

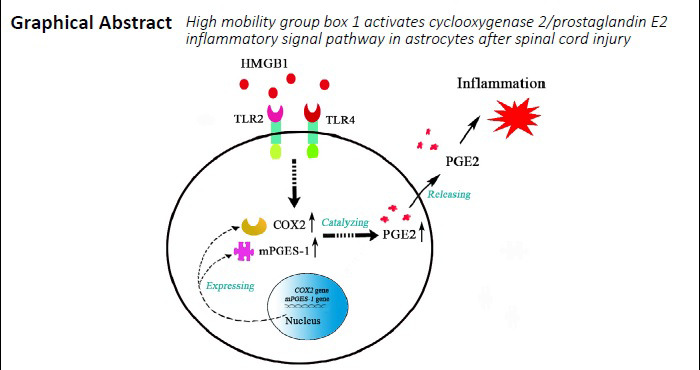
Keywords: astrocytes, COX2, HMGB1, inflammation, spinal cord injury
Abstract
High mobility group box 1 (HMGB1) interacts with pattern-recognition receptors of immune cells to activate the inflammatory response. Astrocytes play a positive role in the inflammatory response of the central nervous system by expressing a broad range of pattern-recognition receptors. However, the underlying relationship between HMGB1 and the inflammatory reaction of astrocytes remains unclear. In this study, we established rat models of spinal cord injury via laminectomy at the T8–10 level, and the injured spinal cord was subjected to transcriptome sequencing. Our results showed that the HMGB1/Toll-like receptor 4 (TLR4) axis was involved in the activation of astrocyte inflammatory response through regulation of cyclooxygenase 2 (COX2)/prostaglandin E2 (PGE2) signaling. Both TLR4 and COX2 were distributed in astrocytes and showed elevated protein levels following spinal cord injury. Stimulation of primary astrocytes with recombinant HMGB1 showed that COX2 and microsomal PGE synthase (mPGES)-1, rather than COX1, mPGES-2, or cytosolic PGE synthase, were significantly upregulated. Accordingly, PGE2 production in astrocytes was remarkably increased in response to recombinant HMGB1 challenges. Pharmacologic blockade of TLR2/4 attenuated HMGB1-mediated activation of the COX2/PGE2 pathway. Interestingly, HMGB1 did not impact the production of tumor necrosis factor-α or interleukin-1β in astrocytes. Our results suggest that HMGB1 mediates the astrocyte inflammatory response through regulating the COX2/PGE2 signaling pathway. The study was approved by the Laboratory Animal Ethics Committee of Nantong University, China (approval No. 20181204-001) on December 4, 2018.
Chinese Library Classification No. R446.1; R745.4; R364.5
Introduction
Spinal cord injury (SCI) is a serious public health problem worldwide and often leads to neurological complications or even paraplegia and quadriplegia (Soden et al., 2000; Binder, 2013; Lee et al., 2014; Wahman et al., 2019; Telemacque et al., 2020; Zheng et al., 2020). The complex pathophysiology of SCI is attributed to primary injury along with sequential secondary damage (David and Kroner, 2011; Wilson et al., 2013; Anwar et al., 2016). The inflammatory response is an important part of the pathological series of events that is triggered by activation of immune cells including microglia, astrocytes, and infiltrating leukocytes (Hausmann, 2003; David and Kroner, 2011; Ransohoff and Brown, 2012; Anwar et al., 2016). Accumulative evidence indicates that acute inflammation is an adaptive and homeostatic response of the body to injury, with a controversial neurotoxic or neuroprotective role, depending on the extent of the response and cell subsets involved (Lai and Todd, 2008; Pinteaux et al., 2009; Okada, 2016). The excessive inflammation induced by SCI exacerbates the detrimental microenvironment, leading to axonal and neuronal deficits (Anwar et al., 2016). Therefore, controlling excessive activation of inflammation is a potential strategy for clinical treatment of SCI.
Following SCI, damage-associated molecular pattern molecules, such as high mobility group box 1 (HMGB1), adenosine triphosphate, and heat shock proteins, are passively released by necrotic cells or actively secreted by immune cells. They interact with pattern-recognition receptors (PRRs) of immune cells to activate inflammatory responses (Bloom et al., 2020). Microglia and infiltrating myeloid cells are considered central players of innate immune responses in the injured spinal cord because of their expression of a wide range of PRRs including the Toll-like receptors (TLRs), nucleotide-binding oligomerization domain proteins, and scavenger receptors (Gadani et al., 2015; Vénéreau et al., 2015). However, mounting evidence suggests that astrocytes also play an active role in neuroinflammation associated with traumatic central nervous system (CNS) injury by expressing a similar PRR profile to microglia (Farina et al., 2007; Colombo and Farina, 2016; Liddelow and Barres, 2017). For example, astrocytes upregulate the interleukin (IL)-17 receptor and recruit nuclear factor-κB activator 1 to form a signaling complex, which facilitates the production of pro-inflammatory cytokines, chemokines, and metalloproteinases during neuroinflammation (Qian et al., 2007). Macrophage migration inhibitory factor has been documented to facilitate inflammation of glial cells by interacting with the CD74 receptor (Zhou et al., 2018). Given that astrocytes are subjected to various inflammatory stimuli in the microenvironment, many inflammation-related signaling pathways will thus be activated to affect the neuropathology of the injured CNS. The extent of neuroinflammation contributed by astrocytes relies on the severity, location, and timing of the CNS damage (Colombo and Farina, 2016).
HMGB1 is a ubiquitous and abundant nuclear protein that participates in nucleosome formation and regulation of gene transcription (Chen et al., 2011). HMGB1 is also characterized as a potent proinflammatory cytokine that mediates the progression of multiple inflammatory diseases (Lotze and Tracey, 2005; Harris et al., 2012). Once released from necrotic or inflammatory cells, HMGB1 interacts with surface receptors, including TLR4, TLR2, and the receptor for advanced glycation end products (RAGE), of monocyte/macrophages to induce tumor necrosis factor-α (TNF-α) and IL-1β production (Ulloa and Messmer, 2006; Andersson and Tracey, 2011; Tsung et al., 2014; Paudel et al., 2019a). In traumatic and ischemic SCI models, HMGB1 is rapidly induced to trigger inflammation by ligation to RAGE and TLRs of infiltrating macrophages or resident microglia (Chen et al., 2011; Kigerl et al., 2018), thereby contributing to the induction of neuronal apoptosis (Kawabata et al., 2010). Therefore, HMGB1 is assumed to be a pivotal regulator of post-SCI inflammation and a potential therapeutic target after SCI (Kim et al., 2006). A variety of signaling pathways relevant to small GTPases, mitogen-activated protein kinases, and nuclear factor-κB are activated in response to HMGB1 stimulation in various cell types (Dumitriu et al., 2005). However, the contribution of HMGB1-mediated astrocyte inflammation and the underlying mechanism remain unclear.
