

Abstract
Sepsis represents a life-threatening event often mediated by the host’s response to pathogens such as gram-negative organisms, which release the pro-inflammatory lipopolysaccharide (LPS). Within the endothelium, the mitogen-activated protein kinase (MAPK) pathway is an important driver of endothelial injury during sepsis, of which oxidant-sensitive apoptosis signal-regulating kinase 1 (ASK1) is postulated to be a critical upstream regulator. We hypothesized that ASK1 would play a key role in endothelial inflammation during bacterial challenge. Utilizing RNA sequencing data from patients and cultured human microvascular endothelial cells (HMVECs), ASK1 expression was increased in sepsis and after LPS challenge. Two ASK1 inhibitors, GS444217 and MSC2023964A, reduced cytokine production in HMVECs following LPS stimulation, but had no effect on permeability as measured by transendothelial electrical resistance (TEER) and intercellular space. MAPKs are known to interact with endothelial nitric oxide synthase (eNOS) and ASK1 expression levels correlated with eNOS expression in patients with septic shock. In addition, eNOS physically interacted with ASK1, though this interaction was not altered by ASK1 inhibition, nor did inhibition alter MAPK p38 activity. Instead, among MAPKs, ASK1 inhibition only impaired LPS-induced JNK phosphorylation. The reduction in JNK activation caused by ASK1 inhibition impaired JNK-mediated cytokine production without affecting permeability. Thus, LPS triggers JNK-dependent cytokine production that requires ASK1 activation, but both its effects on permeability and activation of p38 are ASK1-independent. These data demonstrate how distinct MAPK signaling pathways regulate endothelial inflammatory outputs during acute infectious challenge.
INTRODUCTION:
Sepsis and its associated shock represent a common cause of morbidity and mortality in modern intensive care units [1, 2]. Sepsis, which is the host’s response to a pathogenic challenge, including gram-positive and gram-negative organisms, results in a systemic inflammatory response through the propagation of multiple inflammatory cascades [3]. This results in significant injury to the host and is a critical underlying driver of the severity of illness. A final common pathway of this injury is inflammation within the endothelium, resulting in maldistribution of blood flow, capillary leak and cytokinemia [4]. These derangements contribute to impaired oxygen delivery, reduced clearance of metabolites and tissue edema [5]. Identifying and understanding the pathways that drive endothelial dysfunction serves as a prelude to the identification of future therapeutic interventions for managing this disease.
In mammalian physiology, innate immunity plays a key role in the initial response of the body to infection by recognition of pathogen-associated molecular patterns (PAMPs). PAMPs are recognized by members of the toll-like receptor (TLR) family and result in downstream activation of mitogen-activated protein kinase (MAPK) pathways including c-Jun N-terminal kinase (JNK) and p38 mitogen-activated kinase (p38) [6, 7]. While various components of infectious agents can activate the TLR family including lipoproteins, peptidoglycans, and even nucleic acids [8], lipopolysaccharide (LPS) has been abundantly studied. It is a component of the outer membrane of gram-negative bacteria and leads to significant pro-inflammatory cytokine release which can cause endotoxin-associated septic shock [9, 10]. The interaction of LPS with LPS binding protein and CD14 lead to the eventual activation of TLR4 and its associated downstream signaling pathways [11]. One such pathway is the oxidant-dependent activation of apoptosis signal-regulated kinase 1 (ASK1) [12] that has been suggested to be a critical activator of both p38 and JNK [13, 14]. JNK and p38 are pro-inflammatory MAPKs that regulate cell death, cytokine production, and endothelial integrity [15, 16]. Direct inhibition of these proteins, either pharmacologically or genetically, have shown significant mortality/morbidity benefit in humans and mice [17–21]. Therefore, mechanisms to modulate, but not completely inhibit the signaling of p38 and JNK, have been sought for therapeutic purposes in patients with both acute and chronic inflammatory conditions.
In comparison to p38 and JNK inhibition, ASK1 inhibition has been found to be both safe and effective in humans (for a review, see Ogier et al [22]). ASK1 knockout mice are resistant to LPS-induced endotoxic shock by virtue of reduced reactive oxygen species-dependent activation of ASK1 and subsequent signaling through p38 [14]. Beyond direct downstream modulation of MAPKs, ASK1 has also been suggested to be involved in the maintenance of endothelial homeostasis through modulation of endothelial nitric oxide synthase (eNOS), a key regulator of nitric oxide and redox signaling [23, 24]. These effects could be via alterations in oxidative stress, however it could also be postulated that ASK1 activation would cause disruption in eNOS-MAPK protein-protein interactions, which have been demonstrated to alter the endothelial phenotype during infectious challenge [25, 26]. Therefore, we hypothesized that ASK1, being upstream of key members of the MAPK signaling pathway, would be an integral regulator of the host response to bacterial challenge, specifically in endothelial cells. By understanding the role of ASK1 in endothelial TLR4 signaling, we hoped to determine whether it could be considered a reasonable therapeutic target in sepsis-mediated endothelial dysfunction.
MATERIAL AND METHODS:
Cells and culture:
Pooled neonatal dermal human microvascular endothelial cells (HMVECs) were purchased from Lonza (Basel, Switzerland). HMVECs were grown in Endothelial Growth Media-2 (Lonza) supplemented with 5% fetal bovine serum (FBS). Cells were plated at a density of approximately 30,000 cells/cm2 and grown to confluence. Experiments were conducted between the 2nd and 5th passages. Media was exchanged at least every 3 days.
Transcript profiles of human samples:
Gene array profiles of blood samples were obtained from the publicly available National Centers for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database. Values were obtained from GEO Dataset (GSE13904) initially collected by Wong HR, et al. “Expression profiling across the pediatric systemic inflammatory response syndrome, sepsis, and septic shock spectrum” [27]. The database was queried for MAP3K5 (ASK1), MAPK14 (p38), MAPK8 (JNK1) and NOS3 (eNOS) from samples run on Affymetrix Human Genome U133 Plus 2.0 Array.
Agonist and inhibitor reagents:
The following reagents and concentrations were used in experiments: 1–25 μM GS444217 (Gilead Sciences, Foster City, CA, USA), 1–25 μM MSC2032964A (Tocris Biosciences Bristol, United Kingdom), 10 μM SB203580 (Cayman Chemical, Ann Arbor, MI, USA), 10 μM SP600125 (Cayman Chemical) and 100 ng/ml Ultra-Pure LPS (List Biological Laboratories, Campbell, CA, USA). Agonists, inhibitors, and vehicle controls were provided to cells in the presence of culture media.
RNA isolation and real-time polymerase chain reaction:
RNA was isolated from cultured cells 16 h after LPS exposure via GenElute Mammalian Total RNA Miniprep Kit (MilliporeSigma, St. Louis, MO, USA) following the manufacturer’s instructions. Afterwards, 1.5 μg of total RNA was reverse transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Life Technologies, Grand Island, NY, USA) according to the manufacturer’s protocol. Efficiency of the PCR reactions was tested by amplification of the target from serially diluted cDNA generated from reverse transcription to achieve an efficiency of 95% + 5%. Real-time PCR was performed using the TaqMan Fast Advanced Master Mix on a StepOnePlus Real Time PCR System (Applied Biosystems, Life Technologies). Prime Time® qPCR 5’ Nuclease Assays (Integrated DNA Technologies, Inc., Coralville, IA, USA) were used to amplify the target mRNAs containing the following sequences: 5′-GATGAAGACACTATAAGCCGGTT and 5’-GTCAATGATAGCCTTCCACAGT for ASK1; 5′-ACATCGCTCAGACACCATG and 5′-TGTAGTTGAGGTCAATGAAGGG for GAPDH. Data were normalized as a ratio of threshold cycle of target mRNA to GAPDH and corrected for efficiency using the StepOne software.
Western blot and electrophoresis:
Prior to treatment with LPS, cells were incubated with either ASK1 inhibitor or control for 30 minutes prior to LPS exposure. Cell lysates were collected at 90 min or 6 h (as indicated) after LPS exposure. Protein extracts (50μg/sample) were separated by SDS electrophoresis on a polyacrylamide gel (10%) and transferred to nitrocellulose membranes. Membranes were blocked with Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE, USA) for 1 hr at room-temperature. Membranes were incubated with primary antibodies overnight at 4 °C on a rocker. Antibodies were as follows: ASK1 (Novus Biologicals, Centennial, CO, USA), p-ASK1, IKKαβ, p-IKKβ, p-p38, p38, p-JNK, JNK, p-ERK, ERK, α-tubulin (Cell Signaling Technology, Danvers, MA, USA). Membranes were then incubated with fluorescent secondary antibodies and analyzed on the Odyssey Imaging System (LI-COR Biosciences). Protein quantification was performed via densitometry and normalized as a ratio of phosphorylated protein to respective total protein or ASK1 protein to tubulin.
siRNA transfection:
HMVECs were treated with siRNA (scrambled siControl, sieNOS) according to the manufacturer’s recommendations. In brief, siRNA was procured from Dharmacon (Lafayette, CO, USA). siRNA (25 nmol/L) was incubated with Dharmafect (Dharmacon) in serum-free medium for 20 min. The resultant complex of siRNA-Dharmafect was added to the cells in 5% FBS media without antibiotics for 6 h. Afterwards, the transfection media was replaced with complete media including antibiotics for another 66 h for a total of 72 h siRNA incubation time prior to agonist exposure or imaging.
Cytokine and chemokine production:
Supernatants from cell cultures were collected at the completion of agonist exposure (6 h) based on the timing of cytokine secretion from previously published data [28]. Collected supernatants were stored at −80 °C until analyzed. Supernatant IL-6 (eBioScience, San Diego, CA, USA), IL-8, G-CSF, and soluble VCAM (sVCAM) (R&D Systems, Minneapolis, MN, USA) concentrations were assessed using a commercially available enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s specifications.
