

SUMMARY
We study a patient with the human papilloma virus (HPV)-2-driven “tree-man” phenotype and two relatives with unusually severe HPV4-driven warts. The giant horns form an HPV-2-driven multifocal benign epithelial tumor overexpressing viral oncogenes in the epidermis basal layer. The patients are unexpectedly homozygous for a private CD28 variant. They have no detectable CD28 on their T cells, with the exception of a small contingent of revertant memory CD4+ T cells. T cell development is barely affected, and T cells respond to CD3 and CD2, but not CD28, costimulation. Although the patients do not display HPV-2- and HPV-4-reactive CD4+ T cells in vitro, they make antibodies specific for both viruses in vivo. CD28-deficient mice are susceptible to cutaneous infections with the mouse papillomavirus MmuPV1. The control of HPV-2 and HPV-4 in keratinocytes is dependent on the T cell CD28 co-activation pathway. Surprisingly, human CD28-dependent T cell responses are largely redundant for protective immunity.
In brief
By studying a family of individuals with cutaneous protrusions and severe warts driven by human papilloma virus (HPV) infection, Beziat et al. discover that human CD28 is largely dispensable for protective immunity against most infections. Inherited CD28 deficiency only slightly impairs T cell development and function, but control of HPV in keratinocytes is dependent on the T cell CD28 co-activation pathway.
Graphical Abstract
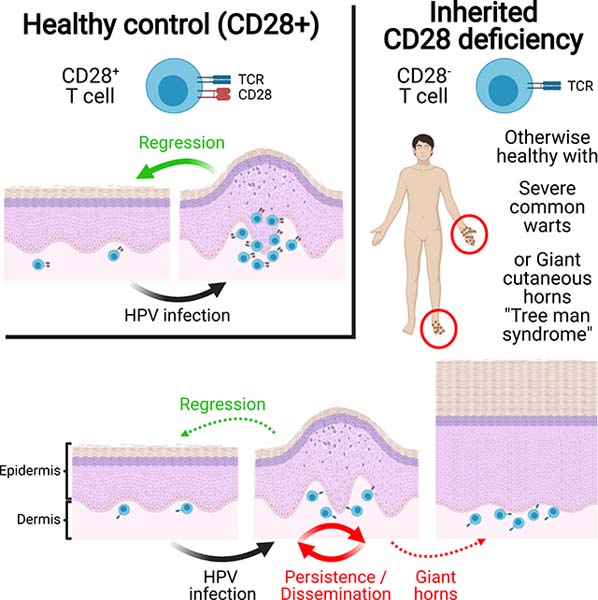
INTRODUCTION
Human papillomavirus (HPV) infections of cutaneous keratinocytes cause various types of lesions, including flat and common warts (Cubie, 2013). Common warts are typically caused by α-papillomavirus HPV-2 and γ-papillomavirus HPV-4. They are seen in the general population, in which they are typically localized and benign (Cubie, 2013). However, they can persist and spread in patients with various inherited or acquired T cell immunodeficiencies, who are also prone to a number of other infections (Béziat, 2020). “Cutaneous horns” are protrusions of keratotic material of various sizes and shapes that may arise from single common warts (Mantese et al., 2010; Yu et al., 1991). These “cutaneous horns” are often both giant and disseminated. This generalized severe hyperkeratotic cutaneous papillomatosis has been named “tree man syndrome” (TMS) (Alisjahbana et al., 2010; Uddin et al., 2018; Wang et al., 2007b). Only four unrelated TMS cases have been reported, and the α-papillomavirus HPV-2, a classic cause of common warts (Orth et al., 1977), was demonstrated to be the cause of three of these cases (Alisjahbana et al., 2010; Uddin et al., 2018; Wang et al., 2007b). These viruses have not been analyzed in depth, and the genomes of the keratinocytes in these lesions were not sequenced. Remarkably, TMS patients have severe skin lesions despite being normally resistant to other infections. TMS should not be confused with epidermodysplasia verruciformis (EV), the cutaneous warts of which are actually flat and caused by β-HPVs (de Jong et al., 2018a). The flat warts of EV often progress to non-melanoma skin cancers. “Typical” or “isolated EV” is due to biallelic null mutations of TMC6 (encoding EVER1), TMC8 (encoding EVER2), or CIB1, selectively disrupting keratinocyte-intrinsic immunity to these viruses (de Jong et al., 2018b; Ramoz et al., 2002). Patients with “atypical” or “syndromic EV” display a number of other infections, due to various inborn errors of T cells (de Jong et al., 2018a). Consistently, T cell responses against commensal β-HPVs may protect against skin cancers in the general population (Strickley et al., 2019).
The rarity and lack of contagiousness of TMS are intriguing, given the ubiquitous and benign nature of the underlying HPV-2 infection. By analogy with EV, it is tempting to speculate that TMS may result from inborn errors of immunity to HPV-2. However, by contrast to most EV kindreds, the four reported cases of TMS were sporadic, as opposed to familial, and parental consanguinity was not reported (Alisjahbana et al., 2010; Uddin et al., 2018; Wang et al., 2007b). Nevertheless, TMS lesions are strikingly reminiscent of those caused by natural infection with Shope virus, the cottontail rabbit papillomavirus (CRPV), in wild rabbits (Breitburd et al., 1997; Giri et al., 1985). In 1933, Richard E. Shope showed that some cottontail rabbits from the US Midwest displayed giant horns caused by a filterable virus (Shope and Hurst, 1933). Peyton Rous performed experimental infections in 1934 and showed that warts resolved in less than 10% of wild cottontail rabbits (between 2–8 weeks after wart appearance), with 65% of rabbits displaying persistent warts, sometime overlaid by giant horns and 25% going on to develop malignant lesions affecting internal organs within 6–12 months (Rous and Beard, 1934). Remarkably, it was shown in 1991 that, in outbred New Zealand White rabbits, the persistence of skin lesions segregated with MHC class II genes, indicating that these CRPV-driven lesions are genetically controlled and due to inadequate CD4+ T cell immunity (Han et al., 1992). The genetic and immunological basis of these lesions has not been characterized further in wild or domestic rabbits. These observations suggest that human TMS might result from an inherited immunodeficiency. Three of the TMS patients had an apparently normal T cell compartment (Wang et al., 2007a, 2007b; Uddin et al., 2018), but the fourth displayed idiopathic CD4+ T cell lymphopenia (Alisjahbana et al., 2010). We thus hypothesized that TMS might result from inborn errors of immunity to HPV-2, a causal agent of common warts.
RESULTS
A rare homozygous CD28 variant segregates with severe verrucosis in three relatives
We studied a 30-year-old Iranian patient with TMS (P1) and two of his relatives with a history of disseminated common warts (P2 and P3, aged 40 and 12 years, respectively; see case reports in the STAR Methods) (Figures 1A and 1B). A detailed clinical description is provided in the STAR Methods. We performed single-nucleotide polymorphism (SNP) array-based genome-wide linkage (GWL) analysis on the three patients and 13 relatives, testing the hypothesis of an autosomal recessive (AR) trait with complete penetrance. Highly significant evidence of linkage was obtained for a single 20.5 Mb region on chromosome 2 (LOD score = 5.12) (Figure 1C). The high percentage of homozygosity in P1 (6%), P2 (5%), and P3 (10%) on microarray analysis confirmed the consanguinity of their parents (Figures 1A and S1A). We also analyzed whole-exome sequencing (WES) results for P1, P2, and P3 (Figure S1B). None of the known genes underlying Mendelian disorders carried heterozygous or homozygous rare non-synonymous or splice variants. Within the linked region, only three protein-coding genes, including CD28, displayed nonsynonymous or splice variants, which were also homozygous and rare (minor allele frequency [MAF] <0.01; Figures S1B and S1C). CD28 is a major costimulatory molecule of T cells that can be engaged by CD80 or CD86 on antigen-presenting cells (Rudd et al., 2009). The private variant of CD28 in this kindred affects the last nucleotide of exon 1 (c.52G>A). It is predicted to replace codon 18, encoding the glycine residue in the last position of the signal peptide, with a codon encoding arginine (p.Gly18Arg) (Figure 1D). Moreover, the c.52G>A variant is computationally predicted to disrupt the donor site for splicing located between exons 1 and 2 (Desmet et al., 2009). The c.52G>A variant has a CADD score of 13.8, above the MSC of 9.2 (Figure 1E) (Kircher et al., 2014; Itan et al., 2016). The segregation of this mutant allele is consistent with a fully penetrant AR trait (Figure 1A). Moreover, there are only five homozygous missense CD28 variants, all with a MAF >0.00002, in public databases (gnomAD, TOPMed, ATAVDB, GME) (Figure 1E). The other two homozygous variants were predicted (KANSL1L) or shown to be neutral (USP37) (Figures S1D–S1H). Moreover, we showed CD28−/− mice to be vulnerable to MmuPV1 (Figure S2). Collectively, these findings suggest that AR CD28 deficiency is exceedingly rare in the general population and disease-causing in this kindred.
Figure 1. Autosomal recessive CD28 deficiency and “tree man” syndrome.
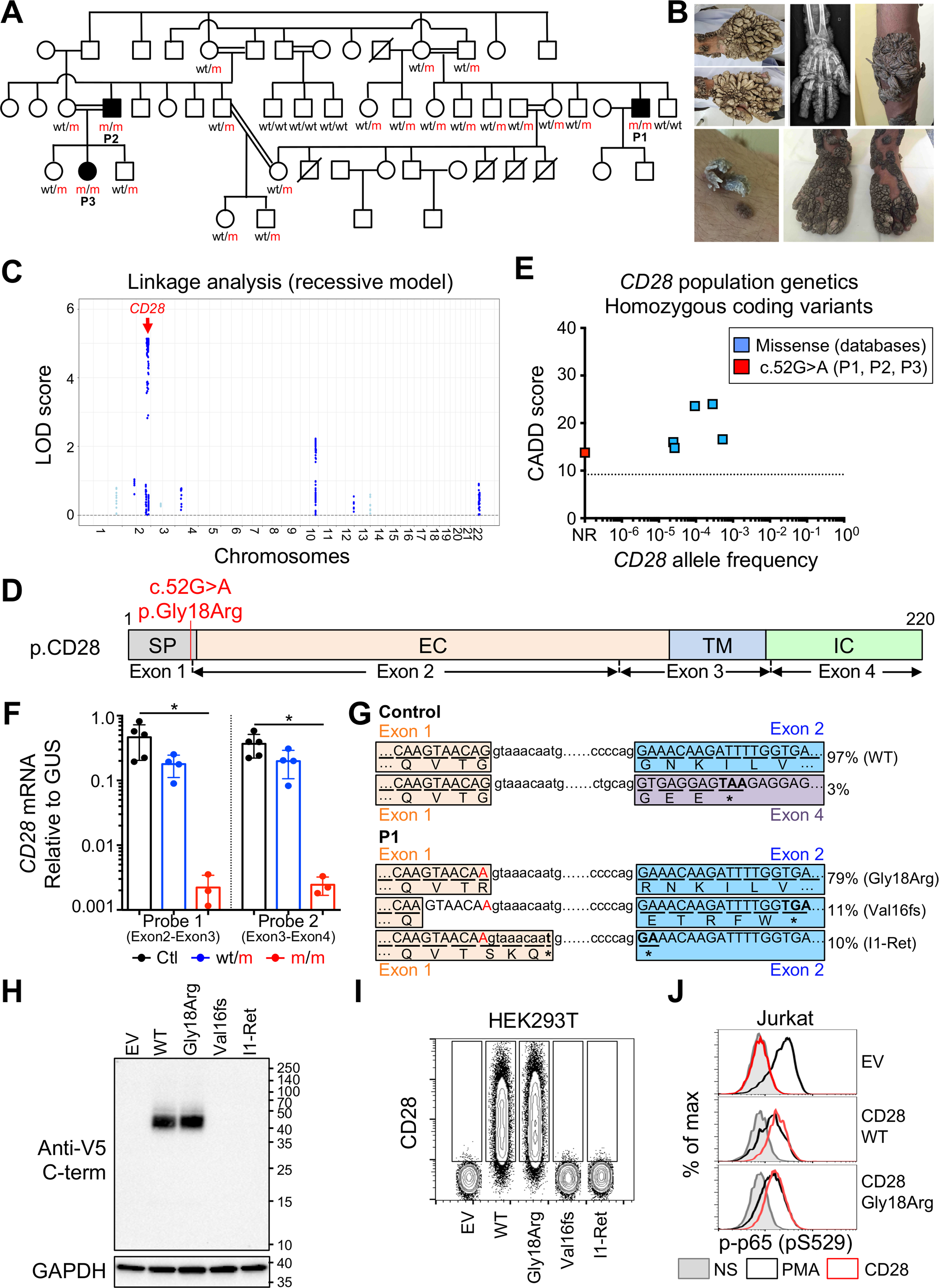
(A) Pedigree showing familial segregation of the c.52G>A mutant CD28 allele.
(B) Images of the TMS phenotype of P1 and X-ray of the right hand of P1 showing that the lesions have not extended to the bones.
(C) Genome-wide linkage analysis on DNA from 16 members of the kindred, assuming AR mode of inheritance, with complete penetrance. The linkage region with the highest LOD score was on chromosome 2 and contained CD28 (red arrow).
(D) Schematic representation of CD28 protein. SP, signal peptide; EC, extracellular domain; TM, transmembrane domain; IC, intracellular domain. Exons and exon boundaries are depicted below. The mutation is indicated with a red line.
(E) Frequency and CADD score for all CD28 variants reported homozygous in the public databases. The dotted line corresponds to the MSC. NR, not reported. The private c.52G>A mutation appears in red.
(F) Reverse transcription qPCR for total CD28 with two different probes representative of three independent experiments. Bars represent the mean and the SD. Mann-Whitney tests were used for comparisons.
(G) Reverse transcription PCR from PHA blasts from one control and P1 was performed to amplify the sequence between the 5′ and 3′ UTRs of the CD28 cDNA for insertion into a vector. We sequenced 190 colonies for the control and 106 for P1. The splice variants involving the splice donor site of exon 1, and their respective frequencies are shown. The amino acid sequence encoded by each transcript is shown below the nucleotide sequence.
(H and I) HEK293T cells were transfected with an empty vector (EV) or with vectors encoding the indicated CD28 transcripts. (H) Immunoblotting with a monoclonal antibody against the V5 tag and GAPDH. (I) Cell surface CD28 levels.
(J) Jurkat cells were transduced with an EV or a plasmid encoding the indicated CD28 cDNA. Phospho-p65 (p-p65) was detected by flow cytometry after the crosslinking of CD28 or PMA stimulation. (H–J) Data representative of three independent experiments.
See also Figures S1, S2, and S3 and Data S1.
The c.52G>A variant greatly decreases CD28 mRNA levels in T lymphocytes
Human CD28 is a transmembrane protein expressed on the surface of T cells (Esensten et al., 2016). We investigated the impact of the c.52G>A variant on endogenous CD28 mRNA levels, by performing reverse transcription PCR and reverse transcription qPCR on ex vivo PHA-expanded primary T cells. Reverse transcription qPCR with two different probes showed that CD28 mRNA levels were 150- to 200-fold lower in PHA-driven T cell blasts from the patients than in those of the controls, whereas the levels in heterozygous cells were half those in control cells (Figure 1F). Reverse transcription PCR detected products of similar molecular weight (MW) in controls, heterozygotes, and patients (Figure S3A). The full-length product was detected in much smaller amounts by reverse transcription PCR in the patients’ cells than in cells from all other individuals tested (Figure S3A). We showed, by TA cloning of the reverse transcription PCR products from a control and P1, that all mRNAs in control T cells used the canonical splicing donor site of exon 1 (Figure 1G). Only 79% of CD28 transcripts in P1’s T cells used the canonical mRNA splicing donor site of exon 1 containing the missense nucleotide substitution (p.Gly18Arg) (Figure 1G). The remaining 21% of the transcripts used alternative donor splice sites corresponding to a truncated and frameshift transcript (referred to hereafter as p.Val16fs) or a transcript retaining the first nine nucleotides of intron 1 (referred to hereafter as I1-Ret). Both aberrant transcripts have a premature stop codon upstream of the segment encoding the transmembrane domain. The same aberrant splice transcripts were obtained by exon trapping (Figures S3B and S3C). These data demonstrate that the c.52G>A variant strongly decreases CD28 mRNA levels in the primary T cells of patients, probably by nonsense-mediated decay of the two aberrant transcripts, p.Val16fs and I1-Ret.
Characterization of CD28 splice variants resulting from the c.52G>A variant
We investigated whether the novel transcripts resulting from the c.52G>A variant encoded functional CD28 isoforms. We transfected HEK293T cells with pcDNA-V3.1 expression vectors containing CD28 cDNAs encoding the wild-type (WT), p.Gly18Arg, p.Val16fs, and I1-Ret isoforms fused to a C-terminal V5 tag. Western blotting with an anti-V5 tag monoclonal antibody (mAb) revealed the presence of the WT and p.Gly18Arg isoforms at the expected MW (~40 kDa) (Figure 1H). No p.Val16fs or I1-Ret proteins were detected, suggesting that the translation of CD28 was not reinitiated downstream of the premature stop codons. The p.Gly18Arg mutant isoform, in which the last amino acid of the CD28 signal peptide is affected (Figure 1D), was predicted to result in a relocation of the signal peptide cleavage site without impairment of protein trafficking to the cell surface (Figure S3D). We transfected HEK293T cells with the WT, p.Gly18Arg, p.Val16fs, and I1-Ret cDNAs and monitored the extracellular expression of CD28 by flow cytometry (Figure 1I). The WT and p.Gly18Arg isoforms of CD28 were normally expressed on the cell surface, whereas the p.Val16fs and I1-Ret isoforms were not detected. Despite normal expression of p.Gly18Arg on the cell surface, the alteration to the signal peptide cleavage site may have affected the primary structure of the protein at the cell surface, and, ultimately, its function. We used a lentiviral vector to transduce Jurkat T cells, which lack CD28 expression, stably with the WT and p.Gly18Arg CD28 isoforms, to determine whether p.Gly18Arg retained the ability to induce nuclear factor κB (NF-κB) signaling upon binding to an anti-CD28 mAb (Figures 1J and S3E) (Rudd et al., 2009). CD28-mediated phosphorylation of p65 (p-p65) was observed with this variant. Stimulation with PMA was used as a positive control. These findings suggested that the homozygous CD28 c.52G>A allele generates extremely low levels (<1% normal levels) of functional p.Gly18Arg proteins in the patients’ T cells because the aberrant transcripts (p.Val16fs and I1-Ret) are degraded and their protein products are not detected.
The c.52G>A variant is loss-of-expression and loss-of-function in T lymphocytes
We assessed the expression and function of CD28, by flow cytometry, on primary CD4+ and CD8+ T cell populations, expanded by PHA ex vivo (Figure 2A). No CD28 expression was detected on PHA-driven T cell blasts from the three CD28-deficient patients, and CD28 levels on heterozygous T cells were 50% lower than those in controls. We tested the functional consequences of the lack of detection of CD28. We crosslinked CD28 and/or CD3 with specific mAbs and monitored NF-κB p65 phosphorylation (p-p65) after 30 min, with PMA stimulation used as a positive control (Figure 2B). PMA stimulation induced high levels of p-p65 in CD4+ and CD8+ T cells from the patients, heterozygotes, and controls. CD3 stimulation alone weakly induced the production of p-p65 in both T cell subsets in all individuals. In the heterozygotes and healthy controls, stimulation via CD28 alone induced strong p65 phosphorylation in CD4+ T cells, and CD3/CD28 costimulation induced strong p65 phosphorylation in CD8+ T cells (Figure 2B). The patients’ CD4+ and CD8+ T cells displayed no detectable p65 phosphorylation following CD28 stimulation alone, and no increase in p65 phosphorylation following CD3/CD28 costimulation, relative to CD3 stimulation alone (Figure 2B). We used a lentiviral vector to express the WT CD28 cDNA in T cells PHA blasts from P1. This rescued p65 phosphorylation in CD4+ T cells upon CD28 crosslinking, and in CD8+ T cells upon CD3/CD28 crosslinking (Figure 2C). Even though the c.52G>A allele generates very low levels of a CD28 mRNA potentially encoding a functional p.Gly18Arg isoform, it is a loss-of-expression and loss-of-function allele in the homozygous primary T cells of the patients, which display complete CD28 deficiency.
Figure 2. Abolished CD28 response in the patients’ primary T cells.
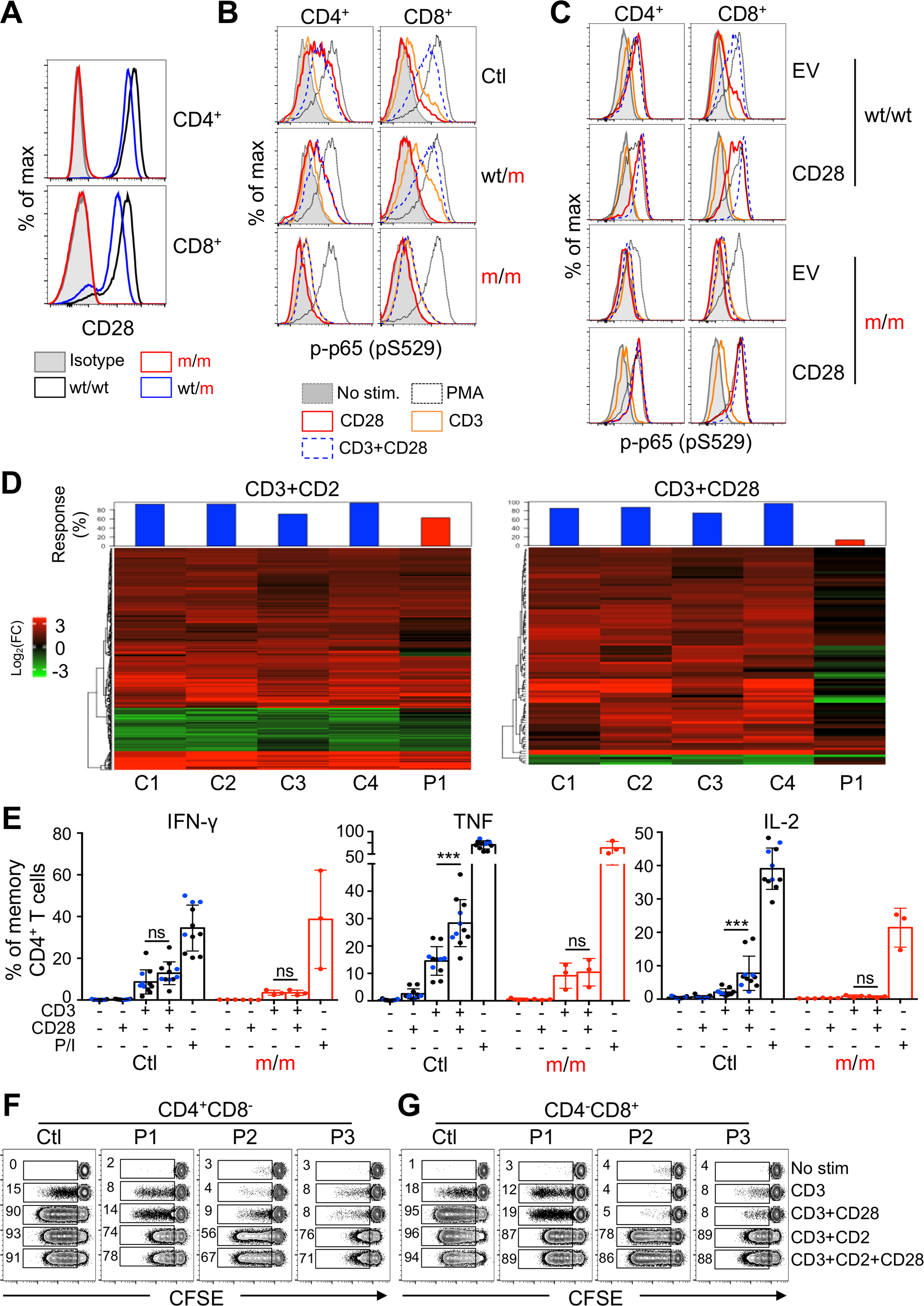
(A) Representative fluorescence-activated cell sorting (FACS) plots showing CD28 expression on PHA blasts from patients, heterozygotes, and controls.
(B) Phospho-p65 (p-p65) detection in CD4+ (left) and CD8+ (right) PHA blasts from P1 (m/m), a heterozygote (wt/m) and a healthy control (wt/wt), after crosslinking of the indicated cell surface receptors. Representative of two independent experiments.
(C) PHA blasts from one control (top) and P1 (bottom) were transduced with an empty vector (EV) or a vector encoding the wild-type CD28. Phospho-p65 (p-p65) detection by flow cytometry in CD4+mCherry+ (left) and CD8+mCherry+ (right) PHA blasts after crosslinking of the indicated cell surface receptors. (B and C) PMA stimulation was used as a positive control.
(D) RNA-seq analysis of primary CD4+ T cells. Compared to CD3 stimulation alone, 368 and 91 genes were significantly up- or downregulated in control’s cell stimulated by CD3+CD2 (left) or CD3+CD28 (right), respectively. The residual responses of the CD28-deficient patient are shown as percentage in the red bar charts for each condition.
(E) Frequency of indicated cytokine in CD4+ memory T cells from healthy controls and patients after stimulation with PMA and ionomycin, or P815 cells in the presence of anti-CD3 and/or anti-CD28 mAbs. Graphs show the mean ± SD. Wilcoxon tests were used for comparisons.
(F and G) CFSE dilution of CD4+ (F) and CD8+ (G) T cells from a control, P1, P2, and P3 after 5 day stimulation with beads coated with the indicated combination of CD2, CD3, and CD28 mAbs. Numbers indicate frequencies of proliferating cells in each condition. Data representative of 2–3 independent experiments.
CD28-dependent responses are abolished in the patients’ T lymphocytes
We used RNA sequencing (RNA-seq) to analyze the induction of CD28 target genes. We sorted CD4+ naive T cells from P1 and four healthy controls, and stimulated them with PMA or anti-CD2, anti-CD3, anti-CD28, anti-CD3 plus anti-CD2, or anti-CD3 plus anti-CD28 mAb-coated beads for 2 h before the extraction and sequencing of mRNA. We first compared the different stimulations with the unstimulated baseline. The patient’s cells responded normally to CD3, CD3 and CD2 costimulation, and to stimulation with PMA (Figure S4A). The stimulation of CD2 or CD28 alone induced only a few genes in T cells from healthy controls relative to unstimulated cells (Figure S4A) (Riley et al., 2002). The weak response to CD28 stimulation alone was abolished in the patient. CD3 and CD28 costimulation in the patient’s naive CD4+ T cells was impaired relative to that in controls (Figure S4A). CD28 stimulation essentially amplifies CD3 responses (Riley et al., 2002). We compared the costimulation of CD3 and CD2, or of CD3 and CD28, with that of CD3 alone (Figure 2D; Table S1). We found that the response to CD3 and CD2 costimulation was similar in the patient and controls, while that to CD3 and CD28 costimulation was abolished in the patient. Levels of mRNA for IL2, a prototypic CD28 target gene (Esensten et al., 2016), displayed an induction of ~24-fold relative to CD3 stimulation alone in control cells, but not in the patient’s cells, after CD3 and CD28 costimulation. We assessed the induction of the interferon (IFN)-γ, tumor necrosis factor (TNF), and interleukin (IL)-2 proteins in resting primary memory CD4+ T cells, following stimulation with a combination of anti-CD3 and anti-CD28 mAbs (Figure 2E). The costimulation of CD3 and CD28 induced a greater increase in TNF and IL-2 levels, but not IFN-γ levels, than CD3 stimulation alone, in heterozygotes and controls. In CD4+ memory T cells from the patients, costimulation with CD3 and CD28 did not increase the production of either cytokine. PMA plus ionomycin, used as a control, induced the production of high levels of all the cytokines tested in controls, heterozygotes, and patients. Costimulation with CD28 plus CD3 strongly increased the proliferation of control CD4+ and CD8+ T cells relative to CD3 stimulation alone (Figured 2F and 2G). CD28 costimulation failed to increase the CD3-mediated proliferation of the patients’ CD4+ and CD8+ T cells. CD2 costimulation, used as a control, increased CD3-mediated proliferation similarly in CD4+ and CD8+ T cells from patients and controls. Consistent with the proximal signaling impairment downstream of CD28, more distal CD28-dependent responses were thus abolished in the patients’ T lymphocytes.
