

Abstract
Human African trypanosomiasis (HAT) is a neglected tropical disease caused by infection with either of two subspecies of the parasite Trypanosoma brucei. Due to a lack of economic incentive to develop new drugs, current treatments have severe limitations in terms of safety, efficacy, and ease of administration. In an effort to develop new HAT therapeutics, we report the structure-activity relationships around T. brucei for a series of benzoxazepinoindazoles previously identified through a high-throughput screen of human kinase inhibitors, and the subsequent in vivo experiments for HAT. We identified compound 18, which showed an improved kinase selectivity profile and acceptable pharmacokinetic parameters, as a promising lead. Although treatment with 18 cured 60% of mice in a systemic model of HAT, the compound was unable to clear parasitemia in a CNS model of the disease. We also report the results of cross-screening these compounds against T. cruzi, L. donovani and S. mansoni.
Graphical Abstract
Introduction
Human African trypanosomiasis (HAT) is a parasitic infection caused by two subspecies of the organism Trypanosoma brucei (T.b. gambiense and T.b. rhodesiense). Prevalent in sub-Saharan Africa, HAT is one of 20 neglected tropical diseases (NTDs) designated by the World Health Organization.1 The disease proceeds in two stages. In the first stage, the parasite is present in the blood and lymph and causes mild, flu-like symptoms.2 A transition to stage 2 occurs when the parasite crosses the blood-brain barrier; this results in more severe symptoms such as disrupted sleeping patterns and mood fluctuation.2 There are five compounds currently in use for the treatment of HAT: pentamidine, suramin, melarsoprol, and an eflornithine/nifurtimox combination therapy, referred to as NECT. These treatments are reasonably efficacious but cause a variety of adverse effects and can be challenging to administer.2 Recent developments include the approval of fexinidazole as an oral treatment for HAT in the Democratic Republic of Congo, and the progression of acoziborole to Phase II/III clinical trials.3 However, given the attrition rate of compounds in clinical trials and the possibility that T. brucei could develop resistance to current drugs, we believe it is prudent to continue to seed the drug discovery pipeline for HAT.
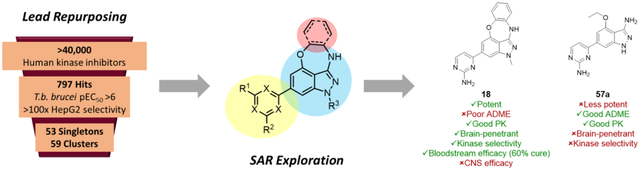
As part of our ongoing NTD drug discovery efforts, we employed a lead repurposing strategy to find new chemical matter to use as a starting point for HAT therapeutics. Lead repurposing entails the screening of sets of targeted chemical matter based on homology between humans and parasites.4 As there are many examples of kinase essentiality in T. brucei,5 we undertook a high-throughput screen (HTS) of human kinase inhibitors against T.b. brucei cultures.6 A set of 42,444 known human kinase inhibitors were screened against T.b. brucei at 4 μM; those that showed >50% inhibition of parasite growth were progressed to dose-response assays against T.b. brucei and HepG2 cells. This screening process yielded 797 compounds with a T.b. brucei pEC50 >6 that were >100x selective against HepG2 cells. These compounds were grouped by structural similarity into 59 clusters and 53 singletons.
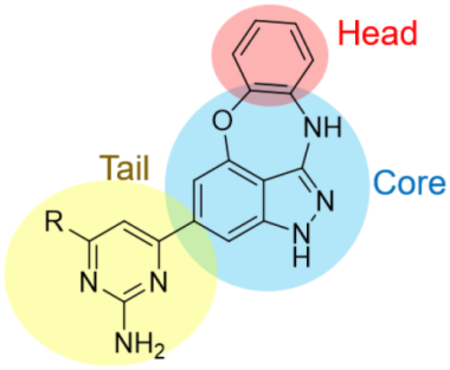
One of the overall most promising clusters to emerge from this HTS comprised a series of substituted benzoxazepinoindazoles (BOXIs). Structural features of note are highlighted in Table 1 and include an aromatic “head” (red), the oxazepinoindazole “core” (blue) and an appended heterocyclic “tail” (yellow). The average potency of compounds in this cluster is a pEC50 of 7.6, and the three hits highlighted in Table 1 (NEU-1117, -1118, and -1119) all have pEC50 >8 while maintaining a good margin of selectivity against mammalian cells. The physicochemical properties of these compounds are also promising: topological polar surface area (TPSA) and molecular weight fall well within the targeted range for lead compounds, and while the cLogP and LogD of these compounds are on average higher than desired, the lipophilic ligand efficiencies (LLEs)7 are high.
Table 1.
Targeted, cluster average, and individual cluster member values for properties of interest. Values highlighted in green meet or exceed targeted values; yellow highlighting indicates mid-range values, and red highlighting indicates values that are well outside the targeted value. nd = no data.
 |
|||||
|---|---|---|---|---|---|
| Targeted Value | Cluster Average | NEU-1117 | NEU-1118 | NEU-1119 | |
| R = CH2OCH3 | R = NH2 | R = H | |||
| T.b.b. pEC50 | ≥7 | 7.6 | 8.1 | 8.9 | 8.4 |
| HepG2 pTC50 | ≤pEC50−2 | 4.8 | 5.1 | 5.5 | 4.0 |
| cLogP | ≤3 | 3.9 | 2.2 | 3.7 | 3.7 |
| TPSA (Å2) | 40<x≤90 | 106 | 110 | 130 | 100 |
| MW (Da) | ≤360 | 332 | 360 | 330 | 320 |
| LLE | ≥4 | 3.7 | 5.8 | 5.2 | 4.7 |
| CNS-MPO Score | ≥4 | 3.9 | 4.4 | 3.9 | 4.5 |
| Fast | Yes | 85% | Yes | Yes | Yes |
| Cidal | Yes | 69% | Yes | Yes | No |
| Aq. Sol. (μM) | >10 | -- | 11 | 4 | 8 |
| PPB (%) | <95 | -- | 98 | 97 | nd |
Additional parameters were also calculated or measured with the end goal of producing a HAT drug in mind. Central nervous system multiparameter optimization (CNS-MPO)8 scores were calculated for the HTS hits in order to provide an indication of whether they were likely to be brain-penetrant, essential for any treatment of stage 2 HAT. On average, the CNS-MPO scores of these compounds are consistent with likely blood-brain barrier penetration. The rate of action and trypanosome cidality of hits were also assessed; fast-acting, trypanocidal compounds are preferred to develop an effective treatment. Most compounds in this cluster are fast-acting and cidal, including NEU-1117 and NEU-1118, though which structural features gave rise to this property was not yet understood.
Despite their promising characteristics, these compounds were not without room for optimization; Table 1 highlights some challenges to be overcome in this cluster of compounds. In particular, in vitro absorption, distribution, metabolism, and excretion (ADME) properties, such as aqueous solubility and plasma protein binding (PPB), were outside of the desired range. As our end goal is to develop an orally available HAT therapeutic, it was essential that these properties be improved. Additionally, a representative compound was selected for assessment against a panel of human kinases. Given the cluster’s origin as human kinase inhibitors, it is perhaps unsurprising that this compound, NEU-1328, is a potent (<100 nM) inhibitor of at least five human kinases (Table S1). Our overall goal, therefore, was to improve the ADME properties of BOXIs while maintaining their potency and favorable physicochemical properties, and to assess how these changes impacted human kinase selectivity.
Results
Initial SAR studies focused on exploring the effect of various nitrogen-containing heterocyclic tails coupled to the BOXI core. The synthesis of such compounds is shown in Scheme 1. Synthesis began with the conversion of 4-bromo-2,6-difluorobenzoic acid 1 to the acid chloride 2; this intermediate was coupled with 2-aminophenol to produce amide 3. The benzoxazepine ring was then formed upon treatment with potassium carbonate. Compound 4 was converted to the thioamide 5 using Lawesson’s reagent, which then yielded benzoxazepinoindazole 6 upon treatment with hydrazine. Protection with acetic anhydride and subsequent borylation gave boronic ester 8, which was then coupled via a Suzuki reaction with the desired aryl halide to yield compounds 9b-s. Analog 9a was synthesized using 2,6-difluorobenzoic acid and following steps a-e.
Scheme 1.
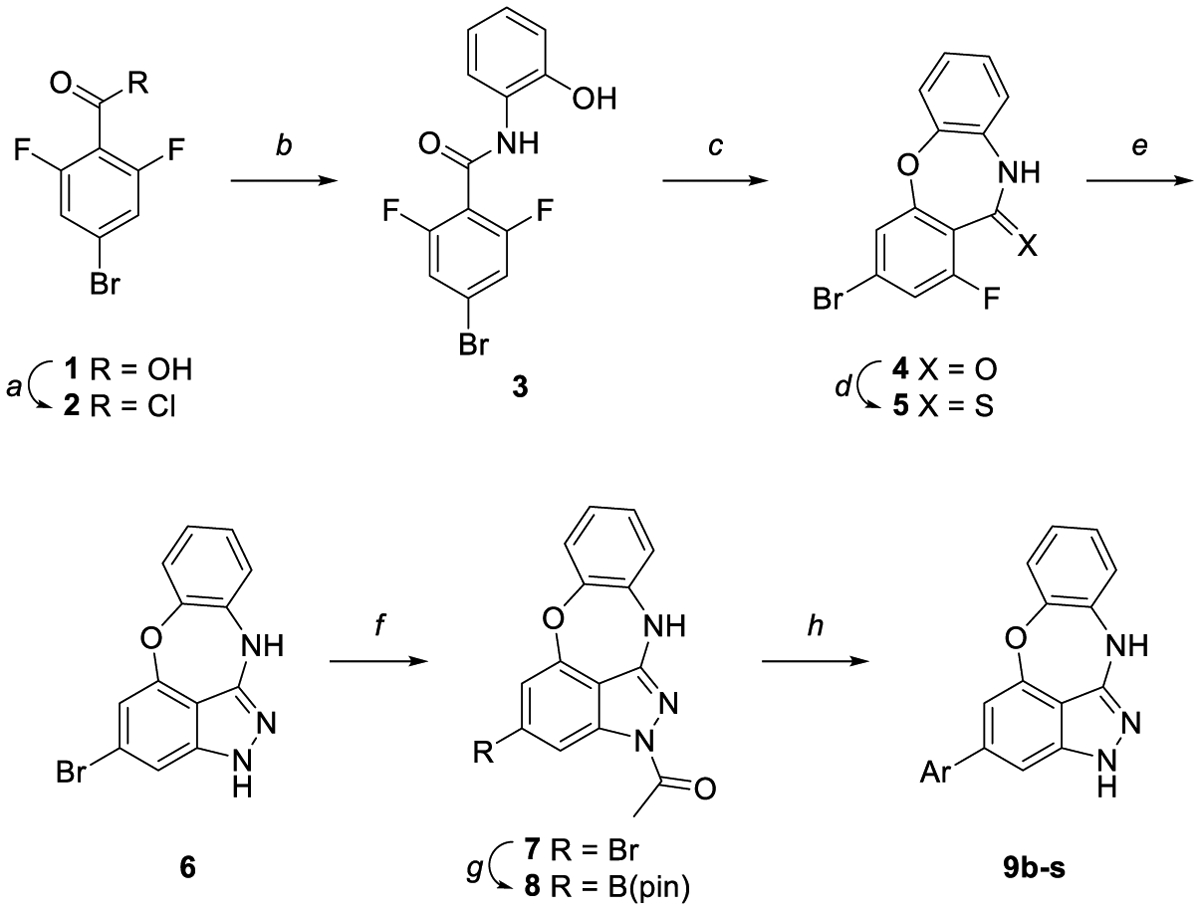
Synthesis of compounds 9a-s. Reagents and reaction conditions: a) SOCl2, 75 °C, 4 h (quant.). b) 2-aminophenol, TEA, DCM, 0 °C to rt, 12 h (87%). c) K2CO3, DMF, rt, 36 h (72%). d) Lawesson’s reagent, toluene, 100 °C, 12 h (68%). e) Hydrazine, dioxane, 85 °C, 3 h (89%). f) Acetic anhydride, 100 °C, 3 h (93%). g) B2(pin)2, KOAc, PdCl2(dppf)·CH2Cl2, dioxane, 145 °C, μw, 1 h (65%). h) Aryl halide, K2CO3, Pd(PPh3)4, 3:1 dioxane:water, 100 °C, 4 h (10–74%).
The biological activity data of the initial tail replacement analogs is shown in Table 2. Of these, the 2-aminopyrimidine motif remained the best option in terms of potency. Excising the tail altogether (9a) led to a drop in potency of ~2 log units; although notably, this compound still retained sub-micromolar activity. Removal of the −NH2 of the tail group resulted in a compound (9b) that was more than 10-fold less active than NEU-1119. Analogs where the 2-aminopyrimidine was replaced with other aminopyrimidines (9c, 9d), aminopyridines (9e, 9f) or a phenyl ring (9g) all lost 1–2 log units of potency compared to NEU-1119.
Table 2.
Biological activity and LLE of tail replacement analogs.
| ID | R | T.b.b. pEC50 | pTC50 | LLE | ID | R | T.b.b. pEC50 | pTC50 | LLE |
|---|---|---|---|---|---|---|---|---|---|
| 9a | H | 6.2 | 4.0 | 2.1 | 9k | 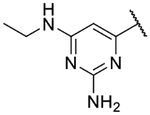 |
7.8 | 4.6† | 5.1 |
| 9b | 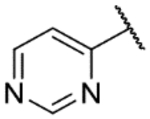 |
6.9 | <4.3† | 4.2 | 9l | 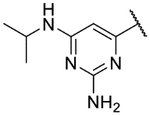 |
6.9 | <4.3† | 3.7 |
| 9c | 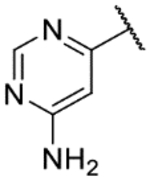 |
7.2 | 4.8 | 3.5 | 9m* | 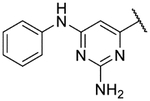 |
7.2 | 5.0 | 0.85 |
| 9d | 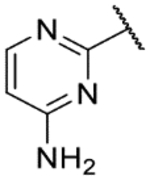 |
6.6 | <4.3† | 3.5 | 9n* | 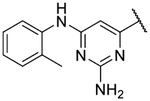 |
7.5 | 5.3 | 0.66 |
| 9e | 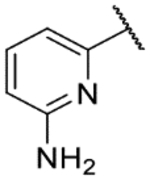 |
6.7 | <4.3† | 3.6 | 9o | 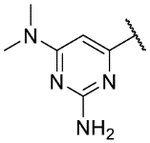 |
6.3 | 4.8† | 3.2 |
| 9f | 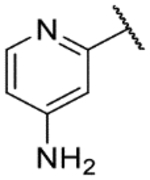 |
6.9 | nd | 4.8 | 9p | 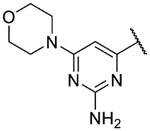 |
6.3 | <4.3† | 3.4 |
| 9g | 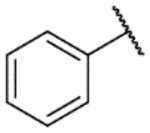 |
5.7 | <4.3† | 1.5 | 9q | 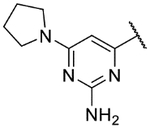 |
6.1 | 4.7† | 2.7 |
| 9h | 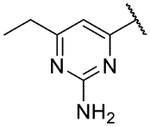 |
7.9 | 4.8 | 3.2 | 9r | 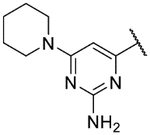 |
4.8 | <4.3† | 0.91 |
| 9i* | 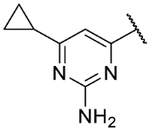 |
7.7 | 4.6 | 3.1 | 9s | 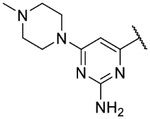 |
5.7 | <4.3† | 2.3 |
| 9j | 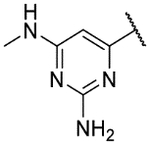 |
7.7 | 5.3 | 3.2 |
HTS hit, not resynthesized.
Data obtained against MRC5 cells are indicated with †; otherwise, HepG2 data is presented.
All SD within ± 0.20.
Small substitutions at the 6-position of the pyrimidine, such as the ethyl of 9h, the cyclopropyl of 9i, the methylamine of 9j, and the ethylamine of 9k were well-tolerated, with only a slight loss in potency as compared to the original hits. Bulkier amines such as the isopropylamine, aniline, and methylaniline of 9l, 9m, and 9n, respectively, were also tolerated from a potency standpoint but showed signs of toxicity in mammalian cells. Tertiary amines at the 6-position, including a dimethylamine (9o), morpholine (9p), pyrrolidine (9q), piperidine (9r), and N-methylpiperazine (9s), were not tolerated and consistently showed a drop in potency by 1–2 log units as compared to the primary amines or the unsubstituted 2-aminopyrimidine. In general, compounds that were active at sub-micromolar concentrations maintained LLEs between 3 and 4. Moving forward, analogs were primarily synthesized with an unsubstituted 2-aminopyrimidine as the “tail” group with the idea that, once an improved core was identified, it could be matched with other “tails” identified in this early SAR exploration if necessary.
The importance of each potential hydrogen bond donor on the 2-aminopyrimidine-substituted core was explored by systematically installing methyl groups at each position. These compounds were synthesized in a manner similar to the other BOXIs in the series (Scheme S1–S2). Methylation of the benzoxazepine nitrogen (15), as well as bis-methylation of the aminopyrimidine (19) resulted in complete loss of activity; however, methyl groups on the indazole nitrogen (18), as well as mono-methylation of the aminopyrimidine 20) were reasonably well-tolerated.
All analogs with a pEC50 >6 were assessed in a speed-to-kill assay.6 Figure 1 shows the rate of action profile of three slow-acting compounds (9a, 9b, and 9f) and three fast-acting compounds (9j, 9l, and 18), where fast-acting compounds are defined as those that reach a pEC50 >6 in less than 18 h of incubation time. Although all six analogs eventually reached a pEC50 >6.5, slow-acting compounds required incubation times of up to 72 h. Interestingly, all fast-acting compounds were analogs that retained the benzoxazepine core and 2-aminopyrimidine tail, while slow-acting compounds were analogs where the 2-aminopyrimidine was either removed or changed to a different heterocycle altogether. These data illustrate the importance of the 2-aminopyrimidine motif to producing not only potent, but fast-acting analogs, and this tail group was prioritized moving forward.
Figure 1.
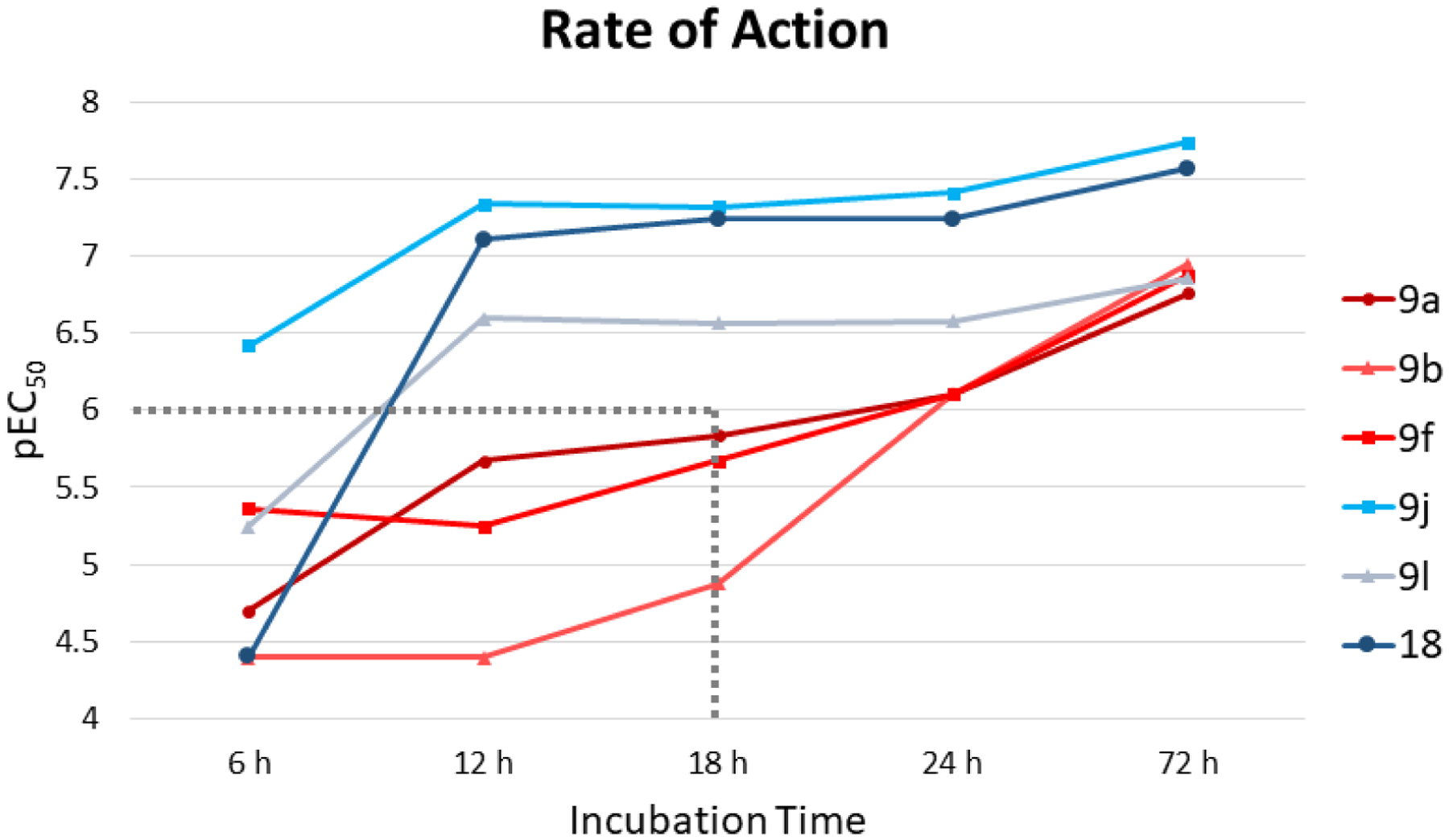
Rate of action of selected analogs. Slow-acting compounds 9a, 9b, and 9f are represented in red and fast-acting compounds 9j, 9l, and 18 are represented in blue. Dotted gray lines show the cutoff of pEC50 of 6 at 18 h incubation time.
Additionally, we assessed the in vitro ADME parameters of these compounds. The properties of tail replacement and methylated analogs for which ADME data was available are shown in Table 4. As was typical for all analogs synthesized as this point in the campaign, these compounds show moderate to high clogP and high logD, as well as low solubility and high PPB. In addition, we observed moderate-to-high microsomal and hepatocyte clearance. With this in mind, despite the high LLE of the BOXI analogs, we felt that further reduction of the lipophilicity would serve the overall goal of developing a potent analog with a good ADME profile. Thus, considering metabolic stability and lipophilicity, we turned our attention to the benzoxazepine core as the next site of modification.
Table 4.