Cyclooxygenase (COX) 2 is a key enzyme in arachidonic acid metabolism that mediates prostanoid production (Font-Nieves et al., 2012). Distinct with the constitutive tissue expression of COX1, COX2 is strongly induced by pro-inflammatory challenges in the CNS under pathological conditions (Gopez et al., 2005; Zhang et al., 2019). COX2 has been shown to be involved in neuronal death and neuroinflammatory responses of the CNS, and inhibition of COX2 provides beneficial effects against damage (Gopez et al., 2005; Candelario-Jalil and Fiebich, 2008). Interestingly, astrocytes are a primary source of COX2 production following CNS injury, suggesting a potential cell target for COX2-mediated inflammation (Font-Nieves et al., 2012; Zhang et al., 2019). HMGB1 can efficiently induce COX2 expression in vascular smooth muscle cells and synovial fibroblasts (Jaulmes et al., 2006; Leclerc et al., 2013). It is postulated that HMGB1 activates COX2 expression in astrocytes, which might mediate the pathology of the injured spinal cord. In this study, we performed transcriptome sequencing with a rat SCI model to gain insight into the potential relationship between extracellular expression of HMGB1 and the inflammatory response in astrocytes. Then, we further investigated the mechanism of HMGB1-mediated prostaglandin E2 (PGE2) production in astrocytes in vitro.
Materials and Methods
Establishment of a rat SCI model
All animal experiments were performed in accordance with the US National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (National Research Council, 1996, USA) and approved by the Laboratory Animal Ethics Committee of Nantong University, China (Approval No. 20181204-001) on December 4, 2018. A total of 60 adult, clean, male Sprague-Dawley rats, weighing 180–220 g and aged 2 months, were provided by the Experimental Animal Center of Nantong University (License Nos. SCXK (Su) 2014-0001 and SYXK (Su) 2012-0031). Based on the post-operative time, rats were randomly divided into four groups, 0, 1, 4, and 7 days, and housed in individual cages in a temperature- and light/dark cycle-controlled environment. All experiments were designed and reported according to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
The SCI surgery performed on animals was previously described (Su et al., 2017). In brief, after deep anesthesia with an intraperitoneal injection of 10% chloral hydrate (3 mg/kg; Richjoint, Shanghai, China), the spinal cord of the rat was exposed by laminectomy at vertebral level T8–10. Contusion injury was performed at the exposed spinal segment using the Infinite Horizons Impactor (Precision Systems, Kentucky, IL, USA) with a force of 150 kilodynes. The impact rod was removed immediately. After the operation, the muscle and skin incision were sutured and disinfected. Antibiotics were applied intraperitoneally to avoid post-operative infection. At the indicated time point (i.e., 0, 1, 4, and 7 days post injury), 1-cm-length segments centered at the injury epicenter were collected for transcriptome analysis, western blot assays, and immunofluorescence staining.
Transcriptome sequencing
Twenty-four injured spinal cord segments (1 cm in length) were collected at 0, 1, 4, and 7 days (n = 6 rats per group) after injury. Transcriptome sequencing was performed as described previously (Zhou et al., 2018). In brief, total RNA was extracted using the mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA) according to the manufacturer’s instructions. The poly(A) mRNA was then isolated by beads with oligo (dT), followed by fragmentation with the fragmentation buffer. The short fragments were used as templates to synthesize first-strand cDNA by random hexamer primers prior to synthesis of the second-strand cDNA using a buffer mixture containing DNA polymerase I, dNTPs, and RNase H. These short fragments were purified with QiaQuick extraction kits (Qiagen, Duesseldorf, Germany) and end repaired by the addition of poly(A), followed by a connection with sequencing adaptors. Libraries were sequenced using an Illumina HiSeq2000 sequencer (Illumina, San Diego, CA, USA). Clean reads were aligned to the reference genomes or sequences of genes using the SOAP program. Less than five mismatches were allowed in the performed alignment. The sequencing quality was assessed by the proportions of the clean reads mapped back to the genome and genes. Gene coverage was defined by the percentage of a gene covered by the reads. Calculation of gene expression was performed by the reads per kb per million mapped reads method (Mortazavi et al., 2008).
Bioinformatic analysis
By comparison with the control (0 days), differentially expressed genes (DEGs) at different time points were designated according to the criteria of greater or less than a two-fold change. Functions of genes were annotated by Blastx against the NCBI or AGRIS database with an E-value threshold of 1 × 10–5. To create heatmaps, DEGs were clustered using the Jensen-Shannon Divergence method. Gene Ontology (GO) classification of DEGs was based on WEGO (http://wego.genomics.org.cn) via the GO id annotated by Perl (https://www.perl.org/) and the R program (https://www.r-project.org/). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using the KEGG Automatic Annotation Server (KAAS) online (https://www.genome.jp/tools/kaas/). The gene regulatory network was constructed using Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, Redwood City, CA, USA) on the basis of DEGs to predict their regulatory relationships.
Western blot assay
Protein was extracted with tissue lysis buffer (Beyotime, Shanghai, China) from spinal segments (n = 6) of 1 cm in length centered on the injury epicenter at 0, 1, 4, and 7 days post-surgery. Alternatively, protein was extracted from primary astrocytes treated with different concentrations of recombinant HMGB1 (rHMGB1; 0, 1, 10, 100, 1000, 2000 ng/mL) using cell lysis buffer (Beyotime). Proteins were quantified and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then were electro-transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked with 5% non-fat milk in Tris-buffered saline for 2 hours, followed by incubation with an appropriate primary antibody at 4°C overnight. The following primary antibodies were used: rabbit anti-HMGB1 (1:200; Cat# 11515; Cayman, Ann Arbor, MI, USA), human anti-TLR4 (1:100; Cat# AF1478; R&D, Minneapolis, MN, USA), rabbit anti-COX1 (1:1000; Cat# 4841S; CST, Danvers, MA, USA), rabbit anti-cytosolic PGE synthase (cPGES; 1:2000; Cat# ab92503; Abcam, Cambridge, UK), rabbit anti-COX2 (1:2000; Cat# 160126; Cayman), rabbit anti-microsomal PGE synthase-1 (mPGES-1; 1:200; Cat# 160140; Cayman), rabbit anti-mPGES-2 (1:200; Cat# 160145; Cayman), and mouse anti-β-actin (1:5000; Cat#:66009-1; Proteintech, Rosemont, IL, USA). The binding antibodies were then detected by horseradish peroxidase-labeled goat anti-rabbit IgG (1:1000; Cat# A0208; Beyotime), horseradish peroxidase-labeled goat anti-mouse IgG (1:1000; Cat# A0216; Beyotime), or horseradish peroxidase-labeled goat anti-human IgG (1:1000; Cat# A0201; Beyotime) prior to chemiluminescence enhancement (Beyotime). Protein bands were analyzed using PDQuest 7.2.0 software (Bio-Rad). β-Actin was used as an internal control. Assays were performed three times using triplicate wells.