CD28 loss has a modest impact on the homeostasis of leukocyte subsets
The global distribution of leukocyte subsets in peripheral blood was normal in the three patients (Table S2). Within the myeloid compartment, the monocyte (P1–P3) and dendritic cell (P1) subsets were normal (Figures S4B and S4C). Within the lymphoid compartment, B cell subset counts and frequencies were normal, and we detected near-normal frequencies of total and class-switched memory B cells in the patients (Figures S4D and S4E; Table S2). Natural killer (NK) cell counts were low (Table S2), but the frequencies of the immature CD56bright and mature CD56dim NK subsets were normal, with the possible exception of an unusually large proportion of NKG2C+ cells, a population previously shown to expand upon CMV infection (Figures S4F–S4H) (Béziat et al., 2013). CD8+ T counts were high in P1 and P3, and CD4+ T cell count was high only in P3 (Table S2). The frequency of naive CD4+ T cells was high, and the frequencies of central memory CD4+ and CD8+ T cells were low (Figures 3A and 3B). The proliferation of T cells from P1 was normal in response to anti-CD3 antibody or PHA stimulation, weak upon tuberculin stimulation, and absent upon tetanus toxin stimulation (in the absence of a booster dose of vaccine since infancy) (Table S2). Within CD3+ T cells, the frequencies of iNKT and γδ T cells were normal, whereas that of MAIT cells was low in all patients (at levels similar to that in heterozygotes) (Figure 3C). Within CD4+ T cells, regulatory T (Treg) cell frequency was ~70% lower than the mean value for controls (Figure 3D), consistent with CD28 requirement and redundancy in mice for natural and induced Treg development and homeostasis, respectively (Salomon et al., 2000; Semple et al., 2011; Tai et al., 2005; Tang et al., 2003). The frequencies of Th subsets (Th1, Th2, Th1*, Th17, and Tfh) were normal (Figure 3E). We then performed single-cell RNA sequencing (scRNA-seq) on peripheral blood mononuclear cells (PBMCs) from three controls and the three patients (Figures 3F–3H and S5A). This analysis confirmed the largely normal distribution of lymphocyte subsets. It did not identify major transcriptomic abnormalities in T cell subsets, including naive and memory T cells, Tregs, and γδ T cells, other than extremely low levels of CD28 mRNA in all T cell compartments tested (Figure S5B). The TCR-Vα and Vβ repertoires of the patients’ CD28− CD4+ T cells displayed normal diversity (Data S1). The induction of costimulatory (OX40, CD40L, and ICOS) and coinhibitory molecules (CTLA4, PD1, Tigit, BTLA, Tim3, and LAG3), as well as CD25 and ICAM1, upon stimulation with PMA plus ionomycin was normal in patients’ CD4+ and CD8+ T cells (Figure S6A; Data S1). The induction of OX40, CD40L, ICOS, Tim3, LAG3, CD25, and ICAM1 upon costimulation with CD2/CD3/CD28 mAb-coated beads was partially impaired in the patients, whereas that of CTLA4, PD1, Tigit, and BTLA was normal. The patients displayed no clinical signs of autoimmunity or autoinflammation. Low levels of EBV (P1, P2, and P3) and HCMV (P1-P2) replication were detected in the patients’ blood, without clinical manifestations (Table S2). We detected IFN-γ+ CD4+ and CD8+ T cells in P1 and P2 upon stimulation with overlapping HCMV peptides in vitro (Figure S6B) and IFN-γ+ CD4+ T cells in all BCG-vaccinated patients following stimulation with tuberculin (PPD) in vitro. Thus, the distribution and function of the various leukocyte subsets were normal or subnormal in the three patients, probably accounting for the lack of infectious phenotypes other than severe verrucosis, and, more generally, the lack of other overt immunopathological phenotypes.
Figure 3. CD28 deficiency has a mild impact on lymphocyte subset development.
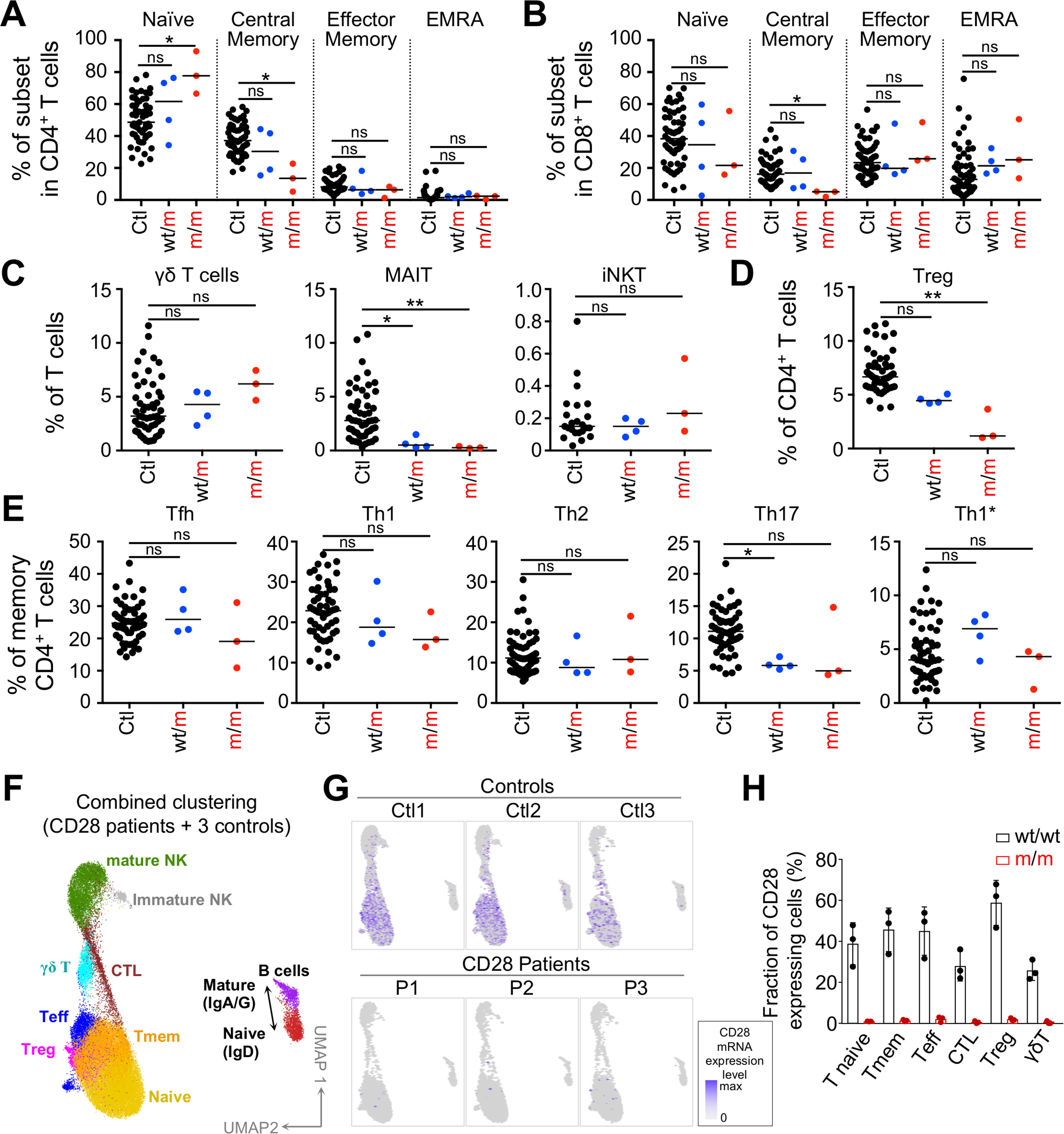
(A–E) Frequency of indicated T cell subsets of controls (n = 23–57), heterozygotes (n = 4), and patients (n = 3). Kruskal-Wallis tests were used for all comparisons. The bars represent the median.
(F) scRNA-seq UMAP clustering of PBMC from 3 CD28 homozygous patients together with a set of 3 controls. CTL, cytotoxic T cells; Teff, effector T cells; Tmem, memory T cells.
(G) CD28 expression level superimposed on the scRNA-seq UMAP clustering.
(H) Fraction of CD28+ cells in T cell subsets indicated and as defined by single-cell RNA-seq UMAP clustering. Graphs show the mean ± SD.
See also Figures S4, S5, and S6, Table S2, and Data S1.
Evidence of somatic mosaicism in CD4+ memory T cells
We then analyzed CD28 expression in the various T cell subsets from the patients. Consistent with our findings for bulk CD4+ and CD8+ T cells (Figure 2A), we detected no CD28 expression on circulating naive CD4+ and CD8+ T cells or on memory CD8+ T cells from the three patients, by flow cytometry (Figure 4A). Surprisingly, we found that 0.8%, 2.1%, and 2.9% of the memory CD4+ T cells of P3 (aged 12 years), P1 (30 years), and P2 (40 years), respectively, expressed CD28, with fluorescence levels similar to those for heterozygotes, but lower than those for controls (Figure 4A). This suggested the possible reversion of one CD28 allele in the progenitors of these patients’ T cells and, possibly, the expansion of the revertant pool with age. The sorting of CD28+ T cells from all three patients confirmed a reversion of one CD28 allele to the WT form, because some cells were heterozygous for the c.52G>A variant, whereas CD28− cells were homozygous (Figures 4B and S7A). We also detected the c.52+5A>G somatic mutation (Figure 4B), which, as predicted (Figure S7B) and confirmed by exon trapping (Figures S3B and S3C), restored normal splicing despite the presence of the original c.52G>A variant. We showed that the revertant cells were autologous, and not maternally derived, by genotyping the rare variant of USP37 (Figures 4B and S7A). We sorted CD4+ CD28+ revertants from P1, P2, and P3 and found their TCR-Vα and -Vβ repertoires to be highly diverse, suggesting that the reversions occurred before T cell Vβ rearrangement in the thymus (Data S1). We also analyzed the identified TCR-Vβ sequences using the GLIPH software that clusters TCRs predicted to bind the same MHC-restricted antigenic peptide (Glanville et al., 2017). This predicted that almost all TCR-Vβ from the revertant cells share specificity with CD28-deficient CD4+ cells. The vast majority of MHC-restricted peptides recognized by the CD28+ revertants are also probably recognized by the patients’ CD28− CD4+ cells. An analysis of >12 million PBMCs from P3 showed that 0.00034% of CD31+CD4+ T cells, which are known to be recent thymic emigrants (RTEs), expressed CD28 (Figure 4C). This implies an ~1,500-fold expansion of revertant cells between RTEs and memory CD4+ T cells. The revertant memory CD4+ T cells were phenotypically diverse, with Treg and Th CD4+ subsets present in proportions close to those observed in the CD28− compartment (Figures 4D and 4E). We then performed cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) on sorted memory CD4+ T cells from one control, P1, and P2 (Figures 4F–4H, S7C, and S7D). Using stringent filtering criteria, we identified a total of 38 CD28+ revertant cells from P1 or P2. Using uniform manifold approximation and projection (UMAP) for dimensions reduction, we found that the CD28+ revertants and the CD28− non-revertants from the two patients were scattered all over controls’ CD4+ memory T cells (Figures 4G, S7C, and S7D). In line with our scRNA-seq on total PBMCs (Figure S5), this confirms that the transcriptome of patients’ CD4+CD28− non-revertant memory T cells is not significantly different from that of controls’ CD4+ memory cells. This also shows that the transcriptome of the CD28+ revertants is not significantly biased toward a specific cluster or subset of memory CD4+ T cells. The only mRNA that was significantly different between patients’ CD28+ revertants and CD28− memory CD4+ T cells was CD28 mRNA level itself, which was, as expected, highly significantly increased in the former (Figure 4H). Overall, the revertant T cells were highly enriched in memory CD4+ T cells, a compartment in which they probably increased in frequency with age (their frequency paralleling the age of the three patients). They were highly diverse in terms of their TCR and functional repertoire, and they occurred in the three patients following at least two types of somatic mutation.
Figure 4. Evidence for somatic mosaicism in the patients’ memory CD4+ T cells.
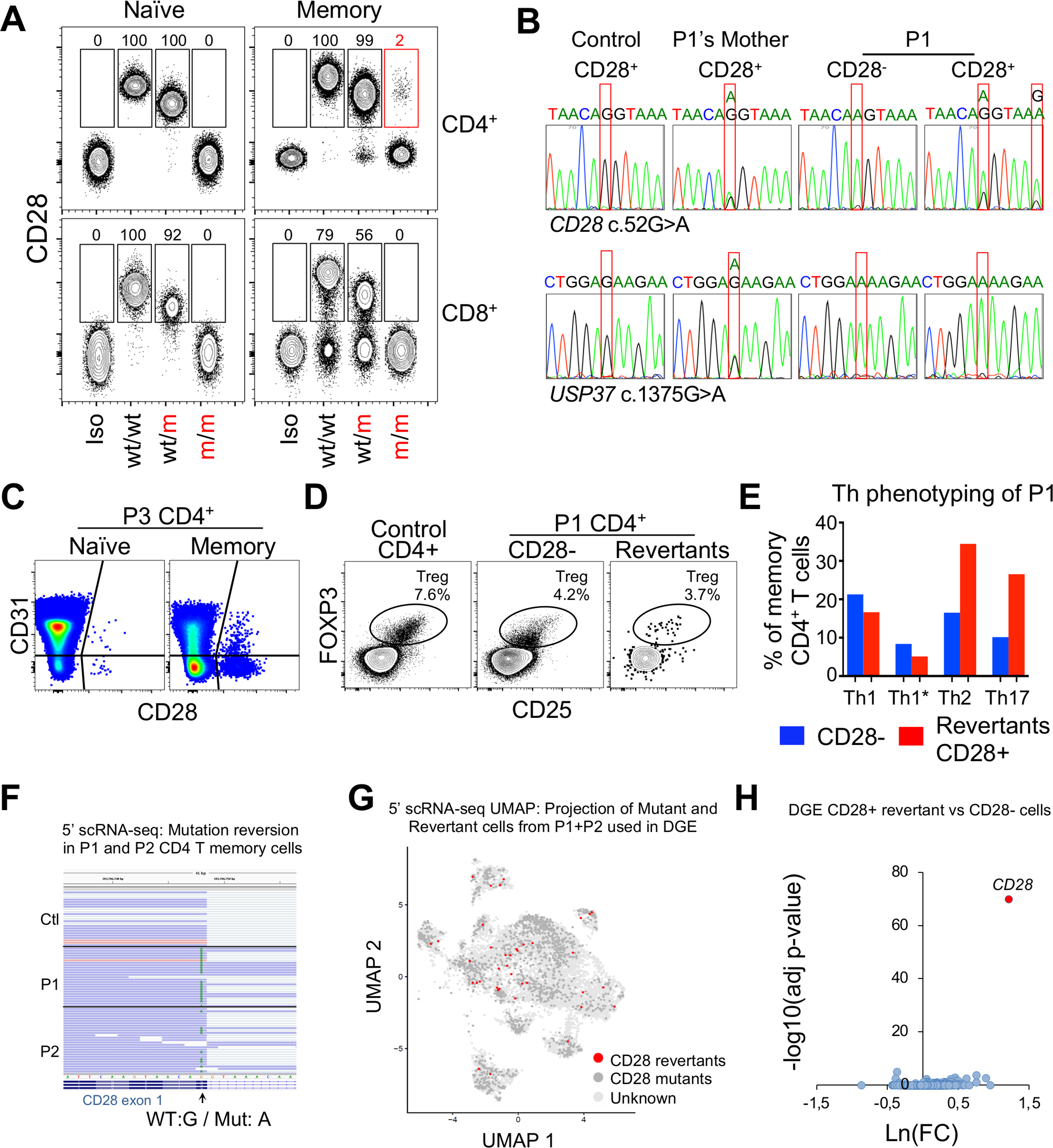
(A) CD28 expression on the indicated T cell subsets from P1 (m/m), a heterozygote (wt/m), and a healthy control (wt/wt). Isotype control (Iso) was used as a negative control. Representative data from the 3 patients and 4 heterozygotes. Numbers are frequencies of CD28+ cells in the indicated subset of each donor.
(B) Sanger sequencing chromatograms of the c.52G>A mutation in gDNA from a control, P1’s mother, and CD28− and CD28+ CD4+ memory T cells from P1 (upper line). A rare mutation of USP37 (c.1375G>A) was used to demonstrate that the CD28+CD4+ memory T cells sorted from P1 were not of maternal origin (bottom line).
(C) FACS plots of CD28 expression on recent thymic emigrants (CD31+CD4+ T cells).
(D) FACS plots showing Tregs in CD4+ T cells of one control or CD28− or CD28+ (revertant) CD4+ T cells from P1.
(E) Comparison of Th subset frequencies in CD28− and CD28+ (revertant) memory CD4+ T cells from P1.
(F–H) CITE-seq analysis of CD4 memory T cells from a control, P1, and P2.
(F) Sequence reads showing the presence or absence of the reversion in RNA from P1 and P2 cells.
(G) UMAP clustering of patients’ CD4 memory T cells with a projection of CD28 revertant identified using strict selection criteria. Cells with no detectable CD28 RNA or protein were considered mutants (dark gray).
(H) Volcano plot for a differential gene expression (DGE) analysis comparing CD28 revertant with CD28− cells, as shown in (G).
CD28 loss results in a moderate impairment of humoral responses, including those against HPVs
CD28-deficient mice produce only small amounts of immunoglobulin G (IgG) antibody (Ab) in response to peptides or viruses and have low serum IgG1 levels (Shahinian et al., 1993). We investigated the humoral responses of the patients. The patients had normal serum Ig classes and IgG subclasses (Table S2), with the exception of slightly high serum IgE levels in P1, who also has food allergy and asthma. Standard immunological assays showed that the patients had detectable Abs against numerous pathogens, including Abs directed against T cell-dependent (e.g., herpesviruses and influenza A) and T cell-independent microbial antigens (pneumococcus and Haemophilus influenzae B) in the absence of vaccination (Table S2). We performed VirScan (Figure 5A). We compared the three CD28-deficient patients with 20 aged-matched healthy controls and found that P1, P2, and P3 had been infected with at least 9, 6, and 7 common viruses, respectively. VirScan is a powerful tool for providing an overall picture of the virome encountered by a subject, but its resolution remains limited for infections due to some common viruses, in particular specific HPV subtypes (Isnard et al., 2019; Xu et al., 2015). In this analysis, none of the patients or controls satisfied the stringent positivity cutoff for HPV-2 and HPV-4 used, despite the reported prevalence of 5%–15% and 20%–50%, respectively, for these viruses in the general population (Antonsson et al., 2010; Waterboer et al., 2009). We performed a Luminex assay detecting antibodies against 38 different HPV L1 virus-like particles, including HPV-2, HPV-4, and all HPVs present in the nonavalent HPV vaccine Gardasil (HPV-6, HPV-11, HPV-16, HPV-18, HPV-31, HPV-33, HPV-45, HPV-52, and HPV-58) (Figures 5B and 5C). We found that P1 was seropositive for HPV-2, HPV-6, HPV-11, HPV-36, HPV-50, HPV-58, HPV-92, HPV-96, and HPV-101; P2 was seropositive for HPV-1 and HPV-4; and P3 was seropositive for HPV-2, HPV-96, and HPV-101. These Ab responses, including those for HPV-2 and HPV-4, remained stable over a period of 14–21 months (Figure 5B). The mother of P1, who lives in the same household, was seropositive for HPV-1, HPV-4, HPV-6, and HPV-80. These data suggest that the patients were exposed to different HPV subtypes and developed lasting Ab responses. However, the lack of significant anti-HPV-4 antibodies in P3 after several years of recurrent infection suggests a suboptimal anti-HPV Ab response in this patient. The presence of anti-HPV-2 Abs in P3 further suggests that the clinical penetrance of unusually severe HPV-2 disease is not complete in humans with CD28 deficiency, possibly due to the development of an effective T cell response, as demonstrated by wart regression in P2 (see case reports). Finally, we tested the T cell-dependent vaccinal response of patients to the nonavalent HPV vaccine (P1 and P3) and DPT (diphtheria, pertussis, and tetanus; P1, P2, and P3) vaccines (Figure 5C; Table S2). Other than for HPV-6 and HPV-11 in P1, who was already seropositive for these subtypes before vaccination, we detected no response in P1 and only a very weak response in P3. No response to tetanus toxin vaccination was observed in any of the three patients 5 months after recall boost (Table S2). No response, a weak response, and a normal response to diphtheria toxin vaccination were observed in P1, P2, and P3, respectively, 5 months after the recall boost (Table S2). Overall, these data suggest that CD28-deficient patients respond poorly to vaccination, but generate good T cell-dependent and -independent IgG responses to numerous pathogens, including HPVs, in natural conditions, although this Ab response is frequently quantitatively weaker than that in controls. Consistently, the patients do not suffer from any of the many clinical manifestations of patients with inborn errors of B cell and Ab immunity, including recurrent bacterial infections of the respiratory and digestive tracts (Notarangelo et al., 2020).
Figure 5. Anti-HPV B and T cell responses.
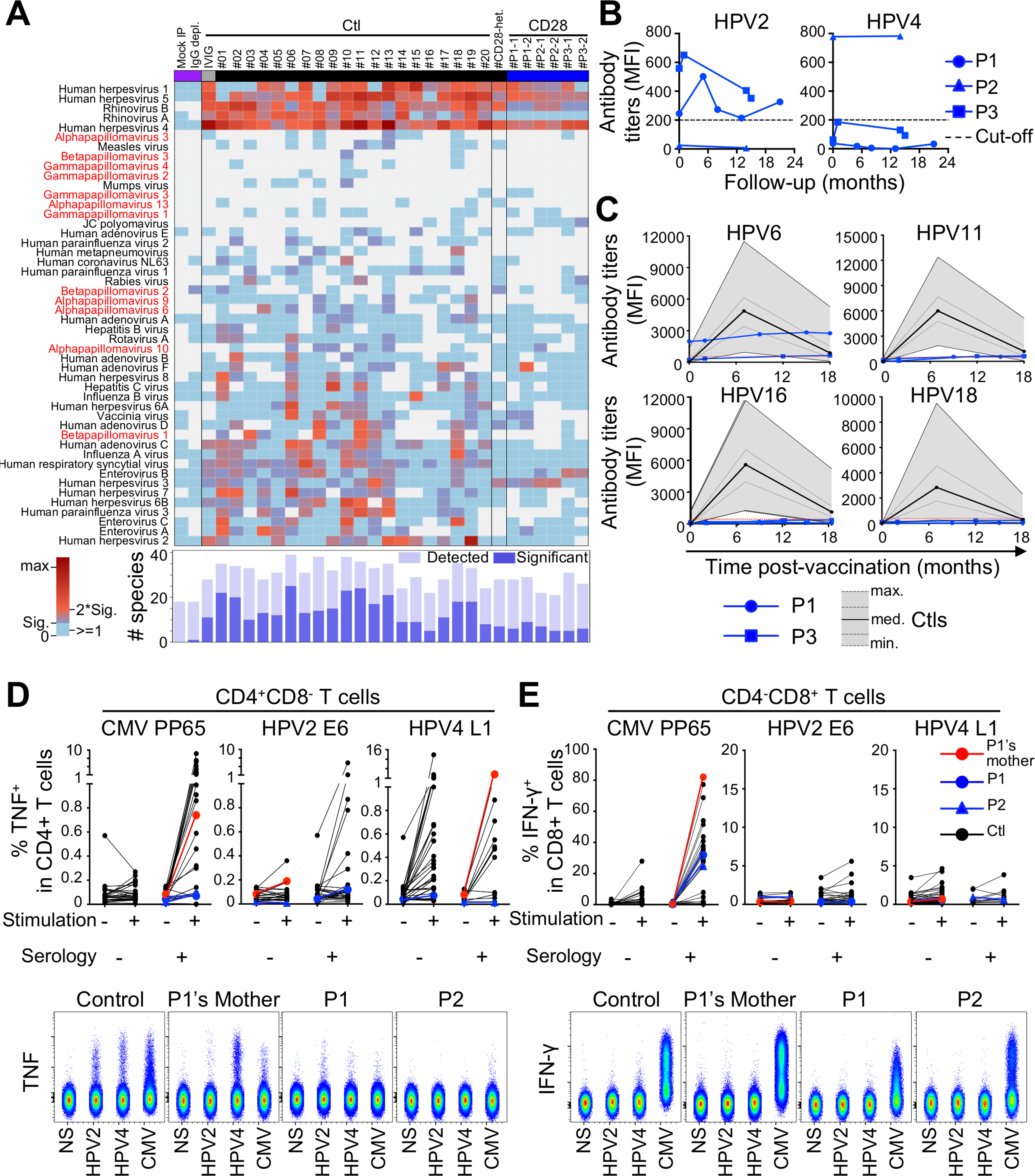
(A) Virscan. Adjusted virus scores for indicated samples from patients, a heterozygote and controls, mock immunoprecipitate (IP) samples and-IgG depleted serum, and IVIg. Shown are virus species for which we found at least one sample to be seropositive. The heatmap shows adjusted virus score values for each sample as a color gradient from blue if antibodies were detected but below our significance cutoff values, through purple to red if the adjusted virus score values were above our significance cutoff values. The bar plot (bottom) illustrates the size of the Ab repertoire for a given sample, indicating the precise number of different species for which peptides were enriched (light blue) and the number of different species for which the adjusted virus score values exceeded the cutoff values for significance (dark blue).
(B) Ab titers against HPV-2 and HPV-4 in patients at multiple time points; sensitivity threshold is indicated by a dashed line.
(C) Titers of antibodies against indicated HPVs in P1 and P3 pre and post Gardasil 9 vaccination (blue lines). The sensitivity threshold is indicated by a red dotted line. The black line represents the median value for a published control group (Bhatla et al., 2018). The gray area and the gray lines indicate the minimum, interquartile range and maximum responses of the control group.
(D and E) PBMCs were stimulated for 14 days with overlapping peptides from pp65 (HCMV), E6 (HPV-2), or L1 (HPV-4). Intracellular production of TNF and IFN-γ was measured in CD4+ (D) and CD8+ (E) T cells, respectively. Controls (n = 46), heterozygote, and patients were categorized on the basis of seropositivity for HCMV, HPV-2, and HPV-4 (top panels). Representative images are shown in the lower panels.
Anti-HPV CD4+ T cell responses are not detected in the patients
We generated overlapping peptides of E6 from HPV-2 and L1 from HPV-4 for functional assessments of the T cell responses to HPVs developed by patients and controls. We incubated PBMCs from 46 healthy controls, P1, P1’s mother, and P2 with overlapping HPV-2, HPV-4, or HCMV peptides for 14 days to induce the expansion of antigen-specific T cells (Bhatt et al., 2020). After 14 days, we stimulated the cells for a further 4 h with the overlapping peptides and then assessed intracellular IL-2, IL-4, IL-17A, IFN-γ, and TNF levels in CD4+ and CD8+ T cells (Figures 5D and 5E and data not shown). Data were stratified on the basis of seropositivity for the three viruses. We detected no IL-2, IL-4, and IL-17A in the controls or patients in response to HPV peptides (data not shown). In response to overlapping peptides from HCMV, we observed high levels of TNF and IFN-γ production in CD8+ T cells, and, to lesser extent, in CD4+ T cells from controls and patients. The anti-HCMV response was correlated with positive results in serological tests for HCMV. A clear CD4+ T cell response to HPV-2 and HPV-4 peptides was found in 7 and 27 controls, respectively (Figure 5D). No robust CD8+ T cell response was detected in controls or patients for either of the two sets of overlapping peptides for HPV proteins (Figure 5E). A robust anti-HPV-2 CD4+ T cell response in controls was associated with high anti-HPV-2 E6 serum Ab titers. The CD4+ T cell anti-HPV-4 response was not correlated with anti-HPV-4 L1 Ab titers, suggesting either that the observed anti-L1-HPV-4 T cell response is not entirely specific for HPV-4, possibly due to cross-reaction with different HPV types, or the serological assay is not sensitive enough to detect all individuals infected with HPV-4. Neither P1 nor P2 presented robust T cell responses to E6-HPV-2 or L1-HPV-4 peptides. P1’s mother, who lives in P1’s household and was seropositive for HPV-4 but seronegative for HPV-2, presented a strong response to L1-HPV-4 peptides. These data show that anti-HPV T cell responses are detectable only in CD4+ T cells in these experimental conditions. They also suggest that, despite prolonged exposure, P1 and P2 did not mount robust T cell responses to HPV-2 or HPV-4, by contrast to P1’s mother, whose cells expressed CD28. This absence of a detectable T cell response to HPV-2 and HPV-4 is consistent with the verrucosis in these patients and may have contributed to their development.
