

Abstract
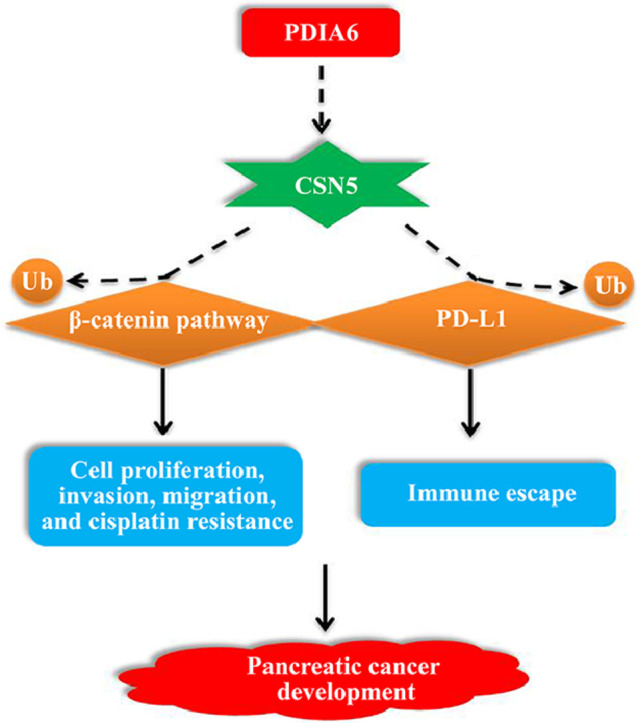
Protein Disulfide Isomerase Family A Member 6 (PDIA6) is an endoplasmic reticulum protein that is capable of catalyzing protein folding and disulfide bond formation. Abnormally elevated expression of PDIA6 has been reported to predict poor outcomes in various cancers. Herein, gain-of- and loss-of-function experiments were performed to investigate how PDIA6 participated in the carcinogenesis of pancreatic cancer (PC). By analyzing the protein expression of PDIA6 in 28 paired PC and para carcinoma specimens, we first found that PDIA6 expression was higher in PC samples. Both the overall survival and disease-free survival rates of PC patients with higher PDIA6 expression were poorer than those with lower PDIA6 (n = 178). Furthermore, knockdown of PDIA6 impaired the malignancies of PC cells — suppressed cell proliferation, invasion, migration, cisplatin resistance, and xenografted tumor growth. PDIA6-silenced PC cells were more sensitive to cytotoxic natural killer (NK) cells. Overexpression of PDIA6 had opposite effects on PC cells. Interestingly, COP9 signalosome subunit 5 (CSN5), a regulator of E3 ubiquitin ligases known to promote deubiquitination of its downstream targets, was demonstrated to interact with PDIA6, and its expression was increased in PC cells overexpressing PDIA6. Additionally, PDIA6 overexpression promoted deubiquitination of β-catenin and PD-L1 and subsequently upregulated their expression in PC cells. These alterations were partly reversed by CSN5 shRNA. Collectively, the above results demonstrate that PDIA6 contributes to PC progression, which may be associated with CSN5-regulated deubiquitination of β-catenin and PD-L1. Our findings suggest PDIA6 as a potential target for the treatment of PC.
Keyword: Pancreatic cancer, PDIA6, CSN5, β-catenin signaling pathway
Graphical Abstract
Introduction
Pancreatic cancer (PC) is regarded as one of the most common malignant tumors and is the fourth leading cause of cancer death [1]. The most common form of PC is pancreatic ductal adenocarcinoma (PDAC), which accounts for about 85% of all PC [2]. According to the GLOBOCAN 2012 estimates, the incidence and mortality of PC in China will increase year by year over the next 20 years [3]. Due to the lack of screening and early diagnosis methods, most patients are already at an advanced stage when they are diagnosed. Despite great advances have been made in cancer treatment, the 5-year survival rate for PC patients is still less than 10% [4,5]. Therefore, a better understanding of the molecular mechanisms underlying PC progression and an exploration of potential therapeutic targets are urgently needed.
Protein Disulfide Isomerase Family A, Member 6 (PDIA6) belongs to the protein disulfide isomerase (PDI) family and mainly exists in the endoplasmic reticulum [6]. PDIA6 can catalyze the formation of disulfide bonds, promote protein folding, and suppress the aggregation of unfolded substrates [6,7]. Earlier studies have shown that PDIA6 exerts a vital role in cancer progression. However, the function of PDIA6 in cancer progression is contradictory. PDIA6 plays a catalytic role in the development of several types of cancer, including non–small cell lung cancer (NSCLC), bladder cancer, and giant cell tumor [8], [9], [10]. On the contrary, Kim et al. [11] demonstrated that after inhibition of PDIA6, the migration and invasion of glioblastoma cells increased significantly. So far, the function of PDIA6 in PC progression has not been reported. The Cancer Genome Atlas (TCGA) data analyzed by GEPIA (Gene Expression Profiling Interactive Analysis, http://gepia.cancer-pku.cn/) showed that the transcripts of PDIA6 genes are more abundant in PC patients with poorer prognosis [12]. Therefore, we speculate that PDIA6 may exert a vital role in the occurrence and development of PC.
COP9 signalosome subunit 5 (CSN5) is a key subunit of the COP9 signal complex and regulates multiple cellular processes, such as proliferation and apoptosis [13,14]. Existing studies have shown that abnormally elevated expression of CSN5 in tumor tissues predicts poor outcomes in various types of cancer, including PC [15]. CSN5, a regulator of E3 ubiquitin ligases, is demonstrated to affect gene stability by regulating the ubiquitination of genes in cancer progression. Specifically, CSN5 has been found to promote hepatocellular carcinoma progression via suppressing the ubiquitination of β-catenin [16]. Furthermore, it has been reported that CSN5 can stabilize programmed death-ligand 1 (PD-L1) in colorectal cancer cells, thus participating in immune evasion of cancer cells [17]. Interestingly, by using HitPredict (http://www.hitpredict.org/) and GEPIA, we found that there was a certain correlation between expression levels of PDIA6 and CSN5 in PC. Taken together, we hypothesized that PDIA6 could interact with CSN5 to regulate its expression, thus affecting the downstream signaling and ultimately affecting PC progression.
Methods and materials
Clinical samples and cell culture
Twenty-eight patients diagnosed with PDAC in The First Affiliated Hospital of Zhengzhou University were enrolled in the present study, including 8 females and 20 males aged from 44 to 80 with a median age of 59 years old. The samples were obtained from January 2015 to May 2017. The clinic-pathology data of the PC patients were presented in Table 1.
PANC-1, BxPC-3, AsPC-1, CFPAC-1, SW1990, and MIA PaCa-2 cells and pancreatic ductal epithelial cells (hTERT-HPNE) were cultured in a humidified incubator with 5% CO2 at 37°C in RPMI-1640 medium (Gibco Life Technologies, Grand Island, NY, USA), IMDM medium (Procell, Wuhan, China), L-15 medium (Procell) or DMEM + 2.5% HS (horse serum) medium (Gibco Life Technologies) supplemented with 10% fetal bovine serum (FBS, Doublehelix, Wuhan, China). The PC cell lines were obtained from Procell, and the hTERT-HPNE cell line was purchased from Zhong Qiao Xin Zhou (Shanghai, China).