Transendothelial electrical resistance (TEER) assay:
TEER analysis was performed utilizing the CellZScope2 (nanoAnalytics GmbH, Münster, Germany). Experiments were performed as previously reported [25]. Briefly, HMVECs were plated on ThinWell Cell Culture inserts (0.4μm pore diameter, Greiner Bio-One, Monroe, NC, USA), coated with poly-L-lysine (2x) for 20 min then washed with Dulbecco’s phosphatebuffered saline (D-PBS). Inserts were then coated with glutaraldehyde 50% (1000x) for 15 min, washed with D-PBS, and lastly coated with gelatin (40x), then washed again with D-PBS. Afterwards, 70% ethanol was applied for 30 min followed by an aldehyde scavenger for another 30 min in medium and then removed. Cells were plated at 40,000 cells/transwell and allowed to grow over 24 h until resistance stabilized. Afterwards, cells were pre-treated with 5 or 25 μM of an ASK1 inhibitor for 30 minutes and concurrently with exposure to LPS (100 ng/mL). TEER was measured continuously in 1 hour increments. Prior to analysis, data were normalized to resistance prior to inhibitor application. Area under the curve was then calculated relative to pre-inhibitor resistance. Averages for AUC with 95% CI were then calculated and compared per statistical analysis methods.
Intercellular space assay:
Assessment of intracellular space was completed as previously reported [26]. Briefly, glassbottom 96-well plates were used (Cellvis, Mountain View, CA, USA) with wells pre-treated with poly-l-lysine, glutaraldehyde, and biotinylated gelatin similar to as described above. HMVECs were seeded at a density of 30,000 cells/well and incubated for 1 day to achieve a confluent monolayer. Cells were treated with the inhibitors or vehicle control as indicated for 30 min prior to treatment with 100 ng/ml LPS. Following 7 h of incubation, cells were washed with 120 μl of Live Cell imaging solution (Thermo Fisher Scientific, Waltham, MA, USA). Cells were then fixed with 4% formaldehyde for 15 minutes. Cells were placed in Live Cell imaging solution and exposed to neutravidin–FITC (1:1000 dilution) for 15 minutes. Following, cells were blocked with 5% goat serum in PBS for 2 h and stained with VE-cadherin rabbit antibody (clone #D87F2, Cell Signaling Technology, Danvers, MA, USA) at 1:100 dilution overnight. Secondary antibody was then applied (Anti-rabbit IgG, 1:200, MilliporeSigma, St. Louis, MO, USA). Nucblue was added and allowed to incubate for 15 minutes. Cells were then washed and mounted using ibidi mounting medium (ibidi GmbH, Gräfelfing, Germany) and imaged using a Zeiss 710 confocal microscope with a 20X objective (Carl Zeiss AG, Oberkochen, Germany). Cell count was obtained via quantification of nuclear staining using ImageJ (National Institutes of Health, Bethesda, MD, USA) software, which ensured a consistent density of the endothelial layers, and monolayer integrity and VE-cadherin staining were assessed via quantification of the total area of fluorescein staining (ImageJ) within the field.
Proximity ligation assay (PLA):
Proximity ligation assays were performed using a commercially available kit according to the manufacturer’s instructions (Duolink PLA, MilliporeSigma, St. Louis, MO, USA). In brief, 96-well glass bottom plates were pretreated with poly-L-lysine, glutaraldehyde 50%, gelatin and an aldehyde scavenger in the same concentrations and times as described for TEER. If indicated, HMVECs were then plated and exposed to siRNA for 72 h total incubation time as previously described. Cells were exposed to GS444217 30 minutes prior to LPS stimulation. Following the indicated times, cells were washed with PBS and exposed to 4% formaldehyde fixative in PBS for 15 min then permeabilized with 0.1% TritonX-100 in PBS for 15 min. Following solution removal, 40 μL of Duolink blocking solution (MilliporeSigma) was placed in each well and incubated at 37 °C for 1 h. The blocking solution was then replaced with Duolink antibody diluent containing the following primary antibodies at a 1:100 dilution: eNOS rabbit mAb (clone #D9A5L, Cell Signaling Technology, Danvers, MA, USA), p-p38αmouse mAb (clone #28B10, Cell Signaling Technology, Danvers, MA, USA) and ASK1 mouse mAb (clone #2E4, Novus Biologicals, Littleton, CO, USA). Plates were then sealed and incubated overnight at 4 °C. The next day, the primary antibodies were removed, and the cells were washed with Duolink Wash Buffer. Duolink PLA probes (anti-rabbit secondary antibody with plus oligonucleotides and antimouse secondary antibody with minus oligonucleotides) were diluted in a 1:5 ratio in Duolink Antibody Diluent and 40 μL of probe containing solution was added to each well for 1 h at 37 °C. Kit ligase was added at a 1:40 dilution to Duolink Ligation Buffer, which contained premixed concentrations of bridging oligonucleotides, and added to each well for 30 min at 37 °C. Next, cells were washed and kit-provided polymerase was added in a 1:80 dilution in Duolink Amplification Buffer and applied to each well for 100 min at 37 °C. Duolink Orange fluorescent probes (ex 554/em 576) were present in the amplification buffer. Cells were then washed and Duolink Wash Buffer containing 1 drop NucBlue (ex 360/em 460, Thermo Fischer Scientific, Waltham, MA, USA) per mL of buffer was added to each well at room temperature for 10 min. The wash buffer was then removed and replaced with 120 μL of Live Cell Imaging Solution (Thermo Fischer Scientific) and imaged at the appropriate wavelength using an inverted confocal microscope (Zeiss LSM 880, Carl Zeiss AG, Oberkochen, Germany). Fluorescent reactions and cell number were counted by averaging two random fields per well using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis:
For cell culture experiments, data are expressed as means ± SE of multiple, individual experiments. Comparisons of treatment groups, controls, and other conditions were done via unpaired t-test for single comparisons and one-way ANOVA, with Bonferroni correction, for multiple-group comparisons. For RNA transcript expression of blood samples, data are expressed as medians ± 75% interquartile range for each data subset. Comparisons of groups were done via a Mann-Whitney test and Spearman correlation for direct expression comparison with 95% CI. All analysis was done using GraphPad Prism 9 statistical software (GraphPad Software Inc., La Jolla, CA USA). A p-value cutoff of < 0.05 was used for statistical significance.
RESULTS:
ASK1 RNA expression is increased in sepsis and after lipopolysaccharide challenge
ASK1 has previously been demonstrated to modulate pro-inflammatory p38 and JNK activity through its MAPK kinase kinase (MAPKKK) function [29], but its role in sepsis is poorly defined. In order to assess the potential role of ASK1 signaling in acute inflammation, the GEO database was queried for pediatric patients diagnosed with sepsis or septic shock as previously collected by Wong, H, et al. “Genomic expression profiling across the pediatric systemic inflammatory response syndrome, sepsis, and septic shock spectrum” [27]. Samples were collected from healthy children under age 10 and children with the clinical diagnosis of sepsis or septic shock as defined by the “International pediatric sepsis consensus conference: Definitions for sepsis and organ dysfunction in pediatrics” [30]. Analysis showed there was an approximate 2-fold increase in ASK1 mRNA levels assessed at day 1 of illness (Figure 1A) in both patients with shock and sepsis compared to non-septic controls. The data further demonstrated that this increased level of expression was maintained through day 3 of illness, though there was a differential expression pattern between the two groups where expression was higher in those with septic shock, and therefore by definition cardiovascular compromise, compared to those with a less severe phenotype. As that data was obtained from whole blood, we compared expression patterns in neonatal human microvascular endothelial cells (HMVECs) exposed to LPS for 16 hours. Once again, TLR4 activation significantly enhanced ASK1 expression in HMVECs by approximately 5-fold compared to unchallenged cells (Figure 1B). This demonstrates that ASK1 expression is strongly upregulated by septic shock in whole blood and within endothelial cells after LPS challenge.
Figure 1: ASK1 expression is increased in pediatric sepsis and following LPS-stimulation.
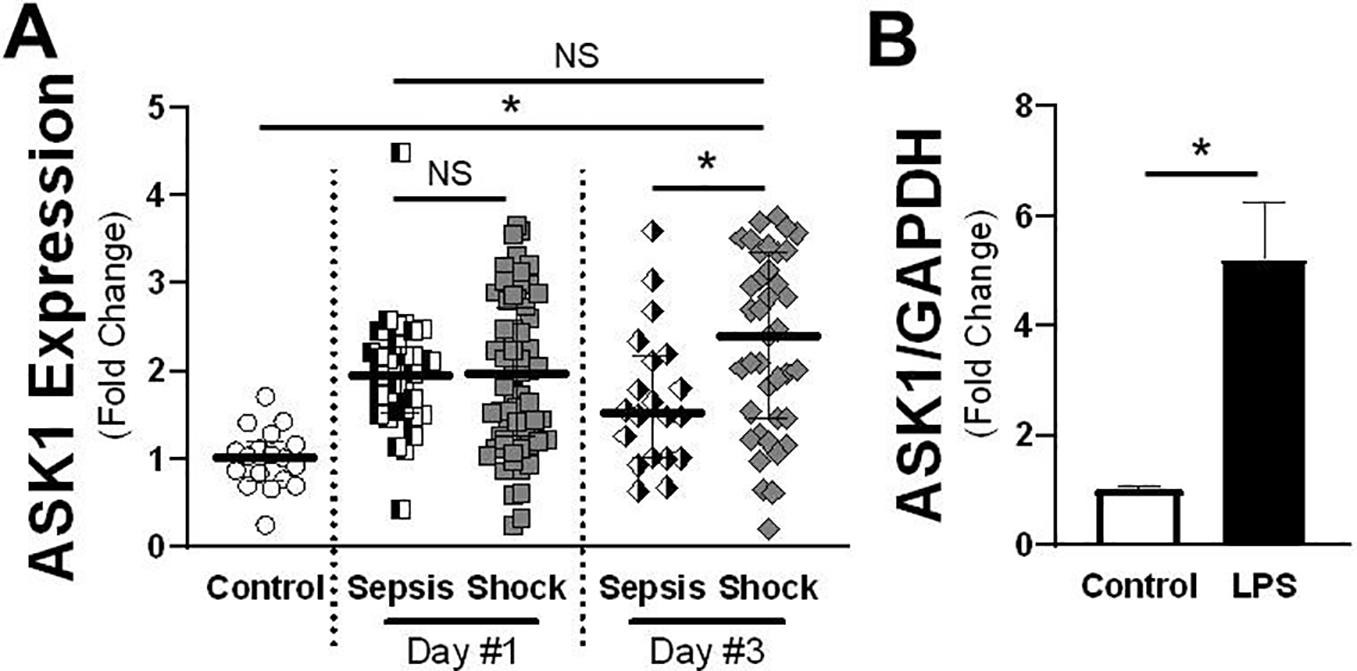
A) ASK1 mRNA relative values are shown for patients with either sepsis or septic shock on days 1 and 3 of illness obtained from GEO Dataset (GSE13904). The median expression value is shown with a horizontal bar per condition with 75% interquartile range normalized to control conditions (Control n = 18; Sepsis & Shock Day 1: n = 84; Day 3: n = 75). B) qRT-PCR values of ASK1 mRNA normalized to GAPDH control either with vehicle or LPS treatment for 16 hours (n = 5 individual replicates). * = p < 0.05 between designated groups. NS = non-significant.