Infected keratinocytes do not express CD28 ligands
We assessed the expression of CD28, CD80, and CD86 in skin biopsy specimens from the patients. We detected CD28+ T cells in the dermis below the lesion in control warts. We found that the lymphocytic infiltrate in the dermis below the lesions in the three patients displayed no CD28 expression on immunohistochemistry (Data S1). This suggests that the few revertant memory CD4+ T cells are not specifically recruited to lesions. Consistent with the lack of CD80 and CD86 expression on keratinocytes in healthy skin and cultured keratinocytes (Black et al., 2007; Orlik et al., 2020), we detected no CD80 and CD86 on HPV-infected keratinocytes within the lesions of the patients (Data S1). We detected CD80 expression only in a few CD207− cells in the dermis. Within the epidermis, we detected only a few CD86+ cells, all co-expressing CD207, a Langerhans cells marker. Most CD86+ cells were within the dermis, below the lesions, and did not co-express CD207, suggesting that CD86 was expressed on CD207− dermal antigen-presenting cells. This analysis suggests that impaired interaction between the rare epidermal T cells and keratinocytes does not underlie HPV-2- and HPV-4-driven lesions in patients with CD28 deficiency. The lesions are more likely to result from impaired interactions between T cells and antigen-presenting cells in the epidermis, dermis, or draining lymph nodes.
The histology of TMS lesions is not typical of viral papillomas
CD28 deficiency explains the susceptibility to HPVs common to these three patients, but it cannot account for their different phenotypes, with TMS in P1 and severe warts in P2 and P3. Because HPV-2 has been detected in all TMS patients tested (Wang et al., 2007b; Alisjahbana et al., 2010), one plausible explanation is that persistent HPV-2 infection is a prerequisite for the development of TMS. According to this hypothesis, HPV-2 infection in P1 drove the formation of common warts, which subsequently progressed to TMS, whereas HPV-4 infection in P2 and P3 drove the formation of common warts with no potential to develop into TMS. We analyzed the skin lesions of the patients. The warts of P2 and P3 had histological features typical of lesions caused by HPV-4 (Figure 6A; Data S1) (Jablonska et al., 1985); three separate HPV-2+ cutaneous horns from P1 displayed an unusual viral papilloma structure different from that of a control HPV-2+ wart (Figure 6A; Data S1). They presented no parakeratosis or vacuolated cells with pyknotic nuclei (koilocytes) (Figure 6A; Data S1). They were not stained with pan-HPV L1 antibody on immunohistochemistry, unlike the lesions of P2, P3, and a control HPV-2+ common wart (Figure 6A; Data S1). We detected strong E4 and minichromosome maintenance (MCM) staining in the HPV-4+ lesions of P2 and P3, and in a control HPV-2+ wart (Data S1). There was no E4 or MCM staining in the lesion of P1, except for a restricted area at the periphery of the lesion (Data S1). Vacuolated cells, expressing L1, MCM, and E4, normally accompany the production of infectious viral particles, suggesting that HPV-2 infection may be non-productive in the lesions of P1. Non-productive infection is reminiscent of cutaneous CRPV infections in rabbit (Breitburd et al., 1997) and cervical intraepithelial neoplasia (CIN) in humans (Doorbar, 2006; Schiffman et al., 2016), although CIN is typically characterized by cytonuclear abnormalities.
Figure 6. Histological and virological studies of TMS lesions reveal an atypical papilloma.
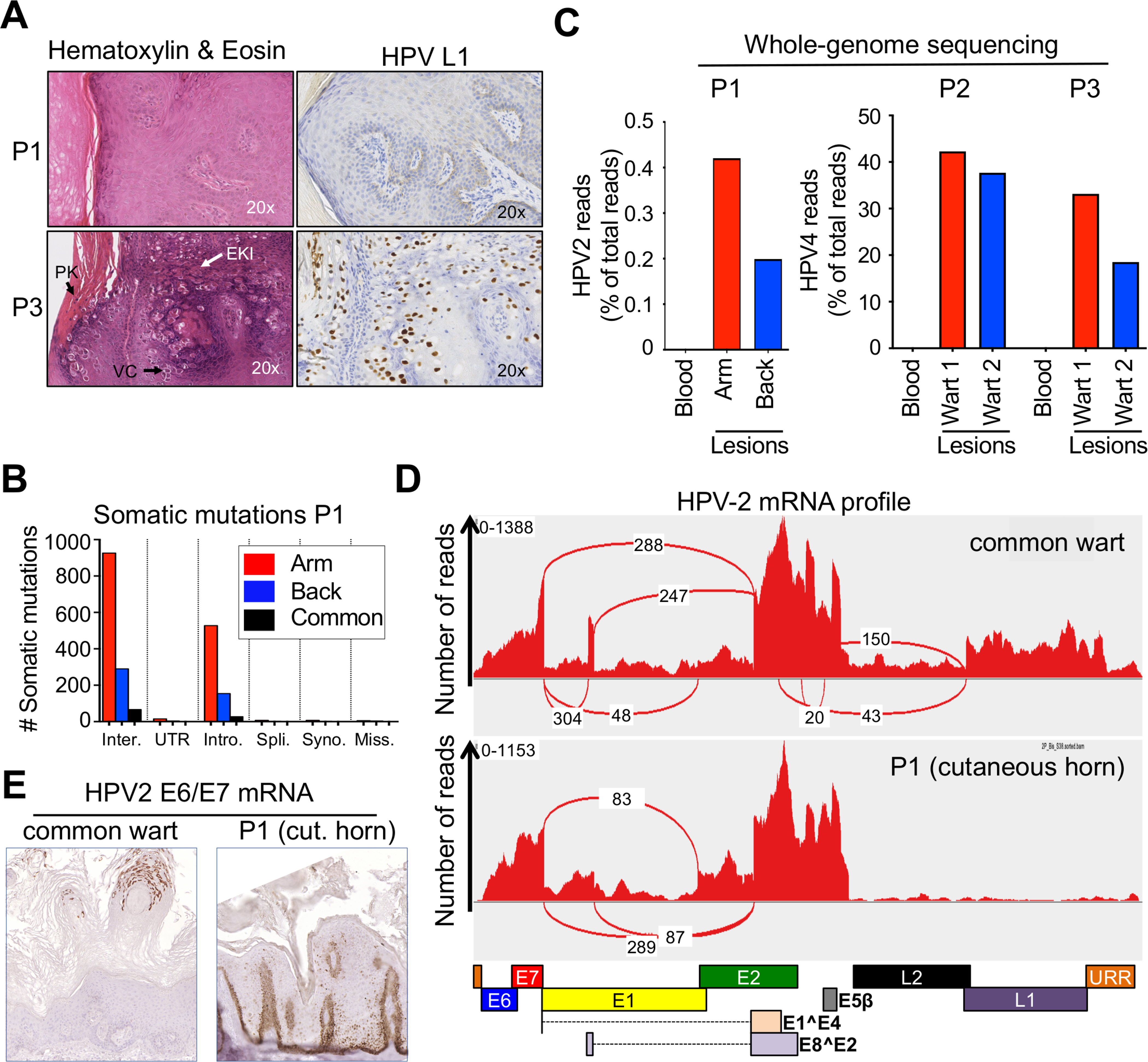
(A) H&E staining (left), immunohistochemical staining of the L1 protein of HPV (right) in a skin lesion from P1 (top) and a common wart from P3 (bottom). Vacuolated cells (VC), parakeratosis (PK), and large eosinophilic keratohyalin inclusions (EKI), suggesting productive viral replication is not found in P1’s lesions.
(B) Bar graph summarizing the number of somatic mutations in P1’s lesions, sorted by mutation type: intergenic (inter.), untranslated region (UTR), intronic (intro.), splicing region (spli.), synonymous (syno.), and missense (miss.). The numbers of somatic mutations common to the 2 skin lesions are shown for each type of mutation.
(C) Bar graph showing HPV-2 (P1) and HPV-4 (P2 and P3) read counts normalized to total read counts extracted from WGS data for two separate lesions (arm and back) and the blood of the patients.
(D) Representative RNA sequencing profiles for a control wart (top) and one TMS lesion from P1 (bottom). The graph shows the number of reads detected for each position. The red connecting lines indicate the most frequent splice events detected; the number of detected reads is indicated. Boxes below the graph indicate the position of the main HPV-2 open reading frames.
(E) In situ hybridization showing E6/E7 mRNA levels in an HPV-2+ common wart and a cutaneous horn from P1.
HPV-2 is episomal in TMS lesions
We performed whole-genome sequencing (WGS) on two separate skin lesions and blood from each patient (P1, P2, and P3). We detected no copy number variations (duplications or deletions) in the genomic DNA (gDNA) from P1’s lesions relative to the blood. We detected 545 and 1,581 somatic substitutions in the two biopsy samples relative to the blood sample (Figure 6B). None of these substitutions was an obvious candidate driver (data not shown). As reported for the somatic mutations detected in the common warts of P2 and P3 (Data S1), only a few mutations were common to the two lesions of P1, suggesting that these lesions did not arise from a common ancestor clone. We obtained 0.5–2.0 million HPV-2 (P1) or HPV-4 (P2 and P3) reads by WGS on the patients’ lesions, none of which was detected in the blood (Figure 6C). No other HPVs and no insertion of HPV-2 into the patient’s genome were detected. These data suggest that episomal and non-productive HPV-2 drive the benign, multifocal TMS lesions of P1, as for other HPVs in many cases of human cervical intra-epithelial neoplasia (CIN) and squamous cell carcinoma (SCC) (Arias-Pulido et al., 2006; Gray et al., 2010; Vinokurova et al., 2008), and in rabbit CRPV-induced horns (Breitburd et al., 1997). The detection of E4 and MCM staining in the periphery of the lesions of P1 suggests that some areas remain productive, perhaps contributing to the spread of TMS lesions in the patient.
Virological studies reveal viral oncogene overexpression in TMS lesions
We analyzed the viruses found in the lesions in greater detail. The HPV-2 strain of P1 was the same in both lesions tested, and 30 variants relative to the reference sequence were detected, 29 of which were also found in a set of 24 HPV-2 strains from conventional HPV-2+ common warts (Table S3). The remaining variant (c.1828G>A) was a synonymous mutation in E1, and was not reported in viruses from two other TMS patients (Wang et al., 2007b). We nevertheless tested the hypothesis that the HPV-2 viral life cycle was abnormal in TMS lesions, by performing RNA-seq on two separate lesions from P1 and comparing the results with RNA-seq results for three separate HPV-2+ common warts from two unrelated donors with severe warts (Figure 6D). We determined the relative abundance of virus transcripts and, consistent with the lack of detection of L1 on immunohistology (Figure 6A), we found that L1 and L2 transcript levels were lower than in the control warts (p < 10−51 and p < 10−10, respectively). Transcripts for the E6 and E7 oncogenes were present at higher levels (p < 10−4 and p = 10−2, respectively). Other transcripts assessed (E1, E2, and E5β) were present in similar amounts in the lesions of P1 and control warts. The splicing junctions identified within the HPV-2 mRNA occurred at the same position in the TMS and common wart lesions. Consistent with the very low levels of L1 and L2 transcripts, splicing involving late transcripts were absent or barely detectable (Table S3). We performed in situ hybridization, with the staining of HPV-2+ lesions from a control and P1 for the E6/E7 mRNA, to assess the potential spatiotemporal deregulation of viral oncogenes (Figure 6E; Data S1). E6/E7 expression in the control HPV-2+ common wart was maximal in the late epidermis. In the lesions of P1, E6/E7 was strongly overexpressed in the basal and parabasal layers of the epidermis. The peripheral area of the lesion from P1 displaying normal MCM and E4 expression also displayed normal E6/E7 expression in the late epidermis, supporting the notion that some areas of TMS lesions may remain productive. Our data demonstrate that the TMS observed in P1 is a slowly spreading, multifocal, benign epithelial tumor driven by a regular episomal HPV-2 strain lacking late transcripts but overexpressing the E6 and E7 oncogenes in the basal and parabasal epidermis in a context of CD28 deficiency impairing the T cell-mediated clearance of HPV-2-infected keratinocytes.
DISCUSSION
We report AR CD28 deficiency in three patients with isolated susceptibility to cutaneous α- and γ-HPVs. One patient (P1) had giant horns and TMS resulting from long-term HPV-2 infection, whereas the other two had disseminated common warts caused by HPV-4. CD28 has been studied since its initial description, in 1985, as a surface receptor specifically activating T cells upon stimulation with a mAb in the presence of phorbol esters (Hara et al., 1985). The human CD28 cDNA was isolated in 1987 (Aruffo and Seed, 1987). The first CD28-deficient mouse was produced in 1993, confirming the major costimulatory role of CD28 in TCR signaling (Shahinian et al., 1993). These mice had normal CD4+ and CD8+ T cell numbers, suggesting that CD28 was not required for the development and maintenance of these subsets in vivo. T cells from CD28-deficient mice became anergic and proliferated poorly in response to transient stimulation with viral peptide antigens or a virus displaying abortive replication, but repeated antigen stimulation or sustained viral infection bypassed the requirement for CD28 (Kündig et al., 1996). Nevertheless, these mice were found to be vulnerable to multiple pathogens, including Listeria monocytogenes, Salmonella typhimurium, and Trypanosoma cruzi (Martins et al., 2004; Mittrücker et al., 1999, 2001). Here, more than 30 years after the discovery of CD28, we report CD28-deficient humans. Surprisingly, these three patients are highly susceptible to cutaneous HPV-2 and HPV-4, but they are otherwise healthy (Casanova and Abel, 2018). They did not even display other HPV infections, despite being seropositive for many other HPVs, including HPV-1 and HPV-6, two skin-tropic HPVs driving plantar warts and condyloma (Doorbar et al., 2015). P1 and P2 are adult men aged 30 and 40 years that have been exposed to, and infected with, various microorganisms. It is unknown whether P3, the only adolescent and the only female CD28-deficient patient described here, will be prone to HPV-driven uterine lesions, including CIN, despite vaccination with Gardasil 9, which did not improve her skin lesions or those of P1.
Causality between CD28 deficiency and skin HPV-2 and HPV-4 lesions in the patients was established by genetic, biochemical, and immunological means. It was replicated in CD28-deficient mice, which were susceptible to skin infections with MmuPV1. The selective expansion of CD28-expressing “revertant” memory CD4+ T cells, following at least two reversion events in each of the three patients studied here, attests to the selective advantage conferred by CD28 on this T cell subset. Somatic reversions in patients with inherited T cell disorders can underlie milder clinical presentations (Pillay et al., 2021). The revertants identified in the CD28-deficient patients are unlikely to have made a major contribution to the control of a broad range of infectious agents. Their TCR repertoire is predicted by GLIPH to largely overlap with that of CD28-deficient memory CD4+ T cells. Furthermore, the patients have very few CD28+CD4+ revertant T cells (1–10 cells/mm3 of blood). Patients with acquired immunodeficiency syndrome (AIDS) develop multiple, severe, opportunistic infections, including severe skin HPV infections, when their CD4+ T cell levels fall below 200 CD4+ T cells/mm3 of blood (Deeks et al., 2015). These findings strongly suggest that human CD28 is largely redundant for host defense (Casanova and Abel, 2018). The many infections of patients with CARMIL2 deficiency (Wang et al., 2016; Schober et al., 2017), whose T cells do not respond to CD28 stimulation, suggests that CARMIL2 plays a broader role in immunity.
The TMS phenotype in P1 manifested after a decade of HPV-2-driven disseminated common warts. All TMS lesions in this and other patients tested were positive for HPV-2, suggesting that common warts induced by this virus have a unique potential to progress to TMS lesions (Wang et al., 2007b; Alisjahbana et al., 2010). We also found that E6 and E7, two potent oncogenes, were overexpressed in a lesion from P1. This phenotype is reminiscent of high-grade cervical lesions and cancers (Schiffman et al., 2016). This deregulation could not be explained by viral integration into the patient’s genome, or mutations of the HPV-2 episomal genome. For instance, the 63T>A variant in the upstream regulatory region (URR), which was previously described in another TMS patient and associated with promoter activity three times stronger than that of the reference promoter (Lei et al., 2007), is present in 81% of the “normal” common warts tested. Only one variant identified in the HPV-2 present in the TMS lesions of P1 was private but it was synonymous and not found in other HPV-2 strains from TMS patients. It is thus unlikely that P1 and other patients with TMS were infected with a specific HPV-2 strain. Surprisingly, E6/E7 overexpression in TMS lesions was restricted to the basal layer of the epidermis. To our knowledge, such a pattern of deregulation has never been reported before and may account for the giant cutaneous horns in the patients. The event driving this deregulation remains unclear but may involve modifier genes in P1 not present in patients with other inborn errors underlying severe HPV-2+ warts without giant horns. However, TMS lesions usually appear in early adulthood, after several years of persistent infection leading to “normal” common warts. The hypothesis of modifier genes thus seems unlikely, and we detected no candidate modifier genes in our patient. The TMS phenotype may, thus, result from the AR CD28 deficiency itself. The initial persistence of warts preceding TMS probably requires a specific T cell defect, of CD28 in P1, but probably other as yet undiscovered inborn errors in other patients (Alisjahbana et al., 2010; Wang et al., 2007b; Uddin et al., 2018).
Finally, our findings provide new insight into the general context of host-PV interaction. They confirm and extend the report of MHC-II-dependence for κ-papillomavirus CRPV-driven warts and tumors in rabbits (Han et al., 1992). Individual rabbits and humans prone to CRPV- and HPV-driven skin warts, respectively, carry genetic variants that weaken adaptive immunity. It would be of interest to determine whether the warts, horns, and tumors that develop in rabbits are dependent on CD28. Generating and testing CD28-deficient rabbits, genetically or with a neutralizing Ab, would answer one of the questions raised by Shope and Rous nearly 100 years ago. In humans, AR EVER1, EVER2, and CIB1 deficiencies underlie isolated EV, which is characterized by flat warts and non-melanoma skin cancer caused by β-HPVs in otherwise healthy individuals (de Jong et al., 2018b; Ramoz et al., 2002), through the disruption of keratinocyte-intrinsic immunity to β-HPVs. By contrast, inborn errors of T cells cause atypical forms of EV (de Jong et al., 2018a). The affected patients are not selectively prone to EV and display a number of other infectious diseases. Their T cell deficits do not impair the CD28 response pathway. There are also many other inherited T cell deficits that underlie severe common warts (Béziat, 2020). These two groups overlap, with some patients having both flat and common warts. It is intriguing that AR CD28 deficiency underlies a selective susceptibility to common warts caused by α- and γ-HPVs in otherwise healthy individuals, through an exclusive and specific impairment of T cell adaptive immunity. The selective susceptibility to α- and γ-HPVs of CD28-deficient patients mirrors the susceptibility to β-HPVs of CIB1-EVER-deficient patients. The cellular basis of these two disorders differs, because CIB1-EVER-dependent keratinocyte-intrinsic immunity to β-HPVs is disrupted in EV patients with disseminated flat warts and skin cancer, whereas CD28-dependent T cell adaptive immunity to α/γ-HPVs is disrupted in patients with disseminated common warts or giant horns.
Limitations of the study
One limitation of our study is that we only studied three relatives living in Iran. Another limitation is that we did not study them from infancy onward.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jean-Laurent Casanova (casanova@mail.rockefeller.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement with Inserm or the Rockefeller University.
Data and code availability
The whole-exome sequencing and whole genome sequencing datasets generated during this study are available at the Sequence Read Archive (SRA: PRJNA715377). The scRNA-seq and CITE-seq datasets generated during this study are available at the EGA European Genome-Phenome Archive (EGA: EGAS00001004837). High-throughput sequencing (HTS) of T cell receptor β (TRB) and T cell receptor ⍺ (TRA) dataset are available from Adaptive Biotechnologies (http://clients.adaptivebiotech.com/login; Email: beziat-review@adaptivebiotech.com; Password: beziat2021review). Primary CD4+ naive T cell RNA-Seq datasets generated during this study are available at the gene expression omnibus: GEO: GSE139299. Lesions RNA-Seq datasets generated during this study are available at the GEO: GSE139259. The assembled genomes are available from GenBank under the accession numbers GenBank: MN605988 and MN605989 for HPV-2 (from P1) and HPV-4 (from P2 and P3), respectively. This study did not generate any unique code. Any other piece of data will be available upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
P1 is a 30-year-old Iranian man with TMS. He was born to Iranian first cousins and he lives in Iran. His medical history until the age of eight years was unremarkable. From the age of eight years, he progressively developed common warts, which eventually entirely covered his hands and feet. At the age of 22 years, large cutaneous horns began to develop over the warts over a period of a few months, despite the application of several topical treatments and the removal of some of the larger warts (Figure 1B). P1 worked as a driver until this time. He weighed more than 60 kg until he was 24 years old. He began losing weight when he was 24, after starting regular opium inhalation. He currently weighs 45 kg. With the exception of a cousin (P2) and his daughter (P3), none of the other members of his family have reported susceptibility to warts. The patient has a family history of atopic dermatitis and diabetes mellitus in his mother, and allergic rhinitis in some sisters and brothers. Dermatological examination showed an extension of multiple horns over the entire body, including the scalp, particularly at the extremities, which were distorted by giant cutaneous horns without normal skin. Histological analysis of two separate skin biopsies revealed a papilloma without parakeratosis or vacuolated cells (Data S1). Multiplex PCR showed HPV-2 to be the only cutaneous HPV present in the warts and common to both biopsy specimens; traces of HPV-3 and HPV-4 were found in one sample each. The presence of HPV-2 in both the lesions of P1 studied was confirmed by sequencing the whole viral genome and by whole-genome DNA sequencing for two separate lesions. No mucosal damage or extension to the bones (Figure 1B), lymph nodes and deep organs from large warts was observed on X-ray. P1 also suffered from food allergies and asthma, with bronchial hyperactivity and superinfections (about one to two per year). A computed tomography scan revealed the presence of kidney stones, and the patient suffered two episodes of renal colic. Standard blood tests were normal, with the exception of high IgE levels (514 kU/L) and vitamin D deficiency. Viremia without clinical manifestations was documented for EBV (4.3 log cp/mL (< 2.0)) and CMV (3.5 log copies/mL (< 2.7)).
P2 is a 40-year-old cousin of P1. He developed his first warts at the age of 10 years. The warts slowly spread until they entirely covered his hands and feet. Unlike P1, P2 did not develop cutaneous horns. At the age of 20 years, the warts suddenly started to regress without therapeutic intervention. Within two years, they had almost completely disappeared, leaving dyspigmentation of the affected skin area. Only two residual warts remained, and these warts were surgically removed for analysis. Histological analyses of these two lesions showed them to have the typical features of common warts (Data S1). Multiplex PCR showed HPV-4 to be the only cutaneous HPV implicated in warts common to two biopsy samples; traces of HPV-2 were found in only one sample. The presence of HPV-4 in both the lesions of P2 was confirmed by whole-viral genome sequencing. P2 suffers from asthma and reported frequent ear infections (several times per year), but has no marked susceptibility to other infections, and has never been hospitalized. Viremia for EBV (3.4 log cp/mL (< 2.0)) and CMV (3.2 log copies/mL (< 2.7)) was documented, without clinical manifestations.
P3 is the 12-year-old daughter of P2. She began to develop common warts at the age of eight years and she currently has 15 warts on her hands. Histological analyses of two lesions showed them to have the typical features of common warts (Data S1). Multiplex PCR showed HPV-4 to be the only cutaneous HPV implicated in warts common to the two biopsy samples. The presence of HPV-4 in both the lesions of P3 was confirmed by whole-viral genome sequencing. P2 and P3 have the same HPV-4 viral strain, suggesting intrafamilial infection. So far, like P2, P3 has not developed the TMS phenotype. She displays no particular susceptibility to other infections, has no allergies or asthma, and has never been hospitalized. EBV (3.2 log cp/mL (< 2.0)) viremia, without clinical manifestations, was documented.
The three patients were otherwise healthy, and normally resistant to other common microorganisms. In particular, they had no other mucocutaneous and/or viral infections, including flat warts, molluscum contagiosum, and laryngeal or anogenital warts.
METHOD DETAILS
Linkage analysis, whole-exome sequencing, whole genome sequencing
We extracted genomic DNA from blood samples collected from the patients, their parents and siblings, with the iPrep PureLink gDNA Blood Kit and iPrep Instruments from Life Technologies. We used the Genome-Wide Human SNP Array 6.0 (Thermo Fisher scientific) for linkage analysis. Multipoint LOD scores were calculated with MERLIN software (Abecasis et al., 2002), assuming that the gene responsible for the defect displayed AR inheritance and complete penetrance. WES was performed for P1, P2 and P3. Exome capture was performed with the SureSelect Human All Exon 71 Mb kit (Agilent Technologies). Paired-end sequencing was performed on a HiSeq 2500 machine (Illumina) generating 100-base reads. We aligned the sequences with the GRCh37 reference build of the human genome, using the BWA (Li and Durbin, 2010). Downstream processing and variant calling were performed with the Genome Analysis Toolkit (McKenna et al., 2010), SAMtools (Li et al., 2009), and Picard tools. Substitution and InDel calls were made with the GATK UnifiedGenotyper. All variants were annotated with an annotation software system developed in-house (Adzhubei et al., 2010; Kircher et al., 2014; Ng and Henikoff, 2001). WGS for P1 (whole blood and warts) were performed as part of the Qatar Genome Programme (QGP). sequencing libraries were generated from whole blood-derived fragmented DNA using the TruSeq DNA Nano kit (Illumina, Inc., San Diego, USA) and sequence reads were generated using a HiSeq X Ten1 system (Illumina, Inc., San Diego, USA). We obtained on average 409×106 (standard deviation: 24.8×106) 2 × 100-bp paired-end reads. We performed WGS for P2 and P3 (whole blood and warts) at Macrogen. Sequencing libraries were generated from whole blood-derived fragmented DNA using the TruSeq DNA PCR-Free kit (Illumina) and sequenced with an Illumina NovaSeq6000 S4. We obtained on average 439×106 (standard deviation: 55.8×106) 2 × 100-bp paired-end reads.
Estimation of homozygosity rate
Homozygosity rate was estimated as previously described (Belkadi et al., 2016). Genomic measurements of individual homozygosity were compared with those for ethnically matched controls with and without consanguinity (self-declared) from our in-house WES database.
Detection and study of somatic mutations in lesions
We used BWA-MEM to align the WGS reads to the GRCh37 reference genome (Li, 2013). For the calling of somatic substitutions in the warts, we used Samtools 1.6 for pileups and then applied Varscan v2.4.3 to the pileups, first with the somatic option, then with the somatic filter with default parameter options (Koboldt et al., 2012; Li et al., 2009). We retained variants for which fewer than two reads were obtained for the blood sample and at least three reads were obtained for one of the warts. Finally, we removed all the substitutions that were present in at least three reads and had a VAF > 0.1 in at least one of 20 private cancer-free whole-genome sequencing samples. We used R v.3.5.2 and the muts.to.sigs.input and whichSignatures functions from the SomaticSignatures package (retrieved from the Bioconductor 3.8 release) to extract the COSMIC signatures (Gehring et al., 2015).
Genomic copy number variation
We identified copy number variations in WGS data by calculating coverage as the number of reads for each 10 kb bin in the genome for the warts and the blood sample. We used Varscan v.2.4.3 to extract log-ratios, which we segmented with the smoothDNA function of the DNACopy package (retrieved from the Bioconductor 3.8 release) (Olshen et al., 2004). We considered each segment with a log-ratio of amplitude greater than 0.2 (i.e., a gain or loss of more than 15%) to constitute a copy number event.
HPV detection and insertion
We quantified HPV viruses in WGS data for the 143 types of HPV from the Papillomavirus Episteme (PaVE) (Van Doorslaer et al., 2013), using HPVDetector v.1.0 (Chandrani et al., 2015) in QuickDetect mode according to the recommendations of the authors who designed this method. We used HPVDetector v.1.0 in Integration mode to search for integration sites, and we removed every integration site present in fewer than five reads.