Tissue immunofluorescence
The injured cord segments were harvested, post-fixed, and sectioned at 10 μm. Sections were co-incubated with mouse anti-glial fibrillary acidic protein (GFAP) polyclonal antibody (1:500; Cat# G3893; Sigma, St. Louis, MO, USA) and human anti-TLR4 polyclonal antibody (1:40; Cat# AF1478; R&D) or rabbit anti-COX2 polyclonal antibody (1:200; Cat# 160126; Cayman) at 4°C for 24 hours. The sections were further reacted with Alexa Fluor®488-conjugated donkey anti-mouse IgG (1:500; Cat# ab150109; Abcam), Cy3-conjugated goat anti-rabbit IgG (1:1000; Cat# AP187C; Sigma), or Cy3-conjugated goat anti-human IgG (1:100; Cat# C2571; Sigma), followed by observation under a fluorescence microscope (Leica, Heidelberg, Germany).
Astrocyte culture
Spinal cords from 10 clean neonatal (1-day old) Sprague-Dawley rats supplied by the Experimental Animal Center of Nantong University were cut into small pieces and digested with 0.25% trypsin for 10 minutes at 37°C. The digestion was neutralized with Dulbecco’s modified Eagle medium complete medium containing 10% fetal bovine serum and 1% penicillin/streptomycin. After centrifugation at 1200 r/minute for 5 minutes, the cells were resuspended and seeded onto poly-L-lysine pre-coated dishes. The culture was kept at 37°C in a 5% CO2 , 95% O2 humidified incubator. The medium was changed at 72 hours after initial seeding and then every other day until the cells reached confluence. The cells were purified by shaking at 200 r/min overnight to remove microglia, followed by purity determination using GFAP antibody and Hoechst 33342 (1:4000, Cat#: B2261; Sigma). Primary astrocytes isolated from the spinal cord were passaged no more than three times prior to use.
Measurement of PGE2 in vitro
Primary astrocytes were stimulated with 500 ng/mL of rHMGB1 (Cat# 1690-HMB; R&D) in the presence or absence of 10 nM atractylenolide I (TLR4 antagonist; Cat# A2737; Sigma) or 10 nM C29 (TLR2 inhibitor; Cat# 363600-92-4, MCE, Monmouth Junction, NJ, USA) dissolved in 0.1% dimethyl sulfoxide for 24 hours. The culture supernatants were then collected, and the cells were subjected to lysis with a buffer (Beyotime). The lysates were centrifuged at 10,000 × g for 15 minutes. The PGE2 concentration in cell supernatants and lysates was measured using a PGE2 high-sensitivity enzyme-linked immunosorbent assay (ELISA) kit (Arbor Assays, Ann Arbor, MI, USA) according to the manufacturer’s instructions.
TNF-α and IL-1β assay in vitro
Primary astrocytes were stimulated with 0–1000 ng/mL of rHMGB1 for 24 hours. The cell supernatants and lysates were collected as described above. Protein levels of TNF-α and IL-1β were measured using appropriate ELISA kits (MultiSciences, Hangzhou, China) according to the manufacturer’s instructions.
Statistical analysis
SPSS 20.0 software (IBM, Armonk, NY, USA) was used to analyze the experimental data. The results are presented as the mean ± standard error of the mean (SEM). Statistical significance of differences among groups was analyzed using one-way analysis of variance followed by Tukey’s post-hoc test. A level of P < 0.05 was considered statistically significant.
Results
Transcriptome profiles of SCI reveal HMGB1 involvement in COX2 signaling
To reveal the potential regulatory mechanism of HMGB1 in the inflammatory response of astrocytes, we analyzed the gene expression profiles of the rat spinal cord following injury at 0, 1, 4, and 7 days by transcriptome sequencing. A total of 1618 DEGs with a greater or less than twofold change were identified (Figure 1A and Additional Table 1 (1.6MB, pdf) ). GO analysis revealed that these DEGs were significantly enriched in T cell immune response, immune cell differentiation, prostaglandin secretion, inflammatory cell aggregation, and chemokine activation (Additional Figure 1 (3.7MB, tif) ). KEGG analysis identified that signaling pathways relevant to TLRs, cytokine-cytokine receptor interaction, and arachidonic acid metabolism were included in the top 30 significant functional enrichment results (Additional Figure 1 (3.7MB, tif) ). We further integrated these DEGs at different time points to narrow the scope of bioinformatic analysis and characterized 204 functional genes, among which 97 DEGs were involved in the inflammatory response and cell chemotaxis (Figure 1B). These 97 DEGs displayed dynamic alteration following SCI, as presented by the heat map (Figure 2). HMGB1 is known as a key pro-inflammatory factor that evokes an inflammatory response through binding with TLR2/4 or RAGE (Park et al., 2004; Paudel et al., 2019b). To understand HMGB1-mediated intracellular signaling in astrocytes, we performed IPA based on inflammation-related DEGs integrated at 1, 4, and 7 days following SCI. A reconstructed gene network was created, identifying that COXs (also known as Ptges), TLR4, and myeloid differentiation factor 88 were exclusively highlighted as the prominent amplifiers of inflammatory signaling with a core regulator for HMGB1 (Figure 3). The transcriptome profile analysis of SCI indicated that the pro-inflammatory factor HMGB1 may be involved in the activation of COX enzymes via TLR4.
Figure 1.
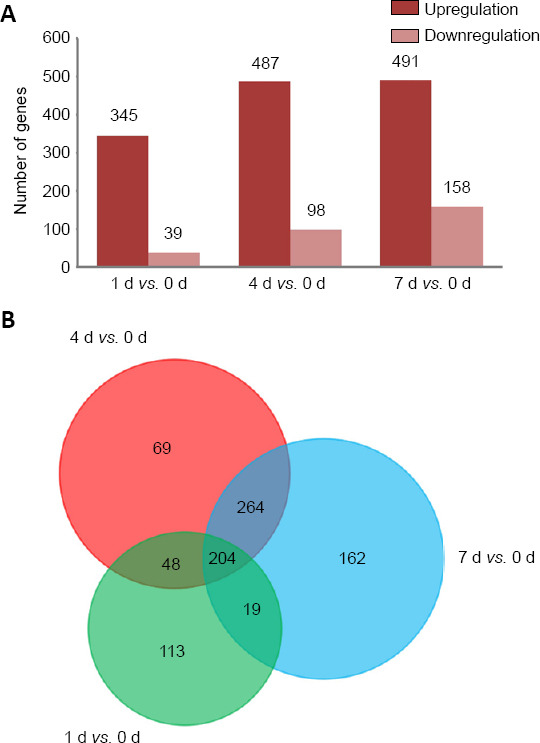
Analysis of mRNA expression profiles detected by transcriptome sequencing following spinal cord injury.