Construction of plasmids
The specific short hairpin RNAs (shRNAs) of PDIA6 were inserted into the region between HpaⅠ and XhoⅠ sites of the lentivirus (LV) plasmid vector to achieve PDIA6 knockdown. shRNA1-PDIA6, 5′…CGATACGGGATTAGAGGATTT…3′ (target sequence); shRNA2-PDIA6, 5′…CCATCGAATTTCAACCGAGAA…3′ (target sequence). shRNA-NC, 5′…TTCTCCGAACGTGTCACGT…3′. To achieve PDIA6 overexpression, the lentivirus vector Pljm1-egfp containing a full-length coding sequence of PDIA6 between the NheⅠ and AgeⅠ sites was constructed. The lentivirus vectors were purchased from Fenghui Biotechnology Co., Ltd (Changsha, China). PANC-1 and AsPC-1 cells were infected with lentivirus shRNAs-PDIA6 or its negative control (shRNA-NC), and BxPC-3 cells were infected with lentivirus-mediated PDIA6 overexpressing vectors (OV-PDIA6) or its NC (OV-NC). After 24 h, cell lines with stable low or high expression of PDIA6 were obtained by the inclusion of puromycin in the culture medium. The viral titer for lentivirus vectors was 108 TU/mL. PC cells were infected with lentiviral vectors at a multiplicity of infection (MOI) of 30.
For CSN5 overexpression in BxPC-3 cells, the full-length coding sequence of CSN5 was cloned into a pcDNA3.1 vector (Genscript, Nanjing, China) for exogenous overexpression. For CSN5 knockdown, shRNAs specifically targeting CSN5 or its NC were inserted into the pRNAH1.1 vector (Genscript) and then transfected into BxPC-3 cells. shRNA1-CSN5, 5′…GGCACTGAAACCCGAGTAAAT…3′ (target sequence); shRNA-NC2, 5′… TTCTCCGAACGTGTCACGT…3′. Cell transfection was performed following the manufacturer's instruction of Lipofectamine 3000 (Gibco Life Technologies). For β-catenin overexpression, β-catenin-S33Y mutant plasmid (Addgene, http://www.addgene.org/19286/) (resistant to proteasomal degradation [18]) and β-catenin overexpressing plasmid (pcDNA3.1, Genscript, Nanjing, China) (not resistant to degradation) were transfected into PANC-1 cells.
For cell administration, the infected cells were exposed to cisplatin (DDP) (Dalian Meilun Biotech, Dalian, China) at a concentration of one-fourth IC50 of control cells for 48 h, and at the time point, the cells were harvested for subsequent testing.
Western blot
RIPA lysis buffer mixed with PMSF was used to obtain total protein from the PC cells and tissues. The protein concentration was then measured using a bicinchoninic acid (BCA) protein assay kit (Solarbio, Beijing, China). The protein (10−20 μg) was separated on SDS-PAGE gels (8%, 10%, or 12% gels) and transferred to the polyvinylidene fluoride (PVDF) membrane. After sealing in skim milk (5%), the membranes were treated with primary antibodies overnight at 4°C. Subsequently, the membranes were treated with corresponding goat anti-mouse/rabbit HRP-conjugated secondary antibodies (Code No. SE131 and SE134, 1:5000 dilution), and those antibodies were obtained from Solarbio (http://www.solarbio.com/). An ECL reagent solution (Solarbio) was used to visualize the protein bands. The expression level of the target protein was standardized by the expression of GAPDH. The primary antibodies utilized for Western blot were shown in Table 2.
Table 1.
Clinic-pathological data of patients with pancreatic adenocarcinoma (n=28).
| Clinical features | Number | |
|---|---|---|
| Age | ≤60 years | 14 (50%) |
| >60 years | 14 (50%) | |
| Gender | Male | 20 (71%) |
| Female | 8 (29%) | |
| Tumor differentiation | Poor | 12 (43%) |
| Moderate | 16 (57%) | |
| Tumor stage | T1-2 | 2 (7%) |
| T3-4 | 26 (93%) | |
| Tumor grade | ≤IIa | 9 (32%) |
| >IIa | 19 (68%) | |
Table 2.
The primary antibodies used for Western blot.
| Antigen | Manufacturer | Host | Clonality Clone number | Dilution |
|---|---|---|---|---|
| cyclin D1 | ABclonal (Code No.A19038) | Rabbit | Monoclonal | 1:1000 |
| c-MYC | ABclonal (Code No. A1309) | Rabbit | Polyclonal | 1:500 |
| PCNA | ABclonal (Code No. A12427) | Rabbit | Monoclonal | 1:1000 |
| pro/cleaved-caspase 3 | ABclonal (Code No. A19654) | Rabbit | Monoclonal | 1:1000 |
| pro/cleaved | ABclonal (Code No. A19596) | Rabbit | Monoclonal | 1:1000 |
| -PARP β-catenin | ABclonal (Code No. A19657) | Rabbit | Monoclonal | 1:1000 |
| CSN5 | ABclonal (Code No. A4087) | Rabbit | Monoclonal | 1:500 |
| PDIA6 | ABclonal (Code No. A7055) | Rabbit | Polyclonal | 1:500 |
| MMP2 | Proteintech (Code No. 10373-2-AP) | Rabbit | Polyclonal | 1:500 |
| MMP9 | Proteintech (Code No. 10375-2-AP) | Rabbit | Polyclonal | 1:1000 |
| GAPDH | Proteintech (Code No. 60004-1-Ig) | Mouse | Monoclonal | 1:10000 |
| PD-L1 | Proteintech (Code No. 17952-1-AP) | Rabbit | Polyclonal | 1:1000 |
| p-β-catenin | Affinity (Ser33/Ser37/Thr41) (Code No. DF2989) | Rabbit | Polyclonal | 1:1000 |
CCK-8 assay
The viability of PDIA6 overexpressing cells or PDIA6-silenced cells was measured by CCK-8 assay. The PC cells were placed into 96-well plates (4000 cells per well). Cells were cultured in an incubator for 0, 24, 48, 72, or 96 h. After that, CCK-8 detection was performed.
Natural killer (NK) cells and PC cells were mixed at mixture ratios of 2:1, 1:1, and 1:2 in a 96-well plate and incubated for 48 h. Five replicates were designed for each group. Thereafter, CCK-8 detection was performed.
For CCK-8 detection, cells were treated with CCK-8 (10 μl, Beyotime, Shanghai, China) for 1 h in an incubator at 37°C with 5% CO2. A microplate reader (BioTek Instruments, Winooski, VT, USA) was used to detect the absorbance value at 450 nm.
Colony formation assay
Cells were inoculated in 35 mm petri dishes with 300 cells per plate when reached about 90% confluence. The petri dishes were placed in an incubator at 37°C with 5% CO2 for 2 weeks. After being washed, cells were treated with paraformaldehyde (4%) for 15 min and then Giemsa solution for 5 min. Finally, the colony formation rate was calculated.
Wound‑healing assay
The cells were cultured to the density of the fusion state. Wounds were made using a pipette tip (200 μl). After washed with serum-free medium, the cells of each group were placed in an incubator at 37°C supplied with 5% CO2 for 0 h and 48 h, respectively, for photo recording (100 ×, magnification).
Transwell assay
Transwell chamber coated with Matrigel was seeded in 24-well plates. In the lower chamber, the culture medium (800 μl) supplemented with 10% FBS was added. The upper chamber was added with cell suspension (200 μl), and the number of cells was 2 × 104 per well. After incubation for 48 h, the invading cells were fixed with paraformaldehyde and treated with crystal violet. Finally, the invading cells were measured under a microscope (200 ×, magnification; Olympus, Tokyo, Japan).
MTT assay
The viability of DDP-treated cells was detected by MTT assay. Briefly, cells (4 × 103/well) were plated into 96-well plates. After incubation with DDP (0, 2, 4, 8, 16, or 32 μM) for 72 h, the MTT assay was performed. Next, cells were treated with 0.5 mg/mL MTT solution (Beyotime) maintained in an incubator at 37°C for 4.5 h. After the addition of DMSO (100 μl) for solubilization of formazan, the cell viability was measured by detecting the optical density (OD) value at 570 nm by a microplate reader (BioTek Instruments).
