

Graphic abstract
Kinetics between 5,10,15,20-tetrakis(N-methylpyridium-4-yl)porphyrin and Ni2+ species were investigated in aqueous solution at 25 ±1 °C in I = 0.10 M (NaNO3). Speciation of Ni2+ was done in I = 0.10 M (NaNO3) for knowing distribution of Ni2+ species with solution pH. Experimental data were compared with speciation diagram constructed from the values of hydrolysis constants of Ni2+ ion. Speciation data showed that hexaaquanickel(II) ions took place in hydrolysis reactions through formation of [Ni(OH2)6-n(OH)n]2-n species with solution pH. According to speciation of Ni2+ and pH dependent rate constants, rate expression can be written as: d[Ni(TMPyP)4+]/dt = (k1[Ni2+(aq)] + k2[Ni(OH)+(aq)] + k3[Ni(OH)2o(aq)] + k4[Ni(OH)3-(aq)])[H2TMPyP4+], where k1, k2, k3 and k4 were found to be k1 = (0.62 ± 0.22) × 10-2; k2 = (3.60 ± 0.40) × 10-2; k3 = (2.09 ± 0.52) × 10-2, k4 = (0.53 ± 0.04) × 10-2 M-1s-1 at 25 ±1 °C, respectively. Formation of hydrogen bonding between [Ni(H2O)5(OH)]+ and [H2TMPyP]4+ causes enhanced reactivity. Rate of formation of [Ni(II)TMPyP]4+ complex was to be 3.99 × 10-2 M-1s-1 in I = 0.10 M, NaNO3 (25 ± 1 °C). UV-Vis and fluorescence data suggested that [Ni(II)TMPyP]4+ and [H2(TMPyP)]4+ interact with DNA via outside binding with self-stacking and intercalation, respectively.
SYNOPSIS

Kinetics of tetrakis(1-methylpyridium-4-yl)porphyrin with Ni2+ were studied in aqueous medium at 25 ±1 °C in I = 0.10 M (NaNO3). Speciation data revealed the stepwise formation of [Ni(H2O)6-n(OH)n]2-n species with solution pH. Kinetic results showed that [Ni(H2O)5(OH)]+ is the most reactive species because of the formation of hydrogen bonding. UV-Vis/fluorescence data suggested that [Ni(II)TMPyP]4+ and [H2TMPyP]4+ interact with DNA via outside binding and intercalation, respectively.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12039-021-01945-y.
Keywords: Speciation of Ni2+; kinetics and mechanism; hydrogen bonding; 5,10,15,20-tetrakis(N-methylpyridium-4-yl)porphyrinatonickel(II) tetracation; outside binding; chemotherapeutic agents
Introduction
Substantial studies by many research groups have been carried out on kinetics and mechanism of formation of metalloporphyrins because of their possible applications as therapeutic agents in medical and biological fields.1-19 The structural similarity of the porphyrins with chlorophylls, green pigments of leaves, has also been attracted by researchers for their potential use in an artificial photosynthetic system.20 In the human body system, the protoporphyrin IX ring is continuously synthesized during biosynthesis of heme, and iron(II) is subsequently coordinated to the porphyrin core. Studies of the kinetics of incorporation of metal ions into the porphyrins’ core provide the mechanistic pathways of the formation of metalloporphyrins. By knowing proper reaction pathways of the formation of metalloporphyrins, it may possible to formulate porphyrin-based new drugs. Hambright and Chock (1974) proposed a general mechanism of formation of metalloporphyrins for the first time, and later that was reviewed by a number of research groups from different kinetic standpoints.21-34 Among the standpoints, the presence of the hydroxo group of the central metal ion enhances the reactivity of macrocyclic porphyrins through the formation of hydrogen bonding between the oxygen atom of the hydroxo-ligand and the pyrrolic hydrogen atom of the free base porphyrin.23,27,34
Nickel is an essential element for humans as well as for other animals in functioning many metabolic reactions. The known multifunctional properties of the porphyrins and metalloporphyrins have been extended the porphyrins’ research in various fields. So, the study of kinetics and mechanism of the formation of Ni(II)porphyrin may open a new research arena of applications of nickel-porphyrin complexes. Nickel is a transition metal having d8 electronic configuration, thus exhibits the least reactivity in complex formation. However, in our previous study, we found enhanced reactivity of Au3+ ion in the formation of complexes with the macrocyclic tetrakis(N-methylpyridinium-4-yl)porphyrin, [H2TMPyP]4+, where Au3+ ion belongs to the d8 electronic configuration.27 According to the speciation diagram of Au3+ ion with solution pH, the monohydroxotrichloroaurate(III), [AuCl3(OH)]-, was found as a predominant species under the experimental condition.27 The negatively charged [AuCl3(OH)]- ion can easily approach the core of the tetracationic porphyrin and the presence of the hydroxo-ligand in the Au3+ species causes enhanced reactivity in the formation of the [Au(III)TMPyP]5+ complex. This is because the hydroxo-ligand of the [AuCl3(OH)]- species forms hydrogen bonding with the pyrrolic hydrogen atom of the porphyrin, which resulted in an enhanced rate of the reaction. Thus, it is suspected that the monohydroxonickel(II), [Ni(H2O)5(OH)]+, species may also exhibit enhance reactivity with the free-base porphyrin, [H2TMPyP]4+. In kinetic studies, the speciation of central metal ion plays a vital role to investigate the reactivity and mechanism of the reactions. Though some attempts have already been paid to study the kinetics of formation of Ni(II)porphyrins,35-38 speciation of Ni(II) ion from the geo- and hydrothermal points of view is available.39,40
Much attention has been paid to explore the interaction between the cationic porphyrins and nucleic acids because of the promising properties of the porphyrins in medical and biological applications.2,3,5-7,9-17,41-51 The potential uses of the porphyrins in medical and biological fields are due to their anticancer,11,13,17 antiviral and/or antibacterial/anti-inflammatory4,6,10,14-16 and antifungal6,52 activities. Porphyrins are also being used as imaging agents in medical imaging systems.6,53-55 Cationic porphyrins interact with DNA in various modes. Three major modes of interaction between porphyrins and DNA are intercalation, outside binding without self-stacking, and outside binding with self-stacking along the DNA surface.41-46 Partial intercalation has also been suggested.45,46 It is noted that potential applications of porphyrins in medical and biological systems depend mainly on the modes of interaction of the porphyrin-DNA adducts.
Very recently Liu and Li (2020) studied the severe health effect of the novel coronavirus (COVID-19) worldwide by applying theoretical models.56,57 They used conserved domain analysis, homology modeling and molecular docking models to compare the biological roles of specific proteins of the COVID-19, and found the novel coronavirus attacks the 1-beta chain of the haemoglobin and captures the protoporphyrin IX to inhibit human heme metabolism. The theoretical results suggest that the coronavirus has a strong affinity for porphyrins. The noble but clinically relevant finding encouraged us to investigate possible applications of the porphyrins as anti-COVID-19 agents.
In this paper, speciation of Ni2+ in an aqueous medium with different solution pH in I = 0.10 M (NaNO3) and 0.10 M NaCl at 25 ±1 °C has been characterized. By applying the distribution of the Ni2+ species with solution pH, the kinetics of the formation of [Ni(II)TMPyP]4+ complex, I (Scheme 1) has been studied to explore the reaction mechanism of the metalation reaction. We have extended our studies on the interaction of DNA with the [Ni(II)TMPyP]4+ complex along the H2TMPyP4+ to investigate their potential applications in medical and biological fields. An investigation of the application of porphyrins, particularly Ni2+-, Zn2+-, Ru2+-, Pt2+-, [Au(III)TMPyP]5+ as anti-COVID-19 agents is now in progress under an international research collaboration.
Scheme 1.

Tetracationic nickel(II)porphyrin, I.
Experimental
Reagents and materials
The tetracationic free-base porphyrin, 5,10,15,20-tetrakis(N-methylpyridinium-4-yl)porphyrin, [H2TMPyP]4+, was purchased as a tosylate from Dojindo Chemical Institute, Kumamoto, Japan. A 50 mL porphyrin solution was prepared by dissolving 68.18 mg of the free base porphyrin, [H2TMPyP]4+, in distilled water. A standard Cu2+ solution was used to standardize the porphyrin solution by using spectrophotometric titration (molar ratio method).27,46,47 Nickel solution was prepared by dissolving the requisite amount of NiCl2.6H2O (Merck, Germany) in an aqueous solution and the concentration was measured by using an atomic absorption spectrophotometer (Perkin Elmer, AAanalyst 200). Sodium nitrate, sodium hydroxide and hydrochloric acid were purchased from Merck, Germany. All the chemicals/reagents were used without further purification. Tetracation nickel(II) porphyrin, [Ni(II)TMPyP]4+, was prepared and absorption spectra were recorded in water at pH 9.50 containing 0.10 M NaNO3. Absorption maximum (λmax) and molar extinction coefficient (ε) of the prepared [Ni(II)TMPyP]4+ complex were 436 nm and 114 × 103 M-1cm-1, respectively (Figure 1).58
Figure 1.