(A) Bar graph of the number of upregulated and downregulated genes in the injured spinal cord at 0, 1, 4, and 7 days post-injury as compared with those at control (0 days). (B) Overlap of differentially expressed genes at 1, 4, and 7 days post spinal cord injury.
Figure 2.
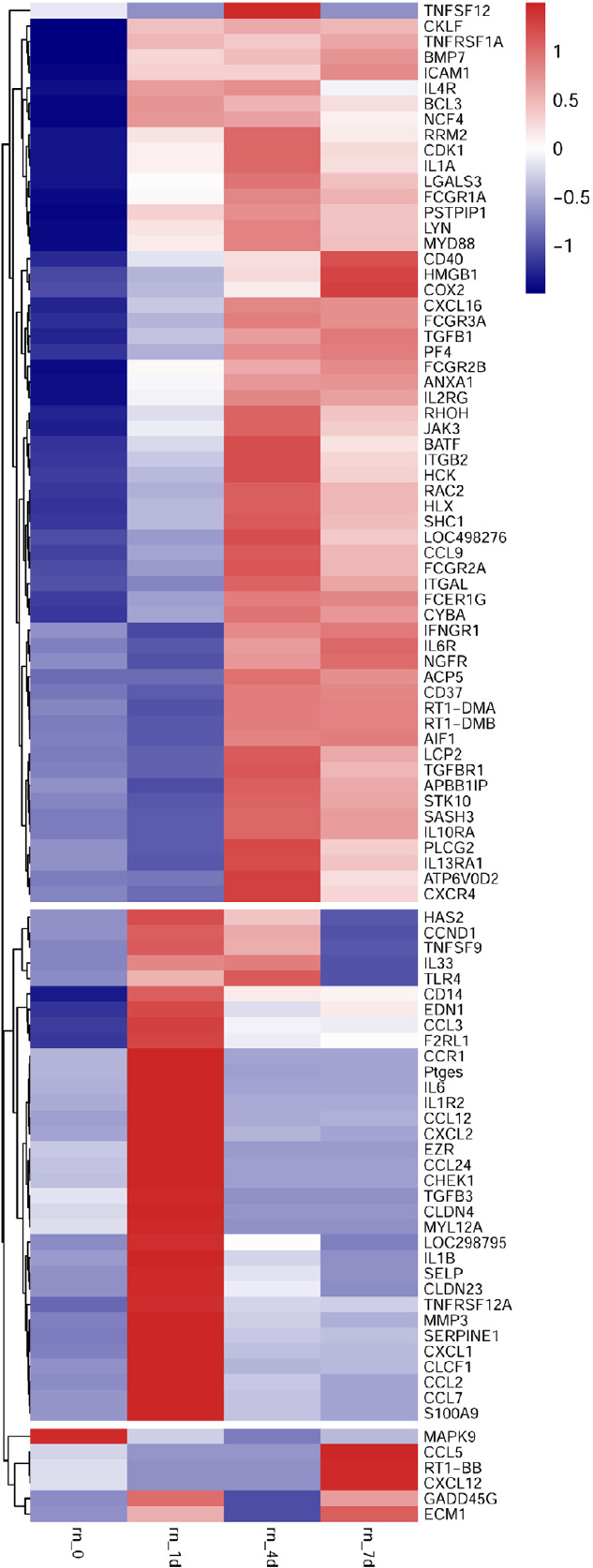
Heatmap and cluster dendrogram of integrated differentially expressed genes involved in inflammatory responses at 0, 1, 4, and 7 days following spinal cord injury.
The color scale shown at the top illustrates the relative expression level of the indicated mRNA across all samples. Red denotes expression > 0, and blue denotes expression < 0.
Figure 3.
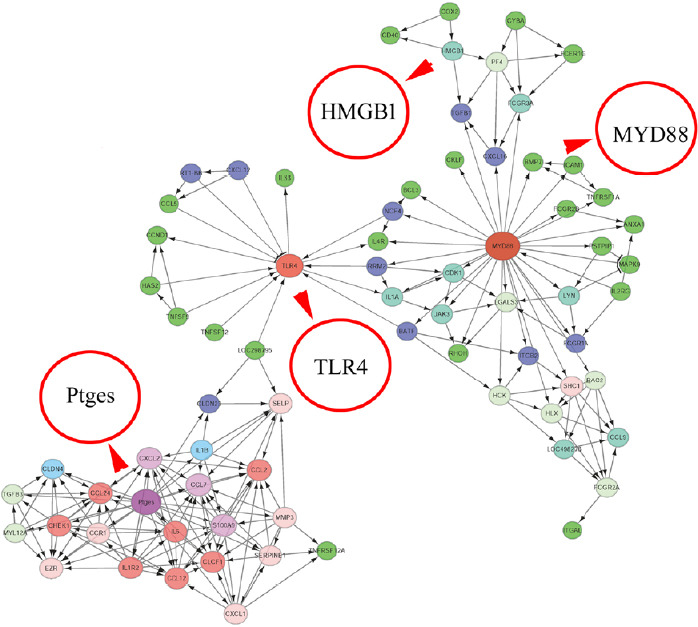
A reconstructed gene network was created using the Ingenuity Pathway Analysis software on the basis of integrated differentially expressed genes involved in inflammatory responses.
The different circles represent gene weights, while the arrows indicate interaction relationships. HMGB1: High mobility group box 1; MYD88: myeloid differentiation factor 88; Ptges: prostaglandin E synthase; TLR4: Toll-like receptor 4.
Expression changes of HMGB1, TLR4, COX2, and PGE2 synthase following SCI
To validate the involvement of the HMGB1/TLR4 axis in the regulation of COX expression in the astrocytes, we first detected the temporal changes in HMGB1, TLR4, COX1, and COX2, as well as the isoforms of PGE2 synthase in the injured spinal cord segments at 0, 1, 4, and 7 days following SCI. Western blot analysis revealed that HMGB1 and TLR4 expression levels were significantly upregulated and peaked at 4 and 7 days, respectively (Figure 4A–C). Meanwhile, COX2 and mPGES-1, but not COX1, mPGES-2, or cPGES, were inducibly expressed following SCI with a peak level at 1 day (Figure 4D–H).
Figure 4.

Protein expression changes in HMGB1, TLR4, COXs, and PGESs in the injured spinal cord of rats following spinal cord injury.
