

Abstract
Objective
Our hypothesis was that the activity of placental nutrient-sensing pathways is associated with adiposity and metabolic health in childhood.
Research design and methods
Using placental villus samples from healthy mothers from the Healthy Start Study, we measured the abundance and phosphorylation of key intermediates in the mTOR, insulin, AMPK, and ER stress signaling pathways. Using multivariate multiple regression models, we tested the association between placental proteins and offspring adiposity (%fat mass) at birth (n = 109), 4–6 months (n = 104), and 4–6 years old (n = 64), adjusted for offspring sex and age.
Results
Placental mTORC1 phosphorylation was positively associated with adiposity at birth (R2 = 0.13, P = 0.009) and 4–6 years (R2 = 0.15, P = 0.046). The mTORC2 target PKCα was positively associated with systolic blood pressure at 4–6 years (β = 2.90, P = 0.005). AMPK phosphorylation was positively associated with adiposity at birth (β = 2.32, P = 0.023), but the ratio of phosphorylated to total AMPK was negatively associated with skinfold thickness (β = −2.37, P = 0.022) and body weight (β = −2.92, P = 0.005) at 4–6 years.
Conclusions
This is the first report of associations between key placental protein activity measures and longitudinal child outcomes at various life stages. Our data indicate that AMPK and mTOR signaling are linked to cardiometabolic measures at birth and 4–6 years, providing novel insight into potential mechanisms underpinning how metabolic signaling in the placenta is associated with future risk of cardiovascular disease.
Introduction
One in five children in the United States are obese, and the prevalence of obesity is increasing in 2–5-year-olds [1], representing a daunting public health problem. Compelling epidemiological and experimental data show a strong link between the intrauterine environment and the risk for developing a range of diseases later in life, including obesity and type 2 diabetes [2–4]. The placenta constitutes the primary interface between mother and fetus, performing a multitude of functions critical for fetal growth and development. Changes in placental structure are associated with the development of chronic disease later in life [5], including an inverse correlation between placental surface area at delivery and hypertension and heart failure in adulthood [6, 7], and an inverse association between placental thickness and sudden cardiac death later in life [8]. It is widely believed that these alterations in structure reflect functional changes in the placenta [5, 9], and recently Chen et al. [10] reported a negative association between placental 11β-HSD2 protein expression and insulin resistance at 1 year of age [10]. However, there remains a paucity of data linking placental function to long-term child outcomes in humans.
Placental function is regulated by complex signaling pathways integrating a multitude of maternal and fetal signals to balance maternal nutrient supply with fetal demand [9] (Fig. 1). Placental insulin signaling activity is decreased in human intrauterine growth restriction (IUGR) [11, 12] and increased in maternal obesity [13, 14], potentially contributing to fetal overgrowth [14]. mTOR Complex 1 (mTORC1) is an important nutrient sensor and regulator of cell growth and metabolism [15], whereas mTOR Complex 2 (mTORC2) plays a key role in regulating insulin action and the activation of downstream signaling pathways including transcription factors that control cellular metabolism via protein kinases B (Akt) and C-α (PKCα). mTORC1 and mTORC2 signaling are positively correlated with birth weight [14] and directly regulate key trophoblast nutrient transporters [16, 17]. However, mTORC1, but not mTORC2, is a positive regulator of placental oxidative phosphorylation mediated by the effects of mitochondrial biogenesis [18]. Thus, by regulating placental ATP production, mTORC1 signaling may indirectly impact all energy-requiring processes. As a master regulator of energy metabolism and a direct ATP energy sensor, AMPK generally acts reciprocally to mTOR, where activated AMPK inhibits mTORC1 and fatty acid synthesis while promoting fatty acid oxidation and glycolysis [19]. Placental AMPK phosphorylation is inversely correlated with maternal BMI and infant birth weight [14]. Finally, ER stress leads to increased phosphorylation of eukaryotic initiation factor 2α (eIF2α), resulting in inhibition of global translation. Restricted fetal growth has been reported to be associated with placental ER stress and inhibition of protein synthesis [12].