Absorption spectra of [Ni(II)TMPyP]4+ complex with time at solution pH = 9.50 (25 °C) in I = 0.10 M (NaNO3). [Ni2+] = 1.00 × 10-3 M; [H2TMPyP]4+ = 1.24 × 10-5 M. Progress of the formation of the [Ni(II)TMPyP]4+ complex was monitored by changing the absorbance of [H2TMPyP]4+ at λmax = 422 nm with time. The formation time are as follows: (1): 0; (2): 5; (3) 10; (4): 20; (5): 30; (6): 50; (7): 70; (8): 100; (9): 140; (10): 200; (11): 260 min, respectively.
A stock solution of salmon fish sperm DNA, purchased from Sigma-Aldrich, was prepared by dissolving in distilled water and the concentration in base pairs was determined by knowing the absorbance at λmax = 260 nm and using the molar extinction coefficient, ε260 =1.32 × 104 M-1cm-1.45,46 Stock solution of the DNA was kept in a refrigerator at -4 °C. The frozen DNA solution was incubated in a water bath at 37 °C for an hour and diluted as required before the experiment. Acetate/sodium acetate and 2-[4-(2-hydroxyethyl)-1-piperazinyl]ethanesulfonic acid (HEPES, Sigma-Aldrich) buffer solution was prepared in 100 mL distilled water as stock solutions and used with required dilution throughout the experiments. pH of the HEPES solution (0.10 M) was adjusted to 7.40 upon addition of either NaOH or HCl. In this work, distilled water was used to perform all the experiments.
Speciation of Ni(II) complexes
Solutions of 5.00 × 10-3 M NiCl2 with changing solution pH from 2.97 to 11.40, were prepared in 50 mL volumetric flasks separately. The requisite volume of sodium nitrate was added to each solution in order to maintain ionic strength (I = 0.10 M). Solution pH was adjusted by the addition of either HCl or NaOH in acetate buffer ([Acetate] = 0.02 M). The UV-Vis spectra of the Ni2+ species were recorded by using a double beam UV-Vis spectrophotometer (SHIMADZU, Model UV-1800) within a range from 350 to 500 nm. A number of Ni2+ solutions (5.00 × 10-3 M) with different concentration of acetate ion ranging from 0 to 1.00 ×10-2 M was prepared under the same experimental conditions to investigate the interaction between Ni2+ and acetate ions and found no formation of Ni(II)-acetate complex. A pH meter (HANNA HI 2211) was used to measure the solution pH.
Kinetics of formation of [Ni(II)TMPyP]4+ complex
Pseudo-first order condition was kept constant throughout the experiment in order to explore the kinetics of the reactions between tetracationic free-base porphyrin and Ni2+ species in I = 0.10 M (NaNO3) at 25 ±1 °C where the pH of the solutions were varied from 2.97 to 11.05. The concentration of Ni2+ was varied from 0.50 × 10-3 to 5.00 × 10-3 M while that for the porphyrin, [H2TMPyP]4+, was kept constant at 1.24 × 10-5 M. The metalloporphyrin was prepared by mixing the porphyrin solution with the Ni2+ solution in a 1-cm cell compartment and pre-equilibrated at 25 ±1 °C. The change in the absorbance was monitored as a function of time at 422 nm (λmax of [H2TMPyP]4+) by using a UV-Vis spectrophotometer (SHIMADZU, Model UV-1800). Formation of the [Ni(II)TMPyP]4+ complex was monitored by observing isosbestic points at 431, 490 and 546 nm in the visible region as the porphyrin reacted with the Ni2+ species. Appearing the isosbestic points is confirming the free-base porphyrin and Ni(II)porphyrin complex are only the absorbing species. Figure 1 shows such a spectral pattern of the formation of the [Ni(II)TMPyP]4+ complex with time. To obtain the observed rate constants (kobs), values of ln(At-Aα) were plotted with time and found linearity over two half-lives. Rate constants for the reactions between the free-base porphyrin and Ni2+ species were determined by varying solution pH, nickel concentrations and ionic strengths. The duplicate runs under the same conditions agreed within a 5% error. A pH meter (HANNA HI 2211) was used to measure the pH of the solution.
Interaction of [Ni(II)TMPyP]4+ complex with DNA
The UV-Vis spectra of the free base porphyrin and [Ni(II)TMPyP]4+ complex upon addition of DNA were recorded by using a double-beam UV-Vis spectrophotometer (UV-1800, Shimadzu, Japan). A fluorescence spectrophotometer (F-7000, Hitachi, Japan) was used to record the luminescence spectra for the free base porphyrin and the Ni(II)porphyrin in the presence of DNA. The fluorescence emission wavelength was scanned from 550 to 800 nm by setting the excitation wavelength at 446 and 431 nm for the [Ni(II)TMPyP]4+ and [H2TMPyP]4+, respectively. This is because the isosbestic points for the binary system of [Ni(II)TMPyP]4+-DNA and [H2TMPyP]4+-DNA were observed at 446 and 431 nm, respectively. Under the present experimental conditions, for 1.14 × 10-5 M of [Ni(II)TMPyP]4+ and [H2TMPyP]4+, the absorbance and luminescence spectra of the porphyrin solutions were not affected by the species adsorbed on the surface of the cell wall. These were confirmed by recording a UV-vis spectrum of ethanol-water after discarding the analyte solution and found no peaks from the ethanol-water. All the experiments were carried out under room light. HEPES solution of 0.02 M (pH 7.40) was used throughout the experiment. A pH meter (HANNA HI 2211) was used to measure the solution pH.
Results and Discussion
Speciation of Ni2+
Speciation of Ni2+ ion in aqueous solution in the presence of 0.10 M of NaNO3 (I) at 25 ±1 °C was carried out to investigate the kinetics of the reaction between the free-base porphyrin, [H2TMPyP]4+, and the Ni2+ species. In order to investigate the kinetics of the metalation reaction, it is highly expected to know the speciation of the relevant metal ion. This is because the speciation diagram provides species distribution that is required to establish the reaction mechanism for the relevant reaction. Figure 2 shows the speciation diagram generated from the hydrolysis constants of Ni2+ species with the solution pH.59 As seen from Figure 2, the hexaaqua Ni2+, [Ni(H2O)6]2+ simply written as Ni2+(aq), species predominantly exists from the acidic to even alkaline pH (~9.50) and is converting to hydroxo species, [Ni(H2O)6-n(OH)n]2-n, through successive replacement of the H2O molecule by the OH- groups with increasing the solution pH. The aqua-monohydroxo Ni2+, [Ni(H2O)5(OH)]+, species is distributed from pH ~7.90 to 11.00 with the maximum distribution that observed at pH 9.50 (Figure 2). Like Ni2+(aq) ions, the monohydroxo Ni2+ ions, [Ni(H2O)5(OH)]+, are also aquatic species, thus they take part in metalation reaction with the water-soluble free-base porphyrin, [H2TMPyP]4+, significantly in the aqueous medium.
Figure 2.

Speciation diagram of Ni2+ species as a function of solution pH. The diagram is reproduced on the basis of hydrolysis constants of Ni2+.59
On the other hand, a small fraction of the dihydroxo Ni2+, [Ni(H2O)4(OH)2]0, species reacts with the free-base porphyrin, [H2TMPyP]4+, because of its very poor existence in the aqueous system under the present experimental conditions. The solubility product for the dihydroxo Ni2+ species is only KbS10 = -15.7, thus it starts to precipitate at pH ~8.15 for 10-3 M of Ni2+ solution.59 This causes the presence of a small fraction of the [Ni(H2O)4(OH)2]0 species in this study (Figure 2). According to the speciation diagram, the [Ni(H2O)4(OH)2]0 species is distributed from pH ~8.25 to the higher pH ( ≥ 12.00) where its maximum distribution is observed at pH ~10.30 (Figure 2). It is expected that the reactivity for the dihydroxo Ni2+, [Ni(H2O)4(OH)2]0, species towards the free-base porphyrin, [H2TMPyP]4+, would be higher than that of the monohydroxo, [Ni(H2O)5(OH)]+, species. This is because the dihydroxo species is electrically neutral, thus the [Ni(H2O)4(OH)2]0 species can easily approach the tetracationic porphyrin’s core without any Coulombic force of repulsion while the unipositive aqua monohydroxo Ni2+, [Ni(H2O)5(OH)]+, species could suffer from the repulsive force. The dihydroxo Ni2+ species exists with a small fraction in the aqueous systems because of its very low solubility product, KbS10 = -15.7, under the present experimental conditions ([Ni2+] = 10-3 M; I = 0.10 M, NaNO3), therefore, it is reasonable to observe the less reactivity for the [Ni(H2O)4(OH)2]0 species towards the [H2TMPyP]4+ as described in the kinetics section. The trihydroxo Ni2+, [Ni(H2O)3(OH)3]- species is distributed from pH 9.4 to the higher pH > 12.00 while its distribution is so small ~1-2% at the experimental solution pH, 9.50 (Figure 2).