ASK1 Inhibition leads to reduced endothelial cytokine production after TLR exposure
TLR4 stimulation causes redox-dependent ASK1 phosphorylation, which reflects activation and correlates with downstream signaling [14]. We therefore anticipated that inhibition of ASK1 would abrogate the endothelial response to LPS challenge. To test this, we utilized two different ASK1-specific inhibitors applied prior to the addition of LPS and assessed changes in downstream cytokine production. Baseline levels of the cytokines measured were nearly undetectable (data not shown). Following administration of LPS, vehicle treated cells demonstrated a robust secretion of IL-6, IL-8, sVCAM, and G-CSF (Figure 2). Pre-treatment with GS444217, an ASK1 inhibitor known to positively impact vascular health via eNOS, significantly inhibited cytokine production at all concentrations [24]. Similarly, 1 μM MSC2032964A, which has been shown to impact ASK1-mediated TLR4-p38 signaling, consistently inhibited all cytokines measured while higher concentrations showed non-significant changes in IL-8 and sVCAM production relative to vehicle control [31]. Additionally, 1 μM demonstrated a higher level of inhibition relative to 10 μM in IL-6 production, suggesting a potent effect of the inhibitor and the potential of off-target effects at the higher concentration. These findings demonstrate that inhibition of ASK1 is sufficient to reduce TLR4-mediated pro-inflammatory cytokine production.
Figure 2: LPS-induced cytokine production is impaired by ASK1 inhibitors.
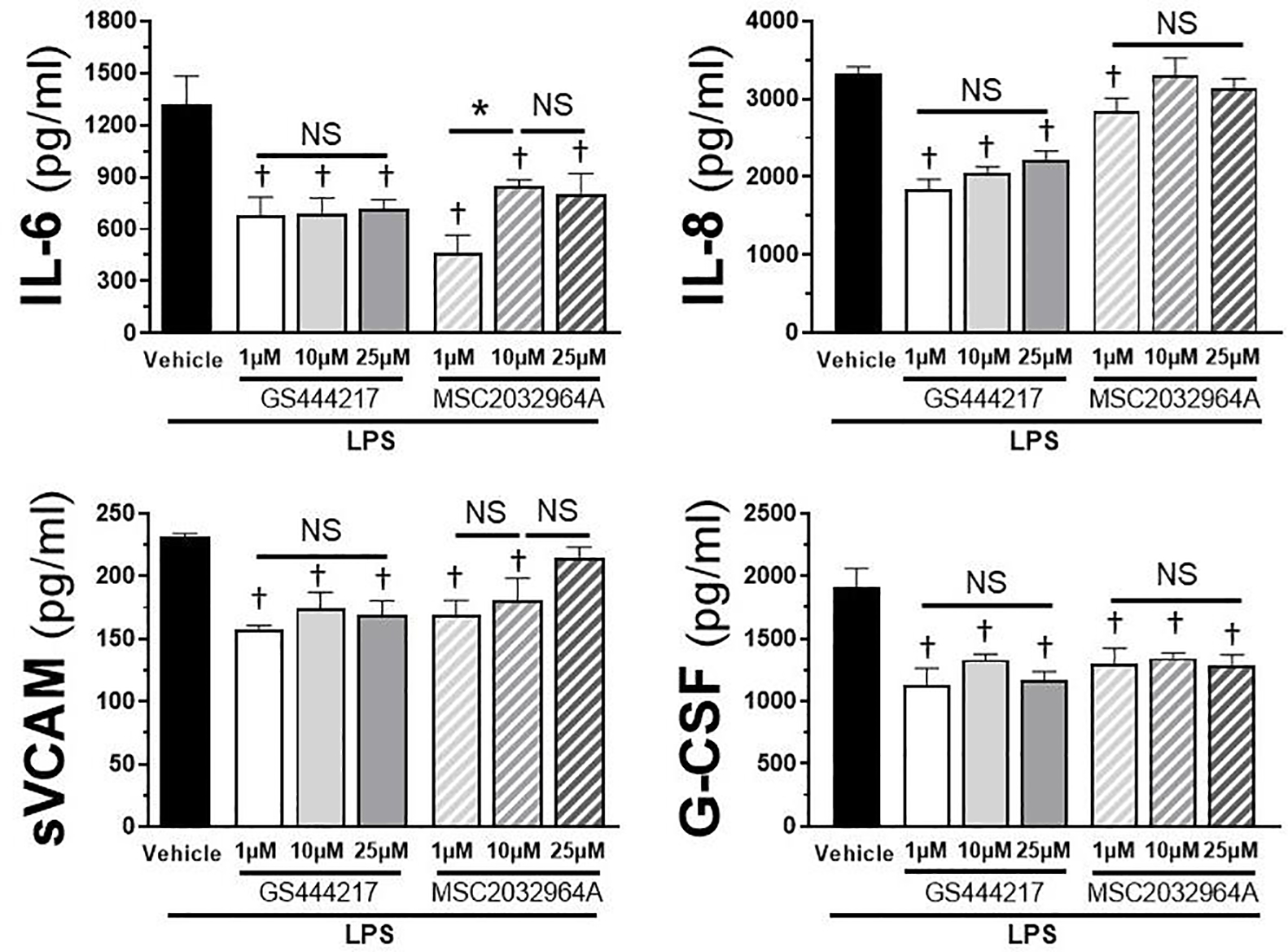
Cytokine concentrations (Il-6, IL-8, sVCAM, G-CSF) shown in pg/ml in HMVEC cells treated with 100 ng/ml LPS in the presence of either GS444217 or MSC2032964A at indicated concentrations (range 1 to 25 μM) following 6 hours of LPS stimulation. Vehicle + LPS is shown as control for all cytokines. Error bars indicate standard error of the mean. n = 4 per treatment. * = p < 0.05 between designated groups. † = p < 0.05 between indicated group and vehicle control. NS = non-significant.
Inhibition of ASK1 does not improve endothelial monolayer resistance
Prior work has demonstrated a strong link between MAPK activation and endothelial dysfunction including the loss of endothelial barrier integrity. Therefore, we postulated that, similar to cytokine production, inhibition of ASK1 would prevent LPS-induced endothelial barrier failure. Using transendothelial electrical resistance (TEER) as a surrogate for endothelial monolayer permeability, we exposed HMVECs to the same conditions as previously: LPS with or without GS444217 or MSC203964A. Data was normalized to baseline resistance prior to inhibitor addition (Figure 3A–B). All treatments, including control conditions, demonstrated a reproducible alteration in resistance during the first two hours post-manipulation and subsequently returned to baseline, related to device-related temperature fluctuations. As expected, the addition of LPS induced a significant reduction in the resistance of the endothelial monolayers. The presence of GS444217 failed to produce any difference in resistance at either a higher (25 μM) or lower (5 μM) concentrations when compared to LPS-treated controls (Figure 3B). In comparison, MSC2032964A displayed no change in the resistance of the monolayer at 5 μM, but potentiated the effect of LPS at 25 μM. Due to the unanticipated findings, the experiments were replicated using an intracellular space assay previously utilized to assess endothelial monolayer integrity [25]. Assessments of intracellular space demonstrated increased gap formation between cells following the administration of LPS and a lack of impact of ASK1 inhibition, similar to as what was observed in the TEER data (Figure 3C–D). A third measure of monolayer integrity (VE-cadherin staining) confirmed that loss of VE-cadherin following LPS that was also not affected by ASK1 inhibition (Figure 3E). These data demonstrate that inhibition of ASK1 has no significant protective effect on endothelial monolayer integrity, and with regards to MSC203964A, may instead impair endothelial integrity to a significant degree in the presence of LPS. Additionally, these experiments confirm that the TEER findings reproducibly and reliably correlate with previously established measures of endothelial integrity.
Figure 3: ASK1 inhibition does not improve monolayer permeability of endothelial cells treated with LPS.
