

Abstract
Background and aims.
The immune compartment is critical for maintaining tissue homeostasis. A weak immune response increases susceptibility to infection, but immune hyperactivation causes tissue damage, and chronic inflammation may lead to cancer development. In the stomach, inflammation damages the gastric glands and drives the development of potentially pre-neoplastic metaplasia. Glucocorticoids are potent anti-inflammatory steroid hormones that are required to suppress gastric inflammation and metaplasia. However, these hormones function differently in males and females. Here, we investigate the impact of sex on the regulation of gastric inflammation.
Methods.
Endogenous glucocorticoids and male sex hormones were removed from mice by adrenalectomy and castration, respectively. Mice were treated with 5α-dihydrotestosterone (DHT) to test the effects of androgens on regulating gastric inflammation. Single-cell RNA sequencing of gastric leukocytes was used to identify the leukocyte populations that were the direct targets of androgen signaling. ILC2s were depleted by treatment with CD90.2 antibodies.
Results.
We show that adrenalectomized female mice develop spontaneous gastric inflammation and spasmolytic polypeptide-expressing metaplasia (SPEM) but that the stomachs of adrenalectomized male mice remain quantitatively normal. Simultaneous depletion of glucocorticoids and sex hormones abolished the male-protective effects and triggered spontaneous pathogenic gastric inflammation and SPEM. Treatment of female mice with DHT prevented gastric inflammation and SPEM development when administered concurrent with adrenalectomy and also reversed the pathology when administered after disease onset. Single cell-RNAseq of gastric leukocytes revealed that type 2 innate lymphoid cells (ILC2s) expressed abundant levels of both the glucocorticoid receptor (Gr) and androgen receptor (Ar). We demonstrated that DHT treatment potently suppressed the expression of the proinflammatory cytokines Il13 and Csf2 by ILC2s. Moreover, ILC2 depletion protected the stomach from SPEM development.
Conclusions.
Here, we report a novel mechanism by which glucocorticoids and androgens exert overlapping effects to regulate gastric inflammation. Androgen signaling within ILC2s prevents their pathogenic activation by suppressing the transcription of proinflammatory cytokines. This work revealed a critical role for sex hormones in regulating gastric inflammation and metaplasia.
Keywords: Stomach, Gastric, SPEM, Macrophage, ILC2
Graphical Abstract
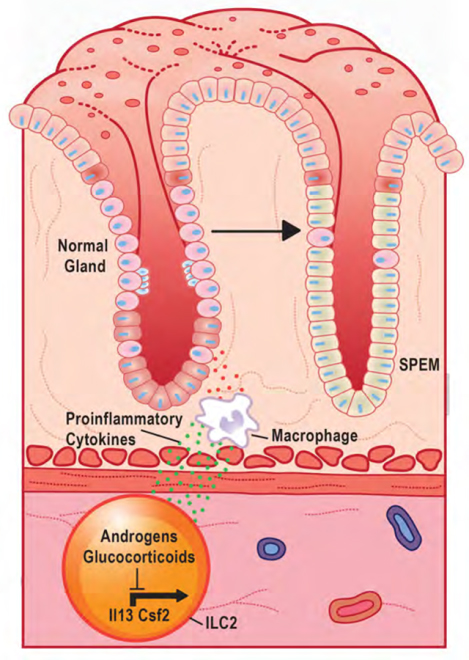
Introduction
The immune system is critical for maintaining tissue homeostasis. While acute hyperactive inflammation can induce tissue damage, chronic inflammation promotes tissue dysfunction and favors cancer development. Inflammation is regulated differently in males and females due, in part, to the actions of sex steroid hormones 1, 2. Women typically are more resistant than men to infection and mount more robust immune responses to vaccination 3, 4. However, women are also more prone to inflammatory diseases, and 80% of autoimmune diseases occur in women 5.
Similar trends are evident within the stomach, where both Helicobacter pylori-independent gastritis and autoimmune gastritis are more common in women 6. While men are more resistant to these inflammatory pathologies, some studies report that men are more susceptible to H pylori infection and harbor higher bacterial loads 7–9. It is clear that sex has a profound role in regulating inflammation; however, the underlying cellular and molecular mechanisms by which sex impacts inflammation are poorly defined.
Within the stomach, immune hyperactivation damages the epithelium and induces the development of spasmolytic polypeptide-expressing metaplasia (SPEM) 10–12. Acute SPEM develops as chief cells transdifferentiate and may function as a healing mechanism 13, 14. However, chronic SPEM is associated with the development of gastric adenocarcinoma 12, 15. Recent studies report that SPEM development is driven by an immune axis of tissue-resident type 2 innate lymphoid cells (ILC2s) and recruited macrophages 10, 11, 16. ILC2s are tissue-resident leukocytes that are widely distributed along mucosal surfaces and respond to epithelial stress to initiate inflammation 17. Activated ILC2s produce large quantities of the cytokines IL4, IL5, IL13, and CSF2 (also known as GM-CSF) 18–20. These cytokines, in turn, promote the recruitment and activation of monocyte-derived macrophages 21. Several recent studies have reported that monocyte-derived macrophages damage the gastric epithelium and induce metaplasia development 10, 16, 22. The exact mechanisms by which inflammation contributes to SPEM development are poorly defined.
Androgens, the chief male sex hormone, signal through the androgen receptor (AR). Macrophages readily respond to androgens but while some tissue-specific macrophage populations, such as alveolar macrophages 23, have been shown to express the AR, it is unclear if macrophages within the GI tract express these receptors. Recently, it has become clear that ILC2s are regulated in a sex-specific manner. Androgens suppress ILC2 proliferation, and male mice have fewer lung ILC2s than females 24. Male mice are also resistant to IL33-induced asthma due to dampened ILC2 activation. However, castration abolishes these male-protective effects, leading to a significant expansion of lung ILC2s and sensitizing males to asthma. Moreover, prolonged treatment of cultured primary ILC2s with the androgen 5α-dihydrotestosterone (DHT) suppresses their production of proinflammatory cytokines, including IL13 25. However, the mechanisms by which androgen signal within ILC2s to regulate proinflammatory gene expression is unknown.
The androgen receptor is a ligand-dependent transcription factor. The AR has high homology to the glucocorticoid receptor (NR3C1, hereafter GR), another steroid hormone receptor with potent anti-inflammatory properties 26, 27. We have previously shown that glucocorticoids suppress pathogenic gastric inflammation, and disruption of glucocorticoid signaling induces spontaneous gastric inflammation and metaplasia in female mice 10. A primary mechanism by which the AR and GR regulate gene transcription is through recognizing specific DNA sequences. The DNA response elements recognized by the AR and GR are nearly identical 28, 29. Indeed, in prostate cancer cells, glucocorticoids can regulate the expression of androgen-dependent genes, providing a route for cancer cells to escape anti-androgen therapies 30. These results raise the possibility that a similar form of regulation might exist within gastric ILC2s where glucocorticoid and androgens may exert redundancies to suppress proinflammatory gene expression. In this study, we report a novel role for androgens in suppressing gastric inflammation and metaplasia. Moreover, we show that ILC2s co-express the AR and GR and that treatment with either androgens or glucocorticoids can suppress transcripts encoding proinflammatory cytokines within ILC2s.
Materials and Methods
Animal care and treatment.
All mouse studies were performed with approval from the animal care and use committees of the National Institute of Environmental Health Sciences and West Virginia University. All mice used in these studies were maintained on a congenic C57BL/6J genetic background except Il33 KO mice which were maintained on a congenic C57BL/6N genetic background. C57BL/6J, Rag1 KO, and Thy1.1 (CD90.1) mice were purchased from the Jackson Laboratories (stock numbers 000664, 002216, and 000406, respectively Bar Harbor, MA). Il33 KO (Il33tm1(KOMP)Vlcg) mice were obtained from the trans-NIH Knock-Out Mouse Project (KOMP) Repository (University of California, Davis). Mice were administered standard chow and water ab libitum and maintained in a temperature and humidity-controlled room with standard 12-hour light/dark cycles. For all experiments, sham, adrenalectomy, and castration surgeries were performed at 8 weeks of age. Following adrenalectomy, mice were maintained on 0.85% saline drinking water to maintain ionic homeostasis. 10 mg DHT 60-day sustained release pellets (Innovative Research of America, Sarasota, FL) or placebo pellets were implanted subcutaneously either during the adrenalectomy procedure (Figure 3) or 1-month post-adrenalectomy (Supplementary Figure 3). To examine if steroid treatment suppresses proinflammatory cytokines within ILC2s, ADX+Cast mice were aged 5 days after surgery to allow clearing of endogenous steroids and then administered a single intraperitoneal injection of dexamethasone or DHT (Steraloids, Newport, RI). The mice were euthanized 3 hours after treatment. Control mice received vehicle (1x PBS). Both steroids were use at a 1 mg/kg concentration.
Figure 3. Androgens suppress gastric inflammation and SPEM following adrenalectomy.