HPV whole-genome sequencing
The HPV-2 genome was fully sequenced from two separate lesions from P1 (from the leg and back). The HPV-4 genome was fully sequenced from the two separate hand lesions from P2 and P3. The HPV-2 and HPV-4 genomes were amplified with the CloneAmp Hifi premix (Takara), using nine and twelve pairs of primers, respectively (See Key resources table), seven of which have been described before (Wang et al., 2007b). The amplicons were subjected to electrophoresis in agarose gels, and Sanger-sequenced as described in the Sanger sequencing section. Sequences were aligned with the HPV-2 and HPV-4 reference sequences (Papillomavirus Episteme database, PaVE) (Van Doorslaer et al., 2013). The HPV-2 sequence from P1 was compared to the HPV-2 sequence of 24 normal HPV-2+ common warts (non TMS) sampled in the United Kingdom (n = 22) and Iran (n = 2).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TotalSeq-C0072 anti-human CD4 Antibody | Biolegend | Cat# 300567; RRID:AB_2800725 |
| TotalSeq-C0386 anti-human CD28 Antibody | Biolegend | Cat# 302963; RRID:AB_2800751 |
| TotalSeq-C0063 anti-human CD45RA Antibody | Biolegend | Cat# 304163; RRID:AB_2800764 |
| TotalSeq-C0049 anti-human CD3 Antibody | Biolegend | Cat# 344849; RRID:AB_2814272 |
| rat monoclonal anti-CD3 | Abcam | Cat# ab11089; RRID:AB_2889189 |
| rabbit polyclonal anti-CD28 | Sigma | Cat# HPA070003; RRID:AB_2686226 |
| rabbit monoclonal anti-CD86 | Cell Signaling | Cat# 91882S; RRID:AB_2797422 |
| mouse monoclonal anti-CD80 | R&D Systems | Cat# MAB140; RRID:AB_2244549 |
| mouse monoclonal anti-CD207 | Novocastra / Leica | Cat# NCL-L-Langerin; RRID:AB_563850 |
| rabbit polyclonal anti-CD207 | Sigma | Cat# HPA011216; RRID:AB_1078453 |
| Cy3-conjugated goat anti-Rat IgG (H+L) | Jackson Immunoresearch | Cat# 112-165-003; RRID:AB_2338240 |
| Alexa Fluor® 488-conjugated goat anti-rat IgG (H+L) | Thermo Fisher Scientific | Cat# A11006; RRID:AB_2534074 |
| Alexa Fluor®488-conjugated goat anti-rabbit IgG (H+L) | Thermo Fisher Scientific | Cat# A11008; RRID:AB_143165 |
| Alexa Fluor® 488-conjugated goat anti-mouse IgG1 | Thermo Fisher Scientific | Cat# A21121; RRID:AB_2535764 |
| Alexa Fluor® 647-conjugated donkey anti-mouse IgG (H+L) | Jackson Immunoresearch | Cat# 715-605-150; RRID:AB_2340862 |
| Alexa Fluor® 594-conjugated donkey anti-rabbit IgG (H+L) | Jackson Immunoresearch | Cat# 711-585-152; RRID:AB_2340621 |
| Anti-FLAG M2 antibody-conjugated agarose beads | Sigma-Aldrich | Cat# A2220; RRID:AB_10063035 |
| Anti-FLAG M2 antibody | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| Brilliant Violet 421 anti-human CD197 (CCR7) Antibody | Biolegend | Cat# 353208; RRID:AB_11203894 |
| APC/Cyanine7 anti-human CD1c Antibody | Biolegend | Cat# 331520; RRID:AB_10644008 |
| BV786 Mouse Anti-Human CD3 Clone UCHT1 | BD Biosciences | Cat# 565491; RRID:AB_2739260 |
| FITC Mouse Anti-Human CD3 Clone UCHT1 | BD Biosciences | Cat# 555916; RRID:AB_396217 |
| CD3-VioBlue, human monoclonal (BW264/56) | Miltenyi Biotec | Cat# 130-094-363; RRID:AB_10831672 |
| CD3d Monoclonal Antibody (7D6), PE | Thermo Fisher Scientific | Cat# MHCD0304; RRID:AB_10376004 |
| CD3 Monoclonal Antibody (OKT3), Functional Grade, eBioscience | Thermo Fisher Scientific | Cat# 16-0037-85; RRID:AB_468855 |
| BV711 Mouse Anti-Human CD4 Clone RPA-T4 | BD Biosciences | Cat# 740769; RRID:AB_2740432 |
| CD4 Antibody, anti-human, APC-Vio 770 (M-T321) | Miltenyi Biotec | Cat# 130-100-355; RRID:AB_2657995 |
| BV650 Mouse Anti-Human CD8 Clone RPA-T8 | BD Biosciences | Cat# 563821; RRID:AB_2744462 |
| CD8 (BW135/80)-FITC, human (clone: BW135/80) | Miltenyi Biotec | Cat# 130-080-601; RRID:AB_244336 |
| PE Mouse Anti-Human CD4 Clone RPA-T4 | BD Biosciences | Cat# 555347; RRID:AB_395752 |
| CD11c APC Clone S-HCL-3 | BD Biosciences | Cat# 333144; RRID:AB_2868645 |
| FITC Mouse Anti-Human CD14 Clone M5E2 | BD Biosciences | Cat# 555397; RRID:AB_395798 |
| FITC Mouse Anti-Human CD16 Clone 3G8 | BD Biosciences | Cat# 555406; RRID:AB_395806 |
| FITC Mouse Anti-Human CD19 Clone 4G7 | BD Biosciences | Cat# 345776; RRID:AB_2868804 |
| APC-H7 Mouse Anti-Human CD19 Clone HIB19 | BD Biosciences | Cat# 560727; RRID:AB_1727437 |
| CD20 Antibody, anti-human, FITC | Miltenyi Biotec | Cat# 130-091-108; RRID:AB_244317 |
| FITC Mouse anti-Human CD56 Clone B159 | BD Biosciences | Cat# 562794; RRID:AB_2737799 |
| BV421 Mouse Anti-Human CD25 Clone M-A251 | BD Biosciences | Cat# 562442; RRID:AB_11154578 |
| Brilliant Violet 421 anti-human CD27 | Sony | Cat# 2114120 |
| FITC Mouse Anti-Human CD27 Clone L128 | BD Biosciences | Cat# 340424; RRID:AB_400031 |
| CD28 Antibody, anti-human, APC | Miltenyi Biotec | Cat# 130-092-923; RRID:AB_871654 |
| PE Mouse Anti-Human CD28 Clone CD28.2 | BD Biosciences | Cat# 555729; RRID:AB_396072 |
| BV421 Mouse Anti-Human CD28 Clone CD28.2 | BD Biosciences | Cat# 562613; RRID:AB_2737676 |
| CD28 Monoclonal Antibody (CD28.2), eBioscience | Thermo Fisher Scientific | Cat# 14-0289-82; RRID:AB_467194 |
| PE-CF594 Mouse Anti-Human CD31 Clone WM59 | BD Biosciences | Cat# 563652; RRID:AB_2738349 |
| PE-CF594 Mouse Anti-Human CD45RA Clone HI100 | BD Biosciences | Cat# 562298; RRID:AB_11154413 |
| PE Mouse Anti-Human CD54 Clone HA58 | BD Biosciences | Cat# 555511; RRID:AB_395901 |
| PE-CF594 Mouse Anti-Human CD56 Clone B159 | BD Biosciences | Cat# 562289; RRID:AB_11152080 |
| CD56 FITC Clone NCAM16.2 | BD Biosciences | Cat# 345811; RRID:AB_2868832 |
| BV711 Mouse Anti-Human Invariant NK T Cell | BD Biosciences | Cat# 747720; RRID:AB_2872199 |
| Alexa Fluor® 488 Mouse anti-Human FoxP3 Clone 259D/C7 | BD Biosciences | Cat# 560047; RRID:AB_1645349 |
| PE/Cyanine7 anti-human CD123 Antibody | Biolegend | Cat# 306010; RRID:AB_493576 |
| PE Mouse Anti-Human CD134 Clone L106 | BD Biosciences | Cat# 340420; RRID:AB_400027 |
| BV711 Mouse Anti-Human CD141 Clone 1A4 | BD Biosciences | Cat# 563155; RRID:AB_2738033 |
| PE Mouse Anti-Human CD152 Clone BNI3 | BD Biosciences | Cat# 555853; RRID:AB_396176 |
| PE anti-human CD154 Antibody | Biolegend | Cat# 310806; RRID:AB_314829 |
| BV711 Mouse Anti-Human CD161 Clone DX12 | BD Biosciences | Cat# 563865; RRID:AB_2738457 |
| FITC anti-human/mouse/rat CD278 (ICOS) Antibody | Biolegend | Cat# 313506; RRID:AB_416330 |
| PE Mouse Anti-Human CD25 Clone M-A251 | BD Biosciences | Cat# 555432; RRID:AB_395826 |
| Alexa Fluor® 700 Mouse Anti-Human IFN-γ Clone B27 | BD Biosciences | Cat# 557995; RRID:AB_396977 |
| Vioblue TCRγ/δ Antibody, anti-human | Miltenyi Biotec | Cat# 130-113-507; RRID:AB_2733977 |
| APC-Vio770 TCR Vα7.2 Antibody, anti-human, REAfinity | Miltenyi Biotec | Cat# 130-100-179; RRID:AB_2653673 |
| CD183 (CXCR3)-VioBright FITC, human (clone: REA232) | Miltenyi Biotec | Cat# 130-118-545; RRID:AB_2734058 |
| CD196 (CCR6)-APC, human (clone: REA190) | Miltenyi Biotec | Cat# 130-100-373; RRID:AB_2655933 |
| CD194 (CCR4)-PE, human (clone: REA279) | Miltenyi Biotec | Cat# 130-103-812; RRID:AB_2655905 |
| CD185 (CXCR5)-PE-Vio770, human (clone: REA103) | Miltenyi Biotec | Cat# 130-105-459; RRID:AB_2655788 |
| PE Mouse anti-NF-κB p65 (pS529) | BD Biosciences | Cat# 558423; RRID:AB_647222 |
| CD159a (NKG2A)-APC, human | Miltenyi Biotec | Cat# 130-098-812; RRID:AB_2655386 |
| CD159c (NKG2C)-PE, human (clone: REA205) | Miltenyi Biotec | Cat# 130-103-635; RRID:AB_2655394 |
| CD158a,h PC7 | Beckman Coulter | Cat# A66899 |
| CD158b1,b2,j PC5.5 | Beckman Coulter | Cat# A66900; RRID:AB_2857331 |
| Alexa Fluor 700 anti-human CD158e1 (KIR3DL1, NKB1) Antibody | Biolegend | Cat# 312712; RRID:AB_2130824 |
| Anti-IgM-APC, human | Miltenyi Biotec | Cat# 130-093-076; RRID:AB_1036084 |
| PE-CF594 Mouse Anti-Human IgG Clone G18-145 | BD Biosciences | Cat# 562538; RRID:AB_2737640 |
| FITC IgA Antibody, anti-human | Miltenyi Biotec | Cat# 130-114-001; RRID:AB_2726443 |
| Pacific Blue anti-human HLA-DR | Biolegend | Cat# 980404; RRID:AB_2632616 |
| CD279 (PD-1) Monoclonal Antibody (MIH4), PE, eBioscience | Thermo Fisher Scientific | Cat# 12-9969-42; RRID:AB_10736473 |
| CD366 (TIM3) Monoclonal Antibody (F38-2E2), PE, eBioscience | Thermo Fisher Scientific | Cat# 12-3109-42; RRID:AB_2572605 |
| CD223 (LAG-3) Monoclonal Antibody (3DS223H), PE, eBioscience | Thermo Fisher Scientific | Cat# 12-2239-42; RRID:AB_2572597 |
| TIGIT Monoclonal Antibody (MBSA43), PE, eBioscience | Thermo Fisher Scientific | Cat# 12-9500-42; RRID:AB_10714831 |
| PE anti-human CD272 (BTLA) Antibody | Biolegend | Cat# 344506; RRID:AB_2065761 |
| HPV Monoclonal Antibody (K1H8) | Thermo Fisher Scientific | Cat# MA5-12446; RRID:AB_10978662 |
| PE Mouse Anti-Human CD15 Clone HI98 | BD Biosciences | Cat# 555402; RRID:AB_395802 |
| PE-Cy7 Mouse Anti-Human CD16 Clone 3G8 | BD Biosciences | Cat# 560918; RRID:AB_10563252 |
| Polyclonal Goat Anti-Mouse Ig Clone Polyclonal | BD Biosciences | Cat# 553998; RRID:AB_395195 |
| PE Mouse IgG2b κ Isotype Control Clone 27–35 | BD Biosciences | Cat# 555743; RRID:AB_396086 |
| Anti-TNF-α-APC, human | Miltenyi Biotec | Cat# 130-091-649; RRID:AB_244201 |
| PE/Cy7 anti-human IL-2 | Sony | Cat# 3101630 |
| V5 Tag Monoclonal Antibody, HRP | Thermo Fisher Scientific | Cat# R961-25; RRID:AB_2556565 |
| GAPDH Antibody (0411) HRP | Santa Cruz Biotechnology | Cat# sc-47724 HRP; RRID:AB_627678 |
| Antibody against mouse papillomavirus E4 | in house | Rabbit serum |
| Antibody against mouse papillomavirus L1 | in house | MPV.B9 |
| Rabbit polyclonal HPVE2 serum | John Doorbar Lab. | N/A |
| Mouse MCM7 antibody | Thermo Fisher Scientific | Cat# MA5-14291; RRID:AB_11009501 |
| Goat anti-rabbit IgG (H+L)Alexa 594 | Thermo Fisher Scientific | Cat# A11012; RRID:AB_141359 |
| Immpress anti-mouse IgG HRP | Vector Labs | Cat# 30026; RRID:AB_2336532 |
| monoclonal mouse IgG1 anti-tag (supernatant from KT3 hybridoma cell line) | (MacArthur and Walter, 1984) | N/A |
| Biotin-SP-conjugated AffiniPure Goat Anti-Mouse IgG + IgM (H+L) | Jackson ImmunoResearch Labs | Cat# 115-065-068; RRID:AB_2338563 |
| Biotin-SP-conjugated AffiniPure Goat Anti-Human IgA + IgG + IgM (H+L) | Jackson ImmunoResearch Labs | Cat# 109-065-064; RRID:AB_2337627 |
| Bacterial and virus strains | ||
| expanded T7 Virscan phage library | S. Elledge (Brigham and Women’s Hospital and Harvard University Medical School, Boston, MA, USA) | VirScan Phage Library, version 3 |
| One Shot TOP10 Chemically Competent E. coli | Thermo Fisher Scientific | Cat# C404003 |
| NEB Stable Competent E. coli (High Efficiency) | New England Biolabs | Cat# C3040H |
| Mouse papillomavirus | HSD:NU mouse tail lesions | MmuPV1 |
| E.coli BL21 | Sehr et al., 2001 | N/A |
| E.coli Rosetta | Michael et al., 2008 | N/A |
| Biological samples | ||
| Peripheral blood mononuclear cells from indicated individuals | This manuscript | N/A |
| Plasma from indicated individuals | This manuscript | N/A |
| Skin scraps of warts | This manuscript | N/A |
| Skin biopsies of patient and control warts | This manuscript | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Polyethylenimine | Polysciences | Cat# 23966 |
| Opti-MEM | Thermo Fisher scientific | Cat# 31985070 |
| Dulbecco’s Modified Eagle Medium | Nacalai | Cat# 08459-64 |
| DMEM, high glucose, GlutaMAX(TM) | Thermo Fisher scientific | Cat# 61-965-026 |
| RPMI 1640 Medium, GlutaMAX Supplement | Thermo Fisher scientific | Cat# 61870010 |
| X-VIVO 20 Serum-free Hematopoietic Cell Medium | Lonza | Cat# BE04-448Q |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat# P8340 |
| FLAG peptide | Sigma-Aldrich | Cat# F3290 |
| Lys48-linked polyubiquitin chains | Boston Biochem | Cat# UC-230-100 |
| 1 M Tris-HCl | Invitrogen | Cat# 15567-027 |
| 0.1 M DTT (dithiothreitol) | Affymetrix | Cat# 70726 150 UL |
| HL-dsDNase | ArcticZymes | Cat# 70800-201 |
| 20 mM Nuclease-free MgCl2 | Thermo Scientific | Cat# AB-0359 |
| EvaGreen for qPCR | Biotium | Cat# 31000 |
| intravenous immunoglobulin (IVIg) | CSL Behring | Privigen |
| IgG-depleted human serum | Molecular Innovations, Inc. | Cat# HPLASERGFA5ML |
| Protein Transport Inhibitor (Containing Brefeldin A) | BD Biosciences | Cat# 555029 |
| Protein Transport Inhibitor (Containing Monensin) | BD Biosciences | Cat# 554724 |
| Tuberculin/PPD | STATEN SERUM INSTITUT | N/A |
| Sephadex G-50 Superfine, Cytiva | Merck | Cat# GE17-0041-01 |
| Phytohemagglutinin PHA-M, lyophilized powder | Sigma | Cat# L2646-10MG |
| Phorbol 12-myristate 13-acetate | Sigma aldrich | Cat# P8139-1MG |
| Ionomycin calcium salt from Streptomyces conglobatus | Sigma aldrich | Cat# I0634-5MG |
| Human IL-2 Recombinant Protein | Thermo Fisher Scientific | Cat# RP-8605 |
| Ficoll® Paque Plus, Cytiva | Merck | Cat# GE17-1440-03 |
| Fetal Bovine serum | Thermo Fisher Scientific | Cat# 16000036 |
| Protamine sulfate | Merck | Cat# P3369-10G |
| Human Serum from human male AB plasma, USA origin, sterile-filtered | Merck | Cat# H4522 |
| PepMix HCMVA (pp65) (> 70%) | JPT technologies | Cat# PM-PP65-1 |
| Proleukin | Novartis | N/A |
| HPV-2E6 overlapping peptides | JPT technologies | Custom order |
| HPV-4L1 overlapping peptides | JPT technologies | Custom order |
| Mouse papillomavirus E4 peptide | https://Chinapeptides.net | PKTTPPRRELFPPTPLTQPP |
| Mouse papillomavirus L1 virus like particles (produced in HEK293T) | In house | MmuPV1-L1-VLPs |
| Tyramide CF 488 | Biotium | Cat# 92171 |
| Dapi: 4’,6-Diamidino-2-phenylindol dihydrochloride | Thermo Fisher Scientific | Cat# 11926621 |
| PE-Streptavidin Conjugate | Moss,Inc. | Cat# SAPE-001 |
| HPV 2, 4, 6, 11, 16, 18 L1 antigens for multiplex serology | (Michael et al., 2008) | N/A |
| SeroMap Microspheres | Luminex (Austin, TX, USA) | Cat# L100-Sxxx-04 |
| Critical commercial assays | ||
| LIVE/DEAD Viability kit | Thermo Fisher Scientific | Cat# L3224 |
| Chromium Single Cell 3′ Reagent Kits (v3.1) | 10X Genomics | Cat# 120237 |
| Chromium Single Cell 5′ Library & Gel Bead kit (V1) | 10X Genomics | Cat# 1000014 |
| Chromium Single Cell 5′ Feature Barcode Library Kit | 10X Genomics | Cat# 1000080 |
| Memory CD4+ T Cell Isolation Kit, human | Miltenyi | Cat# 130-091-893 |
| Dead Cell Removal Kit | Miltenyi | Cat# 130-090-101 |
| Taqman Universal PCR Master Mix for qPCR | Thermo Fisher Scientific | Cat# 4304437 |
| Taqman Gene Expression Assays – CD28-FAM (Probe 1: Hs00174796_m1 and probe 2: Hs01007422_m1) and GUSB | Thermo Fisher Scientific | Cat# 4331182, Cat# 4326320E |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat# 27106 |
| RNeasy Fibrous Tissue Mini Kit | QIAGEN | Cat# 74704 |
| RNeasy Plus Mini Kit | QIAGEN | Cat# 74136 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| HiSpeed Plasmid Maxi Kit | QIAGEN | Cat# 12663 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat# 34096 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat# L34966 |
| Foxp3 / Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat# 00-5523-00 |
| T Cell Activation/Expansion Kit, human | Miltenyi Biotec | Cat# 130-091-441 |
| MasterMix Multiplex Kit | QIAGEN | Cat# 206145 |
| Specific Primer mix (βHPV, γHPV, or warts specific HPV) | International Agency for research on Cancer | N/A |
| Color-coded beads, coupled with specific probe for each HPV | International Agency for research on Cancer | N/A |
| Streptavidin, R-Phycoerythrin Conjugate | Molecular ProbesTM | Cat# S866 |
| Silver Stain Kit | Wako | Cat# 299-58901 |
| Agilent High Sensitivity DNA Kit | Agilent Technologies | Cat# 5067-4626 |
| RNA ScreenTape Analysis | Agilent Technologies | Cat# 5067-5576 |
| RNA ScreenTape Sample buffer | Agilent Technologies | Cat# 5067-5577 |
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 200521 |
| SureSelect Human All Exon V6 | Agilent Technologies | Cat# 5190-8863 |
| Ovation Universal RNA-Seq System 1–16 | TECAN (NuGen) | Cat# 0343 |
| CloneAmp HiFi PCR Premix | Takara Bio | Cat# 639298 |
| SMART-Seq® v4 Ultra® Low Input RNA Kit for Sequencing | Takara Bio | Cat# 634891 |
| In-Fusion® Snap Assembly Master Mix | Takara Bio | Cat# 638948 |
| Nextera XT DNA Library Preparation Kit | Ilumina | Cat# FC-131-1096 |
| Nextera XT Index Kit v2 set A | Ilumina | Cat# TG-131-2001 |
| Protein A Dynabeads | Thermo Fisher Scientific | Cat# 10002D |
| Protein G Dynabeads | Thermo Fisher Scientific | Cat# 10004D |
| QIAquick PCR purification kit (50) | QIAGEN | Cat# 28104 |
| Human IgG TOTAL ELISA | Thermo Fisher Scientific | Cat# 501128669 |
| QIAquick Gel Extraction Kit | QIAGEN | Cat# 28704 |
| Hiseq4000 (300 cycles), paired end kit | Illumina | Cat# FC-410-1003 |
| Nextseq 500 High-output Kit v2 (75 cycles) | Illumina | Cat# FC-4044-2005 |
| Perm Buffer III | BD Biosciences | Cat# 558050 |
| Fix Buffer I | BD Biosciences | Cat# 557870 |
| Bond Polymer Refine Detection | Leica | Cat# DS9800 |
| Invitrogen iPrep PureLink gDNA Blood Kit | Thermo Fisher Scientific | Cat# 10552894 |
| Genome-Wide Human SNP Array 6.0 | Thermo Fisher Scientific | Cat# 901153 |
| BigDye Terminator v3.1 Cycle Sequencing Kit | Thermo Fisher Scientific | Cat# 4337457 |
| Oligo(dT)12-18 Primer | Thermo Fisher Scientific | Cat# 18418012 |
| Reverse Transcriptase SuperScript II | Thermo Fisher Scientific | Cat# 18044-014 |
| Taq DNA polymerase | Thermo Fisher Scientific | Cat# 10342053 |
| TOPO TA Cloning Kit for Sequencing | Thermo Fisher Scientific | Cat# 450030 |
| pcDNA3.1/V5-His TOPO TA Expression Kit | Thermo Fisher Scientific | Cat# K480001 |
| X-tremeGENE 9 DNA Transfection Reagent | Merck | Cat# 6365779001 |
| Calcium Phosphate Transfection Kit | Thermo Fisher Scientific | Cat# K278001 |
| Acrodisc Syringe Filters with Supor Membrane, Sterile - 0.2 μm | Pall | Cat# 4612 |
| CellTrace CFSE Cell Proliferation Kit, for flow cytometry | Thermo Fisher Scientific | Cat# C34570 |
| M.O.M. (Mouse on Mouse) ImmPRESS HRP (Peroxidase) Polymer Kit | Vector | Cat# MP-2400 |
| ImmPRESS anti-rabbit IgG polymer system | Vector | Cat# MP-7801 |
| ImmPACT NovaRED Substrate | Vector | Cat# SK-4805 |
| 50% hematoxylin Gill’s No. 1 solution | Sigma-Aldrich | Cat# GHS132-1L |
| Nuclear Fast Red | American MasterTech, Inc. | Cat# SSKC398 |
| RNAscope® 2.5 VS Probe- V-MusPV-E4 | Advanced Cell Diagnostics, Inc. | Cat# 473289 |
| RNAscope 2.5 HD Assay – BROWN | Advanced Cell Diagnostics, Inc. | Cat# 322300 |
| Megaprime DNA Labeling System | Amersham | Cat# RPN1604 |
| The Brilliant III qPCR kit | Agilent | Cat# 600880 |
| The RevertAid First Strand cDNA synthesis kit | Thermo-Fisher | Cat# K1622 |
| RNAScope 2.5 HD Assay Brown | ACD/Bio-Techne | Cat# 322310 |
| HPV 2 E6/E7 C1 RNAScope probe | ACD/Bio-Techne | Cat# 80660 |
| Human PPIB RNAScope probe | ACD/Bio-Techne | Cat# 313901 |
| Deposited Data | ||
| 3′ Single-cell RNA-seq of the patient PBMC or healthy controls | This manuscript | EGA: EGAS00001004837 |
| CITE-seq (5′) single-cell RNA-seq of P1 and P2 PBMC and 1 healthy control | This manuscript | EGA: EGAS00001004837 |
| High-throughput sequencing (HTS) of T cell receptor β (TRB) and T cell receptor α (TRA) dataset (control and patients) | Adaptive Biotechnologies |
http://clients.adaptivebiotech.com/login |
| Email: beziat-review@adaptivebiotech.com | ||
| Password: beziat2021review | ||
| Whole-Exome and genome Sequencing of the patients (whole blood DNA and warts) | This manuscript | Sequence Read Archive (SRA: PRJNA715377) |
| Primary CD4+ naive T cell RNA-Seq data from the patient and heathy controls | This manuscript | gene expression omnibus (GEO: GSE139299) |
| RNA-Seq of the lesions of P1 and control common warts | This manuscript | gene expression omnibus (GEO: GSE139259) |
| Full HPV2 genome from P1 | This manuscript | GenBank: MN605988 |
| Full HPV4 genome from P2 and P3 | This manuscript | GenBank: MN605989 |
| Experimental models: cell lines | ||
| HEK293T cells | ATCC | Cat# CRL-11268, RRID:CVCL_1926 |
| Expanded CD4+ T cells from healthy controls or Patients’ family | This manuscript | N/A |
| P815 | ATCC | Cat# ATCC TIB-64 |
| Jurkat | In House | N/A |
| Experimental models: mouse strains | ||
| C57BL/6 mice | https://www.jax.org | https://www.jax.org/strain/000664 |
| CD28ko mice | https://www.jax.org | 002666 - B6.129S2-Cd28tm1Mak/J |
| Rag1ko mice | https://www.jax.org | https://www.jax.org/strain/002216 |
| Oligonucleotides | ||
| IS7_HsORF5_2 | IDT Integrated DNA technologies | ACACTCTTTCCCTACACGACTCCAGTC AGGTGTGATGCTC |
| IS8_HsORF3_2 | IDT Integrated DNA technologies | GTGACTGGAGTTCAGACGTGTGCTC TTCCGATCCGAGCTTATCGTCGTCATCC |
| IS4_HsORF5_2 | IDT Integrated DNA technologies | AATGATACGGCGACCACCGAGATCTA CACTCTTTCCCTACACGACTCCAGT |
| T7-Pep2.2_SP_subA | IDT Integrated DNA technologies | CTCGGGGATCCAGGAATTCCGCTGCGT |
| T7-Pep2.2_SP_subB | IDT Integrated DNA technologies | CTCGGGGATCCAGGAATTCGGAGCGGT |
| CD28-gDNA-FOR (sequencing gDNA) | Thermo Fisher Scientific | GCGTCTTTCAGTTCCCCTCA |
| CD28-gDNA-REV (sequencing gDNA) | Thermo Fisher Scientific | ACACATTGCCCTATTACAGCAAAA |
| CD28-cDNA-5UTR-FOR (sequencing cDNA + Exon trapping) | Thermo Fisher Scientific | TGGAACCCTAGCCCATCGTCA |
| CD28-cDNA-3UTR-REV (sequencing cDNA) | Thermo Fisher Scientific | AGCCGGCTGGCTTCTGGATA |
| ACTB-cDNA-FOR | Thermo Fisher Scientific | CCTTCCTGGGCATGGAGTCCT |
| ACTB-cDNA-REV | Thermo Fisher Scientific | AATCTCATCTTGTTTTCTGCG |
| pCMV6-FOR (sequencing insertion in pCMV6) | Thermo Fisher Scientific | ATTCGTCGACTGGATCCGGTA |
| pCMV6-REV (sequencing insertion in pCMV6 + Exon trapping) | Thermo Fisher Scientific | CATTTGCTGCCAGATCCTCTT |
| pLVX-mCherry-CD28-FOR (Cloning in pLVX) | Thermo Fisher Scientific | TATTTCCGGTGAATTATGC TCAGGCTGCTCTTGGC |
| pLVX-mCherry-CD28-REV (Cloning in pLVX) | Thermo Fisher Scientific | GAGAGGGGCGGGATCTCA GGAGCGATAGGCTGCG |
| ExonTrap-CD28-UTR-FOR (Cloning in pCMV6) | Thermo Fisher Scientific | CGAGGAGATCTGCCGCCGCG GCGTCTTTCAGTTCCCCTCAC |
| ExonTrap-CD28-Int1-REV (Cloning in pCMV6) | Thermo Fisher Scientific | TTATCTCTCTCTGCCACC TGTACATCCTTGG |
| ExonTrap-CD28-Int1-FOR (Cloning in pCMV6) | Thermo Fisher Scientific | CAGGTGGCAGAGAGAGAT AACTCCCCTCTGGAGTC |