Fig. 1. Nutrient-sensing pathways investigated in this experiment.
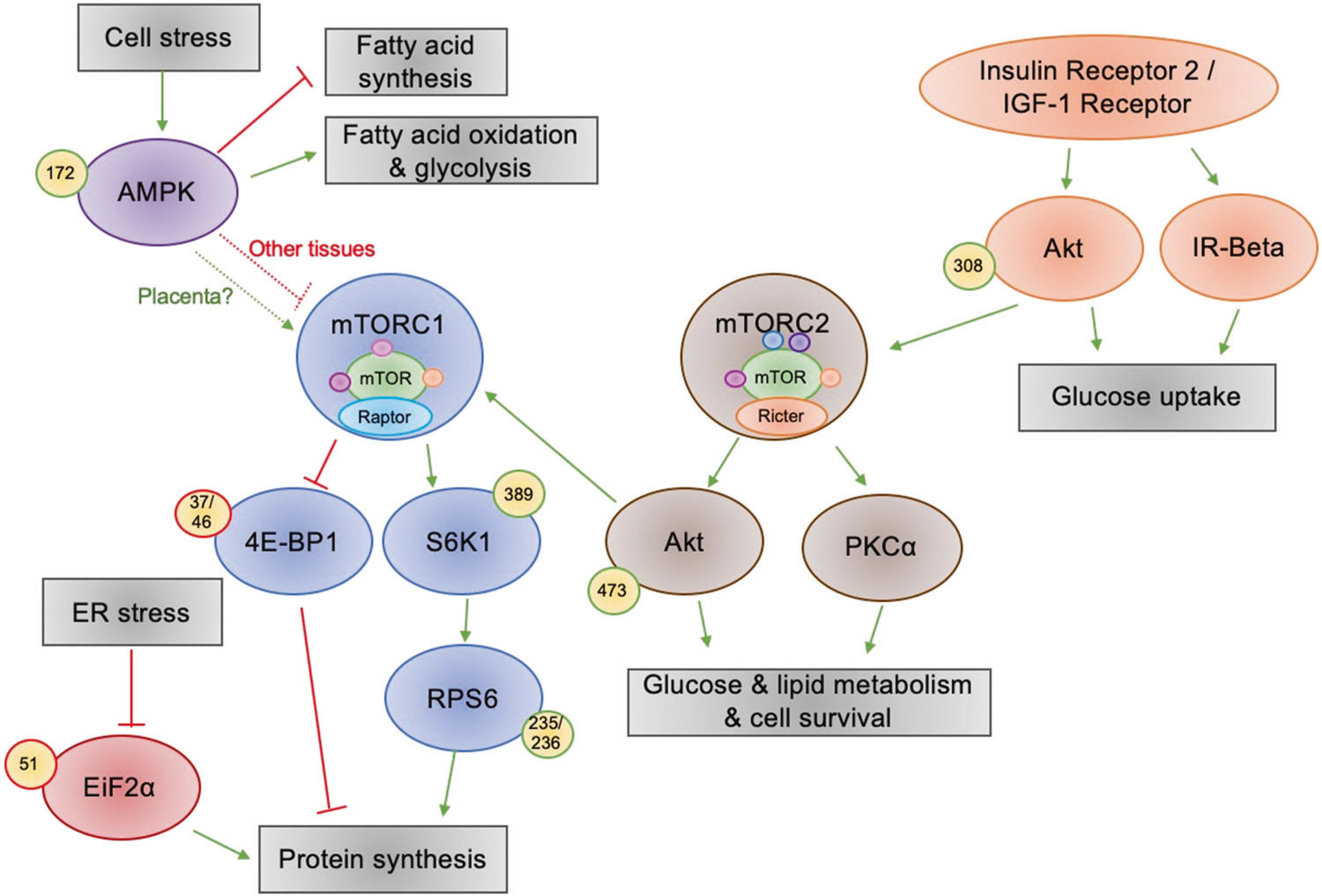
AMPK senses energy in the cell; in low energy conditions it activates fatty acid oxidation and glycolysis and it represses fatty acid synthesis and in most tissues it represses mTORC1; however in this placental tissue, AMPK activation seems to be linked to mTORC1 activation. When activated, mTORC1 promotes protein synthesis by inhibitory phosphorylation of 4E-BP1 and activating phosphorylation of S6K1, which in turn activates RPS6—this promotes protein synthesis. In conditions of ER stress, inhibitory phosphorylation of EiF2α inhibits protein synthesis. Insulin and IGF-1 activate Akt by phosphorylation at Thr-308, which promotes glucose uptake and activates mTORC2. mTORC2 then phosphorylates PKCα and AktSer473, promoting glucose and lipid metabolism as well as cell survival.
Here, we hypothesized that perturbations in these key placental signaling pathways would be associated with infant and child markers of adiposity and future metabolic disease risk. We obtained placentas from 109 mother/infant pairs participating in the larger pre-birth cohort Healthy Start, and measured protein levels to determine associations between the phosphorylation and total content of proteins in these nutrient-sensing pathways and metabolic outcomes measured in the children at various life stages: birth, 4–6 months, and 4–6 years old.
Methods
Ethics statement
This study used placental samples, body composition data, and serum biomarker data collected by the Healthy Start Study (ClinicalTrials.gov; NCT02273297). This study was approved by the Colorado Multiple Institutional Review Board at the University of Colorado Hospital. Written, informed consent was obtained from all participants at enrollment.
Participant measures and sample collection
Pregnant women
The Healthy Start longitudinal pre-birth cohort study enrolled 1410 women aged 16 and older who were ≤23 weeks pregnant at enrollment during 2010–2014. The women were recruited from the University of Colorado Hospital’s obstetrics clinics and were excluded if they currently had a multiple pregnancy or had a prior history of diabetes, premature birth, or serious psychiatric illness. Data collection for Healthy Start has been described in detail previously [20, 21]. Briefly, between 24 and 32 weeks of gestation (median 27 weeks), a fasting blood sample, maternal height, weight, and demographic data were collected.
Placental sample collection
Trophoblast villi samples were collected after delivery in a convenience subsample of the Healthy Start cohort as part of the ancillary Healthy Start BabyBUMP Project (n = 111). Samples were snap frozen in liquid nitrogen following collection and stored at −80 °C until further processing. Placental samples from two of the subjects were excluded because they were from pre-term births, leaving 109 samples to be analyzed.
Placental signaling activity
Frozen placental villus tissue (~20 mg) was homogenized in 75 μL ice cold buffer D (250 mM sucrose, 10 mM HEPES, pH 7.4) containing a 1:100 dilution of protease and phosphatase inhibitors. We performed automated Simple Western assays on WES (ProteinSimple, Santa Clara, CA) to measure the phosphorylation and total protein content of key intermediates in the following pathways: mTORC1 (p70 ribosomal protein S6 kinase [S6K]1, S6K1Thr389, ribosomal protein S6 [RPS6], RPS6Ser235/236, and eukaryotic translation initiation factor 4E-binding protein 1 [4E-BP1], 4E-BP1Thr 37/46), mTORC2 (Akt, AktSer473, PKCα), insulin signaling (insulin receptor [IR]β, AktThr308), energy sensing (AMPK, AMPKThr172), and ER-stress (eIF2α, eIF2αSer51) (Supplementary Fig. S1). WES plates were run according to the manufacturer’s instructions, with minor modification (200 volts, 55 min separation time) with 0.1 mg/mL total protein concentration. To control for variation between plates (24 samples/plate), we included an ‘equalizer’ sample on each plate. This was a sample that had clean, median values for all proteins measured and was run on every single plate. Likewise, a relative ‘loading control’ antibody (vinculin or β-actin) was multiplexed in each capillary assay and all data were normalized to these values. All antibodies were optimized in-house; antibody catalog numbers and dilutions are in Supplementary Table S1.