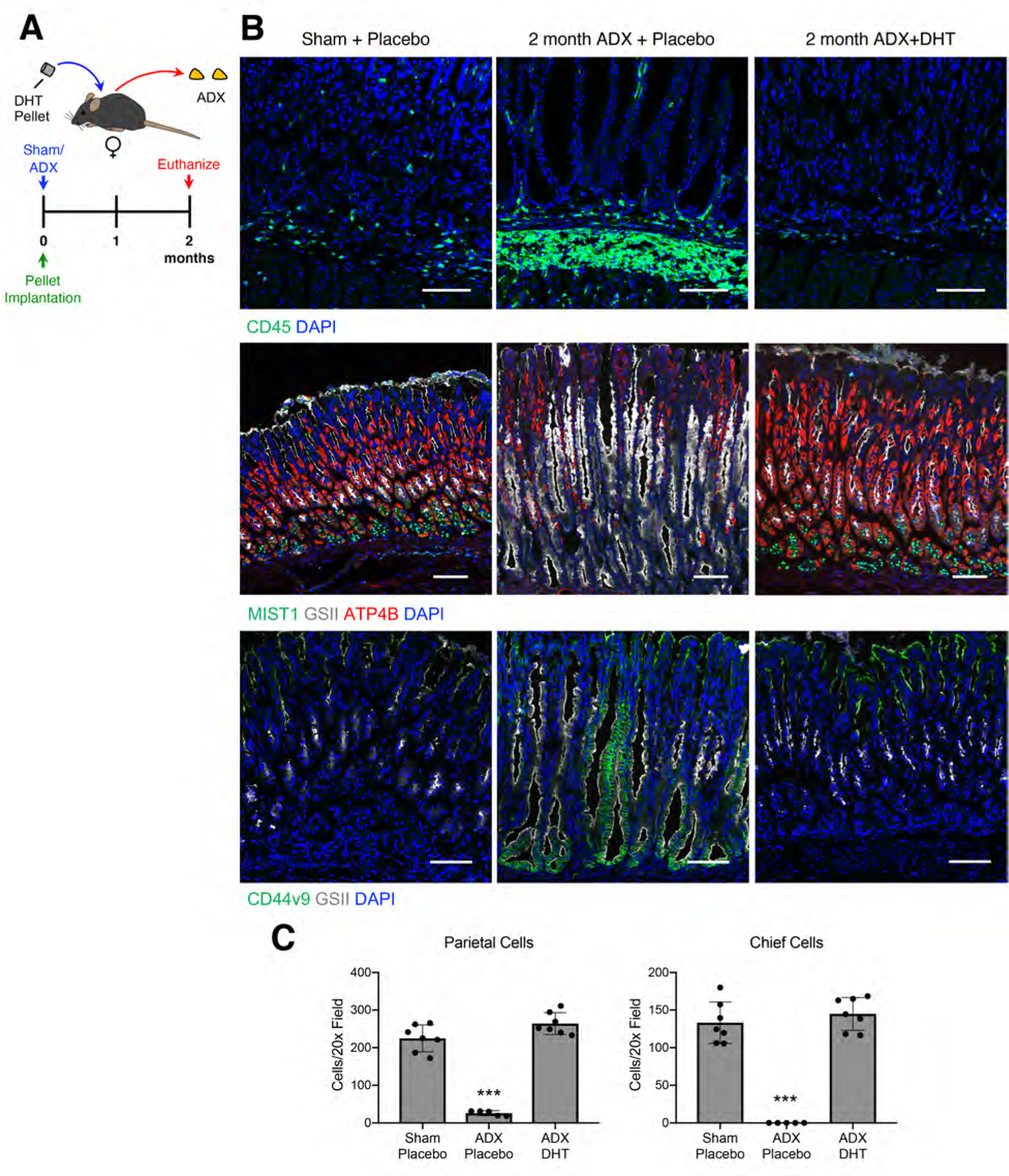
(A) Experimental model: Female mice were adrenalectomized and simultaneously implanted with a sustained-release 5α dihydrotestosterone (DHT) pellet. Stomachs were collected 2 months after surgery. (B) Immunostaining of stomach sections probed for CD45 (pan-leukocytes, green), for ATP4B (parietal cells, red), GSII lectin (mucous neck cells, white), and MIST1(chief cells, green), or for CD44v9 (SPEM, green) and GSII lectin (white). Nuclei are stained with DAPI. Scale bars are 100 µm. (C) Quantitation of the number of parietal cells and chief cells observed per 20x field (n≥5 mice/group). Data are mean ± SD; P-values were determined by one-way ANOVA with post hoc Tukey’s t-test. ***P≤0.0001
Tissue preparation.
The gastric corpus lesser curvature was used for all analyses; a representative schematic is in Supplementary Figure 1A. Stomachs were opened along the greater curvature and washed in phosphate buffered saline to remove the gastric contents. Stomachs used for histology were pinned to cork boards and fixed overnight in 4% paraformaldehyde at 4°C. After fixation, stomachs used for cryosectioning were cryopreserved in 30% sucrose and then embedded in optimal cutting temperature (OCT) media. Stomachs used for paraffin sectioning were processed, embedded, sectioned, and H&E stained according to standard protocols. For RNA isolation, the gastric corpus lesser curvature was collected with a 4 mm biopsy punch and immediately snap-frozen in liquid nitrogen. RNA was extracted in TRIzol (ThermoFisher, Waltham, MA) and precipitated from the aqueous phase using 1.5 volumes of 100% ethanol. The mixture was transferred to a RNeasy column (Qiagen, Hilden, Germany), and the remaining steps were followed according to the RNAeasy kit manufacturers recommendations. RNA was treated with RNase free DNase I (Qiagen) as part of the isolation procedure.
Histology.
Five µm cryosections from the gastric corpus lesser curvature were incubated with ATP4B (clone 2G11, Thermofisher), MIST1 (Cell Signaling Technologies, Danvers, MA), CD45 (clone 104 Bioledgend, San Diego, CA), or CD44v9 (Cosmo Bio, Tokyo, Japan) for 1 hour at room temperature or overnight at 4°C. Primary antibody was omitted as a negative control. Sections were incubated in secondary antibodies for 1 hour at room temperature. Fluorescent conjugated Griffonia simplicifolia lectin (GSII; ThermoFisher) was added with secondary antibodies. Sections were mounted with Vectastain mounting media containing DAPI (Vector Laboratories Burlingame, CA). Images were obtained using a Zeiss 880 confocal laser-scanning microscope (Carl-Zeiss GmbH, Jena, Germany) and running Zen Black imaging software. For AR immunohistochemistry, paraffin sections were probed with anti-AR antibodies (EPR1535(2), Abcam Cambridge, United Kingdom) overnight at 4°C using the Vectastain ABC-HRP kit (Vector Laboratories). 3,3’-diaminobenzidine was used for detection (Vector Laboratories). Staining with isotype IgG was used as a negative control. Human stomach sections were obtained from the West Virginia University tissue bank. Hematoxylin and eosin and IHC stained paraffin sections were imaged by an Aperio AT2 Scanner (Leica Biosystems Inc. Buffalo Grove, IL). Parietal cells and chief cells were quantitated as previously described 10 using confocal micrographs captured using a 20x microscope objective and 1 µm thick optical sections. Cells were counted using the ImageJ (National Institutes of Health, Bethesda, MD) count tool. Cells that stained positive with anti-ATP4B antibodies were identified as parietal cells while cells that stained positive with anti-MIST1 antibodies and were GSII negative were identified as mature chief cells. Counts were reported as the number of cells observed per 20x field. Images were chosen that contain gastric glands cut longitudinally. The first 5–6 glands adjacent to the limiting ridge were excluded from quantitation because they often exhibit an abnormal morphology.
Flow cytometry.
To isolate leukocytes from the stomach, mice were euthanized, and the stomachs were removed as described above. The corpus was washed in Hanks Balanced Salt Solution without Ca2+ or Mg2+ containing 5 mM HEPES, 5 mM EDTA, and 5% FBS at 37°C. Tissue was then washed in Hanks Balanced Salt Solution with Ca2+ and Mg2+ for 10 minutes at 37°C and then digested in 1 mg/ml type 1 collagenase and 0.5 mg/ml DNase I (Worthington Biochemical, Lakewood, NJ) for 20 minutes at 37°C. After digestion, the tissue fragments were pushed through a 100 µm strainer. Fc receptors were blocked with TruStain (BioLegend) and then stained with the antibodies listed in Supplementary Table 1 for 20 minutes at 4°C. Actinomycin D (ThermoFisher) was used to label dead cells. Cells were analyzed on a BD Fortessa (BD Bioscience). For cells sorted from the small intestines, the small intestines were removed and flushed with PBS. The intestines were disassociated using the Lamina Propria Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer’s instructions. Intestinal cells were stained with the antibodies found in Supplementary Table 1 as described above and sorted on a FACS Aria III cell sorter (BD Bioscience) using the gating strategy in Supplementary Figure 6. The data was analyzed by Cytobank software (Cytobank Inc., Santa Clara, CA). RNA was isolated using a NucleoSpin RNA kit (Takara Bio USA Inc. Mountain View, CA).
T cell transfer and ILC2 depletion.
Prior to ILC2 depletion, Rag1 KO mice received an intravenous injection of 5 million splenic T cells isolated from Thy1.1 mice through the lateral tail vein. ILC2s were depleted from Rag1 KO mice using CD90.2 antibodies. The mice received 7 intraperitoneal injections containing 200 µg of CD90.1 antibodies (Clone 30H12, BioxCell, Lebanon, NH) or isotype IgG control antibodies (clone MPC-11) every 2 days for 14 days. Four days after T cell transfer and the first antibody dose, the mice underwent sham surgery or adrenalectomy + castration. The mice were euthanized for analysis 10 days after surgery (schematic in Figure 6A).
Figure 6. ILC2 depletion protects from SPEM development.