Apoptosis analysis
An Apoptosis Detection Kit (KeyGen, Nanjing, China) was used to analyze cell apoptosis. The cells (1 × 105/well) were plated into 6-well plates and cultured for 48 h. Cells were then stained with Annexin V-PE (5 μl) and 7-AAD (5 μl) for 15 min. Cell apoptosis was analyzed using a flow cytometer (ACEA Biosciences, San Diego, CA, USA), and the apoptotic rate was calculated.
Mouse experiments
Healthy male BALB/c nude mice (16 ± 1 g) aged 6 weeks were purchased from Beijing Huafukang Biotechnology Co., Ltd. (Beijing, China). The details of housing and husbandry conditions were: 12:12 h light/dark cycle, 22 ± 1°C (temperature), and 45% to 55% (humidity). Mice were given free access to water and food. Six mice were kept in a cage. The cancer model was established after 1 week of adaptive feeding. Forty-eight mice were randomly divided into the following groups (n = 6 each group): shRNA-NC (injected with PANC-1 or AsPC-1 cells transfected with shRNA-NC), shRNA1-PDIA6 (injected with PANC-1 or AsPC-1 cells with stable knockdown of PDIA6), shRNA2-PDIA6 (injected with PANC-1 or AsPC-1 cells with stable knockdown of PDIA6), OV-NC (injected with BxPC-3 cells transfected with OV-NC), and OV-PDIA6 (injected with BxPC-3 cells with stable overexpression of PDIA6). shRNA-NC and OV-NC served as control groups. PC cells in the logarithmic growth phase (a total of 2 × 106 cells in 50 μl of Matrigel per cell type) were subcutaneously injected into the nude mice. Every 3 days, the 2 perpendicular diameters of tumors were detected, and the tumor volume was calculated. After 21 days, the mice were euthanized by intraperitoneal injection of sodium pentobarbital (200 mg/kg), and tumor tissues were collected for the following experiments.
Immunohistochemistry (IHC)
The tumor tissues were embedded and dissected into sections (5 μm). After dewaxing and hydration, the sections were treated with H2O2 (3%) for 15 min to eliminate the activity of endogenous peroxidase, followed by goat serum incubation for 15 min to prevent nonspecific binding. Immunostaining was conducted using the primary antibody purchased from ABclonal, followed by the incubation of goat anti-rabbit secondary antibody (Code No. #31460, ThermoFisher Scientific, Pittsburgh, PA, USA, 1:500 dilution). Before being counterstained with hematoxylin, sections were incubated with DAB color development reagent (Solarbio) to develop the brown reaction color. Finally, after dehydration and sealing, the sections were observed under the microscope (400 ×, magnification, Olympus). The primary antibodies used for IHC were shown in Table 3.
Table 3.
The primary antibodies used for Immunohistochemistry.
| Antigen | Manufacturer | Host | Clonality Clone number | Dilution |
|---|---|---|---|---|
| Ki67 | ABclonal | Rabbit | Polyclonal | 1:100 |
| MMP2 | (Code No. A2094)ABclonal (Code No. A11144) | Rabbit | Polyclonal | 1:100 |
| β-catenin | ABclonal (Code No. A19657) | Rabbit | Monoclonal | 1:100 |
| PD-L1 | ABclonal (Code No. A1645) | Rabbit | Polyclonal | 1:100 |
Immunofluorescence (IF)
Cell sections were fixed in paraformaldehyde (4%) for 15 min and then treated with 0.1% TritonX-100 for 30 min. The cells were sealed with goat serum (Solarbio) at room temperature for 15 min and incubated with an antibody against β-catenin (Code No. A19657, ABclonal, Wuhan, China, 1:100 dilution) at 4°C overnight. Subsequently, the sections were incubated with the secondary antibody, Cy3-labeled anti-rabbit IgG (Code No. A0516, Beyotime, 1:200 dilution) and counterstained with DAPI (Aladdin, Shanghai, China). The images were captured under a fluorescence microscope (400 ×, magnification, Olympus).
β-catenin activity detection
The β-catenin activity was analyzed using the luciferase reporter assay. In brief, cells were co-transfected with pRL-TK, M50 Super8 × TOP Flash, and shRNAs/plasmids, and luciferase activities were detected by using a Luciferase Detection Kit (Promega, Madison, WI, USA). The β-catenin activity was assessed by the ratio of Firefly luciferase activity to Renilla luciferase activity.
NK cell activation assay
NK cell activation was evaluated by detecting the concentration of Granzyme B and the pro-inflammatory cytokines MIP-1α, IFN-γ, and TNF-α. Their levels were detected by enzyme-linked immunosorbent assay (ELISA) using Human ELISA detection kits (USCN, Wuhan, China) as per the manufacturer's protocols.
Co-immunoprecipitation assay and ubiquitination assay
The co-immunoprecipitation assay was conducted to determine the relationship between PDIA6 and CSN5. The ubiquitination assay was performed to detect the ubiquitination of β-catenin and PD-L1. Briefly, total protein was obtained from PC cells as described in the Western blot section. The protein extract was incubated with 1 μg primary mouse-anti CSN5 antibody (Code No. sc-13157; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or mouse-anti PDIA6 antibody (Code No. sc-365260; Santa Cruz) or its corresponding negative control IgG overnight at 4°C. Subsequently, the antigen-antibody complexes were captured by incubation with Protein A Agarose beads (60 μl) for 2 h at 4°C. Agarose bead-antigen antibody complexes were collected by centrifugation, dissolved in 60 μl protein loading buffer, and boiled for 5 min, followed by Western blot. The primary rabbit antibodies against CSN5 and PDIA6 used for Western blot were purchased from ABclonal (https://abclonal.com.cn/) and presented in Table 2.
GST-pull down assay
Full-length cDNA of PDIA6 was synthesized by PCR amplification using primers with EcoRⅠ and XhoⅠ restriction sites. The primers were: F: 5′…AACGTCGACATGGCTCTCCTGGTGCTCGGT…3′; R: 5′…CCAGCGGCCGCCAACTCATCTTTCCCTAAGT…3′. The PDIA6 cDNA was then cloned into the pGEX-4T-1 vector (Unibio, Chongqing, China) with glutathione S-transferase (GST) tag to obtain GST-PDIA6 fusion protein. The pGEX-4T-1-PDIA6 vector was then transformed into Escherichia coli (E. coli). GST-PDIA6 was overexpressed in E. coli, and GST-agarose resin was used for the affinity purification of the GST-PDIA6 fusion protein from cell lysates. Subsequently, bait-prey protein complexes contained in eluted samples were corrected by using elution buffer and analyzed by SDS-PAGE and Western blot.
Statistical analysis
GraphPad Prism 7 software was used for data analysis. Data were expressed as mean ± standard deviation (SD). For comparisons between 2 groups, an unpaired Student's t test was performed. For comparisons among 3 or more groups, one-way ANOVA or two-way ANOVA with Tukey's post-tests were performed. P < 0.05 was considered statistically significant.