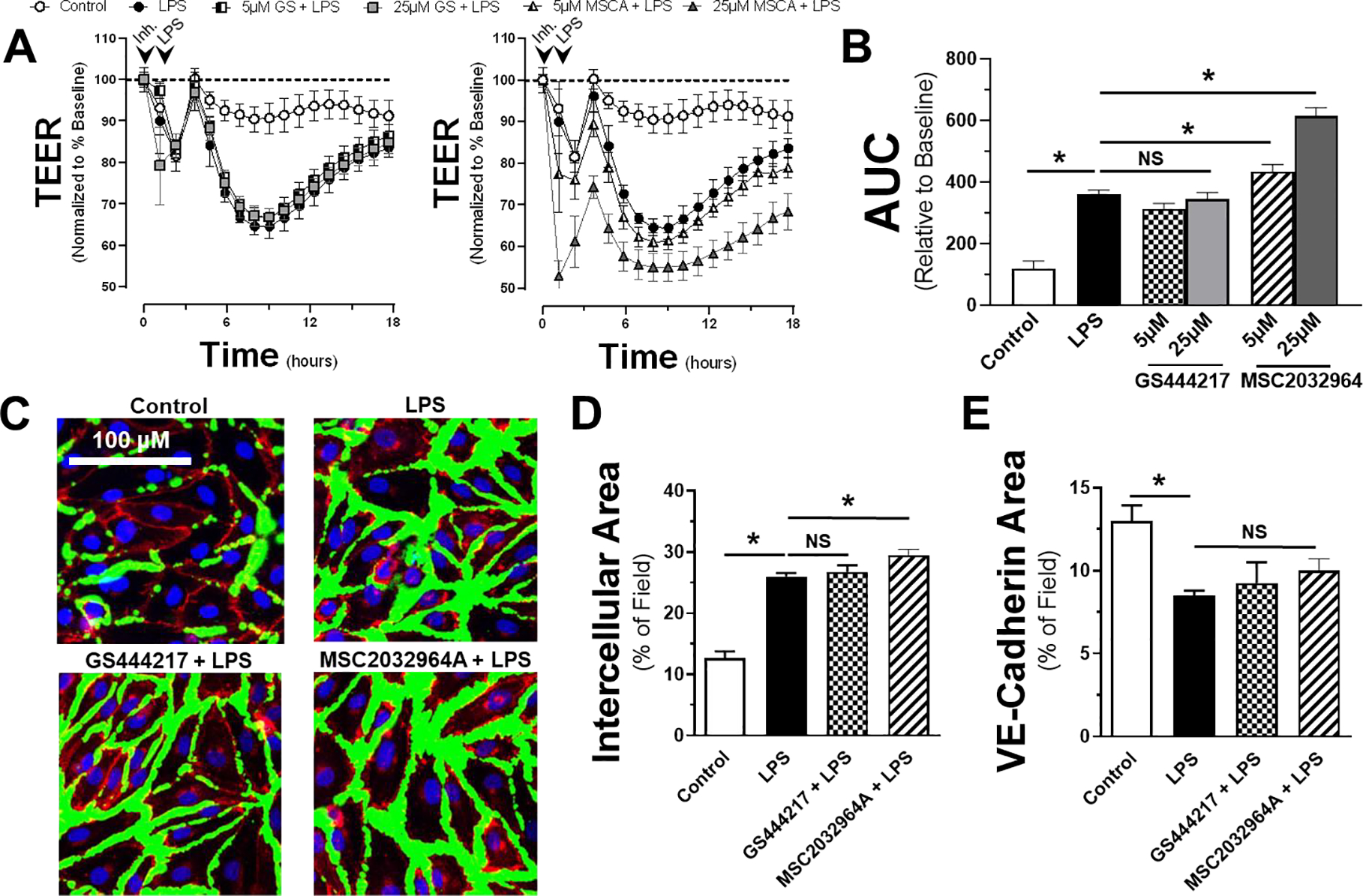
Transendothelial electrical resistance (TEER) of HMVECs pretreated with either GS444217 or MSC2032964A (A) at 5 or 25 μM prior to addition of LPS (100 ng/ml) or vehicle control. All data is normalized to baseline resistance per condition prior to addition of LPS. Arrows indicate time of inhibitor (or vehicle) and LPS (or control) given. B) Area under the curve relative to change from baseline resistance over time calculated for all conditions following LPS addition out to 18h of data (n = 5). C) Representative immunofluorescence images used for quantification of intercellular area (D) and VE-cadherin (E) staining following LPS in the presence or absence of either GS444217 or MSC2032964A (green = intercellular space, red = VE-cadherin, blue = nuclei). * = p < 0.05 between designated groups. NS = non-significant.
ASK1, p38 and eNOS expression correlate and have direct protein-protein interactions
ASK1 activation occurs after TLR4 stimulation and has been suggested to exert important effects via p38 [14, 32]. Likewise, eNOS also regulates TLR4-mediated p38 activation and eNOS expression can be altered in an ASK1-dependent manner [24, 26]. Therefore, we sought to determine whether a similar relationship existed between ASK1 and eNOS, JNK or p38 in patients with sepsis and septic shock. Expression levels of ASK1 (MAP3K5) in patients with sepsis and shock where matched against the respective expression levels of p38 (MAPK14), JNK1- (MAPK8) and eNOS (NOS3) for each patient. In both patient populations (Figure 4A–B), ASK1 expression was directly correlated with expression of p38, with increased ASK1 occurring in concordance with increased p38. There was surprisingly no correlation between JNK and ASK1 expression during sepsis or septic shock showing a Spearman r of −0.053 and 0.015, respectively, likely due to the fact that JNK transcript levels did not change during sepsis compared to controls (data not shown). Lastly, while there was no significant correlation between ASK1 and eNOS expression in patients with sepsis, there was an inverse correlation between the two proteins in patients with the more severe condition of septic shock.
Figure 4: ASK1 expression correlates with p38 and eNOS expression but not JNK in patients with septic shock but not sepsis.
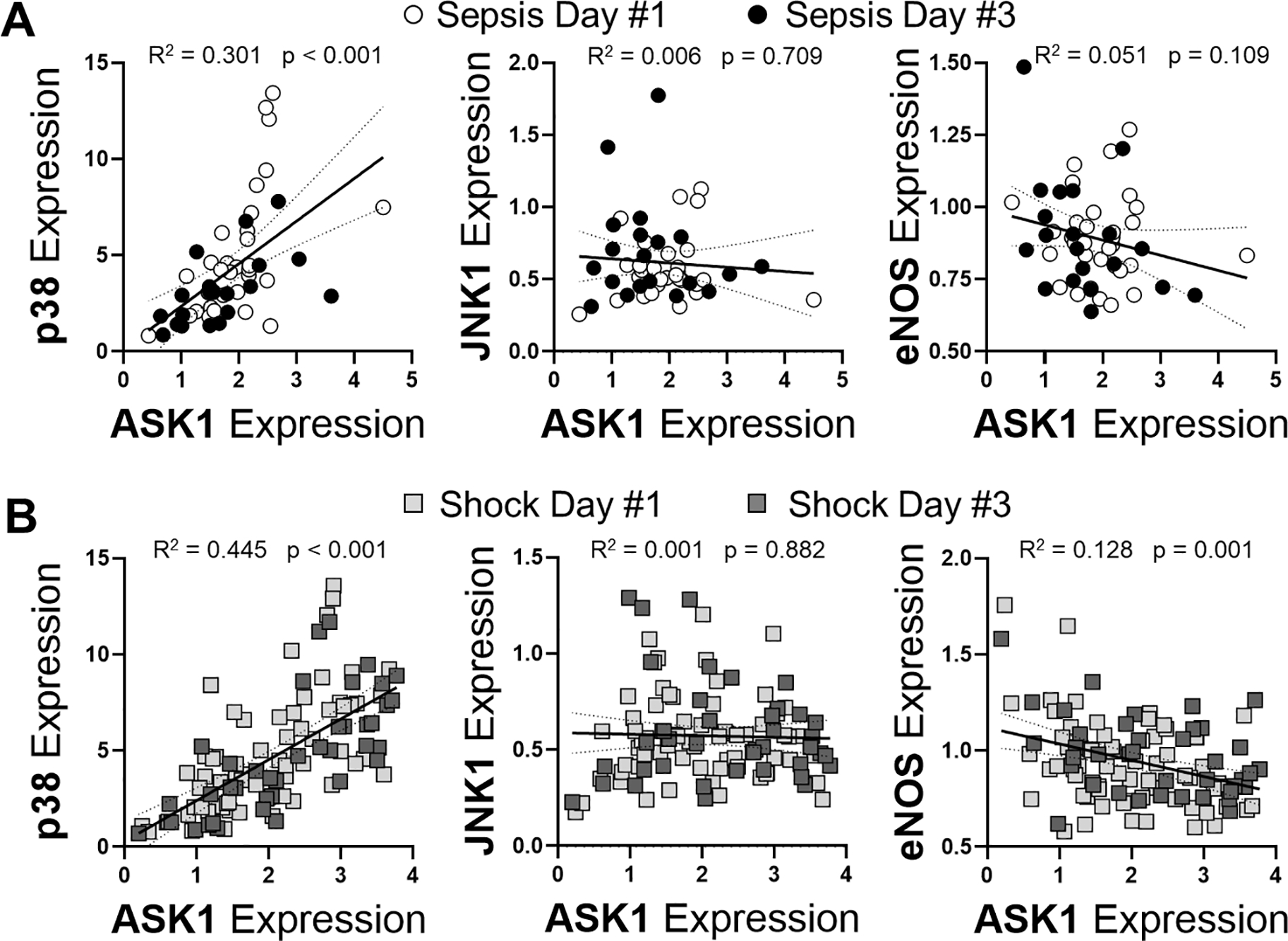
Expression values for ASK1 in comparison to p38, JNK1 and eNOS for patients with sepsis (A) or shock (B) were performed using Spearman r correlation coefficient was calculated with p values displayed. Day 1 of sepsis is shown as open circle, day 3 of sepsis is shown as closed circle, day 1 of shock is shown as light grey box and day 3 of shock is shown as dark grey box. Linear regression (solid lines) with 95% CI (dotted lines) are displayed with associated R2 values of variance.
Having found an inverse correlation between eNOS and ASK1 expression we postulated that the reduction in eNOS expression could be partly responsible for enhanced ASK1-dependent inflammation. Direct protein-protein interaction has been shown by demonstration of eNOS docking to MAPKs through a positively charged arginine-arginine-lysine-arginine-lysine (R-R-K-R-K) MAPK binding domain that binds to negatively charged residues on MAPKs [33]. For p38, the primary MAPK that interacts with eNOS, this sequence is aspartic acid-proline-aspartic acid-aspartic acid (D-P-D-D). On ASK1, a similar acidic motif has the sequence of aspartic acid-valine-glutamic acid-aspartic acid (D-V-E-D). These acidic sequences on p38 and ASK1 are referred to as CD domains [34]. Using a newer, more specific protein-protein interaction technique of proximity ligation assays (PLA) [35], we found that eNOS and ASK1 did indeed have direct interactions throughout the cell. The fluorescent PLA signal was attenuated by siRNA targeting eNOS, indicating specificity (Figure 5A). Having validated the PLA with siRNA targeting eNOS, we then assessed whether the interaction was affected either by activation via LPS (Figure 5B) or by addition of an ASK1 inhibitor (Figure 5C) for 1 hour. Here, we used only GS444217, given the unclear off-target effects of the other ASK1 inhibitor, and only at the lower dose (1 μM) that did not hinder endothelial resistance, but still reduced cytokine production. These experiments revealed no significant impact of LPS stimulation or ASK1 inhibition on association of eNOS with ASK1. We next sought to determine whether eNOS/p-p38 interactions are affected, which might reflect downstream effects of ASK1 modulation. p38 interaction with eNOS has previously been demonstrated to have a significant effect on endothelial inflammation and function [25]. Thus, an ASK1-dependent change in eNOS/p38 binding might provide a novel mechanism of controlling eNOS/p38-dependent inflammation. As expected, there was a time-dependent increase in eNOS/p-p38 interaction following LPS exposure. However, pretreatment with GS444217 had no effect on this interaction as measured by PLA when compared to untreated controls (Figure 5D). While these data show a clear interaction of eNOS with ASK1, this relationship was not significantly altered by LPS or interference with ASK1 activity at the time points measured. More importantly, inhibition of ASK1 did not alter downstream eNOS/p-p38 interactions despite the postulated mechanism of ASK1 being an upstream modulator of p38 function.