| ExonTrap-CD28-ex2 -REV (Cloning in pCMV6) | Thermo Fisher Scientific | CTCGAGCGGCCGCGTACGCGTTTC ACATGGATAATGGTTCCATTG |
| CD28-cDNA-ATG-directional-FOR (Cloning in pcDNA3.1) | Thermo Fisher Scientific | CACCATGCTCAGGCTGCT CTTGGCTCTCA |
| CD28-cDNA-3′w/o.stop-REV (Cloning in pcDNA3.1) | Thermo Fisher Scientific | GGAGCGATAGGCTGCGAAGT |
| MUTsplice-CD28-ExonTrap-FOR (Mutagenesis c.52G>A Exon Trapping) | Thermo Fisher Scientific | CAATTCAAGTAACAAGTAAACAATG |
| MUTsplice-CD28-ExonTrap-REV (Mutagenesis c.52G>A Exon Trapping) | Thermo Fisher Scientific | CATTGTTTACTTGTTACTTGAATTG |
| MUTsplice-CD28-ExonTrap-FOR (Mutagenesis c.52G>A +5A>G Exon Trapping) | Thermo Fisher Scientific | GTAACAAGTAAGCAATGTTAATG |
| MUTsplice-CD28-ExonTrap-REV (Mutagenesis c.52G>A +5A>G Exon Trapping) | Thermo Fisher Scientific | CATTAACATTGCTTACTTGTTAC |
| MUT CD28 Gly18Arg For (Mutagenesis Gly18Arg) | Thermo Fisher Scientific | ATTCAAGTAACAAGAAACAAGAT |
| MUT CD28 Gly18Arg Rev (Mutagenesis Gly18Arg) | Thermo Fisher Scientific | ATCTTGTTTCTTGTTACTTGAAT |
| HPV-2 F1 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-AACCGCTCAGGATGACGAAG-3′ |
| HPV-2 R1 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TCTAAGCGGGACCACGACCT-3′ |
| HPV-2 F2 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-CCCTAATGATAACAACCAACAC-3′ |
| HPV-2 R2 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TACGCTGTTCAACGCTGGT-3′ |
| HPV-2 F2 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-GCAGCCTCAGCAGAATCAA-3′ |
| HPV-2 R3 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TCAACTACAGTGGTGGGTCGC-3′ |
| HPV-2 F4 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-GTGGAACAGAACACTTTAGCAG-3′ |
| HPV-2 R4 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-GCGGGTAACTCATCAAGCAAT-3′ |
| HPV-2 F5 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-GTTTTAGTAGGTTGGGACGC-3′ |
| HPV-2 R5 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TACGCAGCGAGAAGAACATAG-3′ |
| HPV-2 F6 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TAACACAACTATTGAGGACGGG-3′ |
| HPV-2 R6 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TTCATACGGGAGGGGATAC-3′ |
| HPV-2 F7 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-GTATCCCCTCCCGTATGAAT-3′ |
| HPV-2 R7 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TTCGTCATCCTGAGCGGTTT-3′ |
| HPV-2 F8 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-GCTGGGGCAATAGGGTCTTT-3′ |
| HPV-2 R8 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-AGTTGGCTGCAAAAACGACG-3′ |
| HPV-2 F9 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TAAGTGTGGCAGAACCGTCC-3′ |
| HPV-2 R9 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TTACTCAGCAACGTCGCCTT-3′ |
| HPV-4 F1 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-AAAAGGGACAGTGCATTTCTACT-3′ |
| HPV-4 R1 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-GTGCAGCAAAACGTCAGCTT-3′ |
| HPV-4 F2 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-GAGGAGTCGGTGGTTCCATT-3′ |
| HPV-4 R2 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TGTATGAGACCCCATACCATTC-3′ |
| HPV-4 F2 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-GGCTGGACCTTCTAGCCAAG-3′ |
| HPV-4 R3 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TGGCTTCCAATCTCCTTCTTCA-3′ |
| HPV-4 F4 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-GTACGCAGAGGAGGATGCAA-3′ |
| HPV-4 R4 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-AAACGTGCGACTAGGGACTC-3′ |
| HPV-4 F5 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TACTGACGGTACTTGGAAATCTT-3′ |
| HPV-4 R5 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-CCTCAGTGTCGAAGGAGGAG-3′ |
| HPV-4 F6 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-CAGACCATACGGGAGAAAGAGC-3′ |
| HPV-4 R6 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TTCCTTCTACTCAAGCTTTGCAT-3′ |
| HPV-4 F7 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-GCACTGACCAAAGAGACGCT-3′ |
| HPV-4 R7 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-CTCCCCTAGTTGCCAATGCT-3′ |
| HPV-4 F8 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-GCTAGCAGCCACCCTACAAT-3′ |
| HPV-4 R8 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TGCTTTGTTCTCCTGAATGTTGAC-3′ |
| HPV-4 F9 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-AAGTGGTGTGCAAATTGGGC-3′ |
| HPV-4 R9 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-CAAATCTGTTTGGGTCTGGCA-3′ |
| HPV-4 F10 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TTCCACGCTGGTACAGAAAGG-3′ |
| HPV-4 R10 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TAGAGCATCTCCCATGGTGC-3′ |
| HPV-4 F11 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TGCTCCTTTGGATGTCATTGC-3′ |
| HPV-4 R11 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-AAACCGCCTGCCTAAGGAAA-3′ |
| HPV-4 F12 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-CCTACACAGACCCCTGCAAC-3′ |
| HPV-4 R12 (Primer for whole-HPV genome sequencing) | Thermo Fisher Scientific | 5′-TGCAGAAGTCGTCCAAGGTT-3′ |
| USP37-MUT-FOR (Mutagenesis Glu459Lys) | Thermo Fisher Scientific | GAACCTGTTTCTGGAAAAGAAAATTCAC |
| USP37-MUT-REV (Mutagenesis Glu459Lys) | Thermo Fisher Scientific | GTGAATTTTCTTTTCCAGAAACAGGTTC |
| KANSL1L-MUT-FOR (Mutagenesis Ser577Thr) | Thermo Fisher Scientific | GTACAGATTGACCCCTA CTTTTTATTGGAC |
| KANSL1L-MUT-REV (Mutagenesis Ser577Thr) | Thermo Fisher Scientific | GTCCAATAAAAAGTAGG GGTCAATCTGTAC |
| Primers for mouse papillomavirus | IDT | 5′-TAGCTTTGTCTGCCCGCACT-3′ |
| Primers for mouse papillomavirus | IDT | 5′-GTCAGTGGTGTCGGTGGGAA-3′ |
| Probe for mouse papillomavirus | IDT | 5′FAM-CGGCCCGAAGACAACAC CGCCACG-3′TAMRA |
| Recombinant DNA | ||
| pCMV6-Empty vector | Origene | Cat# PS100001 |
| pcDNA3.1/V5-His-Empty vector | Thermo Fisher Scientific | Cat# V81020 |
| pCMV6-CD28_WT-Myc-DDK | Origene | Cat# RC211318 |
| pCMV6-CD28_Gly18Arg-V5-His | This manuscript | N/A |
| pCMV6-CD28_Val16fs-V5-His | This manuscript | N/A |
| pCMV6-CD28_I1-Ret-V5-His | This manuscript | N/A |
| pcDNA3.1-CD28_WT-V5-His | This manuscript | N/A |
| pcDNA3.1-CD28_Gly18Arg-V5-His | This manuscript | N/A |
| pcDNA3.1-CD28_Val16fs-V5-His | This manuscript | N/A |
| pcDNA3.1-CD28_I1-Ret-V5-His | This manuscript | N/A |
| pCMV6-KANSL1L_WT-Myc-DDK | Origene | Cat# RC215502 |
| pCMV6-KANSL1L_Ser577Thr-Myc-DDK | This manuscript | N/A |
| pCMV6-USP37_WT-Myc-DDK | Origene | Cat# RC223407 |
| pCMV6-USP37_Glu459Lys -Myc-DDK | This manuscript | N/A |
| pLVX-EF1α-IRES-mCherry | Clontech | Cat# 631987 |
| pLVX-EF1α- CD28_WT-IRES-mCherry | This manuscript | N/A |
| pLVX-EF1α-CD28_Gly18Arg-IRES-mCherry | This manuscript | N/A |
| psPAX2 | Addgene | Cat# 12260 |
| pCMV-VSV-G | Addgene | Cat# 8454 |
| pHXB2-Env | NIH-AIDS Reagent Program | Cat# 1069 |
| pCMV6-CD28_WT-ExonTrapping | This manuscript | N/A |
| pCMV6-CD28_c.52G>A-ExonTrapping | This manuscript | N/A |
| pCMV6-CD28_c.52+5A>G-ExonTrapping | This manuscript | N/A |
| Software and algorithms | ||
| Cell Ranger | 10X Genomics | v5.0.1 |
| DoubletFinder | McGinnis et al., 2019 | v2.0.3 |
| Seurat R package | Stuart et al., 2019 | V4.0.0 |
| Uniform Manifold Approximation and Projection (UMAP) | Becht et al., 2018 | v.0.3.5 |
| MAST | Finak et al., 2015 | v1.16.0 |
| BWA | Li and Durbin, 2010 | https://github.com/lh3/bwa |
| RSeQC | Wang et al., 2012 | http://rseqc.sourceforge.net |
| DESeq2 | Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| HPVDetector | Chandrani et al., 2015 | N/A |
| HTSeq | Anders et al., 2015 | https://github.com/htseq/htseq |
| IGV | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
| R | The R Project for Statistical Computing | https://www.r-project.org |
| STAR aligner | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| VarScan | Koboldt et al., 2012 | http://dkoboldt.github.io/varscan/ |
| Fiji Software | Schindelin et al., 2012 | https://fiji.sc/ |
| ImmunoSEQ ANALYZER | Adaptive Biotechnologies | http://clients.adaptivebiotech.com/login |
| ComplexHeatmap | Gu et al., 2016 | http://www.bioconductor.org/packages/devel/bioc/html/ComplexHeatmap.html |
| phip-stat | Mohan et al., 2019 | https://github.com/lasersonlab/phip-stat |
| Bowtie for alignment of PhIP-Seq raw reads | Mohan et al., 2019 | http://bowtie-bio.sourceforge.net/index.shtml |
| MERLIN 1.1.2 software | Abecasis et al., 2002 | http://csg.sph.umich.edu/abecasis/merlin/download/ |
| GATK HaplotypeCaller | McKenna et al., 2010 | https://www.broadinstitute.org/gatk |
| SAMtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Picard | http://broadinstitute.github.io/picard/ | http://broadinstitute.github.io/picard/ |
| Alamut Visual 2.15 | https://www.interactive-biosoftware.com/alamut-visual/ | N/A |
| BioRender | https://biorender.com/ | N/A |
HPV typing by multiplex PCR
HPV genotyping was performed with type-specific PCR bead-based multiplex genotyping assays, as previously described (Schmitt et al., 2006; Smelov et al., 2018). Briefly, after DNA extraction from wart lesions, multiplex PCRs were run with specific primers for the detection of: 46 βHPVs (HPV-5, 8, 9, 12, 14, 15, 17, 19, 20, 21, 22, 23, 24, 25, 36, 37, 38, 47, 49, 75, 76, 80, 92, 93, 96, 98, 99, 100, 104, 105, 107, 110, 111, 113, 115, 118, 120, 122, 124, 143, 145, 150, 151, 152, 159, 174); 7 wart-specific HPVs (HPV-1, 2, 3, 4, 10, 27, 57); and 52 γHPVs (HPV-4, 48, 50, 60, 65, 88, 95, 101, 103, 108, 109, 112, 116, 119, 121, 123, 126, 127, 128, 129, 130, 131, 132, 133, 134, 148, 149, 156, SD, 161, 162, 163, 164, 165, 166, 167, 168, 169, 170, 171, 172, 173, 175, 178, 179, 180, 184, 197, 199, 200, 201, 202). As a positive control, to assess DNA quality, we also included primers for the amplification of β-globin in each assay. Multiplex PCR was performed with 5 μl of DNA sample mixed with MasterMix from the QIAGEN Multiplex PCR Kit (QIAGEN) according to the manufacturer’s instructions. We used 10 μL of the PCR products for bead-based genotyping on a Luminex 200 machine (Schmitt et al., 2006).
Sanger sequencing
Genomic DNA was obtained from whole blood or from CD28+ or CD28− CD4+ T cells obtained from patients by FACS (purity > 98%). The CD28 mutation was amplified from genomic DNA by PCR; the PCR products were purified by centrifugation through Sephadex G-50 Superfine resin (Merck) and sequenced with the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems). Sequencing products were purified by centrifugation through Sephadex G-50 Superfine resin, and sequences were analyzed with an ABI Prism 3500 apparatus (Applied Biosystems). The sequences obtained were aligned with the genomic sequence of CD28 (Ensembl), with Serial Cloner 2.6 software.
Cell culture
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Hypaque centrifugation (Merck) from cytopheresis or whole-blood samples obtained from healthy volunteers and patients, respectively. PHA blasts were generated by culturing PBMCs in RPMI-1640 medium supplemented with 10% FCS, 1 μg/mL PHA (Sigma) and 10 ng/mL rIL-2 (Thermo Fisher Scientific). We added rIL-2 to the culture medium every 48 to 72 hours. The Jurkat and P815 cell lines were cultured in RPMI-1640 medium supplemented with 10% FCS. HEK293T cells were cultured in DMEM supplemented with 10% FCS.
mRNA purification, RT-PCR and RT-qPCR
Total RNA was extracted from the indicated cells with the RNeasy Extraction Kit (QIAGEN). RNA was reverse-transcribed with oligo-dT primers (Thermo Fisher Scientific) and SuperScript II reverse transcriptase (Thermo Fisher Scientific). The cDNA for the CD28 transcript was amplified with a recombinant Taq polymerase (Thermo Fisher Scientific) and the following primers binding to the UTRs: forward primer: 5′-TGGAACCCTAGCCCATCGTCA-3′, reverse primer: 5′-AGCCGGCTGGCTTCTGGATA-3′. The ACTB transcript cDNA, used as a positive control, was amplified with the following primers: forward primer 5′-CCTTCCTGGGCATGGAGTCCT-3′, reverse primer: 5′-AATCTCATCTTGTTTTCTGCG-3′. RT-qPCR was performed with the Applied Biosystems Assays-on-Demand probes/primers specific for CD28-FAM (Hs00174796_m1 and Hs01007422_m1), and 13-glucuronidase-VIC (GUS; 4326320E), which was used for normalization. Data are displayed as 2−ΔCt after normalization relative to GUS (endogenous control) expression (ΔCt).
Identification of CD28 mRNA splice variants by TA cloning
Bulk CD28 splice variants were amplified from PHA blast cDNA with recombinant Taq polymerase (Thermo Fisher Scientific) and primers binding to the UTRs, as described in the “mRNA purification, RT-PCR and RT-qPCR” section. PCR products were cloned with the TOPO TA cloning kit (Thermo Fisher Scientific), and used for the one-shot transformation of TOP10 chemically competent E. coli cells (Thermo Fisher Scientific), which were then spread on LB-agar plates containing X-gal and ampicillin. CD28 splice variants were amplified from individual colonies with the M13 forward and reverse primers from the TA cloning kit. We subjected the PCR products to Sanger sequencing, as described in the Sanger sequencing section, and then aligned the sequences with that of the CD28 cDNA (NM_001243077), using SnapGene software to identify alternative splicing variants.
Exon trapping
We cloned CD28 gDNA from P1 and a control, as described in Figure S3B. Briefly, CD28 gDNA containing exon 1 and the first 998 nucleotides of intron 1 gDNA was amplified with CloneAmp Hifi premix (Takara), the forward primer 5′- CGAGGAGATCTGCCGCCGCGGCGTCTTTCAGTTCCCCTCAC-3′, and the reverse primer 5′- TTATCTCTCTCTGCCACCTGTACATCCTTGG-3′. Similarly, CD28 gDNA containing the last 954 nucleotides of intron 1 and exon 2 was amplified with the forward primer 5′- CAGGTGGCAGAGAGAGATAACTCCCCTCTGGAGTC-3′ and the reverse primer 5′- CTCGAGCGGCCGCGTACGCGTTTCACATGGATAATGGTTCCATTG −3′. The Infusion Cloning kit (Clontech) was used to insert both PCR products into the pCMV6 entry vector (Origene) between the ASIS1 and Mlu1 cloning sites by homologous recombination. The c.52+5A>G mutation was inserted by mutagenesis. The exon-trapping plasmid obtained was used to transfect HEK293T cells, from which mRNA was purified after 24 hours, and reverse-transcribed as described in the “mRNA purification, RT-PCR and RT-qPCR” section. Variants for splicing between exons 1 and 2 were amplified with a recombinant Taq polymerase (Thermo Fisher Scientific), the forward primer 5′-TGGAACCCTAGCCCATCGTCA-3′, and the reverse primer 5′- CATTTGCTGCCAGATCCTCTT-3′. PCR products were subsequently cloned with the TOPO TA cloning kit (Thermo Fisher Scientific) and analyzed as described in the “Identification of CD28 mRNA splice variants by TA cloning” section.
Plasmids and transient transfection
The C-terminal Myc/DDK-tagged pCMV6 empty vector and the human CD28 (NM_006139), KANSL1L (NM_152519) and USP37 (NM_020935) expression vectors were purchased from Origene. Constructs carrying mutant alleles were generated by direct mutagenesis with the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies), according to the manufacturer’s instructions. The full-length wild-type and mutant human CD28 cDNAs were inserted into the V5/His-tagged pcDNA3.1 plasmid, with the directional TOPO expression kit (Thermo Fisher Scientific). HEK293T cells were transiently transfected with the various constructs, with a calcium phosphate kit (Thermo Fisher Scientific) or in the presence of X-tremeGENE 9 DNA Transfection Reagent (Roche), according to the manufacturers’ instructions.
Cell lysis and immunoblotting
Total protein extracts were prepared by mixing cells with lysis buffer (50 mM Tris pH7.4, 150 mM NaCl, 2 mM EDTA, 0.5% Triton X-100) and incubating for 30 minutes at 4°C. The cells were centrifuged for 10 minutes at 16000 x g, and the supernatant was collected for immunoblotting. A mixture of protease and phosphatase inhibitors was added to the buffers immediately before use: aprotinin (Sigma, 10 μg/mL), PMSF (Sigma, 1 mM), leupeptin (Sigma, 10 μg/mL), phosSTOP (Sigma, 1x) di-isopropylfluorophosphate (DFP, Sigma, 5 mM). The proteins were separated by SDS-PAGE and immunoblotting was performed with Abs against the V5 tag (R962–25, Thermo Fisher Scientific) and GAPDH (FL335, Santa Cruz).
Flow cytometry
Immunophenotyping was performed by flow cytometry, with mAbs against CCR7 (G043H7, Biolegend), CD1c (L161, Biolegend), CD3 (7D6, Thermo Fisher Scientific; UCHT1, BD), CD4 (RPA-T4, BD), CD8 (RPA-T8, BD), CD11c (S-HCL-3, BD), CD14 (M5E2, BD), CD15 (HI98, BD), CD16 (3G8, BD), CD19 (HIB19 and 4G7, BD), CD20 (LT20, Miltenyi Biotec), CD25 (MA-251, BD), CD27 (O323, Sony; L128, BD), CD28 (15E8, Miltenyi Biotec; CD28.2, BD), CD31 (WM-59, BD), CD45RA (Hl100, BD), CD54 (HA58, BD), CD56 (B159, BD; NCAM16.2, BD), CD123 (6H6, Biolegend), CD134 (L106, BD) CD141 (1A4, BD), CD152 (BNI3, BD), CD154 (24–31, Biolegend), CD161 (DX12, BD), CD183 (REA232, Miltenyi Biotec), CD185 (REA103, Miltenyi Biotec), CD194 (REA279, Miltenyi Biotec), CD196 (REA190, Miltenyi Biotec), CD223 (3DS223H, Thermo Fisher Scientific), CD272 (MIH26, Thermo Fisher Scientific), CD278 (C398.4A, Biolegend), CD279 (MIH4, Thermo Fisher Scientific), CD366 (F38–2E2, Thermo Fisher Scientific), FOXP3 (259D/C7, BD), IFN-γ (B27, BD), IgA (IS11–8E10, Miltenyi Biotec), IgG (G18–145, BD), IgM (PJ2–22H3, Miltenyi Biotec), KIR2DL1/S1 (EB6, Beckman Coulter) KIR2DL2/S2/L3 (GL183, Beckman Coulter), KIR3DL1 (DX9, Biolegend), NKG2A (REA110, Miltenyi Biotec), NKG2C (REA205, Miltenyi Biotec), TCR-iNKT (6B11, BD), TCR-γδ (11F2, Miltenyi Biotec), and TCR-Vα7.2 (REA179, Miltenyi Biotec), TIGIT (MBSA43, Thermo Fisher Scientific). Cells were also stained with the Aqua Live/Dead Cell Stain Kit (Thermo Fisher Scientific). When required, cells were fixed and permeabilized with a fixation/permeabilization kit (eBioscience) after extracellular staining, for intracellular staining. Alternatively, cells were fixed with Fix Buffer 1 (BD) and permeabilized with Perm buffer III (BD) after extracellular staining, for intracellular staining. Samples were analyzed with a FACS Aria II cytometer or a Fortessa X20 (BD) or Gallios (Beckman Coulter) machine, depending on the experiment. Data were then analyzed with FlowJo 10.1r5 software. The terminal differentiation profile of the CD56dim compartment was assessed by determining the distribution of NKG2A and KIR (Béziat et al., 2010; Björkström et al., 2010). T cell subsets were defined as naive (CD45RA+CCR7+), central memory (CD45RA−CCR7+), effector memory (CD45RA−CCR7−) and TEMRA (CD45RA+CCR7−) cells among the CD4+ and CD8+ T cells. γδ T cells (CD3+TCR-γδ+), MAIT (CD3+CD161+TCR-vα7.2+) and iNKT (CD3+TCR-iNKT+) were defined among the total CD3+ T cells. Treg (CD3+CD4+CD25hiFoxP3+) were defined among the CD4+ T cell compartment. T-helper (Th) subsets within the CD4+ memory compartments were defined as follows: Tfh (CXCR5+), Th1 (CXCR5−CXCR3+CCR4−CCR6−), Th2 (CXCR5−CXCR3−CCR4+CCR6−), Th1* (CXCR5−CXCR3+CCR4−CCR6+) and Th17 (CXCR5−CXCR3−CCR4+CCR6+).
Lentivirus production and transduction
Lentiviruses were produced as follows. Two days before transduction, 0.5–1.0×106 HEK293T cells were used to seed a six-well plate. Patient and control PBMCs were thawed and cultured in RPMI supplemented with 10% FCS with PHA (Sigma, 1 μg/mL). The following day, HEK293T cells were transfected with pCMV-VSV-G (0.2 μg) (Stewart et al., 2003), pHXB2-env (0.2 μg; NIH-AIDS Reagent Program; #1069), psPAX2 (1 μg; gift from Didier Trono; Addgene plasmid # 12260), pLVX-CD28_WT-IRES-mCherry (1.6 μg) or pLVX-CD28_Gly18Arg-IRES-mCherry (1.6 μg) or pLVX-EF1α-IRES-mCherry (1.6 μg) (empty vector; Takara) in the presence of X-tremeGENE HP (Sigma Aldrich), in accordance with the manufacturer’s protocol. The medium was replaced after 8 hours of incubation. In parallel, PHA blasts were used to seed 96-well round-bottomed plates at a density of 2×105 cells/well and were stimulated with beads coated with antibodies against CD2 and CD3 (T cell activation/expansion kit, Miltenyi Biotec). Anti-CD28 antibodies were omitted from the bead coating. On day 0, 24 hours after the HEK293T cell medium was changed, the viral supernatant was recovered and passed through a filter with 0.2 μm pores. Protamine sulfate (8 μg/mL) was added to the viral supernatant, which was then added to the activated T cells or Jurkat cell line (immediately after seeding), which were spinoculated for 2 hours at 1200 x g and 25°C. After spinoculation, cells were cultured for 48 hours at 37°C under an atmosphere containing 5% CO2. On day +2, the cells were transferred to a 24-well plate containing RPMI supplemented with 2% human serum AB (Sigma), penicillin/streptomycin (1/1000) and r-IL2 (10 ng/mL, Thermo Fisher Scientific) for primary T cells and RPMI supplemented with 10% FCS for Jurkat cells. The medium was replaced on day +5 and the p-p65 experiment was performed on day +6.
Histological analysis and immunohistochemistry
Formalin-treated paraffin-embedded sections of each specimen were stained with hematoxylin and eosin for histological analysis. Immunohistochemistry (IHC) studies were carried out on 3 μm-thick sections of formalin-fixed, paraffin-embedded tissue, with standard techniques. Pan HPV detection was performed with the K1H8 antibody (Thermo Fisher Scientific), which recognizes the highly conserved L1 protein of the viral capsid. The slides were developed with the Bond Polymer Refine Detection kit (Menarini/Leica).
Immunohistofluorescence analyses
Paraffin-embedded 5 μm-thick skin sections were processed for immunohistofluorescence analyses after heat-induced epitope retrieval in citrate buffer pH 6. Sections were blocked by incubation for 1 h in 5% FCS in PBS and incubated with primary antibodies overnight at 4°C. The following primary antibodies were diluted in 5% FCS in PBS and used at the appropriate dilution: rat monoclonal anti-CD3 (1:100, ab11089, Abcam), rabbit polyclonal anti-CD28 (1:100, HPA070003, Sigma), rabbit monoclonal anti-CD86 (1:50, 91882S, CST), mouse monoclonal anti-CD80 (1:50, MAB140, R&D Systems), mouse monoclonal anti-CD207 (1:50, NCL-L-Langerin, Novocastra) when used with antibodies against CD3 and CD86 or rabbit polyclonal anti-CD207 (1:50, HPA011216, Sigma) when used with anti-CD80 antibody.