Results
PDIA6 is overexpressed in PC tissues and cells and is connected with the poor prognosis of patients with PC
At first, the PDIA6 expression in PC tissues and the adjacent normal tissues was measured. Western blot results showed that 26 of 28 pairs of PC tissues showed higher levels of PDIA6 than their adjacent non–cancer tissues (Fig. 1A and C). According to the data analyzed by GEPIA (based on RNA-seq results of tumor samples and corresponding normal samples in TCGA and the GTEx databases) [12], PC patients with high PDIA6 expression had a worse prognosis than those with low PDIA6 expression (Fig. 1C). Moreover, PDIA6 overexpression was also observed in PC cell lines (CFPAC-1, SW1990, AsPC-1, MIA PaCa-2, BxPC-3, and PANC-1) relative to the hTERT-HPNE cells (Fig. 1D). Among them, PANC-1 and AsPC-1 cells with the highest PDIA6 expression level and BxPC-3 cells with the lowest PDIA6 expression level were selected for subsequent experiments.
Fig. 1.

PDIA6 is overexpressed in PC tissues and cells and is connected with the poor prognosis of patients with PC. (A and B) Relative protein levels of PDIA6 in 28 pairs of PC tissues and paracarcinoma tissues. (C) Overall survival rate and disease-free survival rate of PC patients with high/low PDIA6 expression obtained from the TCGA database. (D) Western blot was conducted to assess PDIA6 protein levels in human PC cell lines (PANC-1, BxPC-3, AsPC-1, CFPAC-1, SW1990, and MIA PaCa-2) and human pancreatic ductal epithelial cell line hTERT-HPNE. N = 3. Data were mean ± SD. Paired t test, one-way ANOVA with Tukey's post-tests.
PDIA6 promotes the growth and metastasis of PC cells
To further investigate the function of PDIA6 in PC cells, the PC cell lines with stable overexpression/knockdown of PDIA6 were established, and the expression levels of PDIA6 were verified by Western blot (Fig. 2A). Results showed that the cell viability and colony formation were dramatically disrupted by PDIA6 silencing but promoted by PDIA6 overexpression (Fig. 2B and C). Meanwhile, PCNA, an essential protein that participates in a variety of processes of DNA metabolism [19], c-MYC and cyclin D1, drivers of cell cycle progression [20,21], were used to confirm cell proliferation. The protein expression levels of PCNA, c-MYC, and cyclin D1 were decreased upon PDIA6 knockdown but increased by PDIA6 overexpression (Fig. 2D). Similarly, the migration and invasion ability of PC cells was significantly weakened by PDIA6 silencing but enhanced by PDIA6 overexpression (Fig. 3A and B). Meanwhile, PDIA6 knockdown decreased MMP2 and MMP9 protein levels, while PDIA6 upregulation showed the opposite effects (Fig. 3C). These data suggested that PDIA6 promoted PC progression by promoting cell proliferation, invasion, and migration.
Fig. 2.

PDIA6 promotes proliferation of PC cells. (A) The knockdown/overexpression efficiency of PDIA6 in PC cells was assessed by Western blot. (B) CCK-8 detection of cell viability in cells with PDIA6 overexpression or knockdown. (C) Analysis of cell colony formation capacity in cells with PDIA6 overexpression or knockdown. (D) PCNA, c-MYC, and cyclin D1 protein levels were assessed by Western blot after PDIA6 inhibition or overexpression. N = 3. *P < 0.05, **P < 0.01, and ***P < 0.001. Data were mean ± SD. Unpaired Student's t test, one-way ANOVA and two-way ANOVA with Tukey's post-tests.
Fig. 3.

PDIA6 promotes migration and invasion of PC cells. (A and B) Cell migration abilities and invasive potential were determined in response to PDIA6 knockdown or overexpression. (C) Relative protein levels of MMP2 and MMP9 after knockdown or overexpression of PDIA6 in PC cells. N = 3. *P < 0.05, **P < 0.01, and ***P < 0.001. Data were mean ± SD. Unpaired Student's t test and one-way ANOVA with Tukey's post-tests.
PDIA6 reduces the chemotherapy sensitivity of PC cells to DDP
We next explored the effect of PDIA6 on the chemotherapy sensitivity of PC cells to DDP. As demonstrated by the MTT assay, all the cells exhibited a marked decrease in cell viability with the increase of DDP concentration, which was further enhanced by PDIA6 knockdown and attenuated by PDIA6 overexpression (Fig. 4A). Compared to relative control cells, the IC50 values of DDP were significantly lower in PDIA6-silenced cells and higher in PDIA6-overexpressing cells (Fig 4B). DDP-induced apoptosis of PC cells was enhanced by PDIA6 downregulation and weakened by PDIA6 overexpression (Fig. 4C). Western blot analysis showed that DDP administration increased cleaved/pro-caspase 3/PARP ratios (apoptosis markers), and the ratios were further increased by PDIA6 silencing but decreased by PDIA6 overexpression (Fig. 4D). These findings indicated that PDIA6 expression was negatively associated with the chemotherapy sensitivity of PC cells to DDP.
Fig. 4.

PDIA6 reduces the chemotherapy sensitivity of PC cells. (A) The cell viability of PDIA6-silenced or -overexpressing cells treated with different concentrations of DDP (0, 2, 4, 8, 16, and 32 μM) was determined by MTT assay. (B) The IC50 value of DDP was calculated in cells with PDIA6 overexpression or knockdown. (C) The apoptotic rate was detected by flow cytometry in PDIA6-silenced or -overexpressing cells treated with or without DDP. (D) Western blot assays detected the protein expression of pro/cleaved caspase 3 and pro/cleaved PARP in PDIA6-downregulated or -overexpressing cells in the presence or absence of DDP. N = 3. *P < 0.05, **P < 0.01, and ***P < 0.001. Data were mean ± SD. Unpaired Student's t test and one-way ANOVA with Tukey's post-tests.
PDIA6 promotes tumor growth in vivo
To determine whether PDIA6 influences tumor growth in vivo, sh-PDIA6 or OV-PDIA6 stably transfected cells were injected into mice. After injection for 21 days, the volume and weight of tumors in mice bearing PDIA6-silenced cells were markedly decreased in comparison to mice bearing control cells (Fig. 5A-C). Conversely, the volume and weight of tumors in nude mice bearing PDIA6-overexpressing cells were markedly increased (Fig. 5A-C). In addition, the protein levels of PDIA6, c-MYC, PCNA, MMP2, MMP9, PD-L1, and β-catenin were notably reduced in tumor tissues of mice bearing PDIA6-silenced cells, while overexpression of PDIA6 had the opposite effects (Fig. 5D). The changes in cell proliferation, migration, cellular immunity, and β-catenin pathway in tumor tissues were further confirmed by IHC staining (SF. 1A). Moreover, results of the TUNEL assay showed that PDIA6 knockdown significantly increased the cell apoptosis in tumor tissues, whereas PDIA6 overexpression decreased the cell apoptosis (SF. 1B). These data revealed that PDIA6 played an important role in tumor growth in vivo, in which the β-catenin signaling pathway and immune evasion might be involved.
Fig. 5.

PDIA6 promotes tumor growth in vivo. (A-C) The images, volume, and weight of tumor tissues were shown after mice were injected with PC cells with stable high/low expression of PDIA6. (D) PDIA6, c-MYC, PCNA, MMP2, MMP9, PD-L1, and β-catenin protein levels in tumor tissues were measured by Western blot. N = 6. *P < 0.05, **P < 0.01, and ***P < 0.001. Data were mean ± SD. Unpaired Student's t test and one-way ANOVA and two-way ANOVA with Tukey's post-tests.
