

Abstract
C4 photosynthesis is a remarkable complex trait, elucidations of the evolutionary trajectory of C4 photosynthesis from its ancestral C3 pathway can help us better understand the generic principles of the evolution of complex traits and guide the engineering of C3 crops for higher yields. Here, we used the genus Flaveria that contains C3, C3–C4, C4-like and C4 species as a system to study the evolution of C4 photosynthesis. We first mapped transcript abundance, protein sequence and morphological features onto the phylogenetic tree of the genus Flaveria, and calculated the evolutionary correlation of different features; we then predicted the relative changes of ancestral nodes of those features to illustrate the major events during the evolution of C4 photosynthesis. We found that gene expression and protein sequence showed consistent modification patterns in the phylogenetic tree. High correlation coefficients ranging from 0.46 to 0.9 among gene expression, protein sequence and morphology were observed. The greatest modification of those different features consistently occurred at the transition between C3-C4 species and C4-like species. Our results show highly coordinated changes in gene expression, protein sequence and morphological features, which support evolutionary major events during the evolution of C4 metabolism.
Subject terms: Computational biology and bioinformatics, Evolution
Introduction
Elucidating the evolutionary and developmental processes of complex traits formation is a major focus of current biological and medical research. Most health-related issues, including obesity and diabetes, as well as agricultural challenges, such as flowering time control, crop yield improvements, and disease resistance, are related to complex traits1–3. Currently, genome-wide association studies are applied in the study of complex traits. Putative genes or molecular markers are then evaluated by a reverse genetics approach to identify those influence the complex traits. C4 photosynthesis is a complex trait that evolved from C3 photosynthesis. When compared with C3 plants, C4 plants have higher water, nitrogen and light use efficiencies4. Interestingly, C4 photosynthesis has evolved independently more than 66 times, representing a remarkable example of convergent evolution5. Accordingly, C4 evolution is an ideal system for the investigation of the mechanisms of convergent evolution of complex traits.
Change to C4 photosynthesis is associated with a number of biochemical, cellular and anatomical modifications when compared with the ancestral C3 photosynthesis6,7. In C3 photosynthesis, CO2 is fixed by ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco), whereas in dual-cell C4 photosynthesis, CO2 is initially fixed into a four-carbon organic acid in mesophyll cells (MCs) by phosphoenolpyruvate carboxylase (PEPC)8. The resulting four-carbon organic acid then diffuses into the bundle-sheath cells (BSCs)9, where CO2 is released and fixed by Rubisco. Hence, C4 photosynthesis requires additional enzymes in CO2 fixation in addition to those already functioning in C3 photosynthesis, including PEPC, NADP-dependent malic enzyme (NADP-ME), and pyruvate, orthophosphate dikinase (PPDK)8. In dual-cell C4 photosynthesis, CO2 is concentrated in BSCs that are surrounded by MCs, forming the so-called Kranz anatomy10–12. Compared with C3 leaf anatomy, Kranz anatomy requires a spatial rearrangement of MCs and BSCs, cell size adjustment for increased numbers of organelles, larger organelles and metabolite transfer between the two cell types, and a reduction in distance between leaf veins.
Much of the current knowledge regarding the evolution of C4 photosynthesis was gained through comparative studies in terms of physiology and anatomy by using genera that have both C3 and C4 species, as well as species performing intermediate types of photosynthesis7,13,14. Among these, the genus Flaveria has been promoted as a model for C4 evolution studies15, because the genus includes 23 known species which represent different photosynthetic types, including C3, C4 and different intermediate photosynthetic types16,17. Flaveria C3-C4 intermediate species are characterized by reduced photorespiration, lower CO2 compensation points compared to C3 species and partial to complete Kranz anatomy15,17,18. C3-C4 intermediate species are further divided into type I C3-C4 and type II C3-C4, the former reduces carbon loss solely relying on the photorespiratory CO2 concentration cycle as a result of reallocation of glycine decarboxylase in the BS tissues, whereas the later performs a partial C4 cycle as a result of increased enzyme activities of PEPC and NADP-malic enzyme19,20. C4-like intermediate species are featured by assimilating the majority of CO2 through the C4 cycle, but lacking a strict compartmentation of C4 enzymes between MC and BSC, and a small fraction of CO2 is initially fixed by Rubisco20,21.
The evolution of C4-related morphological, anatomical and physiological features has been well studied in this genus over the last 40 years15,22–24. The molecular evolution of several key C4 enzymes have been reported in this genus25–27, however, the molecular evolution of most C4 related genes is largely unknown. Besides, the evolutionary relationship between the C4 related genes and morphology features is not clear so far. In this study, we combined transcriptome data and published morphological data, together with the recent phylogenetic tree of the genus Flaveria28, to systematically investigate the key molecular events and evolutionary paths during the C4 evolution.
Results
Transcriptome assembly and quantification
RNA-Seq data of 31 samples of 16 Flaveria species were obtained from the public database Sequence Read Achieve (SRA) of the National Center for Biotechnology Information (NCBI) (Table S1). The 16 species represented two C3 species, seven C3-C4 intermediate species, three C4-like species and four C4 species19,21 (Table S1). On average, 42,132 contigs (from 30,968 to 48,969) were assembled with N50 ranging from 658 to 1208 bp among the 16 species (Table S2). The distribution of the contig length is similar in the 16 species with a peak at 360 bp (Fig. S1).
Since Flaveria is a eudicot genus, we used Arabidopsis thaliana (Arabidopsis) as a reference to annotate Flaveria transcripts. On average, 58.91% of Flaveria contigs had orthologous genes in Arabidopsis. Considering the large evolutionary divergence between Arabidopsis and Flaveria, which was estimated to be 120 million years (http://timetree.org/), we then estimated the accuracy of annotation by examining whether the transcripts annotated to be the same genes were from same orthologous groups. Specifically, we used OrthoFinder29 to predict the orthologous groups based on annotated genes of Flaveria species and then calculated the consistence between our gene annotation and orthologous groups. Specifically, for each orthologous group, we calculated the percentage of genes with the same annotation. For example, for the orthologous group of PPDK(AT4G15530), 29 transcripts from Flaveria and one gene (AT4G15530) with six transcripts from Arabidopsis were clustered in this orthologous group. All of the 29 transcripts from Flaveria were annotated as AT4G15530 (Fig. S2A). Therefore, the consistency of our annotation is 100% (29/29) for this orthologous group. Our result showed that 80% of the total 28,164 orthologous groups has a consistency higher than 90% (Fig. S2).
Transcript abundance was calculated as fragments per kilobase of transcript per million mapped reads (FPKM) (see Methods). The total transcriptome-level comparison revealed higher Pearson correlations in overall transcript abundance in leaf samples from the same species than those of different organs from the same species, regardless of sources (Fig. S3). Specifically, leaves from different developmental stages or from different labs are more closely correlated than leaf samples from different species, or than mean values of pair-wise correlations across all 27 leaf samples (T-test, P < 0.05) (Fig. S4). As a result, 13,081 Arabidopsis orthologs were detected in at least one of the 16 Flaveria species, and 12,215 were kept with the maximum FPKM in 16 species ≥ 1 FPKM.
Investigate the possibility of samples used in this study being hybrid
Considering that intermediacy of traits in the intermediate species may result from a possible hybridization between species with one parent being C3 and another parent being C4 or intermediate species30, we investigated whether the intermediate species used in this study are intermediate species or a hybrid offspring. The hybrid offspring is characterized as expressing different alleles at one DNA site; therefore, we calculated the percentage of DNA sites that expressed different alleles, which is termed as mixed site. The percentage of mixed site was then compared to the positive background generated by pair-wisely mixing of RNA-Seq data of 16 Flaveria species. Our results showed that the known hybrid sample F. pringlei* originated from F. pringlei × F. angustiflolia in28 showed significantly higher percentage of mixed site than background (Binomial test, P < 0.001), whereas, other species showed significantly lower percentages of mixed sites than background (Binomial test, P < 0.001) (Fig. 1). Thus, our data showed that species used in this study are not hybrids and can be used for evolutionary study.