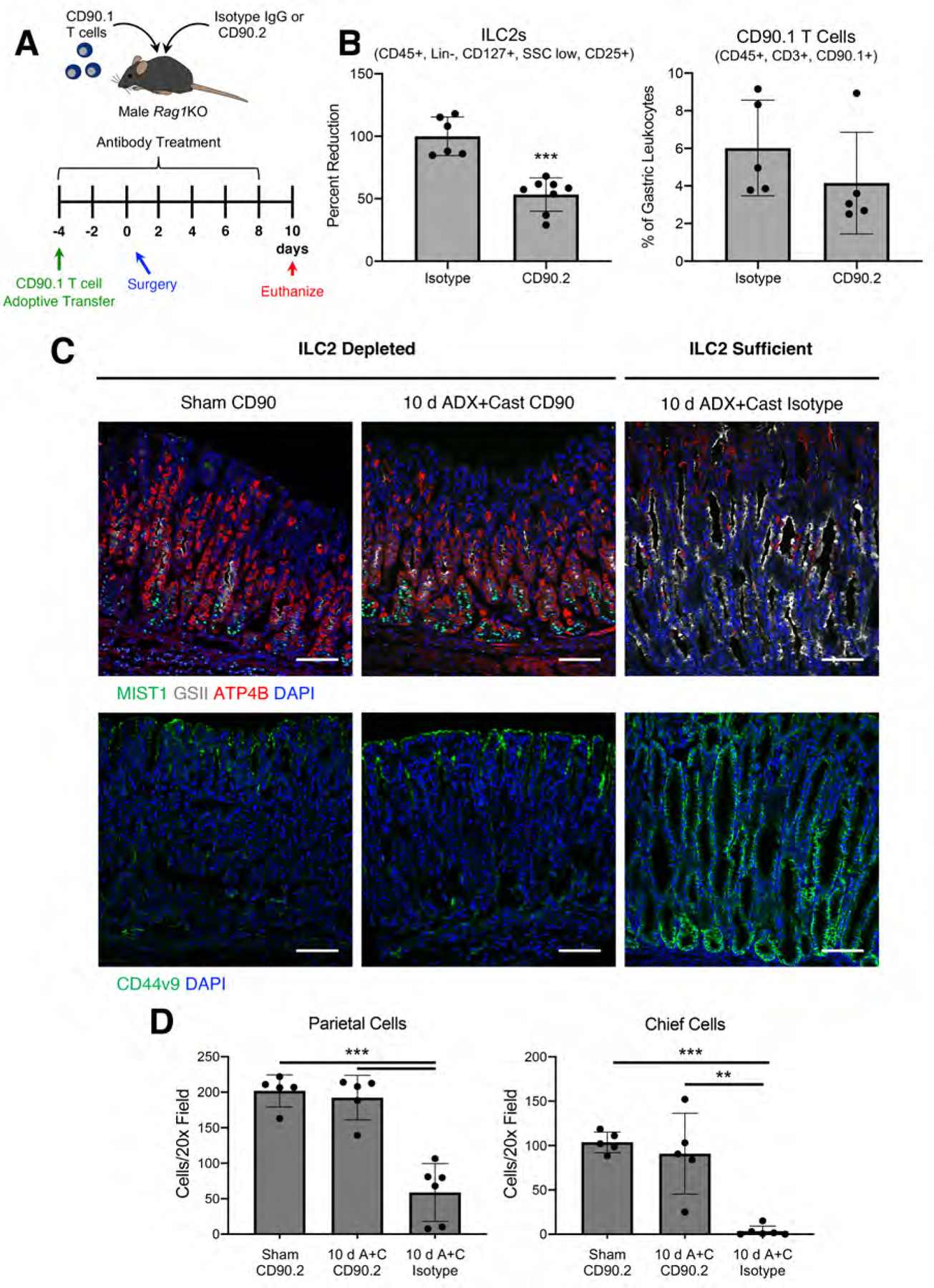
(A) Experimental model: CD90.1 T cells were adoptive transferred into Rag1 KO mice 4 days before surgery. In addition, mice were treated with CD90.2 or isotype control antibodies every 2 days throughout the experiment beginning 4 days before surgery. Stomachs were collected for analysis 10 days after sham surgery or adrenalectomy + castration. (B) Flow cytometry analysis of the gastric corpus from sham mice treated with CD90.2 or isotype control antibodies. (C) Immunostaining of stomach sections probed for (C) ATP4B (parietal cells, red), GSII lectin (mucous neck cells, white), and MIST1(chief cells, green) or for (D) CD44v9 (SPEM, green). Nuclei are stained with DAPI. Scale bars are 100 µm (D) Quantitation of the number of parietal cells and chief cells observed per 20x field (n≥5 mice/group). Data are mean ± SD; P-values were determined by unpaired T test (B) or by one-way ANOVA with post hoc Tukey’s t-test (D). ***P≤0.0001
scRNAseq.
Single-cell RNAseq was performed on pooled leukocytes isolated from the gastric corpus of four mice per treatment group. Viable CD45+ cells were sorted using a FACS Aria II (BD Bioscience, San Jose, CA) running FACS Diva software and resuspended at a concentration of 1.0 x 106 cells/ml in RPMI 1640 + 10% FBS. Single cell libraries were prepared on a 10x Genomics Chromium controller (10x Genomics, Pleasanton, CA) using the version 3 reagent kit according to the manufacturer’s instructions. The reagent mixture was calculated for capturing 6000 cells. The libraries were sequenced on a NovaSeq (Illumina, San Diego, CA) by the NIEHS Epigenomics core laboratory. Raw read processing was carried out using the Cell Ranger Single-Cell Software Suite (version 3.0.1, 10X Genomics). Demultiplexed FASTQ files (paired-end reads 1: 30bp, read 2:100bp) were generated using the CellRanger mkfastq command. The primary data analyses which included alignment, filtering, barcode counting and UMI quantification, quality control, clustering and statistical analysis were performed using CellRanger count command. Gene positions were annotated using Ensembl build 93 and filtered for biotype. Raw gene expression matrices were imported into R (version 3.6.1) and converted to a Seurat object using the Seurat R package (version 3.1.0)31. To remove dead cells and doublets, the total number of UMIs and genes and the percentage of UMIs derived from the mitochondrial genome for each cell were counted. The upper and lower bounds were defined as the mean +/− two standard deviations for both the total UMIs and genes, respectively. Cells which had over 10% UMIs derived from the mitochondrial genome were discarded. Finally, cells with total UMIs or genes outside of the upper and lower bounds were removed. For the remaining cells, gene expression matrices were normalized to the total cellular read count and to the mitochondrial read count. Highly variable genes (HVG) were selected from the normalized data using the Seurat SCTransform function. The top 3000 HVGs were used as features for dimensionality reduction and clustering. The Seurat RunPCA and JackStraw functions were performed to calculate and to select significant (P-value <0.05) principal components (PCs), respectively. The RunUMAP function was then applied to plot the selected significant PCs. The FindNeighbors constructed a Shared Nearest Neighbor (SNN) graph, and FindClusters function with “resolution = 0.1” parameter was carried out to cluster cells into different groups. Marker genes were defined based on the following criteria: 1) the average expression value in the cluster of interest was at least 1.2-fold higher than the average expression in the rest of the clusters; 2) greater than 10% of cells in the cluster of interest which were detectable; and 3) marker genes had the highest mean expression in the cluster of interest compared to the rest of the clusters. Canonical marker genes were used to annotate cell clusters.
qRT-PCR.
Reverse transcription followed by qPCR was performed in the same reaction using the Universal Probes One-Step PCR kit (Bio-Rad Laboratories) and the Taqman primers Cftr (Mm00445197_m1), Olfm4 (Mm01320260_m1), Wfdc2 (Mm00509434_m1), Il13 (Mm00434204_m1), Csf2 (Mm01290062_m1), Ar (Mm00442688_m1), and Nr3c1 (Mm00433832_m1) (ThermoFisher) on a CFX96 Real Time PCR system (Bio-Rad Laboratories). Messenger RNA levels were normalized to the reference gene Ppib (Mm00478295_m1).
Statistical analysis.
All error bars are ± the standard deviation of the mean. The sample size for each experiment is indicated in the figure legends. Experiments were repeated a minimum of two times. Statistical analyses were performed using one-way ANOVA with post-hoc Tukey’s t-test when comparing 3 or more groups or by unpaired t-test when comparing 2 groups. Statistical analysis was performed by Graphpad Prism 9 software. Statistical significance was set at p≤0.05. Specific p values are listed in the figure legends.
Results
Glucocorticoids are required to suppress gastric inflammation in females but are dispensable in males.
We previously reported that glucocorticoids are master regulators of gastric inflammation and that global disruption of glucocorticoid signaling triggers spontaneous gastric inflammation and SPEM 10. To determine how sex impacts gastric inflammation, we adrenalectomized C57B6/J mice and aged them for 2 months after surgery. Female mice exhibited prominent mucosal thickening and chronic inflammation through the entire gastric corpus lesser curvature (Figure 1A and Supplementary Figure 1B). In contrast, the stomachs of adrenalectomized male mice were histologically normal. To determine if male resistance to ADX-induced gastric inflammation involved male sex hormones, male mice were either castrated alone or simultaneously adrenalectomized + castrated (ADX+Cast). The stomachs of castrated males did not exhibit gross histological abnormalities (Figure 1A). However, ADX+Cast mice developed prominent histological lesions that appeared identical to adrenalectomized female mice. Further analysis of discrete cell populations within the gastric glands demonstrated that 86% of parietal cells and 97% of chief cells were lost in adrenalectomized females, but these cell populations were not significantly reduced in adrenalectomized or castrated males (Figure 1B–C). In contrast, 80% of parietal cells and 99% of chief cells were lost in ADX+Cast males.
Figure 1. Glucocorticoids are dispensable in males for suppressing gastritis.