ADME properties of selected analogs.
| ID | T.b.b. PEC50 | cLogP | LogD | Aq. sol. (μM) | PPB (%) | HLM Clint (μg/min/mg protein) | Rat Hepatocyte Clint (μg/min/106 cells) |
|---|---|---|---|---|---|---|---|
| 9a | 6.2 | 2.9 | 4.1 | 19 | >99 | 7.6 | 47 |
| 9b | 6.9 | 3.0 | 3.5 | <1 | >96 | 130 | 68 |
| 9c | 7.2 | 2.8 | 3.8 | 0.6 | >99 | 52 | 73 |
| 9d | 6.6 | 3.4 | 4.1 | 4 | >99 | 34 | 56 |
| 9e | 6.7 | 3.5 | 3.7 | 9 | >81 | 99 | 220 |
| 9j | 7.7 | 2.9 | >4.2 | 2 | >95 | 9.5 | 45 |
| 9k | 7.8 | 3.3 | 4.6 | 2 | >99 | 9 | 26 |
| 9l | 6.9 | 3.7 | 4.9 | 9 | >99 | 6.7 | 32 |
| 9o | 6.3 | 3.6 | >4.4 | 0.4 | >99 | 63 | 44 |
| 9r | 4.8 | 4.4 | 4.6 | 0.3 | >99 | 17 | 25 |
| 9s | 5.7 | 3.4 | 3.4 | 26 | 97* | 41 | 16 |
| 18 | 7.6 | 2.7 | >4.3 | 3 | nd | 110 | 12 |
| 19 | 5.3 | 3.8 | >3.9 | 0.7 | <99 | 51 | 29 |
| 20 | 7.0 | 3.2 | 3.8 | 1.9 | 99 | 55 | 45 |
Predicted PPB.
nd = no data.
Initial analogs targeted a truncated oxazepinoindazole core, the synthesis of which is shown in Scheme 2. Reaction of 2 with PMB-ethanolamine facilitated cyclization to the desired amide 21; direct deprotection of the oxazepine ring allowed the use of Lawesson’s reagent to convert amide 23 to thioamide 24. Subsequent transformations proceeded in an analogous manner to those required to make the benzoxazepine core, with thioamide 24 converted to oxazepinoindazole 25 using hydrazine. This reaction was followed by acetylation and conversion of the bromide 26 to the boronic ester 27 through a Miyaura borylation using bis(pinacolato)diboron. Finally, a Suzuki reaction of this intermediate with the requisite chloropyrimidines yielded the desired products 28a-b.
Scheme 2.
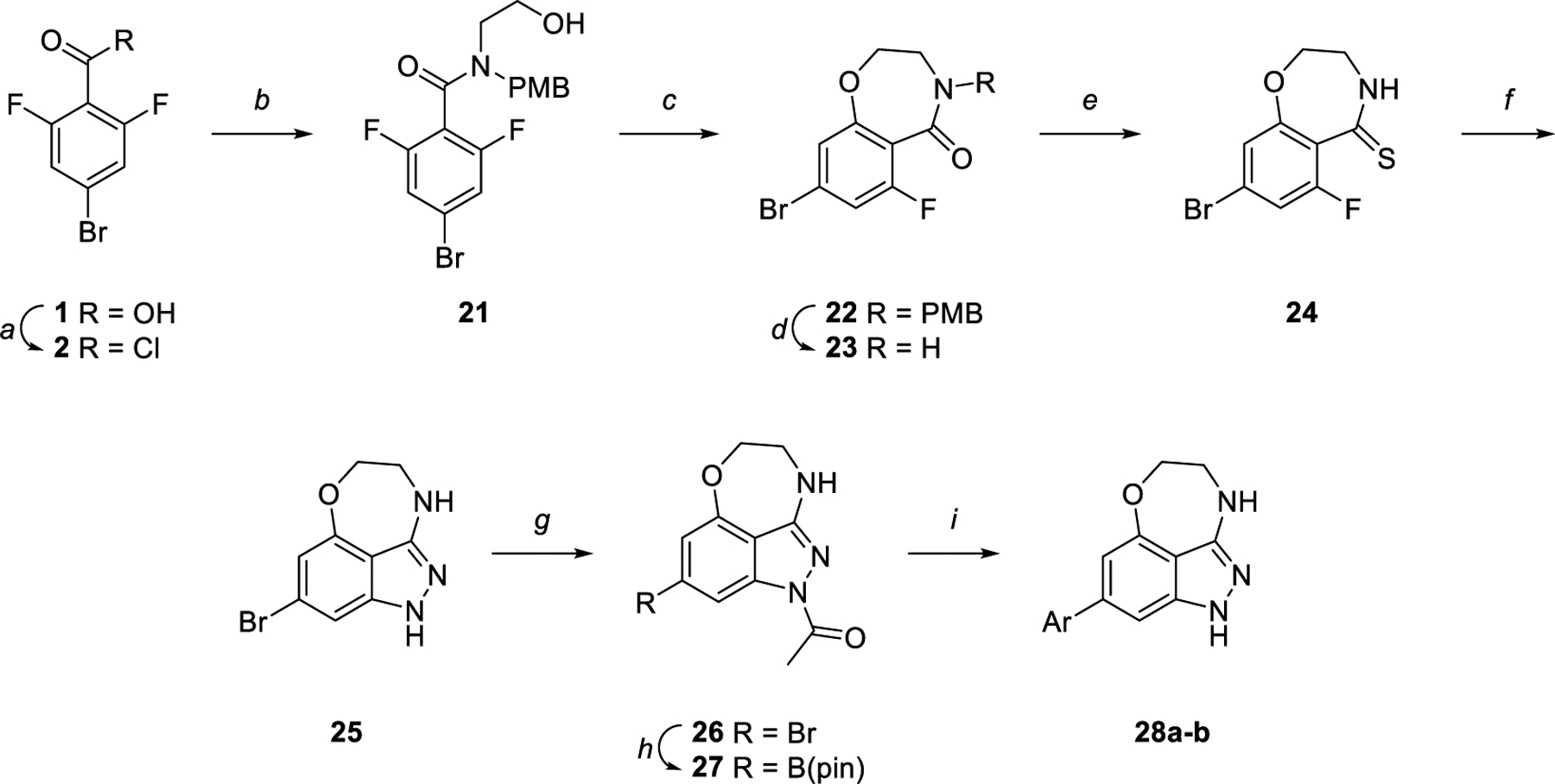
Synthesis of oxazepinoindazole core. Reagents and reaction conditions: a) SOCl2, 75 °C, 4 h (quant.). b) 2-((4-Methoxy-benzyl)amino)ethan-1-ol or ethanolamine, TEA, DCM, 0 °C to rt, 12 h (54%). c) NaH, DMF, rt, 12 h (96%). d) CAN, 3:1 acetonitrile:water, rt, 3 h (36%). e) Lawesson’s reagent, toluene, 100 °C, 12 h (56%). f) Hydrazine, dioxane, 85 °C, 3 h (95%). g) Acetic anhydride, 100 °C, 3 h (60%). h) B2(pin)2, KOAc, PdCl2(dppf)·CH2Cl2, dioxane, 145 °C, μw, 1 h (79%). i) Aryl halide, K2CO3, Pd(PPh3)4, 3:1 dioxane:water, 100 °C, 4 h (12–34%).
We also explored truncation to substituted indazoles and aminoindazoles (Scheme 3). The aminoindazole core was constructed from (4-cyano-3-fluorophenyl)boronic acid 29, which was subjected to a Suzuki reaction with the desired aryl chlorides to form biaryl compounds 30. These intermediates were then reacted with hydrazine to form final compounds 31a-c. To synthesize indazole analogs, 4-bromoindazole was subjected to the same Miyaura and Suzuki conditions as previously described to provide final compounds 34a-c. The syntheses of other core or headgroup replacement analogs, such as 40 and 49, were completed in an analogous route to that described in Scheme 1, and are described in detail in Scheme S3–S4.
Scheme 3.
Synthesis of indazoles and aminoindazoles. Reagents and reaction conditions: a) Aryl halide, NaHCO3 (sat. aq.), Pd(PPh3)4, dioxane, 95 C, 3 h (43–97%). b) Hydrazine, EtOH, 95 °C, 12 h (71–87%). c) B2(pin)2, KOAc, PdCl2(dppf)·CH2Cl2, dioxane, 145 °C, μw, 30 min – 1.5 h. d) Aryl halide, K2CO3, PdCl2(dppf)·CH2Cl2, 3:1 dioxane:water, 150 °C, μw 30 min, (6–26%). Where no yield is reported, crude material was progressed without further purification.
The biological activity and ADME properties of these core replacement analogs are presented in Table 5 (additional ADME data in Table S2). Systematic truncation of the benzoxazepine core to an oxazepinoindazole (28a-b), aminoindazole (31a-c), or indazole (34a-c) core uniformly resulted in a loss in potency of ~3 log units. However, these analogs were much more soluble than the original hits. Interestingly, despite their low potency, they also retained LLEs of around 4, suggesting that the lipophilic benzoxazepine group is contributing to potency in proportion to its lipophilicity. Excision of the indazole ring, as in compound 49, also led to a drop in potency, but without a corresponding lowering of clogP or boost in solubility.
Table 5.
Biological activity and selected ADME data for headgroup truncation and replacement analogs.
| ID | Core | R | T.b.b. pEC50 | MRC5 pTC50 | LLE | Aq. sol. (μM) | cLogP |
|---|---|---|---|---|---|---|---|
| 28a | 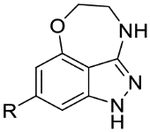 |
 |
4.8 | <4.3 | 4.1 | 65 | 0.61 |
| 28b |  |
5.4 | <4.3 | 4.7 | nd | 0.76 | |
| 31a | 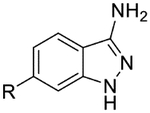 |
 |
4.8 | <4.3 | 4.2 | 58 | 0.64 |
| 31b | 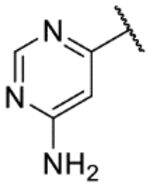 |
4.8 | <4.3 | 4.0 | 5 | 0.79 | |
| 31c | 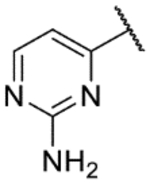 |
5.5 | <4.3 | 4.6 | <13 | 0.87 | |
| 34a | 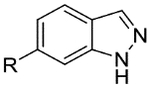 |
 |
5.3 | <4.3 | 3.9 | 660 | 1.4 |
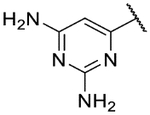
| 34b | 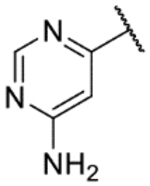 |
4.3 | <4.3 | 2.8 | 332 | 1.5 | |
| 34c |  |
5.2 | <4.3 | 3.6 | 215 | 1.6 | |
| 40 | 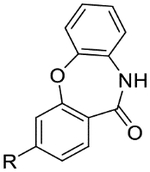 |
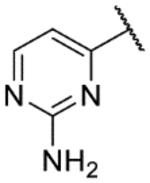 |
5.9 | <4.3 | 3.4 | 0.3 | 2.6 |
| 49 | 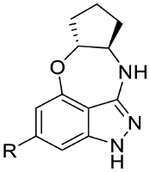 |
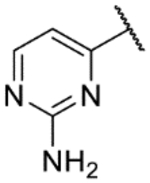 |
7.1 | <4.3 | 5.3 | 13 | 1.8 |
| 50* | 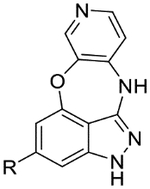 |
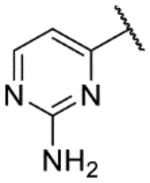 |
8.2 | 5.9 | 5.1 | 10 | 1.4 |
| 51* | 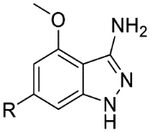 |
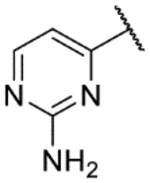 |
6.2 | 4.0 | 5.2 | >100 | 0.71 |
Kinetic aqueous solubility.
Compound and data provided by GSK; HepG2 toxicity and kinetic aqueous solubility.
All SD within ± 0.19.
Given the importance of the full BOXI core to potency and its detrimental effect on solubility, we sought to replace the benzene “headgroup” of this core with other features that would improve solubility while filling the space occupied by the aromatic ring. We hypothesized that increased sp3 content or more polarity might increase solubility without a loss in potency. However, while analogs with either a saturated cyclopentane ring (49) or a heterocyclic pyridine (50) did indeed retain sub-100 nM activity, we did not observe an increase in solubility. Interestingly, the methoxy-substituted aminoindazole 51 retained sub-micromolar potency with solubility of >100 μM; its reduced clogP also translated into an LLE of 5.2.
Capitalizing on the reasonable potency and excellent solubility of 51, we pursued a series substituted aminoindazoles, the synthesis of which is shown in Scheme 4. Starting with 4-bromo-2,6-difluorobenzonitrile 52, desired alkoxybenzonitriles 53 were synthesized by substitution with the corresponding alcohol. The indazole ring was closed using hydrazine to produce 54, then reacted with acetic anhydride to yield the protected indazoles 55. Miyaura borylation of the core and subsequent Suzuki coupling with 2-amino-4-chloropyrimidine gave the final ring-opened products 57a-j. The syntheses of compounds 63 and 68 are presented in Scheme S5–S6.
Scheme 4.
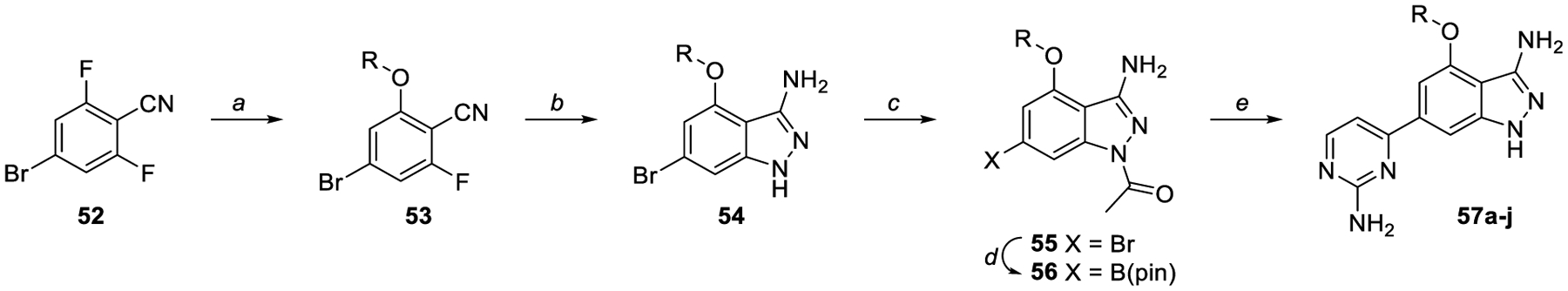
Synthesis of ring-opened analogs. Reagents and reaction conditions: a) Alcohol, LHMDS (1 M in THF), THF, 0 °C to rt, 2 days (55–86%). b) Hydrazine, ethanol, 95 °C, 12 h (71–87%). c) Boc2O, DMAP, DCM, rt, 12 h (75%); or Ac2O, pyridine, 100 C, 3.5 h (24–75%). d) B2(pin)2, KOAc, PdCl2(dppf)·CH2Cl2, dioxane, 145 °C, μw, 30 min – 1.5 h. e) Aryl halide, K2CO3, PdCl2(dppf)·CH2Cl2, 3:1 dioxane:water, 150 °C, μw 30 min, (6–26%). Where no yield is reported, crude material was progressed without further purification.
The biological activity of these compounds is shown in Table 6. Although we were able to slightly improve upon the potency of 51 by introducing bulkier alkoxy substituents at R1 (57a-f), the LLE either remained the same or dropped, suggesting that more lipophilic substituents such as the t-butyl or phenyl were, on balance, making the compound more lipophilic without a corresponding boost in potency. Furthermore, the phenyl substituent 57e introduced some toxicity to mammalian cells.
Table 6.
Biological activity and selected ADME data for ring-opened analogs.
| ID | R1 | R2 | T.b.b. pEC50 | MRC5 pTC50 | LLE | Aq. sol. (μM) | cLogP |
|---|---|---|---|---|---|---|---|
| 31c | −H | −NH2 | 5.5 | <4.3 | 4.4 | <13 | 0.87 |
| 51 | −OMe | −NH2 | 6.2 | <4.3 | 5.2 | 196 | 0.71 |
| 57a | −OEt | −NH2 | 6.4 | 5.0 | 5.3 | 861 | 1.1 |
| 57b* | −OiPr | −NH2 | 6.4 | 5.0 | 4.9 | 354 | 1.5 |
| 57c | −OtBu | −NH2 | 6.2 | 4.6 | 4.4 | 68 | 1.8 |
| 57d | −OcyBu | −NH2 | 6.6 | 5.0 | 5.0 | nd | 1.6 |
| 57e* | −OPh | −NH2 | 5.7 | 5.7 | 3.3 | 117 | 2.4 |
| 57f* | −OBn | −NH2 | 5.9 | 5.0 | 3.4 | 85 | 2.4 |
| 57g | 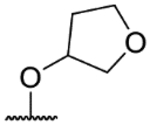 |
−NH2 | 5.5 | <4.3 | 4.9 | 751 | 0.62 |
| 57h | 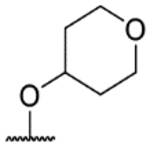 |
−NH2 | 5.8 | <4.3 | 5.1 | 770 | 0.68 |
| 57i | 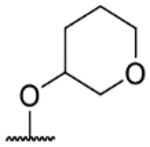 |
−NH2 | 6.1 | <4.3 | 5.0 | 1000 | 1.13 |
| 57j | 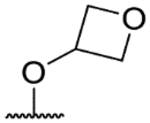 |
−NH2 | 5.7 | <4.3 | 5.1 | 383 | 0.56 |
| 63 | −H | −NHPh | 5.4 | <4.3 | 2.1 | 3 | 3.1 |
| 68 | −H | −NHBn | 6.1 | <4.3 | 2.2 | 17 | 2.9 |
Compound and data provided by GSK; HepG2 toxicity and kinetic aqueous solubility.
All SD within ± 0.16.
We hypothesized that some potency could be gained by introducing substituents containing a hydrogen bond acceptor which could interact with the aminoindazole. This potential intramolecular hydrogen bond could position the substituent in such a way as to mimic the orientation of the benzene ring of the full BOXI core. Compounds 57g-j were synthesized in order to test this hypothesis; unfortunately, none of these analogs showed increased activity. However, they displayed some of the best overall ADME data of the series (Table S2). Finally, compounds with a phenyl- (63) or benzyl- (68) substituted amine were tested; these compounds displayed potency in the micromolar range and did not show significant improvement in ADME profile.
At this point, we assessed the overall profile of several promising compounds. Table 7 represents a heat map of an original HTS hit, NEU-1117, as well as three analogs, 18, 49, and 57a. Values highlighted in green meet or exceed targeted values; yellow highlighting indicates mid-range values, and red highlighting indicates values that are well outside the targeted range. All three compounds show an improved overall profile over NEU-1117. Compounds 18 and 49 both maintain high potency against T.b. brucei and are less toxic against MRC5 cells than NEU-1117, although 49 is highly cleared in both rat hepatocytes and human liver microsomes. These limitations were deemed sufficient to preclude 49 from further assessment. Compound 57a, although approximately 1.5 log units less potent than NEU-1117, nevertheless, exhibits excellent ADME properties including high aqueous solubility, low clearance, and low PPB. Based on these data, we selected 18 and 57a for progression to pharmacokinetic (PK) studies.
Table 7.
Heat maps of NEU-1117, 18, 49, and 57a.
| Targeted Value | NEU-1117 | 18 | 49 | 57a | |
|---|---|---|---|---|---|
| T.b.b. pEC50 | >7.5 | 8.1 | 7.6 | 7.1 | 6.4 |
| MRC5 pTC50 | <5 | 5.1 | 4.3 | 4.3 | 4.3 |
| MW | ≤360 | 360.4 | 330.34 | 308.3 | 270.3 |
| cLogP | ≤3 | 2.6 | 2.7 | 2.0 | 1.1 |
| LogD (7.4) | ≤2 | >4 | >4.3 | 2.8 | 1.7 |
| LLE | ≥4 | 5.5 | 4.9 | 5.1 | 5.3 |
| MPO Score | ≥4 | 3.9 | 3.9 | 4.2 | 4.1 |
| Aq. sol. (μM) | >10 | 1 | 3 | 13 | 861 |
| HLM Clint (μg/min/mg protein) | <9 | 23.6 | 108 | nd* | 25 |
| Rat Hepatocyte Clint (μg/min/106 cells) | <5 | 41.4 | 12.4 | 52 | 20.3 |
| PPB (%) | ≤95 | >99 | nd | 88 | 89 |
nd = no data.
Compound was cleared too rapidly for detection.
The results of the mouse PK studies of 18 and 57a are shown below. Figure 2 shows PK results for two separate experiments: the green line represents blood concentration over time, while the red and blue dots represent sparse sampling of blood and brain concentrations. Parameters calculated from these curves, including Cmax, tmax, and t1/2, are presented in Table 8. Both compounds are cleared from the blood within 4 h after injection, although 18 has a slightly longer half-life. Additionally, both compounds achieve a Cmax >30× their T.b. brucei EC50. Of the two compounds, 18 demonstrates higher brain penetration and maintains its blood/brain ratio for 4 h.
Figure 2.
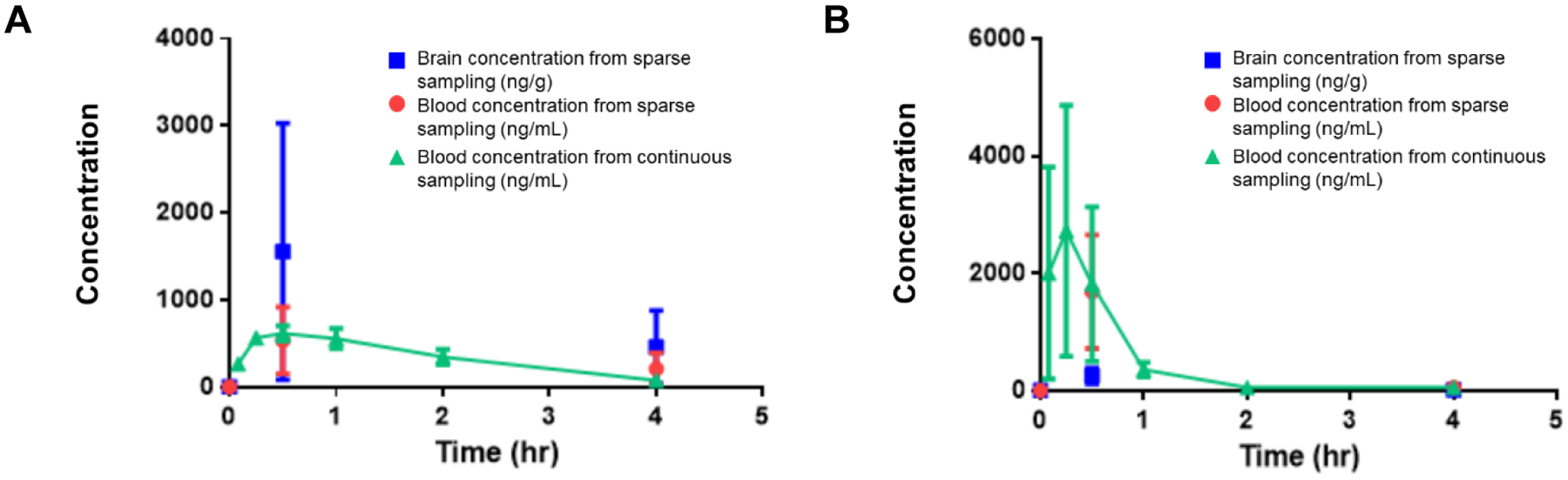
Brain and blood concentrations of (A) 18 and (B) 57a over time after a 10 mg/kg intraperitoneal dose.