Figure 1.
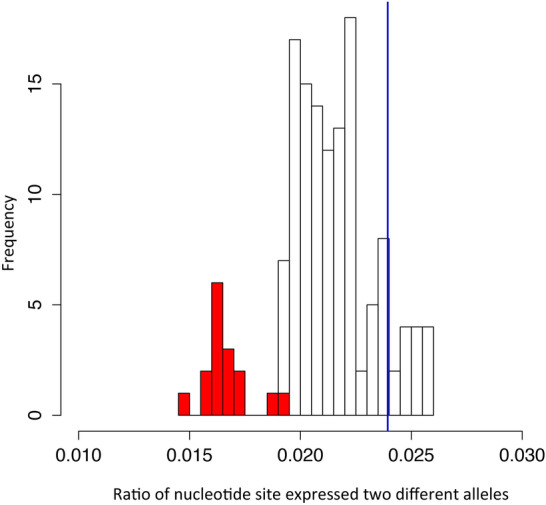
Estimation of the probability of RNA-Seq data from hybrid species. The bars show the distribution of ratio of nucleotide sites expressing two different alleles (mixed site). Mixed RNA-Seq samples are generated by pair-wise mixing RNA-Seq data of 16 Flaveria species, which mimics the case of hybridization. The ratio of mixed site in the mixed RNA-Seq samples is showed in grey bars (positive control). The ratio of mixed site of 16 Flaveria species is showed in red bars (real causes). The ratio of hybrid sample F. pringlei*, was represented in blue vertical line.
We used the phylogenetic tree of the genus Flaveria28 to illustrate the molecular evolution of C4 photosynthesis. We numbered each node of the phylogenetic tree in an order as showed in Fig. 2, usually, the more ancient a node is, the lower number it will be given. The number begins with N1 which refers to the common ancestor of all Flaveria species in the phylogenetic tree, N3 refers to the common ancestor on all intermediate and C4 species, at which intermediate species first evolved. N7 is the common ancestor of C4-like and C4 species in clade A, where a completed C4 cycle evolved. The nodes of clade B were numbered sequentially following clade A (Fig. 2).
Figure 2.

Modifications in genes in C4 pathway in predicted protein sequences and transcript abundances mapped to the Flaveria phylogeny. The predicted amino acid changes and the transcript abundance (FPKM) of the genes encoding the enzymes in C4 pathway are shown. Only the amino acid residues predicted to be different between C3 and C4 species are superimposed on the recent published phylogeny of Flaveria. The colors of amino acid residues have no meaning and are only for visualization purposes. Numbers below the amino acids indicate the location sites in the multiple sequence alignments. FPKM values with standard errors are shown to the right of the amino acid changes as red bars. (A) phosphoenolpyruvate carboxylase (PEPC); (B) pyruvate orthophosphate dikinase (PPDK); (C) NADP-malic enzyme (NADP-ME); (D) pyruvate orthophosphate dikinase regulatory protein (PPDK-RP); (E) phosphoenolpyruvate protein kinase A (PPCKA). Protein sequences from UniprotKP are: F. trinervia PEPC, P30694; F. bidentis PPDK, Q39735; F. brownii PPDK, Q39734; and F. trinervia PPDK, P22221. Sequence alignments are available in Additional file 2.
The modified genes: genes showed differences in gene expression and protein sequence between C3 species and C4 species
We first identified the modified genes, which were defined as genes show differences in both gene expression and protein sequence between C3 and C4 species. We first calculated the differentially expressed (DE) genes between C3 and C4 species in the way of comparing three C3 samples with 8 C4 samples (see Methods), which resulted in 896 DE genes (“BH” correlated P < 0.05) (Additional file 3). We next investigated transcriptome-wide amino acid changes predicted from orthologues of C3 and C4 Flaveria species using the process shown in Fig. S5. Briefly, an amino acid difference was classified as a change if the orthologous from the two C3 species (F. robusta and F. cronquistii) contained the same predicted amino acid at a position, but was different from the corresponding position in orthologs from at least two C4 species (among F. kochiana, F. bidentis, F. trinervia and F. australasica), the detailed process of identifying amino acid changes were described in Supplemental methods. As a result, we obtained 1,018 genes encoding at least one amino acid change between C3 and C4 Flaveria species. 56 out of these 1,018 genes also showed significantly differentially expression between C3 and C4 species, which was termed as modified genes.
The modified genes showed coordination in major changes in the C4 evolutionary pathway in the genus Flaveria
In addition to C4 pathway, cyclic electron transport chain (CET) and photorespiratory pathway are reported to be related to the evolution of C4 photosynthesis. We manually selected genes from these three pathways from the literatures23,24,31 and then tested whether genes from these pathways were significantly enriched in the 56 modified genes (Table S4). Results showed that genes related to C4 photosynthesis pathway and genes related to CET were significantly enriched in the 56 modified genes. (“BH” correlated P < 0.05, Fisher’s exact test, Supplemental methods). We systematically discussed these genes and their changes during C4 evolution in Flaveria with gene expression and predicted protein sequences.
Genes encoding proteins associated with the C4 pathway
Nine genes encoding proteins associated with the C4 pathway were identified, including those encoding three C4 cycle enzymes, PEPC, PPDK and NADP-ME, two regulatory proteins, PPDK regulatory protein (PPDK-RP) and PEPC protein kinase A (PPCKA), two aminotransferases, Alanine aminotransferase (AlaAT) and aspartate aminotransferase 5 (AspAT5), and two transporters, BASS2 and sodium: hydrogen (Na+/H+) antiporter 1 (NHD1) (Table 1). In terms of protein sequence, on average, 66.0% (from 33.33% to 86.7%) of amino acid changes in C4 species occurred at N7 for all of the nine genes (Fig. 2, Figs. S6, Table 1). For example, PEPC in the C4 Flaveria species had 41 predicted amino acid changes compared with those in the C3 species, which were mapped onto the Flaveria phylogeny determined by Lyu et al.28. One of the predicted changes occurred at N6 (D396 in C4 species, hereafter D396), and 34 occurred at N7 (Fig. 2A). The six other predicted amino acid changes occurred at N7 or after N7, although the incomplete assembly of PEPC transcripts from F. palmeri and F. vaginata did not allow resolution of the predicted amino acid sequences. These results suggest a major evolutionary event in the protein sequence at N7 for C4 enzymes.
Table 1.
Proteins showing differences in amino acid sequence between C3 and C4 Flaveria species and the relative changes in their cognate transcripts.