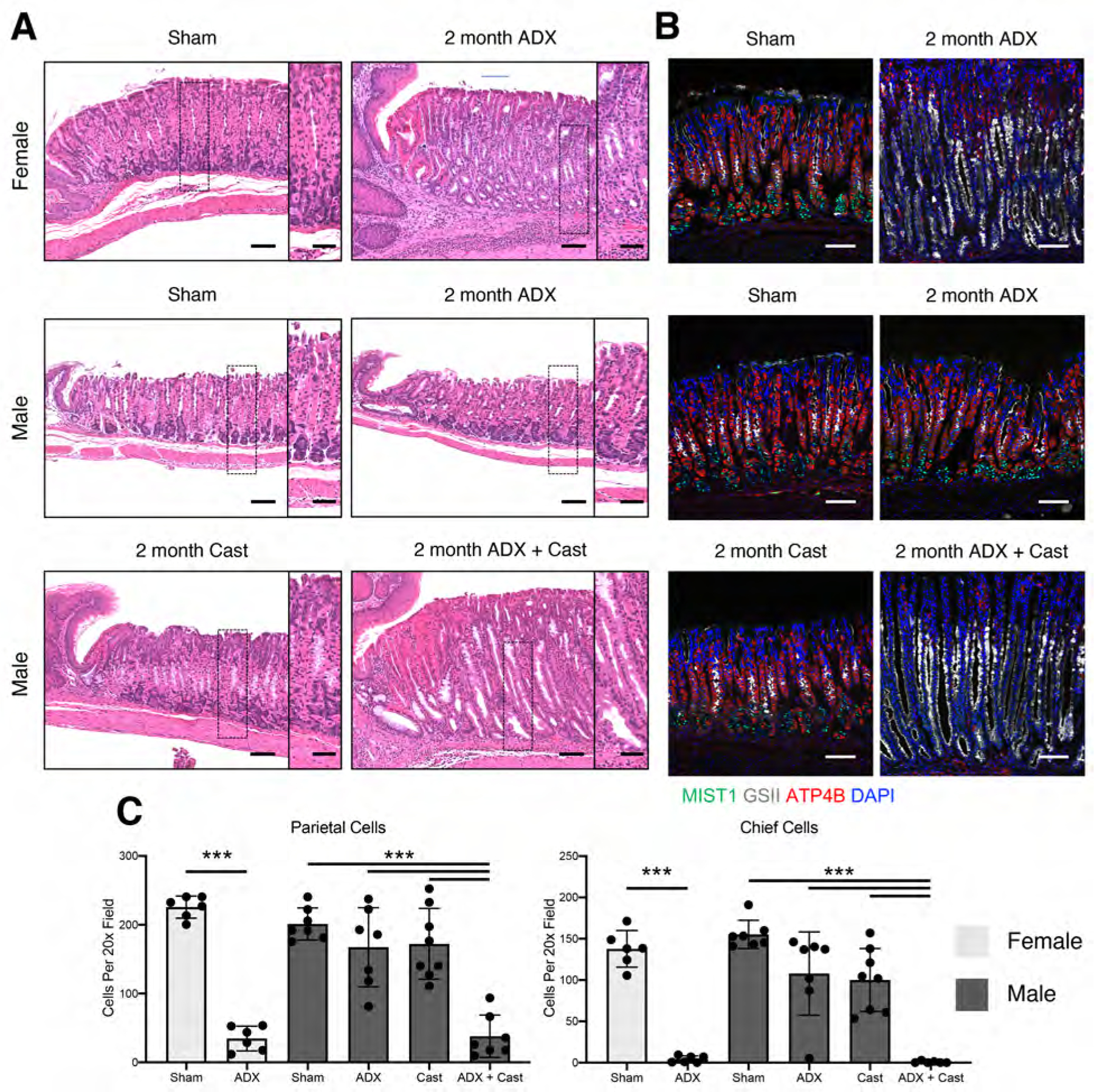
Stomachs were collected from mice 2 months after surgery. (A) Representative micrographs of H&E-stained sections of the gastric corpus. Scale bars are 100 µm and 50µm in the inset. (B) Immunostaining of stomach sections probed for ATP4B (parietal cells, red), GSII lectin (mucous neck cells, white), and MIST1(chief cells, green). Nuclei are stained with DAPI. Scale bars are 100 µm. (C) Quantitation of the number of parietal cells and chief cells observed per 20x field (n≥6 mice/group). Data are mean ± SD; P-values were determined by one-way ANOVA with post hoc Tukey’s t-test. ***P≤0.0001
Chronic gastric inflammation and oxyntic atrophy are linked to SPEM development. SPEM development was confirmed by immunostaining for the SPEM marker CD44 variant 9 (CD44v9), demonstrating that SPEM only developed in adrenalectomized females and ADX+Cast males (Figure 2A). In addition, qRT-PCR of RNA isolated from the gastric corpus revealed that the characteristic SPEM markers Cftr, Olfm4, and Wfdc2 were significantly induced in adrenalectomized females and ADX+Cast males (Figure 2B), but these genes were not significantly increased in either adrenalectomized or castrated males. Together, these results suggest that male sex hormones have an essential role in suppressing pathogenic gastric inflammation and metaplasia.
Figure 2. Glucocorticoids are required to suppress spontaneous SPEM in females but are dispensable in males.
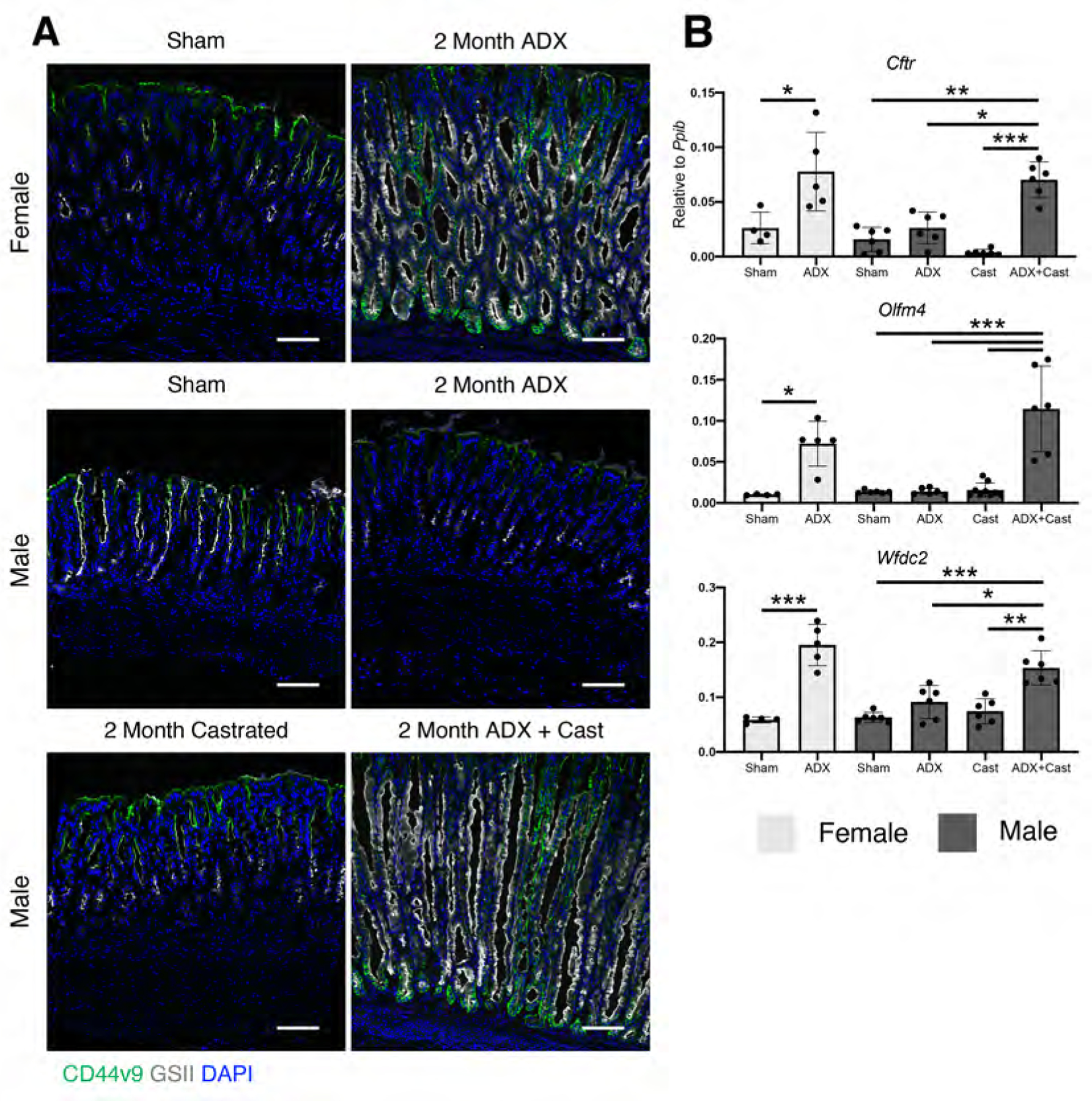
(A) Immunostaining for the SPEM marker CD44 variant 9 (green), mucous neck cells are stained with GSII lectin (white). Nuclei are stained with DAPI. Scale bars are 100 µm. (B) Quantitative RT-PCR of the indicated SPEM marker genes using RNA isolated from the gastric corpus (n≥4 mice/group). Data are mean ± SD; P-values were determined by one-way ANOVA with post hoc Tukey’s t-test. * P≤0.01 **P≤0.001 ***P≤0.0001
Androgens suppress gastric inflammation and metaplasia.