Table 8.
PK parameters for 18 and 57a.
| ID | T.b.b. EC50 (ng/mL) | Cmax (ng/mL) | tmax (h) | t1/2 (h) | Brain/blood ratio (t = 0.5 h) | Brain/blood ratio (t = 4 h) |
|---|---|---|---|---|---|---|
| 18 | 8.9 | 615 ± 85.8 | 0.25–0.5 | 1.06 ± 0.235 | 2.39 ± 1.12 | 2.12 ± 0.52 |
| 57a | 96.2 | 3603 ± 636 | 0.25–0.5 | 0.893 ± 0.500 | 0.155 ± 0.021 | 0.214 |
Because the compounds included in the HTS were designed as human kinase inhibitors, and a compound in this cluster had known human kinase activity, we also assessed the activity of both 18 and 57a against a panel of 46 human kinases at a 1 μM concentration; Figure 3 shows the percent inhibition of each kinase for both compounds. In general, 18 is a much more selective compound than 57a, inhibiting only three kinases >50% (Syk, MAP4K4, and JAK3), and two additional kinases (PKA and IRAK4) when the cutoff is lowered to 30% inhibition. In contrast, at 1 μM 57a is a potent inhibitor of 19 kinases included in the panel and a moderate inhibitor of an additional nine (Table S3).
Figure 3.
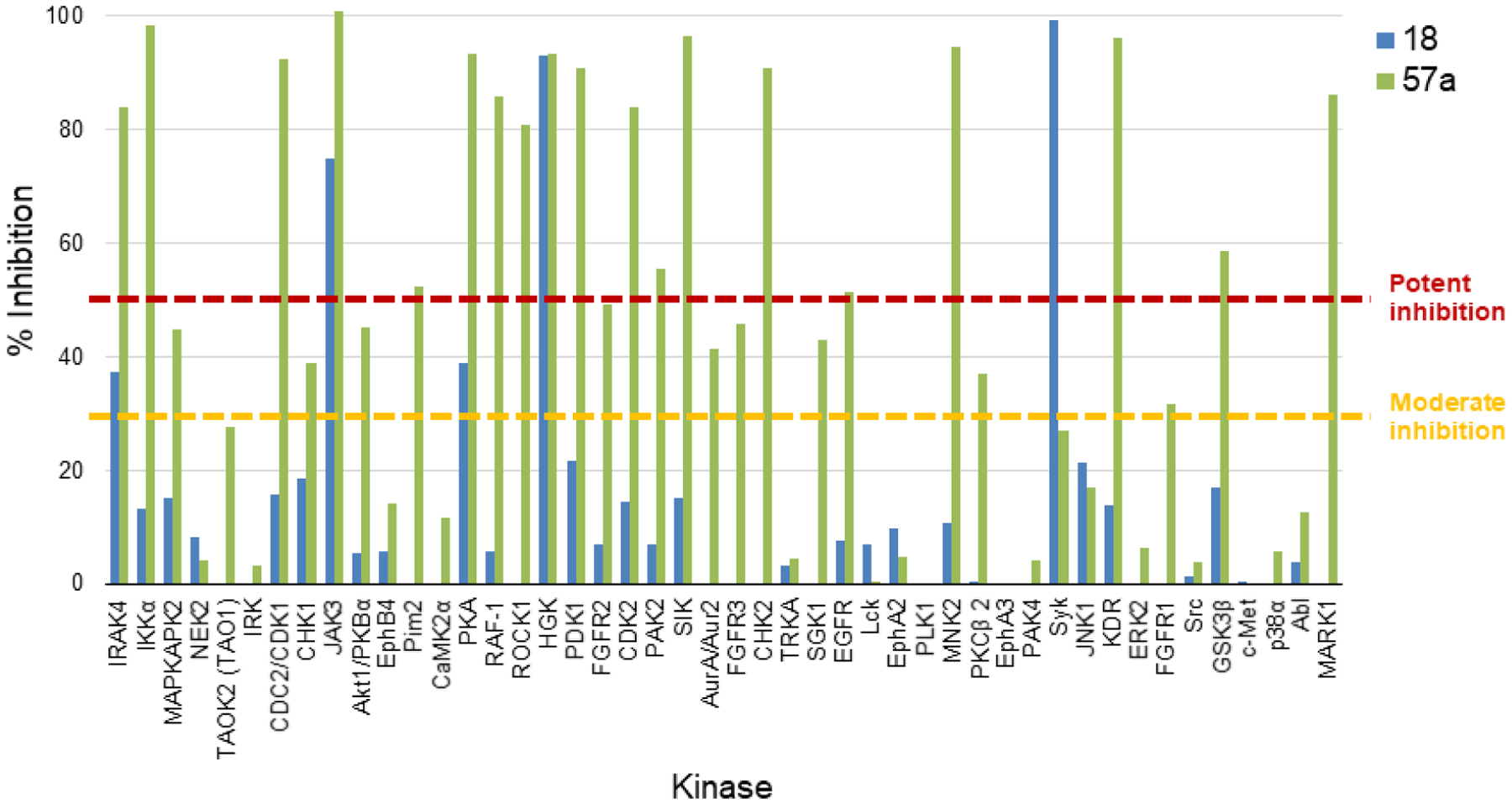
Percent inhibition of a human kinase panel for 18 (blue) and 57a (green). Compounds were tested at a 1 μM concentration.
Given its superior brain penetration and more selective kinase panel results as compared to 57a, compound 18 was progressed to an in vivo efficacy model of blood-stage HAT. Infected mice with T. brucei brucei STIB795 were treated with 18 once daily at 30 mg/kg intraperitoneally (IP) for five days; the results are shown in Figure 4. Compared to vehicle control mice, which did not survive past day 5, all of the mice treated with 18 survived up to day 23, and 60% of mice were cured of infection (undetectable parasitemia on day 30). Furthermore, 18 is a fast-acting, trypanocidal compound: 80% of treated mice had undetectable parasitemia 24 h after treatment began (Table S6).
Figure 4.
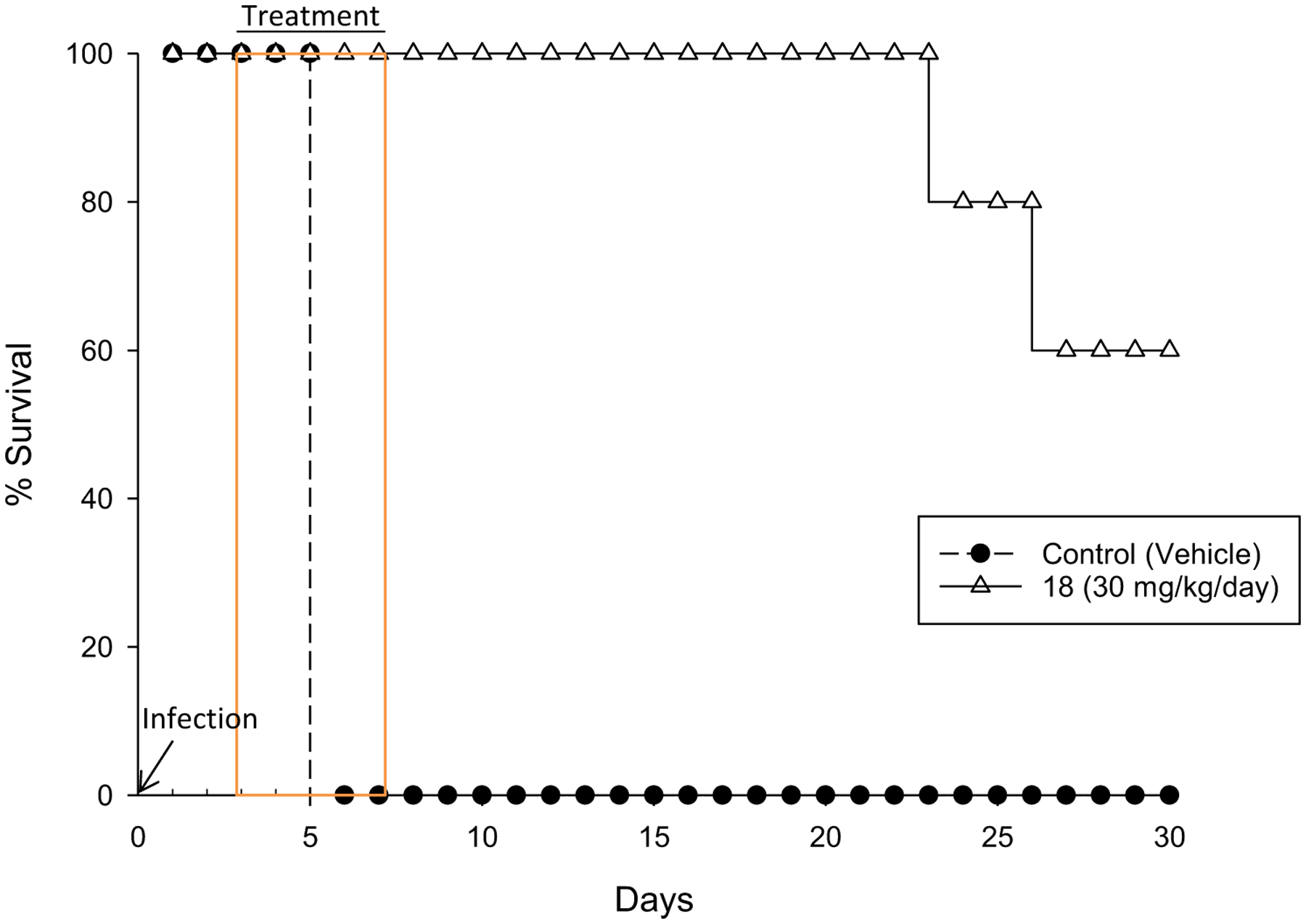
Efficacy model results. Control mice are represented by closed circles and mice treated with 30 mg/kg/day 18 (IP dosing) are represented by open triangles.
Based on the promising results of this experiment, compound 18 was further progressed to a CNS efficacy model to assess its potential as a stage 2 HAT therapeutic. Mice infected with T. brucei brucei GVR35 were divided into three groups: control (Berenil 40 mg/kg pretreatment + vehicle), Treatment 1 (Berenil 40 mg/kg pretreatment + 25 mg/kg BID 18), and Treatment 2 (25 mg/kg BID 18). Berenil®, an anti-trypanosomal therapeutic used in livestock, was administered to control and Treatment 1 groups on day 18 to clear blood stage parasitemia. Compound 18 was administered to Treatment 1 and 2 groups on day 21–25, while vehicle was administered to the control group.
Figure 5 shows the relative survival and relative survival means of the three groups. Treatment with 18 in combination with Berenil did not significantly extend life expectancy over the control group, and the Treatment 2 group (18 alone) showed a lower relapse mean than either the control or Treatment 1 groups. In addition, toxicity was observed when dosing 18 at 50 mg/kg QD with 50% of animals experiencing inflammation after dosing, which resolved with time, and 50% of animals dying within 30 minutes of treatment. For these reasons, 18 was not progressed for further evaluation.
Figure 5.
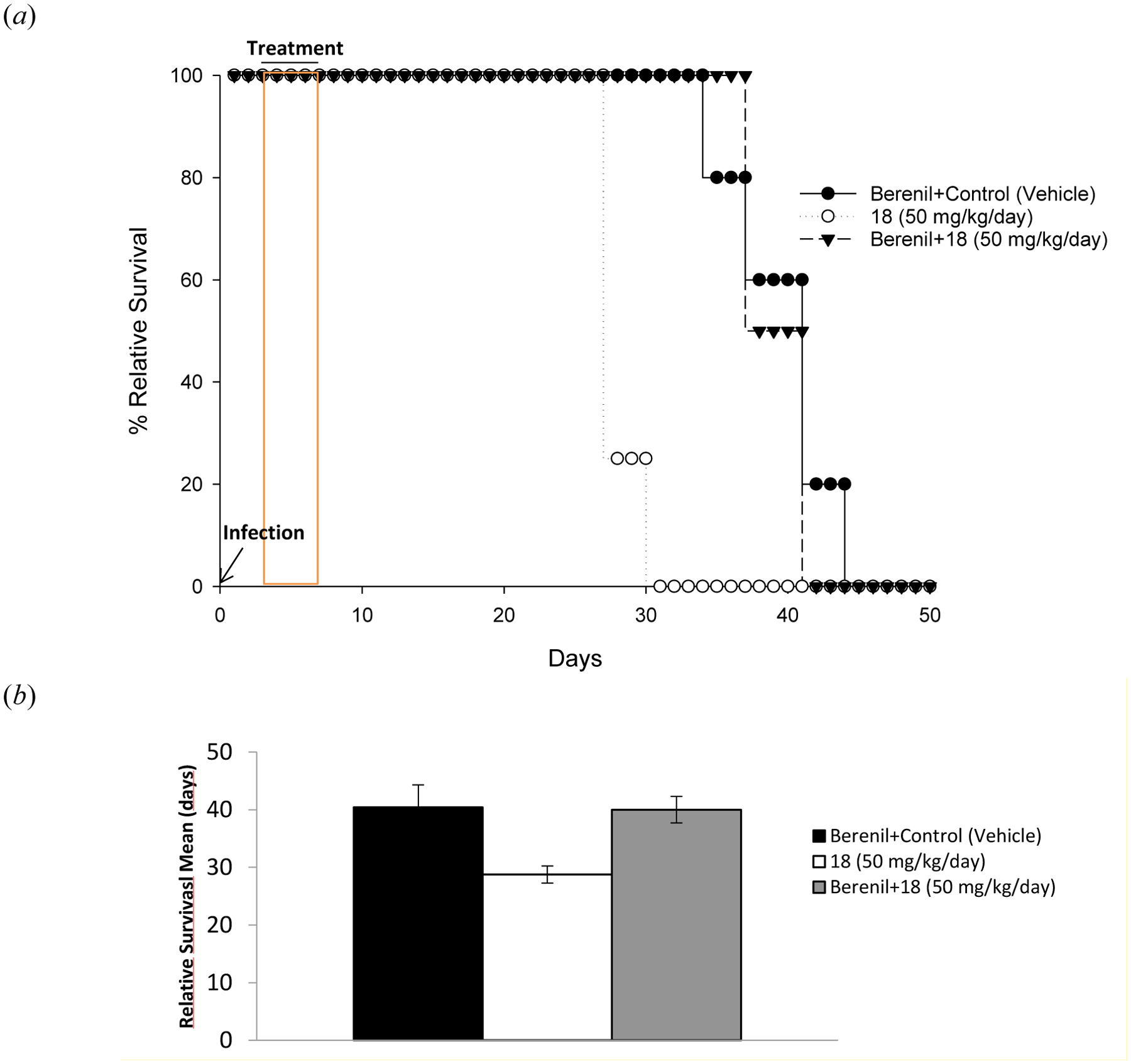
CNS efficacy model results. (a) Relative survival of control mice (closed circles), Treatment 1 mice (closed triangles) and Treatment 2 mice (open circles). (b) Relative survival mean of the control (black), Treatment 1 (white), and Treatment 2 (gray) groups.
In addition to assessing T. brucei activity, we cross-screened selected compounds against other parasites, including Trypanosoma cruzi (the causative agent of Chagas disease), Leishmania donovani (one of the causative agents of leishmaniasis) and Schistosoma mansoni (a trematode flatworm and a causative agent of schistosomiasis). The detailed activity data for T. cruzi and L. donovani are presented in Table S4. Although only 9o showed a modest activity against L. donovani intracellular amastigotes with an acceptable selectivity over host cells, two compounds (NEU-1117 and 18) had pEC50 >7.0 against T. cruzi, and three additional analogs (9j, 9k, and 20) had pEC50 ~ 6.0. Further work is ongoing to elaborate the SAR of this series and assess its potential as anti-T. cruzi agents. Overall, the data highlight the importance of the fully elaborated BOXI core and the 2-aminopyrimidine moiety for activity.
The data for four of the BOXIs vs. adult S. mansoni are presented in Table S5. When these compounds were screened at 10 μM over 48 h against S. mansoni adults, a variety of phenotypic responses and associated severity scores were elicited (Table S5). Thus, 9c, at the earliest observation time point of 3 h, induced uncoordinated muscular activity and an inability of the oral and ventral suckers to adhere to the substratum, which decreased over time; 9d produced a pronounced and sustained flaccid paralysis at 3 h that was reversible upon removal of the compound; 63 elicited an uncoordinated response at 3 h that was then replaced by decreased motility, darkening and an inability of the oral and ventral suckers to adhere to the substratum; finally, 31c was inactive. Investigations into the anti-schistosomal activity of other molecules in this series is continuing.
Discussion and Conclusion
Through a lead repurposing approach, we have evaluated a series of benzoxazepinoindazoles and related compounds as potential HAT therapeutics. Detailed structure-activity and structure-property relationships were established that enabled us to select two compounds, 18 and 57a, for further evaluation. Given its high potency, favorable PK profile, and improved selectivity against human kinases, 18 was assessed for in vivo efficacy. Promising results were obtained with 18 in the systemic T. brucei infection model. This, coupled with a high brain-blood ratio in pharmacokinetic experiments, led to the progression of 18 into the CNS model, though it was not successful in clearing parasitemia and toxicity was observed. Further investigation to understand these findings is ongoing. In addition, several BOXIs have been identified as potent inhibitors of T. cruzi, the causative agent of Chagas disease, and S. mansoni, the causative agent of Schistosomiasis. Investigation of this class of compounds as anti-Chagas agents and their potential as schistosomiasis therapeutics is likewise ongoing.
Experimental Section
In vitro Biology.
In order to determine the T. b. brucei EC50 values, 4 μL per well from compound master plates were dispensed into a new plate and 96 μL of HMI-9 per well were added to generate a 4% DMSO intermediate plate. Mid-log phase growth T. b. brucei was diluted to a working cell density of 2,750 cells/mL and 90 μL/well dispensed into 96-well flat-bottom transparent assay plates (Nunc®). Ten μL/well from intermediate plates were added so final cell concentration was 2,500 cell/mL, and final top concentration of compounds was 40 μM in 0.4% DMSO per well. Assay plates were incubated for 72 h at 37 °C and 5% CO2. Four hours prior to the end of the incubation, 20 μL of a 440 μM resazurin solution in prewarmed HMI-9 was added to each well and incubated for another 4 h. Fluorescence was then measured in an Infinite F200 plate reader (Tecan®) at 550 nm (excitation filter) and 590 nm (emission filter). A four-parameter equation was employed to fit the dose-response curves and determine EC50 using the SigmaPlot® 13.0 software. Assays were performed in duplicate at least twice, to achieve a minimal n=2 per dose response.
Pharmacokinetics Protocols.
Animals and ethical statement:
All animal studies were ethically reviewed and carried out in accordance with European Directive 2010/63/EEC and the GSK Policy on the Care, Welfare and Treatment of Animals. Compound was administered intraperitoneally (IP) to two groups of female NMRI mice (Group 1 n=3; Group 2 n=6) supplied by Charles River (Germany) Ltd. The compound was prepared in 1% (v/v) DMSO:99% (v/v) 20% (w/v) sulfobutyl ether-beta-cyclodextrin (SBE-β-CD/Captisol®, used as a solubilizing agent) in water and the dosing volume was 10 mL/kg for a total dose of 10 mg/kg. Food and tap water were available ad libitum. Following IP dosing, Group 1 blood samples were collected from the tail vein into capillary tubes containing K2EDTA at the following time-points: 0.0833, 0.25, 0.5, 1, 2, 4, 6, 8 and 24 h.
In order to obtain simultaneous blood and brain samples, Group 2 mice were placed under terminal anaesthetic (isoflurane) and blood samples (0.3 mL) collected from the retro-orbital sinus into K2EDTA tubes at 0.5 h (n= 3) and 4 h (n=3) after compound administration. Immediately following blood sample collection, death was confirmed by cervical dislocation and the brain removed. Aliquots of each blood sample were diluted in an equal volume of water. Mouse brain samples were weighed, water was added at a ½ (w/v) ratio (brain/water), and then homogenized. Both blood and brain samples were stored −80 °C until analysis.
Diluted blood and brain homogenates were processed under standard liquid-liquid extraction procedures using CAN containing an internal standard (Nifedipine) and analysed by LC-MS/MS. Non-compartmental analysis was performed using the Phoenix pharmacokinetic software version 1.4 (Certara) and Cmax, tmax, AUClast, AUC, and t1/2 were estimated.
In vivo Efficacy Experiment Protocols.
Animals and ethical statement:
All procedures were approved by ethical committee of Institute of Parasitology and Biomedicine Lopez-Neyra (Spanish National Research Council, CSIC), code MNC.2/2015. This Institute has joined the Agreement on Transparency in Animal Experimentation, promoted by the Confederation of Scientific Societies of Spain (COSCE), with the collaboration of the European Association for Animal Research released on September 20, 2016. The Animal Experimentation Unit is under the control of the competent authority, registered in the national register as Center of Breeding and User of experimental animals code ES-180210000022, according with the European and Spanish regulations. Female NMRI mice (Charles River Laboratories) were provided with sterilized water and commercial pellets ad libitum and maintained under the standard conditions in a conventional room at 20–24 °C with a 12/12-h light/dark cycle. Compound 18 solution for treatment was resuspended in vehicle: 5 % DMSO in 20 %Captisol® sulfobutyl ether β-cyclodextrin that improves solubility and stability for drug dosing.
Blood-stage efficacy model.
Infection was performed by i.p. injection of 104 bloodstream forms in 0.2 mL TDB glucose of T. brucei brucei (STIB795) from a cryopreserved stock. Three days later, 9 infected animals with confirmed parasitemia were divided into two groups: control (infected mice treated with vehicle, n=4) and treated (infected mice treated with 30 mg/kg/day of 18, n=5). For treatment, both control and 18 were heated at 50 °C for 10 min to solubilize. Control and drug-treated mice received a 0.2 mL i.p. injection at the 3rd day from infection during 5 consecutive days. Parasitemia was individually checked by direct microscopic counting of parasites in a Neubauer chamber using 2 μL of blood from infected mice tail, diluted in 100 μL of TDB glucose. Parasitaemia of all mice were periodically checked by tail blood examination up to day 31th. The day of death was recorded.
CNS efficacy model.
Infection was performed by i.p. injection of 2·104 bloodstream forms in 0.2 mL TDB glucose of T. brucei brucei (GVR25 strain) from a cryopreserved stock. Fourteen days later, 13 infected animals with confirmed parasitemia were divided into three equal groups: control (infected mice pretreated with a single dose of 40 mg/kg diminazene aceturate -Berenil®- and treated with vehicle BID, n=5); treatment 1 (infected mice pretreated with 40 mg/kg Berenil® + 25 mg/kg /day BID 11, n=4) and treatment 2 (infected mice just treated with 25 mg/kg /day BID 18, n=4): the total concentration of 18 administered to mice was 50 /mg/kg/day. For treatment, vehicle and 18 were heated at 50 °C for 10 min to solubilize. Mice received a 0.2 mL i.p. injection of vehicle or 18 at the 21th day from infection during 5 consecutive days. Parasitemia was individually checked by direct microscopic counting of parasites in a Neubauer chamber using 2 μL of blood from infected mice tail, diluted in 100 μL of TDB glucose. Parasitaemia of all mice were checked twice a week by tail blood examination and thereafter mice with parasitaemia relapses were euthanized and the day of parasitaemia relapse was recorded. Results are expressed as percentage of accumulative mortality and in Mean Survival Days (MSD).