| Ortholog in A. thaliana | Genes encoding proteins involved in | Mean FPKM (C4 )/mean FPKM (C3 ) | FDR (EdgeR) | Length in Fcro (Frob)α | Protein length in A. thaliana (aa) | aa changes | Stage of key change(s) in sequence | Stage of key change(s) in FPKMβ | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total aa change(s) | Before N 5 | At N5 | At N6 | At N7 | After N7 | ||||||||
| Gene in C4 pathway | |||||||||||||
| AT3G14940 | PEPC | 85.58 | 2.78E−06 | 966 | 968 | 41 | 1 | >=34 | N7 | N7 | |||
| AT4G15530 | PPDK | 123.6 | 9.10E−09 | 958 | 963 | 31 | 2 + 6-aa REP | >=15 | N7 | N7 | |||
| AT1G79750 | NADP-ME | 26.64 | 6.64E−08 | 647 | 646 | 27 | 1 | 8 | 18 | N7 | N3 and N6 | ||
| AT4G21210 | PPDK-RP | 7.57 | 1.63E−03 | 402 | 403 | 13 | 1 | 4 | 7 | N7 | N7 | ||
| AT3G04530 | PEPC-k | 88.78 | 2.93E−03 | 281 | 278 | 12 | 3 | 2 | 7 | N7 | N7 | ||
| AT1G72330 | AlaAT | 9.63 | 1.57E−04 | 544 | 553 | 9 | 2 | 7 | N7 | N3 an N6 | |||
| AT4G31990 | AspAT5 | 36.67 | 4.34E−06 | 459 | 453 | 3 | 1 | 1 | 1 | N7 | N3 and N7 | ||
| AT2G26900 | BASS2 | 39.12 | 5.30E−07 | 415 | 409 | 14 | 2 | 12 | N7 | N7 | |||
| AT3G19490 | NHD1 | 51.19 | 8.49E−07 | 576 | 576 | 15 | 2 | 13 | N7 | N7 | |||
| Gene related to electron transport chain | |||||||||||||
| AT4G22890 | PGR5-like | 7.1 | 4.70E−02 | 328 | 324 | 10+17-aa INS | 1 | 2+17-aa INS | 7 | N6 | N7 | ||
| AT1G14150 | NdhL2/PnsL2 | 3.71 | 2.13E−02 | 190 | 190 | 4 | 2 | 1 | 1 | before N5 | N7 | ||
| AT2G04039 | NdhV | 9.17 | 1.73E−02 | 227 | 199 | 8 | 1 | 6 | 1 | N6 | N3 | ||
| AT5G43750 | Ndh18/PnsB5 | 6.8 | 6.27E−02 | 224 | 212 | 3 | 2 | 1 | N6 | N8 | |||
| AT5G21430 | NdhU/CRRL | 8.55 | 3.11E−03 | 215 | 218 | 4 | 4 | N6 | N7 | ||||
| AT4G37925 | NdhM | 7.01 | 5.24E−02 | 209 | 217 | 3 | 1 | 2 | N7 | N8 | |||
| AT1G15980 | Ndh48/PnsB1 | 8.6 | 1.77E−02 | 465 | 461 | 7 | 1 | 4 | 2 | N6 | N7 | ||
| AT1G18730 | NdhB4/PnsB4 | 8.8 | 9.57E−03 | 182 | 174 | 5 | 1 | 2 | 2 | N6 and N7 | N7 | ||
| AT5G52100 | CRR1 | 4.05 | 6.78E−03 | 302 | 298 | 5 | 1 | 1 | 3 | after N7 | N3 | ||
| Gene in photorespiration pathway | |||||||||||||
| AT5G35630 | GSL1 | 0.08 | 2.15E−02 | 430 | 430 | 8 | 3 | 1 | 2 | 1 | N7 | N7 | |
| AT1G32470 | GDC-H | 0.23 | 7.29E−01 | 162 | 166 | 6+2-aa INS + 1-aa INS | 5 + 2-aa INS + 1-aa INS | 1 | N7 | N7 | |||
| AT4G37930 | SHM | 0.16 | 4.08E−01 | 517 | 517 | 8 | 3 | 5 | 1 | N7 | N7 | ||
| AT1G80380 | GLYK | 0.49 | 3.61E−01 | 443 | 456 | 8 | 2 | 6 | 1 | N7 | N7 | ||
| AT5G04140 | GOGAT | 0.57 | 1.32E−01 | 1616 | 1648 | 18 | 4 | >=1 | >=5 | 2 | N7 | N7 | |
In terms of gene expression, all nine genes showed higher transcript abundance in C4 species than in C3 species and a comparable level in C4-like and C4 species (Table 1). To calculate the relative gene expression changes of each ancestral node, the FPKM values of each ancestral node were predicted and the relative difference were calculated (see Methods). In general, C4 species showed a 7.6-fold to 123.6-fold of FPKM values compared with C3 species. Similar to the pattern of changes of protein sequences, seven of the nine genes showed that the biggest relative changes in gene expression occurred at N7, whereas, both NADP-ME and AlaAT showed the biggest relative changes at two nodes of N3 and N6 with comparable levels (Fig. 2C and Fig. S6A). Our results therefore suggest that the genes encoding proteins associated with C4 pathway showed highly coordinated modification patterns in protein sequence and gene expression at N3, N6 and N7 during the evolutionary change to C4 photosynthesis, while the majority of the predicted amino acid changes occurs at the N7.
Genes encoding proteins involved in CET chain
We identified genes encoding nine proteins that function in the CET chain, namely, proton gradient regulation 5 like (PGR5-like), the chloroplast NAD(P)H dehydrogenase complex (Ndh) L2-2 (NdhL2-2), NdhV, Ndh18, NdhU, NdhM, Ndh48, NdhB4, and chlororespiratory reduction 1 (CRR1). The transcripts encoding all the nine proteins showed higher abundances in C4 species than C3 species (Fig. 3 and Table 1). Compared to the genes encoded protein in the C4 pathway, the genes encoding proteins involved in CET chain showed the biggest changes at diverse nodes rather than at a single node. Specifically, the major changes of predicted protein sequences occurred at N6 and N7 and that of FPKM occurred at N3, N7 and N8 (Table1). Besides this, the modification of protein sequence and FPKM appears to be less coordinated in genes involved in CET chain, for example, the major change of PGR5-like occurred at N6 in predicted protein sequence and at N7 in FPKM (Fig. 3A).
Figure 3.

Modifications in the predicted amino acid sequences of proteins involved in cyclic electron transport and transcript abundances of the cognate transcripts mapped to the Flaveria phylogeny. Changes in predicted amino acid sequence in proteins involved in cyclic electron transport chain and abundances (FPKM) of their cognate transcripts in C4 and C3 Flaveria species are shown. Only the amino acid residues predicted to be different between C3 and C4 species are superimposed recent published phylogeny of Flaveria. The marked colors of amino acid residues have no meaning and are only for visualization purposes. Numbers below the amino acids indicate the location site in the multiple sequence alignments. FPKM values with standard errors are represented to the right of the amino acid changes as red bars. (A) Protein gradient regulation 5 like protein (PGR5-like); (B) NADH dehydrogenase-like (Ndh) L2 subunit (Ndh L2); (C) NdhV; (D) Ndh16; (E) NdhU; (F) NdhM; (G) Ndh48; (H) NdhB4; (I) chlororespiration reduction 1 (CRR1). The sequence alignments are available in Additional file 2.
Genes encoding proteins in the photorespiratory pathway
The establishment of the photorespiratory pump (C2 photosynthesis) is reported to be a prerequisite for the evolution of C4 photosynthesis based on theoretical modeling32. Therefore, we also investigate genes involved in the photorespiratory pathway in terms of predicted protein sequence and FPKM.
One protein involved in photorespiration was included in the 56 modified genes, namely, glutamine synthetase-like 1 (GSL1). Moreover, four other proteins in this pathway showed abundant amino acid changes between C3 and C4 species, namely, glycine decarboxylase complex (GDC) H subunit (GDC-H), serine hydroxymethyltransferase (SHM), glycerate kinase (GLYK), glutamine synthetase and glutamine oxoglutarate aminotransferase (GOGAT) (Fig. 3, Table 1). In general, the predicted amino acid substitution patterns of these five proteins were similar to those observed in the above-described proteins in C4 pathways, with the major predicted amino acid changes in C4 species occurring at N7 (Fig. 4, Table 1), e.g., 16 of 18 in GOGAT occurred at N7 (Fig. 4D). Generally, proteins in the photorespiratory pathway showed fewer predicted amino acid changes than those in the C4 pathway.
Figure 4.