We next asked whether androgens can prevent ADX-induced gastric inflammation and metaplasia. Female mice were adrenalectomized and simultaneously implanted with a 5α-dihydrotestosterone (DHT) sustained-release pellet or a placebo pellet (Figure 3A). Two months after surgery, adrenalectomized mice implanted with placebo pellets exhibited chronic inflammation, loss of parietal and chief cells, and chronic SPEM (Figure 3B–C and Supplementary Figure 2). DHT treatment completely blocked the development of gastric inflammation in adrenalectomized females. Moreover, DHT treatment prevented loss of parietal cells and chief cells and blocked SPEM development. Next, we asked if androgens could reverse ADX-induced gastric inflammation and SPEM. Female mice were aged 1 month after ADX or sham surgery to allow chronic inflammation and SPEM development. Mice were then implanted with a sustained-release DHT pellet and aged an additional month (Figure 4A). One month after surgery, ADX mice had oxyntic atrophy and SPEM (Figure 4B–C). However, 1 month of DHT treatment completely reversed these pathologies. The stomachs of DHT-treated ADX mice were histologically indistinguishable from Sham mice (Figure 4B) and exhibited the normal complement of parietal and chief cells (Figure 4C). These data demonstrate that androgens have potent anti-inflammatory effects within the stomach. Moreover, these data show that androgens can compensate for glucocorticoid insufficiency by controlling gastric inflammation.
Figure 4. Androgen treatment reverses chronic SPEM in female mice.
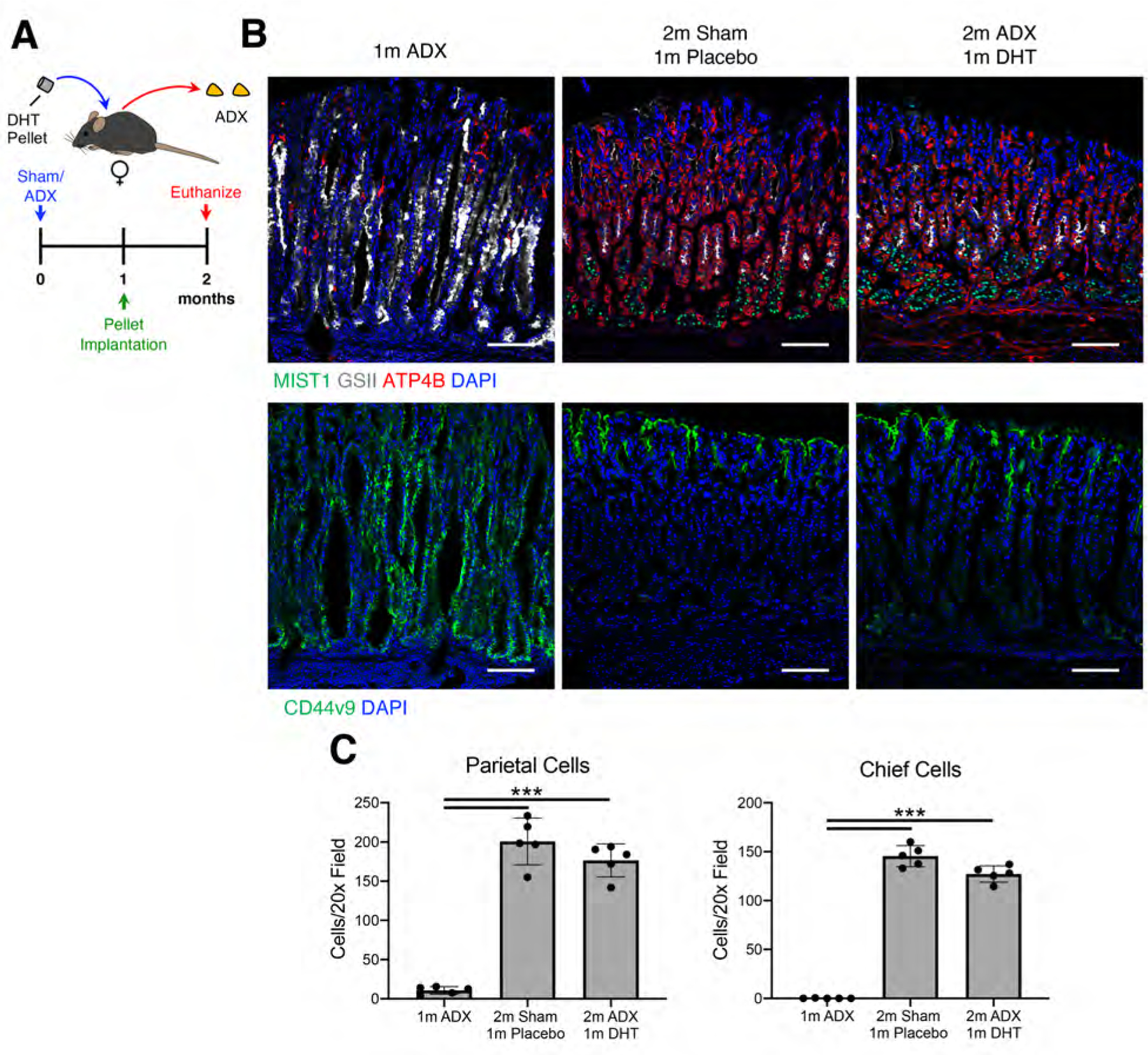
(A) Experimental design. Female mice were aged 1 month after sham surgery or adrenalectomy. They were then implanted with a sustained release dihydrotestosterone pellet or placebo controls and then were aged for an additional month. (B) Immunostaining of stomach sections probed for (B) ATP4B (parietal cells, red), GSII lectin (mucous neck cells, white), and MIST1(chief cells, green) or for (C) CD44v9 (SPEM, green). Nuclei are stained with DAPI. Scale bars are 100 µm (D) Quantitation of the number of parietal cells and chief cells observed per 20x field (n≥5 mice/group). Data are mean ± SD; P-values were determined by one-way ANOVA with post hoc Tukey’s t-test. ***P≤0.0001
Androgens signal through type 2 innate lymphoid cells.
Because DHT treatment suppressed gastric inflammation, we next sought to identify the cell populations within the stomach that respond to androgens. Immunoblot analysis of protein lysates from the gastric corpus exhibited equivalent expression of the AR within male and female stomachs (Figure 5A). We then performed immunohistochemistry (IHC) to identify AR-positive cell populations within the stomach. IHC revealed that the AR was not detectable within the gastric epithelial cells (Figure 5B). However, AR-positive cells were scattered between the gastric glands and within the lamina propria that were morphologically similar to leukocytes. A similar AR-expression pattern was observed in gastric biopsies from human patients (Supplementary Figure 3). Based on these observations, we performed single-cell RNAseq (scRNAseq) of CD45-positive cells isolated from the gastric corpus by flow cytometry. Leukocytes were isolated from male and female mice euthanized 2 months after sham surgery, ADX, and ADX+Cast. After sequencing, cells were dimensionally reduced into clusters using the unbiased Uniform Manifold Approximation and Projection (UMAP) algorithm, which identified 9 distinct cell clusters (Figure 5C). Each cell cluster was annotated based on the unique expression of characteristic marker genes, and representative genes were visualized as a dot plot (Figure 5D). Approximately 50% of stomach leukocytes in sham mice were macrophages (Figure 5E). However, the proportion of macrophages decreased in ADX and ADX+Cast mice due to expansion of the granulocyte, mast cell, and B cell populations. Several recent studies, including our previous work, have demonstrated that macrophages are required to induce SPEM development. Surprisingly, our scRNAseq revealed that Ar mRNA was not detected within the macrophage cluster (Figure 5F). Rather, Ar expression was restricted to T cells, NK cells, ILC2s, and fibroblasts. In contrast, the GR transcript (Nr3c1) was ubiquitously expressed by all of the cell populations (Figure 5F). Interestingly, the relative expression of both the Ar and Gr was highest within the ILC2 cluster.
Figure 5. Gastric ILC2s express abundant AR and GR.