General Chemistry.
Reagents purchased were used as received, unless otherwise noted. Purification of intermediates and final compounds was performed using silica gel chromatography using the Biotage® Isolera™One flash purification system. When required, preparative HPLC was conducted for final compounds on Waters FractionLynx system using acetonitrile/water and 0.1% formic acid gradient and collected based on UV monitoring at 254 nm. LCMS analysis was performed using a Waters Alliance reverse phase HPLC (columns Waters SunFire C18 4.6 × 50 mm, 3.5 μm, or Waters SunFire C8 4.6 × 50 mm, 3.5 μm), using a multi-wavelength photodiode array detector from 210 nm to 600 nm and Waters Micromass ZQ detector (electrospray ionization). All compounds tested had a purity of > 95% as measured by LCMS. 1H NMR spectra were obtained with Varian NMR systems, operating at either 400 or 500 MHz at room temperature, using solvents from Cambridge Isotope Laboratories. Chemical shifts (δ, ppm) are reported relative to the solvent peak (CDCl3: 7.26 [1H]; DMSO-d6: 2.50 [1H]; Acetone-d6: 2.05; or CD3OD: 3.31 [1H]). Data for 1H NMR spectra are reported as follows: chemical shift (ppm), multiplicity (s for singlet, d for doublet, t for triplet, dd for doublet of doublet, m for multiplet), coupling constant (Hz), and integration. Compounds obtained from GSK in-house library were not resynthesized unless otherwise noted.
General Procedure A.
Desired boronate, desired aryl halide (1.5 equiv.), and potassium carbonate (3.5 equiv.) were suspended in 3:1 dioxane:water (0.08 M), and the reaction vial was degassed for ~10 minutes. Pd(PPh3)4 (5 mol%) was added and the reaction was run under nitrogen at 100 °C until completion as indicated by LCMS analysis (~4 h). The reaction mixture was diluted with EtOAc or MeOH, filtered through celite, and concentrated under reduced pressure. The resulting crude residue was purified by the stated method to afford the desired product.
General Procedure B.
The desired alcohol (1.2 equiv.) was dissolved in THF (0.64 M) and 1 M LHMDS in THF (1.2 equiv.) was added dropwise. The reaction was stirred at room temperature for ~1 h, then cooled to 0 °C. 4-Bromo-2,6-difluorobenzonitrile 52 was added and the reaction was allowed to warm to room temperature and stirred until completion by TLC. The reaction mixture was diluted with EtOAc, poured over water, and extracted three times. The combined organic layers were washed once with brine, dried with sodium sulfate and rotovapped. The crude material was purified by the stated method to afford the title compound.
General Procedure C.
The desired 4-bromo-2-alkoxy-6-fluorobenzonitrile 53 was dissolved in ethanol (0.10 M) and hydrazine monohydrate (10 equiv.) was added. The reaction was refluxed overnight at 95 °C. The reaction was cooled to room temperature and the solvent was removed under reduced pressure. The crude material was purified by the stated method to afford the title compound.
General Procedure D.
The desired 6-bromo-4-alkoxy-1H-indazol-3-amine 54 was dissolved in pyridine (0.09 M) and acetic anhydride (3.5 equiv.) was added. The reaction was refluxed at 100 °C for ~4 h. The reaction mixture was diluted with DCM and extracted three times with 1M HCl. The combined aqueous layers were extracted once with DCM, and the combined organic layers were washed once with water, once with brine, dried with sodium sulfate, and concentrated under reduced pressure. The crude material was purified by the stated method to afford the title compound.
General Procedure E.
The desired protected 6-bromo-4-alkoxy-1H-indazol-3-amines 55 (1.0 equiv.), potassium acetate (3.5 equiv.), bis(pinacolato)diboron (1.5 equiv.), and PdCl2(dppf)·CH2Cl2 (0.5 equiv.) were combined in a reaction vial that was filled with nitrogen and evacuated three times. Dry, degassed dioxane (0.10 M) was added and the reaction was run in the microwave (145 °C) for 0.5 h. The reaction mixture was diluted with EtOAc, filtered through celite, and concentrated under reduced pressure. The crude material was either taken forward without further purification or purified by the stated method and used as a mixture of mono- and di-protected boronic acid and ester 56.
Potassium carbonate (3.0 equiv.), 2-amino-4-chloropyrimidine (1.2 equiv.), and PdCl2(dppf)·CH2Cl2 (0.5 equiv.) were combined in a microwave vial that was filled with nitrogen and evacuated three times. The requisite boronic ester 56 or mixture of mono- and di-protected boronic acid and ester was dissolved in 3:1 dioxane:water (0.10 M) and added to the reaction mixture. The reaction was degassed for ~10 minutes and run in the microwave (150 °C) for 0.5–1.5 h. The reaction mixture was diluted with MeOH, filtered through celite, and concentrated under reduced pressure. The resulting crude residue was purified by the stated method to afford the desired product.
4-Bromo-2,6-difluorobenzoyl chloride (2).
4-Bromo-2,6-difluorobenzoic acid 1 (3.00, 12.66 mmol) was suspended in thionyl chloride (15 ml, 207 mmol) and the reaction was refluxed at 75 °C for approximately 4 h. Excess thionyl chloride was removed by distillation as the product was azeotroped with toluene three times, affording the title compound as a yellow oil which was used in the next reaction without further purification.
4-Bromo-2,6-difluoro-N-(2-hydroxyphenyl)benzamide (3).
2-Aminophenol (1.38 g, 12.65 mmol) was dissolved in DCM (30 ml, 0.42 M), and TEA (3.5 ml, 25.11 mmol) was added. Compound 2 (3.23 g, 12.64 mmol) was dissolved in DCM (15 ml, 0.22 M), and this solution was added to the reaction mixture dropwise at 0 °C. The reaction was allowed to warm to room temperature and stirred overnight. After pouring the reaction mixture over 1M HCl, the title compound was isolated by vacuum filtration as an off-white solid (3.62 g, 87%). LCMS [M+H]+ 327.95 m/z (79Br), 329.96 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 10.03 (s, 1 H) 9.88 (br. s., 1 H) 7.88 (dd, J=8.1, 1.2 Hz, 1 H) 7.60 (d, J=7.3 Hz, 2 H) 6.97 – 7.03 (m, 1 H) 6.91 (dd, J=8.1, 1.2 Hz, 1 H) 6.78 – 6.84 (m, 1 H).
3-Bromo-1-fluorodibenzo[b,f][1,4]oxazepin-11(10H)-one (4).
Compound 3 (3.62 g, 11.04 mmol) was dissolved in DMF (30 ml, 0.37 M) and potassium carbonate (3.05 g, 22.06 mmol) was added. The reaction was stirred at room temperature for two days and was then poured over water. The title compound was isolated by vacuum filtration as an off-white solid (2.44 g, 72%). LCMS [M+H]+ 307.99 m/z (79Br), 309.96 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 10.74 (br. s., 1 H) 7.61 (s, 1 H) 7.57 (dd, J=9.8, 1.5 Hz, 1 H) 7.37 – 7.41 (m, 1 H) 7.22 (dd, J=8.3, 1.5 Hz, 1 H) 7.14 – 7.19 (m, 2 H).
3-Bromo-1-fluorodibenzo[b,f][1,4]oxazepine-11(10H)-thione (5).
Compound 4 (2.44 g, 7.91 mmol) was suspended in toluene (160 ml, 0.05M) and Lawesson’s reagent (3.49 g, 8.63 mmol) was added. The reaction was refluxed at 100 °C overnight. Upon cooling to room temperature, a yellow precipitate was observed and collected by vacuum filtration to afford the title compound as a yellow solid (1.75 g) which was carried forward without further purification. LCMS [M+H]+ 323.93 m/z (79Br), 325.95 m/z (81Br).
4-Bromo-2,11-dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazole (6).
Compound 5 (1.00 g, 3.09 mmol) was suspended in dioxane (10 ml, 0.3 M) and hydrazine monohydrate (0.30 ml, 6.11 mmol) was added. The reaction was refluxed for 3 h at 85 °C. After cooling to room temperature, the reaction mixture was diluted with DCM and concentrated under reduced pressure. The residue was purified by flash chromatography (0–5% MeOH:DCM) to afford the title compound as an off-white solid (833 mg, 89%). LCMS [M+H]+ 301.97 m/z (79Br), 303.99 m/z (81Br); 1H NMR (400 MHz, DMSO-d6) δ ppm 12.29 (s, 1 H) 9.52 (s, 1 H) 7.31 (d, J=1.5 Hz, 1 H) 7.26 (dd, J=8.1, 1.5 Hz, 1 H) 7.23 (dd, J=8.1, 1.5 Hz, 1 H) 7.04 – 7.09 (m, 1 H) 6.85 – 6.91 (m, 2 H).
1-(4-Bromobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-11(2H)-yl)ethan-1-one and 1-(4-bromobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-2(11H)-yl)ethan-1-one (7).
Acetic anhydride (12.0 ml, 127.18 mmol) was added to 6 (833 mg, 2.76 mmol) and the reaction was refluxed at 100 °C for ~3 h. Upon cooling to room temperature, a bright yellow precipitate was observed and collected by vacuum filtration to afford the title compounds as a mixture of isomers. (Bright yellow solid, 885 mg, 93%). LCMS [M+H]+ 343.99 m/z (79Br), 345.96 m/z (81Br).
1-(4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)benzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-11(2H)-yl)ethan-1-one (8).
Compound 7 (401 mg, 1.17 mmol), potassium acetate (405 mg, 4.13 mmol), bis(pinacolato)diboron (443 mg, 1.74 mmol), and PdCl2(dppf)·CH2Cl2 (46 mg, 0.06 mmol) were combined in a reaction vial that was filled with nitrogen and evacuated three times. Dry, degassed dioxane (8.0 ml, 0.15 M) was added and the reaction was run in the microwave (145 °C) for 1 h. The reaction mixture was then diluted with EtOAc, filtered through celite, and concentrated under reduced pressure. The residue was purified by flash chromatography (0–50% EtOAc:hexanes) to afford the title compound as an off-white solid (297 mg, 65%). LCMS [M+H]+ 392.13 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 10.03 (s, 1 H) 8.25 (s, 1 H) 7.27 (dd, J=7.6, 3.2 Hz, 2 H) 7.26 (s, 1 H) 7.11 (t, J=7.6 Hz, 1 H) 6.98 (t, J=7.3 Hz, 1 H) 2.62 (s, 3 H) 1.34 (s, 12 H).
4-(Pyrimidin-4-yl)-2,11-dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazole (9b).
The title compound was prepared according to General Procedure A on a 40-mg scale using 8 and 4-chloropyrimidine·HCl. Upon cooling to room temperature, a precipitate was observed and collected by vacuum filtration. The precipitate was suspended in DME (0.75 ml, 0.06 M) and concentrated HCl (0.25 ml, 3.00 mmol) was added. This suspension was stirred at 85 °C for ~1 h, then cooled to room temperature. The resulting precipitate was collected to afford the title compound as an orange solid (12 mg, 40%). LCMS [M+H]+ 302.09 m/z; 1H NMR (400 MHz, DMSO-d6) δ ppm 12.55 (br. s., 1 H) 9.59 (s, 1 H) 9.28 (s, 1 H) 8.89 (d, J=5.9 Hz, 1 H) 8.24 (dd, J=5.1, 1.5 Hz, 1 H) 8.00 (s, 1 H) 7.58 (s, 1 H) 7.29 (dd, J=8.1, 1.5 Hz, 2 H) 7.08 (t, J=7.7 Hz, 1 H) 6.90 (t, J=6.6 Hz, 1 H).
6-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)pyrimidin-4-amine (9c).
The title compound was prepared according to General Procedure A on a 41-mg scale using 8 and 4-amino-6-chloropyrimidine. The crude material was purified preparative HPLC (5–50% water:acetonitrile) to afford the title compound as a yellow solid (15 mg, 45%). LCMS [M+H]+ 317.11 m/z; 1H NMR (400 MHz, DMSO-d6) δ ppm 12.36 (s, 1 H) 9.52 (s, 1 H) 8.46 (s, 1 H) 7.74 (s, 1 H) 7.23 – 7.32 (m, 3 H) 7.06 (t, J=7.3 Hz, 1 H) 6.99 (s, 1 H) 6.92 (s, 2 H) 6.89 (t, J=8.1 Hz, 1 H).
2-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)pyrimidin-4-amine (9d).
The title compound was prepared according to General Procedure A on a 41-mg scale using 8 and 4-amino-2-chloropyrimidine. The crude material was purified by preparative HPLC (5–50% acetonitrile:water) to afford the title compound as a yellow solid (17 mg, 51%). LCMS [M+H]+ 317.11 m/z; 1H NMR (400 MHz, DMSO-d6) δ ppm 12.32 (s, 1 H) 9.49 (s, 1 H) 8.20 (d, J=5.9 Hz, 1 H) 8.07 (s, 1 H) 7.71 (s, 1 H) 7.28 (d, J=8.1 Hz, 2 H) 7.06 (t, J=6.6 Hz, 1 H) 6.96 (br. s., 2 H) 6.88 (t, J=7.3 Hz, 1 H) 6.39 (d, J=5.9 Hz, 1 H).
6-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)pyridin-2-amine formate (9e).
The title compound was prepared according to General Procedure A on a 75-mg scale using 8 and 2-amino-6-chloropyridine. The crude material was purified by flash chromatography (50% EtOAc:hexanes – 33%/33%/33% EtOAc:hexanes:acetone – 33% EtOAc:acetone), further purified by flash chromatography (10% MeOH:DCM), and finally purified by preparative HPLC (5–95% acetonitrile:water to afford the title compound as a colorless solid (45 mg, 74%). LCMS [M+H]+ 315.99 m/z; 1H NMR (399 MHz, DMSO-d6) δ ppm 12.23 (s, 1 H) 9.48 (s, 1 H) 8.15 (s, 1 H) 7.70 (s, 1 H) 7.47 (t, J=8.1 Hz, 1 H) 7.38 (s, 1 H) 7.27 (d, J=8.1 Hz, 2 H) 7.15 (d, J=7.3 Hz, 1 H) 7.05 (t, J=7.0 Hz, 1 H) 6.88 (t, J=8.1 Hz, 1 H) 6.44 (d, J=8.1 Hz, 1 H) 6.06 (s, 2 H).
2-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)pyridin-4-amine (9f).
The title compound was prepared according to General Procedure A on a 51-mg scale using 8 and 4-amino-2-bromopyridine. The crude material was dissolved in MeOH (2.0 ml, 0.10 M) and potassium carbonate (54 mg, 0.391 mmol) was added before stirring at room temperature for ~1 h. The reaction mixture was diluted with EtOAc and washed with water; the aqueous layer was then extracted twice with EtOAc. The combined organic layers were concentrated under reduced pressure and purified by flash chromatography (3–20% MeOH:1% TEA/DCM) to afford the title compound as a dark gold solid (4.8 mg, 8%). LCMS [M+H]+ 315.99 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 12.67 (s, 1 H) 9.65 (s, 1 H) 8.14 (d, J=6.8 Hz, 1 H) 7.96 (br. s, 2 H) 7.54 (s, 1 H) 7.25 – 7.34 (m, 2 H) 7.18 (d, J=2.0 Hz, 1 H) 7.07 – 7.13 (m, 2 H) 6.91 (t, J=8.3 Hz, 1 H) 6.83 (d, J=8.3 Hz, 1 H).
4-Phenyl-2,11-dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazole (9g).
The title compound was prepared according to General Procedure A on a 40-mg scale using 8 and chlorobenzene. The crude material was purified by flash chromatography (40% EtOAc:hexanes) to afford the title compound as a tan solid (14 mg, 46%). LCMS [M+H]+ 300.06 m/z; 1H NMR (400 MHz, DMSO-d6) δ ppm 12.23 (s, 1 H) 9.48 (s, 1 H) 7.75 (d, J=7.3 Hz, 2 H) 7.49 (t, J=7.3 Hz, 2 H) 7.40 (d, J=7.3 Hz, 1 H) 7.24 – 7.32 (m, 3 H) 7.06 (t, J=8.1 Hz, 1 H) 7.01 (s, 1 H) 6.88 (dd, J=8.1, 6.60 Hz, 1 H).
6-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)-N4-methylpyrimidine-2,4-diamine formate (9j).
The title compound was prepared according to General Procedure a on a 50-mg scale using 8 and 6-chloro-N4-methylpyrimidine-2,4-diamine. The crude material was purified by flash chromatography (20–100% EtOAc:hexanes – 0–20% MeOH:EtOAc), then repurified by preparative HPLC (5–95% acetonitrile:water) to afford the formate salt of the title compound as a solid (6 mg, 13%). LCMS [M+H]+ 346.09 m/z; 1H NMR (500 MHz, METHANOL-d4) δ ppm 8.47 (br. s., 1 H) 7.46 (br. s., 1 H) 7.26 (d, J=7.81 Hz, 1 H) 7.16 (d, J=8.30 Hz, 1 H) 7.01 – 7.11 (m, 2 H) 6.91 (t, J=7.81 Hz, 1 H) 6.34 (s, 1 H) 3.00 (s, 3 H).
6-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)-N4-ethylpyrimidine-2,4-diamine (9k).
The title compound was prepared according to General Procedure A on a 40-mg scale using 8 and 6-chloro-N4-ethylpyrimidine-2,4-diamine. The crude material was purified by flash chromatography (0–20% MeOH:DCM) to afford the title compound as a solid (20 mg, 50%). LCMS [M+H]+ 360.39m/z; 1H NMR (500 MHz, METHANOL-d4) δ ppm 7.52 (s, 1 H) 7.26 (dd, J=7.8, 1.5 Hz, 1 H) 7.10 – 7.17 (m, 2 H) 7.03 (td, J=7.6, 1.5 Hz, 1 H) 6.89 (td, J=7.8, 1.5 Hz, 1 H) 6.26 (s, 1 H) 3.40 (q, J=7.2 Hz, 2 H) 1.24 (t, J=7.3 Hz, 3 H).
6-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)-N4-isopropylpyrimidine-2,4-diamine formate (9l).
The title compound was prepared according to General Procedure A on a 50-mg scale 8 and using 6-chloro-N4-isopropylpyrimidine-2,4-diamine. The crude material was purified by flash chromatography (20–100% EtOAc:hexanes – 0–20% MeOH:DCM), then repurified by preparative HPLC (5–95% acetonitrile:water) to afford the formate salt of the title compound as a solid (8 mg, 17%). LCMS [M+H]+ 374.09 m/z; 1H NMR (500 MHz, METHANOL-d4) δ ppm 8.50 (s, 1 H) 7.46 (s, 1 H) 7.27 (dd, J=8.8, 1.46 Hz, 1 H) 7.17 (dd, J=8.3, 1.5 Hz, 1 H) 7.03 – 7.10 (m, 2 H) 6.92 (t, J=6.4 Hz, 1 H) 6.31 (s, 1 H) 4.21 – 4.39 (m, 1 H) 1.27 (d, J=6.8 Hz, 6 H).
6-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)-N4,N4-dimethylpyrimidine-2,4-diamine (9o).
The title compound was prepared according to General Procedure A on a 35-mg scale using 8 and 6-chloro-N4,N4-dimethylpyrimidine-2,4-diamine. The crude material was purified by flash chromatography (4–20% MeOH:DCM) to afford the title compound as a solid (16 mg, 35%). LCMS [M+H]+ 360.39 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 12.35 (s, 1 H) 9.53 (s, 1 H) 7.79 (s, 1 H) 7.41 (s, 1 H) 7.24 – 7.31 (m, 2 H) 7.06 (td, J=6.8, 1.5 Hz, 1 H) 6.90 (td, J=7.8, 1.5 Hz, 1 H) 6.55 (s, 1 H) 6.25 (br. s, 2 H) 3.11 (s, 6 H).
4-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)-6-morpholinopyrimidin-2-amine (9p).
The title compound was prepared according to General Procedure A on a 50-mg scale using 8 and 4-chloro-6-morpholinopyrimidin-2-amine. The crude material was purified by flash chromatography (4–20% MeOH:DCM), then re-purified by preparative HPLC (5–95% acetonitrile:water) to afford the title compound as a solid (5 mg, 10%). LCMS [M+H]+ 402.09 m/z; 1H NMR (500 MHz, Acetone) δ ppm 11.42 (br. s, 1 H) 8.52 (s, 1 H) 7.91 (d, J=1.0 Hz, 1 H) 7.49 (d, J=1.0 Hz, 1 H) 7.33 (ddd, J=13.7, 7.8, 1.5 Hz, 2 H) 7.09 (ddd, J=8.3, 7.1, 1.7 Hz, 1 H) 6.93 (td, J=7.3, 2.0 Hz, 1 H) 6.71 (s, 1 H) 5.69 (br. s., 2 H) 3.70 – 3.76 (m, 4 H) 3.66 – 3.70 (m, 4 H).
4-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)-6-(pyrrolidin-1-yl)pyrimidin-2-amine formate (9q).
The title compound was prepared according to General Procedure A on a 50-mg scale using 8 and 4-chloro-6-(pyrrolidin-1-yl)pyrimidin-2-amine. The crude material was purified by flash chromatography (4–20% MeOH:DCM), then re-purified by preparative HPLC (5–95% acetonitrile:water) to afford the formate salt of the title compound as a solid (9 mg, 18%). LCMS [M+H]+ 386.09 m/z; 1H NMR (500 MHz, Acetone-d6) δ ppm 11.38 (br. s, 1 H) 8.50 (s, 1 H) 8.12 (s, 1 H) 7.88 (s, 1 H) 7.48 (d, J=0.98 Hz, 1 H) 7.34 (t, J=7.81 Hz, 2 H) 7.08 (t, J=7.32 Hz, 1 H) 6.94 (t, J=6.84 Hz, 1 H) 6.39 (s, 1 H) 5.53 (br. s., 2 H) 3.52 (t, J=5.37 Hz, 4 H) 2.00 (br. s., 4 H).
4-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)-6-(piperidin-1-yl)pyrimidin-2-amine formate (9r).
The title compound was prepared according to General Procedure A on a 50-mg scale using 8 and 4-chloro-6-(piperidin-1-yl)pyrimidin-2-amine. The crude material was purified by flash chromatography (20–100% EtOAc:hexanes – 0–20% MeOH:EtOAc), then repurified by preparative HPLC (5–95% acetonitrile:water) to afford the formate salt of the title compound as a solid (7 mg, 14%). LCMS [M+H]+ 400.13 m/z; 1H NMR (500 MHz, METHANOL-d4) δ ppm 8.48 – 8.52 (m, 1 H) 7.52 (s, 1 H) 7.28 (dd, J=8.8, 1.46 Hz, 1 H) 7.14 – 7.19 (m, 2 H) 7.06 (td, J=7.3, 1.5 Hz, 1 H) 6.91 (td, J=7.3, 2.0 Hz, 1 H) 6.62 (s, 1 H) 3.80 (t, J=5.4 Hz, 4 H) 1.73 – 1.79 (m, 1 H) 1.63 – 1.71 (m, 4 H).
