

Abstract
Background and aims:
Atherosclerosis is the most prominent underlying cause of cardiovascular disease (CVD). It is initiated by cholesterol deposition in the arterial intima, which causes macrophage recruitment and proinflammatory responses that promote plaque growth, necrotic core formation, and plaque rupture. Lipin-1 is a phosphatidic acid phosphohydrolase for glycerolipid synthesis. We have shown that lipin-1 phosphatase activity promotes macrophage pro-inflammatory responses when stimulated with modified low-density lipoprotein (modLDL) and accelerates atherosclerosis. Lipin-1 also independently acts as a transcriptional co-regulator where it enhances the expression of genes involved in β-oxidation. In hepatocytes and adipocytes, lipin-1 augments the activity of transcription factors such as peroxisome proliferator-activated receptor (PPARs). PPARs control the expression of anti-inflammatory genes in macrophages and slow or reduce atherosclerotic progression. Therefore, we hypothesize myeloid-derived lipin-1 transcriptional co-regulatory activity reduces atherosclerosis.
Methods:
We used myeloid-derived lipin-1 knockout (lipin-1mKO) and littermate control mice and AAV8-PCSK9 along with high-fat diet to elicit atherosclerosis.
Results:
Lipin-1mKO mice had larger aortic root plaques than littermate control mice after 8 and 12 weeks of a high-fat diet. Lipin-1mKO mice also had increased serum proinflammatory cytokine concentrations, reduced apoptosis in plaques, and larger necrotic cores in the plaques compared to control mice.
Conclusions:
Combined, the data suggest lipin-1 transcriptional co-regulatory activity in myeloid cells is atheroprotective.
Keywords: Lipin-1 transcriptional co-regulatory activity, Macrophage, Atherosclerosis, IL-23, Necrotic core
1. Introduction
Atherosclerosis is an immunometabolic and chronic inflammatory disorder initiated by hypercholesterolemia [1–6]. Hypercholesterolemia promotes lipid deposition (in the form of low-density lipoprotein (LDL)) into the arterial intima. Within the arterial intima, LDLs are modified and cause immune cell recruitment and activation [7,8]. Macrophages attempt to clear excess modified low-density lipoproteins (modLDLs) from the arterial intima.
Plaque macrophages process and manage the excess lipids and free fatty acids from modLDLs through either storage, transport, or breakdown. Excess free fatty acids can be converted into triacylglycerols (TAG) and phosphatidylcholine (PC) by the glycerolipid synthetic pathway and stored in lipids droplets along with cholesterol [2,9,10]. We demonstrated that glycerolipid synthesis promotes macrophage proinflammatory responses and accelerates atherosclerosis [2]. Macrophages can also break down free fatty acids by β-oxidation in the mitochondria [11]. Additionally, free fatty acids bind to and activate numerous transcription factors, including PPARs and LXRs, that increase macrophage β-oxidation and subsequent pro-resolving responses and atheroprotection [12–14]. Pro-resolving responses reduce atherosclerosis progression [12]. Lipid metabolic pathways within macrophages are critical determinants of atherosclerosis progression; therefore, the regulation of lipid metabolic pathways in plaque macrophages is also essential.
Lipin-1 is a phosphatidic acid phosphohydrolase that also independently acts as a transcriptional co-regulator. Lipin-1 phosphatase (enzymatic) activity converts phosphatidic acid into diacylglycerol during glycerolipid synthesis [2,15–20]. In macrophages, lipin-1 enzymatic activity promotes pro-inflammatory responses [2,17,19]. We previously showed that myeloid-associated lipin-1 enzymatic activity is atherogenic [2]. However, lipin-1 is found in the nucleus of macrophages within atherosclerotic plaques [17]. In hepatocytes and adipocytes, lipin-1 transcriptional co-regulatory activity increases transcription factors PPAR-α, PGC-1α, and PPAR-γ activity, leading to increased β-oxidation [21–24]. Lipin-1 transcriptional co-regulatory activity promotes macrophage continuing efferocytosis [25]. Efferocytosis and PPAR activation are atheroprotective, partially due to promoting pro-resolving and wound-healing macrophage phenotypes [11,14,21,23,24,26]. Mice with a global deletion of lipin-1 have an increased atherosclerotic burden in a cholate model, providing evidence that lipin-1 is atheroprotective [27]. These results suggest that lipin-1 transcriptional co-regulatory activity in macrophages may be atheroprotective, and led us to hypothesize that myeloid-derived lipin-1 transcriptional co-regulatory activity reduces atherosclerosis.
2. Materials and methods
2.1. Generation of bone marrow-derived macrophages (BMDMs)
BMDMs were generated as described previously [2]. Briefly, bone marrow cells were flushed from mice femurs with Dulbecco’s modified Eagle’s medium (DMEM; Gibco, 10829). Red blood cells were lysed with ammonium chloride-potassium carbonate lysis. Cells were grown in bone marrow differentiating media (DMEM; (Gibco 10829) Supplementaryed with 30% L-cell conditioned medium [2], 20% fetal bovine serum (Atlanta biologicals S11150), 2 mM glutamax (Thermofisher 35050–061), 100U/ml penicillin-streptomycin (ATCC), 1 mM sodium pyruvate (HyClone), and 0.2% sodium bicarbonate and placed in non-treated T-75 flasks in incubator (5% CO2, 37 °C) until confluent. Once confluent, BMDMs were collected and resuspended in D-10 (DMEM (Gibco: 11965–092), 10% fetal bovine serum (Atlanta biologicals S11150), 2 mM glutamax (Thermofischer 35050–061), 100U/mL penicillin/streptomycin (ATCC), and 1 mM sodium pyruvate (HyClone).
2.2. Peritoneal macrophages
Lipin-1mKO and littermate control mice were intraperitoneally injected with 0.1 mg of zymosan. Six days post zymosan injection, mice were sacrificed, and peritoneal lavages were collected; isolated cells were blocked (anti-CD16/32) for 20 min. Cells were then stained with PECy7 conjugated anti-CD11b (1:4000) (e-Biosciences, 25-0112-81, clone M1/70), AF700 conjugated anti-CD45.2 (1:2000) (Biolegend, 109821,clone104), FITC conjugated anti-Ly6G (1:800) (BD Biosciences, 551460,clone1A8), and PECy5 conjugated anti-F4/80 (1:400) (Invitrogen, 15-4801-80, clone (BM8). Macrophages (CD45+, CD11b+, F4/80+, and Ly6G−) were sorted for total protein isolation using FACSAria Ill Cell Sorter (BD Biosciences San Diego CA).
2.3. Tissue processing for Western blot analysis
Tissues were harvested from mice and flash-frozen in liquid nitrogen. 50 mg tissue samples were lysed in 1 mL RIPA buffer (150 mM Sodium Chloride, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH8) containing protease inhibitor cocktail (Thermo Scientific), phosphatase inhibitor cocktail 2 (Sigma Aldrich) and phosphatase inhibitor cocktail 3 (Sigma Aldrich) using Bioject Inc tissue homogenizer. Samples were then centrifuged at 16,000×g for 35 min at 4 °C. Supernatant below the lipid layer, but above the cell pellet, was collected and stored at −20 °C until use.
2.4. Western blot analysis
All tissue and cell lysate samples were sonicated and heat denatured. Protein concentration was determined by Pierce® 660 nm Protein Assay (Thermo Scientific). 40–50 μg total protein of tissue lysate sample and 20 μg of cell lysate sample was added to 3.75μL4x NuPAGE buffer, 0.75 μL β - mercaptoethanol (Fisher Scientific). Samples were loaded into a 4–12% polyacrylamide NuPAGE Novex gel (Invitrogen). 1x MES (2-ethanesulfonic acid) running buffer was used for tissue samples, and 1x MOPs buffer was used for cell lysate samples. Proteins were transferred onto a polyvinylidene difluoride (Immobilon-FL) membrane (EMD Millipore). Li-Cor Odyssey® blocking buffer (Li-Cor) was used to block membranes. Primary antibodies were diluted 1:5000 (GADPH) or 1:1000 (Lipin-1) in 1% bovine serum albumin in phosphate buffered saline (PBS) with 0.01% sodium aside and incubated at 4 °C overnight on a rocker. The membranes were then washed with 1x Tris-buffered saline and 0.1% Tween 20 (TBST). Goat anti-Rabbit or goat-anti mouse secondary antibody was diluted 1:2000 in 5% dry milk with TBST plus 0.01% SDS. Secondary antibodies were incubated on membranes for 2 h at room temperature with rocking. The membranes were then washed. ImmunoCruz Western blotting luminol reagent (Santa Cruz sc-2048) was added to blots for 1 min prior to exposure. Densitometry was performed using Image Quant TL Toolbox v8.2.0. Bands of interest were normalized to GAPDH for statistical analysis. Primary antibodies: Lipin-1 (Cell signaling technology 14906S), GAPDH (Cell signaling technologies 2118S), pMLKL (Cell signaling technologies 37333S) MLKL (Cell signaling technologies 37705S); Secondary antibodies: Rabbit Anti-Goat IgG (H + L) (Jackson ImmunoResearch 305-035-003) Goat anti-mouse IgG (Rockland 613–1319)).