Figure 5: ASK1 directly interacts with eNOS without affecting LPS-induced eNOS-p38 interaction.
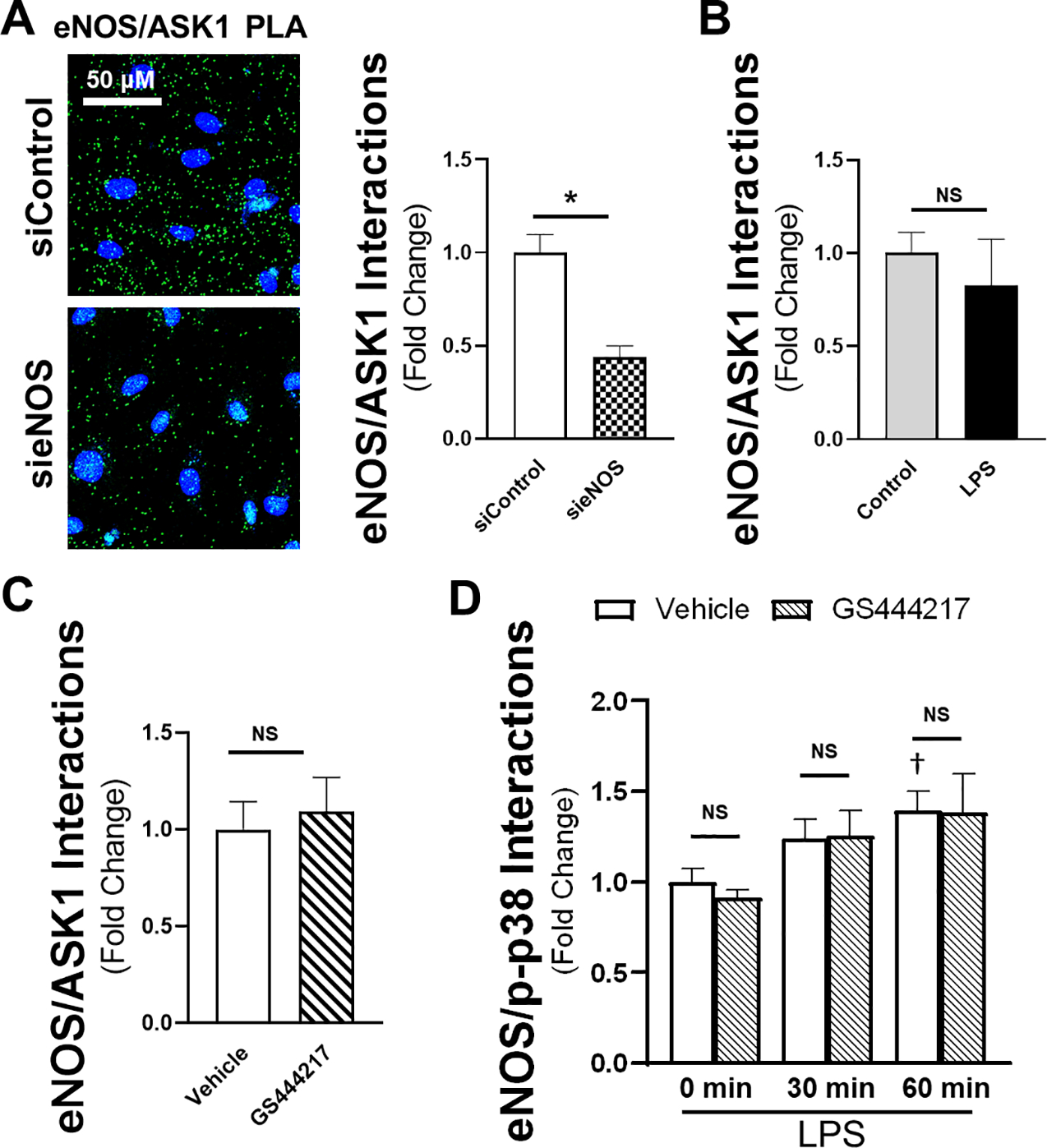
A) PLA for eNOS/ASK1 interaction with indicated siRNA in representative confocal images (40x, line bar represents a distance of 50 μm, green = PLA signal, blue = nuclei). Calculation of relative eNOS/ASK1 PLA signal (as determined by total fluorescence intensity) either in the presence or absence of 100 ng/ml of LPS (B) or 1 μM of GS444217 (C) for 1 hour (n = 5 individual replicate per group). D) eNOS/p-p38 PLA signal at 0, 30, and 60 minutes calculated relative to vehicle control at the time of LPS addition (n = 6 per group). * = p < 0.05 between respective groups. † = p < 0.05 compared to vehicle control at time 0. NS = non-significant.
ASK1 signals through JNK but not p38 in endothelial cells
Previous investigation has demonstrated a varied response to ASK1 inhibition on the downstream signaling cascade of the p38 and JNK pathways [22]. However, the lack of an effect of ASK1 inhibition on eNOS/p38 interactions or permeability suggested that p38 may not be regulated by ASK1 in endothelial cells. Therefore, we sought to confirm the impact of ASK1 inhibition on the phosphorylation fingerprint of downstream MAPK signaling. Cells were treated with LPS for either 90 minutes or 6 hours in either the presence or absence of GS444217 and examined by western blot to determine relative phosphorylation of ASK1 and its downstream targets. As expected, ASK1 phosphorylation was significantly reduced relative to control by pretreatment with 1 μM of GS444217 after 90 minutes of LPS exposure (Supplemental Figure 1A). In addition, given the increase in ASK1 transcript demonstrated earlier (Figure 1B), we saw ASK1 protein expression was significantly increased in vehicle-exposed LPS samples at 6 hours, though the presence of GS444217 enhanced baseline total ASK1 expression, possibly as part of a feedback loop (Supplemental Figure 1B). In examining MAPK pathway activation, p38 phosphorylation was unaffected by the presence of an ASK1 inhibitor at both 90 minutes (Figure 6A) and 6 hours (Figure 6B) of LPS stimulation, confirming an absent impact of ASK1 inhibition on p38. In contrast, ASK1 inhibited JNK phosphorylation both 90 minutes and 6 hours after LPS, showing a sustained suppression of that MAPK pathway. As expected, ASK1 inhibition had no effect on LPS-mediated extracellular signal-regulated kinase (ERK) phosphorylation over time, as that MAPK is regulated by a different MAPKKK. To assess off-target effects of the ASK1 inhibitor, IKK phosphorylation was examined and demonstrated no effective change in phosphorylation at 90 minutes, but there was a significant decreased in phosphorylation at 6 hours when pretreated with the ASK1 inhibitor, possibly due to sustained JNK activity suppression by inhibition of ASK1. Original western blot images are shown in Supplemental Figure 2. Thus, in endothelial cells, treatment with GS444217 reduces ASK1 phosphorylation with subsequent reduction in phosphorylation of JNK without appreciable changes in p38 phosphorylation.
Figure 6: ASK1 inhibition decreases phosphorylation of JNK without effect on p38.
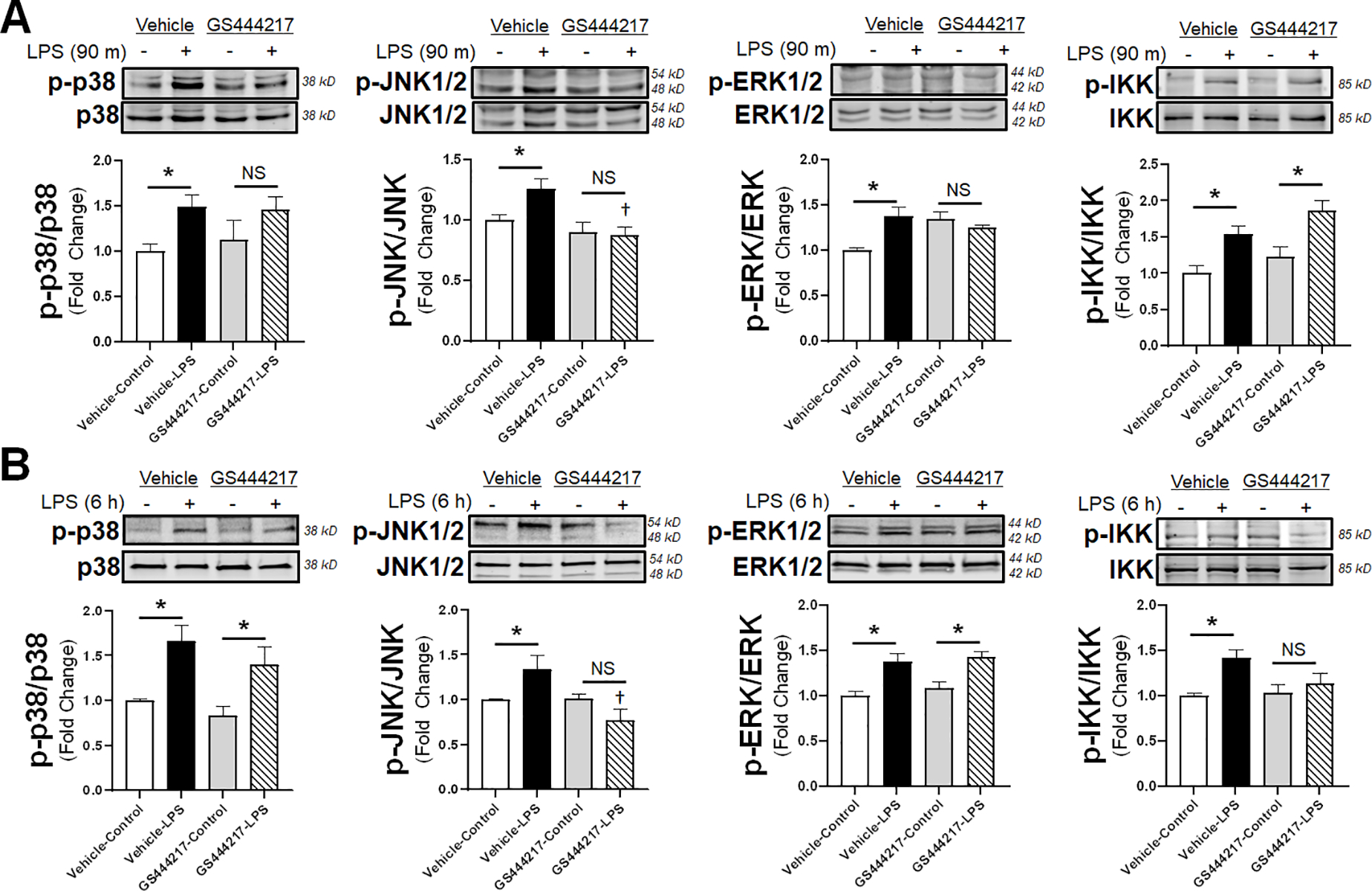
Representative WB images are shown following 90 minutes (A) or 6 hours (B) of LPS incubation in the presence or absence of 1 μM GS444217 for 30 minutes prior to LPS. Densitometry analysis of whole cell lysate western blots calculated for the indicated antibodies (n = 5 individual replicates per group). Phosphorylation signal is normalized to total non-phosphorylated protein within each analysis. * = p < 0.05 between designated groups. † = p < 0.05 between indicated group and vehicle control. NS = non-significant.