The UV-Vis spectral data also confirm the stepwise formation of the hydroxo Ni2+, [Ni(H2O)6-n(OH)n]2-n, species as a function of the solution pH (Figure S1, Supplementary Information). As mentioned above, the Ni2+ exists in an aqueous system as hexaaqua, [Ni(H2O)6]2+, species. The [Ni(H2O)6]2+ species shows the ligand to metal charge transfer (LMCT) transition and the absorption maximum is centered at λmax = 391 nm in the UV region (Figure S1, Supplementary Information). The LMCT transitions have been assigned due to the charge transfer from bonding or nonbonding p-orbital of ligand to high energy antibonding dp*-orbital of the metal ion.60 The UV-Vis absorption spectra for Ni2+ (1.0×10-3 M) in 0.10 M NaNO3 solutions with different solution pH are shown in Figure S1, Supplementary Information. As seen from Figure S1, Supplementary Information, the intensity of the peak centered at λmax 391 nm that gradually decreases as a function of solution pH and reaches at a flat with the higher pH value, ~10.30. This result suggests that the hexaaqua Ni2+, [Ni(H2O)6]2+, species are converting to hydroxo, [Ni(H2O)6-n(OH)n]2-n, species through stepwise replacement of the H2O by the OH- with increasing the solution pH from 2.97 to 11.40. It is noted that the solution does not show any clear absorption maxima between 350 and 450 nm at pH ≥ 10.30. This is because of the formation of the dihydroxo Ni2+, [Ni(H2O)4(OH)2]0, species and then phases out from the aqueous solution as Ni(OH)20 (KbS10 = -15.7), thereby resulting in presence of an insignificant amount of the UV-active species in the aqueous system (Figure S1, Supplementary Information).59 Therefore, it may conclude that the hexaaqua Ni2+, [Ni(H2O)6]2+, species co-exists with the [Ni(H2O)6-n(OH)n]2-n [n = 1, ….,6] within a pH range from ~2 to 14 according to the following equations:59
| 1 |
| 2 |
| 3 |
| 4 |
Kinetics of formation of [Ni(II)TMPyP]4+ complex
Changing the absorbance of the free base porphyrin, [H2TMPyP]4+, at λmax = 422 nm with time is a signature to monitor the progress of formation of the [Ni(II)TMPyP]4+ complex. Thus, this event has been taken into account to investigate the kinetics of Ni2+ into the free base porphyrin, [H2TMPyP]4+, in an aqueous medium at 25 ±1 °C in I = 0.10 M (NaNO3). The spectral pattern of the formation of the [Ni(II)TMPyP]4+ complex is shown in Figure 1. The variation of the absorbance depicted as ln(At-Aα) was plotted with time in order to achieve the observed rate constants for the reactions between the [H2TMPyP]4+ and [Ni(H2O)6-n(OH)n]2-n species as a function of solution pH.
The rate of formation of the [Ni(II)TMPyP]4+ complex is first order with respect to the free base porphyrin that can be written by the following equation:
| 5 |
where kobs is the observed first-order rate constant and kf is the second order formation rate constant.
From the reactions between the [H2TMPyP]4+ and Ni2+ species at different solution pH, the observed rate constants (kobs) were measured to explore the reactivity of the various [Ni(H2O)6-n(OH)n]2-n species. The values of the observed rate constants for the reactions of the free base porphyrins with the Ni2+ species as a function of solution pH are shown in Figure 3. As seen from Figure 3, the observed rate constant increases as a function of the solution pH and goes to its maximum value at pH 9.50 and then slows down as pH is being increased. The rising trend for the rate constants almost remains constant until pH 6.60 and then increases sharply with pH. These results suggest that the reacting species of the Ni2+ ion is mostly hexaaqua Ni2+ ion, [Ni(H2O)6]2+ written as Ni2+(aq), within the pH range from 2.97 to ~8.00 which is one of the less reactive among the [Ni(H2O)6-n(OH)n]2-n [n = 1, ….,6] species (Figure 2).
Figure 3.
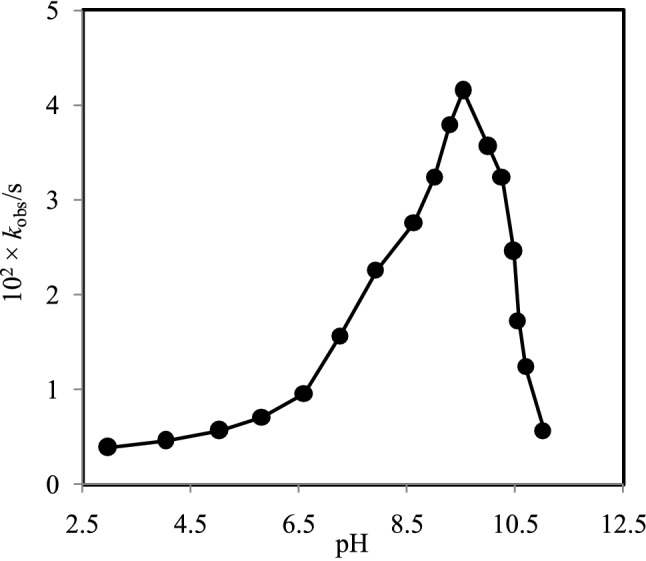
Observed rate constants (kobs) with solution pH within a range from 2.97 to 11.02 in I = 0.10 M (NaNO3) at 25 ±1 °C. [Ni2+] = 1.00 × 10-3 M; [H2TMPyP4+] = 1.24 × 10-5 M.
Thus, at low pH (3.00-6.60) and 0.10 M NaNO3, the main Ni2+ species is hexaaqua, [Ni(H2O)6]2+, and changes to monohydroxo [Ni(H2O)5(OH)+], dihydroxo [Ni(H2O)2(OH)20], trihydroxo [Ni(H2O)3(OH)3-] and tetrahydroxo [Ni(H2O)2(OH)42-] species as a function of solution pH that stated in equations 1–4. In our previous study, it has also been reported that Zn2+ ion exists predominantly as a hexaaqua, [Zn(H2O)6]2+, species at low pH (~2-5) in 0.10 M NaNO3, and changes to hydroxo species stepwise and finally converts to tetrahydroxo [Zn(OH)4-] species at higher solution pH.34
As mentioned above, Ni2+ ion predominantly exists as hexaaquanickel(II), [Ni(H2O)6]2+, species at solution pH 3.0-8.0 and showed less reactivity in incorporation into the free base porphyrin, H2TMPyP4+. As the distribution of aqua-monohydroxo, [Ni(H2O)5(OH)]+, species increases, the reactivity of the Ni2+ ion also increases. As shown in Figure 3, the maximum observed rate constant is found at pH 9.50. This result is confirming the maximum distribution of the [Ni(H2O)5(OH)]+ species at pH ~9.50 and thus exhibited the highest reactivity towards the free-base porphyrin, [H2TMPyP]4+. With increasing the solution pH, the distribution of the aqua-dihydroxo species of Ni2+, [Ni(H2O)4(OH)2]0, is being increased. With further increase the solution pH, the presence of aqua-trihydroxonickelate(II), [Ni(H2O)3(OH)3]-, species is also increased and thus Ni2+ becomes less reactive towards the free-base porphyrin, [H2TMPyP]4+. Aqua-tetrahydroxonickelate(II), [Ni(H2O)2(OH)4]2-, species will be eventually the predominant species at the higher solution pH (~12.00).
Therefore, the rate of incorporation of Ni2+ species into the [H2TMPyP]4+ (written H2P4+ is the simplest form of the [H2TMPyP]4+) is expressed by the following equations:
| 6 |
| 7 |
| 8 |
| 9 |
| 10 |
Since the reactivity of the aqua-tetrahydroxonickelate(II), [Ni(H2O)2(OH)4]2-, species towards the [H2TMPyP]4+ is very poor and its negligible presence at pH 9.50, thus, eqs. (10) can be ignored. Therefore, eqs. (6) to (9) are taken into account in order to calculate the observed rate constants, kobs.