Modifications in photorespiratory protein predicted amino acid sequences and cognate transcript abundances mapped to the Flaveria phylogeny. The predicted amino acid changes in photorespiratory proteins between C4 and C3 Flaveria species and the transcript abundance (FPKM) of genes encoding the proteins are shown. Only the amino acid residues that are predicted to be different between C3 and C4 species are superimposed on the recent published phylogeny of Flaveria. The marked colors of amino acid residues have no meaning and are only for visualization purposes. Numbers below the amino acids indicate the location site in the multiple sequence alignments. FPKM values with standard errors are represented to the right of the amino acid changes as red bars. (A) Glutamine synthetase like 1 (GSL1); (B) glycine decarboxylase complex H subunit (GDC-H); (C) serine hydroxymethyltransferase (SHM); (D) glycerate kinase (GLYK); (E) glutamine synthetase and glutamine oxoglutarate aminotransferase (GOGAT).
The abundance of transcripts encoding these five photorespiratory enzymes was comparable to those in C3 and C3-C4 species, and higher than that in C4 species (Fig. 4A–E). When compared with genes encoding C4 pathway proteins, those encoding photorespiratory proteins showed larger differences between C4-like and C4 species in clade A in terms of gene transcript abundance and protein sequence. The greatest reduction of FPKM in these five genes was observed at N7 (Fig. 4, Table 1). Thus, this suggested that the genes encoded proteins associated with the photorespiratory pathway also showed coordinated changes in protein sequence and gene expression during the evolution of C4 photosynthesis, and with the largest number of changes occurring at N7.
Physiological and anatomical characteristics related to C4 photosynthesis show coordinated changes along the C4 evolutionary pathway in Flaveria
To investigate whether C4 related physiological characteristics also underwent coordinated changes during the evolution of C4 photosynthesis in Flaveria, physiological characteristics taken from the literature18,21,33 were mapped onto the Flaveria phylogeny (Fig. 5). The results revealed a step-wise change for most of the characteristics along the phylogenetic tree as previously suggested15,18,21,33 (Fig. 5). However, coordinated and abrupt changes were observed for a number of features. A major change in CO2 compensation point (Γ) in Flaveria was first seen at N3, where the most ancestral C3-C4 species, F. sonorensis emerging showing a decrease in Γ from 62.1 μbar of its closest C3 relative F. robusta to 29.6 μbar (Fig. 5). The greatest changes in Γ in clade A occurred at N6, which showed a decrease in Γ from 24.1 μbar in F. angustifolia (C3-C4) to 9.0 μbar in F. ramosissima (C3-C4), followed by N7, where a decrease in Γ from 9.0 μbar in F. ramosissima (C3-C4) to 4.7 μbar in F. palmeri (C4-like) was observed. The greatest decrease of Γ in clade B was observed between the two C3-C4 species, F. floridana and F. chloraefolia (C3-C4), where there was a decrease from 29 μbar to 9.5 μbar (Fig. 5). For photosynthetic water use efficiency (PWUE), photosynthetic nitrogen use efficiency (PNUE) and the slope of the response of the net CO2 assimilation rate (A) versus Rubisco, the biggest changes occurred at N7 with increases of around twofold. In contrast, the percentage of 14C fixed into four carbon acids showed no clear trend across the phylogenetic tree, although 3.91-fold and 1.76-fold increases were seen at N6 and N7, respectively. Interestingly, changes in all of these traits uniformly occurred at F. brownii in clade B, the only C4-like species within this clade. Consequently, those data suggest that although there were gradual changes in physiological features along the C3, C3-C4, C4-like and C4 trajectory, there are apparent major modifications at N3, N6 and N7 in these physiological traits across the Flaveria phylogeny (Fig. 5).
Figure 5.

Changes in physiological and anatomical traits mapped onto the Flaveria phylogeny. Overall, C4-related physiological (green bars) and anatomical traits (orange bars) showed a step-wise change along the Flaveria phylogenetic tree; however, a number of the traits showed greater more significant changes at certain nodes. *Grana index: total length of grana/total length of thylakoid membrane X 100. (Γ CO2 compensation point, A CO2 assimilation rate, PWUE instantaneous photosynthetic water use efficiency, PNUE instantaneous photosynthetic nitrogen use efficiency, response slope slope of the response of net CO2 assimilation rate versus leaf Rubisco content, M mesophyll, BS bundle sheath.) Data are from references as given in the Methods.
Anatomical traits15,34 were mapped onto the Flaveria phylogeny to investigate how these features were modified along the evolution of C4 (Fig. 5). For both the area of MCs and the ratio of the area of MCs to that of BSCs (M: BS), the greatest modifications across the phylogeny were found between F. brownii (C4-like) and F. floridana (C3-C4), with a similar degree of change for both characteristics (2.7-fold, Fig. 5). Anatomical data for F. palmeri (C4-like) in clade A are not available, however, large differences in anatomical features were found between the C4-like F. vaginata and C3-C4 F. ramosissima15. The modification of MC area first occurred at N2 which showed a 1.9-fold difference between F. robusta and F. cronquistii followed by a 2.1-fold of difference between F. ramosissima and F. vaginata. A major modification of the ratio of MC and BSC occurred at N2 with a 2.4-fold difference and followed by N6 with a 1.6-fold difference and N7 with a twofold difference. Therefore, similar to the evolutionary pattern of physiological features, large changes in anatomical features also emerged at N3, N6 and the transition between C3-C4 species and C4-like species. Interestingly, the ultrastructure of BSCs chloroplasts showed an abrupt change at N7, with a dramatic decrease in grana thylakoid length and an increase in stroma thylakoid length, whereas these features were comparable in the species at the base of tree and in clade B34.
Coordinated changes of protein sequence, gene expression and morphology with a major evolutionary event at the transition between C3-C4 and C4-like species during the evolution of C4 species
Our above analysis showed that C4 related genes and morphological features showed coordinated changes with an obvious major change at N7. Next, we asked whether species evolution also showed evolutionary coordination and major event during the evolution of species in protein sequence, gene expression and morphology. To answer this question, we calculated the divergence matrices for protein sequence, gene expression, and morphological features between F. cronquistii (at the most basal position on the Flaveria phylogenetic tree) and other Flaveria species. The protein divergence was calculated as the rate of non-synonymous substitutions (dN) of all the genes that were used to construct the Flaveria phylogenetic tree from28, the expression divergence was calculated as Euclidean distance of total expressed genes (see Methods), and the morphology divergence was calculated as Euclidean distance using previously coded morphology value from16, which includes 30 types of morphological traits, such as life history, leaf shape, head types. Our result showed a high linear correlation between the protein divergence, gene expression divergence and morphology divergence, in particular between gene expression divergence and morphology divergence (R2 = 0.9) (Fig. 6A). Thus, our results suggest a coordinated evolution of protein sequence, gene expression and morphology during species evolution. The linear correlation of gene expression divergence vs morphology divergence and protein divergence vs morphology divergence reflects that both gene expression changes and protein sequence changes are related to morphological changes during evolution35,36. Moreover, gene expression changes may be more directly related to morphological changes than protein sequence changes37. It is likely that changes of developmental programs might be mainly due to changes in gene expression levels while changes in the protein sequences might contribute more to changes in metabolism.
Figure 6.

Coordinated evolution of protein sequence, gene expression and morphological traits with apparent major changes. Significant linear correlation between protein divergence, gene expression divergence and morphology divergence were showed in (A). Protein divergence was calculated as non-synonymous mutation (dN). Expression divergence and morphology divergence were calculated as Euclidean distance based on quantile normalized FPKM values calculated here and coded morphology values from Mckown et al. 16, respectively. All the relative divergences were the divergence between F. cronquistii and other Flaveria species. (B) The schema of Flaveria phylogenetic tree modified from Lyu, et al. 28, branch length between ancestral nodes were labeled in grey. (C) The relative changes of each ancestral node compared with its earlier ancestral node in protein sequence, gene expression and morphological traits. P values are from One-way ANOVA analysis followed by Tukey’s Post Hoc test and adjusted by Benjamin-Hochberg correction. The significant levels are: *: P < 0.05; **: P < 0.01; ***: P < 0.001. The bar colors in grey/blue/orange represent species from basal/clade A/clade B of phylogenetic tree, respectively. (D) Pearson correlations between changes of ancestral nodes in protein sequence, gene expression and morphological traits.