Direct inhibition of JNK reduces both cytokine expression and endothelial permeability
Given that inhibition of ASK1 impacted JNK phosphorylation without any appreciable effect on p38, we sought to determine the direct effects of JNK and p38 inhibition on cytokine production and endothelial monolayer integrity for comparison relative to ASK1 inhibition. Cells were treated with either a JNK inhibitor (10 μM SP600125) or a p38 inhibitor (10μM SB203580) and both TEER (Figure 7A) and IL-6 cytokine production were assessed (Figure 7B). Inhibition of p38 or JNK both improved the endothelial monolayer resistance to LPS stimulation, with a much greater effect produced by JNK inhibition. In comparison, though both p38 and JNK inhibition reduced IL-6 production, the reduction was profound after p38 inhibition while more modest after JNK inhibition. When comparing the relative impact of the three MAPK pathway inhibitors utilized, the inhibition of ASK1 on LPS-mediated IL-6 production was similar that of JNK inhibition (Figure 7C). In these normalized comparisons, inhibition of JNK or ASK1 reduced IL-6 secretion after LPS exposure by 49.5% or 48.4%, respectively, with no statistical difference in percent reduction between the two inhibitors. Together, these data demonstrate that while direct inhibition of either downstream MAPK can reduce endothelial inflammation in cells treated with LPS, the effect of ASK1 inhibition seem to be primarily on JNK-mediated cytokine production. Furthermore, in view of the potent effect of JNK inhibition on TEER and the relative lack of effect of ASK1 inhibition, it appears that JNK-dependent regulation of barrier integrity following LPS exposure is ASK1-independent.
Figure 7: Direct JNK and p38 inhibition improves endothelial barrier integrity and decreases IL-6 production.
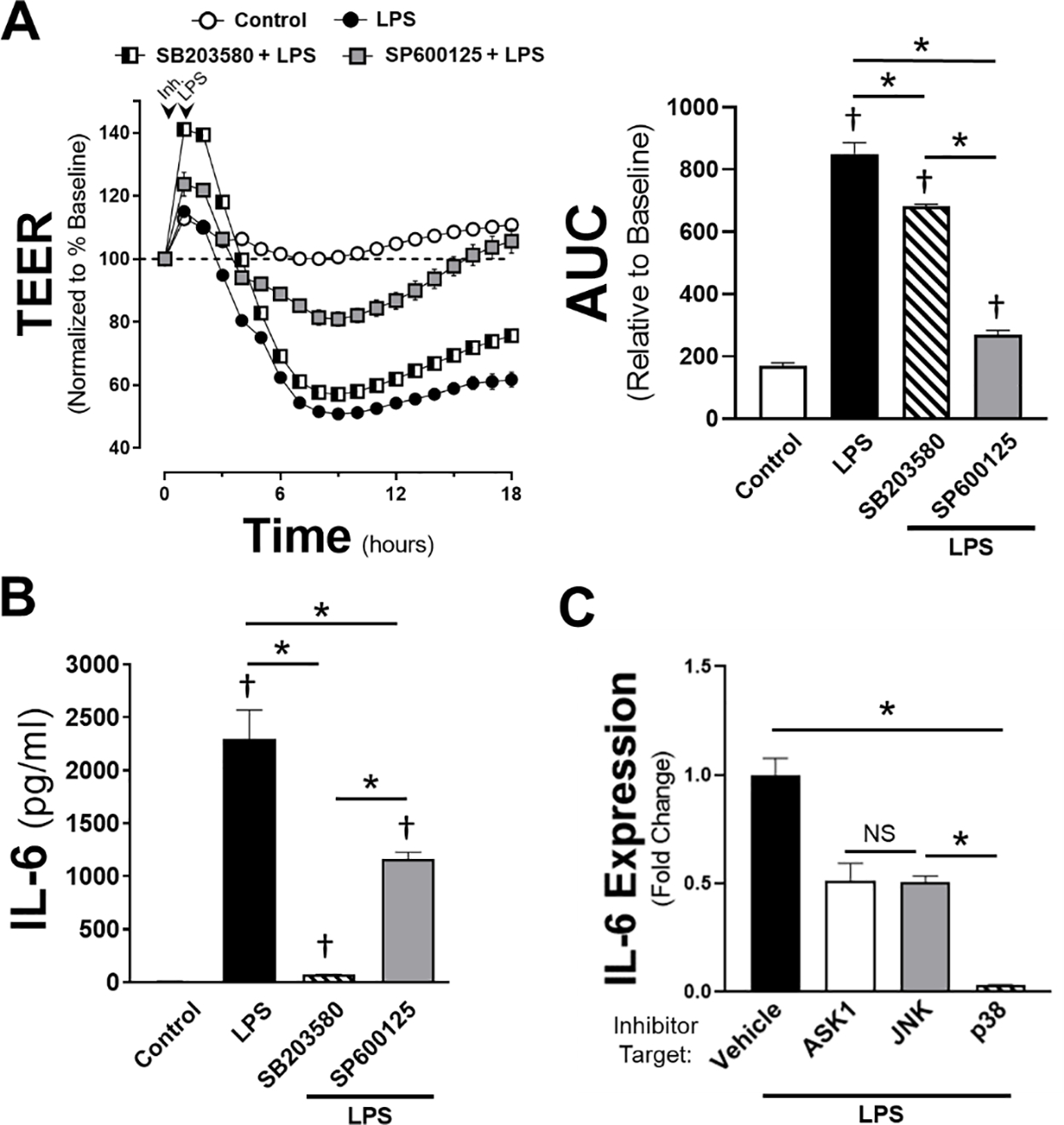
A) TEER data collection of HMVEC cells pretreated with either 10 μM SP600125 (JNK inhibitor) or 10 μM SB203580 (p38 inhibitor) prior to the addition of LPS (100 ng/ml) or vehicle control. Arrows indicate time of inhibitor (or vehicle) and LPS (or control) given. Area under the curve relative to change from baseline resistance over time calculated for all conditions following LPS addition out to 18h of data (n = 5 per treatment). B) IL-6 concentration shown in pg/ml in HMVEC cells treated with 100 ng/ml LPS in the presence of either SB203580 or SP600125 following 6 hours of LPS stimulation. Vehicle + LPS is shown as control. Error bars indicate standard error of the mean (n = 4 per treatment). C) Comparisons of IL-6 expression across all three inhibitors (ASK1: 1 μM GS444217, JNK: 10 μM SP600125, p38: 10 μM SB203580) relative to their respective, normalized groups of vehicle + LPS. * = p < 0.05 between designated groups. † = p < 0.05 between indicated group and control.
DISCUSSION:
We observed a clear increase in ASK1 expression in children with sepsis and septic shock as well as in endothelial cells exposed to LPS. We further demonstrated a decrease in pro-inflammatory cytokine production with selective blockade of ASK1 in endothelial cells. The reduction in secreted cytokines appeared primarily dependent on inhibition of JNK activity as ASK1 inhibition had no effect on p38 phosphorylation or on its interaction with eNOS. Surprisingly though, using two models of endothelial barrier integrity, we showed that blockade of ASK1 signaling offered no improvement in monolayer resistance following LPS. Thus, while there was strong upregulation of ASK1 after infectious challenge and ASK1 inhibitors blunted endothelial cytokine production via impaired JNK activation, barrier function, a critical functional endpoint with high relevance to clinical sepsis, was not protected These findings as a whole indicate both the promise and caution that should be applied to ASK1 directed therapies and their potential role in treating the vascular pathology of sepsis.