(A) Western blot analysis of HMGB1, TLR4, COX1, COX2, mPGES-1, mPGES-2, and cPGES following spinal cord contusion at 0, 1, 4, and 7 days. β-Actin was used as an internal control. (B–H) Quantification data of relative expression of HMGB1 (B), TLR4 (C), COX1 (D), COX2 (E), mPGES-1 (F), mPGES-2 (G), and cPGES (H). Relative expression is expressed as the optical density ratio to β-actin. Experiments were performed in triplicate. Data are expressed as the mean ± SEM (n = 6). *P < 0.05 (one-way analysis of variance followed by Tukey’s post hoc test). COX: Cyclooxygenase; cPGES: cytosolic prostaglandin E synthase; HMGB1: high mobility group box 1; mPGES: microsomal prostaglandin E synthase; TLR4: Toll-like receptor 4.
Subsequently, immunofluorescence staining was used to clarify that COX2 upregulation was astrocyte-related and a response to HMGB1 stimulation. As shown in Figure 5, both TLR4 and COX2 colocalized with GFAP-positive cells in the injured spinal cord, indicating a potential correlation of the HMGB1/TLR4 axis and COX2 expression in astrocytes.
Figure 5.

Co-localization of TLR4 and COX2 with astrocytes in the injured spinal cord of rats following SCI.
(A) Illustration of section sites showing immunofluorescence. The rectangle indicates the observed field. (B) Immunofluorescence analysis of TLR4 (red, stained by Cy3) co-localization with GFAP-positive cells (green, stained by fluorescein isothiocyanate) in the injured spinal segments following SCI at 0 and 4 days. (C) Immunofluorescence analysis of COX2 (red, stained by Cy3) co-localization with GFAP-positive cells (green, stained by fluorescein isothiocyanate) in injured spinal segments following SCI at 0 and 4 days. The rectangle indicates the magnified region. Scale bars: 50 μm. COX2: Cyclooxygenase 2; GFAP: glial fibrillary acidic protein; SCI: spinal cord injury; TLR4: Toll-like receptor 4.
The HMGB1/TLR4 signaling axis facilitates COX2 expression and PGE2 production in primary cultured astrocytes
Next, we sought to confirm the regulatory relationship between HMGB1 and COX2 in astrocytes. Primary cultured astrocytes with a purity of over 90% were stimulated with 0, 1, 10, 100, 1000, or 2000 ng/mL of rHMGB1 (Figure 6A). After exposure to rHMGB1 for 24 hours, the protein levels of COX1, COX2, and the isoforms of PGE2 synthase were detected by western blots. As shown in Figure 6B–G, rHMGB1 significantly induced COX2 and mPGES-1 expression without affecting COX1, mPGES-2, or cPGES expression, which was consistent with the in vivo results.
Figure 6.

Effects of rHMGB1 on the expression of PGE2 synthesis-related proteins in astrocytes.
(A) Purified primary astrocytes stained with GFAP (green, stained by fluorescein isothiocyanate) and Hoechst 33342 (blue). (B) Western blot analysis of COX1, COX2, mPGES-1, mPGES-2, and cPGES in astrocytes following incubation with different concentrations of rHMGB1 for 24 hours. β-Actin was used as an internal control. (C–G) Quantification data of relative expression of COX1 (C), COX2 (D), mPGES-1 (E), mPGES-2 (F), and cPGES (G). The relative expression is expressed as the optical density ratio to β-actin. Experiments were performed in triplicate. Data are expressed as the mean ± SEM. *P < 0.05 (one-way analysis of variance followed by Tukey’s post hoc test). COX: Cyclooxygenase; cPGES: cytosolic prostaglandin E synthase; GFAP: glial fibrillary acidic protein; mPGES: microsomal prostaglandin E synthase; PGE2: prostaglandin E2; rHMGB1: recombinant high mobility group box 1; TLR4: Toll-like receptor 4.
To ascertain whether HMGB1-mediated COX2 elevation is TLR-dependent, astrocytes were treated with 10 nM atractylenolide I or 10 nM C29 for 24 hours in the presence of 500 ng/mL of rHMGB1. The COX2 protein level was determined, and the results showed that the addition of atractylenolide I or C29 efficiently attenuated rHMGB1-induced activation of COX2 (Figure 7A and B). An ELISA was subsequently performed to detect PGE2 production in astrocytes. As shown in Figure 7C and D, rHMGB1 could facilitate PGE2 production in astrocytes, and application of 10 nM atractylenolide I or 10 nM C29 accordingly abrogated the stimulatory effects of rHMGB1. The results indicate that HMGB1 activates the COX2/mPGES1/PGE2 cascade in astrocytes via TLR2/4.
Figure 7.

Effects of TLR2/4 inhibitors on HMGB1-induced COX2 activation and PGE2 production in astrocytes.
Primary cultured astrocytes were stimulated with 500 ng/mL rHMGB1 in the presence of 10 nM AO-I (TLR4 antagonist) or 10 nM C29 (TLR2 inhibitor) for 24 hours. The vehicle (0.1% dimethyl sulfoxide) was used as a control. (A) The COX2 protein level was determined by a western blot assay. (B) Quantification data of COX2 as shown in A. (C, D) PGE2 production in the lysate (C) and supernatant (D) was determined by an enzyme-linked immunosorbent assay. Experiments were performed in triplicate. Data are expressed as the mean ± SEM. *P < 0.05 (one-way analysis of variance followed by Tukey’s pos hoc test). AO-I: Atractylenolide I; COX2: cyclooxygenase 2; HMGB1: high mobility group box 1; PGE2: prostaglandin E2; rHMGB1: recombinant high mobility group box 1; TLR: Toll-like receptor.
HMGB1 fails to activate the classical inflammatory signaling pathway in primary astrocytes
HMGB1, a key proinflammatory cytokine following SCI, has been demonstrated to participate in promoting the release of TNF-α and IL-1β from microglia/macrophages through interaction with TLR2/4 or RAGE (Park et al., 2004; Kigerl et al., 2018). Whether similar effects are shared by astrocytes that express TLR2/4 remains unknown. To address such a possibility, primary astrocytes were incubated with 0–1000 ng/mL rHMGB1 for 24 hours, and the protein levels of TNF-α and IL-1β in the supernatant and lysates were subsequently determined by ELISA. The results demonstrated that HMGB1 did not impact TNF-α and IL-1β production in astrocytes (Figure 8). The data indicate that HMGB1 exclusively mediates astrocyte inflammation through activation of COX2/PGE2 signaling.
Figure 8.

HMGB1 fails to promote TNF-α and IL-1β production in astrocytes.
An enzyme-linked immunosorbent assay was used to determine TNF-α (A, B) and IL-1β (C, D) production in the supernatants and lysates of astrocytes following treatment with 0, 1, 10, 100, or 1000 ng/mL rHMGB1 for 24 hours. Experiments were performed in triplicate. Data are expressed as the mean ± SEM and analyzed by one-way analysis of variance followed by Tukey’s post hoc test. HMGB1: High mobility group box 1; IL-1β: interleukin-1β; rHMGB1: recombinant high mobility group box 1; TNF-α: tumor necrosis factor-α.
