Holt-Oram syndrome (heart-hand syndrome). N Engl J Med. 1994;330:885–891. [DOI] [PubMed] [Google Scholar]
- 21.Meneghini V, Odent S, Platonova N, Egeo A, Merlo GR. Novel TBX3 mutation data in families with ulnar-mammary syndrome indicate a genotype-phenotype relationship: Mutations that do not disrupt the T-domain are associated with less severe limb defects. Eur J Med Genet. 2006;49:151–158. [DOI] [PubMed] [Google Scholar]
- 22.Bamshad M, Lin RC, Law DJ, et al. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat Genet. 1997;16:311–315. [DOI] [PubMed] [Google Scholar]
- 23.Linden H, Williams R, King J, Blair E, Kini U. Ulnar Mammary syndrome and TBX3: expanding the phenotype. Am J Med Genet. 2009;149A:2809–2812. [DOI] [PubMed] [Google Scholar]
- 24.Hoogaars WM, Engel A, Brons JF, et al. Tbx3 controls the sinoatrial node gene program and imposes pacemaker function on the atria. Genes Dev. 2007;21:1098–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoogaars WM, Tessari A, Moorman AF, de Boer PA, Hagoort J, Soufan AT, Campione M, Christoffels VM. The transcriptional repressor Tbx3 delineates the developing central conduction system of the heart. Cardiovasc Res. 2004;62:489–499. [DOI] [PubMed] [Google Scholar]
- 26.Frank DU, Carter KL, Thomas KR, Burr RM, Bakker ML, Coetzee WA, Tristani-Firouzi M, Bamshad MJ, Christoffels VM, Moon AM. Lethal arrhythmias in Tbx3-deficient mice reveal extreme dosage sensitivity of cardiac conduction system function and homeostasis. Proc Natl Acad Sci USA. 2011;109:154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bakker ML, Boink GJ, Boukens BJ, et al. T-box transcription factor TBX3 reprogrammes mature cardiac myocytes into pacemaker-like cells. Cardiovasc Res. 2012;94:439–449. [DOI] [PubMed] [Google Scholar]
- 28.Verheule S, Kaese S. Connexin diversity in the heart: insights from transgenic mouse models. Front Pharmacol. 2013;4:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greener ID, Monfredi O, Inada S, et al. Molecular architecture of the human specialized atrioventricular conduction axis. J Mol Cell Cardiol. 2011;50:642–651. [DOI] [PubMed] [Google Scholar]
- 30.Schram G, Pourrier M, Melnyk P, Nattel S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res. 2002;90:939–950. [DOI] [PubMed] [Google Scholar]
- 31.Remme CA, Verkerk AO, Hoogaars WM, et al. The cardiac sodium channel displays differential distribution in the conduction system and transmural heterogeneity in the murine ventricular myocardium. Basic Res Cardiol. 2009;104:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Graham V, Zhang H, Willis S, Creazzo TL. Expression of a two-pore domain K+ channel (TASK-1) in developing avian and mouse ventricular conduction systems. Dev Dyn. 2006;235:143–151. [DOI] [PubMed] [Google Scholar]
- 33.Donner BC, Schullenberg M, Geduldig N, et al. Functional role of TASK-1 in the heart: studies in TASK-1-deficient mice show prolonged cardiac repolarization and reduced heart rate variability. Basic Res Cardiol. 2011;106:75–87. [DOI] [PubMed] [Google Scholar]
- 34.Yu G, Wang L, Yan G, He Q. DOSE: an R/Bioconductor package for Disease Ontology Semantic and Enrichment analysis. Bioinformatics. 2015;31:608–609. [DOI] [PubMed] [Google Scholar]
- 35.Janse MJ, D’Alnoncourt CN. Reflections on reentry and focal activity. Am J Cardiol. 1987;60:21F–26F. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sloan CA, Chan ET, Davidson JM, et al. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016;44:D726–D732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van den Boogaard M, Wong LY, Tessadori F, et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest. 2012;122:2519–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res. 2011;108:884–897. [DOI] [PubMed] [Google Scholar]
- 40.Papadatos GA, Wallerstein PM, Head CE, et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci USA. 2002;99:6210–6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cerrone M, Napolitano C, Priori SG. Catecholaminergic polymorphic ventricular tachycardia: A paradigm to understand mechanisms of arrhythmias associated to impaired Ca(2+) regulation. Heart Rhythm. 2009;6:1652–1659. [DOI] [PubMed] [Google Scholar]
- 42.Bhuiyan ZA, van den Berg MP, van Tintelen JP, et al. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation. 2007;116:1569–1576. [DOI] [PubMed] [Google Scholar]
- 43.Decher N, Wemhöner K, Rinné S, et al. Knock-out of the potassium channel TASK-1 leads to a prolonged QT interval and a disturbed QRS complex. Cell Physiol Biochem. 2011;28:77–86. [DOI] [PubMed] [Google Scholar]
- 44.Donner BC, Schullenberg M, Geduldig N., et al. Functional role of TASK-1 in the heart: studies in TASK-1-deficient mice show prolonged cardiac repolarization and reduced heart rate variability. Basic Res Cardiol. 2011;106:75–87. [DOI] [PubMed] [Google Scholar]
- 45.van den Boogaard M, Smemo S, Burnicka-Turek O, et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J Clin Invest. 2014;124:1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moskowitz IP, Pizard A, Patel VV, et al. The T-Box transcription factor Tbx5 is required for the patterning and maturation of the murine cardiac conduction system. Development. 2004;131:4107–4116. [DOI] [PubMed] [Google Scholar]
- 48.Hatcher CJ, Basson CT. Specification of the cardiac conduction system by transcription factors. Circ Res. 2009;105:620–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bakker ML, Boukens BJ, Mommersteeg MT, Brons JF, Wakker V, Moorman AF, Christoffels VM. Transcription factor Tbx3 is required for the specification of the atrioventricular conduction system. Circ Res. 2008;102:1340–1349. [DOI] [PubMed] [Google Scholar]