| 11 |
As described above, the speciation diagram exhibited the distribution of the Ni2+ species, [Ni(H2O)6-n(OH)n]2-n, with solution pH, accordingly, their kinetics were also different. According to equ (11), the rate constants such as k1, k2, k3 and k4 belong to hexaaquanickel(II), dihydroxonickel(II), trihydroxonickelate(II) and tetrahydroxonickelate(II) species, respectively. Thus, the individual rate constant was calculated by taking as a mean of the observed rate constants. The calculated observed rate constants are as follows: k1 = (0.62 ± 0.22) × 10-2; k2 = (3.60 ± 0.40) × 10-2; k3 = (2.09 ± 0.52) × 10-2, k4 = (0.53 ± 0.04) × 10-2 M-1s-1 at 25 ±1 °C in I = 0.10 M (NaNO3) where k2 > k3 > k1 > k4. From this sequence, it is concluded that the aqua-monohydroxo Ni2+, [Ni(H2O)5(OH)]+, species exhibited the highest reactivity among the Ni2+ species in reacting with the [H2TMPyP]4+ where k2 = 3.60 ± 0.40 × 10-2 M-1s-1. It has been reported that monohydroxozinc(II), [ZnOH]+, species exhibited the enhanced reactivity with non-N-substituted porphyrins where kZnOH+ > kZn2+(aq), which is comparable to that found in this study.21,60-62 The enhanced rate constant has been ascribed to the formation of hydrogen bonding between the active oxygen atom of the hydroxo ligands and the pyrrolic hydrogen atom of the [H2TMPyP]4+. The [ZnOH]+ species also exhibited enhanced reactivity towards N-p-nitrobenzyl-5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin.31 It is noted that the formation of hydrogen bonding between the oxygen atom of the [ZnOH]+ species and the pyrrolic hydrogen atom of the free base porphyrin is responsible for the enhanced reactivity. In the incorporation of Zn2+ ion into the [H2TMPyP]4+, the presence of hydroxo ligands of the Zn2+ species enhances the reactivity that obeys the following sequence: kZn(H2O)4(OH)20 > kZn(H2O)3(OH)- > kZn(H2O)5(OH)+ > kZn(H2O)62+.34 The monohydroxotrichloroaurate(III) species, [AuCl3(OH)]-, also showed enhanced reactivity towards the [H2TMPyP]4+ where kAuCl3(OH)- > kAuCl4- > kAuCl2(OH)2- > kAuCl(OH)3-.27 Schneider (1975) reported that monohydroxocopper(II), [Cu(OH)]+, species exhibited much more reactivity towards the same free base porphyrin, [H2TMPyP]4+, compared to that of aquacopper(II), Cu2+(aq), species, i.e., kCu(OH)+ >> kCu2+.63 The oxygen atom of the [Cu(OH)]+ species forms H-bonding with the pyrrolic hydrogen atom of the porphyrin core. This is the reason for its substantial reactivity with the free base porphyrin. It is, therefore, concluded that the presence of the OH- group of the aqua-monohydroxo, [Ni(H2O)5(OH)]+, species make possible for the formation of hydrogen bonding with the pyrrolic hydrogen atom, thus the Ni2+ species can easily approach to the porphyrin’s core. The formation of the hydrogen bonding and fast displacement of H2O ligands from the Ni2+ species enhance the reactivity of the aqua-monohydroxo, [Ni(H2O)5(OH)]+, species. However, the aqua Ni2+, [Ni(H2O)6]2+, species lack the hydroxo (OH-) ligands, thus it might have less approaching ability to the porphyrin’s core. This is the reason for less reactivity of the Ni2+(aq) species towards the [H2TMPyP]4+ (k1 = 0.62 ± 0.22 × 10-2 M-1s-1). The hexaaqua Ni2+ ion, [Ni(H2O)6]2+, carries double positive charge that also decreases the reactivity of this species towards the tetracationic porphyrin, [H2TMPyP]4+. This is because of the development of Coulombic force of repulsion between the two positively charged species. It was expected to observe the highest reactivity for the aqua-dihydroxo, [Ni(H2O)4(OH)2]0, towards the [H2TMPyP]4+, however, Ni(OH)20 begins to precipitate at pH ~8.20 for 10-3 M of Ni2+ solution using the log KbS10 of -15.7 for the active Ni(OH)2 [Baes and Mesmer, 1976]. In contrast, the aqua-dihydroxo species of Zn2+, [Zn(H2O)4(OH)2]0, showed the highest reactivity among the [Zn(H2O)6-n(OH)n]n-2 species towards the free base porphyrin, [H2TMPyP]4+.34 Batinić-Haberle et al. (1999) used Marcus plot in order to determine the self-exchange rate constants for monohydroxoiron(III) porphyrins and aquamanganese(III) porphyrins, and they found that to be approximately 1 order of magnitude higher for the monohydroxoiron(III) porphyrins than those of aquamanganese(III) porphyrins.4
However, the hydroxospecies having a higher number of OH- groups showed less reactivity towards the [H2TMPyP]4+ and the reactivity of the [Ni(H2O)6-n(OH)n]2-n species follows the decreasing sequence: kNi(H2O)5(OH)+ > kNi(H2O)4(OH)20 > kNi(H2O)3(OH)3- (Figure 3). Habib et al. (2004) reported that AuCl3(OH)- exhibited the highest reactivity towards the free-base porphyrin, [H2TMPyP]4+, among the [AuCl4-n(OH)n]- (n = 0,---,4) species according to the following sequence: kAuCl3(OH)- > kAuCl2(OH)2- > kAuCl(OH)3-.27 Paquette and Zador (1978) also reported that the reactivity of Zn2+ ion towards hematoporphyrin IX decreases with an increasing number of the OH- group coordinated to the Zn2+ ion and the reactivity order follows the sequence: kZn(OH)2- > kZn(OH)3- > kZn(OH)4-.60 Cabbiness and Margerum (1969) reported that trihydroxocuprate(II), [Cu(OH)3]-, species shows higher reactivity towards the hematoporphyrin IX than the tetrahydroxocuprate(II), [Cu(OH)4]2-, i.e., kCu(OH)3- > kCu(OH)42-.32
It is noteworthy to mention that the OH- group coordinated to the metal ion plays a crucial role in enhancing the reactivity of the metal species towards the [H2TMPyP]4+. This is because the oxygen atom of the aqua-monohydroxo, [Ni(H2O)5(OH)]+, species forms hydrogen bonding with the pyrrolic hydrogen atom of the free base porphyrin, thus the aqua-monohydroxo species can easily approach the porphyrin’s core. The easy approach of the aqua-monohydroxonickel(II) species enhances its reactivity. On the other hand, the aqua Ni2+, [Ni(H2O)6]2+, species lacks hydroxo ligand, so the aqua-species is incapable to form hydrogen bonding with the pyrrolic hydrogen atom. This is the reason for its less and/or least reactivity towards the [H2TMPyP]4+. However, the aqua-dihydroxo, [Ni(H2O)4(OH)2]0, species exhibited less reactivity with the free-base porphyrin, [H2TMPyP]4+. This is because the aqua-dihydroxonickel(II) species take part in hydrolysis reaction at solution pH 8.20 and then phases out from the aqueous system through precipitation reaction at higher pH, e.g., 9.50. The precipitation reaction causes lesser distribution of the dihydroxonickel(II) species compared to the aqua-monohydroxonickel(II) species at solution pH 9.50 (Figure 2). The anionic trihydroxonickelate(II), [Ni(H2O)3(OH)3]-, species seems to be exhibited better reactivity towards the cationic porphyrin ([H2TMPyP]4+), however, the presence of the higher number of the hydroxo groups slows down its kinetics.27, 32,34,60 These results suggest that though the first OH- ligand is responsible for the formation of hydrogen bonding with the pyrrolic hydrogen atom, however, displacement of the remaining OH- seems slow. It has been reported that the OH- group is strongly coordinated to the metal ion having d8 electronic configuration like Pt2+ ion.64 The electronic configuration of Ni2+ is also d8, so their chemical properties are supposed to be similar; hence the OH- groups are also strongly coordinated with the Ni2+ ion. Bailey and Hambright (2003) reported that Cu2+ ion exhibited the highest reactivity among the other first transition metal ions, such as Zn2+, Co2+ and Ni2+ towards the free base H2-BrP(4)4+ and tricationic H-BrP(4)3+ porphyrins at 25 °C in I = 0.10 M (NaNO3) and the reactivity order was found to be Cu2+ > Zn2+ > Co2+ > Ni2+.65 It is expected that the reactivity of Ni2+ among the divalent metal ions towards the porphyrins would be less because of its d8 electronic configuration. However, the presence of hydroxo-ligands with the Ni2+ species enhances its reactivity in incorporation with the free-base porphyrin, [H2TMPyP]4+. Similar results have also been observed for Au3+ ion (d8 electronic configuration) towards the [H2TMPyP]4+.27
Observed rate constants (kobs) as a function of the concentration of Ni2+
Kinetics of the incorporation of Ni2+ ion into the [H2TMPyP]4+ with a variation of the concentration of Ni2+ (I = 0.10 M, NaNO3; pH 9.50) at 25 ±1 °C has also been studied in order to ascertain the formation rate constant for the metalation reaction. The observed rate constants (kobs) with a concentration of Ni2+ were obtained by plotting the ln(At-Aα) vs time. The formation rate constant was obtained by plotting the observed rate constants (kobs) with the concentration of Ni2+ as depicted in Figure 4. As seen from Figure 4, the rate constant for the metalation reaction increases with increasing the concentration of Ni2+ and passes through the origin (r2 = 0.999). This result suggests that the metalation reaction depends on concentrations of both reacting species and follows the first-order kinetics. The formation rate constant (kf) for the [Ni(H2O)5(OH)]+/[H2TMPyP]4+ was found to be 3.99 × 10-2 M-1s-1 in I = 0.10 M (NaNO3) at 25 ±1 °C.
Figure 4.