Infant and child outcomes
Infant birth weight was obtained from medical records. Within 72 h after birth, the length and weight of the infants (n = 107) were determined and their body composition (fat mass [FM] and fat-free mass [FFM]) was measured using whole body air plethysmography (PEA POD, COSMED, Inc.). Adiposity was calculated as FM/(FM + FFM)*100. Adiposity was again measured between 4–6 months in the infants (PEA POD, n = 104). At 4–6 years of age, children returned for a follow-up visit (n = 64, median age = 4.54 years). At this visit, body composition was measured (BOD POD, COSMED, Inc., n = 62), as described [20]. In addition, triceps, subscapular, and midthigh skinfold thicknesses were each measured twice with skinfold calipers, with a third measurement if the first two measures were off by >1 cm. Blood pressure was measured with a digital sphygmomanometer and circulating insulin and triglyceride levels were measured in a fasted venous blood sample.
Statistical analysis
The protein data were analyzed as the phosphorylated form (e.g., AktSer473) or as the ratio of phosphorylated/total (e.g., AktSer473/Akt). Total protein was not analyzed separately, except in cases where the phosphorylated form was not measured (PKCα, IRβ), to avoid collinearity with the ratios of the phosphorylated to total proteins. All statistical analyses were performed in RStudio (version 1.1.442). The Shapiro-Wilk test was used to test the normality of the data. Box-Cox transformations were used to normalize the protein data, and in the four cases where that did not suffice, the inverse normal transformation was used. All statistical tests were performed on the transformed values (Table S2), and graphs are presented with the untransformed values, divided by 10,000. The following identified protein outliers (R, Grubbs test, cutoff p value of 0.05) were removed from different subjects: two values of PKCα, one of eIF2αSer51/eIF2α, one of eIF2αSer51, and one of RPS6Ser235/236/RPS6.
To account for multiple independent and dependent variables, with multiple covariates, multivariate multiple regression (MMR) models were used (R, stats package). We tested the effect of maternal BMI, infant sex, and gestational age on each signaling pathway of interest using MMR. Because infant sex and gestational age were significantly associated with the protein pathways, we included them as covariates in the subsequent analyses.
We then used MMR to test the effect of each pathway (mTORC1 and 2, insulin, AMPK, and ER stress) on offspring body composition (adiposity and body weight). Child sex and age were included as covariates in all models, with gestational age used for the birth and 4–6 months timepoints and child age used at 4–6 years (Table S3). MMR testing was performed separately for each age grouping (birth, 4–6 months, 4–6 years), where adiposity and body weight were dependent variables and placental proteins, sex, and age were independent variables. Blood pressure, skinfolds, and metabolic markers in the serum were only measured at age 4–6 years and were tested in separate models: (1) systolic and diastolic blood pressure; (2) midthigh, triceps, and subscapular skinfold thicknesses; (3) triglycerides; and (4) insulin. Triglycerides and insulin were analyzed separately because of strong collinearity, and as these models each contained only one dependent variable, the analysis was a multiple regression rather than an MMR. We corrected for multiple comparisons using an FDR adjustment (R, Stats package).
Based on evidence demonstrating a U-shaped relationship between birth weight as an index of fetal growth and incidence of metabolic syndrome [22, 23], we anticipated that both low and high activity of mTOR, insulin, AMPK, and ER-stress pathways may be linked to poor child metabolic health outcomes. We used polynomial regression to test whether the relationships were nonlinear. We tested four polynomial models (ranging from 1 to 4) with the extra sum of squares test and selected the polynomial with the lowest Akaike information criterion (AIC), a measure of statistical model quality, that was significantly different from the preceding model. After observing that the AMPK and mTOR pathways seemed to be related in the opposite direction we expected relative to one another, we evaluated the associations between all the protein pathways by testing for significant correlations (R, cor.test).
Results
Subject characteristics
We included 109 mother/infant pairs at birth, 104 of them returned for the 4–6 months measures, and 64 have returned for the 4-year-old visit (Table 1).
Table 1.
Participant characteristics. Data are presented as mean with range in parentheses, unless otherwise stated.
| Maternal characteristics (n = 109) | |
| Pre-pregnancy BMI (kg/m2) | 25.0 (17.9–45.8) |
| Gestational weight gain (kg) | 13.5 (−5.9–47.2) |
| Cesarean delivery, n (%) | 23 (21.1) |
| Newborn characteristics (n = 109) | |
| Gestational age at delivery (weeks) | 39.5 (37–42) |
| Sex, n (female/male) | 58/51 |
| Fat mass at birth (%) | 9.0 (1–17.7) |
| Body weight at birth (kg) | 3.1 ± (2.2–4.3) |
| Infant characteristics (4–6 months of age, n = 104) | |
| Fat mass at 4–6 months (%) | 24.38 (12.9–36.1) |
| Body weight at 4–6 months (kg) | 6.6 (5.2–8.9) |
| Child characteristics (4 years of age, n = 64) | |
| Fat mass (%) | 18.1 (6.3–36.8) |
| Body weight (kg) | 17.2 (12.9–29.6) |
| Systolic blood pressure (mmHg) | 97.8 (77–126) |
| Diastolic blood pressure (mmHg) | 57.1 (41–76) |
| Midthigh Skinfold (mm) | 14.6 (6–27) |
| Triceps Skinfold (mm) | 10.2 (5–24) |
| Subscapular Skinfold (mm) | 5.1 (3–12) |
| Triglycerides (mg/dL) | 56.7 (32–137) |
| Insulin (uIU/mL) | 6.2 (2–35) |
Infant sex, gestational age, and maternal BMI are associated with placental signaling
Three proteins were associated with infant sex: PKCα (P = 0.035), S6K1Thr389 (P = 0.034), and IRβ (P = 0.026) (Fig. S2A; Table S4), where placentas of female infants had higher levels of IRβ and lower levels of PKCα and S6K1Thr389 than placentas of male infants. There was a negative association between gestational age and both S6K1Thr389 (β = −2.83, P = 5.77 × 10−3) and S6K1Thr389/S6K1 (β = −3.81, P = 2.51 × 10−4) (Fig. S2B). Sex and age had a significant effect on the child outcomes, so they were included as covariates in all the models, even in pathways where the proteins themselves were not affected by sex or age. Because it has been previously shown that maternal BMI affects placental function [24, 25], we also tested for its effect in each pathway. Maternal BMI was negatively associated with AktSer473 (β = −3.57, P = 5.38 × 10−4) and AktSer473/Akt (β = −3.41, P = 9.02 × 10−4) (Fig. S2C) but was not associated with any of the other proteins.