2.5. Bone marrow-derived macrophage apoptosis analysis
Wild type and lipin-1mKO bone marrow-derived macrophages were collected and resuspended in D1 media (DMEM; (Gibco 10829) 1% fetal bovine serum (Atlanta biologicals S11150), 2 mM glutamax (Thermofisher 35050–061), 100U/ml penicillin-streptomycin (ATCC), 1 mM sodium pyruvate (HyClone). Cells were placed in flow tubes (VWR 60818–500) and treated with 25 or 50 μg/ml of oxidized LDL for 24 h. LDL was oxidized as described previously [17]. After 24hrs, cells were centrifuged, washed with FACs wash, and stained with APC Annexin V Apoptosis Detection Kit with PI following the kit protocol (Biolegend 640932) and analyzed via ACEA NovoCyte Quanteon Flow Cytometer.
2.6. In vivo mouse studies to analyze atherosclerotic progression
2.6.1. AAV8-PCSK9 viral vector preparation
Jacob Bentzon gifted DNA for the pAAV/D377Y-mPCSK9 (Addgene plasmid #58376) and it was packaged into adeno-associated virus serotype 8 (AAV8) using the helper and capsid plasmids from the University of Pennsylvania described in Ref. [2]. Viral stocks were sterilized via a Millipore Millex-GV syringe filter (Billerica, MA). Post sterilization, viral stocks were titered via dot blot assay and then aliquoted and stored at −80 °C until use.
2.6.2. Animals and tissue harvest
Animal protocols were approved by the LSU Health Sciences Center-Shreveport institutional animal care and use committee (Protocol approval number P-19-029). Animals were cared for according to the National Institute of Health guidelines for the care and use of laboratory animals. Brian Finck generously provided mice with a Lpin1 allele with exon 7 of the Lpin1 gene floxed with LoxP sites. To generate mice lacking lipin-1 in myeloid-derived cells only, the Lpin1 floxed mice were crossed with LysM-Cre transgenic mice purchased from Jackson Laboratory (Bar Harbor, ME). The resulting offspring lacked lipin-1 within myeloid-derived cells (Lipin-1mKO mice). Lipin-1mKO mice were compared with the lipin-1m floxed littermate controls. Lipin-1mEnzyKO mice used are as described previously [2]. 7–14 week old male mice were used for all studies and received retro-orbital injections of 3 × 1010 vector genomes of AAV8-PCSK9(28). Immediately after AAV8-PCSK9 inoculation, mice were fed a high fat, Western diet (TD 88137; Harlan-Teklad, Madison, WI) that contained 21% fat by weight (0.15% cholesterol and 19.5% casein without sodium cholate) for 8 or 12 weeks (duration of the study). Mice were weighed once a week after beginning a high fat diet. After 8 or 12 weeks, mice were anesthetized and subsequently euthanized via exsanguination and pneumothorax. Blood was collected as described previously [2]. After blood isolation, mice were perfused with 1x PBS. Following perfusion, the heart, aorta, and carotids were isolated and stored in 10% formalin until analysis.
2.6.3. Blood analysis
Plasma from mice was analyzed for total cholesterol (Wako 999–02601), triglycerides (Pointe Scientific INC T7532-120), and high-density lipoprotein (Wako 431–52501) using commercially available kits. Low-density lipoproteins were calculated using the Friedwald equation. Any mice with total cholesterol under 500 mg/dL were not considered hypercholesterolemic and were eliminated from analysis.
2.6.4. Histology and image quantification
Hearts and aortic roots from mice were fixed in 10% neutral buffered formalin and embedded in paraffin. Aortic roots were sectioned into 5 μm sections. All sections were equally taken from anatomical landmarks of the initiation of inner leaflet valves. For plaque area analysis, aortic root tissue sections were stained via Russel-Movat pentachrome stain. Collagen composition within the plaque was determined using picrosirius red staining from established protocols.
2.6.5. Nanostring gene expression analysis
RNA was isolated from 12 to 48 5 μm aortic root tissue sections using the RNAstorm™ kit. The final elution was 15–20μL. A minimum of 400 ng total RNA at a minimum concentration of 20ng/ul was required with a minimum basepair region of 300 or more. Canopy Biosciences (St. Louis, MO) analyzed RNA gene expression using the predesigned myeloid innate immunity panel. The resultant data was further analyzed by multiple T-Test with assumption of constant standard deviation followed by Bonferroni-sidak correction for multiple T-tests using Graph Pad Analysis 8.0.
2.6.6. Serum cytokine analysis
Serum cytokine concentration was determined using the BioLegend LEGENDplex Mouse Inflammation Panel (13 plex) (catalog number: 740446) according to the manufacturer’s instructions and ran on the ACEA NovoCyte Quanteon Flow Cytometer. Data were analyzed using the LEGENDplex Data Analysis Software.
2.6.7. Immunofluorescence
Aortic root tissue sections described in 2.6.4 were stained with immunofluorescent antibodies as described previously [2]. For apoptosis within plaques, Millipore Sigma In Situ Cell Death Detection Kit TMR red (Cat # 12156792910) was used with kit instructions with the following modifications: After proteinase K incubation, tissues were blocked with 10% horse serum in PBS with 1% BSA and subsequently incubated with anti-MAC2 conjugated with Alexa Fluor 488 (Biolegend 125410)for 2 h. The rest of the kit protocol was followed. Primary antibodies: Rat-anti-Mac2 antibody (1:100) (Accurate Chem. CL8942AP)), Rabbit-anti-smooth muscle heavy chain II (1:400) (Abcam ab53219), and donkey-anti-smooth muscle actin-Cy3 (1:200) (Sigma Aldrich C6198)). Secondary antibodies: Alexa Fluor® 647 donkey anti-rabbit IgG (A31573) and Alexa Fluor® 488 donkey anti-rat IgG (A21208) purchased from Life Technologies.
2.6.8. Necrotic core formation analysis
Aortic root tissue sections were immunofluorescently stained as described above. Necrotic cores were quantified as areas of negative space (no staining) within the plaques.
2.6.9. Aortic digestion
Aortas were collected, cleaned of fat, and placed in a tube with 400 units/ml Collagenase (Sigma C7657). Incubate aorta/collagenase 10 min 37 °C for 10 min with agitation. Remove the aorta and place in dish with collagenase digestion mix. (Collagenase digestion mix: 450 U/ml collagenase Type I (Sigma SCR103), 125 U/mL Collagenase (Sigma C7657), 60 units DNAse I, 60 U/mL Hyaluronidase. Mince the aorta into small pieces. Collect collagenase digestion mix and minced aorta into large Eppendorf tube and incubate at 37 °C with agitation for 30 min. Once digested, add 7 mL FACs wash, centrifuge, resuspend pellet in FACs wash, count cells, and stain for Flow cytometry. Briefly, cells were blocked with CD16/32 for 20 min at 4 °C. Cells were washed with FACs wash and incubated with BV staining buffer for 5 min. Cells were then washed with FACs wash and stained with antibody for 30 min in the dark on ice. Cells washed with FACs wash and resuspended at 1 × 106/mL and analyzed via flow cytometry. Antibodies used include: CD45.2 AF700, CD3 BV605, CD11c BV786, CD11b PECy7, CD19 PEe 610, Ly6G FITC, CD4 BV 510, CD8a BV570, NK1.1 PECy5. F minus one controls and compensation beads were used for all antibodies.
2.7. Statistical analysis
GraphPad Prism 8.0 (La Jolla, CA) was used for statistical analyses. Student t-test analysis was used for comparison between two data sets. All other statistical significance was determined using a one-way ANOVA with a Dunnett’s post-test. All experiments were performed a minimum of two times.