PDIA6 promotes PC cells to evade immune surveillance
To explore the role of PDIA6 in immune escape, NK-92 cells were used as the effector cell population. As a marker of immune escape, PD-L1 protein level was significantly decreased by PDIA6 silencing but increased by PDIA6 overexpression (Fig. 6A). After being co-cultured with NK-92 cells, the cell viability of PDIA6-silenced or -overexpressing PC cells were detected by MTT assay to assess the NK-92 cell cytotoxicity. The results showed that knockdown of PDIA6 sensitized PC cells to NK-92 cell cytolysis, while PDIA6 overexpression reduced the sensitivity of PC cells to NK-92 cell cytotoxicity (Fig. 6B). Granzyme B, MIP-1α, IFN-γ, and TNF-α are part of the innate immune response and are mainly produced by NK and natural killer T (NKT) cells [22,23]. Interestingly, it was shown that PDIA6 knockdown alone had no significant effect on Granzyme B, MIP-1α, IFN-γ, and TNF-α levels in PC cells. After co-incubation with NK-92 cells, we observed a sharp increase in the concentration of Granzyme B, MIP-1α, IFN-γ, and TNF-α in co-culture supernatants when PDIA6 was downregulated, while PDIA6 overexpression dramatically decreased the release of these cytokines (Fig. 6C). These data supported that PDIA6 reduced the susceptibility of PC cells to NK cell-mediated cytotoxicity.
Fig. 6.

PDIA6 promotes PC cells to evade immune surveillance. (A) PD-L1 protein levels were detected through western blot experiments after the downregulation or overexpression of PDIA6. (B) The cytolytic activity of NK-92 cells against PC cells with PDIA6 knockdown or overexpression was assessed using the CCK-8 assay. (C) Effects of PDIA6 knockdown or overexpression on the concentration of Granzyme B, MIP-1α, IFN-γ, and TNF-α in co-culture supernatants of NK cells and PC cells. N = 3. *P < 0.05, **P < 0.01, and ***P < 0.001. Data were mean ± SD. Unpaired Student's t test and one-way ANOVA and two-way ANOVA with Tukey's post-tests.
PDIA6 activates the β-catenin pathway by regulating β-catenin stability
We further investigated the underlying mechanism of PDIA6 in PC progression. As shown by results from Western blot analysis, the protein level of β-catenin in PDIA6-silenced cells was significantly decreased, and its phosphorylation level was increased compared with those in the control group, while PDIA6 overexpression had the opposite effects (Fig. 7A). IF staining indicated that the translocation of β-catenin to the nucleus was markedly inhibited by PDIA6 downregulation and promoted by PDIA6 overexpression (Fig. 7B). In parallel, the β-catenin activity was significantly decreased in PDIA6-silenced cells, while PDIA6 overexpression increased the β-catenin activity (Fig. 7C). The changes of the β-catenin pathway in PDIA6 or CSN5-silenced cells treated with proteasome inhibitor, MG132 were also measured. As presented in SF. 2A-D, PDIA6 or CSN5 knockdown significantly reduced β-catenin protein levels and activity but increased its phosphorylated form in the DMSO-treated cells. However, after the addition of MG132, β-catenin protein levels, its phosphorylation level, and activity were markedly increased (SF. 2A-D). These results suggested that PDIA6 activated the β-catenin pathway, which might be associated with the stability of β-catenin.
Fig. 7.

PDIA6 activates the β-catenin pathway. (A) P-β-catenin (Ser33/Ser37/Thr41) and β-catenin protein expression was determined by Western blot after the downregulation or overexpression of PDIA6. (B) Immunofluorescence analysis of β-catenin expression and location in PDIA6-downregulated or -overexpressing cells. (C) The β-catenin activity was assessed by luciferase reporter assay after the downregulation or overexpression of PDIA6. (D-F) The effects of β-catenin overexpression on viability, migration, and invasion were detected in PDIA6-downregulated cells. (G) The c-MYC, PCNA, MMP2, and MMP9 protein expression was detected in PDIA6-downregulated cells transfected with β-catenin overexpression plasmids. N = 3. *P < 0.05, **P < 0.01, and ***P < 0.001. Data were mean ± SD. Unpaired Student's t test and one-way ANOVA and two-way ANOVA with Tukey's post-tests.
To confirm this possibility, a β-catenin-S33Y mutant plasmid, resistant to ubiquitin degradation [18], was used to show whether the increase in the stability and expression of β-catenin reversed the effects of PDIA6 knockdown on PC cells. It was shown that the transfection of β-catenin-S33Y mutant plasmid increased the viability, invasion, and migration capability of PDIA6-silenced cells (Fig. 7D-F). Accordingly, the protein levels of c-MYC, PCNA, MMP2, and MMP9 were visibly increased in PDIA6-silenced cells transfected with β-catenin-S33Y plasmids compared with cells transfected with empty vectors (Fig. 7G). Meanwhile, after transfection of the β-catenin overexpressing plasmid, which was not resistant to degradation, phenotypic changes of PDIA6 under-expressing cells were also detected in the presence of MG132. The results showed that β-catenin overexpression reversed the effects of PDIA6 knockdown on cellular malignant phenotype, and MG132 treatment further enhanced the effects mediated by β-catenin overexpression (SF. 2E-G). However, compared with the β-catenin-S33Y plasmid, the β-catenin overexpressing plasmid, which was not resistant to degradation, has a weaker effect (Fig. 7D-F and SF. 2E-G). These data suggested that PDIA6 might activate the β-catenin pathway by regulating the stability of β-catenin, and PDIA6 promoted the development of PC through the β-catenin signaling.
PDIA6 inhibits the ubiquitination of β-catenin and PD-L1 by up-regulating the expression of CSN5
The mechanism by which PDIA6 regulated the expression of PD-L1 and β-catenin was further explored. As presented in Fig. 8A, PDIA6 knockdown enhanced the ubiquitination of β-catenin and PD-L1, whereas PDIA6 overexpression decreased their ubiquitination. Moreover, the decreased ubiquitination of β-catenin and PD-L induced by PDIA6 overexpression were reversed by CSN5 inhibition (Fig. 8B). Furthermore, results of co-immunoprecipitation detection showed that the PDIA6 protein was pulled down by the anti-CSN5 antibody, and CSN5 was also co-immunoprecipitated with PDIA6 by an anti-PDIA6 antibody (Figure 8C), indicating that there was an interaction between PDIA6 and CSN5. GST-pull down assay also confirmed the interaction between PDIA6 and CSN5 (Figure. 8D). Western blot and RT-PCR analysis further confirmed that PDIA6 silencing decreased CSN5 expression at both protein and mRNA levels, whereas PDIA6 overexpression exerted the opposite effects (Fig. 8E and SF. 3C). Those data indicated that PDIA6 promoted CSN5 protein expression and further inhibited the ubiquitination of β-catenin and PD-L1.
Fig. 8.

PDIA6 inhibits the ubiquitination degradation of β-catenin and PD-L1 by up-regulating CSN5 expression. (A) Co-IP and Western blot were used to detect the ubiquitination levels of β-catenin and PD-L1 after the knockdown or overexpression of PDIA6. (B) The ubiquitination levels of β-catenin and PD-L1 were detected in PDIA6-overexpressing cells transfected with sh-CSN5. (C) Co-IP assay and Western blot were conducted to detect the relationship between PDIA6 and CSN5. (D) GST-pull down assay and Western blot was performed to determine the interaction between PDIA6 and CSN5. (E) CSN5 protein levels were assessed through western blot experiments after the downregulation or overexpression of PDIA6. N = 3. **P < 0.01, and ***P < 0.001. Data were mean ± SD. Unpaired Student's t test and one-way ANOVA with Tukey's post-tests.