Next, we predicted the protein sequence, transcript abundance and coded morphology value of ancestral nodes, which were then used to calculate the relative change of the three parameters at each node (see Methods). Surprisingly, protein sequence and gene expression showed significantly more changes from N6 to N7 than changes during transitions between other nodes (P < 0.001, Tukey’s test, “BH” adjusted, the same as following), and the morphology showed the most changes from N6 to N7 with a marginal significant P value (P = 0.06) (Fig. 6B,C). We found high correlations between divergences in protein sequence, gene expression and morphological traits (Fig. 6D), implying that evolutionary coordination of major events on whole transcriptomic level also occurred in species evolution.
We then asked whether the major events were results of a long evolutionary time. We found positive correlations between the divergence (in protein sequence, gene expression and morphology) and branch length. For protein sequence, gene expression and morphological traits, their Pearson correlation coefficients with branch length are 0.35, 0.66 and 0.59, respectively (Fig. S7A). We then normalized the divergence between nodes to branch length to calculate change rate by assuming modification of protein sequence, gene expression and morphology have a linear relationship with evolutionary time. Results showed that N7 does not show the highest change rate, whereas, younger nodes tend to have higher change rates (Fig. S7B). For example, N14 showed higher modification rates than other nodes in gene expression and morphology. This is reasonable, because recovery mutations occurred during evolution, and modification rate was likely to be underestimated within a long evolutionary time. Besides, the relationship between amino acid substitution and evolutionary time may be non-linear but follow complex models38. Consistently, we found that change rates of protein sequence are neither correlated with that of gene expression nor with that of morphological traits. Whereas, change rates of gene expression are positively correlated with that of morphological traits (R2 = 0.91, P value < 0.001) (Fig. S7C). The large number of changes between N6 and N7 are likely a result of a long evolutionary time.
Discussion
Evolutionary coordination of different features towards a functional C4 metabolism
Compared to C3 photosynthesis, the evolution of C4 photosynthesis resulted in the acquisition of many new features in gene expression, protein sequence, morphology and physiology (Figs. 2, 3, 4, 5)39. Coordinated changes on these features were required at key transitions. This is because although C4 photosynthesis can gain higher photosynthetic energy conversion efficiency, highly specialized leaf and cellular anatomical features and biochemical properties of the involved enzymes are required. For example, increased cell wall thickness at the bundle sheath cell and decreased sensitivity of PEPC to malate inhibition are needed for C4 plants to gain higher photosynthetic rates40,41. Furthermore, to gain higher photosynthetic efficiency in C4 plants, the ratio of the quantities of Rubisco content in BSCs and MCs is also critical42. In theory, if the C4 decarboxylation evolves before all of the other accompanying changes required or C4 photosynthesis, leaves will experience high leakage, i.e., costing ATP for a futile cycle without benefit to CO2 fixation. This will inevitably lead to lower quantum yield and a potential driving force for purifying selection. Further evidence for possible purifying selection comes from the observation that genes with cell-specific expression, such as PEPC, PPDK, and NADP-ME, displayed more changes in their predicted protein sequences than ubiquitously expressed genes, such as NDH components (Table 1, Additional file 3). This is because, as discussed earlier, the redox environments between BSCs and MCs might have changed dramatically during the evolution of the C4 cycle, with one of the most likely changes being a more acidic environment due to increased production of Oxaloacetic acid (OAA) and malate. Under such conditions, it is required for enzymes to alter their amino acid sequences to adapt to the new cellular environments. Concurrent changes of gene expression and protein sequence have also been demonstrated previously in animals36,43.
The identified changes in the protein sequences, including amino acid changes, insertions, and deletions (Table 1, Figs. 2, 3, 4), may enable the enzymes or proteins to improve biochemical and regulatory properties to meet the demands of an altered cellular environment, for example, the increased fluxes through the C4 cycle44. It is worth mentioning that some of these predicted amino acid changes have been reported to be functional, such as that the S774 and G884 residues in C4 PEPC determines the high substrate affinity and low inhibitor affinity of this enzyme, respectively25,45. Besides this, many of the predicted amino acid changes are in residues that can be post-translationally modified, for example, six residues in PPDK changed to Serine (S) in C4 species, which can all be target for phosphorylation and hence functional modification.
Major evolutionary events along the C4 evolution in the Flaveria genus
Among the ancestral nodes leading to the C4 emergence in clade A, N7, which is the most recent common ancestral node of C4-like and C4 species in clade A, shows the biggest change in protein sequence, gene expression and morphology in both C4 specific features and also non-C4 specific features (Table 1, Figs. 2, 3, 4, 5, 6). There were also apparent changes in these features at N3 and N6. These three nodes reflect three critical stages in the emergence of C4 metabolism. Firstly, at N3, i.e., during the emergence of C3-C4 species, there were many changes in gene expression, protein sequence and morphology. One of the most important events during this phase is the re-location of GDC from MSCs to BSCs based on earlier western blot data13,46. Here we found that SHM showed decreased expression while most of other photorespiratory related enzymes showed little changes (Fig. 4). Similarly, at this step, the majority of the C4 related genes showed little changes (Fig. 2). However, modification of gene expression levels and protein sequences on transcriptome level and no-C4 morphology features suggests that there are a large number of changes at N3 (Fig. 6), and there is also greater decrease of CO2 compensation point at this stage (Fig. 5).
C3-C4 species were also reported in several other genera from both monocotyledonous and dicotyledonous plants, such as the monocotyledonous Alloteropsis47, Homolepis, Neurachne and the dicotyledonous Steinchisma48 from monocot; Moricandia49, Heliotropium50 and Mollugo51. These C3-C4 species usually have a reduced CO2 compensation point, enlarged BSC, and their GDC-P subunit is predominately expressed in BSC. C3-C4 species in Moricandia do not show enhanced C4 cycle, besides, 14C labeling patterns for photosynthesis related metabolites are comparable to those in C3 species49, which may be equivalent to Flaveria C3-C4 species derived from N3.
N6, i.e., during the emergence of type II C3-C4 F. ramosissima and C4-like species. is the stage where we found the third largest number of changes in C4 related features had occurred. At this stage, we observed large increase in transcript abundance in C4 genes (Fig. 2 & Fig. S5) and photorespiratory genes (Fig. 4), and that a dramatic increase in the percentage of 14C incorporated into the four carbon acids occurred (Fig. 5). The modification of photorespiratory genes might be related to the optimization of C2 cycle to decrease CO2 concentrating point, which can increase fitness of plants under conditions favoring photorespiration52. The concurrent modifications of C4 enzymes, such as PEPC, NADP-ME, PEPCKA, amongst others, which are also involved in nitrogen rebalancing, is consistent with the notation that C4 cycle might be evolved as a result of rebalancing nitrogen metabolism after GDC moving from MC to BSC24. The fact that there is little change in the δ13C in the C3-C4 intermediate as compared to that of C3 species suggests that the contribution of CO2 fixation following evolution of the C4 pathway is relatively minor, i.e., less than 15%, estimated based on an δ13C value of -27.6 in F. ramosissima (Fig. 5), again supporting that the initial role of increased C4 enzymes is not for enhancing CO2 fixation. It is worth pointing out here that the measured initial carbon fixation in the form of C4 compound was 46% (Fig. 5); higher than those estimated based on the δ13C value. This is possibly because although malate releases CO2 into BSCs as a result of the nitrogen rebalancing pathway, most of the CO2 was not fixed by Rubisco, either due to the lack of sufficient Rubisco activity in BSCs or due to a lack of required low BSCs cell wall permeability to maintaining high CO2 concentration in BSCs.