Placental mTORC1 associated with body composition at birth and at age 4–6 years
Phosphorylation of the mTORC1 downstream targets was used as functional readouts of mTORC1, where phosphorylation of S6K1, RPS6, and 4E-BP1 indicates increased mTORC1 activity. MMR analyses revealed significant associations between the mTORC1 proteins and offspring adiposity (R2 = 0.13, F = 2.77, df = 84, adj-P = 0.045) and body weight at birth (R2 = 0.26, F = 5.05, df = 84, adj-P = 0.001, Fig. S3A). At age 4–6 years, there was a significant association between the mTORC1 proteins’ phosphorylation and child adiposity (R2 = 0.15, F = 2.19, df = 84, P = 0.046). Within these analyses, RPS6Ser235/236 was positively associated with body weight at birth (β = 2.27, P = 0.026; Fig. 2a) and 4E-BP1Thr 37/46 was negatively associated with birth weight (β = −2.51, P = 0.014; Fig. 2b). In addition, RPS6Ser235/236/RPS6 was positively associated with adiposity (β = 2.52, P = 0.014; Fig. 2c) at birth. To explore the potential nonlinear relationship between the mTORC1 proteins and offspring outcomes in addition to the linear associations, we then used post-hoc polynomial regression for selecting the best of four polynomial models, which revealed that the relationship between RPS6Ser235/236/RPS6 and adiposity at birth was nonlinear, best represented by a third degree polynomial (Table S5). However, the nonlinear relationship seemed to be dictated by just a few points (Fig. 2c). The 4E-BP1Thr 37/46/4E-BP1 ratio was negatively associated with adiposity at 4–6 years of age (β = −3.00, P = 0.004; Fig. 2d). However, S6K1Thr389 was positively associated with adiposity at that age (β = 2.67, P = 0.010; Fig. 2e). None of the placental protein measures were significantly related to body composition at 4–6 months (Table S6).
Fig. 2. RPS6 and S6K1 phosphorylation are positively associated with offspring adiposity while 4EBP1 phosphorylation is negatively associated, and PKCα is associated with higher systolic blood pressure at age 4–6.
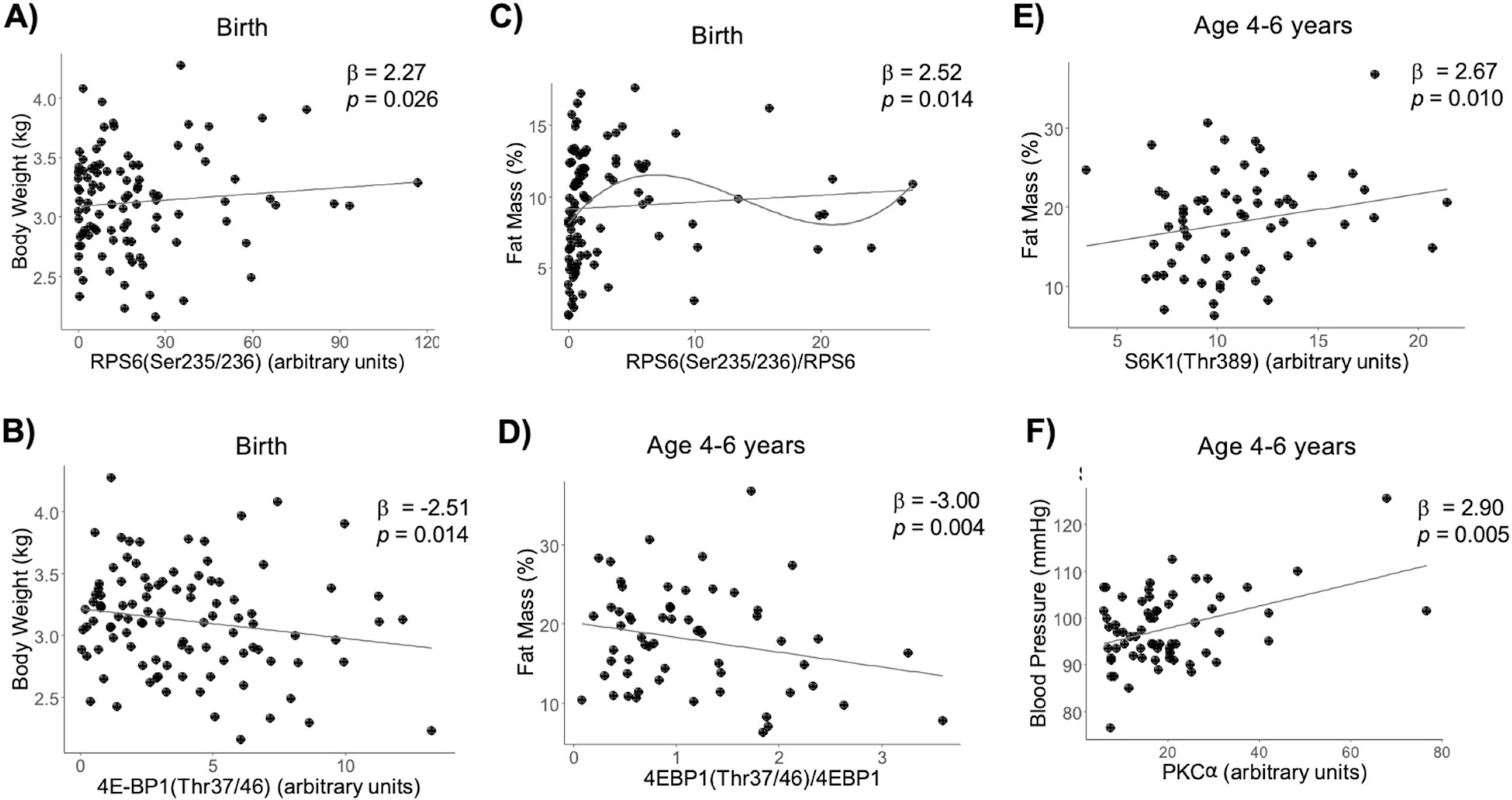
Multivariate multiple regression was performed for placental proteins in the mTORC1 pathway vs. adiposity and body weight at birth, 4–6 months, and 4–6 years of age, while adjusting for child sex and age. a −log10 P values for each MMR analysis where dotted line indicates P < 0.05. b–d Scatter plots are shown for significant MMR results.