Discussion
Acute traumatic injury to the spinal cord usually initiates a cascade of secondary pathophysiological changes characterized by tissue edema, hemorrhage, influx of inflammatory cells, axonal degeneration and oligodendrocyte death, leading to a further loss of tissue and function (Rouanet et al., 2017). Multiple cell events and molecular mechanisms including regulation of non-coding RNAs contribute to such pathological processes (Ning et al., 2014; Zhang et al., 2020). A variety of strategies are being developed to avert specific secondary insults, among which early interventions aimed at the excessive inflammatory response have been shown to be beneficial for preventing deterioration of the injured cord. However, many animal experiments and clinical trials have shown that it is difficult to obtain satisfactory outcomes in reducing CNS inflammation by pharmacological intervention of immune cells (Gorio et al., 2005; Singh et al., 2012; Kigerl et al., 2018; Orr and Gensel, 2018). For example, depletion of microglia or macrophages at the lesion site of cord tissue with clodronate liposomes could not completely attenuate axonal degeneration (Horn et al., 2008). Neutralization of TNF-α and IL-6 is insufficient to improve the inflammatory microenvironment of the injured spinal cord (Mukaino et al., 2010; Esposito and Cuzzocrea, 2011). These results suggest that a plethora of inflammatory cytokines derived from multiple cell types contributes to the excessive inflammation following SCI.
Astrocytes, the main CNS-resident glial cells that normally maintain homeostasis of the CNS, are rapidly activated by a variety of insults to form the reactive phenotype involved in scar formation and inflammatory modulation (Brambilla et al., 2005; Carpentier et al., 2005; Sofroniew, 2015; Liddelow and Barres, 2017). Reactive astrocytes express a wide range of PRRs including TLRs, which exhibit responses to the innate immune system by releasing inflammatory cytokines and chemokines that exacerbate SCI (Jack et al., 2005; Gorina et al., 2011; Sofroniew, 2015). Following SCI, the release of HMGB1 has been found to activate inflammatory responses of microglia/macrophages, monocytes, dendritic cells, and endothelial cells by inducing robust production of TNF-α, IL-1β, and IL-6. Pharmacological inhibition or genetic interference of HMGB1 can attenuate inflammation and improve functional recovery after SCI (Bi et al., 2017; Nakajo et al., 2019; Sun et al., 2019). In the present study, we demonstrated that HMGB1 failed to induce TNF-α and IL-1β production in spinal astrocytes and instead facilitated COX2 activation and proinflammatory PGE2 production, suggesting a novel regulatory mechanism of HMGB1 on astrocyte inflammation.
Classically, the HMGB1/TLR2/4 axis initiates inflammatory responses by regulating downstream myeloid differentiation factor 88, IL-1-receptor-associated kinases 1/4, TNF receptor-associated factor 6, and receptor-interacting protein, leading to activation of the inflammasome and the production of inflammatory cytokines such as TNF-α and IL-1β (Paudel et al., 2019a). Among these effectors, receptor-interacting protein has been shown to be specifically expressed in inflammatory cells and oligodendrocytes, but not in astrocytes of the CNS (Daston and Ratner, 1994). Therefore, it is reasonable that the HMGB1/TLR4 axis of astrocytes inefficiently triggers TNF-α- and IL-1β-related pathways in microglia/macrophages. Whether an enforced expression of receptor-interacting protein in astrocytes can restore HMGB1-mediated production of TNF-α and IL-1β deserves further study. The HMGB1/TLRs axis has also been shown to mediate the inflammatory process by inducing COX2 activation and PGE2 production in vascular smooth muscle cells and synovial fibroblasts (Jaulmes et al., 2006; Leclerc et al., 2013). In the present study, we demonstrated that HMGB1/TLR4 promoted PGE2 production in astrocytes, suggesting a conserved mechanism of HMGB1 in activation of the COX2/PGE2 signaling pathway. We previously found that PGE2 production in astrocytes induced by migration inhibitory factor contributed to fine-tuning of the inflammatory microenvironment of the injured spinal cord (Zhang et al., 2019). COX2-derived PGE2 also participates in excitotoxic and ischemic neuronal cell death by engaging neuronal PGE2 type 1 receptors (Shimamura et al., 2013). Therefore, HMGB1-induced release of COX2-derived PGE2 might play a similar role after SCI. As a variety of inflammatory mediators are induced following SCI, clinical efforts to administer anti-inflammation therapy are usually unsuccessful. The application of a COX2 inhibitor and/or neutralizing antibody of HMGB1 after acute SCI may provide an alternative for neurological recovery.
In conclusion, the HMGB1 protein level was significantly elevated following SCI, which in turn activated the COX2/PGE2 inflammatory signaling pathway of astrocytes, rather than promoting TNF-α and IL-1β production, to mediate neuropathological progression. These findings provide new insight into the immune response mechanism of astrocytes after SCI.
Additional files:
Additional Table 1 (1.6MB, pdf) : The transcriptome profiles of rat injured spinal cord.
The transcriptome profiles of rat injured spinal cord
Additional Figure 1 (3.7MB, tif) : GO and KEGG analysis for inflammation-related differentially expressed genes in injured spinal cord at 1 (A), 4 (B), and 7 (C) days following spinal cord injury.
GO (left) and KEGG (right) analysis for inflammation-related differentially expressed genes in injured spinal cord at 1 (A), 4 (B), and 7 (C) days following spinal cord injury.
GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes.
Additional file 1: Open peer review report 1 (85.6KB, pdf) .
Footnotes
Conflicts of interest: The authors have declared that no competing interests exist.
Financial support: This study was supported by the National Key Research and Development Program of China, No. 2018YFC1105603 (to YJunW), the National Natural Science Foundation of China, No. 31871211 (to YJunW), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) (to YJunW), the China Postdoctoral Science Foundation, No. 2020M681689 (to YMH), and the Basic Scientific Research Projects of Nantong of China, No. JC2018065 (to HHS). The funder had no roles in the study design, conduction of experiment, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional review board statement: The study was ethically approved by the Laboratory Animal Ethics Committee of Nantong University, China (approval No. 20181204-001) on December 4, 2018.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Xavier P. Gaudin, NYU Grossman School of Medicine, USA.
Funding: This study was supported by the National Key Research and Development Program of China, No. 2018YFC1105603 (to YJunW), the National Natural Science Foundation of China, No. 31871211 (to YJunW), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) (to YJunW), the China Postdoctoral Science Foundation, No. 2020M681689 (to YMH), and the Basic Scientific Research Projects of Nantong of China, No. JC2018065 (to HHS).