The sections were washed in 5% FCS in PBS and incubated for 1 h at room temperature with the appropriate secondary antibody: CD3 was detected with Cy3-conjugated goat anti-rat IgG (H+L) (112–165-003, Jackson Immunoresearch) when used with CD28 or with Alexa Fluor® 488-conjugated goat anti-rat IgG (H+L) (A11006, Life Technologies) when used with CD86 and CD207; CD28 was detected with Alexa Fluor® 488-conjugated goat anti-rabbit IgG (H+L) (A11008, Life Technologies); CD80 was detected with Alexa Fluor® 488-conjugated goat anti-mouse IgG1 (A21121, Life Technologies). Mouse CD207 was detected with Alexa Fluor® 647-conjugated donkey anti-mouse IgG (H+L) (715–605-150, Jackson Immunoresearch) and rabbit CD207 and CD86 were detected with Alexa Fluor® 594-conjugated donkey anti-rabbit IgG (H+L) (711–585-152, Jackson Immunoresearch). Slides were mounted in Fluoromount-G® (Clinisciences) supplemented with DAPI (1:1000, Cell Signaling). Laser scanning confocal microscopy was performed with a LEICA SP8 SMD microscope with a 40 x Plan-Apochromat objective and a magnification of 1 x or 0.75 x. Stacks of images were processed with Fiji software (Schindelin et al., 2012). Purified HPV-2 E1Ê4-MBP (maltose binding protein) fusion protein was immunized into rabbits as described previously (Doorbar et al., 1989; Peh et al., 2002). The detection of the E4 and MCM biomarkers was performed as previously described (Middleton et al., 2003 J Virol), with rabbit polyclonal anti-HPV-2 or −4 E4 serum at a 250-fold dilution. Detection was performed with Alexa Fluor 594-conjugated goat anti-rabbit igG (H+L) (Thermo Fisher Scientific) diluted 150-fold. The MCM7 antibody (47DC141, Thermo Fisher Scientific) was incubated with the slides at a 200-fold dilution for 1 hour at room temperature, and the slides were then stained by incubation for 1 hour at room temperature with the ImmPRESS horse anti-mouse IgG HRP polymer (Vector Laboratories). The slides were developed with a 500-fold dilution of Tyramide-488 (Cambridge BioScience), and the nuclei were counterstained with DAPI. Immunofluorescence was recorded by scanning with a Pannoramic digital slide scanner (3DHISTECH, Budapest, Hungary).
In situ hybridization assay for HPV-2 E6/E7
In situ hybridization for HPV-2-specific RNA was performed manually with the RNAscope 2.5 HD detection assay kit (Bio-Techne) – Brown with an HPV-2 E6/E7 probe (Cat. No 806601), according to the manufacturer’s instructions. Sections were lightly counterstained with Carazzi’s hematoxylin and were then classified as positive or negative. Positive sections displayed brown granular cytoplasmic and/or nuclear staining stronger than the signal obtained for the negative control slide. RNA integrity was confirmed with the PPIB control probe (Cat. No 313901). An HPV-2 positive wart was used as a positive control for E6/E7 mRNA levels, with HPV-negative normal skin used as a negative control.
Assays of redirected T cell function
Thawed PBMCs were rested overnight in complete medium and dispensed, at a final concentration of 5×106 cells/mL, into 96-well round-bottomed plates. P815 murine mastocytoma target cells were added to PBMCs at a final concentration of 2.5×106 cells/mL. Anti-CD3-PE (7D6, 1/100), and/or anti-CD28 (CD28.2, 5 μg/mL) antibodies were added alone or in combination to the indicated wells. PMA (40 ng/mL) and ionomycin (10−5M) were used to stimulate the cells in separate wells, as a positive control. The cells were incubated for 6 hours (37°C, 5% CO2) after the addition of monensin (GolgiStop, BD Biosciences, 1/1500 final concentration), and brefeldin A (GolgiPlug, BD Biosciences, 1/1000 final concentration). The cells were washed and stained for CD3, CD4, CD8, CCR7, CD45RA and with a viability stain. They were then permeabilized (fixation/permeabilization buffer, eBioscience) and stained for intracellular TNF (cA2, Miltenyi Biotec), IL-2 (MQ1–17H12, Sony) and IFN-γ (B27, BD) before flow cytometry.
Phospho-NF-κB p65 in PHA blasts
PHA blasts were stained by incubation for 10 minutes at 37°C with the Aqua Live/Dead kit (Thermo Fisher Scientific). We dispensed 106 PHA blasts into each well of a 96-well V-bottomed plate, and incubated them on ice for 10 minutes with anti-CD28 (CD28.2, eBioscience) or anti-CD3 (OKT3, eBioscience) mAb, as indicated (5 μg/mL each). Cells were washed twice with cold medium and a polyclonal goat anti-mouse Ig (BD Biosciences) was added to each well (5 μg/mL) to crosslink activating receptors. PMA (40 ng/mL) was used, in separate wells, as a positive control. After 20 minutes of incubation at 37°C, cells were fixed by incubation for 10 minutes at 37°C with Fix buffer I (BD Biosciences) and stained by incubation for 30 minutes with mAb against CD3 (BW264/56, Miltenyi Biotec), CD4 (M-T321, Miltenyi Biotec) and CD8 (BW135/80, Miltenyi Biotec). The cells were permeabilized by incubation for 20 minutes at room temperature with Perm buffer III (BD Biosciences) and stained by incubation for three hours at room temperature with an anti-NF-κB p65-(pS259)-PE antibody (BD Biosciences). Cells were then acquired on a FACS Gallios (Beckman Coulter) flow cytometer and analyzed with FlowJo v10.
Induction of costimulation and coinhibitory molecules
Freshly thawed PBMCs were cultured in complete medium (RPMI, glutamine, 10% FCS) supplemented with 2% human serum (Sigma), at a final concentration of 2×106 cells/mL in a 24-well plate. Cells were left unstimulated or were stimulated with CD2/CD3/CD28 mAb-coated beads (Miltenyi Biotec) or PMA (40 ng/mL) and ionomycin (10−5M). After 72 hours of incubation (37°C, 5% CO2), the cells were stained for extracellular receptors and with the Aqua Live/Dead Cell Stain Kit (Thermo Fisher Scientific), and analyzed by flow cytometry on a FACS Gallios machine. For CTLA4 analysis, cells were fixed and permeabilized (Fixation/permeabilization buffer, eBioscience), stained for intracellular CTLA4 and analyzed by flow cytometry on a FACS Gallios machine.
Ex vivo CFSE proliferation assay
Freshly thawed PBMCs were stained with CFSE (Thermo Fisher Scientific) and cultured in complete medium (RPMI, glutamine, 10% FCS) supplemented with 2% human serum (Sigma), at a final concentration of 2×106 cells/mL in a 24-well plate. Cells were left unstimulated or were stimulated with CD3 antibody-coated, CD3/CD2 antibody-coated, CD3/CD28 antibody-coated or CD2/CD3/CD28 antibody-coated beads (Miltenyi Biotec). After 5 days of incubation (37°C, 5% CO2), the cells were stained for extracellular receptors (CD3, CD4 and CD8) and with the Aqua Live/Dead Cell Stain Kit (Thermo Fisher Scientific), and analyzed by flow cytometry on a FACS Gallios machine.
Short-term stimulation with overlapping peptides or tuberculin
Freshly thawed PBMCs were cultured in complete medium (RPMI, glutamine, 10% FCS) at a final concentration of 5×106 cells/mL in a 96-well U-bottomed plate. Cells were left unstimulated or were stimulated with overlapping CMVpp65 peptides (JPT Technology, 1 μg/mL final concentration for each peptide), tuberculin PPD (5 μg/mL final concentration; Statens Serum Institut) or CD2/CD3/CD28 mAb-coated beads (Miltenyi Biotec) in the presence of brefeldin A (GolgiPlug, BD Biosciences, 1/1000 final concentration). After 16 hours of incubation (37°C, 5% CO2), the cells were stained for extracellular receptors and with the Aqua Live/Dead Cell Stain Kit (Thermo Fisher Scientific), permeabilized (fixation/permeabilization buffer, eBioscience), stained for intracellular IFN-γ-AF700 (B27) and analyzed by flow cytometry on a FACS Gallios machine.
Long-term stimulation with overlapping peptides
Freshly thawed PBMCs from controls and patients were cultured in complete medium (RPMI, glutamine, 10% FCS) at a final density of 2–4×106 cells/mL. Cells were left unstimulated or were stimulated by incubation for one hour with overlapping CMVpp65, HPV-2E6 or HPV-4L1 peptides (all from JPT Technology, 1 μg/mL final concentration for each peptide), washed and transferred to a 24-well plate. After 24 hours, IL-2 was added to each well (100 IU, Proleukin, Novartis). The medium and IL-2 were renewed every two to three days. After 14 days of culture, overlapping peptides were added to each well at a final concentration of 1 μg/mL for each peptide, and the cells were incubated for four hours. The cells were then washed, stained for extracellular epitopes (CD3, CD4, CD8) and with the Aqua Live/Dead Cell Stain Kit (Thermo Fisher Scientific), permeabilized (fixation/permeabilization buffer, eBioscience), stained for intracellular IFN-γ-AF700 (B27) and TNF-APC (cA2) and analyzed by flow cytometry on a FACS LSR-Fortessa machine.
Single-cell RNA-seq
Single-cell RNA-seq was performed on PBMCs obtained from the three patients, and from three wild-type controls. The frozen PBMCs were quickly thawed at 37°C and gently resuspended by serial additions of DMEM + 10% heat-inactivated FBS, to obtain a final volume of 14 mL. The cells were collected by centrifugation for 5 minutes at 300 x g, cells and were washed twice with 5 mL DMEM + 10% HI-FBS to remove cell debris. Cells were counted and viability was assessed with the LIVE/DEAD Viability kit (Thermo Fisher Scientific), according to the kit manufacturer’s guidelines. The cell preparation was enriched in viable cells, by resuspending one million cells in 200 μL of 1 x PBS, 2% HI-FBS 1 mM CaCl2, passing the resulting suspension through a strainer with 40 μm pores and removing the dead cells with the EasySep Dead Cell Removal Kit (Stem Cell Technologies). The cells were collected by centrifugation for five minutes at 300 x g and resuspended at a concentration of 1000 cells/μL in 0.04% BSA in PBS. Cell viability was ≥ 80% for all samples. The samples were loaded onto a 10X Genomics Chromium chip for single-cell capture. Reverse transcription and library preparation were performed with Chromium Single Cell 3′ Reagent Kits (v3.1), in accordance with the manufacturer’s guidelines, and library quality was assessed with a Bioanalyzer DNA chip. The libraries were sequenced on one lane (S4 flowcell) of an Illumina NovaSeq 6000 sequencer.
Sequence read quality in individual sequencing lanes was assessed with BVAtools. Cell Ranger v3.0.1 was used for mapping reads to the hg38 human reference genome assembly, filtering, and counting barcodes and UMIs, resulting in a list of UMI counts for every gene in every cell. Based on these counts, we extracted the summary statistics for the number of genes expressed per cell and for the number of UMIs per cell. Cells with > 20% mitochondrial genes were excluded. Low-quality cells and doublets were filtered out by excluding cells falling outside the [−1 SD;+2.5 SD] interval for UMI and gene count distributions. The DoubletFinder package was used to identify and filter out the remaining cell doublets (McGinnis et al., 2019). Once dead cells and doublets had been removed, the CD28-deficient patient and control samples were analyzed jointly with the Seurat v3 R package (Stuart et al., 2019) and cell clustering was performed by the uniform manifold approximation and projection dimension reduction method (Becht et al., 2018) with the most variable genes, but excluding mitochondrial and ribosomal protein genes. This analysis identified 10 distinct clusters of lymphoid cells, based on the marker genes for each cluster determined by the MAST approach (Finak et al., 2015). A pseudobulk differential gene expression analysis was performed by extracting the sum of UMI counts for each cluster in each sample, followed by normalization against the total number of counts per cluster and per sample, and quantile normalization. The patients and controls were then compared for each CD28-expressing cluster, with the edgeR package (Robinson and Oshlack, 2010). Genes displaying a two-fold difference in expression, with a Benjamini-Hochberg adjusted p-value < 10−3 were considered to display significant differential expression. For the figures, cluster annotations and expression values for the genes of interest were projected onto the UMAP clustering.
Primary CD4+ naive T cell RNA-Seq analysis
Total RNA from primary naive CD4+ T cells was extracted with the RNeasy Plus Micro Kit (QIAGEN), according to the manufacturer’s instructions. We generated cDNA with the SMART-Seq RNA kit (Takara Bio) for low-input RNA and DNA library preparation, and used the Nextera XT DNA Library Preparation Kit (Illumina) and the Nextera XT Index Kit v2 set A (Illumina), respectively, for indexing with low DNA input requirements. The RSeQC package was used for quality evaluation of the raw high throughput sequence data (Wang et al., 2012). Raw RNA-seq reads were aligned with UCSC human genome assembly version hg38, with STAR aligner (Dobin et al., 2013). HTseq-count (Anders et al., 2015) was used to count the reads mapping to each gene feature. Downstream analysis and heatmap generation were performed with an in-house script in R (R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/), with DESEq2 (Love et al., 2014), and ComplexHeatmap (Gu et al., 2016). In brief, differentially expressed genes (DEGs) were identified in controls by performing Student’s t tests to compare four non-stimulated and stimulated control samples for each of the conditions, with a p-value cutoff of 0.01, and then filtering to select genes displaying at least a two-fold up- or downregulation in at least three of the four controls. The residual responses for the CD28–deficient patient were evaluated by determining the number of responsive genes passing the above filter in healthy controls, as follows: The total count of responsive genes in a subject for a particular set of in vitro stimulation conditions and time point was divided by the mean number of responsive genes in all healthy controls and multiplied by 100. Alternatively, fold-changes in expression after costimulation with CD2+CD3 or CD3+CD28 were calculated relative to stimulation with CD3 alone in certain conditions, as indicated in the figure legends.
High-throughput sequencing (HTS) of T cell receptor β (TRB) and T cell receptor α (TRA)
CD4+CD28− and CD4+CD28+ (revertants) T cells from P1, P2 and P3, and CD4+CD28+ T cells from a control were sorted with a FACSAria cell sorter (Becton Dickinson, San Jose, CA). The TCRβ (TRB) and TCRα (TRA) rearranged genomic products were amplified by multiplex PCR, with DNA from these cells as the template (Adaptive Biotechnologies Seattle, WA). We ensured that all possible VDJ combinations were amplified, by using an adequate set of forward primers for the TRBV and TRAV gene segments and reverse primers for the TRBJ and TRAJ gene segments. Adaptive Biotechnologies use assay-based and computational techniques to minimize PCR amplification bias. The assay is quantitative, and the frequency of a given TRB or TRA sequence is representative of the frequency of that clonotype in the original sample. The PCR products were sequenced on an Illumina HiSeq platform. Custom algorithms were used to filter the raw sequences for errors and to align the sequences with reference genome sequences. The data were then analyzed with ImmunoSeqTM online tools. The frequency of productive and non-productive TRB and TRA rearrangements was analyzed within both unique and total TRB or TRA sequences obtained from sorted T cell subsets. The frequency distributions for individual clonotypes (including TRBV to TRBJ pairing and TRAV to TRAJ pairing) were analyzed within unique sequences. Heatmap representations of the frequencies of individual TRAV to TRAJ gene pairs and TRBV to TRBJ gene pairs were produced with R software version 3.6.3 (2020–02-29). TCR-Vβ sequence of the revertants and CD28- CD4+ T cells from P1, P2 and P3 were also analyzed using the GLIPH software that clusters TCRs that are predicted to bind the same MHC-restricted peptide antigen (Glanville et al., 2017).
CITE-Seq Analysis
PBMCs from P1, P2 and one control were thawed and cultured overnight in X-Vivo 20 (Lonza). Dead cells were removed using the dead cell removal kit (Miltenyi Biotec). CD4+ memory T cells were negatively sorted using the Memory CD4+ T cell isolation kit (Miltenyi Biotec). Purity and cell viability were measure by flow cytometry and were above 90% and 95%, respectively. Cells were stained for CD3 (C0049), CD4 (C0072), CD45RA (C0063) and CD28 (C0386) using Total-seq C antibodies for CITE-seq from Biolegend. Cell suspensions were used for single cell isolation using 10X Genomics Single cell protocols. 5′ expression and TCR Libraries were generated using Chromium Single Cell 5′ Library & Gel Bead kit Version 1 (10x genomics # 1000014). Hashtag libraries were generated using Chromium Single Cell 5′ Feature Barcode Library Kit (# 1000080). Standard protocol from 10X genomics was followed to generate the libraries. After QC, Three expression libraries were pooled together and sequenced on Novaseq using 100 cycle SP flowcell, the TCR and hashtag libraries were pooled together and sequenced on Nextseq. Sequence reads quality of individual sequencing library was assessed using BVAtools (https://bitbucket.org/mugqic/bvatools). Cell Ranger v5.0.1 was used to map the reads to the hg38 human reference genome assembly and detect antibody feature barcodes, perform filtering, and count barcodes and UMIs; resulting in two list of UMI counts: one for every gene in every cell and one for the antibody detection. Based on these counts, we filtered out cells which were not detected in both scRNA and antibody analyses. We extracted the summary statistics for the number of expressed genes per cell along with the summary statistics for the number UMI per cell. Cells with > 20% of mitochondrial genes were excluded. To filter out low quality cells and doublets, cells falling outside the [−1SD;+2.5SD] interval for the UMI and gene count distribution were excluded. Further, the DoubletFinder package was used to identify and filter out remaining cell doublets (McGinnis et al., 2019). After dead cells and doublet removal, the CD28 patient (P1 and P2) and control samples were jointly analyzed using the Seurat v4 R package (Stuart et al., 2019) and cell clustering was performed using the Uniform Manifold Approximation and Projection dimension reduction method (Becht et al., 2018) using the most variable genes, but excluding mitochondrial and ribosomal protein genes. This analysis identified sixteen distinct clusters of cells, based on the marker genes for each cluster determined using the MAST approach (Finak et al., 2015). We identified 12 different CD4+ memory cell populations, and four small additional cluster corresponding to remaining activated T cells, Treg, gdT and NKT, and CD8+ T cells; these four non-CD4 memory cell clusters were excluded from further analyses. We looked at the level of CD28 antibody in controls and determine the standard antibody detection for cell expressing normal CD28 genes expression correspond to a signal between 0.5 and 2. By combining the antibody signal and the gene expression signal, we annotated cells carrying the CD28 mutation if no expression of the gene CD28 is detected and if the antibody signal is lower than 0.25. We annotated the CD28 revertant cells if the expression of the CD28 gene is detected and the signal of antibody is measured between 0.5 and 2 as observed in controls. We thus identify 1438 cells CD28 mutant and 38 revertant cells for P1 and P2 together. A differential gene expression analysis was performed comparing CD28 revertants to mutants using the MAST approach in the twelve CD4+ memory populations. For the figures, cluster annotations, the expression of genes of interest and the antibody signal were projected on the UMAP clustering.
RNA-Seq analysis of the lesions of P1 and control common warts
Two TMS lesions from P1, and three HPV-2+ common warts immersed in RNA-Later were snap-frozen in liquid nitrogen and crushed with a tissue pulverizer kit (Cell Crusher) according to the manufacturer’s instructions. Total RNA was extracted with the RNeasy Fibrous Tissue Mini Kit (QIAGEN), according to the manufacturer’s instructions. The procedure included a DNase digestion step or an additional column to remove contaminating DNA. The concentration and purity of the total RNA extracted were measured by spectrometry with an Xpose (Trinean). RNA integrity was evaluated by capillary electrophoresis with a Tape Station (Agilent). The Ovation Universal RNA-Seq System from NUGEN was used to prepare the RNA-seq libraries from 100 ng of total RNA, as recommended by the manufacturer. This type of RNaseq kit is less sensitive to total RNA degradation (the RNA integrity number (RIN) of the two samples from P1 was quite low: 3.9 for one sample and 5 for the other; for the common warts, all the RIN were > 8). This kit constructs strand-specific RNA-Seq libraries from 10 to 100 ng of total RNA and uses Insert-Dependent Adaptor Cleavage (InDA-C) technology to remove the ribosomal RNA transcripts. The RNA is subjected to reverse transcription, the second strand is synthesized, and a fragmentation step is then performed before Illumina-compatible indexed adaptator ligation. This ligation is followed by a strand-selection enzymatic reaction to provide information about the orientation of the transcripts. Insert-dependent adaptor cleavage (InDA-C)-specific primers were then used for the targeted depletion of human ribosomal RNA sequencing transcripts before PCR enrichment. We ensured that no excess amplification occurred during the final PCR step, by evaluating the number of PCR cycles to be applied to each sample in a preliminary Q-PCR test with EvaGreen. An equimolar pool of the final indexed RNA-Seq libraries was sequenced on an Illumina NovaSeq6000 (paired-end reads, 100 bases + 100 bases) and ~50 million paired-end reads per library were produced. We used STAR 2.7.2b (Dobin et al., 2013) with standard parameters to align RNA-seq reads with a chimeric genome of concatenated GRCH37.87 and HPV-2 downloaded from the PAVE website (Van Doorslaer et al., 2013) (http://pave.niaid.nih.gov). We used IGV 2.4.19 (Robinson et al., 2011) to extract the splicing junction track from the aligned reads.
Animals and MmuPV1 infection
All work on mice was approved by the Institutional Animal Care and Use Committee of Pennsylvania State University’s College of Medicine (COM) and all procedures were performed in accordance with guidelines and regulations. Infectious mouse papillomavirus (MmuPV1) was isolated from lesions on the tails of mice from our previous studies (Cladel et al., 2015, 2019b). In brief, papillomas scraped from the tails of the mice were homogenized in phosphate-buffered saline (1 × PBS) with a Polytron homogenizer (Brinkman PT10–35) at maximum speed for three minutes, with chilling in an ice bath. The homogenate was spun in a bench centrifuge at 10,000 rpm and the supernatant was decanted into Eppendorf tubes for storage at −20°C. For these experiments, the suspension of the mouse papillomavirus was stored at −80°C in glycerol (1:1,V/V). We obtained 27 C57BL/6, six Rag1 KO mice, and 26 female Cd28−/− (6–8 weeks old) mice from Jackson Laboratories. All mice were housed (2–3 mice/cage) in sterile cages within sterile filter hoods and were fed sterilized food and water in the COM BL2 animal core facility. Mice were sedated i.p. with 0.1 mL/10 g body weight of a ketamine/xylazine mixture (100 mg/10 mg in 10 mL double-distilled H2O). The muzzle and tail skin sites were wounded with a scalpel blade. Twenty-four hours after wounding, the mice were again anesthetized and challenged with infectious virus (containing 1 × 109 viral DNA genomes) at the pre-wounded muzzle and tail sites, with a TB needle used to scrape the pre-wounded inoculation sites superficially. Infection of the muzzle and tail was monitored weekly, with a photographic documentation of progression for each animal (Cladel et al., 2013). Cd28−/− mice were killed in weeks 2, 3, 4 and 5 after viral infection. C57BL/6 and Rag1 KO mice were killed in week 6 post infection. The infected tissues were harvested for additional analyses.
Detection of MmuPV1 by in situ hybridization (ISH), RNA in situ hybridization (RNA-ISH), and immunohistochemistry
Biopsy specimens were fixed in 10% neutral buffered formalin and embedded in paraffin. Adjacent sequential sections were cut for hematoxylin and eosin (H&E), in situ hybridization (ISH), RNA in situ hybridization (RNA-ISH), and immunohistochemistry (IHC), as described in previous studies (Cladel et al., 2016, 2017; Hu et al., 2015). For ISH, a biotin-labeled 3913 bp EcoRV/ BamH1 subgenomic fragment of MmuPV1 was used as an in situ hybridization probe for the detection of MmuPV1 DNA in tissues (Cladel et al., 2015). Access to target DNA was obtained by incubation with 0.2 mg/mL pepsin in 0.1 N HCl at 37°C for 8 min. The sections were thoroughly washed and the biotinylated probe was added. The sections were heated at 95°C for 5 min to dissociate the target and probe DNA. Re-annealing was allowed to occur for 2 hours at 37°C. Target-bound biotin was detected by incubation with a streptavidin AP conjugate followed by colorimetric development in BCIP/NBT. ISH, counterstaining was performed with Nuclear Fast Red (American MasterTech, Inc.). Viral RNA was detected in formalin-fixed, paraffin-embedded (FFPE) tissues with RNAscope technology (Advanced Cell Diagnostics, Inc.), using custom probes mapping within the second exon of the E1Ê4 ORF (nt 3139–3419), according to the manufacturer’s protocol. After hybridization, the bound probes were detected by colorimetric staining in the RNAscope 2.5 HD Assay - BROWN. For IHC, an in-house anti-MmuPV1 E4 polyclonal antibody (from Dr. John Doorbar) or anti-MmuPV1 L1 antibody (MPV.B9, in-house) was used on FFPE sections. Before mounting, the sections were counterstained with 50% hematoxylin Gill’s No. 1 solution (Sigma-Aldrich) and 0.02% ammonium hydroxide solution (Sigma-Aldrich) (Cladel et al., 2017).
MmuPV1 viral load assessment by RT-qPCR
We collected tail tissues from the mice killed at different time points after infection. Total RNA was extracted from these tissues and reverse-transcribed to generate cDNA for quantitative PCR analysis. Copy numbers were assessed with 200 ng RNA from each sample (Viral RNA E1Ê4 transcripts were quantified with the primers 5′-TAGCTTTGTCTGCCCGCACT-3′ and 5′-GTCAGTGGTGTCGGTGGGAA-3′ and probe 5′FAM-CGGCCCGAAGACAACACCGCCACG-3′TAMRA. We reverse-transcribed 200 ng of RNA with the RevertAid First-Strand cDNA synthesis kit (Thermo-Fisher) and used 1 μL of the resulting cDNA for Q-PCR analysis. We used 500 nM of each primer and 250 nM probe with the Brilliant III Q-PCR kit (Agilent), under the following qPCR conditions: 95°C for 3 min, followed by 50 cycles of 95°C for 5 s and 60°C for 10 s on an Agilent AriaMx Q-PCR machine.
Anti-MmuPV1 antibody detection by ELISA
Serum was collected from the mice at the end of the experiment. MmuPV1 VLP and KLH-conjugated MmuPV1 E4 peptide (PKTTPPRRELFPPTPLTQPP) were used as antigens for ELISA. MPV.A4 was used as the positive control for VLP. Serum from naive animals was used as the negative control. ELISA was performed as previously described (Cladel et al., 2019b).
USP37 in vitro deubiquitination assays
HEK293T cells were transfected with a plasmid encoding the WT or Glu459Lys USP37, with polyethylenimine (Linear, Molecular Weight 25,000) (Polysciences, Warrington, PA, USA). We mixed 10 μg plasmid with 30 μL polyethylenimine solution (1 mg/mL) in 1 mL Opti-MEM (Life Technologies). The mixture was incubated for 15 minutes and added to subconfluent cells in 100 mm dishes (in 10 mL of 10% FBS-DMEM). Two days later, cells were lysed in ice-cold lysis buffer [50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM NaF, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1 mM DTT, protease inhibitor cocktail (Sigma-Aldrich, #P8340)]. The lysates were centrifuged at 15,000 x g for 10 min at 4°C. The supernatants were subjected to immunoprecipitation with anti-FLAG M2 antibody-conjugated agarose beads (Sigma-Aldrich), followed by elution with FLAG peptide (Sigma-Aldrich), according to the protocols recommended by the manufacturer. Immunopurified USP37 proteins (wild-type or mutant, 50 ng) were incubated with 0.4 μg of Lys48-linked polyubiquitin chains (2–7-mer, Boston Biochem) in 20 μL of 5 mM MgCl2 and 2 mM DTT in TBS at 37°C, for 0, 10, 30, or 120 min. Reaction products were separated by SDS-PAGE and detected by silver staining.
VirScan - phage immunoprecipitation-sequencing (PhIP-Seq)
Antibody profiling by phage immunoprecipitation-sequencing (PhIP-Seq) was performed on plasma samples from patients and controls using an expanded version of the original VirScan library, and the data were analyzed as previously described (Drutman et al., 2020; Kerner et al., 2020; Mina et al., 2019; Mohan et al., 2019), but with the following modifications. We calculated species-specific significance cutoff values to estimate the minimum number of enriched, non-homologous peptides required to consider a sample seropositive, as previously described with an in-house dataset and a generalized linear model (Hasan et al., 2021; Khan et al., 2021). For each sample, we calculated virus-specific scores, by dividing the counts of enriched, non-homologous peptides by the estimated score cutoff. These adjusted virus scores were further depicted in heatmap plots. We randomly selected an age-matched subset of 19 individuals from a larger cohort of 800 individuals of Arab ancestry (representing general adult population) from our inhouse database, for comparison with the patients and initial controls. Pooled human plasma used for IVIg (Privigen® CSL Behring AG), and human IgG- depleted serum (Molecular Innovations, Inc.) were used as additional controls. All research on human subjects was performed with informed written consent, and the procedures were approved by the Institutional Research Ethics Boards of Sidra Medicine and Qatar Biobank.