Protein expression of PKCα, an mTORC2 target, is associated with systolic blood pressure at age 4–6 years
Placental mTORC2 activation, as determined by increased AktSer473 or AktSer473/Akt, was not significantly associated with any of the child outcomes at any age (Table S7). Placental expression of PKCα was positively correlated with systolic blood pressure at 4–6 years of age (β = 2.90, P = 0.005; Fig. 2f, Fig. S3B). Polynomial regression confirmed that the relationship was linear. However, this result must be interpreted with caution as the mTORC2 pathway was not significantly related to blood pressure at the multivariate level.
No association of placental insulin signaling proteins or ER stress activation with child outcomes
AktThr308, AktThr308/Akt, and IRβ abundance were used as functional readouts of placental insulin signaling. eIF2αSer51 and eIF2αSer51/eIF2α were measured as indicators of ER stress signaling. In our MMR analysis, we found no significant association between placental insulin signaling readouts or ER stress activation and child outcomes, although offspring sex and age had significant effects on the child outcomes (Table S8, S9; Fig. S3C).
Placental AMPK activation is associated with offspring body composition
AMPKThr172 and AMPKThr17/AMPK were used as a measure of AMPK activation. The MMR models testing for a relationship between the placental AMPK pathway and offspring outcomes revealed a significant overall effect of the pathway on adiposity at birth (R2 = 0.10, F = 3.75, df = 97, adj-P = 0.038). At age 4–6 years, there was an association between the AMPK pathway and midthigh skinfold thickness (R2 = 0.24, F = 4.19, df = 53, adj-P = 0.030), with a relationship in the same direction between AMPK and body weight when not adjusting for multiple testing (R2 = 0.17, F = 2.93, df = 56, P = 0.029, adj-P = 0.110; Fig. S3D). Specifically, AMPK Thr172 was positively associated with adiposity at birth (β = 2.32, P = 0.023; Fig. 3a). In contrast, AMPKThr172/AMPK was negatively associated with midthigh skinfold thickness (β = −2.92, P = 0.005; Fig. 3b) and body weight at 4–6 years of age (β = −2.37, P = 0.022; Fig. 3c). Polynomial regression indicated that the relationship between AMPK Thr172/AMPK and midthigh skinfold thickness was nonlinear, the best model being a second-degree polynomial (R2 = 0.22, F = 4.83, df = 53, P = 0.002; Table S10). No other relationships with the AMPK pathway were significant (Table S11).
Fig. 3. Placental AMPK activation associated with offspring body composition.
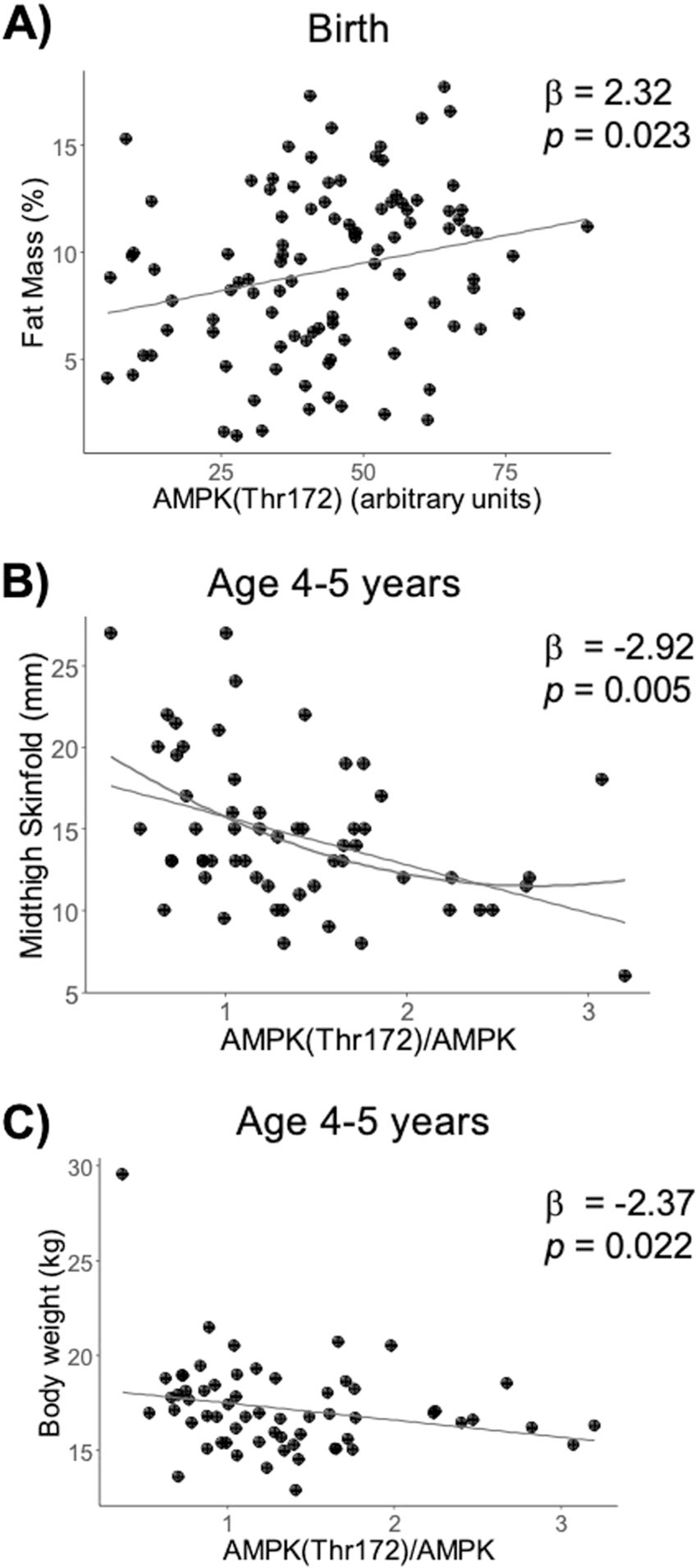
Multivariate multiple regression was performed for the placental AMPK pathway vs. adiposity and body weight at birth, 4–6 months, and 4–6 years of age, and vs. skinfold measures at age 4–6 years, while adjusting for child sex and age. a–c Scatter plots are shown for significant MMR results.