N7, i.e., during the emergence of C4-like and C4 species, witnesses abrupt changes for both the gene expression and proteins sequence and morphology (Figs. 2, 3, 4, 6). The majority of the C4 related genes showed the most modification in gene expression and protein sequence at N7, especially for genes in C4 cycle and photorespiratory pathway. Moreover, N7, at which C4-like species (clade A) appear, represents a dramatic shift of CO2 fixation from being dominated by a C2 concentrating mechanism to being dominated by a C4 concentrating mechanism. Based on the δ13C value in F. palmeri, the fixation through the C4 concentrating mechanism is up to 93%, which is consistent with the measured proportion of initial carbon fixation in the form of C4 compound (Fig. 5), suggesting at this step, the released CO2 in the BSCs can be largely fixed by Rubisco, whereas, the transition between C4-like to C4 process is an evolutionarily "down-hill" process and most optimization occurred through fine-tuning gene expression.
Genes from C4 pathway and photorespiration displayed the major evolutionary changes at N7 both in gene expression and protein level (Table 1). At the same time, we found morphological features, protein sequence and gene expression, which were not necessarily related to C4 photosynthesis also showed major changes at that stage (Fig. 6). This coincidence suggests that the C4 and photorespiratory pathway may be a main driving force in the evolution in Flaveria.
The genus Flaveria has been used as a model system to study the evolution and regulation of C4 photosynthesis15–17. Over the past 40 years, many labs conducted studies related to metabolism, physiology, anatomy, morphology, transcriptome and transcript regulation for different groups of Flaveria species15,22–24. Here, we combined data from different aspects for 16 Flaveria species with a purpose to examine whether the different C4 related traits evolve in a coordinated manner and at the same time to study the relationship between species evolution and C4 photosynthesis evolution. Here we caution that Flaveria plants from different labs were grown under different conditions, data from different labs may be not in complete accord. For examples, the RNA-seq data used here were originally generated from two labs (Supplemental methods), plants were grown under different conditions and RNA-seq data were sequenced from different strategies. Hence, we performed additional normalization to the FPKM to make gene expression comparable among different samples. For DE genes between C3 and C4 species, we applied calcNormFactors function embed in edgeR53 to calculate scaling factors and convert raw library sizes into effective library sizes. DE genes called here may be not exactly consistent with that called based on RNA-seq data from each one of the labs, but should reflect the major DE genes between C3 and C4 species.
Materials and methods
Data retrieval
RNA-Seq data of Flaveria species were downloaded from the Sequence Read Achieve (SRA) of the National Center for Biotechnology Information (NCBI). The source of RNA-seq data and plant grown conditions are detailed in Supplementary Methods. All accession numbers for RNA-Seq data are shown in Table S1.
CO2 compensation points (Γ) (except for F. kochiana), δ13C (except for F.kochiana), %O2 inhibition of Pmax (except F. kochiana), and CO2 assimilation rates were from21. Γ, δ13 and %O2 inhibition of F. kochiana were from33. Data for % initial C4 products in total fixed carbon were from54. Data for PWUE, PNUE, and net CO2 assimilation rate (A) versus Rubisco content were from33. Data for M area, M:BS ratio, vein density and number of ground tissue layers were from15. The values of M area, M:BS ratio and vein density were measured from figures in McKown and Dengler15 with GetData (http://www.getdata-graph-digitizer.com). Mean values from 20 measurements were used. Ultrastructural data of BS cell chloroplasts were from Nakamura et al.34.
Transcriptome assembly and quantification
Transcripts of Flaveria species generated with Illumina sequencing were assembled using Trinity (version 2.02)55 with default parameters (Table S1). Contigs of four Flaveria species from 454 sequencing data were assembled using CAP356 with default parameters. In all cases, only contigs of at least 300 bp in length were saved. Transcript abundances of 31 Flaveria samples were analyzed by mapping Illumina short reads to assembled contigs of corresponding species and then normalized to the fragment per kilobase of transcript per million mapped reads (FPKM) using the RSEM package (version 1.2.10)57. Functional annotations of Flaveria transcripts were determined by searching for the best hit in the coding sequence (CDS) dataset of Arabidopsis thaliana (Arabidopsis) in TAIR 10 (http://www.arabidopsis.org) by using BLAST in protein space with E-value threshold 0.001. If multiple contigs shared the same best hit in CDS reference of Arabidopsis, then the sum FPKM of those contigs was assigned to the FPKM value of the gene in Flaveria.
To estimate the consistence of Flaveria gene annotation, we used OrthoFinder29 to predict the orthologous group based on the annotated Flaveria gene together with gene of Arabidopsis from TAIR10, and then calculated the consistence between gene annotation and orthologous group in two ways. (1) If the orthologous group contains Arabidopsis gene(s), the consistency was calculated as the percentage of genes that have the same annotation with the Arabidopsis gene(s). (2) If there the orthologous group does not contain Arabidopsis gene, we calculated the percentage of genes for each gene ID in this group, and the highest percentage was assigned to the consistency.
To make the FPKM comparable across different samples, we normalized the FPKM value by a scaling strategy as used by Brawand et al.35. Specifically, among the transcripts with FPKM values ranking in 20–80% region in each sample, we identified the 1000 genes that had the most-conserved ranks among 29 leaf samples, which were then used as an internal reference, and the transcript of each sample was normalized according to the mean value of these 1000 genes in the sample. We then multiplied all the FPKM values in all samples by the mean value of 1000 genes in the 29 leaf samples. The three samples from C3 species and eight samples from C4 species (Table S1) were used to recall differentially expressed (DE) genes applying edgeR53, and the Benjamini-Hochberg (“BH”) procedure was used in multiple testing correction with a threshold of P (“BH” corrected) to be 0.05.
Investigation the species used in this study being from hybrid of two species
To investigate whether the intermediate species used in this study are from hybrid offspring of two species, DNA sites that expressed different alleles were identified, which termed as mixed sites. The mixed sites were identified based on RNA-Seq data as described in28. Hybrid offspring from two different species are expected to have higher percentage of mixed sites among all expressed sites than no hybrid species. To create a positive background of hybrid samples, RNA-Seq data of 16 species were pair-wisely mixed and their mixed sites were also identified. The mixed sites of the known hybrid sample F. pringlei* originated from F. pringlei × F. angustiflolia in28 were also identified. The percentage of mixed sites was calculated as the ratio of mixed sites to the total expressed DNA sites in a certain sample.
Protein divergence, gene expression divergence and morphology divergence
Pair-wise protein divergence (dN) was calculated by applying codeml program in PAML package58 by using F3X4 condon frequency. The input super CDS sequence was from the linked coding sequences (CDS) as used in construct phylogenetic tree of Flaveria genus28, which contains 2,462 genes. Gene expression divergence was calculated as Euclidean distance applying R package based on gene expression values (FPKM) of 12,218 genes. Encoded morphology values of 30 morphology traits were from16. The morphology divergence was calculated as Euclidean distance of morphology values. Expression and morphology values were normalized using quantile normalization applying preprocessCore package in R. Linear regression of pair-wise correlation was inferred apply lm function in R package.
Relative difference of each ancestral node in the phylogenetic tree
The protein sequences at the whole transcriptomic scale of ancestral node were predicted using FASTML59. The protein alignment was from28. Gene expression abundance and morphological characteristics of all ancestral nodes were predicted by applying ape package of R which uses a maximal likelihood method. For all C4 related gene expression, protein sequences and physiological data, their values of the ancestral nodes were assigned to those of the most recent species derived from the node.
Relative difference of protein sequence at each ancestral node was inferred by comparing the sequence at this node (N) with the nearest preceding node of N (N[pre]), e.g., the number of different amino acid between N2 with N1 is the number of changed amino acid at N2. The number of different amino acid changes divided by the aligned length of the protein was calculated as relative protein difference for each gene. Relative difference of gene expression and morphology were calculated as (N -N[pre])/N[pre]. In most cases, the nearest preceding node of N[i] is N[i-1], there are two exceptions: the ancestral node of N11 is N5, and N10 is N8. One-way ANOVA analysis followed by Tukey’s Post Hoc test was used to calculate the significance of relative difference between any two ancestral nodes. P values were adjusted by Benjamin-Hochberg (BH) correction.