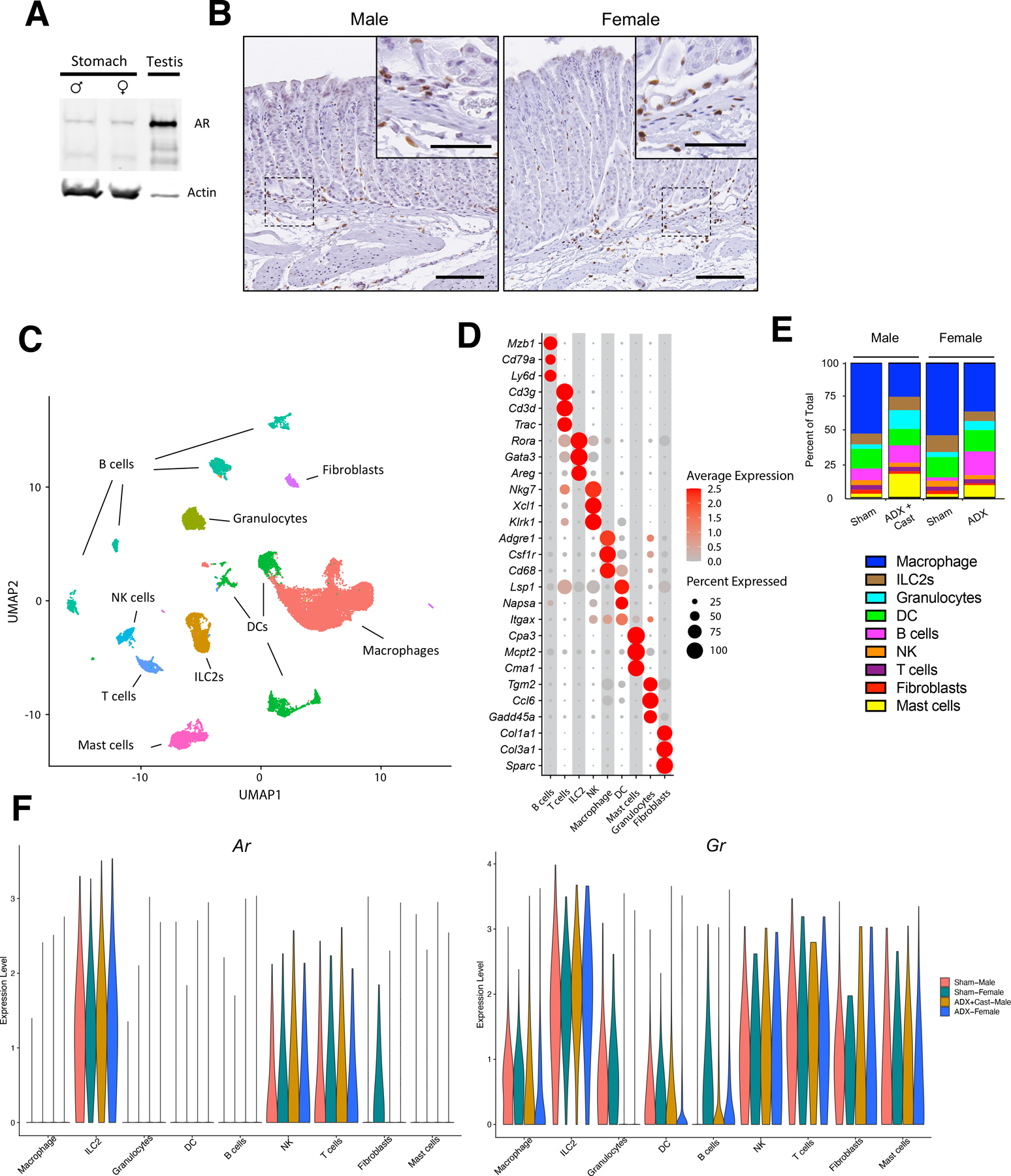
(A) Immunoblot for the androgen receptor using protein isolated from the gastric corpus of male or female mice or the testis probed. (B) Immunohistochemistry of the gastric corpus stained with anti-AR antibodies. Scale bars are 100 µm and 50 µm in the inset. (C) UMAP unbiased clustering of single cell RNAseq performed using CD45+ cells isolated from the gastric corpus from sham males, ADX+Cast males, sham females, and ADX female euthanized 2 months after surgery. n=4. (D) Cell clusters annotated based on their expression of characteristic marker genes. Dot size represents the percentage of positive cells within a cluster and dot color represents the relative gene expression. (E) Ratios of each cell population in each treatment group. Total=100%. (F) Violin plots of relative Ar and Gr expression within cell clusters stratified by treatment group.
ILC2s are critical regulators of gastric inflammation.
Pathogenic macrophage activation drives SPEM development 10, 22. However, our scRNAseq data demonstrated that macrophages do not express detectable Ar mRNA. Recently, it was shown that ILC2s are required for SPEM development in L365 treated mice 11. Because ILC2s express abundant Ar and Gr, we hypothesized that ILC2s regulate gastric inflammation and SPEM development. To test this hypothesis, we depleted ILC2s by injecting CD90.2 antibodies (Figure 6A). CD90 is expressed by both ILC2s and T cells. Therefore, we utilized Rag1 KO mice which lack mature B and T cells. We previously showed that ADX-induced SPEM development occurs normally in Rag1 KO mice 10. However, because gastric T cells express Ar, we replace the gastric T cell compartment by adoptive transfer of CD90.1 T cells which were spared by CD90.2 antibody treatment 32. CD90.2 antibody treatment significantly reduced the gastric ILC2 population by 50% relative to mice treated with isotype control antibodies (Figure 6B), but the transferred CD90.1 T cells and macrophages, eosinophils, and NK cells were not affected by antibody injection (Figure 6B and Supplementary Figure 4). Ten days after surgery, ADX+Cast mice exhibited a significant 71 % loss of parietal and a 91% loss of chief cells (Figure 6C–D) and widespread induction of the SPEM marker CD44v9 (Figure 6C). In contrast, ILC2 depletion suppressed these ADX+Cast-induced pathologies preventing parietal and chief cell loss and CD44v9 induction.
To further test the role of ILC2s in gastric inflammation and SPEM development, we evaluated if impaired ILC2 function could protect from pathogenic inflammation-induced SPEM. To achieve this, we utilized Il33 KO mice. IL33 is an alarmin which drives ILC2 proliferation and activation 33 . In WT mice, IL33 is constitutively expressed by surface mucous (pit) cells within the gastric corpus (Supplementary Figure 5A). We found that during steady-state conditions, Il33 KO males had significantly fewer tissue-resident ILC2s within the gastric corpus (Supplementary Figure 5B). Ten days after ADX+Cast, Il33 KO mice were resistant to parietal and chief cell loss while WT controls exhibited a significant 47% loss of parietal cells and 87% loss of chief cells (Supplementary Figure 5C–D). Moreover, Il33 KO mice had limited induction of the SPEM marker CD44v9 compared to widespread SPEM development in ADX+Cast WT controls (Supplementary Figure 5C). These data further demonstrate that ILC2s are important for initiating pathogenic gastric inflammation and SPEM.
Androgens signal within ILC2s to suppress cytokine production.
Because ILC2s are required to induce SPEM, we next asked if androgens could suppress the expression of transcripts encoding proinflammatory cytokines within ILC2s. Analysis of the scRNAseq data revealed that Il13 and Csf2 were abundant within ILC2s (Figure 7A). These transcripts are well-known direct targets of glucocorticoids 34, 35 and the receptors for these cytokines are widely expressed by gastric leukocytes (Figure 7A). Moreover, IL13 has previously been shown to be required to induce SPEM development in L635 treated mice 16. Therefore, we utilized the cytokines Il13 and Csf2 to assess the ability of androgens to suppress the expression of proinflammatory genes within ILC2s. Quantitative RT-PCR demonstrated a significant increase in Il13 and Csf2 mRNA within the gastric corpus of adrenalectomized females and ADX+Cast males, but not in adrenalectomized or castrated males (Figure 7B). Next, we asked if Il13 and Csf2 were regulated by androgens or glucocorticoids. We utilized intestinal ILC2s for this procedure due to the relatively few ILC2s present within the stomach. RNA was isolated from macrophages, B cells, T cells, and ILC2s sorted from the small intestine (see Supplementary Figure 6 for gating strategy) and analyzed by qRT-PCR. Similar to stomach leukocytes, Ar mRNA was undetectable in macrophages and B cells, expressed at relatively low levels in T cells, but was abundant within ILC2s (Figure 7C). In contrast, Gr expression was equivalent in all of the cell populations. Next, ADX+Cast males were treated with a single intraperitoneal injection of either DHT or dexamethasone (Figure 7D). Three hours after injection, the mice were euthanized, and ILC2s were collected from the small intestines. Immunostaining for the AR in isolated intestinal ILC2s demonstrated that AR was predominantly within the cytoplasm of vehicle-treated mice but markedly translocated to the nucleus upon treatment with DHT (Figure 7E), demonstrating the normal nuclear translocation of activated steroid hormone receptors. Quantitative RT-PCR of RNA isolated from intestinal ILC2s revealed that treatment with either DHT or dexamethasone significantly suppressed expression of both Il13 and Csf2 after 3 hours (Figure 7F). These results demonstrate a novel role for androgens in suppressing the expression of mRNAs encoding proinflammatory cytokines within ILC2s.
Figure 7. Androgens and glucocorticoids suppress proinflammatory cytokine production by ILC2s.