P-Reviewer: Gaudin XP; C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Kreiner L, Yu J, Song LP; T-Editor: Jia Y
References
- 1.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anwar MA, Al Shehabi TS, Eid AH. Inflammogenesis of secondary spinal cord injury. Front Cell Neurosci. 2016;10:98. doi: 10.3389/fncel.2016.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bi Y, Zhu Y, Zhang M, Zhang K, Hua X, Fang Z, Zhou J, Dai W, Cui Y, Li J, You T. Effect of shikonin on spinal cord injury in rats via regulation of HMGB1/TLR4/NF-kB signaling pathway. Cell Physiol Biochem. 2017;43:481–491. doi: 10.1159/000480474. [DOI] [PubMed] [Google Scholar]
- 4.Binder H. Traumatic spinal cord injury. Handb Clin Neurol. 2013;110:411–426. doi: 10.1016/B978-0-444-52901-5.00035-6. [DOI] [PubMed] [Google Scholar]
- 5.Bloom O, Herman PE, Spungen AM. Systemic inflammation in traumatic spinal cord injury. Exp Neurol. 2020;325:113143. doi: 10.1016/j.expneurol.2019.113143. [DOI] [PubMed] [Google Scholar]
- 6.Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Candelario-Jalil E, Fiebich BL. Cyclooxygenase inhibition in ischemic brain injury. Curr Pharm Des. 2008;14:1401–1418. doi: 10.2174/138161208784480216. [DOI] [PubMed] [Google Scholar]
- 8.Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49:360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]
- 9.Chen KB, Uchida K, Nakajima H, Yayama T, Hirai T, Rodriguez Guerrero A, Kobayashi S, Ma WY, Liu SY, Zhu P, Baba H. High-mobility group box-1 and its receptors contribute to proinflammatory response in the acute phase of spinal cord injury in rats. Spine (Phila Pa 1976) 2011;36:2122–2129. doi: 10.1097/BRS.0b013e318203941c. [DOI] [PubMed] [Google Scholar]
- 10.Colombo E, Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. 2016;37:608–620. doi: 10.1016/j.it.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Daston MM, Ratner N. Amphoterin (P30, HMG-1) and RIP are early markers of oligodendrocytes in the developing rat spinal cord. J Neurocytol. 1994;23:323–332. doi: 10.1007/BF01188500. [DOI] [PubMed] [Google Scholar]
- 12.David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 2011;12:388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- 13.Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere-Querini P. HMGB1: guiding immunity from within. Trends Immunol. 2005;26:381–387. doi: 10.1016/j.it.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 14.Esposito E, Cuzzocrea S. Anti-TNF therapy in the injured spinal cord. Trends Pharmacol Sci. 2011;32:107–115. doi: 10.1016/j.tips.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Font-Nieves M, Sans-Fons MG, Gorina R, Bonfill-Teixidor E, Salas-Pérdomo A, Márquez-Kisinousky L, Santalucia T, Planas AM. Induction of COX-2 enzyme and down-regulation of COX-1 expression by lipopolysaccharide (LPS) control prostaglandin E2 production in astrocytes. J Biol Chem. 2012;287:6454–6468. doi: 10.1074/jbc.M111.327874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gadani SP, Walsh JT, Lukens JR, Kipnis J. Dealing with danger in the CNS: the response of the immune system to injury. Neuron. 2015;87:47–62. doi: 10.1016/j.neuron.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gopez JJ, Yue H, Vasudevan R, Malik AS, Fogelsanger LN, Lewis S, Panikashvili D, Shohami E, Jansen SA, Narayan RK, Strauss KI. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery. 2005;56:590–604. doi: 10.1227/01.NEU.0000154060.14900.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59:242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- 20.Gorio A, Madaschi L, Di Stefano B, Carelli S, Di Giulio AM, De Biasi S, Coleman T, Cerami A, Brines M. Methylprednisolone neutralizes the beneficial effects of erythropoietin in experimental spinal cord injury. Proc Natl Acad Sci U S A. 2005;102:16379–16384. doi: 10.1073/pnas.0508479102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol. 2012;8:195–202. doi: 10.1038/nrrheum.2011.222. [DOI] [PubMed] [Google Scholar]
- 22.Hausmann ON. Post-traumatic inflammation following spinal cord injury. Spinal Cord. 2003;41:369–378. doi: 10.1038/sj.sc.3101483. [DOI] [PubMed] [Google Scholar]
- 23.Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci. 2008;28:9330–9341. doi: 10.1523/JNEUROSCI.2488-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- 25.Jaulmes A, Thierry S, Janvier B, Raymondjean M, Maréchal V. Activation of sPLA2-IIA and PGE2 production by high mobility group protein B1 in vascular smooth muscle cells sensitized by IL-1beta. FASEB J. 2006;20:1727–1729. doi: 10.1096/fj.05-5514fje. [DOI] [PubMed] [Google Scholar]
- 26.Kawabata H, Setoguchi T, Yone K, Souda M, Yoshida H, Kawahara K, Maruyama I, Komiya S. High mobility group box 1 is upregulated after spinal cord injury and is associated with neuronal cell apoptosis. Spine (Phila Pa 1976) 2010;35:1109–1115. doi: 10.1097/BRS.0b013e3181bd14b6. [DOI] [PubMed] [Google Scholar]
- 27.Kigerl KA, Lai W, Wallace LM, Yang H, Popovich PG. High mobility group box-1 (HMGB1) is increased in injured mouse spinal cord and can elicit neurotoxic inflammation. Brain Behav Immun. 2018;72:22–33. doi: 10.1016/j.bbi.2017.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, Lee MH, Han PL, Park JS, Lee JK. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci. 2006;26:6413–6421. doi: 10.1523/JNEUROSCI.3815-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai AY, Todd KG. Differential regulation of trophic and proinflammatory microglial effectors is dependent on severity of neuronal injury. Glia. 2008;56:259–270. doi: 10.1002/glia.20610. [DOI] [PubMed] [Google Scholar]
- 30.Leclerc P, Wähämaa H, Idborg H, Jakobsson PJ, Harris HE, Korotkova M. IL-1β/HMGB1 complexes promote The PGE2 biosynthesis pathway in synovial fibroblasts. Scand J Immunol. 2013;77:350–360. doi: 10.1111/sji.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee BB, Cripps RA, Fitzharris M, Wing PC. The global map for traumatic spinal cord injury epidemiology: update 2011, global incidence rate. Spinal Cord. 2014;52:110–116. doi: 10.1038/sc.2012.158. [DOI] [PubMed] [Google Scholar]