AMPK activation accompanied mTORC1 activation
In many cells, activation of AMPK is associated with inhibition of mTORC1, and it was therefore surprising that mTORC1 activation had a similar relationship with the offspring outcomes as AMPK activation. Thus, we tested the associations of the phosphorylated, total, and phosphorylated to total ratios of the proteins with each other to uncover how the pathways were interacting. As expected, phosphorylation of the insulin signaling pathway was positively correlated with activation of the mTORC2 pathway proteins, with AktSer308 being positively correlated with both AktSer473 (R = 0.31, P = 9.40 × 10−4) and PKCα (R = 0.21, P = 0.027). Interestingly, activation of the AMPK pathway was associated with mTORC1 activation rather than inhibition; AMPKThr172/AMPK was negatively correlated with total 4E-BP1 (R = −0.25, P = 0.010) and also had a borderline significant positive correlation with RPS6Ser235/236/RPS6 (R = 0.19, P = 0.061). Greater activation of PKCα from the mTORC2 pathway was linked to higher levels of 4E-BP1Thr 37/46 (R = 0.238, P = 0.016) and total 4E-BP1 (R = 0.289, P = 0.003). There was a similar positive, but non-significant trend between Akt473 and 4E-BP1Thr 37/46/4E-BP1.
Discussion
A suboptimal nutritional environment in utero is linked to the increased risk for obesity and diabetes in childhood and into adulthood in humans [2–4]. However, the mechanisms underpinning the intrauterine origins of metabolic disease remain to be established. Emerging evidence implicates changes in placental structure and function in the risk of adult disease [5, 9], although the molecular mechanisms are largely unknown. This is the first report of associations between functional protein measures in the placenta and longitudinal metabolic health outcomes in children at several life stages. Our results show that placental AMPK and mTOR signaling are associated with cardiometabolic measures at birth and age 4–6 years old.
Placental insulin and mTOR signaling promotes nutrient transport, oxidative phosphorylation, protein synthesis, and hormone synthesis, affecting fetal nutrient avaiability and growth [26]. This is demonstrated in humans and in animal models, where reduced mTOR activity is observed in the placenta with IUGR [11, 12] and greater activivation of these pathways tracks with maternal obesity and fetal overgrowth [13, 14]. Here, although we did not observe an association with proteins in the insulin signaling pathway and offspring outcomes, we did find that placental RPS6 phosphorylation, an index of mTORC1 activity, was indeed positively correlated with birth weight and adiposity, as we have shown previously in rodents fed a high-fat diet during pregnancy [13]. Longitudinally, we observed that phosphorylation of S6K1 was also positively correlated with offspring adiposity at age 4–6 years. However, contrary to these results, we found an inverse relationship between placental 4E-BP1 phosphorylation and both birth weight and body mass at 4–6 years. One possible explanation for these two mTORC1 signaling proteins showing divergent associations with offspring growth is that they represent two separate arms of mTORC1 signaling, which can be differentially regulated in the human placenta [27] and may have distinct effects on cellular function. For example, although both S6K1/RPS6 and 4E-BP1 pathways are known to regulate translation initiation and protein synthesis [28], some data suggest that mTORC1-mediated protein sythesis primarily occurs via 4E-BP1 inhibition [29], while RPS6 promotes ribosomal biogenesis. The specific effects of S6K1/RPS6 and 4E-BP1 on cellular function in the human placenta are not known. However, mTOR/S6K1 signaling has been shown to be highly sensitive to insulin, the cellular energy state (ATP), and nutrients [30]. Therefore, it may be speculated that 4E-BP1 is the predominant signaling pathway promoting protein synthesis, and therefore placenta growth, while S6K1/RPS6-mediated mTOR signaling may promote nutrient transport and oxidative phosphorylation, and therefore fetal growth. This concept is supported by our results showing that phosphorylation of AMPK, the energy sensor and master regulator of metabolism, was inversely associated with 4E-BP1 and tended to be positively associated with S6K1.
It is generally well accepted that AMPK activation inhibits the mTOR pathway, which has been studied in detail in many tissues. But the role of AMPK in the placenta is less clear. Here, we found that AMPK activation was positively associated with mTORC1 activation, indicating that AMPK may have a different relationship with mTOR in the placenta than it does in other tissues (Fig. 1). Indeed, we have previously reported inhibition of placental mTOR in the absence of AMPK activation in maternal nutrient restricted rats [31] and baboons [32], demonstrating a lack of the expected relationship between AMPK and mTOR. This may be in part explained by the fact that mTORC1 is regulated by a multitude of pathways, which may be more robust than its interaction with AMPK in the placenta.
In the present study, we found that AMPK phosphorylation was positively associated with adiposity at birth. This was unexpected based on previous literature linking AMPK activity with improved metabolic health outcomes [33] and our previous report showing AMPK phosphorylation was inversely associated with birth weight in the context of maternal obesity [14]. AMPKThr172/AMPK was negatively associated with body weight at 4–6 months, however the rest of the proteins were not associated with body composition at that age, suggesting that infant feeding is a stronger contributor to body composition at this age than placental function. By 4–6 years, we found that greater placental AMPK activity was in fact associated with lower child skinfold thickness and body weight. This is more in line with expected results and for the first time demonstrates placental AMPK activity association with long-term metabolic health in the offspring. There are several possible explanations for the discrepancy in associations between birth and age 4–6 years. The relationship between early childhood growth/adiposity and later development of metabolic disease is complex [34]. In sheep, IUGR has been associated with increased placental AMPK phosphorylation [35], and maternal obesity has been associated with decreased placental AMPK gene expression in humans [36]. This is the first large-scale measurement of placental AMPK phosphorylation in a relatively healthy pregnancy cohort, so it is possible that perturbations in AMPK activation may be evident only in these types of pregnancy complications. Furthermore, the positive relationship between AMPK activity and adiposity at birth seems to be driven by a difference in AMPKThr172 that is not evident when AMPKThr172 is normalized to total AMPK protein; however, the negative relationship between AMPK activity with body weight and skinfold thickness at 4–6 years seems to be driven by a difference in AMPKThr172/AMPK, with no difference in AMPKThr172. Together, these observations suggest that the total content of AMPK protein may play a more important role in the relationship with adiposity and body weight. Indeed, we recently reported that total AMPK, but not AMPKThr172, was reduced in umbilical cord-derived mesenchymal stem cells from high adiposity infants born to mothers with obesity [37].