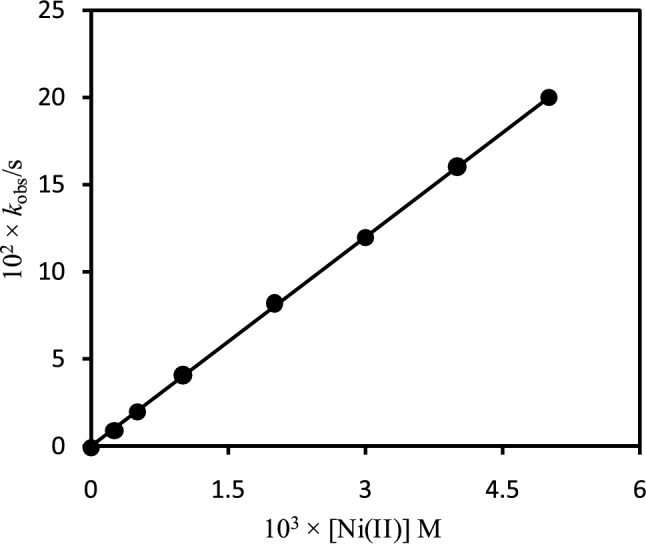
Dependence of the observed rate constants (kobs) for the reaction of [H2TMPyP]4+ with on the concentration of Ni2+ in I = 0.10 M (NaNO3) at 25 ±1 °C. [H2TMPyP4+] = 1.24 × 10-5 M; pH = 9.50.
Observed rate constants (kobs) with ionic strength
The rate constants of a reaction for opposite charged reacting species decrease as the ionic strength increases while that increase for the same charged species.66 Figure 5 shows the dependence of the ionic strength on the rate constants for the reaction between the free-base porphyrin, [H2TMPyP]4+, and Ni2+ species in I = 0 - 10.0 × 10-2 M (NaNO3) at pH 9.50 where the experimental conditions were kept constant. The observed rate constants (kobs) were obtained by plotting the ln(At-Aα) vs time at different ionic strengths (Figure 5). As seen from Figure 5, the observed rate constants (kobs) exponentially decrease with the ionic strengths. These results suggest that the reacting compounds exist as oppositely charged species in solution, however, the speciation diagram is indicating the existence of the monopositive monohydroxo Ni2+, [Ni(H2O)5(OH)]+, species at solution pH 9.50. In our previous study, we also found the retardation of the kinetics between the dihydroxo Zn2+, [Zn(H2O)4(OH)2]0, species and [H2TMPyP]4+ in the presence of NaNO3 (I = 0.10 M) and the net charge of the porphyrin, [H2TMPyP]4+, was calculated to be +3.4.34 The rate constants for the incorporation of monohydroxotrichloroaurate(III), [AuCl3(OH)]-, into the [H2TMPyP]4+ also decrease exponentially with ionic strength and the calculated net charge of the free base porphyrin was found to be +3.4 by using the Fuoss equation.27 However, in this study, we found a less decreasing tendency of the rate constants as the ionic strength increases. This may be due to the existence of monopositive monohydroxo Ni2+, [Ni(H2O)5(OH)]+, species in the aqueous solution at pH 9.50 (Figure 2 and Figure 5). Hambright (2002) also reported that the rate constants for the reactions of the [H2TMPyP]4+ with Zn2+ species decrease as ionic strength increases, and the apparent net charge of the tetracationic porphyrin was found to be +1.4 on the basis of the Bronsted-Bjerrum equation [Hambright, 2002]. It seems that the direct reaction of the dipositive Ni2+ ion with the [H2TMPyP]4+ is strenuous, thus the presence of the nitrate ions causes to reduce the repulsive force between the [Ni(H2O)5(OH)]+ species and the [H2TMPyP]4+ by interacting with the positively charged tetracationic porphyrin that facilitates the formation of aggregates of cation-anion with a lower positive charge. Thus, it is assumed that the tetracationic free-base porphyrin, [H2TMPyP]4+, carries a relatively lower charge than the actual charge, +4.0 and probably both the monopositive monohydroxo, [Ni(H2O)5(OH)]+, and neutral dihydroxo, [Ni(H2O)4(OH)2]0, species of Ni2+ ion took part in the reactions with the free-base porphyrin, [H2TMPyP]4+, at pH 9.50.6,27,34
Figure 5.
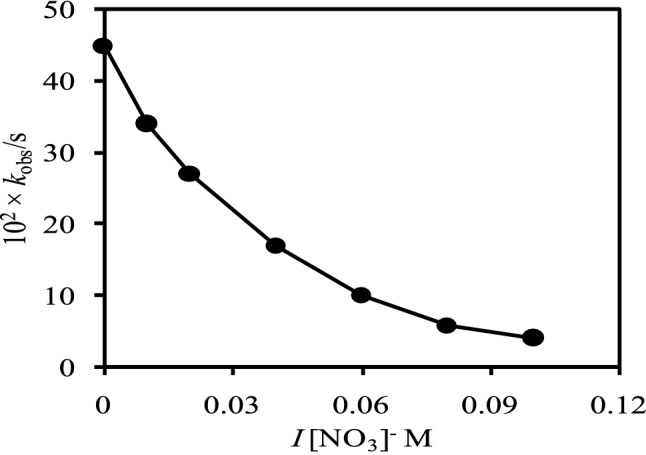
Observed rate constants, (kobs), with ionic strengths (NaNO3) at 25 ± 1 °C. [H2TMPyP4+] =1.24×10-5 M; [Ni2+] = 1.00 × 10-3 M; Solution pH: 9.50.
Figure 6 shows the Brønsted-Bjerrum plot for nickel incorporation into the tetracationic porphyrin, [H2TMPyP]4+, where Debye-Hückel Limiting (DHL) ionic strength function was applied. As seen from Figure 6, regression coefficient (R2), error bars and slope for the plot are 0.974, 1% and -3.59, respectively. It is expected that the intercept for the plot of logkobs vs √I should be zero, however, that is observed as -0.273. This is because of the presence of inherent ions that cause intrinsic ionic strength. Nwaeme and Hambright (1984) studied the effects of ionic strength on the rate of the reactions for both the positive and negative porphyrins with divalent metal ions.67 They reported that the rates of the reactions for positive porphyrins with positive divalent metal ions increase as increasing the ionic strength and that decrease for oppositely charged reacting species with the ionic strength. Williams et al. (1979) also reported the anionic effect on the reaction rate for tetracationic porphyrins in detergent solution.68
Figure 6.
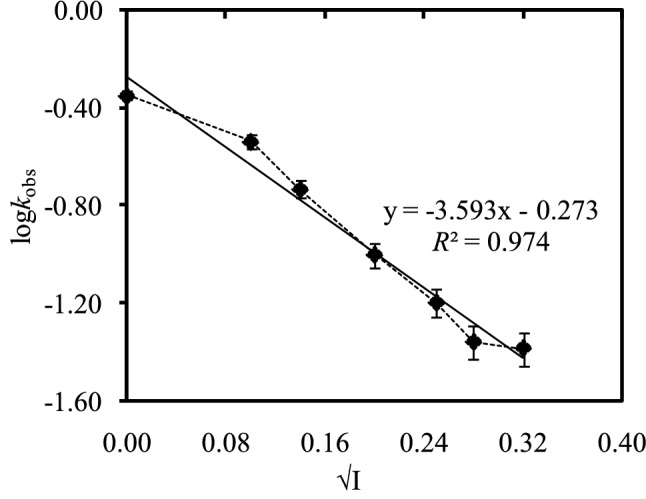
Brønsted-Bjerrum plot for nickel incorporation (1.00 × 10-3 M) into positive porphyrin, [H2TMPyP]4+, using Debye-Hückel Limiting (DHL) ionic strength function in NaNO3 medium at 25 ± 1 °C. Error bars represent a variation of 1%.
According to the speciation diagram (Figure 2), the predominant species of Ni2+ is monopositive monohydroxo, [Ni(H2O)5(OH)]+, at solution pH 9.50. Thus, the rate of reaction of the tetracationic tetrakis(N-methylpyridium-4-yl)porphyrin, [H2TMPyP]4+, with the monopositive monohydroxonickel(II), [Ni(H2O)5(OH)]+, species should be increased with increasing the ionic strength, however, that decreases as a function of ionic strength (Figure 5). These results are suggesting the presence of anionic species of Ni2+ under the experimental conditions, however, speciation diagram (Figure 2) shows the presence of monopositive monohydroxo, [Ni(H2O)5(OH)]+, species at solution pH 9.50. The monopositive Ni2+ species, [Ni(H2O)5(OH)]+, may associate with the inherent anion, thus forms negatively charged inner sphere reacting species in character such as ([Ni(H2O)5(OH)]+-inherent anion)-.63,69 The addition of nitrate ion reduces the positive charge of the cationic porphyrin at the transition state through the formation of anion-cation-porphyrin adducts that causes the decreasing reaction rates for negatively charged nickel species.21,63,70
Interaction of [Ni(II)TMPyP]4+ complex and [H2TMPyP]4+ with DNA
Figures 7a and b show the UV-vis spectral change of the [Ni(II)TMPyP]4+ complex and the free base porphyrin, [H2TMPyP]4+, upon addition of DNA, respectively. As seen from Figure 7a, a small hypochromicity (~ 13%) at λ = 436 nm (λmax of [Ni(II)TMPyP]4+) and a very small Bathochromic shift (Δλ = ~1 nm) were observed upon addition of DNA within a range from 0 to 7.96 × 10-6 M base pairs into the [Ni(II)TMPyP]4+ solution. However, the free-base porphyrin, [H2TMPyP]4+, displayed a substantial hypochromicity (~ 31%) at 422 nm (λmax of [H2TMPyP]4+) and a wide Bathochromic shift (Δλ = 17 nm) upon addition of the same DNA concentration (0 to 7.96 × 10-6 M base pairs) (Figure 6b).