3. Results
3.1. Lipin-1 transcriptional co-regulatory activity is involved in atherosclerosis
To determine the contribution of myeloid-associated lipin-1 transcriptional co-regulatory activity to atherosclerosis, we developed a mouse model in which the mice lack lipin-1 in myeloid-derived cells. At this time, we are unable to generate a mouse model lacking only lipin-1 transcriptional co-regulatory activity because deleting this activity also diminishes enzymatic activity [21]. However, we can compare and contrast results with the previously published data with the lipin-1mEnzyKO mice to infer the contribution of myeloid-associated lipin-1 transcriptional co-regulatory activity to atherosclerosis [2]. To confirm myeloid-specific deletion of lipin-1, protein was isolated from BMDMs, zymosan-elicited peritoneal macrophages, lung tissue, and heart tissue from lipin-1mKO and lipin-1m fl/fl (wild type littermate control) mice. An 85% reduction of lipin-1 protein was observed in lipin-1mKO BMDMs and peritoneal macrophages, while equivalent lipin-1 protein levels in the lung and heart tissue lysates were observed (Fig. 1A). Additionally, since lipin-1 was reduced in zymosan-elicited peritoneal macrophages from lipin-1mKO mice, it suggest lipin-1KO macrophages still have a loss of lipin-1 even during an active inflammatory process (Fig. 1A).
Fig. 1.
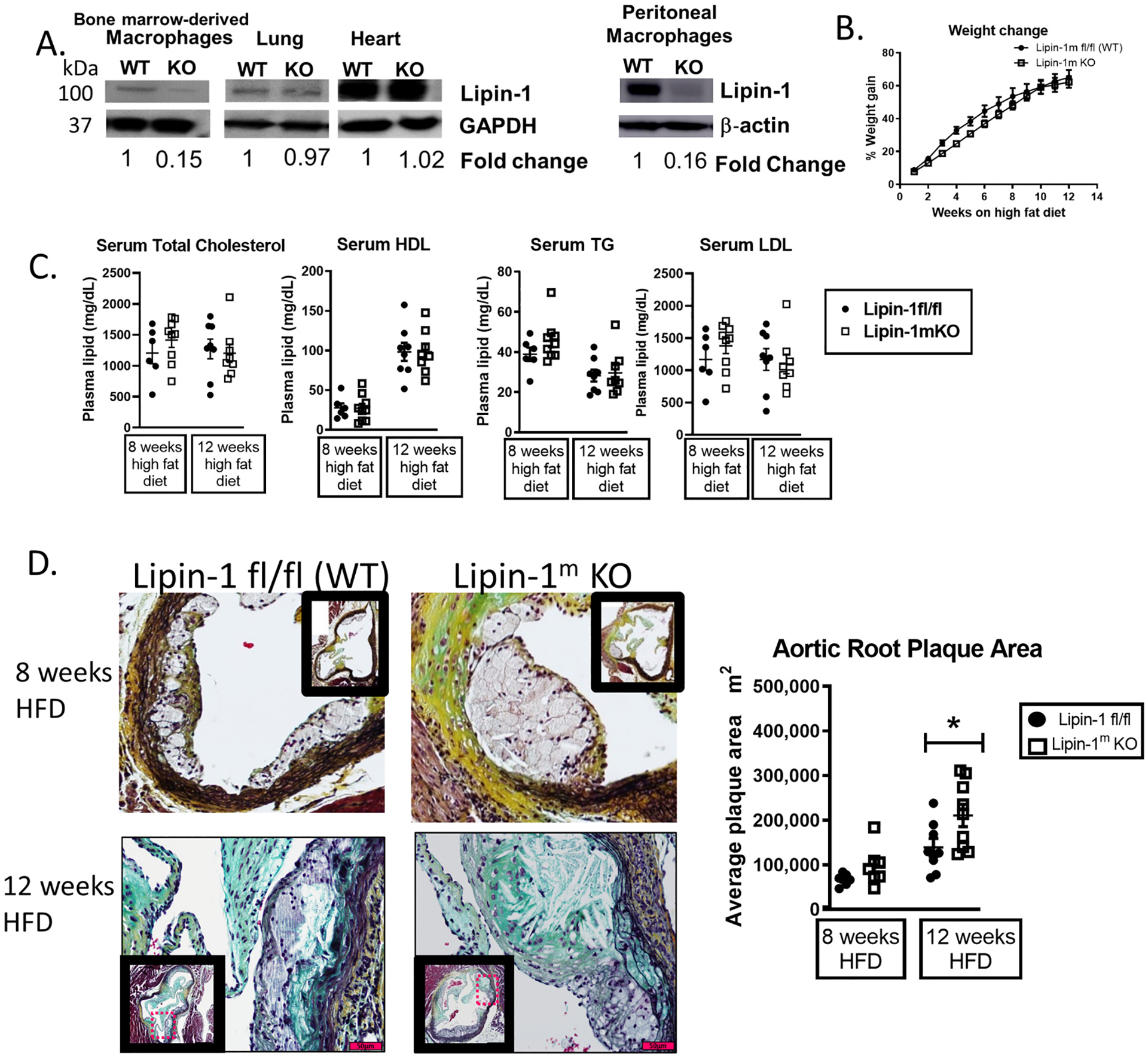
Lipin-1 transcriptional co-regulatory activity reduces atherosclerosis.
(A) Lipin-1 protein levels in macrophages, lung, and heart tissues from wild type (WT) and lipin-1mKO(KO) mice analyzed by Western blot. GAPDH/β-actin = loading control. (B–D): Lipin-1mKO and WT (littermate control) mice were injected with AAV8-PCSK9 to induce hypercholesterolemia and then fed a high fat diet for 8 or 12 weeks. (B) Percentage weight gain on a high fat diet (HFD). (C) Plasma lipid content. TC = total cholesterol; HDL = high density lipoprotein; TG = triglycerides; LDL = low-density lipoprotein. (D) Aortic roots from mice were stained via MOVAT for plaque area analysis. N = 6–9 mice *p < 0.5 Student t-test analysis was used for statistical analysis between wild type and lipin-1mKO mice after either 8 weeks or 12 weeks high fat diet.
Male lipin-1mKO and littermate control mice were injected with 3 × 1010 AAV8-PCSK9 viral genomes and then fed a high fat diet for 8 or 12 weeks to induce hypercholesterolemia [28]. Littermate control and lipin-1mKO mice equally gained weight and had equivalent total cholesterol, low-density lipoproteins, high-density lipoproteins, and triglycerides in their serum, indicating equal hypercholesterolemia (Fig. 1B and C). Therefore, differences in plaque size and composition would likely be due to the presence or absence of myeloid-associated lipin-1 in the plaque. We quantified plaque size of the aortic root as a marker of atherosclerotic severity. There was a slight increase in aortic root plaque area in lipin-1mKO mice after 8 weeks of high fat diet and a significant increase in aortic root plaque area in lipin-1mKO mice after 12 weeks high fat diet as compared to littermate controls (Fig. 1D). These results are the opposite of the lipin-1mEnzyKO mouse studies, as lipin-1mEnzyKO mice had smaller plaques than littermate control mice after 8- and 12-weeks high fat diet feeding [2], suggesting that myeloid-associated lipin-1 transcriptional co-regulatory activity is atheroprotective.
3.2. Increased expression of collagen and proinflammatory genes in plaques of lipin-1mKO mice
We hypothesized myeloid-associated lipin-1 transcriptional co-regulatory activity promotes anti-inflammatory responses within the atherosclerotic plaque. In order to investigate this, we examined differences in plaque gene expression between lipin-1mKO mice and littermate controls. mRNA was isolated from similarly positioned lesions within aortic roots of lipin-1mKO and littermate control mice on a high fat diet for 8 and 12 weeks. Following mRNA isolation, we used Nanostring technology to quantify gene expression. We observed a significant increase in both proinflammatory genes and collagen genes in plaques of lipin-1mKO mice after both 8- and 12-weeks of high-fat diet feeding compared to littermate controls. Collagen 12a1 mRNA and IL-23 mRNA were upregulated in plaques from lipin-1mKO mice after 8- and 12-weeks high-fat diet feeding (Fig. 2A and B). By 12 weeks, collagen 11a1, 10a1, and 1a2 were also upregulated, as were proinflammatory chemokines such as CCL28 and CCL12 along with IL-23. IL-23 can induce apoptosis and necrosis in cells within an atherosclerotic plaque. Apoptosis and necrosis both promote necrotic core formation and thickening of the fibrous cap in the plaque. As such, we concentrated on aspects of IL-23-and collagen-mediated responses.