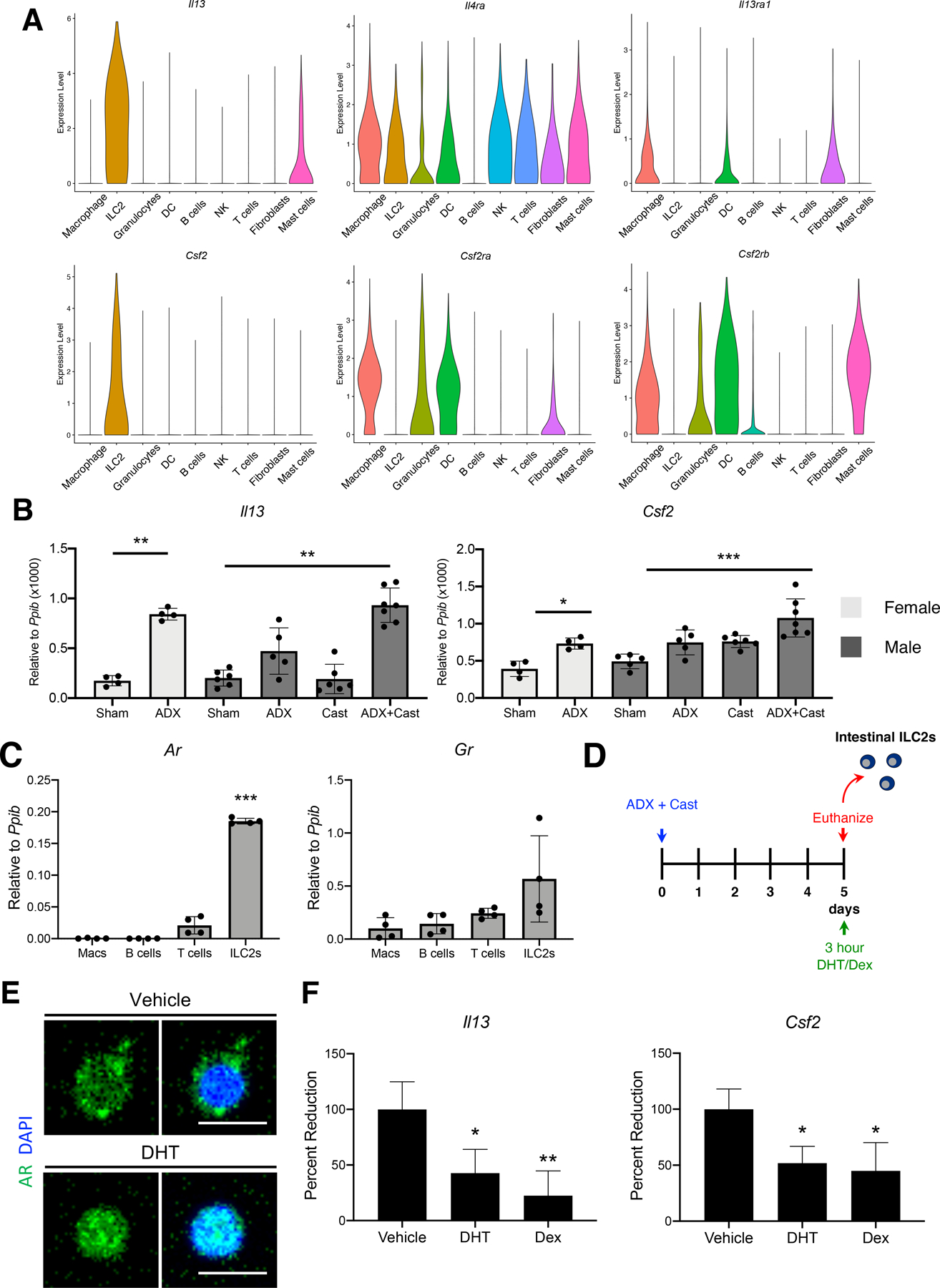
(A) Violin plots of the indicated genes derived from the overlayed scRNAseq data of Sham males, ADX+Cast males, Sham females, and ADX female 2 months after surgery. (B) Quantitative RT-PCR of the indicated genes using RNA isolated from the gastric corpus (n≥4 mice/group). (C) Quantitative RT-PCR of the Ar and Gr using RNA isolated from intestinal macrophages, B cells, T cells, and ILC2s. n=8. (D) Experimental model: Male mice were adrenalectomized + castrated to clear endogenous hormones. Five days after surgery, mice received a single intraperitoneal injection of 5α-dihydrotestosterone (DHT) or dexamethasone (Dex). Three hours after injection, the intestines were removed for isolation of discrete leukocyte populations. (E) Immunostaining of intestinal ILC2s isolated from ADX+Cast mice 3 hours after treatment with vehicle or DHT. Cells were stained with AR antibodies, and nuclei were stained with DAPI. Scale bar 10 µm. (F) Quantitative RT-PCR of Il13 and Csf2 using RNA isolated from intestinal ILC2s. Experiments were repeated 3 times with cells pooled from ≥2 mice/group. All data are mean ± SD; P-values were determined by one-way ANOVA with post hoc Tukey’s t-test. *P≤0.05, **P≤0.001, and ***P≤0.0001.
Discussion
Biological sex has profound effects on inflammation, and a host of inflammatory syndromes and diseases are stratified by sex 5. Females typically elicit a more robust inflammatory response to pathogens and have increased protection from infection 1. Moreover, females develop higher antibody titers in response to vaccines 3. In contrast, males have generally weaker inflammatory reactions and are more likely than women to die from infection 36. The robust inflammatory response in females may come with an increased risk of inflammatory diseases, and 8 of 10 individuals with autoimmune disease are women 1. While many of these inflammatory diseases are multi-factorial, sex hormones play important roles in regulating inflammation. Before puberty, boys and girls exhibit similar asthma rates, but after puberty, the ratio of asthma in males becomes significantly lower, suggesting that increased androgens may be protective 37. A similar trend is observed within the GI tract where pre-puberal boys and girls have similar rates of Crohn’s disease, but women exhibit higher rates than men 38, 39. Within the stomach, non-autoimmune, autoimmune, and eosinophilic gastritis are more common in women, but the etiology underlying these sex-specific effects in inflammatory disease are not clear 40. Studies have found that bacterial infections by H pylori are more common in men and that men have higher bacterial loads than women 7–9. While glucocorticoids are the primary anti-inflammatory steroid hormone, these hormones signal differently in males and females, in part due to interactions with sex steroid receptors 2, 41, 42. Moreover, recent studies have found that androgens also have overt anti-inflammatory effects 24, 25. Indeed, within the lungs, androgens signal in ILC2s to suppress their activation and production of proinflammatory cytokines. However, the exact mechanisms by which sex influences the inflammatory response are poorly understood. Here, we report a novel role for androgens in regulating gastric inflammation. We found that adrenalectomized females develop pathogenic inflammation and SPEM. In contrast, the stomachs of adrenalectomized males remain normal due to the anti-inflammatory effects of androgens, which signaled within ILC2s to suppress the expression of genes encoding proinflammatory cytokines.
Glucocorticoids and androgens predominately signal through binding to the GR and AR, respectively. These receptors are ligand-dependent transcription factors that regulate gene transcription by binding DNA response elements to recruit initiation factors, chromatin modifiers, and RNA polymerase II 43. The GR and AR are structurally similar to each other, and the DNA response elements bound by these receptors are nearly identical 26. The GR and AR can readily bind each other’s response elements in vitro, and GR and AR co-expression in immortalized cell lines demonstrates significant overlap in their target genes 44, 45. Moreover, expression of the GR in prostate cancer is thought to enable cancer cells to escape anti-androgen therapies by regulating the expression of AR target genes 30. We demonstrate that the GR is ubiquitously expressed by all leukocytes, consistent with its established role in regulating inflammation. However, AR expression is much more restricted, suggesting a more nuanced role in regulating inflammation. ILC2s co-express high levels of the GR and AR. Within ILC2s, treatment with either steroid suppressed expression of the proinflammatory cytokines Il13 and Csf2. These mRNAs were significantly suppressed within 3 hours of DHT treatment. The short timeframe for this repression suggests that androgens are directly inhibiting these transcripts. The redundant functions of glucocorticoids and androgens may provide males with an extra layer of protection from inflammatory diseases and partially explain why men have lower rates of inflammatory and autoimmune diseases than women.
Several recent studies report that macrophages induce gastric damage and drive SPEM development 10, 16, 22. Other studies suggest that androgens suppress macrophage activation 46. However, we could not detect Ar mRNA within stomach macrophages by scRNAseq, or within intestinal macrophages by qRT-PCR. While androgens may signal in macrophages through AR independent mechanisms such as interacting with g-protein-coupled receptors, androgens also may impact macrophage activity indirectly by signaling in other cell populations. ILC2s are well-known for their ability to induce and coordinate inflammation. Here, we demonstrate that gastric ILC2s express Il13 and Csf2, cytokines that are known to modulate macrophage activity 47, 48. Recently, it was reported that IL13 activates gastric macrophages and is required to induce SPEM development 16. Th2 T cells can also produce IL13, but ILC2s produce approximately 10-fold more IL13 than CD4+ Th2 T cells 20. We previously showed that T cell-deficient Rag1 KO mice develop SPEM normally 10, 22, and we showed here that ILC2 depletion protects from SPEM development. Thus, ILC2s are ideally positioned within the stomach to regulate inflammation and macrophage activation, and androgen signaling within ILC2s likely shapes the gastric macrophage response.
Our data indicate that males are more resistant than females to gastric inflammation. Helicobacter pylori-induced chronic inflammation is thought to be the primary driver of gastric cancer development 12. Metaplasia is also considered a potential precursor of gastric adenocarcinoma, although SPEM develops in response to a variety of glandular insults and may not progress to cancer. Here, we found that females were more susceptible to gastric inflammation and SPEM than males, but men are nearly two times more likely than women to develop gastric cancer 49. Interestingly, the male bias in H pylori-induced carcinogenesis is not always born out in animal studies. Some studies have found that male mice are more susceptible to H pylori-induced gastric inflammation, mutation, and cancer 50, 51; others have found no male bias 8, 52. The increased rate of gastric cancer in men is partially attributed to lifestyle differences, with men consuming more alcohol and being more likely to smoke than women 53–55. Age is another significant risk factor for gastric cancer 56. Decreased androgen production during male menopause may increase cancer susceptibility later in life. Another possibility is that androgen receptor expression may change within the stomach during cancer development. We show that the gastric epithelium does not express detectable levels of the androgen receptor in samples from healthy mice and humans. However, a recent study found that 43% of gastric cancers were AR-positive 57. AR-positive gastric cancers are associated with increased metastasis and decreased patient survival 58. These features are reminiscent of prostate cancer, where androgens have different roles in healthy tissue and cancer. Thus, androgens may exert dual functions to protect from inflammatory and autoimmune diseases during normal conditions, but they may promote cancer growth and progression when dysregulated. Future studies will assess the role of glucocorticoids and sex hormones in regulating inflammation and cancer development during H pylori infection.
Supplementary Material
Acknowledgments
The authors thank the NIEHS Comparative Medicine Branch, Histology Core Laboratory, Epigenomes Core Laboratory, Flow Cytometry Center, Fluorescence Microscopy Imaging Center, and NIEHS Arts and Graphics for their assistance.