Figure 7.

Changes in UV-vis spectra of (a) [Ni(II)TMPyP]4+ and (b) [H2TMPyP]4+ in the presence of DNA of (1) 0, (2) 0.56 (3)1.12, (4) 3.33, (5) 4.43, (6) 5.52, (7) 6.06, (8) 6.88, (9) 7.42, (10) 7.96 ×10-6 M base pairs at pH 7.40 (HEPES). Total concentration of porphyrin is 1.14 × 10-5 M. Cell path length is 10 mm.
A fluorescence spectrophotometer was also used in order to investigate the interactions of the [Ni(II)TMPyP]4+ and the [H2TMPyP]4+ with DNA (Figure S2, Supplementary Information). As seen from Figure S2a, Supplementary Information [Ni(II)TMPyP]4+ complex exhibited a weak excitation fluorescence spectrum centered at 633 nm while the free-base porphyrin, [H2TMPyP]4+, showed high excitation fluorescence spectrum centered at 660 nm with a hump at ~628 nm in the absence of DNA (Figure S2b, Supplementary Information). The heavy atom effect by the nickel causes weak intensity for the [Ni(II)TMPyP]4+ complex. In our previous study, we also found weak intensities from the Ru2+-, Pd2+-, Pt2+- and [Au(III)TMPyP]5+ porphyrins in an aqueous solution because of the heavy atom effect.45,46
The fluorescence intensity for the [Ni(II)TMPyP]4+ complex is significantly decreased upon addition of a low concentration of DNA, and then increased with further addition of DNA (Figure S2a, Supplementary Information). The porphyrin molecules aggregate on the negatively charged phosphate network of the DNA molecules through self-stacking in the presence of a low concentration of DNA, however, de-aggregation occurs upon the addition of additional DNA. This causes the increasing the fluorescence intensity of the metalloporphyrin.45,46
On the other hand, the intensity of the fluorescence centered at 660 nm did not change but the intensity of the hump appeared at ~628 nm is increased with a low concentration of DNA into the [H2TMPyP]4+ solution. The intensity of the hump increases with the addition of DNA and the fluorescence spectrum is finally centered at 630 nm (Figure S2b, Supplementary Information). These results suggested that the cationic free base porphyrin initially interacts with DNA via a negatively charged phosphate network in the presence of a low concentration of DNA, and then de-stacking occurs upon further addition of DNA.45,46 From the UV-vis and fluorescence spectral results, it is confirmed that both the metalloporphyrin, [Ni(II)TMPyP]4+, and the free base porphyrin interact with DNA but their modes of interaction are different. As seen from Figure 6a, the hypochromicity and Bathochromic shift (Δλ) for the [Ni(II)TMPyP]4+ are only ~ 13% (at λmax 436 nm) and ~1 nm upon addition of high concentration of DNA, respectively. These results suggested that [Ni(II)TMPyP]4+ interacts with DNA via outside binding with self-stacking.41-46 However, the significant hypochromicity (~ 31% at 422 nm) and a wide Bathochromic shift (Δλ = 17 nm) for the free base porphyrin upon addition of the same amount of DNA confirm its interaction with DNA through intercalation.41-46 The presence of metal ion in the porphyrin core is responsible for carrying water molecules as axial ligands that make the bulkiness of the metalloporphyrin molecules. The large size of the metalloporphyrin molecules interact with DNA via outside binding rather than intercalation, however, the free base porphyrin interacts with DNA via intercalation because of its smaller size that facilitates easy excess into the DNA grooves. Metalloporphyrins that are outside binders have catalytic effects to cleave DNA,45,46,71,72 thus it is expected that the [Ni(II)TMPyP]4+ complex can be used as a chemotherapeutic agent in the medical as well as in the biological fields.
Conclusions
In this work, kinetics and mechanism of formation of [Ni(II)TMPyP]4+ have been studied at 25 ±1 °C in I = 0.10 M (NaNO3) within a pH range from 2.97 to 11.40 in an aqueous medium. Speciation of Ni2+ ions in an aqueous medium has also been done in 0.10 M NaNO3 in order to provide the distribution of the Ni2+ species as a function of solution pH for the kinetic study. The experimental data have been compared with the speciation diagram generated from the values of hydrolysis constants of Ni2+ ion. The speciation data exhibited the stepwise formation of [Ni(H2O)6-n(OH)n]2-n species as a function of solution pH. Kinetic studies showed that aqua-monohydroxo, [Ni(H2O)5(OH)]+, species showed the highest reactivity towards the free-base porphyrin, [H2TMPyP]4+. The formation of hydrogen bonding between the oxygen atom of the hydroxo-ligand of the [Ni(H2O)5(OH)]+ species and the pyrrolic hydrogen atom of the [H2TMPyP]4+ enhances the reactivity of the aqu-monohydroxonickel(II) species. Ionic strength effect on the reaction rate is suggested that the tetracationic porphyrin carries a charge less than +4.0. It has been expected that the rate of incorporation of the [Ni(H2O)5(OH)]+ species into the [H2TMPyP]4+ would be slow but showed fast. From the UV-Vis and fluorescence spectroscopic results, it is concluded that the [Ni(II)TMPyP]4+ complex and [H2TMPyP]4+ interact with DNA where UV-Vis data suggested that the metallo-complex follows outside binding with self-stacking with DNA while the free base porphyrin shows intercalation. An investigation of the application of the [Ni(II)TMPyP]4+ complex with other metalloprphyrins, for example, Zn2+-, Ru2+-, Pt2+-, [Au(III)TMPyP]5+ as anti-COVID-19 agents are now in progress under the international collaboration.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors acknowledge the Ministry of Science and Technology, the People’s Republic of Bangladesh for financial support to carry out this work under the project “Photoelectrochemical splitting of water into hydrogen using solar light”.
Declarations
Competing interests
The authors declare no competing financial/commercial interest.
Supplementary Information (SI)
Absorption spectra of Ni(II) in 0.10 M NaNO3 solution at 25 ±1 0C within a pH range from 2.97 to 11.40, and fluorescence spectra of [Ni(II)TMPyP]4+ and [H2TMPyP]4+ in presence of DNA are available at www.ias.ac.in/chemsci.