ASK1 signaling and its potential as a broad therapeutic target in inflammation continues to be thoroughly investigated [22]. Herein, we demonstrated a clear upregulation of ASK1 in pediatric patients experiencing sepsis or shock, an acute inflammatory state (Figures 1 and 4). Given these findings, ASK1 would seemingly represent an ideal target for human therapeutic intervention during infectious challenge given its role as an upstream mediator of MAPK signaling. ASK1 has been studied in clinical trials for its safety and effect on inflammation through the p38 and JNK pathways [36–38]. However, a recent review hypothesized that acute and chronic signaling from the p38 and JNK signaling pathways represent potential divergent clinical responses when providing ASK1 blockade [22]. Our findings somewhat support this hypothesis showing impaired cytokine secretion (Figure 2) without protection from injury to endothelial barrier function (Figure 3). However, this finding was counter to our initial hypothesis given that ASK1 had been previously demonstrated to signal through p38, and LPS and tumor necrosis factor alpha (TNFα) signaling have previously been shown to disrupt endothelial integrity via activation of the p38 pathway [39, 40]. Indeed, increased expression of ASK1 strongly correlated with p38, while having no association with expression of JNK (Figure 4). Yet, further examination of the p38 MAPK signaling through either protein-protein interactions of p38 (Figure 5) or phosphorylation (Figure 6) showed that p38 activity was unperturbed after ASK1 inhibition.
As opposed to the absent effect of ASK1 on p38 activity, we did confirm the impact of ASK1 on JNK signaling that translated into reduced global cytokine production by endothelial cells [13]. Inhibition of JNK has been shown to be protective in various models of infection-mediated inflammation. Non-selective JNK inhibitors have been demonstrated to reduce cytokine production and improve survival during lethal endotoxemia [41]. In sepsis models utilizing cecal ligation and perforation (CLP), use of JNK inhibitors showed reduced inflammation and edema in lung tissue as well as improved survival [42–44]. Interestingly, in one of the studies using CLP [42], septic rodents displayed increased JNK mRNA which was not observed in whole blood from septic children in this study, suggesting unique transcription profiles either between tissues or species. Irrespective though, our observed impairment of cytokine production after reduced ASK1 activation supports a partial explanation as to why JNK inhibition has been shown to be a successful mediator of infection-induced inflammation.
A further observation was the effect of JNK inhibition on endothelial permeability. While the role of p38 in endothelial barrier integrity is well supported [39, 40], the role of JNK is less clear. There is data to suggest that JNK plays no active role in endothelial cytoskeletal rearrangement or endothelial monolayer permeability after TNFα [45, 46]. Alternatively, it has been shown that occludin, which is important in endothelial tight junction formation, can be regulated by JNK after LPS [47]. Though differences in agonists may partly explain the discrepancy, what is clear from our data is that both p38 and JNK impact endothelial permeability and cytokine production, albeit to differing degrees. More interestingly though, is that while the ASK1 inhibition did impair JNK phosphorylation, this effect appeared to only impact cytokine production, as direct JNK inhibition had a robust reduction on endothelial permeability not observed after ASK1 inhibition (Figure 7). Thus, while ASK1 is a modulator of JNK, alterations in endothelial barrier integrity by JNK appear to be ASK1-independent.
Activation of JNK can occur via multiple MAPKKKs. While we focused on ASK1 for its postulated effects on p38 and JNK, it is clear that with regards to endothelial TLR4-induced inflammation, there are additional inputs that impact JNK signaling leading to endothelial pathology. These alternative inputs include MEKK1/4, MLK2/3, TAK1, and Tpl2 [34]. Of these, TAK1 may be responsible for the alterations in endothelial permeability observed after JNK inhibition, as TAK1 deficiency has been demonstrated to impair cell-cell junctions and enhance endothelial permeability [48, 49]. This complexity of MAPK activation is exacerbated by additional modifiers of these pathways. Specific to JNK, non-canonical Wnt signaling has been suggested to modulate endothelial JNK either directly or through Rho GTPases [50]. Additionally, ASK1 inhibition has been shown to impact other mediators of vascular health, such as angiopoietin-2, which is known to increase in sepsis and is well documented in its effects on vascular pathology [51, 52]. Understanding the dynamics of both the additive and reciprocal nature of these pathways in maintaining endothelial barrier homeostasis is crucial for finding potential treatment modalities in sepsis.
Finally, it is worth noting that while we initially hypothesized that ASK1 and eNOS could potentially modulate inflammation either through direct interactions or indirectly via p38 through MAPK-CD domain docking sites, we found that ASK1 inhibition did not affect ASK1/eNOS nor eNOS/p38 interactions (Figure 5). While this finding correlates with the inability of ASK1 inhibitors to impact p38 activity, it raises questions regarding the functional implication of this interaction. ASK1 is known to exhibit redox regulation secondary to its association with thioredoxin [53], and there is a well-recognized interplay between thioredoxin and nitric oxide, the product of eNOS activity [54]. While the functional impact of ASK1 inhibition seems to be independent of its ability to affect eNOS activity or binding with p38, this interaction would benefit from further study to determine if a functional link exists between physiologic ASK1 activity and eNOS regulation in the inflamed state. It also raises further questions of whether the effects of ASK1/eNOS interactions are time dependent given the temporal increase in expression of ASK1 after LPS or whether ASK1/eNOS interactions regulate eNOS interactions with JNK as opposed to p38 to alter endothelial responses during acute pathological condition
CONCLUSION:
ASK1 is an oxidant-sensitive inducer of MAPK signaling that is upregulated in patients with sepsis and in endothelial cells after LPS challenge. Though inhibition of ASK1 tempered TLR4-mediated endothelial cytokine production, it either had no effect or worsened endothelial barrier integrity depending on the ASK1 inhibitor applied. Further, despite prior evidence of ASK1 mediating its inflammatory effects via p38 activation or alterations in eNOS, we were unable to demonstrate any significant role of either of these postulated mechanisms on the acute inflammatory challenge after LPS. Instead, the primary impact of ASK1 activation was through JNK, suggesting that in endothelial cells, ASK1 activation of MAPK signaling seems more specific than previously thought. Additionally, JNK regulating endothelial barrier loss as well as cytokine production, with only the latter attributable to ASK1 activity, highlights the complexity of the multiple, integrated MAPK signaling pathways and their contribution to the inflammatory response and subsequent disease-related outcomes. In total, these data point to a unique role of ASK1 in endothelial cells driving specific activation of JNK-mediated cytokine production that has not been observed consistently in other innate immune cells.
Supplementary Material
Supplemental Figure 1: ASK1 inhibition on LPS-mediated ASK1 phosphorylation and ASK1 protein expression overtime. A) Representative WB images are shown following phosphorylation of ASK1 90 minutes after LPS exposure in the presence or absence of 1 μM GS44421 30 minutes prior to agonist. B) Representative WB images are shown following total ASK1 6 hours after LPS exposure in the presence or absence of GS44421 at the same time and dose as A. Densitometry analysis of whole cell lysate western blots (n = 5 individual replicates per group) calculated for the indicated antibodies. Phosphorylation signal is normalized to total non-phosphorylated protein within each analysis. Total ASK1 is normalized to tubulin as indicated for protein expression. * = p < 0.05 between designated groups. † = p < 0.05 between indicated group and vehicle control. NS = non-significant.
Supplemental Figure 2: Original western blot images with concomitant molecular weight ruler presented in the main body of the text. A) LPS with 90 minutes or B) 6 hours of exposure with or without 1 μM GS44421 30 minutes prior to agonist. Probing for proteins of interest are displayed adjacent to their respective western blot image.
Acknowledgements:
This study was supported by grants from the National Institutes of Health (RJS: K08 GM117367 and R35 GM138191, FSL: R01 HL128386, MRM: T32 HL144446) and a Turner-Hazinski Award from the VUMC Department of Pediatrics (RJS). All authors have read the journal’s authorship agreement and policy on disclosure of potential conflicts of interest and have no conflicts of interest to disclose. MRM performed experiments, wrote and revised the manuscript as well as interpreted data. SRK performed the experiments and acquired and interpreted data. HC revised the manuscript and interpreted data. FSL revised the manuscript, interpreted data, and provided experimental design guidance. RJS performed experiments, interpreted data, revised the manuscript, and provided overall guidance of the project.