- 50.Seymour PA, Shih HP, Patel NA, Freude KK, Xie R, Lim CJ, Sander M. A Sox9/Fgf feed-forward loop maintains pancreatic organ identity. Development. 2012;139:3363–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bort R, Signore M, Tremblay K, Martinez Barbera JP, Zaret KS. Hex homeobox gene controls the transition of the endoderm to a pseudostratified, cell emergent epithelium for liver bud development. Dev Biol. 2006;290:44–56. [DOI] [PubMed] [Google Scholar]
- 52.Sumazaki R, Shiojiri N, Isoyama S, Masu M, Keino-Masu K, Osawa M, Nakauchi H, Kageyama R, Matsui A. Conversion of biliary system to pancreatic tissue in Hes1-deficient mice. Nat Genet. 2004;36:83–87. [DOI] [PubMed] [Google Scholar]
- 53.Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32:128–134. [DOI] [PubMed] [Google Scholar]
- 54.Horsthuis T, Buermans HPJ, Brons JF, et al. Gene expression profiling of the forming atrioventricular node using a novel tbx3-based node-specific transgenic reporter. Circ Res. 2009;105:61–69. [DOI] [PubMed] [Google Scholar]
- 55.Masino AM, Gallardo TD, Wilcox CA, Olson EN, Williams RS, Garry DJ. Transcriptional regulation of cardiac progenitor cell populations. Circ Res. 2004;94:389–397. [DOI] [PubMed] [Google Scholar]
- 56.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 57.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rasband WS. ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2016. [Google Scholar]
- 59.Wheeler MT, Allikian MJ, Heydemann A, Hadhazy M, Zarnegar S, McNally EM. Smooth muscle cell–extrinsic vascular spasm arises from cardiomyocyte degeneration in sarcoglycan-deficient cardiomyopathy. J Clin Invest. 2004;113:668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mitra R, Morad M. A uniform enzymatic method for dissociation of myocytes from hearts and stomachs of vertebrates. Am J Physiol. 1985;249:H1056–1060. [DOI] [PubMed] [Google Scholar]
- 61.Levin MD, Lu MM, Petrenko NB, et al. Melanocyte-like cells in the heart and pulmonary veins contribute to atrial arrhythmia triggers. J Clin Invest. 2009;119:3420–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nadadur RD, Broman MT, Boukens B, et al. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci Transl Med. 2016;8:354ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu F, Levin MD, Petrenko NB, Lu MM, Wang T, Yuan LJ, Stout AL, Epstein JA, Patel VV. Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy independent of angiotensin. J Mol Cell Cardiol. 2008;45:715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laughner JI, Ng FS, Sulkin MS, Arthur RM, Efimov IR. Processing and analysis of cardiac optical mapping data obtained with potentiometric dyes. Am J Physiol Heart Circ Physiol. 2012;303:H753–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boukens BJ, Hoogendijk MG, Verkerk AO, Linnenbank A, van Dam P, Remme CA, Fiolet JW, Opthof T, Christoffels VM, Coronel R. Early repolarization in mice causes overestimation of ventricular activation time by the QRS duration. Cardiovasc Res. 2013;97:182–191. [DOI] [PubMed] [Google Scholar]
- 66.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. and 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc. 2012;7:1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 2016. [Google Scholar]
- 71.Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. [DOI] [PubMed] [Google Scholar]
- 72.Bioconductor Core Team and Bioconductor Package Maintainer, TxDb.Mmusculus.UCSC.mm10.knownGene: Annotation package for TxDb object(s). 2016.
- 73.Zhu LJ. Integrative analysis of ChIP-chip and ChIP-seq dataset. Methods Mol Biol. 2013;1067:105–124. [DOI] [PubMed] [Google Scholar]
- 74.Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, Green MR. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics. 2010;11:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ou J, Wang Y, Zhu LJ. trackViewer: A bioconductor package with minimalist design for drawing elegant tracks or lollipop plot. R package version 1.10.2. 2016. [Google Scholar]
- 77.Lawrence M, Gentleman R, Carey V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics. 2009;25:1841–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Neuwirth E. RColorBrewer: ColorBrewer Palettes. R package version 1.1–2. 2014. [Google Scholar]
- 79.Carlson M. org.Mm.eg.db: Genome wide annotation for Mouse. R package version 3.4.0. 2016. [Google Scholar]
- 80.Yue F, Cheng Y, Breschi A, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Waggott D, Haider S, Lalonde E, Fung C, Boutros PC. Genomic Region Processing using Tools Such as ‘BEDTools’, ‘BEDOPS’ and ‘Tabix’. Version 1.0.7. 2016. https://cran.r-project.org/web/packages/bedr/index.html [Google Scholar]
- 83.Smit AFA, Hubley R, Green P. RepeatMasker Open-4.0. 2013–2015. http://www.repeatmasker.org
- 84.Bailey TL, Machanick P. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res. 2012;40:e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weirauch MT, Yang A, Albu M, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158:1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu G, Wang L, Han Y, He Q. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all supporting data and materials presented within this manuscript and its Online Supplemental Materials are available from the corresponding author upon reasonable request. TBX5 ChIP-seq data has been deposited to the Gene Expression Omnibus (GEO) database, accession number GSE139803.
A detailed description of the methods is provided in the Online Supplemental Materials.
Please see the Major Resources Table in the Online Supplemental Materials