References
- 1.Pasternack RF, Skowronek WR., Jr Catalysis of the disproportionation of superoxide by metalloporphyrins. J. Inorg. Biochem. 1979;11:261. doi: 10.1016/S0162-0134(00)80022-7. [DOI] [PubMed] [Google Scholar]
- 2.Marzilli LG. Medical aspects of DNA-Porphyrin Interaction . New J. Chem. 1990;14:409. [Google Scholar]
- 3.DeCamp DL, Babé LM, Salto R, Lucich JL, Koo MS, Kahl SB, Craik CS. Specific inhibition of HIV-1 protease by boronated porphyrins. J. Med. Chem. 1992;35:3426. doi: 10.1021/jm00096a020. [DOI] [PubMed] [Google Scholar]
- 4.Batinić-Haberle I, Spasojević I, Hambright P, Benov L, Crumbliss AL, Fridovich I. Relationship among Redox Potentials, Proton Dissociation Constants of Pyrrolic Nitrogens, and in Vivo and in Vitro Superoxide Dismutating Activities of Manganese(III) and Iron(III) Water-Soluble Porphyrins. Inorg. Chem. 1999;38:4011. doi: 10.1021/ic990118k. [DOI] [Google Scholar]
- 5.Sasaki K, Yumita N, Nishigaki R, Sakata I, Nakajima S, Umemura SI. Pharmacokinetic study of a gallium-porphyrin photo- and sono-sensitizer, ATX-70, in tumor-bearing mice . Jpn. J. Cancer Res. 2001;92:989. doi: 10.1111/j.1349-7006.2001.tb01190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hambright P 2002 In The Porphyrin Handbook K M Kadish, K M Smith and R Guilard (Eds.) (San Diego: Academic Press) p. 163
- 7.Lin L, Hu J. Inhibition of hepadnavirus reverse transcriptase-epsilon RNA interaction by porphyrin compounds. J. Virol. 2008;82:2305. doi: 10.1128/JVI.02147-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Batinic-Haberle I, Reboucas J, Spasojevic I. Superoxide dismutase mimics: chemistry, pharmacology and therapeutic potential . Antioxid. Redox Signal. 2010;13:877. doi: 10.1089/ars.2009.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng Y, Tsou LK, Cai J, Aya T, Dutschman GE, Gullen EA, Grill SP, Chen AP, Lindenbach BD, Hamilton AD, Cheng YC. A novel class of meso-tetrakis-porphyrin derivatives exhibits potent activities against hepatitis C virus genotype 1b replicons in vitro . Antimicrob Agents Chemother. 2010;54:197. doi: 10.1128/AAC.01206-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo H, Pan X, Mao R, Zhang X, Wang L, Lu X, Chang J, Guo J-T, Passic S, Krebs FC, Wigdahl B, Warren TK, Retterer CJ, Bavari S, Xu X, Cuconati A, Block TM. Alkylated Porphyrins Have Broad Antiviral Activity against Hepadnaviruses, Flaviviruses, Filoviruses, and Arenaviruse . Antimicrob. Agents Chemother. 2011;2:478. doi: 10.1128/AAC.00989-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rapozzi V, Zorzet S, Zacchigna M, Pietra ED, Cogoi S, Xodo LE. Anticancer activity of cationic porphyrins in melanoma tumour-bearing mice and mechanistic in vitro studies . Mol. Cancer . 2014;13:75. doi: 10.1186/1476-4598-13-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.A. Huang H, Song W, Rieffel J and Lovell J F, Emerging applications of porphyrins in photomedicine . Front. Phys. 2015;3:23. doi: 10.3389/fphy.2015.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tovmasyan A, Sampaio RS, Boss MK, Bueno-Janice JC, Bader BH, Thomas M, Reboucas JS, Orr M, Chandler JD, Go YM, Jones DP, Venkatraman TN, Haberle S, Kyui N, Lascola CD, Dewhirst MW, Spasojevic I, Benov L, Batinic-Haberle I. Anticancer therapeutic potential of Mn porphyrin/ascorbate system . Free Radic. Biol. Med. 2015;89:1231. doi: 10.1016/j.freeradbiomed.2015.10.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varchi G, Foglietta F, Canaparo R, Ballestri M, Arena F, Sotgiu G, Fanti S. Engineered porphyrin loaded core-shell nanoparticles for selective sonodynamic anticancer treatment. Nanomed. 2015;10:3483. doi: 10.2217/nnm.15.150. [DOI] [PubMed] [Google Scholar]
- 15.Su S, Ding Y, Li Y, Wu Y, Nie G. Integration of photothermal therapy and synergistic chemotherapy by a porphyrin self-assembled micelle confers chemosensitivity in triple-negative breast cancer. Biomater. 2016;80:169. doi: 10.1016/j.biomaterials.2015.11.058. [DOI] [PubMed] [Google Scholar]
- 16.Malatesti N, Munitic I, Jurak I. Porphyrin-based cationic amphiphilic photosensitisers as potential anticancer, antimicrobial and immunosuppressive agents . Biophys. Rev. 2017;9:149. doi: 10.1007/s12551-017-0257-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Batinic-Haberle I, Tovmasyan A, Spasojevic I. Mn Porphyrin-Based Redox-Active Drugs: Differential Effects as Cancer Therapeutics and Protectors of Normal Tissue Against Oxidative Injury . Antioxid Redox Signal. 2018;29:1691. doi: 10.1089/ars.2017.7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aggarwal A, Samaroo D, Jovanovic IR, Singh S, Tuz MP, Mackiewicz MR. Porphyrinoid-based photosensitizers for diagnostic and therapeutic applications: An update. J. Porphyr. Phthalocyan. 2019;23:729. doi: 10.1142/S1088424619300118. [DOI] [Google Scholar]
- 19.Garcia-Sampedro A, Tabero A, Mahamed I, Acedo P. In Multimodal use of the porphyrin TMPyP: From cancer therapy to antimicrobial applications. J. Porphyr. Phthalocyan. 2019;23:11. doi: 10.1142/S1088424619500111. [DOI] [Google Scholar]
- 20.Tebo A, Herrero C and Aukauloo A 2014 In Porphyrins and Metalloporphrins as Components In Artificial Photosynthesis Research K MKadish, K M Smith and R Guilard (Eds.) (Location: World Scientific) pp. 195-237
- 21.Hambright P, Chock PB. Metal-porphyrin interactions. III. Dissociative-interchange mechanism for metal ion incorporation into porphyrin molecules. J. Am. Chem. Soc. 1974;96:3123. doi: 10.1021/ja00817a018. [DOI] [PubMed] [Google Scholar]
- 22.Hambright P 1975 In Porphyrins and Metalloporphyrins K M Smith (Ed.) (Elsevier) pp.233-278
- 23.Tanaka M. Kinetics of metalloporphyrin formation with particular reference to the metal ion assisted mechanism . Pure Appl. Chem. 1983;55:151. doi: 10.1351/pac198355010151. [DOI] [Google Scholar]
- 24.Lavallee DK. Kinetics and mechanisms of metalloporphyrin reactions . Coord. Chem. Rev. 1985;61:55. doi: 10.1016/0010-8545(85)80002-3. [DOI] [Google Scholar]
- 25.Funahashi S, Ito Y, Kakito H, Inamo M, Hamada Y, Tanaka M. Metal ion incorporation into n-methyl-5,10,15,20-tetrakis (4-sulfonatophenyl) porphine and its differential rates as applied to the kinetic determination of copper(II) and zinc(II) in serum . Microchim. Acta. 1986;88:33. doi: 10.1007/BF01196685. [DOI] [Google Scholar]
- 26.Tabata M and Tanaka M 1983 Kinetics and mechanism of cadmium(II) ion assisted incorporation of manganese(II) into 5,10,15,20tetrakis(4-sulphonatophenyl)porphyrinate(4-) J. Chem. Soc., Dalton Trans. 1955
- 27.Habib A, Tabata M, Wu Y. Kinetics and mechanism of gold(III) incorporation into tetrakis(1-methylpyridium-4-yl)porphyrin in aqueous solution. J. Porphyrins Phthalocyanines. 2004;8:1269. doi: 10.1142/S1088424604000623. [DOI] [Google Scholar]
- 28.Almodôvar VAS, Tomé AC. Porphyrin–diketopyrrolopyrrole conjugates and related structures: Synthesis, properties and applications. J. Porphyr. Phthalocyan. 2020;24:43. doi: 10.1142/S1088424619300271. [DOI] [Google Scholar]
- 29.Margerum D W and Cayley G R 1978 In Coordination Chemistry A E Martell (Ed.) (American Chemical Society) pp.1-194
- 30.Shamim A, Hambright P. Exchange reactions of transition metal ions and labile cadmium porphyrins. J. Inorg. Nucl. Chem. 1980;42:1645. doi: 10.1016/0022-1902(80)80331-9. [DOI] [Google Scholar]
- 31.Tabata M, Ishimi H. Kinetics and mechanism for the formation and dissociation reactions of 21-(4-nitrobenzyl)-5,10,15,20-tetrakis(4-sulfonatophenyl)-23H-porphyrinatozinc(II) and -cadmium(II) . Bull. Chem. Soc. Jpn. 1997;70:1353. doi: 10.1246/bcsj.70.1353. [DOI] [Google Scholar]
- 32.Cabbiness DK, Margerum D. Macrocyclic effect on the stability of copper(II) tetramine complexes. J. Am. Chem. Soc. 1969;91:6540. doi: 10.1021/ja01051a091. [DOI] [Google Scholar]
- 33.Lavellee D K 1987 In The Chemistry and Biochemistry of N-Substituted Porphyrins (VCH Publishers) pp. 1-313.