Supplementary Information
Acknowledgements
The authors thank Haiyang Hu and Yimin Tao for great discussion and suggestion; Lyu thanks Yongyao Zhao for persisting in encouraging her to submit this work and Xinyu Liu for technical assistance. We also thank anonymous reviewers for their kind suggestions in improving the manuscript both in writing and science. This work was sponsored by Strategic Priority Research Program of the General Project of Chinese Academy of Sciences (grant number XDB27020105), Shanghai Sailing Program [17YF421900], National Science Foundation of China [31701139 to Ming-Ju Amy Lyu, 30970213 to Xin-Guang Zhu]. This paper is dedicated to the memory of our dear colleague and friend Udo Gowik who untimely passed way.
Abbreviations
- A
CO2 assimilation rate
- AlaAT
Alanine aminotransferase
- AspAT5
Aspartate aminotransferase 5
- BSCs
Bundle sheath cells
- CET
Cyclic electron transport
- CRR1
Chlororespiratory reduction 1
- DE
Differentially expressed
- FPKM
Fragments per kilobase of transcript per million mapped reads
- GDC
Glycine decarboxylase complex
- GLYK
Glycerate kinase
- GOGAT
Glutamine synthetase and glutamine oxoglutarate aminotransferase
- GSL1
Glutamine synthetase-like 1
- MCs
Mesophyll cells
- NADP-ME
NADP-dependent malic enzyme
- NCBI
National Center for Biotechnology Information
- Ndh
NADH dehydrogenase-like
- NHD1
Sodium: hydrogen (Na + /H +) antiporter 1
- PEPC
Phosphoenolpyruvate carboxylase
- PGR5-like
Proton gradient regulation 5 like
- PIFI
Post-illumina chlorophyll fluorescence increase
- PNUE
Instantaneous photosynthetic nitrogen use efficiency
- PPCKA
PEPC protein kinase A
- PPDK-RP
PPDK regulatory protein
- PPDK
Pyruvate, orthophosphate dikinase
- PWUE
Instantaneous photosynthetic water use efficiency
- SHM
Hydroxymethyltransferase
- SRA
Sequence Read Achieve
- Γ
CO2 compensation point
Authors' Contributions
M.A.L. and X.G.Z. designed the study and wrote the main manuscript, M.A.L. did RNA-seq assembly and quantification, U.G., S.C. conducted PAML positive selection analysis. S.K., J.M.H., R.F.S., M.L., G.K.S. and P.W. wrote the section of Discussion. All authors reviewed the manuscript.
Data availability
All data generated during this study are included in this published article and its supplementary information files. Codes used during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-93381-8.
References
- 1.Pattin KA, Moore JH. Genome-wide association studies for the identification of biomarkers in metabolic diseases. Exp. Opin. Med. Diagn. 2010;4:39–51. doi: 10.1517/17530050903322245. [DOI] [PubMed] [Google Scholar]
- 2.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang X, Han B. Natural variations and genome-wide association studies in crop plants. Annu. Rev. Plant Biol. 2014;65:531–551. doi: 10.1146/annurev-arplant-050213-035715. [DOI] [PubMed] [Google Scholar]
- 4.Zhu X-G, Shan L, Wang Y, Quick WP. C4 rice—an ideal arena for systems biology research. J. Integr. Plant Biol. 2010;52:762–770. doi: 10.1111/j.1744-7909.2010.00983.x. [DOI] [PubMed] [Google Scholar]
- 5.Sage RF, Christin PA, Edwards EJ. The C4 plant lineages of planet Earth. J. Exp. Bot. 2011;62:3155–3169. doi: 10.1093/Jxb/Err048. [DOI] [PubMed] [Google Scholar]
- 6.Hatch MD. C4 photosynthesis: A unique blend of modified biochemistry, anatomy and ultrastructure. Biochem. Biophys. Acta. 1987;895:81–106. [Google Scholar]
- 7.Sage RF. The evolution of C4 photosynthesis. New Phytol. 2003;161:341–347. doi: 10.1111/j.1469-8137.2004.00974.x. [DOI] [PubMed] [Google Scholar]
- 8.Hatch MD, Slack CR. A new enzyme for the interconversion of pyruvate and phosphopyruvate and its role in the C4 dicarboxylic acid pathway of photosynthesis. Biochem. J. 1968;106:141–146. doi: 10.1042/bj1060141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson HS, Hatch MD. The C4-dicarboxylic acid pathway of photosynthesis. Identification of intermediates and products and quantitative evidence for the route of carbon flow. Biochem. J. 1969;114:127–134. doi: 10.1042/bj1140127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dengler, N. & Nelson, T. in C4 plant biology. (Sage, R. F. & Monson, R. K. eds) (Academic Press: San Diego, etc, 1999).
- 11.Hatch MD, Osmond CB. Compartmentation and transport in C4 photosynthesis. Encycl. Plant Physiol. 1976;3:144–184. [Google Scholar]
- 12.Slack CR, Hatch MD, Goodchild DJ. Distribution of enzymes in mesophyll and parenchyma-sheath chloroplasts of maize leaves in relation to the C4-dicarboxylic acid pathway of photosynthesis. Biochem. J. 1969;114:489–498. doi: 10.1042/bj1140489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sage RF, Sage TL, Kocacinar F. Photorespiration and the evolution of C4 photosynthesis. Annu. Rev. Plant Biol. 2012;63:19–47. doi: 10.1146/annurev-arplant-042811-105511. [DOI] [PubMed] [Google Scholar]
- 14.Lauterbach M, et al. Evolution of leaf anatomy in arid environments—A case study in southern African Tetraena and Roepera (Zygophyllaceae) Mol. Phylogenet. Evol. 2016;97:129–144. doi: 10.1016/j.ympev.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 15.McKown AD, Dengler NG. Key innovations in the evolution of Kranz anatomy and C4 vein pattern in Flaveria (Asteraceae) Am. J. Bot. 2007;94:382–399. doi: 10.3732/ajb.94.3.382. [DOI] [PubMed] [Google Scholar]
- 16.McKown AD, Moncalvo J-M, Dengler NG. Phylogeny of Flaveria (Asteraceae) and inference of C4 photosynthesis evolution. Am. J. Bot. 2005;92:1911–1928. doi: 10.3732/ajb.92.11.1911. [DOI] [PubMed] [Google Scholar]
- 17.Ku MS, et al. Photosynthetic characteristics of C3–C4 intermediate flaveria species: I. Leaf anatomy, photosynthetic responses to O(2) and CO(2), and activities of key enzymes in the C(3) and C(4) pathways. Plant Physiol. 1983;71:944–948. doi: 10.1104/pp.71.4.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rumpho ME, Ku MS, Cheng SH, Edwards GE. Photosynthetic characteristics of C3–C4 intermediate flaveria species: III. Reduction of photorespiration by a limited C(4) pathway of photosynthesis in Flaveria ramosissima. Plant Physiol. 1984;75:993–996. doi: 10.1104/pp.75.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards GE, Ku MS. Biochemistry of C3–C4 intermediates. In: Hatch MD, Boardman NK, editors. The Biochemistry of Plants. Academic Press; 1987. pp. 275–325. [Google Scholar]
- 20.Moore BD, Monson RK, Ku MSB, Edwards GE. Activities of principal photosynthetic and photorespiratory enzymes in leaf mesophyll and bundle sheath protoplasts from the C3–C4 Intermediate Flaveria-Ramosissima. Plant Cell Physiol. 1988;29:999–1006. [Google Scholar]
- 21.Ku MS, et al. Photosynthetic and photorespiratory characteristics of flaveria species. Plant Physiol. 1991;96:518–528. doi: 10.1104/pp.96.2.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown NJ, Parsley K, Hibberd JM. The future of C4 research–maize, Flaveria or Cleome? Trends Plant Sci. 2005;10:215–221. doi: 10.1016/j.tplants.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Gowik U, Brautigam A, Weber KL, Weber AP, Westhoff P. Evolution of C4 photosynthesis in the genus Flaveria: How many and which genes does it take to make C4? Plant Cell. 2011;23:2087–2105. doi: 10.1105/tpc.111.086264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mallman J, et al. The role of photorespiration during the evolution of C4 photosynthesis in the genus Flaveria. Elife. 2014;3:e02478. doi: 10.7554/eLife.02478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engelmann S, Blasing OE, Gowik U, Svensson P, Westhoff P. Molecular evolution of C4 phosphoenolpyruvate carboxylase in the genus Flaveria–a gradual increase from C3 to C4 characteristics. Planta. 2003;217:717–725. doi: 10.1007/s00425-003-1045-0. [DOI] [PubMed] [Google Scholar]