We observed a strong association between total PKCα protein abundance in the placenta and systolic blood pressure at 4–6 years of age, although the mTORC2 pathway as a whole was not associated with blood pressure. mTORC2 phosphorylates PKCα at Ser657, activating key downstream targets of mTORC2. Unfortunatley, despite extensive attempts, we were unable to indentify an unambigous phosphorylated PKCα band using simple western. However, increased total expression of the kinase is consistent with an activation of the pathway. The role of PKCα in the human placenta is largely unknown, and the molecular underpinnings linking placental total PKCα and childhood blood pressure remain to be established using mechanistic studies in animals. However, it is of interest that PKCα is associated with cardiomyocyte hypertrophy and atherosclerosis [38]. Observations linking mTORC2 signaling to hypertension includes reports of increased blood pressure following inhibition of mTORC2 in the brain and adipose tissue [39], as well as a relationship between mTORC2 inhibition and pulmonary hypertension [40].
Both low [22] and high birth weight [23] are strongly associated to later development of obesity and diabetes. Placental insulin and mTOR signaling is typically inhibited in intrauterine growth restriction [11, 12, 27] and activated in fetal overgrowth [14], wheareas placental ER Stress and AMPK signaling have been reported to be activated in IUGR [12] and inhibited in placentas of large babies [14]. Taken together, these observations in IUGR and fetal overgrowth suggest that both inhibition and activation of placental nutrient-sensing pathways may be associated with similar long-term outcomes, providing the rationale for testing the hypothesis that there is a U-shaped relationship between placental signaling activity and child metabolic outcomes in our cohort. We did observe a modest but significant U-shaped relationship between the phosphorylated to total ratio of AMPK and skinfold thickness, supporting this hypothesis. However, we did not observe a compelling U-shaped relationship between the rest of the placental functional readouts and child outcomes, likley due to characteristics of the study participants in our cohort, which largley were healthy, term pregnant women.
By design, this study assessed healthy women, introducing some limitations, including a lack of diversity of health outcomes in the women (few had GDM) and their children (most are within a normal BMI range). Strengths of this study include the wide range of maternal BMI, the relatively large sample size, and the 4 years of follow-up with the children. We have demonstrated some robust associations between placental signaling activity and, in particular, cardiometabolic outcomes at 4–6 years of age, which warrants confirmation in other metabolically diverse cohorts with follow-up in older children. These new findings show for the first time in humans that placental mTORC1 activation is associated with adiposity at birth and 4–6 years in a complex way, that PKCα is associated with higher systolic blood pressure at 4–6 years, and that AMPK activation is linked to greater adiposity at birth, but reduced skinfold thickness and body weight at 4–6 years of age. These data indicate that placental AMPK and mTOR signaling are linked to cardiometabolic outcomes from birth to 4–6 years of age, which may provide novel insight into the mechanisms underpinning intrauterine programming of metabolic and cardiovascular disease. If replicated in other cohorts and following establishing cause-and-effect in relevant animal models, this information could provide the foundation for future interventions targeting placental function to prevent fetal programming.
Supplementary Material
Acknowledgements
The authors thank M. Martinez (the Healthy Start Study project coordinator, Colorado School of Public Health, University of Colorado) and the Healthy Start team for their hard work and dedication to this study.
Funding
The Healthy Start BabyBUMP Project is supported by grants from the American Heart Association (predoctoral fellowship 14PRE18230008) and by the parent Healthy Start Study (to DD). The Healthy Start Study was supported by the National Institute of Health (R01 DK076648 to DD) and the Colorado Clinical and Translational Sciences Institute (UL1 TR001082) for maternal visits and collection of birth measures. The funders had no influence on the results of the study.
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Supplementary information The online version of this article (https://doi.org/10.1038/s41366-020-0574-y) contains supplementary material, which is available to authorized users.