4-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)-6-(4-methylpiperazin-1-yl)pyrimidin-2-amine (9s).
The title compound was prepared according to General Procedure A on a 20-mg scale using 8 and 4-chloro-6-(4-methylpiperazin-1-yl)pyrimidin-2-amine. The crude material was purified by flash chromatography (0–60% 10% NH4OH in MeOH:DCM) to afford the title compound as a solid (7 mg, 19%). LCMS [M+H]+ 415.47 m/z; 1H NMR (500 MHz, METHANOL-d4) δ ppm 7.58 (s, 1 H) 7.27 (dd, J=7.8, 1.5 Hz, 1 H) 7.23 (s, 1 H) 7.16 (dd, J=8.8, 1.5 Hz, 1 H) 7.06 (td, J=8.3, 1.5 Hz, 1 H) 6.92 (td, J=8.8, 1.5 Hz, 1 H) 6.55 (s, 1 H) 3.76 (t, J=4.9 Hz, 4 H) 2.54 (t, J=4.9 Hz, 4 H) 2.36 (s, 3 H).
3-Bromo-1-fluoro-10-methyldibenzo[b,f][1,4]oxazepin-11(10H)-one (10).
Compound 4 (482 mg, 1.56 mmol) was dissolved in dry DMF (6 ml, 0.27 M). Sodium hydride (129 mg, 3.23 mmol) was added and the reaction was stirred for 30 min at room temperature. Methyl iodide (0.15 mL, 2.41 mmol) was added dropwise at 0 °C and the reaction was stirred for an additional 4 h, warming to room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted three times with EtOAc. The combined organic layers were washed once with water and once with brine, dried with sodium sulfate, and concentrated under reduced pressure. The crude residue was purified by flash chromatography (10–20% EtOAc:hexanes) to afford the title compound as a white solid (267 mg, 53%). LCMS [M+H]+ 321.98 m/z (79Br), 323.99 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 7.62 (s, 1 H) 7.51 – 7.59 (m, 2 H) 7.43 (d, J=8.3 Hz, 1 H) 7.33 (dd, J=7.8 Hz, 1 H) 7.26 (dd, J=7.8 Hz, 1 H) 3.49 (s, 3 H).
3-Bromo-1-fluoro-10-methyldibenzo[b,f][1,4]oxazepine-11(10H)-thione (11).
Compound 10 (280 mg, 0.869 mmol) was dissolved in toluene (3.5 ml, 0.25 M) and phosphorus pentasulfide (291 mg, 1.31 mmol) was added. The reaction was refluxed at 100 °C overnight, then additional phosphorus pentasulfide (256 mg, 1.15 mmol) was added. The reaction continued to reflux at 100 °C for an additional 24 h. The reaction was cooled to room temperature and the solvent was removed under reduced pressure. The crude residue was purified by flash chromatography (0–25% methyl tert-butyl ether:hexanes) to afford the title compound as a yellow solid (200 mg, 68%). LCMS [M+H]+ 337.94 m/z (79Br), 339.93 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 7.66 (dd, J=7.3, 2.0 Hz, 1 H) 7.58 (t, J=1.7 Hz, 1 H) 7.52 (dd, J=10.0, 2.0 Hz, 1 H) 7.44 – 7.48 (m, 1 H) 7.32 – 7.40 (m, 2 H) 3.93 (s, 3 H).
4-Bromo-11-methyl-2,11-dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazole (12).
Compound 11 (200 mg, 0.620 mmol) was suspended in dioxane (3.1 mL, 0.2 M) and hydrazine monohydrate (0.06 mL, 1.22 mmol) was added. The reaction was refluxed for 3 h at 85 °C. After cooling to room temperature, the solvent was removed under reduced pressure. The resulting crude residue was purified by flash chromatography (20–30% EtOAc:hexanes) to afford the title compound as an orange solid (125 mg, 63%). LCMS [M+H]+ 315.93 m/z (79Br), 317.92 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 12.36 (s, 1 H) 7.31 – 7.35 (m, 3 H) 7.26 (td, J=7.8, 1.5 Hz, 1 H) 7.08 (td, J=6.8, 1.5 Hz, 1 H) 6.91 (d, J=1.0 Hz, 1 H) 3.45 (s, 3 H).
1-(4-Bromo-11-methylbenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-2(11H)-yl)ethan-1-one (13).
Acetic anhydride (1.1 mL, 11.64 mmol) was added to 12 (125 mg, 0.395 mmol). The reaction was refluxed at 100 °C for 3 h, then cooled to room temperature. Upon cooling, a white precipitate was observed and collected by vacuum filtration (washed with water) to afford the title compound as a white solid (126 mg, 89%). LCMS [M+H]+ 357.96 m/z (79Br), 359.93 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 8.08 (s, 1 H) 7.42 (d, J=7.8 Hz, 1 H) 7.39 (s, 1 H) 7.36 (d, J=7.8 Hz, 1 H) 7.32 (t, J=7.3 Hz, 1 H) 7.18 (t, J=8.3 Hz, 1 H) 3.52 (s, 3 H) 2.63 (s, 3 H).
1-(11-Methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzo[2,3][1,4]oxazepino [5,6,7-cd]indazol-2(11H)-yl)ethan-1-one (14).
Compound 13 (95 mg, 0.265 mmol), potassium acetate (92 mg, 0.937 mmol), bis(pinacolato)diboron (102 mg, 0.402 mmol), and PdCl2(dppf) (12 mg, 14.7 μmol) were combined in a reaction vial that was filled with nitrogen and evacuated three times. Dry, degassed dioxane (2.2 mL, 0.12 M) was added and the reaction was run in the microwave (145 °C) for 1 h. The reaction mixture was diluted with EtOAc and filtered through Celite®, and the crude residue was purified by flash chromatography (0–20% EtOAc:hexanes) to afford the title compound as an off-white solid (90 mg, 83%). LCMS [M+H]+ 406.09 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 8.24 (s, 1 H) 7.38 – 7.43 (m, 2 H) 7.26 – 7.32 (m, 2 H) 7.16 (td, J=7.6, 1.5 Hz, 1 H) 3.53 (s, 3 H) 2.64 (s, 3 H) 1.16 (s, 12 H).
4-(11-Methyl-2,11-dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)pyrimidin-2-amine (15).
The title compound was prepared according to General Procedure A on a 54-mg scale using 14 and 2-amino-4-chloropyrimidne. The crude material was purified twice by flash chromatography (20–100% EtOAc:DCM - 0–5% MeOH:EtOAc, then 1–5% 1% NH4OH/MeOH:EtOAc) and once by preparative HPLC (5–95% water:acetonitrile) to afford the title compound as a yellow solid (4 mg, 10%). LCMS [M+H]+ 331.07 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 12.45 (s, 1 H) 8.32 (d, J=5.4 Hz, 1 H) 7.82 (s, 1 H) 7.45 (s, 1 H) 7.32 – 7.38 (m, 2 H) 7.25 (t, J=7.3 Hz, 1 H) 7.22 (d, J=5.4 Hz, 1 H) 7.08 (t, J=8.3 Hz, 1 H) 6.73 (s, 2 H) 3.48 (s, 3 H).
4-Bromo-2-methyl-2,11-dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazole (16).
Compound 5 (199 mg, 0.614 mmol) was suspended in dioxane (2.5 mL, 0.25 M) and methyl hydrazine (0.10 mL, 1.90 mmol) was added. The reaction was refluxed for 4 h at 85 °C, then stopped and cooled to room temperature. Upon cooling, a precipitate was observed and collected by vacuum filtration (washed with water) to afford the title compound as an off-white solid (83 mg, 43%). LCMS [M+H]+ 315.95 m/z (79Br), 317.96 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 9.56 (s, 1 H) 7.55 (s, 1 H) 7.21 – 7.25 (m, 2 H) 7.07 (ddd, J=8.2, 7.0, 1.5 Hz, 1 H) 6.86 – 6.91 (m, 2 H) 3.87 (s, 3 H).
2-Methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,11-dihydrobenzo[2,3][1,4] oxazepino[5,6,7-cd]indazole (17).
Compound S6 (83 mg, 0.262 mmol), potassium acetate (91 mg, 0.927 mmol), bis(pinacolato)diboron (101 mg, 0.398 mmol), and PdCl2(dppf)·CH2Cl2 (12 mg, 0.015 mmol) were combined in a reaction vial that was filled with nitrogen and evacuated three times. Dry, degassed dioxane (2.7 mL, 0.15 M) was added and the reaction was run in the microwave (140 °C) for 30 min. The reaction was diluted with EtOAc, filtered through celite, and concentrated under reduced pressure. The crude residue was purified by flash chromatography (20–60% EtOAc:hexanes) to afford the title compound as a yellow solid (60 mg, 63%). LCMS [M+H]+ 364.18 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 9.49 (s, 1 H) 7.49 (s, 1 H) 7.25 (d, J=7.8 Hz, 2 H) 7.05 (td, J=8.3, 1.5 Hz, 1 H) 6.93 (s, 1 H) 6.87 (td, J=8.3, 1.5 Hz, 1 H) 3.92 (s, 3 H) 1.33 (s, 12 H).
4-(2-Methyl-2,11-dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)pyrimidin-2-amine (18).
The title compound was prepared according to General Procedure A on a 60-mg scale using 17 and 2-amino-4-chloropyrimidne. The crude material was purified by flash chromatography (0–100% EtOAc:hexanes - 0–10% MeOH:EtOAc), then by preparative HPLC (95–30% water:acetonitrile) to afford the title compound as a yellow solid (6 mg, 10%). LCMS [M+H]+ 331.12 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 9.55 (s, 1 H) 8.35 (d, J=5.4 Hz, 1 H) 7.95 (s, 1 H) 7.50 (s, 1 H) 7.30 (d, J=4.9 Hz, 1 H) 7.24 – 7.29 (m, 2 H) 7.06 (t, J=7.8 Hz, 1 H) 6.89 (t, J=7.81 Hz, 1 H) 6.73 (s, 2 H) 3.96 (s, 3 H).
4-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)-N,N-dimethylpyrimidin-2-amine (19).
The title compound was prepared according to General Procedure A on a 100-mg scale using 8 and 4-chloro-N,N-dimethylpyrimidin-2-amine. The crude material was purified by flash chromatography (0–100% EtOAc:hexanes), then repurified by preparative HPLC (95–5% water:acetonitrile) to afford the final compound as a yellow solid (7 mg, 8%). LCMS [M+H]+ 345.00 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 12.38 (s, 1 H) 9.55 (s, 1 H) 8.43 (d, J=4.9 Hz, 1 H) 7.90 (d, J=1.0 Hz, 1 H) 7.49 (d, J=1.0 Hz, 1 H) 7.25 – 7.32 (m, 3 H) 7.07 (td, J=8.3, 1.5 Hz, 1 H) 6.89 (td, J=8.3, 1.5 Hz, 1 H) 3.23 (s, 6 H).
4-(2,11-Dihydrobenzo[2,3][1,4]oxazepino[5,6,7-cd]indazol-4-yl)-N-methylpyrimidin-2-amine (20).
The title compound was prepared according to General Procedure B on a 75-mg scale using 8 and 4-bromo-N-methylpyrimidin-2-amine. The crude material was purified by flash chromatography (70% EtOAc:hexanes) to afford the title compound as a yellow solid (13 mg, 21%). LCMS [M+H]+ 331.12 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 12.38 (s, 1 H) 9.53 (s, 1 H) 8.36 (s, 1 H) 7.87 (s, 1 H) 7.47 (s, 1 H) 7.28 (dd, J=7.8, 1.5 Hz, 2 H) 7.24 (d, J=4.9 Hz, 1 H) 7.13 – 7.20 (m, 1 H) 7.06 (td, J=8.8, 7.3 Hz, 1 H) 6.88 (td, J=9.3, 6.3 Hz, 1 H) 2.90 (br. s., 3 H).
4-Bromo-2,6-difluoro-N-(2-hydroxyethyl)-N-(4-methoxybenzyl)benzamide (21).
2-((4Methoxybenzyl)amino)ethan-1-ol (2.32 g, 12.8 mmol) was dissolved in DCM (30 ml, 0.43 M) and TEA (3.80 ml, 27.3 mmol) was added. Compound 2 (2.83 g, 11.1 mmol) was dissolved in DCM (15 ml, 0.74 M) and this solution was added to the reaction mixture dropwise at 0 °C. The reaction was allowed to warm to room temperature and stirred overnight. The reaction mixture was diluted with DCM and washed once with 1M HCl, once with water, and once with brine. The organic layer was dried with sodium sulfate and concentrated under reduced pressure. The title compound was isolated by flash chromatography (20–50% EtOAc:hexanes) as a yellow oil (3.81 g, 86%). LCMS [M+H]+ 399.86 m/z (79Br), 401.90 m/z (81Br).
8-Bromo-6-fluoro-4-(4-methoxybenzyl)-3,4-dihydrobenzo[f][1,4]oxazepin-5(2H)-one (22).
Sodium hydride (766 mg, 19.2 mmol) was suspended in dry DMF (50 ml, 0.38 M). Compound 14 (3.81 g, 9.53 mmol) was dissolved in dry DMF (50 ml, 0.19 M) and added slowly to the sodium hydride suspension. The reaction was stirred at room temperature overnight. The reaction mixture was diluted with DCM and washed three times with saturate aqueous NaHCO3, once with water, and once with brine. The organic layer was dried with sodium sulfate and concentrated under reduced pressure to afford the title compound as a solid (3.06 g, 84%). LCMS [M+H]+ 379.99 m/z (79Br), 381.99 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 7.46 (dd, J=9.3, 2.0 Hz, 1 H) 7.29 (d, J=8.3 Hz, 2 H) 7.19 (d, J=1.5 Hz, 1 H) 6.93 (d, J=8.3 Hz, 2 H) 4.66 (s, 2 H) 4.11 (t, J=5.6 Hz, 2 H) 3.74 (s, 3 H) 3.50 (t, J=5.6 Hz, 2 H).
8-Bromo-6-fluoro-3,4-dihydrobenzo[f][1,4]oxazepin-5(2H)-one (23).
Compound 22 (1.65 g, 4.33 mmol) was taken up in 3:1 acetonitrile:water (48 ml, 0.09 M) and cerium ammonium nitrate (7.13 g, 13.0 mmol) was added, upon which the reaction mixture turned orange. The reaction was stirred at room temperature and monitored by LCMS. After ~3 h, the reaction mixture was diluted with water and extracted three times with EtOAc. The combined organic layers were washed once with sat. aq. NaHCO3 and once with brine. The combined organic layers were dried with sodium sulfate and concentrated under reduced pressure. The crude residue was purified by flash chromatography (20–80% EtOAc:hexanes) to afford the final product as a solid (410 mg, 36%). LCMS [M+H]+ 259.96 m/z (79Br), 261.96 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 8.56 (br. s., 1 H) 7.43 (dd, J=9.5, 1.71 Hz, 1 H) 7.20 (t, J=1.7 Hz, 1 H) 4.21 (t, J=5.6 Hz, 2 H) 3.26 (q, J=5.5 Hz, 2 H).
8-Bromo-6-fluoro-3,4-dihydrobenzo[f][1,4]oxazepine-5(2H)-thione (24).
Compound 23 (410 mg, 1.58 mmol) was suspended in toluene (30 ml, 0.05 M) and Lawesson’s reagent (702 mg, 1.74 mmol) was added. The reaction was refluxed overnight at 100 °C. The reaction mixture was concentrated under reduced pressure to a yellow residue, which was then purified by flash chromatography (0–50% EtOAc:hexanes) to afford the title compound as a solid (245 mg, 56%). LCMS [M+H]+ 275.94 m/z (79Br), 277.96 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 10.93 (br. s., 1 H) 7.47 (dd, J=9.3, 1.95 Hz, 1 H) 7.22 (t, J=1.5 Hz, 1 H) 4.27 (t, J=5.9 Hz, 2 H) 3.42 (q, J=5.9 Hz, 2 H).
4-Bromo-2,7,8,9-tetrahydro-[1,4]oxazepino[5,6,7-cd]indazole (25).
Compound 24 (245 mg, 0.887 mmol) was dissolved in dioxane (3.0 ml, 0.30 M) and hydrazine monohydrate (0.10 ml, 2.0 mmol) was added. The reaction was refluxed at 85 °C for 3 h. The reaction mixture was concentrated under reduced pressure, leaving an orange residue. The product was purified by flash chromatography (0–5% MeOH:DCM) to afford the title compound as a solid (214 mg, 95%). LCMS [M+H]+ 253.96 m/z (79Br), 255.99 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 11.75 (s, 1 H) 6.99 (d, J=1.5 Hz, 1 H) 6.46 (br. s., 1 H) 6.45 (d, J=1.5 Hz, 1 H) 4.32 – 4.40 (m, 2 H) 3.39 – 3.45 (m, 2 H).
1-(4-Bromo-8,9-dihydro-[1,4]oxazepino[5,6,7-cd]indazol-2(7H)-yl)ethan-1-one (26).
Acetic anhydride (2.5 ml, 27 mmol) was added to 25 (214 mg, 0.842 mmol) and the reaction was refluxed at 100 °C. After ~3 h, the reaction was cooled further to 0 °C and the title compound was isolated as a yellow solid by vacuum filtration (151 mg, 60%). LCMS [M+H]+ 295.99 m/z (79Br), 297.96 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 7.99 (d, J=1.5 Hz, 1 H) 7.45 (br. s., 1 H) 6.99 (d, J=1.5 Hz, 1 H) 4.41 – 4.45 (m, 2 H) 3.51 – 3.55 (m, 2 H) 3.32 (s, 3 H).
1-(4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-8,9-dihydro-[1,4]oxazepino[5,6,7-cd]indazol-2(7H)-yl)ethan-1-one (27).
Compound 26 (151 mg, 0.510 mmol), potassium acetate (175 mg, 1.78 mmol), bis(pinacolato)diboron (193 mg, 0.760 mmol), and PdCl2(dppf) (20 mg, 0.06 mmol) were combined in a reaction vial that was filled with nitrogen and evacuated three times. Dry, degassed dioxane (3.4 ml, 0.15 M) was added and the reaction was run in the microwave (145 °C) for 1 h. The reaction mixture was then diluted with EtOAc, filtered through celite, and concentrated under reduced pressure. The crude material was purified by flash chromatography (20–75% EtOAc:hexanes) to afford the title compound as a solid (139 mg, 79%). LCMS [M+H]+ 344.12 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 8.18 (s, 1 H) 7.40 (br. s., 1 H) 6.97 (s, 1 H) 4.38 – 4.43 (m, 2 H) 3.53 (m, J=4.9 Hz, 2 H) 2.51 (s, 3 H) 1.31 (s, 12 H).
6-(2,7,8,9-Tetrahydro-[1,4]oxazepino[5,6,7-cd]indazol-4-yl)pyrimidine-2,4-diamine (28a).
The title compound was prepared accoridng to General Procedure A on a 29-mg scale using 27 and 2,6-diamino-4-chloropyrimidine. The crude material was purified by flash chromatography (3–10% 15% NH4OH in MeOH:DCM), then re-purified by preparative HPLC (5–95% acetonitrile:water) to afford the title compound as a solid (8 mg, 33%). LCMS [M+H]+ 284.12 m/z; 1H NMR (500 MHz, Acetone) δ ppm 11.10 (br. s, 1 H) 8.14 (s, 1 H) 7.60 (s, 1 H) 6.97 (d, J=1.0 Hz, 1 H) 6.41 (s, 1 H) 5.97 (br. s, 2 H) 5.68 (br. s, 2 H) 4.41 – 4.47 (m, 2 H) 3.60 (m, J=6.3 Hz, 2 H).
6-(2,7,8,9-Tetrahydro-[1,4]oxazepino[5,6,7-cd]indazol-4-yl)pyrimidin-4-amine (28b).
The title compound was prepared accoridng to General Procedure A on a 29-mg scale using 27 and 4-amino-6-chloropyrimidine. The crude material was purified by flash chromatography (3–10% MeOH:DCM), then re-purified by preparative HPLC (5–95% acetonitrile:water) to afford the title compound as a solid (3 mg, 12%). LCMS [M+H]+ 269.11 m/z; 1H NMR (500 MHz, Acetone) δ ppm 11.03 (br. s, 1 H) 8.47 (s, 1 H) 7.67 (s, 1 H) 7.04 (s, 2 H) 6.21 (br. s, 2 H) 5.71 (br. s, 1 H) 4.44 – 4.48 (m, 2 H) 3.59 – 3.63 (m, 2 H).
4-(2,6-Diaminopyrimidin-4-yl)-2-fluorobenzonitrile (30a).
(4-Cyano-3-fluorophenyl)boronic acid 29 (75 mg, 0.46 mmol) and 6-chloropyrimidin-2,4-diamine (67 mg, 0.46 mmol) were combined in a reaction vial and dissolved in dioxane (3.0 mL, 0.15 M), followed by the addition of saturated aqueous NaHCO3 (0.75 mL). The reaction vial was purged with nitrogen for 10 min and Pd(PPh3)4 (28 mg, 0.024 mmol) was added. The reaction was run at 95 °C for 3 h. Upon completion, the reaction mixture was diluted with EtOAc, filtered through Celite®, and concentrated in vacuo. The crude material was purified by flash chromatography (0–7% MeOH:DCM) to afford the title compound as a yellow solid (75 mg, 72%). LCMS [M+H]+ 230.03 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 8.00 (t, J=7.3 Hz, 1 H) 7.95 (d, J=11.2 Hz, 1 H) 7.88 (dd, J=8.8, 1.47 Hz, 1 H) 6.54 (br. s., 2 H) 6.29 (s, 1 H) 6.12 (s, 2 H).
4-(6-Aminopyrimidin-4-yl)-2-fluorobenzonitrile (30b).
(4-Cyano-3-fluorophenyl)boronic acid 29 (75 mg, 0.46 mmol) and 6-chloropyrimidin-4-amine (60 mg, 0.46 mmol) were combined in a reaction vial and dissolved in dioxane (3.0 mL, 0.15 M), followed by the addition of saturated aqueous NaHCO3 (0.75 mL). The reaction vial was purged with nitrogen for 10 min and Pd(PPh3)4 (27 mg, 0.023 mmol) was added. The reaction was run at 95 °C for 3 h. Upon completion, the reaction mixture was diluted with EtOAc, filtered through celite, and concentrated in vacuo. The crude material was purified by flash chromatography (70–100% EtOAc:hexanes) to afford the title compound as an off-white solid (42 mg, 43%). LCMS [M+H]+ 214.95 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 8.49 (s, 1 H) 8.02 – 8.09 (m, 2 H) 7.98 (d, J=7.8 Hz, 1 H) 7.13 (s, 2 H) 6.99 (s, 1 H).
4-(2-Aminopyrimidin-4-yl)-2-fluorobenzonitrile (30c).