PDIA6 promotes PC progression by up-regulating the expression of CSN5
The overexpression efficiency of CSN5 in BxPC-3 cells was verified by Western blot (SF. 4). As presented in Fig. 9A, CSN5 overexpression decreased the phosphorylation of β-catenin but increased β-catenin and PD-L1 levels. Decreased β-catenin and PD-L1 ubiquitination were observed in CSN5-overexpressing cells (Fig. 9B). Whether CSN5 is involved in PDIA6-mediated cellular function was further examined by a series of rescue experiments. It was shown that PDIA6 overexpression significantly increased the cell viability and capability of invasion and migration of PC cells, which were offset by CSN5 downregulation (Fig. 9C-D). Additionally, the increased β-catenin and PD-L1 protein levels and the decreased p-β-catenin level induced by PDIA6 overexpression were also reversed by CSN5 silencing (Fig. 9E). Overall, these data suggested that PDIA6 participated in the development of PC by interacting with CSN5.
Fig. 9.

PDIA6 promotes the development of PC by up-regulating CSN5 expression. (A) Relative protein levels of p-β-catenin (Ser33/Ser37/Thr41), β-catenin, and PD-L1 in BxPC-3 cells after the overexpression of CSN5. (B) Co-IP combined with Western blot was performed to measure the ubiquitination levels of β-catenin and PD-L1 in CSN5-overexpressing cells. (C-D) The cell viability, migration, and invasion of BxPC-3 cells were detected after the PDIA6-overexpressing cells were treated with or without CSN5 shRNA. (E) The protein levels of p-β-catenin (Ser33/Ser37/Thr41), β-catenin, and PD-L1 were measured by Western blot in the PDIA6-overexpressing cells treated with or without CSN5 shRNA. N = 3. *P < 0.05, **P < 0.01, and ***P < 0.001. #P < 0.05 and ###P < 0.001. Data were mean ± SD. Unpaired Student's t test and one-way ANOVA and two-way ANOVA with Tukey's post-tests.
Discussion
The PDI family contains 21 members with different structures and functions [24]. It was reported that members of the PDI protein family were overexpressed in cancer progression, including brain and other central nervous system cancers and male germ cell tumors [25]. PDIA6, one of the PDI family members, was proved to be overexpressed in NSCLC, and higher PDIA6 expression was correlated with a poorer prognosis [8]. Similar to previous studies [9,26], the present study demonstrated that PDIA6 was positively associated with the growth, metastasis, and sensitivity to cisplatin of PC cells. Moreover, bioinformatics analysis showed that PC patients with high PDIA6 expression had a poor prognosis. Altogether, we demonstrate that PDIA6 promotes the development of PC.
NK cells are large granular lymphocytes derived from bone marrow and act as a cancer cell killer [27,28]. When NK cells are activated, cytotoxic particles containing perforin and various Granzyme enzymes, such as Granzyme B, are released, leading to target cell perforation, then the permeable Granzyme induces cell apoptosis and death [29,30]. Another mechanism by which NK cells work is secreting a variety of cytokines and chemokines, including MIP-1α, IFN-γ, and TNF-α [31]. Herein, we found that these factors were affected by PDIA6 expression only after PC cells were co-incubated with NK-92 cells. The findings suggest that PDIA6 may not affect the release of immune factors from PC cells themselves, but affects the binding of cancer cells to NK cells and thus stimulates NK cell activation. Our results are similar to previous studies [32].
CSN5 is a critical subunit of the COP9 signalosome and is essentially involved in diverse types of cancer, including PC [15]. It was analyzed by GEPIA that CSN5 was highly expressed in PC tissues, and the 5-year survival rate of PC patients with high CSN5 expression was lower than those with lower CSN5. Additionally, high expression of CSN5 was positively correlated with TNM staging and distant metastasis of patients with PC [15]. Overall, these results suggest that CSN5 may be a prognostic marker in PC. In the present study, we proved that PDIA6 upregulated CSN5 expression. Rescue experiments further indicated that CSN5 mediated the regulation of PDIA6 in PC progression. For instance, the promoting effect of PDIA6 overexpression on cell viability was offset by CSN5 silencing. PDIA6, known as endoplasmic reticulum sulfhydryl disulfide oxidoreductase, is involved in the formation of disulfide bonds [33]. The formation of disulfide bonds is an important post-translational modification of proteins and is also a key step for the correct folding of new peptides in the endoplasmic reticulum [24,34]. Based on the function of PDIA6, PDIA6 may affect the expression of CSN5 by regulating the formation of disulfide bonds in CSN5 and further increasing the correct folding of mature CSN5 proteins. Interestingly, PDIA6 was also proved to affect the expression of CSN5 at the mRNA level, which provided the possibility for PDIA6 to regulate the transcription of CSN5.
CSN5 has been reported to interact with multiple signaling molecules and regulate various cellular processes, including cell proliferation and apoptosis [35]. In addition, CSN5 exerts deubiquitination activity. For instance, the deubiquitination of HSP70 and Snail induced by CSN5 regulated exosomal protein sorting and enhanced the invasion and migration of cancer cells [36,37]. CSN5 could directly bind to FOXM1 and reduced its ubiquitination to enhance the protein stability of FOXM1 [15]. Furthermore, CSN5 was reported to inhibit the ubiquitination of PD-L1 and β-catenin [16,38], which was confirmed in the present study. β-catenin that is phosphorylated at residues Ser33/Ser37/Thr41 is ultimately recognized by the β-TrCP E3 ubiquitin–ligase complex, ubiquitinylated, and rapidly degraded by the 26S proteasome [39]. Previous studies indicated that CSN5 depletion in K562 cells resulted in a modest reduction of β-TrCP, including mRNA and protein levels [40]. Herein, we demonstrate that PDIA6 may also regulate the β-catenin pathway through the CSN5/β-TrCP axis.
The β-catenin signaling is an evolutionally conserved general pathway and regulates embryonic development, tissue dynamic balance, and a variety of human diseases [41]. Abnormal activation of the β-catenin pathway leads to the accumulation of β-catenin in the nucleus and further promotes the transcription of many oncogenes, such as c-MYC and cyclin D1 [42]. It was reported that the activation of β-catenin signaling led to the treatment resistance in patients with PC and drove PC progression [43 44]. Herein, we demonstrate that PDIA6 activates the β-catenin signaling through upregulating CSN5 and further promoting PC progression. Furthermore, β-catenin was reported to increase nuclear factor kappa B (NF-κB) activity in colorectal cancer cells [45]. NF-κB mediated induction of membrane-type 1 matrix metalloproteinase (MT1-MMP) in human skin [46]. Previous studies indicated that MT1-MMP induced the cleavage of pro MMP2 and pro MMP9 [47]. Therefore, the regulatory effect of β-catenin signaling on cell invasion may be mediated by NF-κB. Besides, DDP, a commonly used chemotherapeutic agent for solid tumors, is effective as a single agent or in combination with other drugs for the treatment of cancers [48]. However, DDP is known to be susceptible to drug resistance. The present study indicates that PDIA6/CSN5/β-catenin axis regulates the sensitivity of PC cells to DDP, which provides new ideas for the clinical treatment of PC patients. Furthermore, the activation of the β-catenin signaling was proved to promote resistance to gemcitabine in PC cell lines [49]. These findings suggest that PDIA6 may also affect the sensitivity of PC cells to gemcitabine, which may be achieved by regulating the β-catenin pathway.
PD-L1 is the ligand of PD-1, a receptor expressed on T cells to restrain effector T-cell functions. By interacting with PD-1, PD-L1 could help tumor cells evading the anti-tumor immune responses. PD-L1 was up-regulated in tumor cells, and the upregulation of PD-L1 led to effective immunosuppression and tumor immune escape [50]. PD-L1 expression in cancer cells resulted in reduced NK cell responses and the generation of more aggressive tumors in vivo [51]. Therefore, the effect of PDIA6 on NK cell activation may be mediated by PD-L1. Overall, we demonstrate that PDIA6 may promote the immune escape of PC cells via increasing PD-L1 expression, and this promotion may be mediated by CSN5. Of note, previous studies proved that the activation of the β-catenin pathway in cancer cells induced PD-L1 expression [52,53]. The findings above demonstrate that CSN5 may not only promote the immune escape of PC cells by directly regulating PD-L1 expression but also increase the expression of PD-L1 by indirectly activating the β-catenin pathway.