Funding: This research was supported by a Postdoctoral Research Associate (PRAT) fellowship from the National Institute of General Medical Sciences 1Fi2GM123974 (J.T.B.), by West Virginia University start-up funds (J.T.B), and by the Intramural Research Program of the NIH/NIEHS 1ZIAES090057 (J.A.C). The West Virginia University Microscope Imaging Facility and Flow Cytometry Core Facility are supported by NIH grants P30GM103488, P20GM103434, and U54GM10942. P30GM121322
Abbreviations:
- AR
androgen receptor
- GR
glucocorticoid receptor
- ILC2
type 2 innate lymphoid cell
- SPEM
spasmolytic polypeptide expressing metaplasia
Footnotes
Transcript Profiling: GSE147177
Disclosures: The authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fish EN. The X-files in immunity: sex-based differences predispose immune responses. Nat Rev Immunol 2008;8:737–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quinn M, Ramamoorthy S, Cidlowski JA. Sexually dimorphic actions of glucocorticoids: beyond chromosomes and sex hormones. Ann N Y Acad Sci 2014;1317:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischinger S, Boudreau CM, Butler AL, et al. Sex differences in vaccine-induced humoral immunity. Semin Immunopathol 2019;41:239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol 2016;16:626–38. [DOI] [PubMed] [Google Scholar]
- 5.Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol 2014;35:347–69. [DOI] [PubMed] [Google Scholar]
- 6.El-Zimaity H, Riddell RH. Beyond Helicobacter: dealing with other variants of gastritis-an algorithmic approach. Histopathology 2021;78:48–69. [DOI] [PubMed] [Google Scholar]
- 7.de Martel C, Parsonnet J. Helicobacter pylori infection and gender: a meta-analysis of population-based prevalence surveys. Dig Dis Sci 2006;51:2292–301. [DOI] [PubMed] [Google Scholar]
- 8.Sheh A, Lee CW, Masumura K, et al. Mutagenic potency of Helicobacter pylori in the gastric mucosa of mice is determined by sex and duration of infection. Proc Natl Acad Sci U S A 2010;107:15217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ibrahim A, Morais S, Ferro A, et al. Sex-differences in the prevalence of Helicobacter pylori infection in pediatric and adult populations: Systematic review and meta-analysis of 244 studies. Dig Liver Dis 2017;49:742–749. [DOI] [PubMed] [Google Scholar]
- 10.Busada JT, Ramamoorthy S, Cain DW, et al. Endogenous glucocorticoids prevent gastric metaplasia by suppressing spontaneous inflammation. The Journal of Clinical Investigation 2019;129:1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer AR, Engevik AC, Madorsky T, et al. Group 2 Innate Lymphoid Cells Coordinate Damage Response in the Stomach. Gastroenterology 2020;159:2077–2091.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer AR, Goldenring JR. Injury, repair, inflammation and metaplasia in the stomach. J Physiol 2018;596:3861–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teal E, Dua-Awereh M, Hirshorn ST, et al. Role of metaplasia during gastric regeneration. Am J Physiol Cell Physiol 2020;319:C947–c954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miao ZF, Sun JX, Adkins-Threats M, et al. DDIT4 Licenses Only Healthy Cells to Proliferate During Injury-induced Metaplasia. Gastroenterology 2021;160:260–271.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sáenz JB, Mills JC. Acid and the basis for cellular plasticity and reprogramming in gastric repair and cancer. Nat Rev Gastroenterol Hepatol 2018;15:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petersen CP, Meyer AR, De Salvo C, et al. A signalling cascade of IL-33 to IL-13 regulates metaplasia in the mouse stomach. Gut 2017. [DOI] [PMC free article] [PubMed]
- 17.Germain RN, Huang Y. ILC2s - resident lymphocytes pre-adapted to a specific tissue or migratory effectors that adapt to where they move? Curr Opin Immunol 2019;56:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoyler T, Klose CS, Souabni A, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 2012;37:634–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gour N, Smole U, Yong HM, et al. C3a is required for ILC2 function in allergic airway inflammation. Mucosal Immunol 2018;11:1653–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halim TY, Steer CA, Matha L, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 2014;40:425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mindt BC, Fritz JH, Duerr CU. Group 2 Innate Lymphoid Cells in Pulmonary Immunity and Tissue Homeostasis. Front Immunol 2018;9:840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petersen CP, Weis VG, Nam KT, et al. Macrophages promote progression of spasmolytic polypeptide-expressing metaplasia after acute loss of parietal cells. Gastroenterology 2014;146:1727–38.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becerra-Díaz M, Strickland AB, Keselman A, et al. Androgen and Androgen Receptor as Enhancers of M2 Macrophage Polarization in Allergic Lung Inflammation. J Immunol 2018;201:2923–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laffont S, Blanquart E, Savignac M, et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J Exp Med 2017;214:1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cephus JY, Stier MT, Fuseini H, et al. Testosterone Attenuates Group 2 Innate Lymphoid Cell-Mediated Airway Inflammation. Cell Rep 2017;21:2487–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claessens F, Joniau S, Helsen C. Comparing the rules of engagement of androgen and glucocorticoid receptors. Cell Mol Life Sci 2017;74:2217–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol 2017;17:233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaffer PL, Jivan A, Dollins DE, et al. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci U S A 2004;101:4758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Tilborg MA, Bonvin AM, Hard K, et al. Structure refinement of the glucocorticoid receptor-DNA binding domain from NMR data by relaxation matrix calculations. J Mol Biol 1995;247:689–700. [DOI] [PubMed] [Google Scholar]
- 30.Arora VK, Schenkein E, Murali R, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013;155:1309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Butler A, Hoffman P, Smibert P, et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rafei-Shamsabadi DA, van de Poel S, Dorn B, et al. Lack of Type 2 Innate Lymphoid Cells Promotes a Type I-Driven Enhanced Immune Response in Contact Hypersensitivity. J Invest Dermatol 2018;138:1962–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McHedlidze T, Waldner M, Zopf S, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 2013;39:357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newton R, King EM, Gong W, et al. Glucocorticoids inhibit IL-1beta-induced GM-CSF expression at multiple levels: roles for the ERK pathway and repression by MKP-1. Biochem J 2010;427:113–24. [DOI] [PubMed] [Google Scholar]
- 35.Paranjape A, Chernushevich O, Qayum AA, et al. Dexamethasone rapidly suppresses IL-33-stimulated mast cell function by blocking transcription factor activity. J Leukoc Biol 2016;100:1395–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owens IP. Ecology and evolution. Sex differences in mortality rate. Science 2002;297:2008–9. [DOI] [PubMed] [Google Scholar]
- 37.Fröhlich M, Pinart M, Keller T, et al. Is there a sex-shift in prevalence of allergic rhinitis and comorbid asthma from childhood to adulthood? A meta-analysis. Clin Transl Allergy 2017;7:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greuter T, Manser C, Pittet V, et al. Gender Differences in Inflammatory Bowel Disease. Digestion 2020;101 Suppl 1:98–104. [DOI] [PubMed] [Google Scholar]
- 39.Severs M, Spekhorst LM, Mangen MJ, et al. Sex-Related Differences in Patients With Inflammatory Bowel Disease: Results of 2 Prospective Cohort Studies. Inflamm Bowel Dis 2018;24:1298–1306. [DOI] [PubMed] [Google Scholar]
- 40.Cabrera de León A, Almeida González D, Almeida AA, et al. Factors associated with parietal cell autoantibodies in the general population. Immunol Lett 2012;147:63–6. [DOI] [PubMed] [Google Scholar]
- 41.Quinn MA, Cidlowski JA. Endogenous hepatic glucocorticoid receptor signaling coordinates sex-biased inflammatory gene expression. Faseb j 2016;30:971–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quinn MA, Xu X, Ronfani M, et al. Estrogen Deficiency Promotes Hepatic Steatosis via a Glucocorticoid Receptor-Dependent Mechanism in Mice. Cell Rep 2018;22:2690–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 2013;132:1033–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Vos P, Claessens F, Peeters B, et al. Interaction of androgen and glucocorticoid receptor DNA-binding domains with their response elements. Mol Cell Endocrinol 1993;90:R11–6. [DOI] [PubMed] [Google Scholar]
- 45.Sahu B, Laakso M, Pihlajamaa P, et al. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res 2013;73:1570–80. [DOI] [PubMed] [Google Scholar]
- 46.Fuseini H, Newcomb DC. Mechanisms Driving Gender Differences in Asthma. Curr Allergy Asthma Rep 2017;17:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamilton JA. GM-CSF-Dependent Inflammatory Pathways. Front Immunol 2019;10:2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shapouri-Moghaddam A, Mohammadian S, Vazini H, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 2018;233:6425–6440. [DOI] [PubMed] [Google Scholar]
- 49.Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108. [DOI] [PubMed] [Google Scholar]
- 50.Fox JG, Rogers AB, Ihrig M, et al. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer Res 2003;63:942–50. [PubMed] [Google Scholar]
- 51.Ohtani M, Garcia A, Rogers AB, et al. Protective role of 17 beta -estradiol against the development of Helicobacter pylori-induced gastric cancer in INS-GAS mice. Carcinogenesis 2007;28:2597–604. [DOI] [PubMed] [Google Scholar]
- 52.Sutton P, Wilson J, Genta R, et al. A genetic basis for atrophy: dominant non-responsiveness and helicobacter induced gastritis in F(1) hybrid mice. Gut 1999;45:335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamaji Y, Watabe H, Yoshida H, et al. High-risk population for gastric cancer development based on serum pepsinogen status and lifestyle factors. Helicobacter 2009;14:81–6. [DOI] [PubMed] [Google Scholar]
- 54.Parascandola M, Xiao L. Tobacco and the lung cancer epidemic in China. Transl Lung Cancer Res 2019;8:S21–s30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Teixido-Compano E, Espelt A, Sordo L, et al. Differences between men and women in substance use: the role of educational level and employment status. Gac Sanit 2018;32:41–47. [DOI] [PubMed] [Google Scholar]
- 56.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 57.Tang W, Liu R, Yan Y, et al. Expression of estrogen receptors and androgen receptor and their clinical significance in gastric cancer. Oncotarget 2017;8:40765–40777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kominea A, Konstantinopoulos PA, Kapranos N, et al. Androgen receptor (AR) expression is an independent unfavorable prognostic factor in gastric cancer. J Cancer Res Clin Oncol 2004;130:253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.