Antibody profiling by phage immunoprecipitation-sequencing (PhIP-Seq) (Xu et al., 2015) was performed on plasma samples from patients and controls, and the data were analyzed as previously described (Drutman et al., 2020; Kerner et al., 2020), but with the following modifications. We calculated species-specific significance cutoff values to estimate the minimum number of enriched, non-homologous peptides required to consider a sample seropositive, as previously described with an in-house dataset and a generalized linear model (Xu et al., 2015). For each sample, we calculated virus-specific scores, by dividing the counts of enriched, nonhomologous peptides by the estimated score cutoff. These adjusted virus scores were further depicted in heatmap plots. We randomly selected an age-matched subset of 19 individuals from a larger cohort of 800 individuals of Arab ancestry (representing general adult population) from our in-house database, for comparison with the patients and initial controls. Pooled human plasma used for IVIg (Privigen® CSL Behring AG), and human IgG-depleted serum (Molecular Innovations, Inc.) were used as additional controls. All research on human subjects was performed with informed written consent, and the procedures were approved by the Institutional Research Ethics Boards of Sidra Medicine and Qatar Biobank.
Anti-HPV Luminex serological tests
Plasma samples were sent to the German Cancer Research Center (DKFZ, Heidelberg, Germany) on dry ice for serological analysis. Antibodies against L1 antigens of HPV types 1, 2, 4, 5, 6, 8, 9, 10, 11, 12, 15, 16, 17, 18, 21, 22, 23, 24, 27b, 31, 33, 36, 38, 41, 45, 48, 50, 52, 58, 60, 75, 80, 88, 92, 93, 96, 101 and 103 were analyzed simultaneously with Luminex-based multiplex serological tests, as previously described (Waterboer et al., 2005). In brief, HPV L1 antigens were expressed as recombinant glutathione S-transferase (GST) fusions proteins, loaded onto polystyrene beads and simultaneously presented to primary serum antibodies. The immunocomplexes formed were detected with a biotinylated secondary antibody and streptavidin-R-phycoerythrin as a reporter dye. Sera were tested at a dilution of 1:100 and antigen-specific seropositivity was determined on the basis of predefined cutoff values (Clifford et al., 2007; Michael et al., 2008; Sankaranarayanan et al., 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
Two-tailed Mann-Whitney tests tests were used for single comparisons of independent groups. Wilcoxon tests were used for single comparisons of paired groups. Kruskal-Wallis tests were used for multiple comparisons of independent groups. In the relevant figures, n.s. indicates not significant; ***p < 0.001; **p < 0.01; and *p < 0.05. Analyses were performed with GraphPad software.
Supplementary Material
Figure S1. Familial genetics, population genetics of KANSL1L and USP37, and functional assay for the pGlu459Lys USP37 allele, related to Figure 1
(A) Homozygosity rates for P1 to P3 relative to consanguineous (open circles) and nonconsanguineous (closed circles) ethnically matched controls. The red line corresponds to the 90% percentile.
(B) An analysis of whole-exome sequencing (WES) data identified three genes as the only genes carrying a homozygous coding mutation common to the three patients and within the linked region.
(C) Rare homozygous variants within the linked region. This region includes CD28 (Gly18Arg), USP37 (p.Glu459Lys) and KANSL1L (p.Ser577Thr). USP37 and KANSL1L have no known role in immunity. USP37 expression is ubiquitous, and involved in the cell cycle and homologous recombination (Huang et al., 2011; Tanno et al., 2014; Typas et al., 2015). The mutation in USP37 results in a glutamic acid-to-lysine exchange at position 459. Glutamic acid is negatively charged, whereas lysine is positively charged. By contrast, KANSL1L mRNA is mostly restricted to the parathyroid gland (The Human Protein Atlas) and has no known function. The mutation in KANSL1L results in a Serine to Threonine exchange, two polar uncharged amino acids (p.Ser577Thr).
(D) HEK293T cells were transfected with an empty pCMV6 plasmid or with pCMV6 plasmids encoding the wild-type (WT) or p.Ser577Thr (c.1730G>C) KANSL1L transcripts (left), or encoding the WT or p.Glu459Lys (c.1375G>A) USP37 transcripts. All constructs contained a C-terminal DDK tag. Total protein extracts were subjected to immunoblotting with a monoclonal antibody against the DDK tag. GAPDH was used as a loading control. The two missense proteins are normally expressed following the transfection of HEK293T cells with the corresponding cDNAs. A western blot representative of three independent experiments is shown. Molecular weights are shown on the left side of the blots.
(E) Frequency and combined annotation–dependent depletion score (CADD), for all homozygous KANSL1L (left) and USP37 (right) variants reported in public databases. The dotted line corresponds to the mutation significance cutoff (MSC). NR: not reported. The mutations of the patients appear in red. The p.Ser577Thr KANSL1L variant was identified four times, in the homozygous state, in the ATAVDB database and its minor allele frequency is as high as 0.004 in South Asians (gnomAD). By contrast, the USP37 variant in our patients was private.
(F) Amino-acid conservation, across species, for the mutated area of the KANSL1L and USP37 proteins. KANSL1L orthologs are found in other taxonomic groups, as far back in evolution as insects (Drosophila melanogaster), but serine 577 is conserved only in mammals, a threonine residue being present in the corresponding position in Drosophila. Together, population genetics, evolutionary genetics, and biochemical analysis suggest that this KANSL1L variant is neutral. USP37 orthologs are found across vertebrates, but the glutamic acid in position 459 is conserved only in mammals. In birds and amphibians, it is replaced by an aspartic acid, an amino acid with a similar negative charge. (G-H) Population genetics, evolutionary genetics, and biochemical analysis suggested that the USP37 variant might be deleterious. We therefore assessed the functional impact of the p.Glu459Lys substitution on USP37 deubiquitination (Huang et al., 2011). We immunoprecipitated and purified WT or p.Glu459Lys mutant USP37 overproduced in HEK293T cells and performed an in vitro deubiquitination assay with K48-linked ubiquitin chains (2–7-mer) as a substrate, followed by SDS-PAGE and silver staining.
(G) Amounts of immunopurified USP37 (WT, MUT) protein used in H. IB, immunoblotting.
(H) In vitro deubiquitination of Lys48-linked polyubiquitin chains (2–7-mer) by immunopurified USP37 (WT, MUT). The p.Glu459Lys mutation did not affect deubiquitination activity. These results suggest that the USP37 variant is unlikely to contribute to the severe verrucosis phenotype observed in the patients studied. The sample in the first lane contained polyubiquitin chains but not USP37. Ub, ubiquitin. Representative results from three independent experiments are shown.
Figure S2. CD28-deficient mice are susceptible to MmuPV1 skin infection, related to Figure 1
In the absence of CD28-deficient rabbits, it was not possible to use the CRPV model, which would have been physiologically relevant (Cladel et al., 2019a). Instead, we used the MmuPV1 infection model in congenic wild-type (WT) and Cd28−/− C57BL/6 mice (Ingle et al., 2011). The MmuPV1 virus causes skin wart lesions in athymic nude or RAG1-deficient C57BL/6 mice but not in C57BL/6 WT mice (Cladel et al., 2013; Wang et al., 2015; Handisurya et al., 2014). MmuPV1 does not cause skin lesions in mice with several other types of immunodeficiency, including C57BL/6 mice, after CD4+ or CD8+ T cell depletion (Handisurya et al., 2014; Wang et al., 2015), or in C57BL/6 mice homozygous for null mutations of Ifnar1, Cd4, Cd40lg (Hu et al., 2017; Wang et al., 2015), Ifng, Sting, or Ccr10 (unpublished data).
(A) Representative images of tail and muzzle lesions (red circles) from RAG1-deficient (middle) and CD28-deficient mice (right) 5 weeks post-infection with MmuPV1 are shown. Images of a wild-type mouse (left) are also shown for comparison. We infected the muzzle and tail of 23 (15 male and 8 female) Cd28−/− mice, six Rag1−/− mice, and 27 WT mice with MmuPV1. Rag-1−/− mice have no T or B cells. We monitored skin lesions between two and seven weeks before the animals were killed. None of the WT C57BL/6 mice developed skin lesions, consistent with the findings of previous studies (Hu et al., 2017). All six Rag1−/− mice developed skin warts three weeks post infection. These lesions persisted until the end of monitoring. Cd28−/− mice were tested for infection and killed at various time points (week 2, 3, 4, and 5) after infection. As many as 72% of Cd28−/− mice began to develop visible lesions three weeks post-infection (of 21 mice with at least three weeks of follow-up). The lesions began to regress in week 4 and disappeared during week 5. The spontaneous regression of the lesions observed in mice six weeks after infection was reminiscent of that seen in P2.
(B) Infected tissues from Cd28−/− mice were harvested at different time points for additional tests, including histology (H&E staining), immunohistochemistry (IHC) and in situ hybridization (ISH). Tail tissues from Cd28−/− mice displayed epidermal hyperplasia, with vacuolated cells and parakeratosis. Strong MmuPV1 DNA and RNA (E1Ê4) signals, equivalent to those observed in infected Rag1−/− mice, were detected in infected Cd28−/− mice by in situ hybridization three weeks after infection. Using antibodies against E4 and L1, we detected strong viral protein signals in the lesions. Viral signals (DNA/RNA/protein) were absent from the tail tissues of Cd28−/− and WT C57BL/6 mice displaying regression after week 5 post viral infection (not shown). Representative images of a wart and healthy skin from the same CD28-deficient animal are shown. The data shown are representative of the three experiments performed.
(C) RT-qPCR measuring MmuPV1 viral load in the skin lesions of CD28-deficient mice post-infection (2–5 weeks). Intralesional viral load was determined by RTqPCR for MmuPV1 E1Ê4 mRNA. The viral mRNA load was high as early as week 2 post-infection, despite the lesions not yet being visible. Viral load peaked three weeks after infection, began to decrease in week 4 and disappeared in week 5.
(D) ELISA measuring anti-MmuPV1 E4 and L1 antibodies in RAG1−/−, WT or CD28−/− mouse plasma sampled 3–6 weeks post-infection. Normal anti-L1 and E4 Ab levels were detected in Cd28−/− but not Rag1−/− mice. These data demonstrate that Cd28−/− mice can mount an anti-MmuPV1 T- and B cell response, but their control and clearance of infection are impaired, consistent with the clearance of the lesions of P2.
Figure S3. Impact of c.52G>A mutation on mRNA and prediction of signal peptide cleavage, related to Figure 1
(A) RNA was extracted from P1, a heterozygote and three controls and reverse-transcribed. Conventional RT-PCR was conducted to detect the full-length CD28 transcript. ACTB cDNA amplification was used as a positive control. Results representative of three independent experiments including the three patients and four heterozygotes. (B-C) Exon trapping.
(B) Schematic representation of the vector used for exon trapping. The blue part corresponds to the CD28 gDNA sequence inserted into the pCMV6 vector. The primers used for RT-PCR are depicted at the bottom. TSS: transcription start site.
(C) HEK293T cells were transfected with the pCMV6 exon trapping vector containing the wt or mutant (c.52G>A) gDNA sequence, as depicted in B. RNA was extracted, reverse-transcribed, and RT-PCR was performed to amplify CD28 splice variants for insertion into the pCR2.1 topo vector. We sequenced 211 colonies for the wild-type, 241 colonies for the c.52G>A mutant and 120 colonies for the c.52G>A plus c.52+5A>G reversion. The identified variants for splicing between exon 1 and 2, and their respective frequencies are shown.
(D) Signal peptide prediction results according to the SignalP 4.1 server (Petersen et al., 2011). Signal peptide cleavage is predicted between amino acids 18 and 19 for the wild-type sequence, and between amino acids 16 and 17 for the p.Gly18Arg mutant CD28 protein.
(E) FACS analysis of CD28 expression on Jurkat cells stably transduced with an empty vector (EV) or with a vector encoding the WT or p.Gly18Arg isoform.
Figure S4. RNA-seq from primary CD4+ naive T cells and immunophenotyping, related to Figures 2 and 3
(A) Primary CD4+ T cells from 4 healthy unrelated donors and P1 were stimulated by incubation for 2 hours with beads coated with antibodies against CD2, CD3, or CD28 alone or in combination (CD3 plus CD2 or CD3 plus CD28). They were then subjected to RNA-seq analysis. PMA stimulation was used as a positive control. Genes were selected on the basis of Student’s t tests comparing four non-stimulated and stimulated control samples for each of the conditions with a p value cutoff of 0.01, followed by filtering to select genes displaying at least two-fold up- (red) or downregulation (green) in at least three of the four controls. The number of genes passing the selection criteria for each condition were 17 for CD2, 13 for CD28, 267 for CD3, 1578 for CD3+CD2, 815 for CD3+CD28 and 2220 for PMA. Residual responses for the CD28–deficient patient are shown as a percentage in the red bar graphs for each condition.
(B) Frequencies of monocyte subsets, as assessed by measuring the expression of CD16 and CD14, for controls (n = 16), heterozygotes (n = 4) and CD28-deficient patients (n = 3).
(C) Frequencies of cDC1 (Lin−HLA-DR+CD11c+CD1c+CD141−), cDC2 (Lin−HLA-DR+CD11c+CD1c−CD141+) and pDCs (Lin−HLA-DR+CD11c−CD123+) among the PBMCs of controls (n = 24), one heterozygote and P1.
(D) Frequency of CD27+ memory cells within the B cell compartment of controls (n = 52), heterozygotes (n = 4) and patients (n = 3).
(E) Frequency of IgM+, IgA+ and IgG+ cells within the memory B cell compartment of controls (n = 31–52), heterozygotes (n = 4) and patients (n = 3).
(F-H) NK cell immunophenotyping for controls (n = 48–52), heterozygotes (n = 4) and patients (n = 3), showing the frequency of CD56bright cells within the NK-cell compartment (F), the terminal differentiation profile of the CD56dim compartment (G), and the frequency of NKG2C+NKG2A− cells within the CD56dim compartment (H).
(B-H) Kruskal-Wallis tests were used for all comparisons. The bars represent the median.
Figure S5. scRNA-seq on patients’ lymphocytes, related to Figure 3
(A) Same UMAP clustering representation as in Figure 4G, but for each patient and control sample separately; the number of cells analyzed for each sample is indicated.
(B) Volcano plot showing the results of a pseudobulk-type differential gene expression analysis, comparing normalized gene expression levels between patients and controls for major T lymphocyte clusters identified in Figure 4G. The genes with a fold change ≥ 2 and an adjusted p-value < 10−3 were considered significant and are annotated in red or blue if up- or downregulated, respectively, in the patients. Only a few genes were dysregulated, and CD28 was consistently the most strongly downregulated in all patient lymphocyte clusters.
Figure S6. Costimulation and coinhibition molecules induction on T cells, and in vitro T cell response to HCMV peptides and tuberculin, related to Figure 3
(A) Freshly isolated PBMCs from the three patients and two controls were stimulated with anti-CD2/CD3/CD28 mAb-coated beads or PMA and ionomycin for 72 hours, and the induction of the indicated coinhibitory or coactivating molecules was measured by flow cytometry on total CD4+ and CD8+ T cells. The bars represent the mean ± SD.
(B) Freshly thawed PBMCs from the three patients and four controls were stimulated for 16 hours with overlapping peptides of pp65 (HCMV) or tuberculin (PPD) or anti-CD2/CD3/CD28 mAb-coated beads, and intracellular IFN-γ production was measured by flow cytometry. CD28 staining shows that responses are confined to the CD28− subset in the patients; no response was detected in the CD4+CD28+ revertant T cell population. Only one representative control is depicted.
Figure S7. Detection of revertants by Sanger sequencing and scRNA-seq analysis, related to Figure 4
(A) Sanger sequencing chromatograms for the c.52G>A mutation in control gDNA, and in gDNA extracted from CD28− and CD28+ CD4+ memory T cells from P2 and P3 (upper line). A rare mutation of USP37 (c.1375G>A) was used to demonstrate that the CD28+CD4+ memory T cells sorted from P1 were not of maternal origin (bottom line).
(B) Splicing prediction (Alamut software) for the different mutant alleles (bottom) relative to the wild-type allele (top). The blue bars show splicing donor site prediction by different programs as a function of the input sequences. The c.52G>A mutation is predicted to disrupt the splicing donor site found in the wt allele. The second site reversion (c.52+5A>G), is predicted to restore normal splicing when simultaneously found with the c.52G>A mutation.
(C) UMAP clustering of 5′ scRNA-seq performed on CD4+ memory T cells enriched from P1 and P2 PBMCs, as well as for a healthy control. Sixteen cell clusters are identified based on cell marker expression analysis. Small residual clusters for CD8+ T, γδ T and NKT cells are identified, as well as Treg and activated CD4+ T cells (labeled in red); they were excluded from the CD28 revertant versus mutant differential gene expression analysis. Peripheral CD4+ memory cell clusters separated based on the TCR chain they expressed as annotated.
(D) UMAP (as in C) projecting the cells from the healthy control, P1 and P2 samples.
Highlights.
Inherited human CD28 deficiency underlies severe HPV-2 and HPV-4 skin lesions
Human CD28 is otherwise largely redundant in protective immunity to infection
Inherited CD28 deficiency only slightly impairs T cell development and function
HPV-2 giant horns overexpress HPV oncogenes in the basal layer of the epidermis
ACKNOWLEDGMENTS
We thank patients and families, members of the laboratory, Stephen Elledge, Genomics Core of Sidra Medicine, Primary Immunodeficiency Foundation of Sedigheh Tahereh, regional Tumor Bank of Franche-Comté (TRFC; BB-0033-00024), Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID and ANR-11-LABX-0021), French National Agency for Research (ANR-10-IAHU-01 and NKIR-ANR-13-PDOC-0025-01), ITMO Cancer of Aviesan and INCa within the framework of the 2021-2030 Cancer Control Strategy, Qatar National Research Fund (NPRP9-251-3-045), Sidra Medicine, American Association for the Study of Liver Diseases, National Health and Medical Research Council of Australia, Office of Health and Medical Research of the New South Wales State Government, National Center for Advancing Translational Sciences, National Institutes of Health Clinical and Translational Science Award program, National Institute of Dental and Craniofacial Research program, NIH, The Jake Gittlen Memorial Golf Tournament, The Gittlen Laboratories for Cancer Research, the Research Fund of Pathology Department and Cancer Institute of Penn State University College of Medicine, and JSPS KAKENHI (19H05289).
Footnotes
DECLARATION OF INTERESTS
L.D.N. receives compensation as Chief Editor of Frontiers in Immunology. T.W. serves on advisory boards for MSD (Merck Sharp and Dohme). J.-L.C. serves on the scientific advisory boards of ADMA Biologics Inc., Kymera Therapeutics, and Elixiron Immunotherapeutics. All other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.06.004.
REFERENCES
- Abecasis GR, Cherny SS, Cookson WO, and Cardon LR (2002). Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet 30, 97–101. [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alisjahbana B, Dinata R, Sutedja E, Suryahudaya I, Soedjana H, Hidajat NN, Soetikno RD, Oktaliansah E, Deng A, Rady P, et al. (2010). Disfiguring generalized verrucosis in an indonesian man with idiopathic CD4 lymphopenia. Arch. Dermatol 146, 69–73. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonsson A, Green AC, Mallitt KA, O’Rourke PK, Pandeya N, Pawlita M, Waterboer T, and Neale RE (2010). Prevalence and stability of antibodies to 37 human papillomavirus types–a population-based longitudinal study. Virology 407, 26–32. [DOI] [PubMed] [Google Scholar]
- Arias-Pulido H, Peyton CL, Joste NE, Vargas H, and Wheeler CM (2006). Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J. Clin. Microbiol 44, 1755–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruffo A, and Seed B (1987). Molecular cloning of a CD28 cDNA by a high-efficiency COS cell expression system. Proc. Natl. Acad. Sci. USA 84, 8573–8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44. [DOI] [PubMed] [Google Scholar]
- Belkadi A, Pedergnana V, Cobat A, Itan Y, Vincent QB, Abhyankar A, Shang L, El Baghdadi J, Bousfiha A, Alcais A, et al. ; Exome/Array Consortium (2016). Whole-exome sequencing to analyze population structure, parental inbreeding, and familial linkage. Proc. Natl. Acad. Sci. USA 113, 6713–6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béziat V (2020). Human genetic dissection of papillomavirus-driven diseases: new insight into their pathogenesis. Hum. Genet 139, 919–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béziat V, Descours B, Parizot C, Debré P, and Vieillard V (2010). NK cell terminal differentiation: correlated stepwise decrease of NKG2A and acquisition of KIRs. PLoS ONE 5, e11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béziat V, Liu LL, Malmberg JA, Ivarsson MA, Sohlberg E, Björklund AT, Retière C, Sverremark-Ekström E, Traherne J, Ljungman P, et al. (2013). NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 121, 2678–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatla N, Nene BM, Joshi S, Esmy PO, Poli URR, Joshi G, Verma Y, Zomawia E, Pimple S, Prabhu PR, et al. ; Indian HPV vaccine study group (2018). Are two doses of human papillomavirus vaccine sufficient for girls aged 15–18 years? Results from a cohort study in India. Papillomavirus Res. 5, 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt KH, Neller MA, Srihari S, Crooks P, Lekieffre L, Aftab BT, Liu H, Smith C, Kenny L, Porceddu S, and Khanna R (2020). Profiling HPV-16-specific T cell responses reveals broad antigen reactivities in oropharyngeal cancer patients. J. Exp. Med 217, e20200389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkström NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, Björklund AT, Flodström-Tullberg M, Michaëlsson J, Rottenberg ME, et al. (2010). Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood 116, 3853–3864. [DOI] [PubMed] [Google Scholar]
- Black APB, Ardern-Jones MR, Kasprowicz V, Bowness P, Jones L, Bailey AS, and Ogg GS (2007). Human keratinocyte induction of rapid effector function in antigen-specific memory CD4+ and CD8+ T cells. Eur. J. Immunol 37, 1485–1493. [DOI] [PubMed] [Google Scholar]
- Breitburd F, Salmon J, and Orth G (1997). The rabbit viral skin papillomas and carcinomas: a model for the immunogenetics of HPV-associated carcinogenesis. Clin. Dermatol 15, 237–247. [DOI] [PubMed] [Google Scholar]
- Casanova J-L, and Abel L (2018). Human genetics of infectious diseases: Unique insights into immunological redundancy. Semin. Immunol 36, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrani P, Kulkarni V, Iyer P, Upadhyay P, Chaubal R, Das P, Mulherkar R, Singh R, and Dutt A (2015). NGS-based approach to determine the presence of HPV and their sites of integration in human cancer genome. Br. J. Cancer 112, 1958–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cladel NM, Budgeon LR, Cooper TK, Balogh KK, Hu J, and Christensen ND (2013). Secondary infections, expanded tissue tropism, and evidence for malignant potential in immunocompromised mice infected with Mus musculus papillomavirus 1 DNA and virus. J. Virol 87, 9391–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cladel NM, Budgeon LR, Balogh KK, Cooper TK, Hu J, and Christensen ND (2015). A novel pre-clinical murine model to study the life cycle and progression of cervical and anal papillomavirus infections. PLoS ONE 10, e0120128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cladel NM, Budgeon LR, Balogh KK, Cooper TK, Hu J, and Christensen ND (2016). Mouse papillomavirus MmuPV1 infects oral mucosa and preferentially targets the base of the tongue. Virology 488, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cladel NM, Budgeon LR, Cooper TK, Balogh KK, Christensen ND, Myers R, Majerciak V, Gotte D, Zheng Z-M, and Hu J (2017). Mouse papillomavirus infections spread to cutaneous sites with progression to malignancy. J. Gen. Virol 98, 2520–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cladel NM, Peng X, Christensen N, and Hu J (2019a). The rabbit papillomavirus model: a valuable tool to study viral-host interactions. Philos. Trans. R. Soc. Lond. B Biol. Sci 374, 20180294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cladel NM, Jiang P, Li JJ, Peng X, Cooper TK, Majerciak V, Balogh KK, Meyer TJ, Brendle SA, Budgeon LR, et al. (2019b). Papillomavirus can be transmitted through the blood and produce infections in blood recipients: Evidence from two animal models. Emerg. Microbes Infect 8, 1108–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford GM, Shin H-R, Oh J-K, Waterboer T, Ju Y-H, Vaccarella S, Quint W, Pawlita M, and Franceschi S (2007). Serologic response to oncogenic human papillomavirus types in male and female university students in Busan, South Korea. Cancer Epidemiol. Biomarkers Prev. 16, 1874–1879. [DOI] [PubMed] [Google Scholar]
- Cubie HA (2013). Diseases associated with human papillomavirus infection. Virology 445, 21–34. [DOI] [PubMed] [Google Scholar]
- de Jong SJ, Imahorn E, Itin P, Uitto J, Orth G, Jouanguy E, Casanova J-L, and Burger B (2018a). Epidermodysplasia Verruciformis: Inborn Errors of Immunity to Human Beta-Papillomaviruses. Front. Microbiol 9, 1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong SJ, Créquer A, Matos I, Hum D, Gunasekharan V, Lorenzo L, Jabot-Hanin F, Imahorn E, Arias AA, Vahidnezhad H, et al. (2018b). The human CIB1-EVER1-EVER2 complex governs keratinocyte-intrinsic immunity to β-papillomaviruses. J. Exp. Med 215, 2289–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks SG, Overbaugh J, Phillips A, and Buchbinder S (2015). HIV infection. Nat. Rev. Dis. Primers 1, 15035. [DOI] [PubMed] [Google Scholar]
- Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, and Béroud C (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorbar J (2006). Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. (Lond.) 110, 525–541. [DOI] [PubMed] [Google Scholar]
- Doorbar J, Coneron I, and Gallimore PH (1989). Sequence divergence yet conserved physical characteristics among the E4 proteins of cutaneous human papillomaviruses. Virology 172, 51–62. [DOI] [PubMed] [Google Scholar]
- Doorbar J, Egawa N, Griffin H, Kranjec C, and Murakami I (2015). Human papillomavirus molecular biology and disease association. Rev. Med. Virol 25 (Suppl 1 ), 2–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drutman SB, Mansouri D, Mahdaviani SA, Neehus A-L, Hum D, Bryk R, Hernandez N, Belkaya S, Rapaport F, Bigio B, et al. (2020). Fatal Cytomegalovirus Infection in an Adult with Inherited NOS2 Deficiency. N. Engl. J. Med 382, 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esensten JH, Helou YA, Chopra G, Weiss A, and Bluestone JA (2016). CD28 costimulation: from mechanism to therapy. Immunity 44, 973–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring JS, Fischer B, Lawrence M, and Huber W (2015). SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics 31, 3673–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri I, Danos O, and Yaniv M (1985). Genomic structure of the cottontail rabbit (Shope) papillomavirus. Proc. Natl. Acad. Sci. USA 82, 1580–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, Ji X, Han A, Krams SM, Pettus C, et al. (2017). Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray E, Pett MR, Ward D, Winder DM, Stanley MA, Roberts I, Scarpini CG, and Coleman N (2010). In vitro progression of human papillomavirus 16 episome-associated cervical neoplasia displays fundamental similarities to integrant-associated carcinogenesis. Cancer Res. 70, 4081–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Han R, Breitburd F, Marche PN, and Orth G (1992). Linkage of regression and malignant conversion of rabbit viral papillomas to MHC class II genes. Nature 356, 66–68. [DOI] [PubMed] [Google Scholar]
- Handisurya A, Day PM, Thompson CD, Bonelli M, Lowy DR, and Schiller JT (2014). Strain-specific properties and T cells regulate the susceptibility to papilloma induction by Mus musculus papillomavirus 1. PLoS Pathog. 10, e1004314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Fu SM, and Hansen JA (1985). Human T cell activation. II. A new activation pathway used by a major T cell population via a disulfide-bonded dimer of a 44 kilodalton polypeptide (9.3 antigen). J. Exp. Med 161, 1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan MR, Rahman M, Khan T, Saeed A, Sundararaju S, Flores A, Hawken P, Rawat A, Elkum N, Hussain K, et al. (2021). Virome-wide serological profiling reveals association of herpesviruses with obesity. Sci. Rep 11, 2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Budgeon LR, Cladel NM, Balogh K, Myers R, Cooper TK, and Christensen ND (2015). Tracking vaginal, anal and oral infection in a mouse papillomavirus infection model. J. Gen. Virol 96, 3554–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Cladel NM, Budgeon LR, Balogh KK, and Christensen ND (2017). The Mouse Papillomavirus Infection Model. Viruses 9, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Summers MK, Pham V, Lill JR, Liu J, Lee G, Kirkpatrick DS, Jackson PK, Fang G, and Dixit VM (2011). Deubiquitinase USP37 is activated by CDK2 to antagonize APC(CDH1) and promote S phase entry. Mol. Cell 42, 511–523. [DOI] [PubMed] [Google Scholar]