Fig. 2.
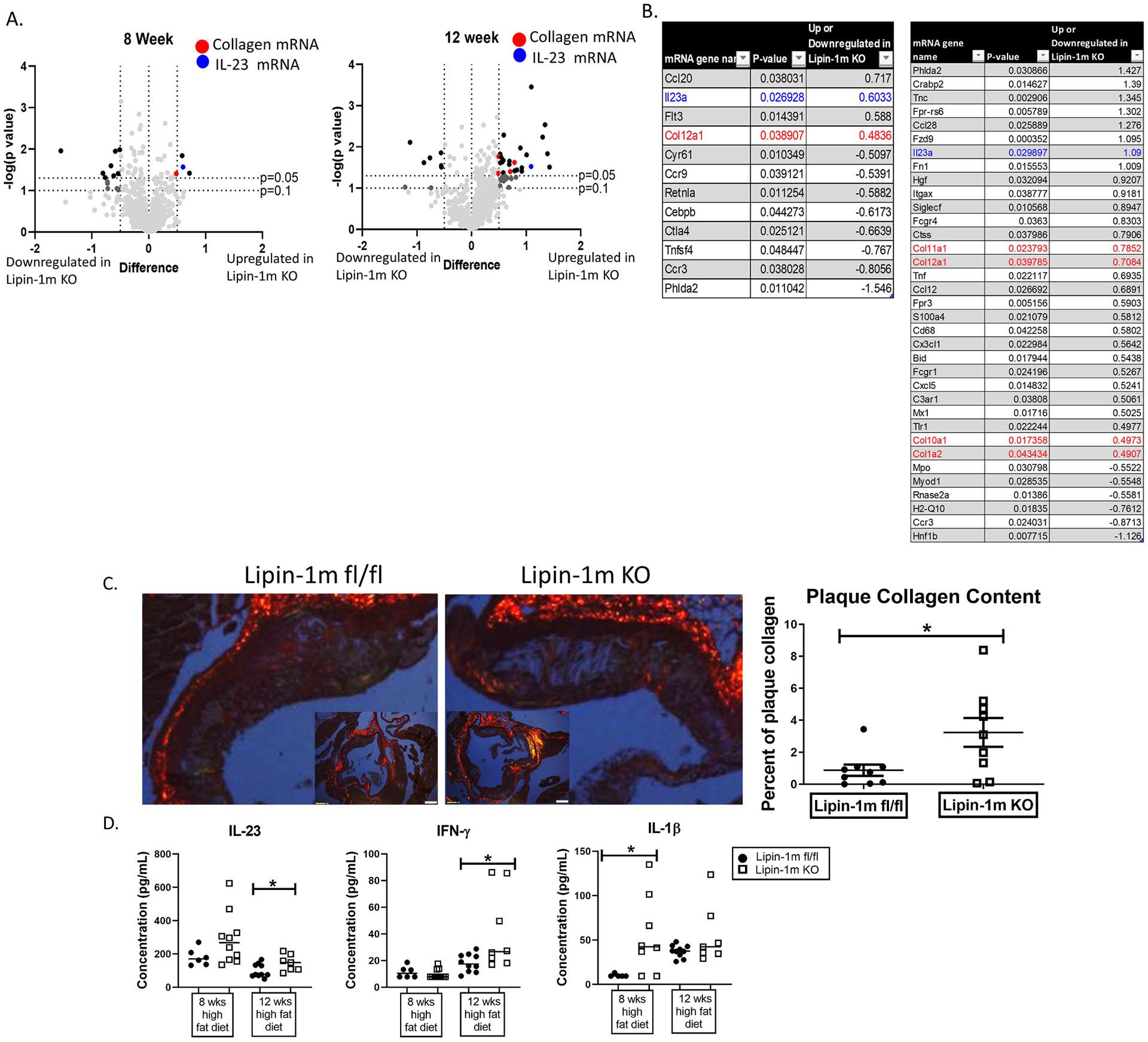
Lipin-1 regulates collagen and IL-23 gene expression.
Mice described in Fig. 1B were used. (A and B) RNA was extracted from aortic roots and Nanostring gene expression analysis was performed. Up and downregulated genes for the lipin-1mKO mice are displayed. (A) All genes plotted, (B) genes that are significantly up or downregulated 0.5 fold or more in the lipin-1mKO mice. (C) Aortic root tissue sections were stained with picrosirus red and analyzed for collagen deposition within the plaques. (D) After euthanasia, serum was collected mice. Cytokines were measured via cytometric bead array. N = 6–10 mice. *p < 0.5 Student t-test analysis was used for statistical analysis between wild type and lipin-1mKO at each time point.
Atherosclerotic plaque growth induces collagen production to stabilize the plaque and prevent rupture [5,6,29,30]. Increased collagen mRNA in the lipin-1mKO mice suggests increased collagen deposition. We stained aortic root tissue sections from lipin-1mKO and littermate control mice with picrosirius red to quantify plaque collagen deposition in plaques. Little to no collagen deposition was observed in plaques from lipin-1mKO and littermate control mice on a high fat diet for 8 weeks. However, by 12 weeks, lipin-1mKO mice had increased plaque collagen compared to littermate control mice (Fig. 2C), supporting our mRNA gene expression data. These data, taken together, suggest the loss of myeloid-associated lipin-1 accelerates plaque growth and corresponding collagen deposition, likely a compensation mechanism to reduce potential plaque rupture.
Gene expression data also showed increased IL-23 mRNA in the lipin-1mKO plaques after 8- and 12-weeks high-fat diet. Increased IL-23 mRNA should result in increased cytokine production that can be detected in the serum. Therefore, we used a cytometric bead array and quantified cytokine concentration in serum from the mice, allowing us to quantify pro-inflammatory cytokines in addition to IL-23. We detected increased IL-23 cytokine concentrations in the serum isolated from lipin-1mKO mice than littermate control mice at 12-weeks high fat diet (Fig. 2D). At 12 weeks high fat diet, we observed increased IFN-γ as well. IL-1β was increased in the serum of lipin-1mKO mice after 8 weeks of high fat diet (Fig. 2D). IL-23, IFN-γ and IL-1β are pro-inflammatory and pro-atherogenic cytokines. There was no significant change in the other cytokines analyzed (Supplementary Fig. 1). Together, these results confirm our gene expression data suggesting a more proinflammatory plaque environment.
3.3. Lipin-1mKO mice have reduced plaque apoptosis and macrophages have increased necroptosis
IL-23 can induce macrophage apoptosis or necrosis. Aortic root tissue sections were stained for macrophage and apoptotic cells using the in situ cell death TMR red kit. We selected this kit due to its ability to specifically quantify apoptosis. Plaques from lipin-1mKO mice had significantly reduced apoptotic cells compared to wild-type mice. The lack of apoptotic cells may suggest that macrophages lacking lipin-1 were resistant to oxLDL induced cell death. To test this, we treated bone marrow-derived macrophages with oxidized LDL for 24 h and monitored cell death using Annexin V and PI. There was no difference in either early apoptosis or late apoptosis/cell death (Fig. 3B), suggesting that loss of lipin-1 did not make macrophages resistant to oxLDL induced cell death.
Fig. 3.
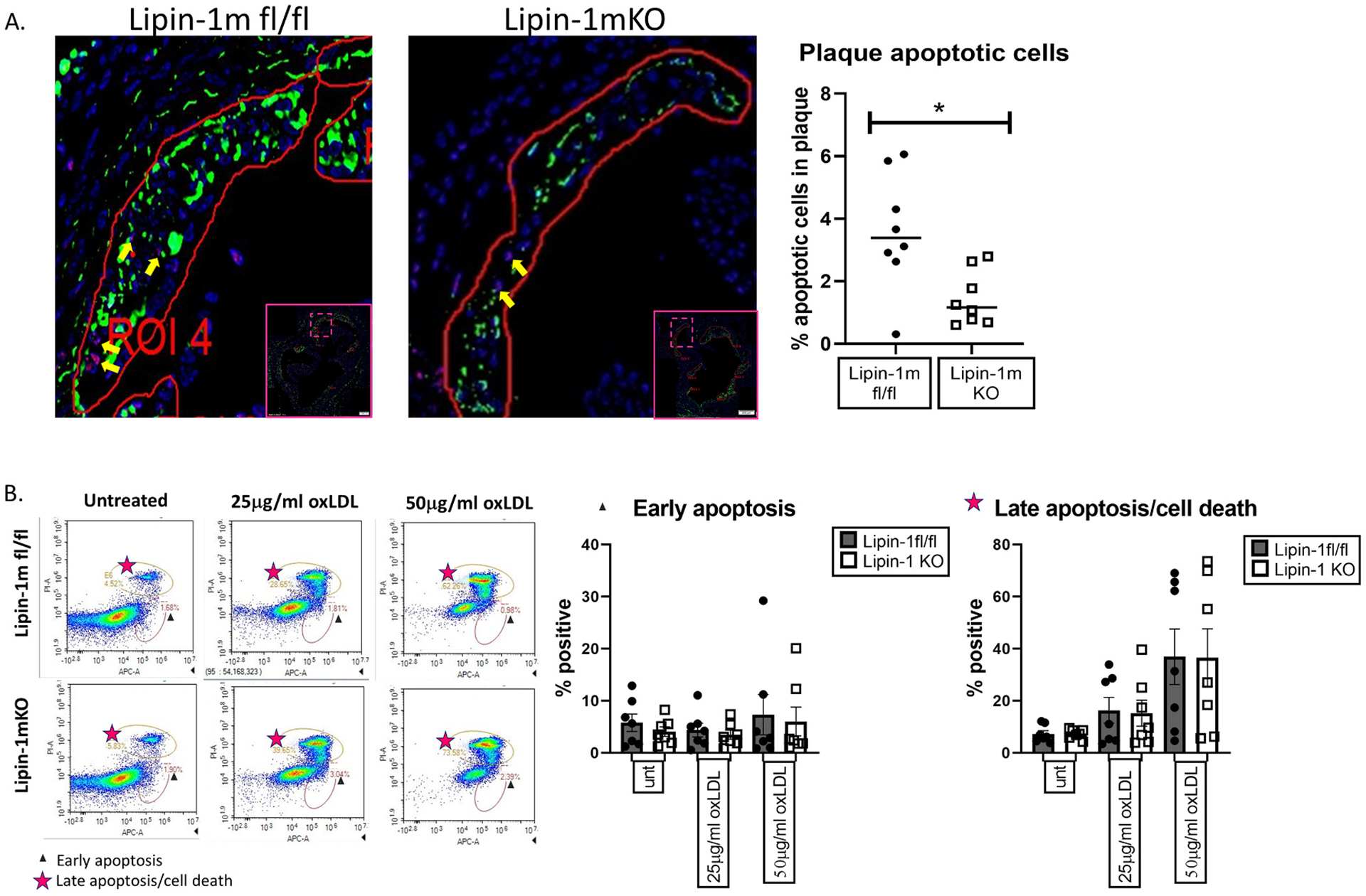
Lipin-1mKO mice have fewer apoptotic cells in plaques.
(A) Aortic root tissue sections from wild-type and lipin-1mKO mice on high fat diet for 12 weeks were stained for apoptotic cells, macrophages, and DAPI. Apoptotic cells within the plaques, shown with yellow arrows, were quantified as cells positive for DAPI (blue) and TUNEL (red) (B) Wild type and lipin- KO bone marrow-derived macrophages were treated with oxLDL for 24hrs. Cells were then stained with Annexin V and PI and analyzed via flow cytometry. Early apoptosis = Annexin V + and PI−. Late apoptosis = Annexin V+ and PI+. *p < 0.5 Student t-test analysis was used for statistical analysis between wild type and lipin-1mKO.
The reduction in apoptotic cells in the plaques of lipin-1mKO mice but no difference in oxLDL induced apoptosis in macrophages suggested that lipin-1KO macrophages may be dying via a mechanism other than apoptosis, potentially necroptosis. Phosphorylation of mixed lineage kinase domain like pseudokinase (MLKL) at Ser 345 is a marker associated with RIPK3 induced necroptosis. We treated wild-type and lipin-1mKO macrophages with 25, 50, or 100 μg/mL oxLDL. 100 μg/mL oxLDL treatment is known to induce necroptosis in macrophages [31]. OxLDL treated lipin-1KO macrophages had increased pMLKL and MLKL protein compared to wild-type macrophages (Fig. 4). 100 μg/ml oxLDL treatment induces necroptosis, so the wild type and knockout macrophages have no difference in pMLKL or MLKL levels. These results suggest that the loss of lipin-1 leads to increased necroptosis.
Fig. 4.
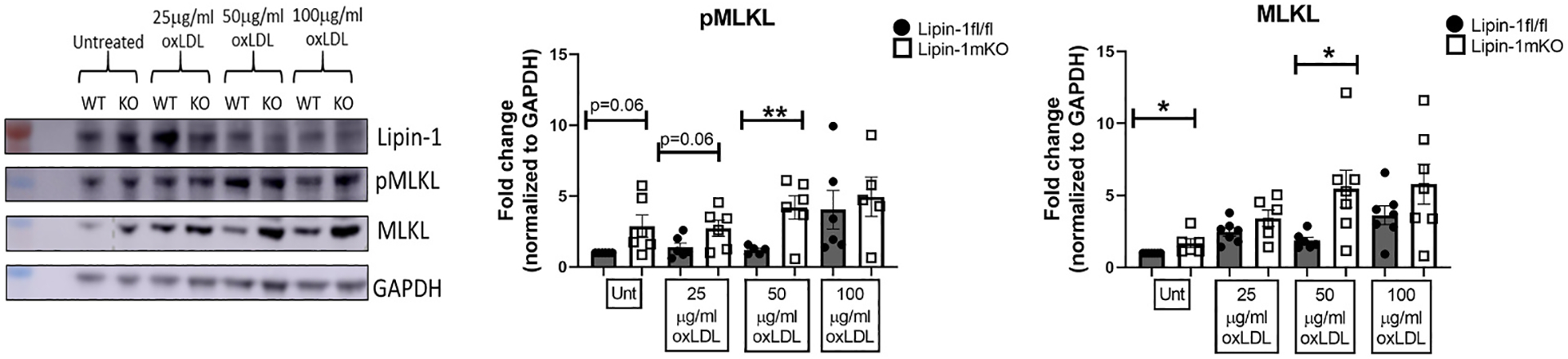
Lipin-1 KO macrophages have increased necroptotic markers pMLKL and MLKL.
(A) Representative Western of wild type and lipin-1KO macrophages treated with oxLDL for 24 h pMLKL and MLKL quantified in lysates. GAPDH was used as a loading control. (B) Quantification of pMLKL and MLKL. n = 5–7. *p < 0.5; **p < 0.01 Student t-test analysis was used for statistical analysis between wild type and lipin-1mKO.
3.4. Lipin-1 is involved in necrotic core formation
Increased necroptosis and reduced efferocytosis are both known to promote necrotic core formation and growth and promote pro-inflammatory responses [1,32–34]. We recently demonstrated that lipin-1 is involved in continuing efferocytosis, i.e., eating multiple dying/apoptotic cells [25]. Additionally, we observed an increase in both IL-23 mRNA and serum IL-23 in the lipin-1mKO mice. Macrophage-associated IL-23 promotes necrotic core formation within plaques [35]. These data led us to hypothesize that there would be larger necrotic cores in the plaques of lipin-1mKO mice.
To determine necrotic core size, aortic root tissue sections from lipin-1mKO and littermate control mice were stained for macrophages, smooth muscle cells, and nuclei. We observed no difference in macrophage or smooth muscle cell content in the plaques between lipin-1mKO and littermate controls (Supplementary Fig. 2A). We also performed aortic digestion on aortas isolated from lipin-1mKO and littermate control mice on high fat diet for 12 weeks. We did observe an increase in leukocytes and a decrease in B cells; however, there was no significant difference in macrophages, T cells, dendritic cells, NK cells, or PMNs (Supplementary Fig. 2B). We next quantified necrotic cores as areas of unstained tissue within the plaques. No difference in necrotic core formation between lipin-1mKO and littermate control mice after 8 weeks high fat diet was observed (Fig. 5). However, after 12 weeks of high fat diet, plaques in lipin-1mKO mice had significantly larger necrotic cores than plaques in littermate control mice (Fig. 5). We quantified necrotic core area in previously isolated lipin-1mEnzyKO mice and their littermate controls [2]. We see that the loss of lipin-1 enzymatic activity does not increase plaque necrotic core area compared to littermate controls (Fig. 5). Together, the data suggest myeloid-associated lipin-1 transcriptional co-regulatory activity reduces necrotic core formation and contributes to a more stable plaque.
Fig. 5.
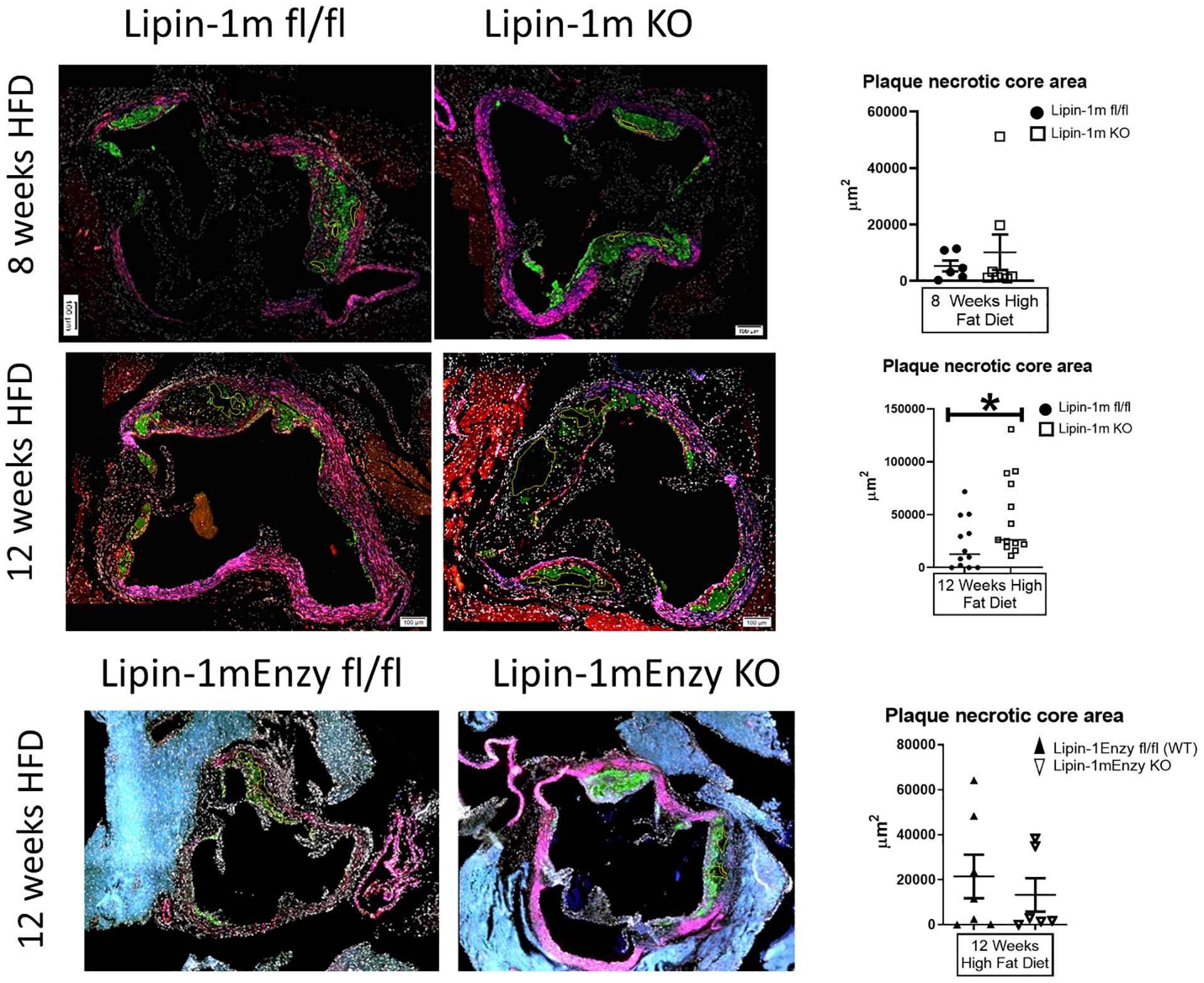
Lipin-1 transcriptional co-regulatory activity reduces necrotic core formation.
Mice described in Fig. 1 plus wild type and lipin-1mEnzyKO mice induced with hypercholesterolemia via AAV8-PCSK9 and high fat diet for 12 weeks (described in Ref. [2]) were stained as described above. Necrotic core areas within plaques were quantified as areas with negative space (no staining) and are outlined in yellow. *p ≤ 0.05 n = 7–13 mice. Student t-test analysis was used for statistical analysis between wild type and lipin-1mKO or wild type and lipin-1mEnzyKO at each time point (either 8 or 12 weeks high fat diet).
4. Discussion
Myeloid associated lipin-1 enzymatic activity is atherogenic [2]. In contrast, we provide evidence that lipin-1 transcriptional co-regulatory activity is atheroprotective and the dominant activity within macrophages in atherosclerotic lesions. Our results suggest that lipin-1 transcriptional co-regulatory activity protects against necroptosis-induced cell death and slows necrotic core growth. Along with previous work that highlights that lipin-1 transcriptional co-regulatory activity promotes pro-resolving polarization and continuing efferocytosis, this study highlights the atheroprotective activity of myeloid associated lipin-1 transcriptional co-regulatory activity.
Single nucleotide polymorphisms in lipin-1 are associated with more severe metabolic syndrome and mice with loss of lipin-1 are known to have increased atherosclerotic severity [27,36]. We provide evidence here that myeloid-associated lipin-1 transcriptional co-regulatory activity is atheroprotective through the control of cell death within the plaque. We observed not only increased plaque size in lipin-1mKO mice, but increased necrotic core size which is not seen in lipin-1mEnzyKO mice.
Humans with advanced atherosclerosis have increased apoptotic and necrotic macrophages in severe plaques [33,37,38]. Defects in the clearance of apoptotic cells lead to a secondary form of cell death called necroptosis [1]. Necroptosis contributes to necrotic core growth and pro-inflammatory responses in the plaque, as evident by the fact that inhibiting necroptosis either genetically or pharmacologically reduces necrotic core size [31,39,40]. We previously showed lipin-1KO macrophages could not perform continuing efferocytosis to the same extent as wild type macrophages [25]. Additionally, we demonstrated that oxLDL treatment of lipin-1KO macrophages led to increased phosphorylation of MLKL, a strong indicator of increased necroptosis.
Furthermore, we observed an increase in pro-inflammatory cytokines in our lipin-1mKO mice. One such cytokine we observed was IL-23. IL-23 produced within the plaque increases macrophage apoptosis and necrosis in mice [35] and is correlated with increased human carotid atherosclerosis progression and mortality [41]. Increased necrotic core formation in our lipin-1mKO mice may be due to increased IL-23 production within the plaques. However, in some studies, reduction of IL-23 did not reduce atherosclerosis progression [42,43]. Thus, IL-23 itself may not be sufficient to promote necrotic core formation and other processes may be involved leading to increased necrotic core formation in our lipin-1mKO mice. We would hypothesize that the increase in necrotic core is the combined effect of increased IL-23, defective efferocytosis [25], and the increased potential of lipin-1KO macrophages to undergo necroptosis.
Additionally, IL-23 promotes Th17 differentiation; however, we did not see increased IL-17 in serum of lipin-1mKO mice. We did observe increased IFN-γ, which can be produced by Th1 cells. Th17 cells are able to convert into Th1 cells, which are pro-atherogenic and found near necrotic cores [44,45]. Together, these data suggest that lipin-1 transcriptional co-regulatory activity promotes atheroprotective responses via modulation of immune responses that reduce necroptosis and necrotic core growth.
In addition to observing increased necrotic cores, we also observed increased collagen deposition, which may seem contradictory. Collagen strengthens the fibrous cap of the plaque, which typically protects the lesion from rupturing and is generally deemed atheroprotective. However, in certain mouse LysMCre atherosclerotic models, fibrous cap thickness was increased in response to larger lesion size with advanced necrotic core formation [46]. Mice lacking myeloid-derived lipin-1 have increased collagen deposition within the plaque compared to littermate control mice. These data, combined with the increase in lesion area and necrotic core area observed in the lipin-1mKO mice, suggest a similar compensation mechanism occurs in the lipin-1mKO mice where the more inflammatory plaque environment with larger plaques and necrotic cores causes increased collagen deposition.
In conclusion, the results from this study begin to highlight the contribution of atheroprotective roles of lipin-1 suggested in earlier work [27]. Future work will seek to better understand how lipin-1 transcriptional co-regulatory activity can repress cell death within the plaque. Lipin-1 augments PPAR activity [21,22]. PPARs are transcription factors that promote macrophage wound healing and β–oxidation responses and induce atheroprotective effects [23,24,47,48]. PPARs reduce IL-23 production, MLKL activation, necroptosis and inflammatory pathways [49,50]. It is likely that lipin-1 regulates PPARs in macrophages and is responsible for the phenotypes we observe. Loss of lipin-1 transcriptional co-regulatory activity may reduce PPAR activity leading to increased necroptosis and necrotic core growth. Future work will examine how lipin-1 transcriptional co-regulatory activity regulates PPAR activity in macrophages.
Supplementary Material
Acknowledgements
We would like to thank Gabrielle Gahn, Faith Saxon, and Tyler Williams for determining genotypes of mice, Deshawn Blankenship for sectioning tissues used in this study, David Custis for his help in running serum samples for cytokine concentrations via flow cytometry, and Camille Abshire for helping verify sample concentrations and purity for the gene expression experiments.
Financial support
This work was supported by the following: National Heart, Lung, and Blood Institute R01 HL131844 to M.D.W.; R01 HL 119225 to B.N.F.; Malcolm Feist Predoctoral Fellowships in house fellowships to C.M.R.B. and A.E.V.; and American Heart Association Predoctoral Fellowship 17PRE33661114 to A.E.V. The content is solely the authors’ responsibility and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.atherosclerosis.2021.06.927.
References
- [1].Yurdagul A, Doran AC, Cai B, Fredman G, Tabas IA, Mechanisms and consequences of defective efferocytosis in atherosclerosis, Front Cardiovasc Med 4 (2017) 86, 10.3389/fcvm.2017.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Vozenilek AE, Navratil AR, Green JM, Coleman DT, Blackburn CMR, Finney AC, Pearson BH, Chrast R, Finck BN, Klein RL, Orr AW, Woolard MD, Macrophage-associated lipin-1 enzymatic activity contributes to modified low-density lipoprotein-induced proinflammatory signaling and atherosclerosis, Arterioscler. Thromb. Vasc. Biol 38 (2) (2018) 324–334, 10.1161/ATVBAHA.117.310455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Group CT, Antiinflammatory therapy with canakinumab for atherosclerotic disease, N. Engl. J. Med 377 (12) (2017) 1119–1131, 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- [4].Ridker PM, Morrow DA, C-reactive protein, inflammation, and coronary risk, Cardiol. Clin 21 (3) (2003) 315–325. [DOI] [PubMed] [Google Scholar]
- [5].Hansson GK, Libby P, Tabas I, Inflammation and plaque vulnerability, J. Intern. Med 278 (5) (2015) 483–493, 10.1111/joim.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Halvorsen B, Otterdal K, Dahl TB, Skjelland M, Gullestad L, Øie E, Aukrust P, Atherosclerotic plaque stability–what determines the fate of a plaque? Prog. Cardiovasc. Dis 51 (3) (2008) 183–194, 10.1016/j.pcad.2008.09.001. [DOI] [PubMed] [Google Scholar]
- [7].Park YM, Febbraio M, Silverstein RL, CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima, J. Clin. Invest 119 (1) (2009) 136–145, 10.1172/JCI35535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pentikäinen MO, Oörni K, Ala-Korpela M, Kovanen PT, Modified LDL - trigger of atherosclerosis and inflammation in the arterial intima, J. Intern. Med 247 (3) (2000) 359–370, 10.1046/j.1365-2796.2000.00655.x. [DOI] [PubMed] [Google Scholar]
- [9].Bozza PT, Magalhães KG, Weller PF, Leukocyte lipid bodies - biogenesis and functions in inflammation, Biochim. Biophys. Acta 1791 (6) (2009) 540–551, 10.1016/j.bbalip.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].McGookey DJ, Anderson RG, Morphological characterization of the cholesteryl ester cycle in cultured mouse macrophage foam cells, J. Cell Biol 97 (4) (1983) 1156–1168, 10.1083/jcb.97.4.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O’Neill LA, Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages, Cell 167 (2) (2016) 457–470, 10.1016/j.cell.2016.08.064, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nomura M, Liu J, Yu ZX, Yamazaki T, Yan Y, Kawagishi H, Rovira II, Liu C, Wolfgang MJ, Mukouyama YS, Finkel T, Macrophage fatty acid oxidation inhibits atherosclerosis progression, J. Mol. Cell. Cardiol 127 (2019) 270–276, 10.1016/j.yjmcc.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, Finkel T, Fatty acid oxidation in macrophage polarization, Nat. Immunol 17 (3) (2016) 216–217, 10.1038/ni.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM, Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma, Cell 93 (2) (1998) 229–240, 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- [15].Reue K, The lipin family: mutations and metabolism, Curr. Opin. Lipidol 20 (3) (2009) 165–170, 10.1097/MOL.0b013e32832adee5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Reue K, Dwyer JR, Lipin proteins and metabolic homeostasis, J. Lipid Res 50 (Suppl) (2009) S109–S114, 10.1194/jlr.R800052-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Navratil AR, Vozenilek AE, Cardelli JA, Green JM, Thomas MJ, Sorci-Thomas MG, Orr AW, Woolard MD, Lipin-1 contributes to modified low-density lipoprotein-elicited macrophage pro-inflammatory responses, Atherosclerosis 242 (2) (2015) 424–432, 10.1016/j.atherosclerosis.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Meana C, García-Rostan G, Peña L, Lordén G, Cubero Á, Orduña A, Győrffy B, Balsinde J, Balboa MA, The phosphatidic acid phosphatase lipin-1 facilitates inflammation-driven colon carcinogenesis, JCI Insight 3 (18) (2018), 10.1172/jci.insight.97506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Meana C, Peña L, Lordén G, Esquinas E, Guijas C, Valdearcos M, Balsinde J, Balboa MA, Lipin-1 integrates lipid synthesis with proinflammatory responses during TLR activation in macrophages, J. Immunol 193 (9) (2014) 4614–4622, 10.4049/jimmunol.1400238. [DOI] [PubMed] [Google Scholar]
- [20].Chen Z, Gropler MC, Norris J, Lawrence JC, Harris TE, Finck BN, Alterations in hepatic metabolism in fld mice reveal a role for lipin 1 in regulating VLDL-triacylglyceride secretion, Arterioscler. Thromb. Vasc. Biol 28 (10) (2008) 1738–1744, 10.1161/ATVBAHA.108.171538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Kelly DP, Lipin 1 is an inducible amplifier of the hepatic PGC-1 alpha/PPARalpha regulatory pathway, Cell Metabol. 4 (3) (2006) 199–210, 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- [22].Kim HE, Bae E, Jeong DY, Kim MJ, Jin WJ, Park SW, Han GS, Carman GM, Koh E, Kim KS, Lipin 1 regulates PPARγ transcriptional activity, Biochem. J 453 (1) (2013) 49–60, 10.1042/BJ20121598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Staels B, PPARgamma and atherosclerosis, Curr. Med. Res. Opin 21 (Suppl 1) (2005) S13–S20, 10.1185/030079905X36440. [DOI] [PubMed] [Google Scholar]
- [24].Reiss AB, Vagell ME, PPARgamma activity in the vessel wall: anti-atherogenic properties, Curr. Med. Chem 13 (26) (2006) 3227–3238. [DOI] [PubMed] [Google Scholar]
- [25].Schilke RS, B CMR, Rao S, Krzywanski DM, Finck BN, Woolard MD, Macrophage-associated lipin-1 promotes β-oxidation in response to proresolving stimuli, in: ImmunoHorizons, 2020, 10.4049/immunohorizons.2000047. October 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nomura M, Liu J, Yu ZX, Yamazaki T, Yan Y, Kawagishi H, Rovira II, Liu C, Wolfgang MJ, Mukouyama YS, Finkel T, Macrophage fatty acid oxidation inhibits atherosclerosis progression, J. Mol. Cell. Cardiol (2019), 10.1016/j.yjmcc.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Reue K, Xu P, Wang XP, Slavin BG, Adipose tissue deficiency, glucose intolerance, and increased atherosclerosis result from mutation in the mouse fatty liver dystrophy (fld) gene, J. Lipid Res 41 (7) (2000) 1067–1076. [PubMed] [Google Scholar]
- [28].Vozenilek AE, Blackburn CMR, Schilke RM, Chandran S, Castore R, Klein RL, Woolard MD, AAV8-mediated overexpression of mPCSK9 in liver differs between male and female mice, Atherosclerosis 278 (2018) 66–72, 10.1016/j.atherosclerosis.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sluijter JP, Pulskens WP, Schoneveld AH, Velema E, Strijder CF, Moll F, de Vries JP, Verheijen J, Hanemaaijer R, de Kleijn DP, Pasterkamp G, Matrix metalloproteinase 2 is associated with stable and matrix metalloproteinases 8 and 9 with vulnerable carotid atherosclerotic lesions: a study in human endarterectomy specimen pointing to a role for different extracellular matrix metalloproteinase inducer glycosylation forms, Stroke 37 (1) (2006) 235–239, 10.1161/01.STR.0000196986.50059.e0. [DOI] [PubMed] [Google Scholar]
- [30].Katsuda S, Okada Y, Minamoto T, Oda Y, Matsui Y, Nakanishi I, Collagens in human atherosclerosis. Immunohistochemical analysis using collagen type-specific antibodies, Arterioscler. Thromb 12 (4) (1992) 494–502, 10.1161/01.atv.12.4.494. [DOI] [PubMed] [Google Scholar]
- [31].Karunakaran D, Geoffrion M, Wei L, Gan W, Richards L, Shangari P, DeKemp EM, Beanlands RA, Perisic L, Maegdefessel L, Hedin U, Sad S, Guo L, Kolodgie FD, Virmani R, Ruddy T, Rayner KJ, Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis, Sci Adv 2 (7) (2016), e1600224, 10.1126/sciadv.1600224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Thorp E, Tabas I, Mechanisms and consequences of efferocytosis in advanced atherosclerosis, J. Leukoc. Biol 86 (5) (2009) 1089–1095, 10.1189/jlb.0209115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Martinet W, Schrijvers DM, De Meyer GR, Necrotic cell death in atherosclerosis, Basic Res. Cardiol 106 (5) (2011) 749–760, 10.1007/s00395-011-0192-x. [DOI] [PubMed] [Google Scholar]
- [34].Fok P-W, Growth of necrotic cores in atherosclerotic plaque, in: Mathematical Medicine and Biology: A Journal of the IMA, 2012. [DOI] [PubMed] [Google Scholar]
- [35].Subramanian M, Thorp E, Tabas I, Identification of a non-growth factor role for GM-CSF in advanced atherosclerosis: promotion of macrophage apoptosis and plaque necrosis through IL-23 signaling, Circ. Res 116 (2) (2015) e13–24, 10.1161/CIRCRESAHA.116.304794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wiedmann S, Fischer M, Koehler M, Neureuther K, Riegger G, Doering A, Schunkert H, Hengstenberg C, Baessler A, Genetic variants within the LPIN1 gene, encoding lipin, are influencing phenotypes of the metabolic syndrome in humans, Diabetes 57 (1) (2008) 209–217, 10.2337/db07-0083. [DOI] [PubMed] [Google Scholar]
- [37].Kolodgie FD, Narula J, Burke AP, Haider N, Farb A, Hui-Liang Y, Smialek J, Virmani R, Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death, Am. J. Pathol 157 (4) (2000) 1259–1268, 10.1016/S0002-9440(10)64641-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Stoneman VE, Bennett MR, Role of apoptosis in atherosclerosis and its therapeutic implications, Clin. Sci. (Lond.) 107 (4) (2004) 343–354, 10.1042/CS20040086. [DOI] [PubMed] [Google Scholar]
- [39].Rasheed A, Robichaud S, Nguyen MA, Geoffrion M, Wyatt H, Cottee ML, Dennison T, Pietrangelo A, Lee R, Lagace TA, Ouimet M, Rayner KJ, Loss of MLKL (mixed lineage kinase domain-like protein) decreases necrotic core but increases macrophage lipid accumulation in atherosclerosis, Arterioscler. Thromb. Vasc. Biol 40 (5) (2020) 1155–1167, 10.1161/ATVBAHA.119.313640. [DOI] [PubMed] [Google Scholar]
- [40].Hosseini Z, Marinello M, Decker C, Sansbury BE, Sadhu S, Gerlach BD, Bossardi Ramos R, Adam AP, Spite M, Fredman G, Resolvin D1 enhances necroptotic cell clearance through promoting macrophage fatty acid oxidation and oxidative phosphorylation, Arterioscler. Thromb. Vasc. Biol 41 (3) (2021) 1062–1075, 10.1161/ATVBAHA.120.315758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Abbas A, Gregersen I, Holm S, Daissormont I, Bjerkeli V, Krog-Sorensen K, Slagen KR, Dahl TB, Russell D, Almas T, Bundgaard D, Alteheld LH, Rashidi A, Dahl CP, Michelsen AE, Biessen EA, Aukrust P, Halvorsen B, Sjelland M, Interleukin 23 levels are increased in carotid atherosclerosis: possible role for the interleukin 23/interleukin 17 axis, in: Stroke, 2015, 10.1161/STROKEAHA.114.006516. [DOI] [PubMed] [Google Scholar]
- [42].Wang J, Zhao P, Gao Y, Zhang F, Yuan X, Jiao Y, Gong K, The effects of anti-IL-23p19 therapy on atherosclerosis development in ApoE, J. Interferon Cytokine Res 39 (9) (2019) 564–571, 10.1089/jir.2019.0050. [DOI] [PubMed] [Google Scholar]
- [43].Engelbertsen D, Depuydt MAC, Verwilligen RAF, Rattik S, Levinsohn E, Edsfeldt A, Kuperwaser F, Jarolim P, Lichtman AH, IL-23R deficiency does not impact atherosclerotic plaque development in mice, J Am Heart Assoc 7 (8) (2018), 10.1161/JAHA.117.008257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT, Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis, Proc. Natl. Acad. Sci. U. S. A 112 (22) (2015) 7061–7066, 10.1073/pnas.1415675112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Frostegård J, Ulfgren AK, Nyberg P, Hedin U, Swedenborg J, Andersson U, Hansson GK, Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines, Atherosclerosis 145 (1) (1999) 33–43, 10.1016/s0021-9150(99)00011-8. [DOI] [PubMed] [Google Scholar]
- [46].Neele AE, Gijbels MJJ, van der Velden S, Hoeksema MA, Boshuizen MCS, Prange KHM, Chen HJ, Van den Bossche J, van Roomen CPPA, Shami A, Levels JHM, Kroon J, Lucas T, Dimmeler S, Lutgens E, de Winther MPJ, Myeloid Kdm6b deficiency results in advanced atherosclerosis, Atherosclerosis 275 (2018) 156–165, 10.1016/j.atherosclerosis.2018.05.052. [DOI] [PubMed] [Google Scholar]
- [47].Fedorova LV, Sodhi K, Gatto-Weis C, Puri N, Hinds TD, Shapiro JI, Malhotra D, Peroxisome proliferator-activated receptor δ agonist, HPP593, prevents renal necrosis under chronic ischemia, PloS One 8 (5) (2013), e64436, 10.1371/journal.pone.0064436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA, Fazio S, Linton MF, Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice, Arterioscler. Thromb. Vasc. Biol 25 (8) (2005) 1647–1653, 10.1161/01.ATV.0000173413.31789.1a. [DOI] [PubMed] [Google Scholar]
- [49].Xu J, Racke MK, Drew PD, Peroxisome proliferator-activated receptor-alpha agonist fenofibrate regulates IL-12 family cytokine expression in the CNS: relevance to multiple sclerosis, J. Neurochem 103 (5) (2007) 1801–1810, 10.1111/j.1471-4159.2007.04875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Peng S, Xu J, Ruan W, Li S, Xiao F, PPAR-y activation prevents septic cardiac dysfunction via inhibition of apoptosis and necroptosis, Oxid Med Cell Longev (2017) 8326749, 10.1155/2017/, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.