Collectively, we demonstrated that PDIA6 promoted the development of PC via 2 pathways. The first pathway is that PDIA6 suppressed the ubiquitination of β-catenin by up-regulating CSN5 and further enhanced the proliferation, migration, invasion, and chemotherapy resistance of PC cells. Another pathway is that the upregulation of CSN5 increased the stability of PD-L1 through the deubiquitination of PD-L1, thereby promoted the immune evasion of PC cells. However, as previous reports have shown, CSN5 regulates the ubiquitination of multiple downstream genes. Therefore, the regulatory effect of CSN5 on the β-catenin pathway and PD-L1 discovered in this study may be one of the important pathways for the PDIA6/CSN5 axis to participate in the development of PC.
Ethics approval and consent to participate
The present study was under the approval of the Ethics Committee of Zhengzhou University. All the enrolled patients signed the informed consent for the acquisition of tissue samples and the study was performed in accordance with the Declaration of Helsinki. The animal experiments were carried out according to the National Institutes of Health Guide for the care and use of laboratory animals.
Consent for publication
Not applicable.
Data availability
The data will be made available from the corresponding author on reasonable request.
Authors' contributions
YM conceived and designed the experiments and wrote the paper. PX, ZW, and JX performed the experiments and analyzed the data. LZ and YJ contributed reagents/materials/analysis tools.
Acknowledgments
Not applicable.
Footnotes
Competing interests: The authors declare no competing interests.
Funding information: Not applicable.
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.neo.2021.07.004.
Appendix. Supplementary materials
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin;69(1):7-34 doi:10.3322/caac.21551. [DOI] [PubMed]
- 2.Elaileh A, Saharia A, Potter L, Baio F, Ghafel A, Abdelrahim M, Heyne K. Promising new treatments for pancreatic cancer in the era of targeted and immune therapies. Am J Cancer Res. 2019;9(9):1871–1888. [PMC free article] [PubMed] [Google Scholar]
- 3.Lin QJ, Yang F, Jin C, Fu DL. Current status and progress of pancreatic cancer in China. World J Gastroenterol. 2015;21(26):7988–8003. doi: 10.3748/wjg.v21.i26.7988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol. 2016;22(44):9694–9705. doi: 10.3748/wjg.v22.i44.9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jooste V, Dejardin O, Bouvier V, Arveux P, Maynadie M, Launoy G, Bouvier AM. Pancreatic cancer: Wait times from presentation to treatment and survival in a population-based study. Int J Cancer. 2016;139(5):1073–1080. doi: 10.1002/ijc.30166. [DOI] [PubMed] [Google Scholar]
- 6.Kikuchi M, Doi E, Tsujimoto I, Horibe T, Tsujimoto Y. Functional analysis of human P5, a protein disulfide isomerase homologue. J Biochem. 2002;132(3):451–455. doi: 10.1093/oxfordjournals.jbchem.a003242. [DOI] [PubMed] [Google Scholar]
- 7.Wang L, Wang X, Wang CC. Protein disulfide-isomerase, a folding catalyst and a redox-regulated chaperone. Free Radic Biol Med. 2015;83:305–313. doi: 10.1016/j.freeradbiomed.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 8.Bai Y, Liu X, Qi X, Liu X, Peng F, Li H, Fu H, Pei S, Chen L, Chi X. PDIA6 modulates apoptosis and autophagy of non-small cell lung cancer cells via the MAP4K1/JNK signaling pathway. EBioMedicine. 2019;42:311–325. doi: 10.1016/j.ebiom.2019.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng HP, Liu Q, Li Y, Li XD, Zhu CY. The inhibitory effect of PDIA6 downregulation on bladder cancer cell proliferation and invasion. Oncol Res. 2017;25(4):587–593. doi: 10.3727/096504016X14761811155298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herr I, Sahr H, Zhao Z, Yin L, Omlor G, Lehner B, Fellenberg J. MiR-127 and miR-376a act as tumor suppressors by in vivo targeting of COA1 and PDIA6 in giant cell tumor of bone. Cancer Lett. 2017;409:49–55. doi: 10.1016/j.canlet.2017.08.029. [DOI] [PubMed] [Google Scholar]
- 11.Kim TW, Ryu HH, Li SY, Li CH, Lim SH, Jang WY, Jung S. PDIA6 regulation of ADAM17 shedding activity and EGFR-mediated migration and invasion of glioblastoma cells. J Neurosurg. 2017;126(6):1829–1838. doi: 10.3171/2016.5.JNS152831. [DOI] [PubMed] [Google Scholar]
- 12.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan Y, Yang H, Claret FX. Emerging roles of Jab1/CSN5 in DNA damage response, DNA repair, and cancer. Cancer Biol Ther. 2014;15(3):256–262. doi: 10.4161/cbt.27823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cope GA, Deshaies RJ. COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell. 2003;114(6):663–671. doi: 10.1016/s0092-8674(03)00722-0. [DOI] [PubMed] [Google Scholar]
- 15.Mao L, Le S, Jin X, Liu G, Chen J, Hu J. CSN5 promotes the invasion and metastasis of pancreatic cancer by stabilization of FOXM1. Exp Cell Res. 2019;374(2):274–281. doi: 10.1016/j.yexcr.2018.10.012. [DOI] [PubMed] [Google Scholar]
- 16.Liu H, Hu J, Pan H, Luo D, Huang M, Xu W. CSN5 Promotes Hepatocellular Carcinoma Progression by SCARA5 Inhibition Through Suppressing beta-Catenin Ubiquitination. Dig Dis Sci. 2018;63(1):155–165. doi: 10.1007/s10620-017-4855-9. [DOI] [PubMed] [Google Scholar]
- 17.Liu C, Yao Z, Wang J, Zhang W, Yang Y, Zhang Y, Qu X, Zhu Y, Zou J, Peng S. Macrophage-derived CCL5 facilitates immune escape of colorectal cancer cells via the p65/STAT3-CSN5-PD-L1 pathway. Cell Death Differ. 2020;27(6):1765–1781. doi: 10.1038/s41418-019-0460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sadot E, Conacci-Sorrell M, Zhurinsky J, Shnizer D, Lando Z, Zharhary D, Kam Z, Ben-Ze’ev A, Geiger B. Regulation of S33/S37 phosphorylated beta-catenin in normal and transformed cells. J Cell Sci. 2002;115(Pt 13):2771–2780. doi: 10.1242/jcs.115.13.2771. [DOI] [PubMed] [Google Scholar]
- 19.Cardano M, Tribioli C, Prosperi E. Targeting proliferating cell nuclear antigen (PCNA) as an effective strategy to inhibit tumor cell proliferation. Curr Cancer Drug Targets. 2020;20(4):240–252. doi: 10.2174/1568009620666200115162814. [DOI] [PubMed] [Google Scholar]
- 20.Lemaitre JM, Buckle RS, Mechali M. c-Myc in the control of cell proliferation and embryonic development. Adv Cancer Res. 1996;70:95–144. doi: 10.1016/s0065-230x(08)60873-8. [DOI] [PubMed] [Google Scholar]