(4-Cyano-3-fluorophenyl)boronic acid 29 (75 mg, 0.46 mmol) and 4-chloropyrimidin-2-amine (59 mg, 0.46 mmol) were combined in a reaction vial and dissolved in dioxane (3.0 mL, 0.15 M), followed by the addition of saturated aqueous NaHCO3 (0.75 mL). The reaction vial was purged with nitrogen for 10 min and Pd(PPh3)4 (27 mg, 0.023 mmol) was added. The reaction was run at 95 °C for 3 h. Upon completion, the reaction mixture was diluted with EtOAc, filtered through Celite®, and concentrated in vacuo. The crude material was purified by flash chromatography (0–50% EtOAc:hexanes) to afford the title compound as an off-white solid (93 mg, 97%). LCMS [M+H]+ 215.01 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 8.42 (d, J=5.4 Hz, 1 H) 8.17 (d, J=11.7 Hz, 1 H) 8.11 (s, 2 H) 7.29 (d, J=4.9 Hz, 1 H) 6.88 (s, 2 H).
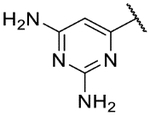
6-(3-Amino-1H-indazol-6-yl)pyrimidine-2,4-diamine (31a).
Compound 30a (50 mg, 0.218 mmol) was suspended in EtOH (3.7 mL, 0.06 M) and hydrazine monohydrate (0.900 mL, 18.33 mmol) was added. The reaction was run overnight at 95 °C. Upon cooling, an off-white precipitate was observed and collected by vacuum filtration to afford the title compound as an off-white solid (29 mg, 55%). LCMS [M+H]+ 242.05 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 11.51 (s, 1 H) 7.81 (s, 1 H) 7.69 (d, J=8.3 Hz, 1 H) 7.39 (d, J=8.8 Hz, 1 H) 6.33 (br. s., 2 H) 6.24 (s, 1 H) 5.95 (br. s., 2 H) 5.37 (s, 2 H).
6-(6-Aminopyrimidin-4-yl)-1H-indazol-3-amine (31b).
Compound 30b (42 mg, 0.196 mmol) was suspended in EtOH (3.3 mL, 0.06 M) and hydrazine monohydrate (0.800 mL, 16.30 mmol) was added. The reaction was run overnight at 95 °C. Upon cooling, a yellow precipitate was observed and collected by vacuum filtration to afford the title compound as a yellow solid (25 mg, 57%). LCMS [M+H]+ 227.01 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 11.58 (s, 1 H) 8.44 (d, J=1.0 Hz, 1 H) 7.88 (s, 1 H) 7.75 (d, J=8.3 Hz, 1 H) 7.48 (dd, J=8.6, 1.22 Hz, 1 H) 6.92 (d, J=1.0 Hz, 1 H) 6.89 (s, 2 H) 5.42 (br. s., 2 H).
6-(2-Aminopyrimidin-4-yl)-1H-indazol-3-amine (31c).
Compound 30c (50 mg, 0.233 mmol) was suspended in EtOH (3.7 mL, 0.06 M) and hydrazine monohydrate (0.900 mL, 18.33 mmol) was added. The reaction was run overnight at 95 °C. Upon cooling, a yellow precipitate was observed and collected by vacuum filtration to afford the title compound as a yellow solid (44 mg, 83%). LCMS [M+H]+ 227.03 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 11.62 (s, 1 H) 8.29 (d, J=4.9 Hz, 1 H) 7.98 (s, 1 H) 7.76 (d, J=8.3 Hz, 1 H) 7.58 (dd, J=8.6, 1.22 Hz, 1 H) 7.16 (d, J=5.4 Hz, 1 H) 6.66 (s, 2 H) 5.44 (br. s., 2 H).
6-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazole (33).
4-Bromoindazole 32 (251 mg, 1.27 mmol), bis(pinacolato)diboron (480 mg, 1.89 mmol), potassium acetate (433 mg, 4.41 mmol) and PdCl2(dppf)·CH2Cl2 (53 mg, 0.065 mmol) were combined in a microwave vial that was filled with nitrogen and evacuated three times. Dry, degassed dioxane (10 mL, 0.13 M) was added and the reaction was run in the microwave (145 °C) for 3 h. The reaction was diluted with EtOAc, filtered through celite, and concentrated under reduced pressure. The crude material was purified by flash chromatography (20–50% EtOAc:hexanes) to afford the title compound as a solid (183 mg, 59%). LCMS [M+H]+ 245.11 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 13.16 (s, 1 H) 8.09 (s, 1 H) 7.86 (s, 1 H) 7.75 (d, J=7.8 Hz, 1 H) 7.36 (d, J=7.8 Hz, 1 H) 1.32 (s, 12 H).
6-(1H-Indazol-6-yl)pyrimidine-2,4-diamine (34a).
The title compound was prepared according to General Procedure A on a 54-mg scale using 33 and 2,6-diamino-4-chloropyrimidine. The crude material was purified by flash chromatography (0–10% 10% NH4OH in MeOH:DCM) to afford the title compound as a solid (16 mg, 43%). LCMS [M+H]+ 227.08 m/z; 1H NMR (399 MHz, DMSO-d6) δ ppm 8.11 (s, 1 H) 8.09 (s, 1 H) 7.79 (d, J=8.8 Hz, 1 H) 7.61 (d, J=9.5 Hz, 1 H) 6.38 (br. s., 2 H) 6.29 (s, 1 H) 6.00 (br. s., 2 H).
6-(1H-Indazol-6-yl)pyrimidin-4-amine (34b).
The title compound was prepared according to General Procedure A on a 66-mg scale using 33 and 4-amino-6-chloropyrimidine. The crude material was purified by flash chromatography (1–10% MeOH:DCM) to afford the title compound as a solid (16 mg, 28%). LCMS [M+H]+ 212.00 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 13.25 (s, 1 H) 8.46 (s, 1 H) 8.18 (s, 1 H) 8.12 (s, 1 H) 7.85 (d, J=8.3 Hz, 1 H) 7.70 (dd, J=8.6, 1.2 Hz, 1 H) 6.97 (s, 1 H) 6.92 (s, 2 H).
4-(1H-Indazol-6-yl)pyrimidin-2-amine (34c).
The title compound was prepared according to General Procedure A on a 44-mg scale using 33 and 2-amino-4-chloropyrimidine. The crude material was purified by flash chromatography (0–10% 10% NH4OH in MeOH:DCM) to afford the title compound as a solid. (16 mg, 43%). LCMS [M+H]+ 212.00 m/z; 1H NMR (399 MHz, DMSO-d6) δ ppm 8.32 (d, J=5.1 Hz, 1 H) 8.27 (s, 1 H) 8.13 (s, 1 H) 8.05 (d, J=2.2 Hz, 1 H) 7.79 – 7.88 (m, 2 H) 7.21 (d, J=5.1 Hz, 1 H) 6.70 (s, 2 H).
4-Bromo-2-fluorobenzoyl chloride (36).
To 35 (1.00 g, 4.57 mmol) was added thionyl chloride (3.3 mL, 45.5 mmol). The reaction was refluxed at 75 °C for 5 h. Excess thionyl chloride was removed by distillation and the product was azeotroped with toluene three times to afford the title compound as a tan solid which was used in the next reaction without further purification.
4-Bromo-2-fluoro-N-(2-hydroxyphenyl)benzamide (37).
2-Aminophenol (253 mg, 2.32 mmol) was dissolved in DCM (6.0 mL) and TEA (0.400 mL, 2.87 mmol) was added. Compound 36 (500 mg, 2.11 mmol) was dissolved in DCM (3.0 mL) and this solution was added to the reaction mixture dropwise at 0 °C. The reaction was allowed to warm to room temperature and stirred overnight. Upon the addition of 1M HCl, an off-white precipitate was observed; this was collected by vacuum filtration to afford the title compound as an off-white solid (341 mg, 52%). LCMS [M+H]+ 310.03 (79Br) m/z, 311.99 (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 10.04 (s, 1 H) 9.47 (d, J=7.3 Hz, 1 H) 8.01 (d, J=7.8 Hz, 1 H) 7.81 (t, J=8.3 Hz, 1 H) 7.76 (dd, J=10.7, 1.5 Hz, 1 H) 7.59 (dd, J=8.3, 1.5 Hz, 1 H) 7.00 (td, J=8.8, 1.0 Hz, 1 H) 6.92 (d, J=7.8 Hz, 1 H) 6.83 (t, J=7.8 Hz, 1 H).
3-Bromodibenzo[b,f][1,4]oxazepin-11(10H)-one (38).
Compound 37 (219 mg, 0.938 mmol) was dissolved in DMF (7.2 mL, 0.13 M) and cesium carbonate (457 mg, 1.40 mmol) was added. The reaction was stirred at 50 °C overnight. The reaction was stopped and cooled to room temperature. Upon addition of water, a gray precipitate was observed; this was collected by vacuum filtration and washed with water to afford the title compound as a gray solid (244 mg, 90%). LCMS [M+H]+ 290.03 m/z (79Br), 219.99 (81Br) m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 10.62 (s, 1 H) 7.67 – 7.72 (m, 2 H) 7.54 (dd, J=8.3, 2.0 Hz, 1 H) 7.37 (dd, J=7.3, 1.5 Hz, 1 H) 7.13 – 7.23 (m, 3 H).
3-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)dibenzo[b,f][1,4]oxazepin-11(10H)-one (39).
Compound 38 (244 mg, 0.841 mmol), potassium acetate (290 mg, 2.95 mmol), bis(pinacolato)diboron (322 mg, 1.23 mmol), and PdCl2(dppf)·CH2Cl2 (33 mg, 0.040 mmol) were combined in a reaction vial that was filled with nitrogen and evacuated three times. Dry, degassed dioxane (9.5 mL, 0.09 M) was added and the reaction was run in the microwave (145 °C) for 30 min. The reaction was diluted with EtOAc, filtered through Celite®, and concentrated under reduced pressure. The crude material was purified by flash chromatography (20–30% EtOAc:hexanes) to afford the title compound as a tan solid (250 mg, 88%). LCMS [M+H]+ 338.16 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 10.59 (s, 1 H) 7.78 (d, J=7.3 Hz, 1 H) 7.53 – 7.59 (m, 2 H) 7.41 (d, J=7.8 Hz, 1 H) 7.10 – 7.20 (m, 3 H) 1.30 (s, 12 H).
3-(2-Aminopyrimidin-4-yl)dibenzo[b,f][1,4]oxazepin-11(10H)-one (40).
Compound 39 (76 mg, 0.225 mmol), K2CO3 (94 mg, 0.680 mmol), and 2-amino-4-chloropyrimidine (48 mg, 0.370 mmol) were combined in a reaction vial and 3:1 dioxane:water (2.2 mL, 0.10 M) was added. The reaction was degassed for 10 min and Pd(PPh3)4 (15 mg, 0.013 mmol) was added. The reaction was refluxed at 100 °C for 3 h, then cooled to room temperature and stirred overnight. The reaction mixture was diluted with EtOAc and the crude material was purified by flash chromatography (50–100% EtOAc:hexanes), then repurified by preparative HPLC (5–95% water:ACN) to afford the title compound as a white solid (4 mg, 6%). LCMS [M+H]+ 305.11 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 10.63 (s, 1 H) 8.54 (s, 1 H) 8.38 (d, J=5.4 Hz, 1 H) 8.02 (d, J=1.5 Hz, 1 H) 7.99 (dd, J=7.3, 2.0 Hz, 1 H) 7.88 (d, J=8.3 Hz, 1 H) 7.37 (d, J=6.8 Hz, 1 H) 7.13 – 7.25 (m, 4 H) 6.82 (s, 1 H).
(1R,2R)-2-((4-Methoxybenzyl)amino)cyclopentan-1-ol (41).
4-Methoxybenzaldehyde (0.46 mL, 3.78 mmol) was dissolved in DCM (14.5 mL, 0.26 M) and (1R,2R)-2-aminocyclohexan-1-ol hydrochloride (701 mg, 5.09 mmol), TEA (1.0 mL, 7.17 mmol), and acetic acid (0.43 mL, 7.52 mmol) were added in that order. The reaction was stirred at room temperature for 30 min. NaHB(OAc)3 (1.47 g, 6.94 mmol) was added and the reaction was stirred at room temperature overnight. The reaction was diluted with DCM and washed twice with 1M NaOH, once with water, and once with brine. The organic layer was dried with sodium sulfate and concentrated under reduced pressure. The crude material was purified by flash chromatography (20–100% EtOAc:hexanes - 0–20% DCM:EtOAc) to afford the title compound as an off-white solid (417 mg, 37%). LCMS [M+H]+ 222.16 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 7.22 (d, J=8.8 Hz, 2 H) 6.86 (d, J=8.3 Hz, 2 H) 4.47 (d, J=4.4 Hz, 1 H) 3.73 – 3.77 (m, 1 H) 3.72 (s, 3 H) 3.61 (q, J=13.2 Hz, 2 H) 2.72 (q, J=6.3 Hz, 1 H) 1.91 (br. s, 1 H) 1.79 (spt, J=6.3 Hz, 2 H) 1.50 – 1.61 (m, 2 H) 1.34 – 1.42 (m, 1 H) 1.26 (sxt, J=12.7 Hz, 1 H).
4-Bromo-2,6-difluoro-N-((1R,2R)-2-hydroxycyclopentyl)-N-(4-methoxybenzyl)benzamide (42).
Compound 41 (417 mg, 1.88 mmol) was dissolved in DCM (4.4 mL) and TEA (0.40 mL, 2.87 mmol) was added. Compound 2 (526 mg, 2.06 mmol) was dissolved in another DCM (2.2 mL) and this solution was added to the reaction mixture dropwise at 0 °C. The reaction was allowed to warm to room temperature and stirred overnight. The reaction mixture was diluted with DCM and washed once with 1M HCl, once with water, and once with brine. The combined organic layers were dried with sodium sulfate and concentrated under reduced pressure. The crude material was purified by flash chromatography (20–50% EtOAc:hexanes) to afford the title compound as an orange oil (764 mg) which was used without further purification. LCMS [M+H]+ 440.13 m/z (79Br), 442.15 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 7.61 (d, J=7.3 Hz, 2 H) 7.25 (d, J=8.8 Hz, 2 H) 6.88 (d, J=8.8 Hz, 2 H) 4.78 (q, J=4.9 Hz, 2 H) 3.94 (quin, J=7.8 Hz, 1 H) 3.73 (s, 3 H) 3.59 (q, J=8.3 Hz, 1 H) 1.55 – 1.78 (m, 3 H) 1.38 – 1.54 (m, 3 H) 1.26 (td, J=7.8, 4.9 Hz, 1 H).
(3aR,10aR)-6-Bromo-8-fluoro-10-(4-methoxybenzyl)-1,2,3,3a,10,10a-hexahydro-9H-benzo[f]cyclopenta[b][1,4]oxazepin-9-one (43).
Compound 42 (764 mg, 1.74 mmol) was dissolved in DMF (12.5 mL, 0.14 M) and cesium carbonate (849 mg, 2.61 mmol) was added. The reaction was stirred at room temperature overnight. The reaction temperature was then increased to 50 °C and the reaction was heated another 24 h. The reaction was stopped and cooled to room temperature. The reaction mixture was diluted with EtOAc and washed three times with water. The combined aqueous layers were extracted once with EtOAc, and the combined organic layers were washed once with brine, dried with sodium sulfate, and concentrated under reduced pressure to afford the title compound as a yellow oil (614 mg, 84%). LCMS [M+H]+ 420.06 m/z (79Br), 422.04 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 7.38 (dd, J=9.8, 2.0 Hz, 1 H) 7.22 (d, J=8.3 Hz, 2 H) 7.07 (s, 1 H) 6.88 – 6.93 (m, 2 H) 4.69 (s, 2 H) 4.51 (q, J=9.0 Hz, 1 H) 4.09 (ddd, J=11.5, 9.8, 7.6 Hz, 1 H) 3.73 (s, 3 H) 1.84 – 1.91 (m, 1 H) 1.75 – 1.84 (m, 1 H) 1.67 – 1.75 (m, 1 H) 1.54 – 1.66 (m, 2 H) 1.44 – 1.54 (m, 1 H).
(3aR,10aR)-6-Bromo-8-fluoro-1,2,3,3a,10,10a-hexahydro-9H-benzo[f]cyclopenta[b][1,4] oxazepin-9-one (44).
To 43 (614 mg, 1.46 mmol) was added TFA (1.1 mL, 14.37 mmol). The reaction was run in the microwave (140 °C) for 30 min. The reaction mixture was then diluted with EtOAc and poured over sat. aq. NaHCO3. The aqueous layer was extracted three times with EtOAc, and the combined organic layers were washed once with brine, dried with sodium sulfate, and concentrated under reduced pressure. The crude material was purified by flash chromatography (20–50% EtOAc:hexanes) to afford the title compound as a pale yellow solid (337 mg, 77%). LCMS [M+H]+ 300.02 m/z (79Br), 302.03 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 8.57 (d, J=5.4 Hz, 1 H) 7.21 (dd, J=10.3, 2.0 Hz, 1 H) 7.02 (t, J=1.7 Hz, 1 H) 4.52 (q, J=8.0 Hz, 1 H) 3.66 – 3.77 (m, 1 H) 2.06 – 2.16 (m, 1 H) 1.96 (m, J=7.8 Hz, 1 H) 1.62 – 1.77 (m, 4 H).
(3aR,10aR)-6-Bromo-8-fluoro-1,2,3,3a,10,10a-hexahydro-9H-benzo[f]cyclopenta[b][1,4] oxazepine-9-thione (45).
Compound 44 (287 mg, 0.956 mmol) was suspended in toluene (19 mL, 0.05 M) and Lawesson’s reagent (777 mg, 1.92 mmol) was added. The reaction was refluxed at 100 °C overnight. The reaction was stopped and cooled to room temperature and the crude material was purified by flash chromatography (20–50% EtOAc:hexanes) to afford the title compound as a yellow solid (171 mg) which was used in next reaction without further purification. LCMS [M+H]+ 315.99 m/z (79Br), 318.03 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 11.07 (d, J=5.4 Hz, 1 H) 7.33 (dd, J=9.8, 2.0 Hz, 1 H) 7.03 (t, J=1.5 Hz, 1 H) 4.61 (q, J=9.8 Hz, 1 H) 3.95 (quint, J=10.3, 10.3, 10.3, 10.3, 8.3, 8.3 Hz, 1 H) 1.91 – 1.97 (m, 1 H) 1.75 – 1.90 (m, 2 H) 1.62 – 1.71 (m, 2 H) 1.51 – 1.61 (m, 1 H).
(6aR,9aR)-4-Bromo-6a,7,8,9,9a,10-hexahydro-2H-cyclopenta[2,3][1,4]oxazepino[5,6,7-cd]indazole (46).
Compound 45 (171 mg, 0.540 mmol) was suspended in dioxane (1.8 mL, 0.30 M), and hydrazine monohydrate (0.15 mL, 3.06 mmol) was added. The reaction was refluxed for 3 h at 85 °C. The reaction mixture was concentrated under reduced pressure and the crude material was purified by flash chromatography (50–80% EtOAc:hexanes) to afford the title compound as a tan solid (149 mg, 94%). LCMS [M+H]+ 294.09 m/z (79Br), 296.06 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 11.80 (s, 1 H) 6.99 (d, J=1.5 Hz, 1 H) 6.58 (s, 1 H) 6.45 (d, J=1.5 Hz, 1 H) 4.31 (q, J=6.8 Hz, 1 H) 3.48 (q, J=8.8 Hz, 1 H) 2.29 (m, J=8.3, 8.3, 6.3, 6.3, 6.3 Hz, 1 H) 2.18 (m, J=7.8 Hz, 1 H) 1.90 (sxt, J=7.3 Hz, 1 H) 1.75 – 1.84 (m, 1 H) 1.61 – 1.75 (m, 2 H).
1-((6aR,9aR)-4-Bromo-6a,7,8,9,9a,10-hexahydro-2H-cyclopenta[2,3][1,4]oxazepino[5,6,7-cd]indazol-2-yl)ethan-1-one (47).
Acetic anhydride (2.7 mL, 28.56 mmol) was added to 46 (130 mg, 0.442 mmol), upon which the reaction mixture turned bright yellow, and the reaction was refluxed at 100 °C for 3 h. The reaction was kept at room temperature for an additional 12 h, after which a yellow precipitate was observed and collected by vacuum filtration (washed with water) to afford the title compound as a yellow solid (89 mg, 60%). LCMS [M+H]+ 336.06 m/z (79Br), 338.08 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 8.03 (d, J=1.5 Hz, 1 H) 7.63 (s, 1 H) 6.99 (d, J=1.5 Hz, 1 H) 4.38 (q, J=8.3 Hz, 1 H) 3.62 (q, J=8.8 Hz, 1 H) 2.51 (s, 3 H) 2.27 – 2.39 (m, 1 H) 2.16 – 2.25 (m, 1 H) 1.87 – 1.98 (m, 1 H) 1.77 – 1.85 (m, 1 H) 1.60 – 1.76 (m, 2 H).
1-((6aR,9aR)-4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-6a,7,8,9,9a,10-hexahydro-2H-cyclopenta[2,3][1,4]oxazepino[5,6,7-cd]indazol-2-yl)ethan-1-one (48).
Compound 47 (79 mg, 0.235 mmol), potassium acetate (81 mg, 0.825 mmol), bis(pinacolato)diboron (93 mg, 0.366 mmol), and PdCl2(dppf)·CH2Cl2 (10 mg, 12.3 μmol) were combined in a reaction vial that was filled with nitrogen and evacuated three times. Dry, degassed dioxane (2.6 mL, 0.09 M) was added and the reaction was run in the microwave (145 °C) for 20 min. The reaction was diluted with MeOH, filtered through Celite®, and concentrated under reduced pressure. The crude material was purified by flash chromatography (20–50% EtOAc:hexanes) to afford the title compound as an off-white solid (56 mg, 62%). LCMS [M+H]+ 384.26 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 8.20 (s, 1 H) 7.58 (s, 1 H) 6.97 (s, 1 H) 4.33 (q, J=7.8 Hz, 1 H) 3.61 (q, J=8.3 Hz, 1 H) 2.27 – 2.38 (m, 1 H) 2.17 – 2.26 (m, 1 H) 1.88 – 1.97 (m, 1 H) 1.78 – 1.88 (m, 1 H) 1.61 – 1.77 (m, 2 H) 1.34 (s, 3 H) 1.31 (s, 12 H).
1-((6aR,9aR)-4-(2-Aminopyrimidin-4-yl)-6a,7,8,9,9a,10-hexahydro-2H-cyclopenta[2,3][1,4]oxazepino[5,6,7-cd]indazol-2-yl)ethan-1-one (49).