- 26.Westhoff P, Gowik U. Evolution of C4 phosphoenolpyruvate carboxylase. Genes and proteins: a case study with the genus Flaveria. Ann. Bot. 2004;93:13–23. doi: 10.1093/aob/mch003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engelmann S, et al. The gene for the P-subunit of glycine decarboxylase from the C4 species Flaveria trinervia: Analysis of transcriptional control in transgenic Flaveria bidentis (C4) and Arabidopsis (C3) Plant Physiol. 2008;146:1773–1785. doi: 10.1104/pp.107.114462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lyu MJ, et al. RNA-Seq based phylogeny recapitulates previous phylogeny of the genus Flaveria (Asteraceae) with some modifications. BMC Evol. Biol. 2015;15:116. doi: 10.1186/s12862-015-0399-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emms DM, Kelly S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kadereit G, Bohley K, Lauterbach M, Tefarikis DT, Kadereit JW. C3–C4 intermediates may be of hybrid origin—a reminder. New Phytol. 2017;215:70–76. doi: 10.1111/nph.14567. [DOI] [PubMed] [Google Scholar]
- 31.Peng LW, Yamamoto H, Shikanai T. Structure and biogenesis of the chloroplast NAD(P)H dehydrogenase complex. BBA-Bioenerg. 1807;945–953:2011. doi: 10.1016/j.bbabio.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 32.Mallmann J, et al. The role of photorespiration during the evolution of C4 photosynthesis in the genus Flaveria. Elife. 2014;3:e02478. doi: 10.7554/eLife.02478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogan PJ, Sage RF. Water-use efficiency and nitrogen-use efficiency of C(3) -C(4) intermediate species of Flaveria Juss. (Asteraceae) Plant Cell Environ. 2011;34:1415–1430. doi: 10.1111/j.1365-3040.2011.02340.x. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura N, Iwano M, Havaux M, Yokota A, Munekage YN. Promotion of cyclic electron transport around photosystem I during the evolution of NADP-malic enzyme-type C4 photosynthesis in the genus Flaveria. New Phytol. 2013;199:832–842. doi: 10.1111/nph.12296. [DOI] [PubMed] [Google Scholar]
- 35.Brawand D, et al. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–348. doi: 10.1038/nature10532. [DOI] [PubMed] [Google Scholar]
- 36.Warnefors M, Kaessmann H. Evolution of the correlation between expression divergence and protein divergence in mammals. Genome Biol. Evol. 2013;5:1324–1335. doi: 10.1093/Gbe/Evt093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hart JC, Ellis NA, Eisen MB, Miller CT. Convergent evolution of gene expression in two high-toothed stickleback populations. PLoS Genet. 2018;14:e1007443. doi: 10.1371/journal.pgen.1007443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 39.Sage RF, Zhu X-G. Exploiting the engine of C4 photosynthesis. J. Exp. Bot. 2011;62:2989–3000. doi: 10.1093/jxb/err179. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Long SP, Zhu XG. Elements required for an efficient NADP-malic enzyme type C4 photosynthesis. Plant Physiol. 2014;164:2231–2246. doi: 10.1104/pp.113.230284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wedding RT, Black MK, Meyer CR. Inhibition of phosphoenolpyruvate carboxylase by malate. Plant Physiol. 1990;92:456–461. doi: 10.1104/pp.92.2.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Virtanen J, Xue Z, Zhang Y. I-TASSER-MR: Automated molecular replacement for distant-homology proteins using iterative fragment assembly and progressive sequence truncation. Nucleic Acids Res. 2017;45:W429–W434. doi: 10.1093/nar/gkx349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hunt BG, Ometto L, Keller L, Goodisman MAD. Evolution at two levels in fire ants: The relationship between patterns of gene expression and protein sequence evolution. Mol. Biol. Evol. 2013;30:263–271. doi: 10.1093/Molbev/Mss234. [DOI] [PubMed] [Google Scholar]
- 44.Stitt M, Zhu X-G. The large pools of metabolites involved in intercellular metabolite shuttles in C4 photosynthesis provide enormous flexibility and robustness in a fluctuating light environment. Plant Cell Environ. 2014;2:2. doi: 10.1111/pce.12290. [DOI] [PubMed] [Google Scholar]
- 45.Paulus JK, Schlieper D, Groth G. Greater efficiency of photosynthetic carbon fixation due to single amino-acid substitution. Nat. Commun. 2013;4:1518. doi: 10.1038/ncomms2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morgan CL, Turner SR, Rawsthorne S. Coordination of the cell-specific distribution of the 4 subunits of glycine decarboxylase and of serine hydroxymethyltransferase in leaves of c3–c4 intermediate species from different genera. Planta. 1993;190:468–473. doi: 10.1007/BF00224785. [DOI] [Google Scholar]
- 47.Lundgren MR, et al. Evolutionary implications of C3–C4 intermediates in the grass Alloteropsis semialata. Plant, Cell Environ. 2016;39:1874–1885. doi: 10.1111/pce.12665. [DOI] [PubMed] [Google Scholar]
- 48.Khoshravesh R, et al. C3–C4 intermediacy in grasses: Organelle enrichment and distribution, glycine decarboxylase expression, and the rise of C2 photosynthesis. J. Exp. Bot. 2016;67:3065–3078. doi: 10.1093/jxb/erw150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schluter U, et al. Photosynthesis in C3–C4 intermediate Moricandia species. J. Exp. Bot. 2017;68:191–206. doi: 10.1093/jxb/erw391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muhaidat R, Sage TL, Frohlich MW, Dengler NG, Sage RF. Characterization of C3–C4 intermediate species in the genus Heliotropium L. (Boraginaceae): Anatomy, ultrastructure and enzyme activity. Plant Cell Environ. 2011;34:1723–1736. doi: 10.1111/j.1365-3040.2011.02367.x. [DOI] [PubMed] [Google Scholar]
- 51.Hylton CM, Rawsthorne S, Smith AM, Jones DA, Woolhouse HW. Glycine decarboxylase is confined to the bundle-sheath cells of leaves of C3–C4 intermediate species. Planta. 1988;175:452–459. doi: 10.1007/Bf00393064. [DOI] [PubMed] [Google Scholar]
- 52.Sage TL, et al. Initial events during the evolution of C4 photosynthesis in C3 species of Flaveria. Plant Physiol. 2013;163:1266–1276. doi: 10.1104/pp.113.221119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moore B, Ku MSB, Edwards G. C4 photosynthesis and light-dependent accumulation of inorganic carbon in leaves of C3–C4 and C4 Flaveria species. Austr. J. Plant Physiol. 1987;14:658–668. [Google Scholar]
- 55.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 59.Ashkenazy H, et al. FastML: A web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Res. 2012;40:W580–584. doi: 10.1093/nar/gks498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated during this study are included in this published article and its supplementary information files. Codes used during the current study are available from the corresponding author on reasonable request.