Abbreviations:
- ASK1
apoptosis signal-regulating kinase 1
- HMVECs
human microvascular endothelial cells
- JNK
c-Jun N-terminal kinase
- eNOS
endothelial nitric oxide synthase
- MAPK
mitogen-activated protein kinase
- MAPKKK
MAPK kinase kinase
- LPS
lipopolysaccharide
- TEER
transendothelial electrical resistance
- TLR
toll-like receptor
- PAMPs
pathogen-associated molecular patterns
- ERK
extracellular signal-regulated kinase
- PLA
proximity ligation assays
- TNFα
tumor necrosis factor alpha
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- [1].Boeddha NP, Schlapbach LJ, Driessen GJ, Herberg JA, Rivero-Calle I, Cebey-Lopez M, et al. Mortality and morbidity in community-acquired sepsis in European pediatric intensive care units: a prospective cohort study from the European Childhood Life-threatening Infectious Disease Study (EUCLIDS). Crit Care. 2018;22:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shankar-Hari M, Harrison DA, Rubenfeld GD, Rowan K. Epidemiology of sepsis and septic shock in critical care units: comparison between sepsis-2 and sepsis-3 populations using a national critical care database. Br J Anaesth. 2017;119:626–36. [DOI] [PubMed] [Google Scholar]
- [3].Cinel I, Opal SM. Molecular biology of inflammation and sepsis: a primer. Crit Care Med. 2009;37:291–304. [DOI] [PubMed] [Google Scholar]
- [4].Joffre J, Hellman J, Ince C, Ait-Oufella H. Endothelial Responses in Sepsis. Am J Respir Crit Care Med. 2020;202:361–70. [DOI] [PubMed] [Google Scholar]
- [5].Orfanos SE, Mavrommati I, Korovesi I, Roussos C. Pulmonary endothelium in acute lung injury: from basic science to the critically ill. Intensive Care Med. 2004;30:1702–14. [DOI] [PubMed] [Google Scholar]
- [6].Medzhitov R Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–45. [DOI] [PubMed] [Google Scholar]
- [7].Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. [DOI] [PubMed] [Google Scholar]
- [8].Jin MS, Lee JO. Structures of the toll-like receptor family and its ligand complexes. Immunity. 2008;29:182–91. [DOI] [PubMed] [Google Scholar]
- [9].Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol. 2003;3:169–76. [DOI] [PubMed] [Google Scholar]
- [10].Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. [DOI] [PubMed] [Google Scholar]
- [11].Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–51. [DOI] [PubMed] [Google Scholar]
- [12].Noh KT, Park YM, Cho SG, Choi EJ. GSK-3beta-induced ASK1 stabilization is crucial in LPS-induced endotoxin shock. Exp Cell Res. 2011;317:1663–8. [DOI] [PubMed] [Google Scholar]
- [13].Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, et al. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2:389–95. [DOI] [PubMed] [Google Scholar]
- [14].Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–92. [DOI] [PubMed] [Google Scholar]
- [15].Dhanasekaran DN, Reddy EP. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer. 2017;8:682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res. 2015;35:600–4. [DOI] [PubMed] [Google Scholar]
- [17].Mudgett JS, Ding J, Guh-Siesel L, Chartrain NA, Yang L, Gopal S, et al. Essential role for p38alpha mitogen-activated protein kinase in placental angiogenesis. Proc Natl Acad Sci U S A. 2000;97:10454–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–76. [DOI] [PubMed] [Google Scholar]
- [19].Greenblatt MB, Shim JH, Zou W, Sitara D, Schweitzer M, Hu D, et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J Clin Invest. 2010;120:2457–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tong C, Yin Z, Song Z, Dockendorff A, Huang C, Mariadason J, et al. c-Jun NH2-terminal kinase 1 plays a critical role in intestinal homeostasis and tumor suppression. Am J Pathol. 2007;171:297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cohen SB, Cheng TT, Chindalore V, Damjanov N, Burgos-Vargas R, Delora P, et al. Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis Rheum. 2009;60:335–44. [DOI] [PubMed] [Google Scholar]
- [22].Ogier JM, Nayagam BA, Lockhart PJ. ASK1 inhibition: a therapeutic strategy with multisystem benefits. J Mol Med (Berl). 2020;98:335–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yamamoto E, Kataoka K, Shintaku H, Yamashita T, Tokutomi Y, Dong YF, et al. Novel mechanism and role of angiotensin II induced vascular endothelial injury in hypertensive diastolic heart failure. Arterioscler Thromb Vasc Biol. 2007;27:2569–75. [DOI] [PubMed] [Google Scholar]
- [24].Liles JT, Corkey BK, Notte GT, Budas GR, Lansdon EB, Hinojosa-Kirschenbaum F, et al. ASK1 contributes to fibrosis and dysfunction in models of kidney disease. J Clin Invest. 2018;128:4485–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Koch SR, Choi H, Mace EH, Stark RJ. Toll-like receptor 3-mediated inflammation by p38 is enhanced by endothelial nitric oxide synthase knockdown. Cell Commun Signal. 2019;17:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stark RJ, Koch SR, Choi H, Mace EH, Dikalov SI, Sherwood ER, et al. Endothelial nitric oxide synthase modulates Toll-like receptor 4-mediated IL-6 production and permeability via nitric oxide-independent signaling. FASEB J. 2018;32:945–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wong HR, Cvijanovich N, Allen GL, Lin R, Anas N, Meyer K, et al. Genomic expression profiling across the pediatric systemic inflammatory response syndrome, sepsis, and septic shock spectrum. Crit Care Med. 2009;37:1558–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Koch SR, Lamb FS, Hellman J, Sherwood ER, Stark RJ. Potentiation and tolerance of toll-like receptor priming in human endothelial cells. Transl Res. 2017;180:53–67 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–4. [DOI] [PubMed] [Google Scholar]
- [30].Goldstein B, Giroir B, Randolph A, International Consensus Conference on Pediatric S. International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med. 2005;6:2–8. [DOI] [PubMed] [Google Scholar]
- [31].Katome T, Namekata K, Guo X, Semba K, Kittaka D, Kawamura K, et al. Inhibition of ASK1-p38 pathway prevents neural cell death following optic nerve injury. Cell Death Differ. 2013;20:270–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mizumura K, Gon Y, Kumasawa F, Onose A, Maruoka S, Matsumoto K, et al. Apoptosis signal-regulating kinase 1-mediated signaling pathway regulates lipopolysaccharide-induced tissue factor expression in pulmonary microvasculature. Int Immunopharmacol. 2010;10:1062–7. [DOI] [PubMed] [Google Scholar]
- [33].Chrestensen CA, McMurry JL, Salerno JC. MAP kinases bind endothelial nitric oxide synthase. FEBS Open Bio. 2012;2:51–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Koos B, Andersson L, Clausson CM, Grannas K, Klaesson A, Cane G, et al. Analysis of protein interactions in situ by proximity ligation assays. Curr Top Microbiol Immunol. 2014;377:111–26. [DOI] [PubMed] [Google Scholar]
- [36].Chertow GM, Pergola PE, Chen F, Kirby BJ, Sundy JS, Patel UD, et al. Effects of Selonsertib in Patients with Diabetic Kidney Disease. J Am Soc Nephrol. 2019;30:1980–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology. 2018;67:549–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nelson CH, Etchevers K, Yi S, Breckenridge D, Hepner M, Patel U, et al. Pharmacokinetics, Safety, and Tolerability of Selonsertib, an Apoptosis Signal-Regulating Kinase 1 (ASK1) Inhibitor, Following First-in-Human Single and Multiple Ascending Doses in Healthy Subjects. Clin Pharmacokinet. 2020;59:1109–17. [DOI] [PubMed] [Google Scholar]
- [39].Adam AP, Lowery AM, Martino N, Alsaffar H, Vincent PA. Src Family Kinases Modulate the Loss of Endothelial Barrier Function in Response to TNF-alpha: Crosstalk with p38 Signaling. PLoS One. 2016;11:e0161975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Usatyuk PV, Vepa S, Watkins T, He D, Parinandi NL, Natarajan V. Redox regulation of reactive oxygen species-induced p38 MAP kinase activation and barrier dysfunction in lung microvascular endothelial cells. Antioxid Redox Signal. 2003;5:723–30. [DOI] [PubMed] [Google Scholar]
- [41].Prasad KD, Trinath J, Biswas A, Sekar K, Balaji KN, Guru Row TN. Anthrapyrazolone analogues intercept inflammatory JNK signals to moderate endotoxin induced septic shock. Sci Rep. 2014;4:7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lou L, Hu D, Chen S, Wang S, Xu Y, Huang Y, et al. Protective role of JNK inhibitor SP600125 in sepsis-induced acute lung injury. Int J Clin Exp Pathol. 2019;12:528–38. [PMC free article] [PubMed] [Google Scholar]
- [43].Pizzino G, Bitto A, Pallio G, Irrera N, Galfo F, Interdonato M, et al. Blockade of the JNK signalling as a rational therapeutic approach to modulate the early and late steps of the inflammatory cascade in polymicrobial sepsis. Mediators Inflamm. 2015;2015:591572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zheng Q, Wang YC, Liu QX, Dong XJ, Xie ZX, Liu XH, et al. FK866 attenuates sepsis-induced acute lung injury through c-jun-N-terminal kinase (JNK)-dependent autophagy. Life Sci. 2020;250:117551. [DOI] [PubMed] [Google Scholar]
- [45].Mong PY, Petrulio C, Kaufman HL, Wang Q. Activation of Rho kinase by TNF-alpha is required for JNK activation in human pulmonary microvascular endothelial cells. J Immunol. 2008;180:550–8. [DOI] [PubMed] [Google Scholar]
- [46].Jamaluddin MS, Yan S, Lu J, Liang Z, Yao Q, Chen C. Resistin increases monolayer permeability of human coronary artery endothelial cells. PLoS One. 2013;8:e84576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Qin LH, Huang W, Mo XA, Chen YL, Wu XH. LPS Induces Occludin Dysregulation in Cerebral Microvascular Endothelial Cells via MAPK Signaling and Augmenting MMP-2 Levels. Oxid Med Cell Longev. 2015;2015:120641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nighot M, Rawat M, Al-Sadi R, Castillo EF, Nighot P, Ma TY. Lipopolysaccharide-Induced Increase in Intestinal Permeability Is Mediated by TAK-1 Activation of IKK and MLCK/MYLK Gene. Am J Pathol. 2019;189:797–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Naito H, Iba T, Wakabayashi T, Tai-Nagara I, Suehiro JI, Jia W, et al. TAK1 Prevents Endothelial Apoptosis and Maintains Vascular Integrity. Dev Cell. 2019;48:151–66 e7. [DOI] [PubMed] [Google Scholar]
- [50].Lutze G, Haarmann A, Demanou Toukam JA, Buttler K, Wilting J, Becker J. Non-canonical WNT-signaling controls differentiation of lymphatics and extension lymphangiogenesis via RAC and JNK signaling. Sci Rep. 2019;9:4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Savira F, Kompa AR, Magaye R, Xiong X, Huang L, Jucker BM, et al. Apoptosis signal-regulating kinase 1 inhibition reverses deleterious indoxyl sulfate-mediated endothelial effects. Life Sci. 2021;272:119267. [DOI] [PubMed] [Google Scholar]
- [52].Ziegler T, Horstkotte J, Schwab C, Pfetsch V, Weinmann K, Dietzel S, et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J Clin Invest. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Benhar M Nitric oxide and the thioredoxin system: a complex interplay in redox regulation. Biochim Biophys Acta. 2015;1850:2476–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: ASK1 inhibition on LPS-mediated ASK1 phosphorylation and ASK1 protein expression overtime. A) Representative WB images are shown following phosphorylation of ASK1 90 minutes after LPS exposure in the presence or absence of 1 μM GS44421 30 minutes prior to agonist. B) Representative WB images are shown following total ASK1 6 hours after LPS exposure in the presence or absence of GS44421 at the same time and dose as A. Densitometry analysis of whole cell lysate western blots (n = 5 individual replicates per group) calculated for the indicated antibodies. Phosphorylation signal is normalized to total non-phosphorylated protein within each analysis. Total ASK1 is normalized to tubulin as indicated for protein expression. * = p < 0.05 between designated groups. † = p < 0.05 between indicated group and vehicle control. NS = non-significant.
Supplemental Figure 2: Original western blot images with concomitant molecular weight ruler presented in the main body of the text. A) LPS with 90 minutes or B) 6 hours of exposure with or without 1 μM GS44421 30 minutes prior to agonist. Probing for proteins of interest are displayed adjacent to their respective western blot image.