Compound 48 (56 mg, 0.164 mmol), K2CO3 (70 mg, 0.507 mmol), and 2-amino-4-chloropyrimidine (34 mg, 0.262 mmol) were combined in a reaction vial and 3:1 dioxane:water (1.6 mL, 0.10 M) was added. The reaction was degassed for 10 min and PdCl2(dppf)·CH2Cl2 (8 mg, 0.010 mmol) was added. The reaction was refluxed at 100 °C overnight. The reaction mixture was diluted with MeOH, filtered through Celite®, and concentrated under reduced pressure. The crude material was purified by flash chromatography (1–15% 10% NH4OH/MeOH:DCM), then repurified by flash chromatography (0–10% MeOH:EtOAc) to afford the title compound as a yellow solid (25 mg, 56%). LCMS [M+H]+ 309.17 m/z; 1H NMR (500 MHz, METHANOL-d4) δ ppm 8.28 (d, J=5.9 Hz, 1 H) 7.64 (s, 1 H) 7.25 (d, J=5.9 Hz, 1 H) 7.12 (s, 1 H) 4.36 – 4.48 (m, 1 H) 3.66 (q, J=8.8 Hz, 1 H) 2.27 – 2.46 (m, 2 H) 2.02 – 2.12 (m, 1 H) 1.89 – 1.98 (m, 1 H) 1.73 – 1.88 (m, 2 H).
4-Bromo-2-ethoxy-6-fluorobenzonitrile (53a).
The title compound was prepared according to General Procedure B on a 302-mg scale using EtOH. The reaction was stirred at room temperature overnight, then heated to 50 °C for another 24 h. The crude material was purified by flash chromatography (0–20% EtOAc:hexanes) to afford the title compound as a white solid (280 mg, 83%). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.48 (d, J=8.8 Hz, 1 H) 7.40 (s, 1 H) 4.26 (q, J=7.3 Hz, 2 H) 1.36 (t, J=7.0 Hz, 3 H).
*Compound does not ionize.
4-Bromo-2-(tert-butoxy)-6-fluorobenzonitrile (53b).
The title compound was prepared according to General Procedure B on a 300-mg scale using tert-butanol. The reaction was stirred at room temperature overnight, then heated to 50 °C for another 24 h. The crude material was purified by flash chromatography (0–20% EtOAc:hexanes) to afford the title compound as a yellow oil (258 mg, 69%). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.60 (d, J=8.8 Hz, 1 H) 7.40 (s, 1 H) 1.45 (s, 9 H).
*Compound does not ionize.
4-Bromo-2-cyclobutoxy-6-fluorobenzonitrile (53c).
The title compound was prepared according to General Procedure B on a 300-mg scale using cyclobutanol. The reaction was stirred at room temperature overnight, then heated to 50 °C for another 24 h. The crude material was purified by flash chromatography (0–20% EtOAc:Hexanes) to afford the title compound as a yellow oil (259 mg, 70%). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.48 (d, J=9.5 Hz, 1 H) 7.18 (s, 1 H) 4.96 (quin, J=7.0 Hz, 1 H) 2.46 (br. s., 2 H) 2.09 (quin, J=10.3 Hz, 2 H) 1.82 (q, J=10.3 Hz, 1 H) 1.63 (tq, J=10.3, 8.8 Hz, 1 H).
*Compound does not ionize.
4-Bromo-2-fluoro-6-((tetrahydrofuran-3-yl)oxy)benzonitrile (53d).
The title compound was prepared according to General Procedure B on a 204-mg scale using tetrahydrofuran-3-ol. The reaction was stirred at room temperature for two days and the crude material was purified by flash chromatography (0–30% EtOAc:hexanes) to afford the title compound as a white solid (183 mg, 68%). 1H NMR (500 MHz, DMSO-d6) δ ppm 7.50 (dd, J=8.8, 1.5 Hz, 1 H) 7.43 (s, 1 H) 5.32 (t, J=5.1 Hz, 1 H) 3.82 – 3.92 (m, 3 H) 3.77 (td, J=8.3, 4.4 Hz, 1 H) 2.28 (sxt, J=7.3 Hz, 1 H) 1.95 – 2.04 (m, 1 H).
*Compound does not ionize.
4-Bromo-2-fluoro-6-((tetrahydro-2H-pyran-4-yl)oxy)benzonitrile (53e).
The title compound was prepared according to General Procedure B on a 51-mg scale using tetrahydro-2H-pyran-4-ol. The reaction mixture was stirred at room temperature overnight and the crude material was purified by flash chromatography (20–30% EtOAc:hexanes) to afford the title compound as an off-white solid (37 mg, 53%). 1H NMR (500 MHz, DMSO-d6) δ ppm 7.57 (s, 1 H) 7.47 (dd, J=8.8, 1.5 Hz, 1 H) 4.94 (tt, J=8.2, 4.0 Hz, 1 H) 3.79 – 3.89 (m, 2 H) 3.52 (ddd, J=11.5, 8.5, 2.9 Hz, 2 H) 1.93 – 2.05 (m, 2 H) 1.63 (dtd, J=12.9, 8.6, 8.6, 3.7 Hz, 2 H).
*Compound does not ionize.
4-Bromo-2-fluoro-6-((tetrahydro-2H-pyran-3-yl)oxy)benzonitrile (53f).
The title compound was prepared according to General Procedure B on a 200-mg scale using tetrahydro-2H-pyran-3-ol. The reaction was stirred at room temperature overnight and the crude material was purified by flash chromatography (0–20% EtOAc:hexanes) to afford the title compound as a yellow solid (143 mg, 52%). 1H NMR (500 MHz, DMSO-d6) δ ppm 7.53 (s, 1 H) 7.47 (dd, J=8.8, 1.5 Hz, 1 H) 4.74 (m, J=2.4 Hz, 1 H) 3.75 (dd, J=12.0, 2.2 Hz, 1 H) 3.53 – 3.65 (m, 3 H) 1.94 – 2.05 (m, 1 H) 1.72 – 1.87 (m, 2 H) 1.49 – 1.58 (m, 1 H).
*Compound does not ionize.
4-Bromo-2-fluoro-6-(oxetan-3-yloxy)benzonitrile (53g).
The title compound was prepared according to General Procedure B on a 200-mg scale using oxetan-3-ol. The reaction was stirred at room temperature for two days and the crude material was purified by flash chromatography (0–50% EtOAc:hexanes) to afford the title compound as a beige solid (121 mg, 49%). 1H NMR (500 MHz, DMSO-d6) δ ppm 7.55 (dd, J=8.8, 1.5 Hz, 1 H) 7.07 (s, 1 H) 5.52 (quin, J=5.4 Hz, 1 H) 4.97 (t, J=6.8 Hz, 2 H) 4.58 (dd, J=7.6, 4.6 Hz, 2 H).
*Compound does not ionize.
6-Bromo-4-ethoxy-1H-indazol-3-amine (54a).
The title compound was prepared according to General Procedure C on a 280-mg scale using 53a. The crude material was purified by flash chromatography (0–60% EtOAc:hexanes) to afford the title compound as a yellow solid (240 mg, 82%). LCMS [M+H]+ 256.00 m/z (79Br), 258.01 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 11.51 (s, 1 H) 6.96 (d, J=1.0 Hz, 1 H) 6.43 (s, 1 H) 5.02 (s, 2 H) 4.13 (q, J=7.0 Hz, 2 H) 1.40 (t, J=6.8 Hz, 3 H).
6-Bromo-4-(tert-butoxy)-1H-indazol-3-amine (54b).
The title compound was prepared according to General Procedure C on a 258-mg scale using 53b. The crude material was purified by flash chromatography (0–50% EtOAc:hexanes) to afford the title compound as a red solid (230 mg, 85%). LCMS [M+H]+ 284.08 m/z (79Br), 286.03 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 11.53 (s, 1 H) 7.03 (s, 1 H) 6.55 (s, 1 H) 4.96 (s, 2 H) 1.45 – 1.48 (m, 9 H).
6-Bromo-4-cyclobutoxy-1H-indazol-3-amine (54c).
The title compound was prepared according to General Procedure C on a 259-mg scale using 53c. The crude material was purified by flash chromatography (0–50% EtOAc:hexanes) to afford the title compound as a light yellow solid (236 mg, 87%). LCMS [M+H]+ 282.07 m/z (79Br), 284.04 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 11.51 (s, 1 H) 6.95 (s, 1 H) 6.25 (s, 1 H) 5.04 (s, 2 H) 4.83 (quin, J=7.1 Hz, 1 H) 2.41 – 2.49 (m, 2 H) 2.16 (ddd, J=9.8, 7.3, 2.4 Hz, 2 H) 1.82 (q, J=10.7 Hz, 1 H) 1.67 (sxt, J=10.3 Hz, 1 H).
6-Bromo-4-((tetrahydrofuran-3-yl)oxy)-1H-indazol-3-amine (54d).
The title compound was prepared according to General Procedure C on a 143-mg scale using 53d. The crude material was purified by flash chromatography (50–100% EtOAc:hexanes) to afford the title compound as an off-white solid (130 mg, 68%). LCMS [M+H]+ 297.94 m/z (79Br), 299.92 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 11.54 (s, 1 H) 6.98 (d, J=1.0 Hz, 1 H) 6.42 (s, 1 H) 5.15 (br. s., 1 H) 5.02 (s, 2 H) 3.92 (d, J=2.9 Hz, 2 H) 3.88 (q, J=7.3 Hz, 1 H) 3.77 (td, J=8.3, 4.4 Hz, 1 H) 2.25 (m, J=7.3 Hz, 1 H) 2.11 (dt, J=13.3, 6.3 Hz, 1 H)
6-Bromo-4-((tetrahydro-2H-pyran-4-yl)oxy)-1H-indazol-3-amine (54e).
The title compound was prepared according to General Procedure C on a 111-mg scale using 53e. The crude material was purified by flash chromatography (50–100% EtOAc:hexanes) to afford the title compound as a tan solid (104 mg, 90%). LCMS [M+H]+ 311.92 m/z (79Br), 313.93 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 11.52 (s, 1 H) 6.95 (d, J=1.0 Hz, 1 H) 6.57 (s, 1 H) 5.03 (s, 2 H) 4.77 (tt, J=8.2, 4.3 Hz, 1 H) 3.81 – 3.89 (m, 2 H) 3.53 (m, J=2.9 Hz, 2 H) 2.00 (m, J=12.2 Hz, 2 H) 1.66 – 1.76 (m, 2 H).
6-Bromo-4-((tetrahydro-2H-pyran-3-yl)oxy)-1H-indazol-3-amine (54f).
The title compound was prepared according to General Procedure C on a 143-mg scale using 4-bromo-2-fluoro-6-((tetrahydro-2H-pyran-3-yl)oxy)benzonitrile 53f. The crude material was purified by flash chromatography (40–70% EtOAc:hexanes) to afford the title compound as a beige solid (123 mg, 82%). LCMS [M+H]+ 311.98 m/z (79Br), 313.99 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 11.52 (br. s., 1 H) 6.96 (d, J=1.0 Hz, 1 H) 6.54 (s, 1 H) 5.07 (s, 2 H) 4.59 (tt, J=3.9, 2.4 Hz, 1 H) 3.77 (dd, J=10.7, 2.0 Hz, 1 H) 3.55 – 3.72 (m, 3 H) 1.94 – 2.02 (m, 1 H) 1.83 – 1.90 (m, 1 H) 1.78 (dtd, J=17.3, 8.8, 8.8, 4.6 Hz, 1 H) 1.53 (m, J=10.1, 3.0, 3.0 Hz, 1 H).
6-Bromo-4-(oxetan-3-yloxy)-1H-indazol-3-amine (54g).
The title compound was prepared according to General Procedure C on a 121-mg scale using 4-bromo-2-fluoro-6-(oxetan-3-yloxy)benzonitrile 53g. The crude material was purified by flash chromatography (100% EtOAc) to afford the title compound as a beige solid (82 mg, 65%). LCMS [M+H]+ 283.96 m/z (79Br), 285.97 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 11.58 (s, 1 H) 7.01 (d, J=1.0 Hz, 1 H) 6.06 (s, 1 H) 5.39 (quin, J=5.4 Hz, 1 H) 5.20 (s, 2 H) 4.95 (t, J=6.8 Hz, 2 H) 4.70 (dd, J=7.1, 5.1 Hz, 2 H).
1-(3-Amino-6-bromo-4-ethoxy-1H-indazol-1-yl)ethan-1-one (55a).
The title compound was prepared according to General Procedure D on a 240-mg scale using 6-bromo-4-ethoxy-1H-indazol-3-amine 54a. The crude material was purified by flash chromatography (0–50% EtOAc:hexanes) to afford the title compound as a light yellow solid (65 mg, 23%). LCMS [M+H]+ 298.03 m/z (79Br), 300.01 m/z (81Br). 1H NMR (500 MHz, DMSO-d6) δ ppm 7.92 (d, J=1.5 Hz, 1 H) 7.00 (d, J=1.0 Hz, 1 H) 5.99 (s, 2 H) 4.23 (q, J=7.2 Hz, 2 H) 2.49 (s, 3 H) 1.41 (t, J=7.1 Hz, 3 H).
1-(3-Amino-6-bromo-4-(tert-butoxy)-1H-indazol-1-yl)ethan-1-one (55b).
The title compound was prepared according to General Procedure D on a 205-mg scale using 54b. The crude material was purified by flash chromatography (0–50% EtOAc:hexanes) to afford the title compound as an orange solid (92 mg, 39%). LCMS [M+H]+ 326.03 m/z (79Br), 328.04 m/z (81Br). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.00 (d, J=1.5 Hz, 1 H) 7.07 (d, J=1.5 Hz, 1 H) 5.89 (s, 2 H) 2.50 (s, 3 H) 1.51 (s, 9 H).
1-(3-Amino-6-bromo-4-cyclobutoxy-1H-indazol-1-yl)ethan-1-one (55c).
The title compound was prepared according to General Procedure D on a 236-mg scale using 54c. The crude material was purified by flash chromatography (0–50% EtOAc:hexanes) to afford the title compound as an off-white solid (65 mg, 24%). LCMS [M+H]+ 324.03 m/z (79Br), 326.02 m/z (81Br). 1H NMR (500 MHz, DMSO-d6) δ ppm 7.92 (d, J=1.5 Hz, 1 H) 6.81 (d, J=1.0 Hz, 1 H) 6.03 (s, 2 H) 4.93 (quin, J=7.1 Hz, 1 H) 2.49 (s, 3 H) 2.41 – 2.48 (m, 2 H) 2.23 (quint, J=9.7, 9.7, 9.7, 9.7, 2.3, 2.3 Hz, 1 H) 1.82 (qt, J=10.3, 2.4 Hz, 1 H) 1.66 (qt, J=10.7, 8.3 Hz, 1 H).
1-(3-Amino-6-bromo-4-((tetrahydrofuran-3-yl)oxy)-1H-indazol-1-yl)ethan-1-one and N-(1-acetyl-6-bromo-4-((tetrahydrofuran-3-yl)oxy)-1H-indazol-3-yl)acetamide (55d).
The title compound was prepared according the General Procedure D on a 130-mg scale using 6-bromo-4-((tetrahydrofuran-3-yl)oxy)-1H-indazol-3-amine 54d. The crude material was not purified but taken forward as a mixture of mono- and di-acetylated products (170 mg orange solid). LCMS [M+H]+ 339.94 m/z (79Br mono-aceylated), 341.81 m/z (81Br mono-aceylated), 381.88 m/z (79Br di-aceylated), 383.89 m/z (81Br di-aceylated).
1-(3-Amino-6-bromo-4-((tetrahydro-2H-pyran-4-yl)oxy)-1H-indazol-1-yl)ethan-1-one and N-(1-acetyl-6-bromo-4-((tetrahydro-2H-pyran-4-yl)oxy)-1H-indazol-3-yl)acetamide (55e).
The title compound was prepared according the General Procedure D on a 104-mg scale using 6-bromo-4-((tetrahydro-2H-pyran-4-yl)oxy)-1H-indazol-3-amine 54e. The crude material was not purified but taken forward as a mixture of mono- and di-acetylated products (148 mg orange solid). LCMS [M+H]+ 353.92 m/z (79Br mono-aceylated), 355.93 m/z (81Br mono-aceylated), 395.92 m/z (79Br di-aceylated), 397.87 m/z (81Br di-aceylated).
1-(3-Amino-6-bromo-4-((tetrahydro-2H-pyran-3-yl)oxy)-1H-indazol-1-yl)ethan-1-one and N-(1-acetyl-6-bromo-4-((tetrahydro-2H-pyran-3-yl)oxy)-1H-indazol-3-yl)acetamide (55f).
The title compound was prepared according the General Procedure D on a 104-mg scale using 6-bromo-4-((tetrahydro-2H-pyran-3-yl)oxy)-1H-indazol-3-amine 54f. The crude material was not purified but taken forward as a mixture of mono- and di-acetylated products (147 mg orange solid). LCMS [M+H]+ 353.98 m/z (79Br mono-aceylated), 356.00 m/z (81Br mono-aceylated), 395.98 m/z (79Br di-aceylated), 398.00 m/z (81Br di-aceylated).
1-(3-Amino-6-bromo-4-(oxetan-3-yloxy)-1H-indazol-1-yl)ethan-1-one and N-(1-acetyl-6-bromo-4-(oxetan-3-yloxy)-1H-indazol-3-yl)acetamide (55g).
The title compound was prepared according the General Procedure D on an 82-mg scale using 6-bromo-4-(oxetan-3-yloxy)-1H-indazol-3-amine 54g. The crude material was not purified but taken forward as a mixture of mono- and di-acetylated products (84 mg yellow solid). LCMS [M+H]+ 325.96 m/z (79Br mono-aceylated), 327.97 m/z (81Br mono-aceylated), 367.96 m/z (79Br di-aceylated), 369.91 m/z (81Br di-aceylated).
6-(2-Aminopyrimidin-4-yl)-4-ethoxy-1H-indazol-3-amine formate (57a).
The title compound was prepared according to General Procedure E on a 348-mg scale using 55a. The crude material of the Miyaura reaction was taken forward without further purification. The crude residue of the Suzuki reaction was purified by flash chromatography (20–100% EtOAc:hexanes – 0–10% MeOH:DCM), then repurified by preparative HPLC (5–50% ACN:water) to afford the formate salt of the title compound as an orange solid (18 mg, 6%). LCMS [M+H]+ 271.17 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 11.67 (br. s., 1 H) 8.52 (s, 1 H) 8.28 (d, J=5.4 Hz, 1 H) 7.52 (s, 1 H) 7.14 (d, J=5.4 Hz, 1 H) 6.96 (s, 1 H) 6.64 (s, 2 H) 5.03 (s, 2 H) 4.21 (q, J=7.2 Hz, 2 H) 1.45 (t, J=7.1 Hz, 3 H).
6-(2-Aminopyrimidin-4-yl)-4-(tert-butoxy)-1H-indazol-3-amine (57c).
The title compound was prepared according to General Procedure E on a 92-mg scale using 55b. The product of the Miyaura reaction was isolated by flash chromatography (20% EtOAc:hexanes) to afford 1-(3-amino-4-(tert-butoxy)-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazol-1-yl)ethan-1-one 56a as an off-white solid (73 mg, 69%). LCMS [M+H]+ 374.19 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 8.19 (s, 1 H) 7.12 (s, 1 H) 5.89 (s, 2 H) 2.50 (s, 3 H) 1.48 (s, 9 H) 1.32 (s, 12 H).
The crude residue of the Suzuki reaction was purified by flash chromatography (5% MeOH:DCM), then repurified by preparative HPLC (5–50% acetronitrile:water) to afford the title compound as a brown solid (8 mg, 14%). LCMS [M+H]+ 299.19 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 11.68 (s, 1 H) 8.28 (d, J=5.4 Hz, 1 H) 7.58 (d, J=1.0 Hz, 1 H) 7.13 (d, J=1.0 Hz, 1 H) 7.09 (d, J=5.4 Hz, 1 H) 6.64 (s, 2 H) 4.97 (s, 2 H) 1.50 (s, 9 H).
6-(2-Aminopyrimidin-4-yl)-4-cyclobutoxy-1H-indazol-3-amine formate (57d).
The title compound was prepared according to General Procedure E on a 65-mg scale using 55c. The product of the Miyaura reaction was isolated by flash chromatography (20% EtOAc:hexanes) to afford 1-(3-amino-4-cyclobutoxy-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazol-1-yl)ethan-1-one 56b as an off-white solid (52 mg, 71%). LCMS [M+H]+ 372.18 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 8.11 (s, 1 H) 6.79 (s, 1 H) 6.00 (s, 2 H) 4.91 (quin, J=7.1 Hz, 1 H) 2.50 (s, 3 H) 2.38 – 2.47 (m, 2 H) 2.17 – 2.29 (m, 2 H) 1.83 (q, J=10.7 Hz, 1 H) 1.72 (q, J=9.8 Hz, 1 H) 1.32 (s, 12 H).
The crude residue of the Suzuki reaction was purified by flash chromatography (5% MeOH:DCM), then repurified by preparative HPLC (5–50% ACN:water) to afford the title compound as a brown solid (5 mg, 12%). LCMS [M+H]+ 297.18 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 11.66 (s, 1 H) 8.52 (s, 1 H) 8.28 (d, J=4.9 Hz, 1 H) 7.52 (s, 1 H) 7.11 (d, J=5.4 Hz, 1 H) 6.80 (s, 1 H) 6.64 (s, 2 H) 5.04 (s, 2 H) 4.91 (quin, J=7.3 Hz, 1 H) 2.53 (m, J=9.8 Hz, 2 H) 2.21 (quint, J=10.3, 10.3, 10.3, 10.3, 2.4, 2.4 Hz, 2 H) 1.84 (q, J=10.3 Hz, 1 H) 1.71 (quin, J=10.7 Hz, 1 H).
6-(2-Aminopyrimidin-4-yl)-4-((tetrahydrofuran-3-yl)oxy)-1H-indazol-3-amine (57g).
The title compound was prepared according to General Procedure E on a 150-mg scale 55d. The crude Miyaura reaction mixture was run through a pad of silica using 100% EtOAc as the eluent to afford a mixture of mono-, bi- and tri-acetylated boronic acid and ester which was taken forward to the Suzuki reaction without further purification. The crude residue of the Suzuki reaction was purified by flash chromatography (5–10% 5% NH4OH/MeOH:DCM, step gradient) to afford a mixture of the title compound and the acetylated product. This crude material was suspended in MeOH (10 mL, 0.04 M) and 12M HCl (1 mL, 12 mmol) was added, upon which the reaction mixture went from a cloudy suspension to a clear solution. The mixture was refluxed at 65 °C overnight. The solvent was removed under reduced pressure and the resulting residue was redissolved in EtOAc. The reaction mixture was poured over saturated aqueous NaHCO3 and the aqueous layer was extracted four times with EtOAc. The combined organic layers were washed once with brine, dried with sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (10% 5%NH4OH/MeOH:EtOAc) to afford the title compound as a yellow solid (26 mg, 19%). LCMS [M+H]+ 313.05 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 11.67 (s, 1 H) 8.29 (d, J=5.4 Hz, 1 H) 7.56 (s, 1 H) 7.16 (d, J=4.9 Hz, 1 H) 6.91 (s, 1 H) 6.64 (s, 2 H) 5.25 (br. s., 1 H) 5.03 (s, 2 H) 3.94 – 4.00 (m, 2 H) 3.91 (q, J=7.8 Hz, 1 H) 3.80 (sxt, J=4.4 Hz, 1 H) 2.30 (quin, J=6.8 Hz, 1 H) 2.13 – 2.21 (m, 1 H).
6-(2-Aminopyrimidin-4-yl)-4-((tetrahydro-2H-pyran-4-yl)oxy)-1H-indazol-3-amine (57h).