- 32.Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46:957–967. doi: 10.1016/j.immuni.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 33.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 34.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 35.Mukaino M, Nakamura M, Yamada O, Okada S, Morikawa S, Renault-Mihara F, Iwanami A, Ikegami T, Ohsugi Y, Tsuji O, Katoh H, Matsuzaki Y, Toyama Y, Liu M, Okano H. Anti-IL-6-receptor antibody promotes repair of spinal cord injury by inducing microglia-dominant inflammation. Exp Neurol. 2010;224:403–414. doi: 10.1016/j.expneurol.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 36.Nakajo M, Uezono N, Nakashima H, Wake H, Komiya S, Nishibori M, Nakashima K. Therapeutic time window of anti-high mobility group box-1 antibody administration in mouse model of spinal cord injury. Neurosci Res. 2019;141:63–70. doi: 10.1016/j.neures.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 37.Ning B, Gao L, Liu RH, Liu Y, Zhang NS, Chen ZY. microRNAs in spinal cord injury: potential roles and therapeutic implications. Int J Biol Sci. 2014;10:997–1006. doi: 10.7150/ijbs.9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okada S. The pathophysiological role of acute inflammation after spinal cord injury. Inflamm Regen. 2016;36:20. doi: 10.1186/s41232-016-0026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orr MB, Gensel JC. Spinal cord injury scarring and inflammation: therapies targeting glial and inflammatory responses. Neurotherapeutics. 2018;15:541–553. doi: 10.1007/s13311-018-0631-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 41.Paudel YN, Angelopoulou E, C BK, Piperi C, Othman I. High mobility group box 1 (HMGB1) protein in multiple sclerosis (MS): mechanisms and therapeutic potential. Life Sci. 2019a;238:116924. doi: 10.1016/j.lfs.2019.116924. [DOI] [PubMed] [Google Scholar]
- 42.Paudel YN, Angelopoulou E, Piperi C, Balasubramaniam V, Othman I, Shaikh MF. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur J Pharmacol. 2019b;858:172487. doi: 10.1016/j.ejphar.2019.172487. [DOI] [PubMed] [Google Scholar]
- 43.Pinteaux E, Trotter P, Simi A. Cell-specific and concentration-dependent actions of interleukin-1 in acute brain inflammation. Cytokine. 2009;45:1–7. doi: 10.1016/j.cyto.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 44.Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, Kordula T, Zhang QW, Vallance B, Swaidani S, Aronica M, Tuohy VK, Hamilton T, Li X. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 45.Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122:1164–1171. doi: 10.1172/JCI58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rouanet C, Reges D, Rocha E, Gagliardi V, Silva GS. Traumatic spinal cord injury: current concepts and treatment update. Arq Neuropsiquiatr. 2017;75:387–393. doi: 10.1590/0004-282X20170048. [DOI] [PubMed] [Google Scholar]
- 47.Shimamura M, Zhou P, Casolla B, Qian L, Capone C, Kurinami H, Iadecola C, Anrather J. Prostaglandin E2 type 1 receptors contribute to neuronal apoptosis after transient forebrain ischemia. J Cereb Blood Flow Metab. 2013;33:1207–1214. doi: 10.1038/jcbfm.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh PL, Agarwal N, Barrese JC, Heary RF. Current therapeutic strategies for inflammation following traumatic spinal cord injury. Neural Regen Res. 2012;7:1812–1821. doi: 10.3969/j.issn.1673-5374.2012.23.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soden RJ, Walsh J, Middleton JW, Craven ML, Rutkowski SB, Yeo JD. Causes of death after spinal cord injury. Spinal Cord. 2000;38:604–610. doi: 10.1038/sj.sc.3101080. [DOI] [PubMed] [Google Scholar]
- 50.Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16:249–263. doi: 10.1038/nrn3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Su Y, Wang Y, Zhou Y, Zhu Z, Zhang Q, Zhang X, Wang W, Gu X, Guo A, Wang Y. Macrophage migration inhibitory factor activates inflammatory responses of astrocytes through interaction with CD74 receptor. Oncotarget. 2017;8:2719–2730. doi: 10.18632/oncotarget.13739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun L, Li M, Ma X, Zhang L, Song J, Lv C, He Y. Inhibiting high mobility group box-1 reduces early spinal cord edema and attenuates astrocyte activation and aquaporin-4 expression after spinal cord injury in rats. J Neurotrauma. 2019;36:421–435. doi: 10.1089/neu.2018.5642. [DOI] [PubMed] [Google Scholar]
- 53.Telemacque D, Zhu FZ, Ren ZW, Chen KF, Drepaul D, Yao S, Yang F, Qu YZ, Sun TF, Guo XD. Effects of durotomy versus myelotomy in the repair of spinal cord injury. Neural Regen Res. 2020;15:1814–1820. doi: 10.4103/1673-5374.280304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsung A, Tohme S, Billiar TR. High-mobility group box-1 in sterile inflammation. J Intern Med. 2014;276:425–443. doi: 10.1111/joim.12276. [DOI] [PubMed] [Google Scholar]
- 55.Ulloa L, Messmer D. High-mobility group box 1 (HMGB1) protein: friend and foe. Cytokine Growth Factor Rev. 2006;17:189–201. doi: 10.1016/j.cytogfr.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 56.Vénéreau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol. 2015;6:422. doi: 10.3389/fimmu.2015.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wahman K, Nilsson Wikmar L, Chlaidze G, Joseph C. J Rehabil Med. Vol. 51. Sweden: Towards developing prevention strategies; 2019. Secondary medical complications after traumatic spinal cord injury in Stockholm; pp. 513–517. [DOI] [PubMed] [Google Scholar]
- 58.Wilson JR, Forgione N, Fehlings MG. Emerging therapies for acute traumatic spinal cord injury. CMAJ. 2013;185:485–492. doi: 10.1503/cmaj.121206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X, Guo H, Xie A, Liao O, Ju F. microRNA-331-3p attenuates neuropathic pain following spinal cord injury via targeting RAP1A. J Biol Regul Homeost Agents. 2020;34:25–37. doi: 10.23812/19-291-A. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y, Zhou Y, Chen S, Hu Y, Zhu Z, Wang Y, Du N, Song T, Yang Y, Guo A, Wang Y. Macrophage migration inhibitory factor facilitates prostaglandin E(2) production of astrocytes to tune inflammatory milieu following spinal cord injury. J Neuroinflammation. 2019;16:85. doi: 10.1186/s12974-019-1468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng Y, Mao YR, Yuan TF, Xu DS, Cheng LM. Multimodal treatment for spinal cord injury: a sword of neuroregeneration upon neuromodulation. Neural Regen Res. 2020;15:1437–1450. doi: 10.4103/1673-5374.274332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou Y, Guo W, Zhu Z, Hu Y, Wang Y, Zhang X, Wang W, Du N, Song T, Yang K, Guan Z, Wang Y, Guo A. Macrophage migration inhibitory factor facilitates production of CCL5 in astrocytes following rat spinal cord injury. J Neuroinflammation. 2018;15:253. doi: 10.1186/s12974-018-1297-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The transcriptome profiles of rat injured spinal cord
GO (left) and KEGG (right) analysis for inflammation-related differentially expressed genes in injured spinal cord at 1 (A), 4 (B), and 7 (C) days following spinal cord injury.
GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes.