

Graphical Abstract
Short Statement
Recent publications have suggested that oxidative DNA damage mediated by hydroxyl radical (•OH) is unimportant in vivo, and that carbonate anion radical (CO3•−) plays the key role. We examine these claims and summarize the evidence that •OH does play a key role as an important member of the reactive oxygen species (ROS) in vivo.
1. Introduction to reactive oxygen species and DNA damage
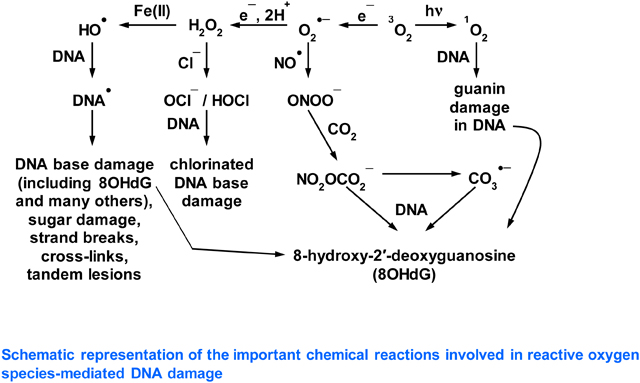
A wide range of reactive oxygen species (ROS) is formed in vivo in the human body and in other living organisms (reviewed in [1]). The term “reactive” covers a broad spectrum: some ROS, such as superoxide anion radical (O2•−), nitric oxide (NO•) and hydrogen peroxide (H2O2) are very selective in their reactions. Others, such as hypochlorous acid (HOCl), carbonate anion radical (CO3•−) and the two singlet states of oxygen (1O2), are fiercer and can attack several biomolecules. By contrast, the hydroxyl radical (•OH) reacts at or near a diffusion-controlled rate with almost every organic biomolecule found in living organisms [1, 2]. Several ROS, generally the ones of lower reactivity such as H2O2 and NO•, play important physiological roles in vivo, but the ones of higher reactivity can cause oxidative damage to biomolecules, resulting in impairment of cellular functions (reviewed in [1, 3]). In particular, oxidative damage to DNA plays an important role in the origin and progression of a number of human diseases, most prominently cancer but also others, such as neurodegenerative diseases and atherosclerosis [1, 4–6]. The ability of several ROS to attack DNA and generate mutagenic end-products plays a key role in cancer development in humans. Much attention has been paid to the mutagenic lesion 8-hydroxy-2′-deoxyguanosine (8OHdG) in this context [1, 7], but many other mutagenic and/or cytotoxic lesions are formed when •OH attacks DNA [1, 5, 8–15]. However, recent articles [16–18] have suggested that •OH is not involved in DNA damage caused by oxidative stress and argue a key role instead for CO3•¯, which attacks guanine residues in DNA to form 8OHdG. We would like to bring two matters to the attention of the journal readership,
that there is much more to biologically-significant oxidative DNA damage than only 8OHdG formation, and
that •OH does play a significant role in causing oxidative DNA damage in vivo.
2. How does hydroxyl radical arise in vivo?
Hydroxyl radical is generated in vivo by several mechanisms, including:
- through the reaction of certain transition metal ions (especially Fe2+ and Cu+ (reaction 1, Fenton reaction) with H2O2 (reviewed in [1, 3]).
The question of the availability, catalytic activity and chemical nature of transition metal ions in vivo has been repeatedly discussed [1, 3, 19–21], but there is no clear consensus as yet, although the recent discovery of ferroptosis, a form of iron ion-induced cell death, has rekindled interest in this topic [3, 22]. For example, Fe2+ ions bound to phosphate, polyphosphate, citrate, ATP etc. have shown variable activities in •OH generation in vitro [1, 21–28], but these simple studies in solution rarely reflect the complex cellular and extracellular environment in vivo (which is enormously rich in proteins, lipids, nucleic acids and hundreds of different metabolites). We return to this question in Section 4 below.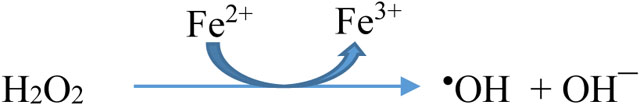
(1) -
The fission of H2O upon exposure to ionizing radiation (to which we have a constant background exposure [1, 9, 31]). Water cation radical (H2O•+) is the primary species formed in the physical stage (~10−15s) due to the interaction of ionizing radiation with water (reviewed in [31]). Subsequently, there is ultrafast proton transfer from H2O•+ in the physicochemical stage (10−15 – 10−12 s) to a surrounding water molecule (reaction 3).
(3) In addition, •OH is formed by homolysis (reaction 4) of the excited water molecule ((H2O)*) [1, 9, 31].(4) Indeed, the damage that •OH causes to DNA helps to explain why exposure to ionizing radiation can lead to cancer development [1, 4, 5, 9].
That •OH is generated in vivo (including by Fenton chemistry) has been demonstrated by a multiplicity of methods, including aromatic hydroxylation and ESR spin trapping [1, 32–42]. Owing to its high electrophilicity and high reactivity [1, 2, 9], •OH reacts at or near a diffusion-controlled rate (rate constant >109 M−1s−1) with almost all organic biomolecules. As a result, when •OH is generated in vivo, it will attack whichever of these organic molecules are adjacent to it [1, 2, 9].
3. The role of bicarbonate in vivo
As mentioned, recent articles [16–18] have argued that CO3•¯ and not •OH plays the major role in causing oxidative DNA damage in vivo. It is well known that bicarbonate anion (HCO3¯) is important in maintaining physiological pH and is indeed present intracellularly at high mM (10–40 mM) concentration [16–18 and references therein]. In vitro studies have suggested that in the presence of HCO3¯ the reaction of Fe2+ and H2O2 does not generate •OH but instead CO3•¯ [16–18, 43]. An alternative explanation is that •OH is generated but immediately reacts with HCO3¯ to give CO3•¯. However, the rate constant for the formation of CO3•¯ via H-atom abstraction from HCO3¯ by •OH (reaction 5) under physiological conditions has been measured by pulse radiolysis and is found to be quite low, 8.5 × 106 M−1s−1 [44].
| (5) |
Molecules such as 2′-deoxyribose phosphate, the purine and pyrimidine bases of DNA and RNA, reduced glutathione (GSH) and proteins, present in vivo also at substantial concentrations, react much faster with •OH, at diffusion-controlled rates (>109 M−1s−1) and so may be preferred targets, depending on the location and environment in which the •OH is generated [1, 2, 6, 8, 9], as we discuss in Section 4. However, CO3•¯ (and possibly some •OH) can also be generated in pathways involving NO•, CO2 and peroxynitrite (reviewed in [1, 45, 46]). The rate constant of the reaction of CO2 with peroxynitrite involved in this process, ranges from 3 × 104 M−1 s−1 to 5.8 × 104 M−1 s−1 [1, 45, 46].
4. The relative reactivities of •OH and CO3•¯ with DNA
Two approaches can throw light on this question, an examination of thermodynamic parameters and direct experimental studies. The absolute reduction potentials (E°) and midpoint potentials (E7) of •OH, CO3•¯, and the DNA components are presented in Table 1 below [8, 47–50].
Table 1.
The absolute reduction potentials (E°) and the midpoint potential (E7) of •OH, CO3•¯ and of base cation radicals. The E7 value of 2′-deoxyribose (dR) is also listed.
| Bases and radical | E vs. SHE (V) | |||
|---|---|---|---|---|
| Couple (E°) | E° in DMF | Couple (E7) | E7 by pulse radiolysis in water | |
| G (Guanine base) | (G•+/G) | 1.49 | (G(N1–H)•)/H+, G) | 1.29 |
| A (Adenine base) | (A•+/A) | 1.96 | (A(N6–H)•)/H+, A) | 1.42 |
| C (Cytosine base) | (C•+/C) | 2.14 | (C(N4–H)•)/H+, C) | 1.6 |
| T (Thymine base) | (T•+/T) | 2.11 | (T(N3–H)•)/H+, T) | 1.7 |
| •OH | •OH, H+/H2O | 2.3 | ||
| CO3•¯ | CO3•¯/CO32¯ | 1.59 | ||
| •CH2CH3 | •CH2CH3, H+/CH3CH3 | 1.9 | ||
| dR• | dR• / H+, dR | >1.8 | ||
From Table 1 and assuming the E7 of •CH2CH3 [48] and of dR [49] as a guide for that of the sugar moiety in DNA, we conclude that CO3•¯ is very unlikely to cause oxidative damage to dR and pyrimidines and should be capable of oxidizing only guanine , and perhaps adenine to a much lesser extent. Following the ionization potentials of the bases and according to Table 1 above, guanine should be the major or only site of oxidative damage by CO3•¯ in DNA. Indeed, a combination of laser flash photolysis and product analysis studies has confirmed that CO3•¯ oxidizes guanine in DNA, to form 8OHdG [45, 51]. We can find no literature evidence of adenine oxidation by CO3•¯. Also, if CO3•¯ were the main player in oxidative DNA damage, as argued in [16–18] and due to the repulsive forces of the highly negative charged polymer (DNA) and CO3•¯, we should not expect CO3•¯ mediated sugar-phosphate damage leading to strand break formation and indeed this is scarcely observed [51, 52].
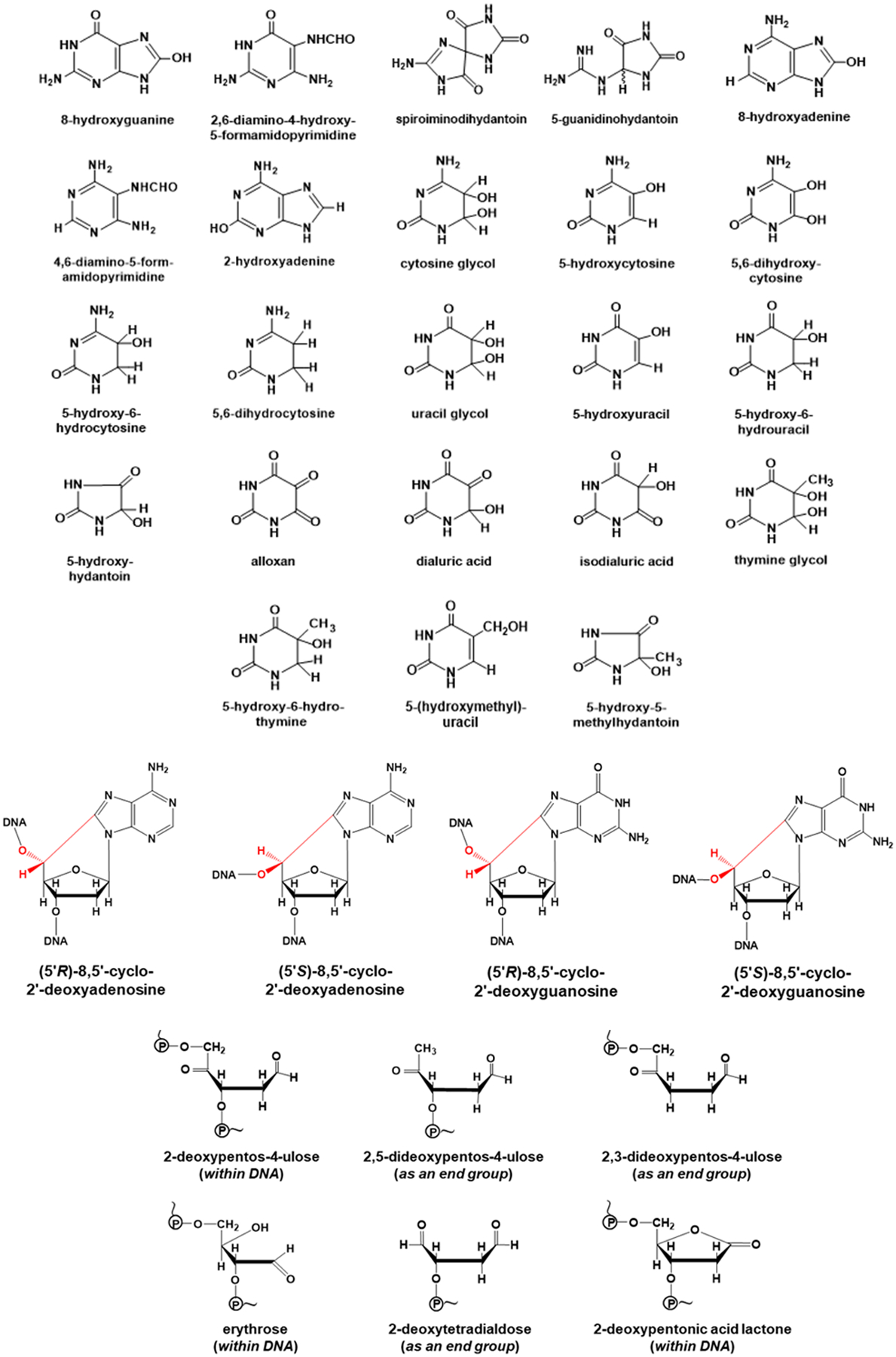
In agreement with the E° values in Table 1, direct experimental results show that when •OH reacts with DNA it forms a multiplicity of damage products (Figure 1) from all four purine and pyrimidine bases and from the dR moiety [1, 8–14, 53, 54]. No other known ROS forms such a wide range of products: some (such as H2O2 and O2•¯) do not react directly with DNA at all whereas others (e.g. CO3•¯, 1O2) target guanine selectively [1, 8, 16]. Hence, the demonstration that this wide range of products (shown in Figure 1) is formed in vivo is excellent evidence that •OH has been generated and has attacked DNA, whatever studies on simplified systems in vitro that do not reflect the complex cellular environment in vivo may suggest. To take one example, when human respiratory tract epithelial cells were exposed to 100 μM H2O2, there was rapid induction of DNA strand breakage and chemical modifications to all 4 DNA bases, diagnostic of attack by •OH [53]. How can this diagnostic damage pattern of •OH attack be explained, since H2O2 does not react with DNA? We have already mentioned our poor knowledge of the availability and distribution of transition metal ions in vivo, but evidence suggests that DNA in vivo has transition metal ions such as Fe2+ and Cu+ bound to it, given its very strong negative charge due to the phosphate groups (reviewed in [1]). Indeed, Fe2+ bound to phosphate is generally agreed (even by Prof. Burrows [17]) to generate •OH from H2O2, and the reasons for this have been recently elucidated [55]. The phosphate levels in the nucleus are very high due to the phosphate residues in DNA and so •OH formation will be favoured. H2O2 crosses plasma and intracellular membranes reasonably freely [1] and, if it reaches the nucleus, H2O2 can react with such metal ions to generate •OH directly upon the DNA, causing immediate oxidative damage, often called “site-specific” damage [1, 2]. This “site-specific” damage by localized •OH generation also occurs with biomolecules other than DNA, such as proteins, again generating multiple products diagnostic of •OH attack [1, 56, 57]. It cannot be prevented by external molecules that scavenge •OH, such as HCO3¯, glucose or GSH [1]. Furthermore, the formation of a thymine-tyrosine crosslink has been observed upon treatment of mammalian cells with Fe(II), and involvement of •OH has been suggested in this crosslink formation [58]. The free radical mechanistic pathways of •OH - mediated formation of multiple guanine and other DNA base damage products that are produced via oxidative damage, have been well documented in the literature [1, 6, 8, 12, 59].
Figure 1. Products resulting from attack of hydroxyl radicals on DNA.
By contrast, carbonate anion radical modifies only guanine residues
The exact molecular ratios of different DNA base and sugar damage products generated by site-specific •OH formation or other modes of •OH attack upon DNA depend on several factors, including where upon the DNA the metal ions are bound [9–12]. This pattern of multiple DNA base damage products is indeed observed in vivo: low levels of multiple base DNA damage products are present in DNA from all human and other animal tissues examined and the levels increase when oxidative stress is imposed by a variety of mechanisms [1, 6, 8–14, 59–64], e.g. in diabetes [65]. For example, 8,5′-cyclopurine-2′-deoxynucleosides in DNA are generated exclusively by •OH attack upon 2′-deoxyribose units generating C5′ radicals, followed by cyclization with the C8 position of the purine base [59, 66, 67]. This vast literature unequivocally demonstrates the formation of •OH-induced DNA base and 2′-deoxyribose products in vivo. In addition, oxidative stress can liberate catalytically-active transition metal ions (especially iron ions) from a range of cellular proteins (such as iron-sulphur proteins, and ferritin) [1, 19, 29, 68, 69], and some of these may bind to DNA, making it a further in vivo target of oxidative damage by site-specific •OH generation [1].
5. There is much more to biologically-significant oxidative DNA damage than 8OHdG formation
Apart from 8OHdG, the importance of many other DNA lesions, some of which are shown in Fig. 1, in cancer development in vivo has been highlighted, and the existence of DNA repair enzymes needed for their removal and whose genetic deletions increase cancer development in animals is further evidence that these mutagenic and/or cytotoxic lesions are formed in vivo and are important in the development of cancer and other diseases [1, 8, 70, 71].
6. Conclusion
There is unequivocal evidence of the •OH-specific pattern of oxidative DNA damage in vivo and in isolated cells subjected to oxidative stress. This, combined with the ability to trap •OH by specific methods in living systems, provides substantial evidence that •OH plays an important role in oxidative DNA damage, and other aspects of oxidative damage, including protein and lipid damage, in vivo [1]. This is in part due to formation of 8OHdG, which can also be generated by attack of 1O2 and of CO3•¯ on DNA, but also due to many other mutagenic and/or cytotoxic lesions, formed from purines, pyrimidines and 2′-deoxyribose by •OH attack (Fig. 1). Carbonate anion radical might also play an important role in vivo [16–18]. Certain other ROS, such as HOCl, can also attack DNA. Hypochlorous acid forms chlorinated base products, which have indeed been detected in vivo [72, 73].
Supplementary Material
Acknowledgements
BH thanks the distinguished Tan Chin Tuan family for support of his Centennial Professorship at NUS. AA is grateful to the National Cancer Institute of the National Institutes of Health (Grant RO1CA045424) and the National Science Foundation (Grant No. CHE- 1920110) for support. AA thanks the Center for Biomedical Research, Research Excellence Fund at Oakland University for support.
References
- 1.Halliwell B and Gutteridge JMC, Free Radicals in Biology and Medicine. 2015, Clarendon Press, Oxford; (fifth edition), UK. [Google Scholar]
- 2.Pryor WA, Free Radic. Biol. Med, 1988, 4, 219. [DOI] [PubMed] [Google Scholar]
- 3.Halliwell B, Free Radic. Biol. Med, 2020, 161, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayes JD, Dinkova-Kostova AT and Tew KD, Cancer Cell, 2020, 38, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halliwell B, Biochem. J, 2007, 401, 1. [DOI] [PubMed] [Google Scholar]
- 6.Dizdaroglu M, Mutat. Res. Rev. Mutat. Res, 2015, 763, 212. [DOI] [PubMed] [Google Scholar]
- 7.Gorini F, Scala G, Cooke MS, Majello B and Amente S, DNA Repair, 2021, 97, 103027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chatgilialoglu C, Ferreri C, Krokidis MG, Masi A and Terzidis MA, Free Radic. Res, 2021, 26, 1. [DOI] [PubMed] [Google Scholar]
- 9.von Sonntag C, Free-Radical-Induced DNA Damage and its Repair, 2006, Springer-Verlag, Berlin, Heidelberg. [Google Scholar]
- 10.Halliwell B and Dizdaroglu M, Free Radic. Res. Commun, 1992, 16, 75. [DOI] [PubMed] [Google Scholar]
- 11.Aruoma OI, Halliwell B and Dizdaroglu M, J. Biol. Chem, 1989, 264, 13024. [PubMed] [Google Scholar]
- 12.Aruoma OI, Halliwell B, Gajewski E and Dizdaroglu M, Biochem. J, 1991, 273, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dizdaroglu M, Rao G, Halliwell B and Gajewski E, Arch. Biochem. Biophys, 1991, 285, 317. [DOI] [PubMed] [Google Scholar]
- 14.Dizdaroglu M and Jaruga P, Free Radic. Res, 2012, 46, 382–419. [DOI] [PubMed] [Google Scholar]
- 15.Cadet J, Davies KJA, Medeiros MH, Di Mascio P and Wagner JR, Free Radic. Biol. Med, 2017, 107, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fleming AM and Burrows CJ, Chem. Soc. Rev, 2020, 49, 6524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fleming AM and Burrows CJ, Chem. Commun, 2020, 56, 9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fleming AM, Redstone SCJ and Burrows CJ, 2021, In DNA Damage, DNA Repair and Disease (Dizdaroglu M, Lloyd RS (), Royal Society of Chemistry, Cambridge, UK, vol. 1, 61. [Google Scholar]
- 19.Halliwell B and Gutteridge JMC, Biochem. J, 1984, 219, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kell DB, BMC Med. Genomics, 2009, 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gutteridge JMC and Halliwell B, Biochem. Biophys. Res. Commun, 2018, 502, 183. [DOI] [PubMed] [Google Scholar]
- 22.Wu H, Wang F, Ta N, Zhang T and Gao W, Life (Basel), 2021, 11, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biaglow JE and Kachur AV, Radiat. Res, 1997, 148, 181. [PubMed] [Google Scholar]
- 24.Adam FI, Bounds PL, Kissner R and Koppenol WH, Chem. Res. Toxicol, 2015, 28, 604. [DOI] [PubMed] [Google Scholar]
- 25.van der Wier B, Balk JM, Haenen GRMM et al. , FEBS Lett. 2013, 587, 2461. [DOI] [PubMed] [Google Scholar]
- 26.Illés E, Patra SG, Marks V, Mizrahi A and Meyerstein D, J. Inorg. Biochem, 2020, 206, 111018. [DOI] [PubMed] [Google Scholar]
- 27.Koppenol WH and Hider RH, Free Radic. Biol. Med, 2019, 133, 3. [DOI] [PubMed] [Google Scholar]
- 28.Flitter W, Rowley DA and Halliwell B, FEBS Lett, 1983, 158, 310. [Google Scholar]
- 29.Halliwell B and Gutteridge JMC, Mol. Asp. Med, 1985, 8, 89. [DOI] [PubMed] [Google Scholar]
- 30.Kachur AV, Manevich Y and Biaglow JE, Free Radic. Res, 1997, 26, 399. [DOI] [PubMed] [Google Scholar]
- 31.Ma J, Denisov SA, Adhikary A and Mostafavi M, Int. J. Mol. Sci, 2019, 20, 4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whiteman M and Halliwell B, Br. J. Pharmacol, 2004, 142, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.B Yan E, Unthank JK, Castillo-Melendez M, Miller SL, Langford SJ and Walker DW, J Appl. Physiol, 1985, 98, 2304. [DOI] [PubMed] [Google Scholar]
- 34.Freinbichler W, Bianchi L, Colivicchi MA, Ballini C, Tipton KF, Linert W and Corte LD, J. Inorg. Biochem, 2008, 102, 1329. [DOI] [PubMed] [Google Scholar]
- 35.Mason RP, Hanna PM, Burkitt MJ and Kadiiska MB, Environ. Health Perspect, 1994, 102, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huycke MM and Moore DR, Free Radic. Biol. Med, 2002, 33, 818. [DOI] [PubMed] [Google Scholar]
- 37.Takeshita K, Fujii K, Anzai K and Ozawa T, Free Radic. Biol. Med, 2004, 36, 1134. [DOI] [PubMed] [Google Scholar]
- 38.Kadiiska MB, Burkitt MJ, Xiang QH and Mason RP, J. Clin. Invest, 1995, 96, 1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grootveld M and Halliwell B, Biochem. J, 1986, 237, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halliwell B, Grootveld M and Gutteridge JM, Methods Biochem. Anal, 1998, 33, 59. [DOI] [PubMed] [Google Scholar]
- 41.Sun JZ, Kaur H, Halliwell B, Li XY and Bolli R, Circ. Res, 1993, 73, 534. [DOI] [PubMed] [Google Scholar]
- 42.Ferger F, Rose S, Jenner A, Halliwell B and Jenner P, NeuroReport, 2001, 12, 1155. [DOI] [PubMed] [Google Scholar]
- 43.Illés E, Mizrahi A, Marks V and Meyerstein D, Free Radic. Biol. Med, 2019, 131, 1. [DOI] [PubMed] [Google Scholar]
- 44.Buxton GV and Elliot AJ, Radiat. Phys. Chem, 1986, 27, 241. [Google Scholar]
- 45.Dedon PC and Tannenbaum SR, Arch. Biochem. Biophys, 2004, 423, 12. [DOI] [PubMed] [Google Scholar]
- 46.Radi R, Proc. Natl. Acad. Sci. USA, 2018, 115, 5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schroeder CA, Pluharova E, Seidel R, Schroeder WP, Faubel M, Slavícek P, Winter B, Jungwirth P and Bradforth SE, J. Am. Chem. Soc, 2015, 137, 201. [DOI] [PubMed] [Google Scholar]
- 48.Buettner G, Arch. Biochem. Biophys, 1993, 300, 535. [DOI] [PubMed] [Google Scholar]
- 49.Khanduri D, Adhikary A and Sevilla MD, J. Am. Chem. Soc, 2011, 133, 4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steenken S, and Jovanovic S, J. Am. Chem. Soc, 1997, 119, 617. [Google Scholar]
- 51.Joffe A, Geacintov NE and Shafirovich V, Chem. Res. Toxicol, 2003, 16, 1528. [DOI] [PubMed] [Google Scholar]
- 52.Roginskaya M, Moore TJ, Ampadu-Boateng D and Razskazovskiy Y, Free Radic. Biol. Med, 2015, 49, 1431. [DOI] [PubMed] [Google Scholar]
- 53.Spencer JP, Jenner A, Aruoma OI, Cross CE, Wu R and Halliwell B, Biochem. Biophys. Res. Commun, 1996, 224, 17. [DOI] [PubMed] [Google Scholar]
- 54.Fleming AM, Muller JG, Ji I and Burrows CJ, Org. Biomol. Chem, 2011, 9, 3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen HY, ACS Omega, 2019, 4, 14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garner B, Davies MJ and Truscott RJ, Exp. Eye Res, 2000, 70, 81. [DOI] [PubMed] [Google Scholar]
- 57.Rykaer M, Svensson B, Davies MJ and Hagglund P, J. Proteome Res, 2017, 16, 3978. [DOI] [PubMed] [Google Scholar]
- 58.Altman SA, Zastawny TH, Randers-Eichhorn L, Cacciuttolo MA, Akman SA, Dizdaroglu M, Rao G, Free Radic. Biol. Med, 1995, 19, 897. [DOI] [PubMed] [Google Scholar]
- 59.Jaruga P and Dizdaroglu M, DNA Repair, 2008, 7, 1413. [DOI] [PubMed] [Google Scholar]
- 60.Kasprzak KS, Diwan BA, Rice JM, Misra M, Riggs CW, Olinski R and Dizdaroglu M, Chem. Res. Toxicol, 1992, 5, 809. [DOI] [PubMed] [Google Scholar]
- 61.Misra M, Olinski R, Dizdaroglu M and Kasprzak KS. Chem. Res. Toxicol, 1993, 6, 33. [DOI] [PubMed] [Google Scholar]
- 62.Toyokuni S, Mori T and Dizdaroglu M, Int. J. Cancer, 1994, 57, 123. [DOI] [PubMed] [Google Scholar]
- 63.Chan W, Chen B, Wang L, Taghizadeh K, Demott MS and Dedon PC, J. Am. Chem. Soc, 2010, 132, 6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muruzabal D, Collins A and Azqueta A, Food Chem. Toxicol, 2021, 147, 111865. [DOI] [PubMed] [Google Scholar]
- 65.Rehman A, Nourooz-Zadeh J, Möller W, Tritschler H, Pereira P and Halliwell B, FEBS Lett, 1999, 448, 120. [DOI] [PubMed] [Google Scholar]
- 66.Chatgilialoglu C, Ferreri C, Geacintov NE et al. , Cells, 2019, 8, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mori T, Nakane H, Iwamoto T, Krokidis MG, Chatgilialoglu C, Tanaka K, Kaidoh T, Hasegawa M and Sugiura S, DNA Repair , 2019, 80, 52. [DOI] [PubMed] [Google Scholar]
- 68.Sobota JM, Gu M and Imlay JA, J. Bacteriol, 2014, 196, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kakhlon O and Cabantchik Z, Free Radic. Biol. Med, 2002, 33, 1037. [DOI] [PubMed] [Google Scholar]
- 70.Chan MK, Ocampo-Hafalla MT, Vartanian V, Jaruga P, Kirkali G, Koenig KL, Brown S, Lloyd RS, Dizdaroglu M and Teebor GW, DNA Repair, 2009, 8, 768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brooks SC, Adhikary S, Rubinson EH and Eichman Brandt F., Biochim. Biophys. Acta, 2013, 1834, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fedeles BI, Freudenthal BD, Yau E, Singh V, Chang S, Li D, Delaney JC, Wilson SH and Essigmann JM, Proc. Natl. Acad. Sci. U S A, 2015, 112, E4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spencer JP, Whiteman M, Jenner A and Halliwell B, Free Radic. Biol. Med, 2000, 28, 1039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.