The title compound was prepared according to General Procedure E on a 120-mg scale using 55e. The crude Miyaura reaction mixture was run through a pad of silica using 100% EtOAc as the eluent to afford a mixture of mono-, bi- and tri-acetylated boronic acid and ester which was taken forward to the Suzuki reaction without further purification. The crude residue of the Suzuki reaction was purified by flash chromatography (1–10% 5%NH4OH/MeOH:EtOAc) to afford a mixture of the title compound and the acetylated product. This crude material was suspended in MeOH (10 mL, 0.04 M) and 12M HCl (1 mL, 12 mmol) was added, upon which the reaction mixture went from a cloudy suspension to a clear solution. The mixture was refluxed at 65 °C overnight, then concentrated under reduced pressure. The resulting residue was redissolved in EtOAc. The reaction mixture was poured over saturated aqueous NaHCO3 and the aqueous layer was extracted four times with EtOAc. The combined organic layers were washed once with brine, dried with sodium sulfate, and concentrated under reduced pressure. The crude material was repurified by flash chromatography (3% 5%NH4OH/MeOH:EtOAc) to afford the title compound as a yellow-green solid (26 mg, 24%). LCMS [M+H]+ 327.09 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 11.65 (s, 1 H) 8.29 (d, J=5.4 Hz, 1 H) 7.54 (s, 1 H) 7.16 (d, J=5.4 Hz, 1 H) 7.02 (s, 1 H) 6.64 (s, 2 H) 5.03 (s, 2 H) 4.87 (m, J=3.9 Hz, 1 H) 3.82 – 3.93 (m, 2 H) 3.57 (td, J=8.8, 2.4 Hz, 2 H) 2.02 – 2.11 (m, 2 H) 1.76 (m, J=8.8 Hz, 2 H).
6-(2-Aminopyrimidin-4-yl)-4-((tetrahydro-2H-pyran-3-yl)oxy)-1H-indazol-3-amine (57i).
The title compound was prepared according to General Procedure E on a 140-mg scale using 55f. The crude Miyaura reaction mixture was run through a pad of silica using 100% EtOAc as the eluent to afford a mixture of mono-, bi- and tri-acetylated boronic acid and ester which was taken forward to the Suzuki reaction without further purification. The crude residue of the Suzuki reaction was purified by flash chromatography (1–10% 5% NH4OH/MeOH:EtOAc) to afford a mixture of the title compound and the acetylated product. This crude material was suspended in MeOH (10 mL, 0.04 M) and 12M HCl (1 mL, 12 mmol) was added, upon which the reaction mixture went from a cloudy suspension to a clear solution. The mixture was refluxed at 65 °C overnight. The solvent was removed under reduced pressure and the resulting residue was redissolved in EtOAc. The reaction mixture was poured over saturated aqueous NaHCO3 and the aqueous layer was extracted three times with EtOAc. The combined organic layers were washed once with brine, dried with sodium sulfate, and concentrated under reduced pressure. The crude material was then purified by flash chromatography (5% 5% NH4OH/MeOH:EtOAc) to afford the title compound as a yellow solid (12 mg, 9%). LCMS [M+H]+ 327.09 m/z; 1H NMR (500 MHz, METHANOL-d4) δ ppm 8.27 (d, J=4.9 Hz, 1 H) 7.51 (s, 1 H) 7.12 (d, J=5.4 Hz, 1 H) 7.09 (s, 1 H) 4.66 – 4.72 (m, 1 H) 3.94 (dd, J=10.7, 2.0 Hz, 1 H) 3.84 (dd, J=14.6, 5.4 Hz, 1 H) 3.74 – 3.80 (m, 1 H) 3.71 (m, J=8.1, 3.2 Hz, 1 H) 2.03 – 2.15 (m, 2 H) 1.97 (m, J=8.7, 4.2, 4.2 Hz, 1 H) 1.58 – 1.67 (m, 1 H).
6-(2-Aminopyrimidin-4-yl)-4-(oxetan-3-yloxy)-1H-indazol-3-amine (57j).
The title compound was prepared according to General Procedure E on an 84-mg scale using 1-(3-amino-6-bromo-4-(oxetan-3-yloxy)-1H-indazol-1-yl)ethan-1-one 55g. The crude Miyaura reaction mixture was run through a pad of silica using 100% EtOAc as the eluent to afford a mixture of mono-, bi- and tri-acetylated boronic acid and ester which was taken forward to the Suzuki reaction without further purification. The crude residue of the Suzuki reaction was purified by flash chromatography (1-5-10% 5% NH4OH/MeOH:EtOAc, step gradient) to afford the title compound as a yellow solid (20 mg, 26%). LCMS [M+H]+ 299.07 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 11.72 (s, 1 H) 8.28 (d, J=5.4 Hz, 1 H) 7.59 (s, 1 H) 7.13 (d, J=4.9 Hz, 1 H) 6.64 (s, 2 H) 6.55 (s, 1 H) 5.48 (quin, J=5.1 Hz, 1 H) 5.18 (s, 2 H) 5.01 (t, J=6.8 Hz, 2 H) 4.75 (dd, J=7.3, 4.9 Hz, 1 H).
4-Bromo-2-fluoro-N-phenylbenzamide (58).
Aniline (0.200 ml, 2.19 mmol) was dissolved in 6.0 ml DCM and TEA (0.400 ml, 2.87 mmol) was added. Compound 36 (500 mg, 2.11 mmol) was dissolved in another 3.0 ml DCM and this solution was added to the reaction mixture dropwise at 0 °C. The reaction was allowed to warm to room temperature and stirred overnight. The reaction was diluted with DCM and washed once with 1M HCl, once with water, and once with brine. The combined organic layers were dried with sodium sulfate and concentrated under reduced pressure to afford the title compound as an off-white solid (485 mg, 78%). LCMS [M+H]+ 294.03 m/z (79Br), 295.99 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 10.45 (s, 1 H) 7.75 (dd, J=9.8, 2.0 Hz, 1 H) 7.70 (d, J=7.8 Hz, 2 H) 7.63 (t, J=8.3 Hz, 1 H) 7.57 (dd, J=7.3, 1.5 Hz, 1 H) 7.36 (t, J=7.8 Hz, 2 H) 7.12 (t, J=7.6 Hz, 1 H).
4-Bromo-2-fluoro-N-phenylbenzothioamide (59).
Compound 58 (365 mg, 1.24 mmol) was suspended in toluene (25 ml, 0.05M) and Lawesson’s reagent (1.01 g, 2.50 mmol) was added. The reaction was refluxed at 100 °C overnight. Upon cooling to room temperature, a pale yellow precipitate was observed and removed by vacuum filtration (washed with hexanes). The filtrate was purified by flash chromatography (5–20% EtOAc:hexanes) to afford the title compound as a yellow solid (283 mg, 74%). LCMS [M+H]+ 310.02 m/z (79Br), 311.99 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 12.11 (s, 1 H) 7.91 (d, J=7.8 Hz, 2 H) 7.69 (dd, J=9.8, 2.0 Hz, 1 H) 7.57 (t, J=7.3 Hz, 1 H) 7.51 (dd, J=6.3, 2.0 Hz, 1 H) 7.45 (t, J=7.8 Hz, 2 H) 7.29 (t, J=7.3 Hz, 1 H).
6-Bromo-N-phenyl-1H-indazol-3-amine (60).
Compound 59 (266 mg, 0.858 mmol) was suspended in dioxane (2.5 ml, 0.34 M) and hydrazine monohydrate (0.25 ml, 5.09 mmol) was added. The reaction was refluxed overnight at 85 °C. The reaction was diluted with MeOH and purified directly by flash chromatography (0–50% EtOAc:hexanes) to afford the title compound as a yellow solid (174 mg, 70%). LCMS [M+H]+ 288.06 m/z (79Br), 290.03 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 12.11 (s, 1 H) 8.93 (s, 1 H) 7.93 (d, J=8.8 Hz, 1 H) 7.66 (d, J=7.8 Hz, 2 H) 7.58 (s, 1 H) 7.26 (t, J=7.8 Hz, 2 H) 7.17 (dd, J=9.0, 1.2 Hz, 1 H) 6.81 (t, J=7.3 Hz, 1 H).
1-(6-Bromo-3-(phenylamino)-1H-indazol-1-yl)ethan-1-one (61).
Acetic anhydride (1.50 ml, 15.9 mmol) was added to 60 (156 mg, 0.541 mmol) and the reaction was refluxed at 100 °C for ~3.5 h (reaction mixture turns bright yellow as acetic anhydride is added). Upon cooling to room temperature, an off-white precipitate was observed and collected by vacuum filtration (washed with water) to afford the title compound as a white solid (134 mg, 75%). LCMS [M+H]+ 330.03 m/z (79Br), 332.06 m/z (81Br); 1H NMR (500 MHz, DMSO-d6) δ ppm 9.57 (s, 1 H) 8.46 (s, 1 H) 8.18 (d, J=8.3 Hz, 1 H) 7.83 (d, J=8.8 Hz, 2 H) 7.65 (dd, J=8.5, 1.7 Hz, 1 H) 7.37 (t, J=7.3 Hz, 2 H) 6.95 – 7.01 (m, 1 H) 2.67 (s, 3 H).
1-(3-(phenylamino)-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazol-1-yl)ethan-1-one (62).
Compound 61 (115 mg, 0.348 mmol), potassium acetate (119 mg, 1.21 mmol), bis(pinacolato)diboron (132 mg, 0.520 mmol), and PdCl2(dppf)·CH2Cl2 (14 mg, 17.14 μmol) were combined in a reaction vial that was filled with nitrogen and evacuated three times. Dry, degassed dioxane (3.9 ml, 0.09 M) was added and the reaction was run in the microwave (145 °C, N abs) for 30 minutes. The reaction was diluted with EtOAc and filtered through celite. The crude material was purified by flash chromatography (20–50% EtOAc:hexanes) to afford the title compound as a white solid (112 mg, 85%). LCMS [M+H]+ 378.22 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 9.55 (s, 1 H) 8.65 (s, 1 H) 8.24 (d, J=7.8 Hz, 1 H) 7.85 (d, J=7.3 Hz, 2 H) 7.72 (d, J=7.8 Hz, 1 H) 7.37 (t, J=8.1 Hz, 2 H) 6.97 (t, J=7.3 Hz, 1 H) 2.68 (s, 3 H) 1.35 (s, 12 H).
6-(2-aminopyrimidin-4-yl)-N-phenyl-2H-indazol-3-amine (63).
Compound 62 (75 mg, 0.199 mmol), K2CO3 (97 mg, 0.702 mmol), and 2-amino-4-chloropyrimidine (54 mg, 0.417 mmol) were combined in a reaction vial and 3:1 dioxane:water (2.0 ml, 0.10 M) was added. The reaction was degassed for ~10 minutes and PdCl2(dppf)·CH2Cl2 (8 mg, 0.011 mmol) was added. The reaction was refluxed at 100 °C overnight. The reaction was diluted with MeOH and filtered through celite and the crude material was purified by flash chromatography (50–100% EtOAc:hexanes) to afford the title compound as a light orange solid (19 mg, 32%). LCMS [M+H]+ 303.14 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 12.24 (s, 1 H) 8.93 (s, 1 H) 8.33 (d, J=5.4 Hz, 1 H) 8.11 (s, 1 H) 8.04 (d, J=8.8 Hz, 1 H) 7.73 (dd, J=8.8, 1.5 Hz, 1 H) 7.68 (d, J=7.8 Hz, 2 H) 7.26 (dd, J=8.5, 7.6 Hz, 2 H) 7.21 (d, J=5.4 Hz, 1 H) 6.81 (t, J=7.3 Hz, 1 H) 6.69 (s, 2 H).
4-(2-Chloropyrimidin-4-yl)-2-fluorobenzonitrile (64).
Compound 29 (400 mg, 2.43 mmol), 2,4-dichloropyrimidine (444 mg, 2.98 mmol), potassium carbonate (1.01 g, 7.31 mmol), and PdCl2(dppf) (91 mg, 0.124 mmol) were combined in a microwave vial that was filled with nitrogen and evacuated three times. 3:1 Dioxane:water (16 ml, 0.15 M) was added and the reaction was degassed for ~10 min. The reaction was run in the microwave (145 °C, H abs) for 30 min. The reaction mixture was diluted with EtOAc, filtered through celite, and concentrated under reduced pressure. The crude material was purified by flash chromatography (0–50% EtOAc:Hex) to afford the title compound as an off-white solid (308 mg, 54%). LCMS [M+H]+ 233.96 m/z (35Cl), 235.93 m/z (37Cl); 1H NMR (399 MHz, DMSO-d6) δ ppm 8.98 (d, J=5.1 Hz, 1 H) 8.29 – 8.35 (m, 2 H) 8.24 (d, J=8.8 Hz, 1 H) 8.17 (dd, J=8.1, 6.6 Hz, 1 H).
4-(2-(Bis(4-methoxybenzyl)amino)pyrimidin-4-yl)-2-fluorobenzonitrile (65).
Compound 64 (308 mg, 1.32 mmol) was suspended in MeOH (6.6 ml, 0.20 M) and bis(4-methoxybenzyl)amine (409 mg, 1.59 mmol) was added, followed by the addtion of Hunig’s base (0.7 m, 4.02 mmol). The reaction was refluxed for 36 hrs at 65 °C. The reaction was concentrated under reduced pressure and redissolved in DCM. The organic layer was washed once with 1M HCl, once with water, and once with brine, then dried with sodium sulfate and concentrated under reduced pressure. The crude matieral was purified by flash chromatography (20–50% EtOAc:Hex) to afford the title compound as a yellow solid (301 mg, 50%). LCMS [M+H]+ 455.21 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 8.58 (d, J=4.9 Hz, 1 H) 8.18 (d, J=10.7 Hz, 1 H) 8.15 (dd, J=8.3, 1.0 Hz, 1 H) 8.07 (m, J=6.8 Hz, 1 H) 7.40 (d, J=5.4 Hz, 1 H) 7.21 (d, J=8.3 Hz, 4 H) 6.87 (d, J=8.8 Hz, 4 H) 4.81 (br. s., 4 H) 3.71 (s, 6 H).
6-(2-(Bis(4-methoxybenzyl)amino)pyrimidin-4-yl)-1H-indazol-3-amine (66).
Compound 65 (301 mg, 0.662 mmol) was suspended in ethanol (11.0 ml, 0.06M) and hydrazine monohydrate (1.0 ml, 20.37 mmol) was added. The reaction was run overnight at 95 °C, then left standing overnight. A bright yellow precipitate was observed and collected by vacuum filtration to afford the title compound as a bright yellow solid (124 mg, 40%). LCMS [M+H]+ 467.15 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 11.59 (s, 1 H) 8.45 (d, J=5.4 Hz, 1 H) 8.03 (s, 1 H) 7.76 (d, J=8.3 Hz, 1 H) 7.64 (d, J=8.8 Hz, 1 H) 7.29 (d, J=5.4 Hz, 1 H) 7.24 (br. s., 4 H) 6.89 (d, J=8.8 Hz, 4 H) 5.43 (s, 2 H) 4.81 (br. s., 4 H) 3.72 (s, 6 H).
N-benzyl-6-(2-(bis(4-methoxybenzyl)amino)pyrimidin-4-yl)-1H-indazol-3-amine (67).
Benzal-dehyde (0.06 ml, 0.588 mmol) was dissolved in DCM (1.9 ml, 0.31 M) and compound 66 (197 mg, 0.422 mmol), and acetic acid (50 μl, 0.874 mmol) were added in that order. Upon addition of acetic acid, the reaction mixture went from cloudy yellow to clear yellow. The reaction was stirred at room temperature for ~40 minutes, after which NaHB(OAc)3 (313 mg, 1.48 mmol) was added. The reaction was stirred overnight at room temperature. The reaction was diluted with DCM and washed twice with 3M NaOH, once with water, and once with brine. The organic layer was dried with sodium sulfate and concentrated under reduced pressure. The crude material was purified three times by flash chromatography (0–50% EtOAc:Hex, then 1–10% MeOH:DCM, then 0–20% EtOAc:DCM) to afford the title compound as a pale yellow solid (77 mg, 33%). LCMS [M+H]+ 557.12 m/z; 1H NMR (500 MHz, DMSO-d6) δ ppm 11.63 (s, 1 H) 8.46 (d, J=5.4 Hz, 1 H) 8.05 (s, 1 H) 7.84 (d, J=8.3 Hz, 1 H) 7.65 (d, J=8.8 Hz, 1 H) 7.41 (d, J=6.8 Hz, 2 H) 7.28 – 7.33 (m, 3 H) 7.18 – 7.27 (m, 5 H) 6.88 (d, J=8.8 Hz, 4 H) 6.65 (t, J=6.1 Hz, 1 H) 4.82 (br. s., 4 H) 4.48 (d, J=5.9 Hz, 2 H) 3.72 (s, 6 H).
6-(2-aminopyrimidin-4-yl)-N-benzyl-2H-indazol-3-amine (68).
To compound 67 (77 mg, 0.138 mmol) was added TFA (0.3 ml, 3.92 mmol). The reaction was run overnight at 95 °C. The reaction mixture was diluted with MeOH and purified by preparative HPLC (5–50% water:acetonitrile), then repurified by flash chromatography (20% EtOAc:Hex - 0–5% MeOH:DCM) to afford the title compound as a yellow solid (9.6 mg, 22%). LCMS [M+H]+ 317.11 m/z; 1H NMR (500 MHz, METHANOL-d4) δ ppm 8.29 (d, J=5.4 Hz, 1 H) 8.00 (s, 1 H) 7.80 (d, J=8.3 Hz, 1 H) 7.63 (dd, J=8.5, 1.2 Hz, 1 H) 7.44 (d, J=6.8 Hz, 2 H) 7.31 (dd, J=7.6 Hz, 2 H) 7.23 (t, J=7.3 Hz, 1 H) 7.17 (d, J=5.4 Hz, 1 H) 4.56 (s, 2 H).
Supplementary Material
Table 3.
Biological activity of N-methylated analogs. All SD within ± 0.08.
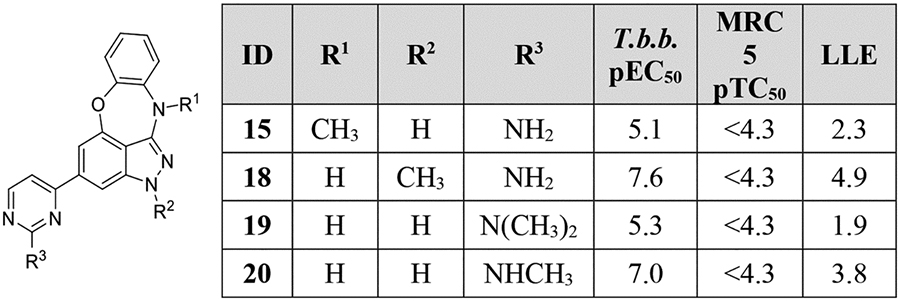
|
Acknowledgements
The authors wish to acknowledge funding from the National Institute of Allergy and Infectious Diseases (MPP & MN: R01AI114685; MPP: 1R21AI127594, R01AI124046; CRC: R21AI126296; https://www.niaid.nih.gov/), the Spanish Ministerio de Economía, Industria y Competitividad, (MN: SAF2015-71444-P; DG-P: SAF2016-79957-R. http://www.mineco.gob.es) and Subdirección General de Redes y Centros de Investigación Cooperativa (RICET) (MN: RD16/0027/0019; DG-P: RD16/0027/0014. https://bit.ly/2OegOnX). RTI2018-097210-B-I00 (MINCIU-FEDER) to FG. CRC thanks Brian M. Suzuki for expert technical assistance with the Schistosoma mansoni screening activities. An ACS MEDI Predoctoral Fellowship for DK is gratefully acknowledged. We thank AstraZeneca and GlaxoSmithKline for the provision of the in vitro ADME and physicochemical properties data.
Abbreviations used
- BID
twice daily
- BOXI
benzoxazepinoindazole
- Clint
intrinsic clearance
- Cmax
maximum concentration
- CNS-MPO
CNS-multiparameter optimization
- GSK
GlaxoSmithKline
- HAT
human African trypanosomiasis
- HLM
human liver microsomes
- NECT
nifurtimox-eflornithine combination therapy
- NTD
neglected tropical disease
- PPB
plasma protein binding
- QD
once daily
- SD
standard error of the mean
- T.b.b.
Trypanosoma brucei brucei
- TEA
triethylamine
- tmax
time at which maximum concentration is achieved
- TPSA
topological polar surface area
Footnotes
Supporting Information Available
1. MS Word document containing: Supplemental biological and ADME data, supplemental synthetic schemes, HPLC chromatograms, cell and whole-organism assay protocols, and ADME experiment protocols.
2. MS Excel document containing: SMILES strings for final compounds with biological activity.
References
- 1.World Health Organization. Neglected Tropical Diseases. http://www.who.int/neglected_diseases/diseases/en/ (accessed November 24, 2015).
- 2.Buscher P; Cecchi G; Jamonneau V; Priotto G, Human African trypanosomiasis. Lancet 2017, 390, 2397–2409. [DOI] [PubMed] [Google Scholar]
- 3.(a) Drugs for Neglected Diseases Initiative. Fexinidazole (HAT). http://www.dndi.org/diseases-projects/portfolio/fexinidazole/ (accessed February 13, 2018);; (b) Mesu VKBKM; Kalonji WM; Bardonneau C; Mordt OV; Blesson S; Simon F; Delhomme S; Bernhard S; Kuziena W; Lubaki J-PF; Vuvu SL; Ngima PN; Mbembo HM; Ilunga M; Bonama AK; Heradi JA; Solomo JLL; Mandula G; Badibabi LK; Dama FR; Lukula PK; Tete DN; Lumbala C; Scherrer B; Strub-Wourgaft N; Tarral A, Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis; a pivotal multicentre, randomised, non-inferiority trial. Lancet 2017, 391 (10116), 144–154; [DOI] [PubMed] [Google Scholar]; (c) Drugs for Neglected Diseases Initiative. Acoziborole. http://www.dndi.org/diseasesprojects/portfolio/scyx-7158/ (accessed February 13, 2017).
- 4.Klug DM; Gelb MH; Pollastri MP, Repurposing strategies for neglected diseases. Bioorg. Med. Chem. Lett 2016, 26, 2569–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merritt C; Silva LE; Tanner AL; Stuart K; Pollastri MP, Kinases as druggable targets in trypanosomatid protozoan parasites. Chem. Rev 2014, 114, 11280–11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diaz R; Luengo-Arratta S; Seixas J. o. D.; Amata E; Devine W; Cordon-Obras C; Rojas-Barros DI; Jimenez E; Ortega F; Crouch S; Colmenarejo G; Fiandor JM; Martin JJ; Berlanga M; Gonzalez S; Manzano P; Navarro M; Pollastri MP, Identification and characterization of hundreds of potent and selective inhibitors of Trypanosoma brucei growth from a kinase-targeted library screening campaign. PLoS Negl. Trop. Dis 2014, 8 (10), e3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shultz MD, The thermodynamic basis for the use of lipophilic efficiency (LipE) in enthalpic optimizations. Bioorg Med Chem Lett 2013, 23, 5992–6000. [DOI] [PubMed] [Google Scholar]
- 8.Wager T; Hou X; Verhoest PR; Villalobos A, Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci 2010, 1, 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.