- Ingle A, Ghim S, Joh J, Chepkoech I, Bennett Jenson A, and Sundberg JP (2011). Novel laboratory mouse papillomavirus (MusPV) infection. Vet. Pathol 48, 500–505. [DOI] [PubMed] [Google Scholar]
- Isnard P, Kula T, Avettand Fenoel V, Anglicheau D, Terzi F, Legendre C, Elledge SJ, and Canaud G (2019). Temporal virus serological profiling of kidney graft recipients using VirScan. Proc. Natl. Acad. Sci. USA 116, 10899–10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, Scott E, Shah I, Stenson PD, Gleeson J, et al. (2016). The mutation significance cutoff: gene-level thresholds for variant predictions. Nat. Methods 13, 109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonska S, Orth G, Obalek S, and Croissant O (1985). Cutaneous warts. Clinical, histologic, and virologic correlations. Clin. Dermatol 3, 71–82. [DOI] [PubMed] [Google Scholar]
- Kerner G, Rosain J, Guérin A, Al-Khabaz A, Oleaga-Quintas C, Rapaport F, Massaad MJ, Ding J-Y, Khan T, Ali FA, et al. (2020). Inherited human IFN-γ deficiency underlies mycobacterial disease. J. Clin. Invest 130, 3158–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T, Rahman M, Ali FA, Huang SSY, Ata M, Zhang Q, Bastard P, Liu Z, Jouanguy E, Béziat V, et al. (2021). Distinct antibody repertoires against endemic human coronaviruses in children and adults. JCI Insight 6, e144499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, and Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kündig TM, Shahinian A, Kawai K, Mittrücker H-W, Sebzda E, Bachmann MF, Mak TW, and Ohashi PS (1996). Duration of TCR stimulation determines costimulatory requirement of T cells. Immunity 5, 41–52. [DOI] [PubMed] [Google Scholar]
- Lei Y-J, Wang C, Gao C, Jiang H-Y, Chen J-M, Han J, Yuan Y-K, and Dong X-P (2007). HPV-2 isolates from patients with huge verrucae vulgaris possess stronger promoter activities. Intervirology 50, 353–360. [DOI] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, arXiv:1303.3997. [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur H, and Walter G (1984). Monoclonal antibodies specific for the carboxy terminus of simian virus 40 large T antigen. J. Virol. 52, 483–491. 10.1128/jvi.52.2.483-491.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantese SA de O, Diogo PM, Rocha A, Berbert ALCV, Ferreira AKM, and Ferreira TC (2010). Cutaneous horn: a retrospective histopathological study of 222 cases. An. Bras. Dermatol 85, 157–163. [DOI] [PubMed] [Google Scholar]
- Martins GA, Campanelli AP, Silva RB, Tadokoro CE, Russo M, Cunha FQ, Rizzo LV, and Silva JS (2004). CD28 is required for T cell activation and IFN-gamma production by CD4+ and CD8+ T cells in response to Trypanosoma cruzi infection. Microbes Infect. 6, 1133–1144. [DOI] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 8, 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael KM, Waterboer T, Sehr P, Rother A, Reidel U, Boeing H, Bravo IG, Schlehofer J, Gärtner BC, and Pawlita M (2008). Seroprevalence of 34 human papillomavirus types in the German general population. PLoS Pathog. 4, e1000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mina MJ, Kula T, Leng Y, Li M, de Vries RD, Knip M, Siljander H, Rewers M, Choy DF, Wilson MS, et al. (2019). Measles virus infection diminishes preexisting antibodies that offer protection from other pathogens. Science 366, 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittrücker H-W, Köhler A, Mak TW, and Kaufmann SHE (1999). Critical role of CD28 in protective immunity against Salmonella typhimurium. J. Immunol 163, 6769–6776. [PubMed] [Google Scholar]
- Mittrücker H-W, Kursar M, Köhler A, Hurwitz R, and Kaufmann SHE (2001). Role of CD28 for the generation and expansion of antigen-specific CD8(+) T lymphocytes during infection with Listeria monocytogenes. J. Immunol 167, 5620–5627. [DOI] [PubMed] [Google Scholar]
- Mohan D, Wansley DL, Sie BM, Noon MS, Baer AN, Laserson U, and Larman HB (2019). Publisher Correction: PhIP-Seq characterization of serum antibodies using oligonucleotide-encoded peptidomes. Nat. Protoc 14, 2596. [DOI] [PubMed] [Google Scholar]
- Ng PC, and Henikoff S (2001). Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notarangelo LD, Bacchetta R, Casanova J-L, and Su HC (2020). Human inborn errors of immunity: An expanding universe. Sci. Immunol 5, eabb1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, and Wigler M (2004). Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572. [DOI] [PubMed] [Google Scholar]
- Orlik C, Deibel D, Küblbeck J, Balta E, Ganskih S, Habicht J, Niesler B, Schröder-Braunstein J, Schäkel K, Wabnitz G, and Samstag Y (2020). Keratinocytes costimulate naive human T cells via CD2: a potential target to prevent the development of proinflammatory Th1 cells in the skin. Cell. Mol. Immunol 17, 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth G, Favre M, and Croissant O (1977). Characterization of a new type of human papillomavirus that causes skin warts. J. Virol 24, 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peh WL, Middleton K, Christensen N, Nicholls P, Egawa K, Sotlar K, Brandsma J, Percival A, Lewis J, Liu WJ, and Doorbar J (2002). Life cycle heterogeneity in animal models of human papillomavirus-associated disease. J. Virol 76, 10401–10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, and Nielsen H (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. [DOI] [PubMed] [Google Scholar]
- Pillay BA, Fusaro M, Gray PE, Statham AL, Burnett L, Bezrodnik L, Kane A, Tong WWY, Abdo C, Winter S, et al. (2021). Somatic reversion of pathogenic DOCK8 variants alters lymphocyte differentiation and function to effectively cure DOCK8 deficiency. J. Clin. Invest 131, 142434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramoz N, Rueda L-A, Bouadjar B, Montoya L-S, Orth G, and Favre M (2002). Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat. Genet 32, 579–581. [DOI] [PubMed] [Google Scholar]
- Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, Gregson BP, June CH, and Linsley PS (2002). Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc. Natl. Acad. Sci. USA 99, 11790–11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rous P, and Beard JW (1934). A Virus-Induced Mammalian Growth with the Characters of a Tumor (the Shope Rabbit Papilloma): I. the Growth on Implantation Within Favorable Hosts. J. Exp. Med 60, 701–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd CE, Taylor A, and Schneider H (2009). CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev 229, 12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, and Bluestone JA (2000). B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12, 431–440. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan R, Prabhu PR, Pawlita M, Gheit T, Bhatla N, Muwonge R, Nene BM, Esmy PO, Joshi S, Poli URR, et al. ; Indian HPV Vaccine Study Group (2016). Immunogenicity and HPV infection after one, two, and three doses of quadrivalent HPV vaccine in girls in India: a multicentre prospective cohort study. Lancet Oncol. 17, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman M, Doorbar J, Wentzensen N, de Sanjosé S, Fakhry C, Monk BJ, Stanley MA, and Franceschi S (2016). Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2, 16086. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M, Bravo IG, Snijders PJF, Gissmann L, Pawlita M, and Waterboer T (2006). Bead-based multiplex genotyping of human papillomaviruses. J. Clin. Microbiol 44, 504–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober T, Magg T, Laschinger M, Rohlfs M, Linhares ND, Puchalka J, Weisser T, Fehlner K, Mautner J, Walz C, et al. (2017). A human immunodeficiency syndrome caused by mutations in CARMIL2. Nat. Commun 8, 14209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehr P, Zumbach K, and Pawlita M (2001). A generic capture ELISA for recombinant proteins fused to glutathione S-transferase: validation for HPV serology. J. Immunol. Methods 253, 153–162. [DOI] [PubMed] [Google Scholar]
- Semple K, Nguyen A, Yu Y, Wang H, Anasetti C, and Yu X-Z (2011). Strong CD28 costimulation suppresses induction of regulatory T cells from naive precursors through Lck signaling. Blood 117, 3096–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahinian A, Pfeffer K, Lee KP, Kündig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, and Mak TW (1993). Differential T cell costimulatory requirements in CD28-deficient mice. Science 261, 609–612. [DOI] [PubMed] [Google Scholar]
- Shope RE, and Hurst EW (1933). Infectious Papillomatosis of Rabbits: With a Note on the Histopathology. J. Exp. Med 58, 607–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smelov V, Muwonge R, Sokolova O, McKay-Chopin S, Eklund C, Komyakov B, and Gheit T (2018). Beta and gamma human papillomaviruses in anal and genital sites among men: prevalence and determinants. Sci. Rep 8, 8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. (2003). Lentivirusdelivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickley JD, Messerschmidt JL, Awad ME, Li T, Hasegawa T, Ha DT, Nabeta HW, Bevins PA, Ngo KH, Asgari MM, et al. (2019). Immunity to commensal papillomaviruses protects against skin cancer. Nature 575, 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai X, Cowan M, Feigenbaum L, and Singer A (2005). CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat. Immunol 6, 152–162. [DOI] [PubMed] [Google Scholar]
- Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, Zheng XX, Strom TB, and Bluestone JA (2003). Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J. Immunol 171, 3348–3352. [DOI] [PubMed] [Google Scholar]
- Tanno H, Shigematsu T, Nishikawa S, Hayakawa A, Denda K, Tanaka T, and Komada M (2014). Ubiquitin-interacting motifs confer full catalytic activity, but not ubiquitin chain substrate specificity, to deubiquitinating enzyme USP37. J. Biol. Chem 289, 2415–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas D, Luijsterburg MS, Wiegant WW, Diakatou M, Helfricht A, Thijssen PE, van den Broek B, Mullenders LH, and van Attikum H (2015). The de-ubiquitylating enzymes USP26 and USP37 regulate homologous recombination by counteracting RAP80. Nucleic Acids Res. 43, 6919–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin KMF, Amin R, Majumder SN, Aleem MA, Rahaman A, Dity NJ, Baqui MDA, Akter H, Rahman MM, Woodbury-Smith M, et al. (2018). An ANKRD26 nonsense somatic mutation in a female with epidermodysplasia verruciformis (Tree Man Syndrome). Clin. Case Rep. 6, 1426–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Doorslaer K, Tan Q, Xirasagar S, Bandaru S, Gopalan V, Mohamoud Y, Huyen Y, and McBride AA (2013). The Papillomavirus Episteme: a central resource for papillomavirus sequence data and analysis. Nucleic Acids Res. 41, D571–D578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinokurova S, Wentzensen N, Kraus I, Klaes R, Driesch C, Melsheimer P, Kisseljov F, Dürst M, Schneider A, and von Knebel Doeberitz M (2008). Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res. 68, 307–313. [DOI] [PubMed] [Google Scholar]
- Wang C, Wang W, Lei YJ, Wang JY, Dong XP, Wang J, Sheng RH, Pan ZA, Zhu WY, You LP, et al. (2007a). Multiple huge cutaneous horns overlying verrucae vulgaris induced by human papillomavirus type 2: a case report. Br. J. Dermatol 156, 760–762. [DOI] [PubMed] [Google Scholar]
- Wang W, Wang C, Xu S, Chen C, Tong X, Liang Y, Dong X, Lei Y, Zheng X, Yu J, and Wang J (2007b). Detection of HPV-2 and identification of novel mutations by whole genome sequencing from biopsies of two patients with multiple cutaneous horns. J. Clin. Virol 39, 34–42. [DOI] [PubMed] [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Wang JW, Jiang R, Peng S, Chang Y-N, Hung C-F, and Roden RBS (2015). Immunologic Control of Mus musculus Papillomavirus Type 1. PLoS Pathog. 11, e1005243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ma CS, Ling Y, Bousfiha A, Camcioglu Y, Jacquot S, Payne K, Crestani E, Roncagalli R, Belkadi A, et al. (2016). Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J. Exp. Med 213, 2413–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterboer T, Sehr P, Michael KM, Franceschi S, Nieland JD, Joos TO, Templin MF, and Pawlita M (2005). Multiplex human papillomavirus serology based on in situ-purified glutathione s-transferase fusion proteins. Clin. Chem 51, 1845–1853. [DOI] [PubMed] [Google Scholar]
- Waterboer T, Neale R, Michael KM, Sehr P, de Koning MNC, Weißenborn SJ, Sampogna F, Abeni D, Green AC, Bouwes Bavinck JN, and Pawlita M; The Epi-Hpv-Uv-Ca Group (2009). Antibody responses to 26 skin human papillomavirus types in the Netherlands, Italy and Australia. J. Gen. Virol 90, 1986–1998. [DOI] [PubMed] [Google Scholar]
- Xu GJ, Kula T, Xu Q, Li MZ, Vernon SD, Ndung’u T, Ruxrungtham K, Sanchez J, Brander C, Chung RT, et al. (2015). Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science 348, aaa0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RCH, Pryce DW, Macfarlane AW, and Stewart TW (1991). A histopathological study of 643 cutaneous horns. Br. J. Dermatol 124, 449–452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Familial genetics, population genetics of KANSL1L and USP37, and functional assay for the pGlu459Lys USP37 allele, related to Figure 1
(A) Homozygosity rates for P1 to P3 relative to consanguineous (open circles) and nonconsanguineous (closed circles) ethnically matched controls. The red line corresponds to the 90% percentile.
(B) An analysis of whole-exome sequencing (WES) data identified three genes as the only genes carrying a homozygous coding mutation common to the three patients and within the linked region.
(C) Rare homozygous variants within the linked region. This region includes CD28 (Gly18Arg), USP37 (p.Glu459Lys) and KANSL1L (p.Ser577Thr). USP37 and KANSL1L have no known role in immunity. USP37 expression is ubiquitous, and involved in the cell cycle and homologous recombination (Huang et al., 2011; Tanno et al., 2014; Typas et al., 2015). The mutation in USP37 results in a glutamic acid-to-lysine exchange at position 459. Glutamic acid is negatively charged, whereas lysine is positively charged. By contrast, KANSL1L mRNA is mostly restricted to the parathyroid gland (The Human Protein Atlas) and has no known function. The mutation in KANSL1L results in a Serine to Threonine exchange, two polar uncharged amino acids (p.Ser577Thr).
(D) HEK293T cells were transfected with an empty pCMV6 plasmid or with pCMV6 plasmids encoding the wild-type (WT) or p.Ser577Thr (c.1730G>C) KANSL1L transcripts (left), or encoding the WT or p.Glu459Lys (c.1375G>A) USP37 transcripts. All constructs contained a C-terminal DDK tag. Total protein extracts were subjected to immunoblotting with a monoclonal antibody against the DDK tag. GAPDH was used as a loading control. The two missense proteins are normally expressed following the transfection of HEK293T cells with the corresponding cDNAs. A western blot representative of three independent experiments is shown. Molecular weights are shown on the left side of the blots.
(E) Frequency and combined annotation–dependent depletion score (CADD), for all homozygous KANSL1L (left) and USP37 (right) variants reported in public databases. The dotted line corresponds to the mutation significance cutoff (MSC). NR: not reported. The mutations of the patients appear in red. The p.Ser577Thr KANSL1L variant was identified four times, in the homozygous state, in the ATAVDB database and its minor allele frequency is as high as 0.004 in South Asians (gnomAD). By contrast, the USP37 variant in our patients was private.
(F) Amino-acid conservation, across species, for the mutated area of the KANSL1L and USP37 proteins. KANSL1L orthologs are found in other taxonomic groups, as far back in evolution as insects (Drosophila melanogaster), but serine 577 is conserved only in mammals, a threonine residue being present in the corresponding position in Drosophila. Together, population genetics, evolutionary genetics, and biochemical analysis suggest that this KANSL1L variant is neutral. USP37 orthologs are found across vertebrates, but the glutamic acid in position 459 is conserved only in mammals. In birds and amphibians, it is replaced by an aspartic acid, an amino acid with a similar negative charge. (G-H) Population genetics, evolutionary genetics, and biochemical analysis suggested that the USP37 variant might be deleterious. We therefore assessed the functional impact of the p.Glu459Lys substitution on USP37 deubiquitination (Huang et al., 2011). We immunoprecipitated and purified WT or p.Glu459Lys mutant USP37 overproduced in HEK293T cells and performed an in vitro deubiquitination assay with K48-linked ubiquitin chains (2–7-mer) as a substrate, followed by SDS-PAGE and silver staining.
(G) Amounts of immunopurified USP37 (WT, MUT) protein used in H. IB, immunoblotting.
(H) In vitro deubiquitination of Lys48-linked polyubiquitin chains (2–7-mer) by immunopurified USP37 (WT, MUT). The p.Glu459Lys mutation did not affect deubiquitination activity. These results suggest that the USP37 variant is unlikely to contribute to the severe verrucosis phenotype observed in the patients studied. The sample in the first lane contained polyubiquitin chains but not USP37. Ub, ubiquitin. Representative results from three independent experiments are shown.
Figure S2. CD28-deficient mice are susceptible to MmuPV1 skin infection, related to Figure 1
In the absence of CD28-deficient rabbits, it was not possible to use the CRPV model, which would have been physiologically relevant (Cladel et al., 2019a). Instead, we used the MmuPV1 infection model in congenic wild-type (WT) and Cd28−/− C57BL/6 mice (Ingle et al., 2011). The MmuPV1 virus causes skin wart lesions in athymic nude or RAG1-deficient C57BL/6 mice but not in C57BL/6 WT mice (Cladel et al., 2013; Wang et al., 2015; Handisurya et al., 2014). MmuPV1 does not cause skin lesions in mice with several other types of immunodeficiency, including C57BL/6 mice, after CD4+ or CD8+ T cell depletion (Handisurya et al., 2014; Wang et al., 2015), or in C57BL/6 mice homozygous for null mutations of Ifnar1, Cd4, Cd40lg (Hu et al., 2017; Wang et al., 2015), Ifng, Sting, or Ccr10 (unpublished data).
(A) Representative images of tail and muzzle lesions (red circles) from RAG1-deficient (middle) and CD28-deficient mice (right) 5 weeks post-infection with MmuPV1 are shown. Images of a wild-type mouse (left) are also shown for comparison. We infected the muzzle and tail of 23 (15 male and 8 female) Cd28−/− mice, six Rag1−/− mice, and 27 WT mice with MmuPV1. Rag-1−/− mice have no T or B cells. We monitored skin lesions between two and seven weeks before the animals were killed. None of the WT C57BL/6 mice developed skin lesions, consistent with the findings of previous studies (Hu et al., 2017). All six Rag1−/− mice developed skin warts three weeks post infection. These lesions persisted until the end of monitoring. Cd28−/− mice were tested for infection and killed at various time points (week 2, 3, 4, and 5) after infection. As many as 72% of Cd28−/− mice began to develop visible lesions three weeks post-infection (of 21 mice with at least three weeks of follow-up). The lesions began to regress in week 4 and disappeared during week 5. The spontaneous regression of the lesions observed in mice six weeks after infection was reminiscent of that seen in P2.
(B) Infected tissues from Cd28−/− mice were harvested at different time points for additional tests, including histology (H&E staining), immunohistochemistry (IHC) and in situ hybridization (ISH). Tail tissues from Cd28−/− mice displayed epidermal hyperplasia, with vacuolated cells and parakeratosis. Strong MmuPV1 DNA and RNA (E1Ê4) signals, equivalent to those observed in infected Rag1−/− mice, were detected in infected Cd28−/− mice by in situ hybridization three weeks after infection. Using antibodies against E4 and L1, we detected strong viral protein signals in the lesions. Viral signals (DNA/RNA/protein) were absent from the tail tissues of Cd28−/− and WT C57BL/6 mice displaying regression after week 5 post viral infection (not shown). Representative images of a wart and healthy skin from the same CD28-deficient animal are shown. The data shown are representative of the three experiments performed.
(C) RT-qPCR measuring MmuPV1 viral load in the skin lesions of CD28-deficient mice post-infection (2–5 weeks). Intralesional viral load was determined by RTqPCR for MmuPV1 E1Ê4 mRNA. The viral mRNA load was high as early as week 2 post-infection, despite the lesions not yet being visible. Viral load peaked three weeks after infection, began to decrease in week 4 and disappeared in week 5.
(D) ELISA measuring anti-MmuPV1 E4 and L1 antibodies in RAG1−/−, WT or CD28−/− mouse plasma sampled 3–6 weeks post-infection. Normal anti-L1 and E4 Ab levels were detected in Cd28−/− but not Rag1−/− mice. These data demonstrate that Cd28−/− mice can mount an anti-MmuPV1 T- and B cell response, but their control and clearance of infection are impaired, consistent with the clearance of the lesions of P2.
Figure S3. Impact of c.52G>A mutation on mRNA and prediction of signal peptide cleavage, related to Figure 1
(A) RNA was extracted from P1, a heterozygote and three controls and reverse-transcribed. Conventional RT-PCR was conducted to detect the full-length CD28 transcript. ACTB cDNA amplification was used as a positive control. Results representative of three independent experiments including the three patients and four heterozygotes. (B-C) Exon trapping.
(B) Schematic representation of the vector used for exon trapping. The blue part corresponds to the CD28 gDNA sequence inserted into the pCMV6 vector. The primers used for RT-PCR are depicted at the bottom. TSS: transcription start site.
(C) HEK293T cells were transfected with the pCMV6 exon trapping vector containing the wt or mutant (c.52G>A) gDNA sequence, as depicted in B. RNA was extracted, reverse-transcribed, and RT-PCR was performed to amplify CD28 splice variants for insertion into the pCR2.1 topo vector. We sequenced 211 colonies for the wild-type, 241 colonies for the c.52G>A mutant and 120 colonies for the c.52G>A plus c.52+5A>G reversion. The identified variants for splicing between exon 1 and 2, and their respective frequencies are shown.
(D) Signal peptide prediction results according to the SignalP 4.1 server (Petersen et al., 2011). Signal peptide cleavage is predicted between amino acids 18 and 19 for the wild-type sequence, and between amino acids 16 and 17 for the p.Gly18Arg mutant CD28 protein.
(E) FACS analysis of CD28 expression on Jurkat cells stably transduced with an empty vector (EV) or with a vector encoding the WT or p.Gly18Arg isoform.
Figure S4. RNA-seq from primary CD4+ naive T cells and immunophenotyping, related to Figures 2 and 3
(A) Primary CD4+ T cells from 4 healthy unrelated donors and P1 were stimulated by incubation for 2 hours with beads coated with antibodies against CD2, CD3, or CD28 alone or in combination (CD3 plus CD2 or CD3 plus CD28). They were then subjected to RNA-seq analysis. PMA stimulation was used as a positive control. Genes were selected on the basis of Student’s t tests comparing four non-stimulated and stimulated control samples for each of the conditions with a p value cutoff of 0.01, followed by filtering to select genes displaying at least two-fold up- (red) or downregulation (green) in at least three of the four controls. The number of genes passing the selection criteria for each condition were 17 for CD2, 13 for CD28, 267 for CD3, 1578 for CD3+CD2, 815 for CD3+CD28 and 2220 for PMA. Residual responses for the CD28–deficient patient are shown as a percentage in the red bar graphs for each condition.
(B) Frequencies of monocyte subsets, as assessed by measuring the expression of CD16 and CD14, for controls (n = 16), heterozygotes (n = 4) and CD28-deficient patients (n = 3).
(C) Frequencies of cDC1 (Lin−HLA-DR+CD11c+CD1c+CD141−), cDC2 (Lin−HLA-DR+CD11c+CD1c−CD141+) and pDCs (Lin−HLA-DR+CD11c−CD123+) among the PBMCs of controls (n = 24), one heterozygote and P1.
(D) Frequency of CD27+ memory cells within the B cell compartment of controls (n = 52), heterozygotes (n = 4) and patients (n = 3).
(E) Frequency of IgM+, IgA+ and IgG+ cells within the memory B cell compartment of controls (n = 31–52), heterozygotes (n = 4) and patients (n = 3).
(F-H) NK cell immunophenotyping for controls (n = 48–52), heterozygotes (n = 4) and patients (n = 3), showing the frequency of CD56bright cells within the NK-cell compartment (F), the terminal differentiation profile of the CD56dim compartment (G), and the frequency of NKG2C+NKG2A− cells within the CD56dim compartment (H).
(B-H) Kruskal-Wallis tests were used for all comparisons. The bars represent the median.
Figure S5. scRNA-seq on patients’ lymphocytes, related to Figure 3
(A) Same UMAP clustering representation as in Figure 4G, but for each patient and control sample separately; the number of cells analyzed for each sample is indicated.
(B) Volcano plot showing the results of a pseudobulk-type differential gene expression analysis, comparing normalized gene expression levels between patients and controls for major T lymphocyte clusters identified in Figure 4G. The genes with a fold change ≥ 2 and an adjusted p-value < 10−3 were considered significant and are annotated in red or blue if up- or downregulated, respectively, in the patients. Only a few genes were dysregulated, and CD28 was consistently the most strongly downregulated in all patient lymphocyte clusters.
Figure S6. Costimulation and coinhibition molecules induction on T cells, and in vitro T cell response to HCMV peptides and tuberculin, related to Figure 3
(A) Freshly isolated PBMCs from the three patients and two controls were stimulated with anti-CD2/CD3/CD28 mAb-coated beads or PMA and ionomycin for 72 hours, and the induction of the indicated coinhibitory or coactivating molecules was measured by flow cytometry on total CD4+ and CD8+ T cells. The bars represent the mean ± SD.
(B) Freshly thawed PBMCs from the three patients and four controls were stimulated for 16 hours with overlapping peptides of pp65 (HCMV) or tuberculin (PPD) or anti-CD2/CD3/CD28 mAb-coated beads, and intracellular IFN-γ production was measured by flow cytometry. CD28 staining shows that responses are confined to the CD28− subset in the patients; no response was detected in the CD4+CD28+ revertant T cell population. Only one representative control is depicted.
Figure S7. Detection of revertants by Sanger sequencing and scRNA-seq analysis, related to Figure 4
(A) Sanger sequencing chromatograms for the c.52G>A mutation in control gDNA, and in gDNA extracted from CD28− and CD28+ CD4+ memory T cells from P2 and P3 (upper line). A rare mutation of USP37 (c.1375G>A) was used to demonstrate that the CD28+CD4+ memory T cells sorted from P1 were not of maternal origin (bottom line).
(B) Splicing prediction (Alamut software) for the different mutant alleles (bottom) relative to the wild-type allele (top). The blue bars show splicing donor site prediction by different programs as a function of the input sequences. The c.52G>A mutation is predicted to disrupt the splicing donor site found in the wt allele. The second site reversion (c.52+5A>G), is predicted to restore normal splicing when simultaneously found with the c.52G>A mutation.
(C) UMAP clustering of 5′ scRNA-seq performed on CD4+ memory T cells enriched from P1 and P2 PBMCs, as well as for a healthy control. Sixteen cell clusters are identified based on cell marker expression analysis. Small residual clusters for CD8+ T, γδ T and NKT cells are identified, as well as Treg and activated CD4+ T cells (labeled in red); they were excluded from the CD28 revertant versus mutant differential gene expression analysis. Peripheral CD4+ memory cell clusters separated based on the TCR chain they expressed as annotated.
(D) UMAP (as in C) projecting the cells from the healthy control, P1 and P2 samples.
Data Availability Statement
The whole-exome sequencing and whole genome sequencing datasets generated during this study are available at the Sequence Read Archive (SRA: PRJNA715377). The scRNA-seq and CITE-seq datasets generated during this study are available at the EGA European Genome-Phenome Archive (EGA: EGAS00001004837). High-throughput sequencing (HTS) of T cell receptor β (TRB) and T cell receptor ⍺ (TRA) dataset are available from Adaptive Biotechnologies (http://clients.adaptivebiotech.com/login; Email: beziat-review@adaptivebiotech.com; Password: beziat2021review). Primary CD4+ naive T cell RNA-Seq datasets generated during this study are available at the gene expression omnibus: GEO: GSE139299. Lesions RNA-Seq datasets generated during this study are available at the GEO: GSE139259. The assembled genomes are available from GenBank under the accession numbers GenBank: MN605988 and MN605989 for HPV-2 (from P1) and HPV-4 (from P2 and P3), respectively. This study did not generate any unique code. Any other piece of data will be available upon reasonable request.