- 34.Habib A, Islam R, Chakraborty M, Serniabad S, Khan MS, Qais DS, Quayum ME, Alam MA, Ismail IMI, Tabata M. Kinetics and mechanism of incorporation of zinc(II) into tetrakis(1-methylpyridium-4-yl)porphyrin in aqueous solution . Arab. J. Chem. 2020;13:6552. doi: 10.1016/j.arabjc.2020.06.011. [DOI] [Google Scholar]
- 35.Bain-Ackerman M J and Lavallee D K 1979 Kinetics of metal-ion complexation with N-methyltetraphenylporphyrin. Evidence concerning a general mechanism of porphyrin metalation Inorg. Chem. 18 3358
- 36.Turay J, Hambright P. Activation parameters and a mechanism for metal-porphyrin formation reactions . Inorg. Chem. 1980;19:562. doi: 10.1021/ic50204a059. [DOI] [Google Scholar]
- 37.Sato T, Ebisawa K, Sue K, Ito S, Saito T, Itoh N. The Kinetics of the incorporation of metals into tetraphenylporphyrin with metal salts in high-temperature water . Indust. Eng. Chem. Res. 2012;51:13908. doi: 10.1021/ie2030294. [DOI] [Google Scholar]
- 38.Peters MK, Herges R. Insertion of Ni(I) into porphyrins at room temperature: preparation of Ni(II)porphyrins, and Ni(II)chlorins and observation of hydroporphyrin intermediates . Inorg. Chem. 2018;57:3177. doi: 10.1021/acs.inorgchem.7b03164. [DOI] [PubMed] [Google Scholar]
- 39.Tan X L, Hu J, Montavon G and Wang X K 2011 Sorption Speciation of Nickel(II) onto Ca-Montmorillonite: Batch, EXAFS Techniques and Modeling Dalton Trans. 41 [DOI] [PubMed]
- 40.Tian Y, Etschmann B, Liu W, Borg S, Mei Y, Testemale D, O'Neill B, Rae N, Sherman DM, Ngothai Y, Johannessen B, Glover C, Brugger C. Speciation of nickel(II) chloride complexes in hydrothermal fluids: In situ XAS study . Chem. Geolog. 2012;334:345. doi: 10.1016/j.chemgeo.2012.10.010. [DOI] [Google Scholar]
- 41.Carvlin MJ, Fiel RJ. Intercalative and nonintercalative binding of large cationic porphyrin ligands to calf thymus DNA . Nucl. Acids Res. 1983;11:6121. doi: 10.1093/nar/11.17.6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dougherty G. Intercalation of tetracationic metalloporphyrins and related compounds into DNA. J. Inorg. Biochem. 1988;34:95. doi: 10.1016/0162-0134(88)85021-9. [DOI] [PubMed] [Google Scholar]
- 43.Fiel RJ. Porphyrin-nucleic acid interactions: a review. J. Biomol. Struc. Dyn. 1989;6:1259. doi: 10.1080/07391102.1989.10506549. [DOI] [PubMed] [Google Scholar]
- 44.Guliaev A B and Leontis N B 1999 Cationic 5,10,15,20-tetrakis(N-methylpyridinium-4-yl)porphyrin fully intercalates at 5'-CG-3' steps of duplex DNA in solution Biochem. 38 15425 [DOI] [PubMed]
- 45.Nyarko E, Hanada N, Habib A, Tabata M. Fluorescence and phosphorescence spectra of Au(III), Pt(II) and Pd(II) porphyrins with DNA at room temperature . Inorg. Chim. Acta. 2004;357:739. doi: 10.1016/j.ica.2003.08.023. [DOI] [Google Scholar]
- 46.Habib A, Sarker AK, Tabata M. Interactions of DNA with H2TMPyP4+ and Ru(II)TMPyP4+: Probable Lead Compounds for African Sleeping Sickness . Bangla Pharma. J. 2014;17:79. doi: 10.3329/bpj.v17i1.22321. [DOI] [Google Scholar]
- 47.B. Xu Z B, Yu F Q, Wu F, Zhang H, Wang K and Zhang X L, Synthesis, DNA photocleavage, singlet oxygen photogeneration and two photon absorption properties of ruthenium-phenanthroline porphyrins. J. Porphyr. Phthalocyan. 2015;19:1046. doi: 10.1142/S1088424615500790. [DOI] [Google Scholar]
- 48.Wang M, Mao Z, Kang T-S, Wong C-Y, Mergny J-L, Leung C-H, Ma D-L. Conjugating a groove-binding motif to an Ir(iii) complex for the enhancement of G-quadruplex probe behavior . Chem. Sci. 2016;7:2516. doi: 10.1039/C6SC00001K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chao X-J, Tang M, Huang R, Huang C-H, Shao J, Yan Z-Y, Zhu B-Z. Targeted live-cell nuclear delivery of the DNA ‘light-switching’ Ru(II) complex via ion-pairing with chlorophenolate counter-anions: the critical role of binding stability and lipophilicity of the ion-pairing complexes . Nucl. Acids Res. 2019;47:10520. doi: 10.1093/nar/gkz152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williamson A, Leiros H-KS. Structural intermediates of a DNA-ligase complex illuminate the role of the catalytic metal ion and mechanism of phosphodiester bond formation . Nucl. Acids Res. 2019;47:7147. doi: 10.1093/nar/gkz596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spence P, Fielden J, Waller ZAE. Beyond solvent exclusion: i-Motif detecting capability and an alternative DNA light-switching mechanism in a ruthenium(II) polypyridyl complex. J. Am. Chem. Soc. 2020;142:13856. doi: 10.1021/jacs.0c04789. [DOI] [PubMed] [Google Scholar]
- 52.Ptaszynska AA, Trytek M, Borsuk G, Buczek K, Rybicka-Jasin´ska K and Gryko D, Porphyrins inactivate Nosema spp. Microsporidia. Sci. Rep. 2018;8:5523. doi: 10.1038/s41598-018-23678-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fazaeli Y, Jalilian AR, Amini MM, Aboudzadeh M, Feizi S, Rahiminezhad A, Yousefi K. Preparation, nano purification, quality control and labeling optimization of [64Cu]-5,10,15,20-tetrakis (penta fluoro phenyl) porphyrin complex as a possible imaging agent. J. Radioanal. Nucl. Chem. 2013;295:255. doi: 10.1007/s10967-012-1885-9. [DOI] [Google Scholar]
- 54.Fazaeli Y, Jalilian AR, Feizi S, Shadanpour N. Development of a radiothallium(III) labeld porphyrin complex as a potential imaging agent . Radiochim. Acta. 2013;101:795. doi: 10.1524/ract.2013.2092. [DOI] [Google Scholar]
- 55.Vahidfar N, Jalilian AR. An Overview of Labeled Porphyrin Molecules in Medical Imaging. Recent Patents Top. Imag. 2015;5:3. doi: 10.2174/2451827105666150522235440. [DOI] [Google Scholar]
- 56.Liu W and Li H 2020 COVID-19 Disease: ORF8 and Surface Glycoprotein Inhibit Heme Metabolism by Binding to Porphyrin ChemRxiv. 10.26434/chemrxiv.11938173.v2.
- 57.Cavezzi A, Troiani E, Corrao S. COVID-19: hemoglobin, iron, and hypoxia beyond inflammation. A narrative review. Clin. Pract. 2020;10:1271. doi: 10.4081/cp.2020.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hambright P 2020 In Chemistry of Water Soluble Porphyins K M Kadish, K M Smith and R Guilard (Eds.) (New York: Academic Press) p. 199
- 59.Baes C F and Mesmer R E 1976 In The Hydrolysis of Cations (New York: Wiley) p. 246.
- 60.Paquette G, Zador M. Kinetics of interaction of Zn(II) with hematoporphyrin IX in basic aqueous solution . Inorg. Chim. Acta. 1978;26:L23. doi: 10.1016/S0020-1693(00)87162-5. [DOI] [Google Scholar]
- 61.Thompson AN, Krishnamurthy M. Peripheral charge effects on the kinetics of Zn(II)-porphyrin system. J. Inorg. Nucl. Chem. 1979;41:1251. doi: 10.1016/0022-1902(79)80497-2. [DOI] [Google Scholar]
- 62.Sutter TPG, Hambright P. The effects of peripheral substituents on the kinetics of zinc ion incorporation and acid catalyzed removal from water soluble sulfonated porphyrins. J. Coord. Chem. 1993;30:317. doi: 10.1080/00958979308022765. [DOI] [Google Scholar]
- 63.Schneider W 1975 Kinetics and mechanism of metalloporphyrin formation. In Biochemistry. Structure and Bonding (Berlin: Springer). 10.1007/BFb0116551.
- 64.Pearson RG. Hard and soft acids and bases. J. Am. Chem. Soc. 1963;85:3533. doi: 10.1021/ja00905a001. [DOI] [Google Scholar]
- 65.Bailey SL, Hambright P. Kinetics of the reactions of divalent copper, zinc, cobalt, and nickel with a deformed water soluble centrally monoprotic porphyrin . Inorg. Chim. Acta. 2003;344:43. doi: 10.1016/S0020-1693(02)01323-3. [DOI] [Google Scholar]
- 66.Castaneda-Agullo M, Del Castillo LM, Whitaker JR, Tappel AL. Effect of ionic strength on the kinetics of trypsin and alpha chymotrypsin. J. Gen. Physiol. 1961;44:1103. doi: 10.1085/jgp.44.6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nwaeme J, Hambright P. Magnitudes of ionic strength effects in porphyrin metalation and acid solvolysis reactions . Inorg. Chem. 1984;23:1990. doi: 10.1021/ic00181a044. [DOI] [Google Scholar]
- 68.Williams G, Williams RFX, Lewis A, Hambright PJ. Synthesis, characterization and copper incorporation into 5-(4-pyridyl)-10,15,20-triphenylporphyrin . Inorg. Nucl. Chem. 1979;41:41. doi: 10.1016/0022-1902(79)80390-5. [DOI] [Google Scholar]
- 69.Longo F 1978 In The Porphyrins D Dolphin (Ed.) (Academic Press) Vol. 5, Ch. 10.
- 70.Rau WG, Longo FR. Study of the aqueous equilibrium system involving meso-tetrapyridylporphine, alkali metal ions, and hydrogen ions . Inorg. Chem. 1977;16:1372. doi: 10.1021/ic50172a024. [DOI] [Google Scholar]
- 71.Tabata M, Nakajima K, Nyarko E. Metalloporphyrin mediated DNA cleavage by a low concentration of HaeIII restriction enzyme. J. Inorg. Biochem. 2000;78:383. doi: 10.1016/S0162-0134(00)00067-2. [DOI] [PubMed] [Google Scholar]
- 72.Habib A, Hara T, Nargis A, Nyarko E and Tabata M 2020 DNA Cleavage and Trypanosomes Death by a Combination of Alamar Blue and Au(III) J. Chem. Soc. Pak. 42 141
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.