- 21.Tchakarska G, Sola B. The double dealing of cyclin D1. Cell Cycle. 2020;19(2):163–178. doi: 10.1080/15384101.2019.1706903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 23.Mulherkar R, Karabudak A, Ginwala R, Huang X, Rowan A, Philip R, Murphy EL, Clements D, Ndhlovu LC, Khan ZK. In vivo and in vitro immunogenicity of novel MHC class I presented epitopes to confer protective immunity against chronic HTLV-1 infection. Vaccine. 2018;36(33):5046–5057. doi: 10.1016/j.vaccine.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galligan JJ, Petersen DR. The human protein disulfide isomerase gene family. Hum Genomics. 2012;6:6. doi: 10.1186/1479-7364-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu S, Sankar S, Neamati N. Protein disulfide isomerase: a promising target for cancer therapy. Drug Discov Today. 2014;19(3):222–240. doi: 10.1016/j.drudis.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 26.Yan C, Song X, Wang S, Wang J, Li L. Knockdown of PDIA6 inhibits cell proliferation and enhances the chemosensitivity in gastric cancer cells. Cancer Manag Res. 2020;12:11051–11062. doi: 10.2147/CMAR.S267711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008;27(45):5932–5943. doi: 10.1038/onc.2008.267. [DOI] [PubMed] [Google Scholar]
- 28.Smyth MJ, Hayakawa Y, Takeda K, Yagita H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat Rev Cancer. 2002;2(11):850–861. doi: 10.1038/nrc928. [DOI] [PubMed] [Google Scholar]
- 29.Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol. 2003;3(5):361–370. doi: 10.1038/nri1083. [DOI] [PubMed] [Google Scholar]
- 30.Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006;6(12):940–952. doi: 10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 31.Dorner BG, Smith HR, French AR, Kim S, Poursine-Laurent J, Beckman DL, Pingel JT, Kroczek RA, Yokoyama WM. Coordinate expression of cytokines and chemokines by NK cells during murine cytomegalovirus infection. J Immunol. 2004;172(5):3119–3131. doi: 10.4049/jimmunol.172.5.3119. [DOI] [PubMed] [Google Scholar]
- 32.AlHossiny M, Luo L, Frazier WR, Steiner N, Gusev Y, Kallakury B, Glasgow E, Creswell K, Madhavan S, Kumar R. Ly6E/K Signaling to TGFbeta promotes breast cancer progression, immune escape, and drug resistance. Cancer Res. 2016;76(11):3376–3386. doi: 10.1158/0008-5472.CAN-15-2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Appenzeller-Herzog C, Ellgaard L. The human PDI family: versatility packed into a single fold. Biochim Biophys Acta. 2008;1783(4):535–548. doi: 10.1016/j.bbamcr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 34.Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11(11):2807–2850. doi: 10.1089/ARS.2009.2466. [DOI] [PubMed] [Google Scholar]
- 35.Wang L, Zheng JN, Pei DS. The emerging roles of Jab1/CSN5 in cancer. Med Oncol. 2016;33(8):90. doi: 10.1007/s12032-016-0805-1. [DOI] [PubMed] [Google Scholar]
- 36.Liu Y, Shah SV, Xiang X, Wang J, Deng ZB, Liu C, Zhang L, Wu J, Edmonds T, Jambor C. COP9-associated CSN5 regulates exosomal protein deubiquitination and sorting. Am J Pathol. 2009;174(4):1415–1425. doi: 10.2353/ajpath.2009.080861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15(5):416–428. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, Chang SS, Lin WC, Hsu JM, Hsu YH. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30(6):925–939. doi: 10.1016/j.ccell.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hart M, Concordet JP, Lassot I, Albert I, del los Santos R, Durand H, Perret C, Rubinfeld B, Margottin F, Benarous R. The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr Biol. 1999;9(4):207–210. doi: 10.1016/s0960-9822(99)80091-8. [DOI] [PubMed] [Google Scholar]
- 40.Pearce C, Hayden RE, Bunce CM, Khanim FL. Analysis of the role of COP9 Signalosome (CSN) subunits in K562; the first link between CSN and autophagy. BMC cell biology. 2009;10:31. doi: 10.1186/1471-2121-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 42.Shang S, Hua F, Hu ZW. The regulation of beta-catenin activity and function in cancer: therapeutic opportunities. Oncotarget. 2017;8(20):33972–33989. doi: 10.18632/oncotarget.15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ram Makena M, Gatla H, Verlekar D, Sukhavasi S, M KP, K CP. Wnt/beta-catenin signaling: the culprit in pancreatic carcinogenesis and therapeutic resistance. Int J Mol Sci. 2019;20(17) doi: 10.3390/ijms20174242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sano M, Driscoll DR, DeJesus-Monge WE, Quattrochi B, Appleman VA, Ou J, Zhu LJ, Yoshida N, Yamazaki S, Takayama T. Activation of WNT/beta-catenin signaling enhances pancreatic cancer development and the malignant potential via up-regulation of Cyr61. Neoplasia. 2016;18(12):785–794. doi: 10.1016/j.neo.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schon S, Flierman I, Ofner A, Stahringer A, Holdt LM, Kolligs FT, Herbst A. Beta-catenin regulates NF-kappaB activity via TNFRSF19 in colorectal cancer cells. Int J Cancer. 2014;135(8):1800–1811. doi: 10.1002/ijc.28839. [DOI] [PubMed] [Google Scholar]
- 46.Han YP, Tuan TL, Wu H, Hughes M, Garner WL. TNF-alpha stimulates activation of pro-MMP2 in human skin through NF-(kappa)B mediated induction of MT1-MMP. J Cell Sci. 2001;114(Pt 1):131–139. doi: 10.1242/jcs.114.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Z, Takino T, Endo Y, Sato H. Activation of MMP-9 by membrane type-1 MMP/MMP-2 axis stimulates tumor metastasis. Cancer Sci. 2017;108(3):347–353. doi: 10.1111/cas.13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cisplatin Ghosh S. The first metal based anticancer drug. Bioorg Chem. 2019;88 doi: 10.1016/j.bioorg.2019.102925. [DOI] [PubMed] [Google Scholar]
- 49.Nagano H, Tomimaru Y, Eguchi H, Hama N, Wada H, Kawamoto K, Kobayashi S, Mori M, Doki Y. MicroRNA-29a induces resistance to gemcitabine through the Wnt/beta-catenin signaling pathway in pancreatic cancer cells. Int J Oncol. 2013;43(4):1066–1072. doi: 10.3892/ijo.2013.2037. [DOI] [PubMed] [Google Scholar]
- 50.Munir S, Andersen GH, Met O, Donia M, Frosig TM, Larsen SK, Klausen TW, Svane IM, Andersen MH. HLA-restricted CTL that are specific for the immune checkpoint ligand PD-L1 occur with high frequency in cancer patients. Cancer Res. 2013;73(6):1764–1776. doi: 10.1158/0008-5472.CAN-12-3507. [DOI] [PubMed] [Google Scholar]
- 51.Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, Azimi CS, Scheer AK, Randolph HE, Thompson TW. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest. 2018;128(10):4654–4668. doi: 10.1172/JCI99317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gan S, Ye J, Li J, Hu C, Wang J, Xu D, Pan X, Chu C, Chu J, Zhang J. LRP11 activates beta-catenin to induce PD-L1 expression in prostate cancer. J Drug Target. 2020;28(5):508–515. doi: 10.1080/1061186X.2019.1687710. [DOI] [PubMed] [Google Scholar]
- 53.Ruan Z, Liang M, Lai M, Shang L, Deng X, Su X. KYA1797K down-regulates PD-L1 in colon cancer stem cells to block immune evasion by suppressing the beta-catenin/STT3 signaling pathway. Int Immunopharmacol. 2020;78 doi: 10.1016/j.intimp.2019.106003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data will be made available from the corresponding author on reasonable request.