References
- 1.Skinner AC, Ravanbakht SN, Skelton JA, Perrin EM, & Armstrong SC (2018). Prevalence of obesity and severe obesity in US children, 1999–2016. Pediatrics, e20173459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barker DJ. The developmental origins of insulin resistance. Hormone Res Paediatrics. 2005;64:2–7. [DOI] [PubMed] [Google Scholar]
- 3.Gluckman PD, Hanson MA. The developmental origins of the metabolic syndrome. Trends Endocrinol Metab. 2004;15:183–7. [DOI] [PubMed] [Google Scholar]
- 4.Gluckman PD, Hanson MA, Cooper C. In utero and early-life conditions and adult health and disease. N Engl J Med. 2008;359:1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burton GJ, Fowden AL, Thornburg KL. Placental origins of chronic disease. Physiol Rev. 2016;96:1509–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barker DJ, Gelow J, Thornburg K, Osmond C, Kajantie E, Eriksson JG. The early origins of chronic heart failure: impaired placental growth and initiation of insulin resistance in childhood. Eur J Heart Failure. 2010;12:819–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barker DJ, Thornburg KL, Osmond C, Kajantie E, Eriksson JG. The surface area of the placenta and hypertension in the offspring in later life. Int J Dev Biol. 2010;54:525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barker DJ, Larsen G, Osmond C, Thornburg KL, Kajantie E, Eriksson JG. The placental origins of sudden cardiac death. Int J Epidemiol. 2012;41:1394–9. [DOI] [PubMed] [Google Scholar]
- 9.Jansson T, Powell TL. Role of placental nutrient sensing in developmental programming. Clin Obstet Gynecol. 2013;56:591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, Guilmette J, Luo Z-C, Cloutier A, Wang W-J, Yang M-N, et al. Placental 11β-HSD2 and cardiometabolic health indicators in infancy. Diabetes Care. 2019;42:964–71. [DOI] [PubMed] [Google Scholar]
- 11.Laviola L, Perrini S, Belsanti G, Natalicchio A, Montrone C, Leonardini A, et al. Intrauterine growth restriction in humans is associated with abnormalities in placental insulin-like growth factor signaling. Endocrinology. 2005;146:1498–505. [DOI] [PubMed] [Google Scholar]
- 12.Yung H-w, Calabrese S, Hynx D, Hemmings BA, Cetin I, Charnock-Jones DS, et al. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am J Pathol. 2008;173:451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaccioli F, White V, Capobianco E, Powell TL, Jawerbaum A, Jansson T. Maternal overweight induced by a diet with high content of saturated fat activates placental mTOR and eIF2alpha signaling and increases fetal growth in rats. Biol Reprod. 2013;89:91–11. 96 [DOI] [PubMed] [Google Scholar]
- 14.Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, et al. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metabol. 2013;98:105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J, Guan K-L. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol. 2019;21:63. [DOI] [PubMed] [Google Scholar]
- 16.Rosario FJ, Kanai Y, Powell TL, Jansson T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J Physiol. 2013;591:609–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosario FJ, Powell TL, Jansson T. Mechanistic target of rapamycin (mTOR) regulates trophoblast folate uptake by modulating the cell surface expression of FR-α and the RFC. Sci Reports. 2016;6:31705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosario FJ, Gupta MB, Myatt L, Powell TL, Glenn JP, Cox L, et al. Mechanistic target of rapamycin complex 1 promotes the expression of genes encoding electron transport chain proteins and stimulates oxidative phosphorylation in primary human trophoblast cells by regulating mitochondrial biogenesis. Sci. Reports. 2019;9:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kola B, Grossman AB, & Korbonits M The role of AMP-activated protein kinase in obesity. In: Obesity and metabolism, Vol. 36, Karger Publishers; 2008. pp. 198–211. [DOI] [PubMed] [Google Scholar]
- 20.Sauder KA, Stamatoiu AV, Leshchinskaya E, Ringham BM, Glueck DH, Dabelea D. Cord blood vitamin D levels and early childhood blood pressure: the healthy start study. J Am Heart Assoc. 2019;8:e011485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Starling AP, Brinton JT, Glueck DH, Shapiro AL, Harrod CS, Lynch AM, et al. Associations of maternal BMI and gestational weight gain with neonatal adiposity in the Healthy Start study. Am J Clin Nutrition. 2014;101:302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barker DJ, Godfrey K, Gluckman PD, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. The Lancet. 1993;341:938–41. [DOI] [PubMed] [Google Scholar]
- 23.Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115:e290–e296. [DOI] [PubMed] [Google Scholar]
- 24.Roberts VH, Smith J, McLea SA, Heizer AB, Richardson JL, Myatt L. Effect of increasing maternal body mass index on oxidative and nitrative stress in the human placenta. Placenta. 2009;30:169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saben J, Lindsey F, Zhong Y, Thakali K, Badger T, Andres A, et al. Maternal obesity is associated with a lipotoxic placental environment. Placenta. 2014;35:171–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta M, & Jansson T Novel roles of mTOR signaling in regulating fetal growth. Biol Reprod. 2018;100:872–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y-Y, Rosario FJ, Shehab MA, Powell TL, Gupta MB, & Jansson T Increased ubiquitination and reduced plasma membrane trafficking of placental amino acid transporter SNAT-2 in human IUGR. Clin Sci. 2015;129:1131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tee AR, & Blenis J mTOR, translational control and human disease. Paper presented at the Seminars in cell & developmental biology. 2005. [DOI] [PubMed] [Google Scholar]
- 29.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. [DOI] [PubMed] [Google Scholar]
- 31.Rosario FJ, Jansson N, Kanai Y, Prasad PD, Powell TL, Jansson T. Maternal protein restriction in the rat inhibits placental insulin, mTOR, and STAT3 signaling and down-regulates placental amino acid transporters. Endocrinology. 2011;152:1119–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kavitha JV, Rosario FJ, Nijland MJ, McDonald TJ, Wu G, Kanai Y, et al. Down-regulation of placental mTOR, insulin/IGF-I signaling, and nutrient transporters in response to maternal nutrient restriction in the baboon. FASEB J. 2014;28:1294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9:407–16. [DOI] [PubMed] [Google Scholar]
- 34.Ó’Grada C Fetal origins, childhood development, and famine: a bibliography and literature review. SSRN Elect J. 2012;1–76. 10.2139/ssrn.1980709. [DOI] [Google Scholar]
- 35.Ma Y, Zhu MJ, Uthlaut AB, Nijland MJ, Nathanielsz PW, Hess BW, et al. Upregulation of growth signaling and nutrient transporters in cotyledons of early to mid-gestational nutrient restricted ewes. Placenta. 2011;32:255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martino J, Sebert S, Segura M, Garcia-Valdes L, Florido J, Padilla M, et al. Maternal body weight and gestational diabetes differentially influence placental and pregnancy outcomes. J Clin Endocrinol. 2016;101:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boyle K, Patinkin Z, Shapiro A, Bader C, Vanderlinden L, Kechris K, et al. Maternal obesity alters fatty acid oxidation, AMPK activity, and associated DNA methylation in mesenchymal stem cells from human infants. Mol Metab. 2017;6:1503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konopatskaya O, Poole AW. Protein kinase Cα: disease regulator and therapeutic target. Trends Pharmacol Sci. 2010;31:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drägert K, Bhattacharya I, Pellegrini G, Seebeck P, Azzi A, Brown SA, et al. Deletion of rictor in brain and fat alters peripheral clock gene expression and increases blood pressure. Hypertension. 2015;66:332–9. [DOI] [PubMed] [Google Scholar]
- 40.Tang H, Wu K, Wang J, Vinjamuri S, Gu Y, Song S, et al. Pathogenic role of mTORC1 and mTORC2 in pulmonary hypertension. JACC Basic Transl Sci. 2018;3:744–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.