

ABSTRACT
Introduction: Enteroviruses are common viruses causing a huge number of acute and chronic infections and producing towering economic costs. Similarly, coronaviruses cause seasonal mild infections, epidemics, and even pandemics and can lead to severe respiratory symptoms. It is important to develop broadly acting antiviral molecules to efficiently tackle the infections caused by thes.
Areas covered: This review illuminates the differences and similarities between enteroviruses and coronaviruses and examines the most appealing therapeutic targets to combat both virus groups. Publications of both virus groups and deposited structures discovered through PubMed to March 2021 for viral proteases have been evaluated.
Expert opinion: The main protease of coronaviruses and enteroviruses share similarities in their structure and function. These proteases process their viral polyproteins and thus drugs that bind to the active site have potential to target both virus groups. It is important to develop drugs that target more evolutionarily conserved processes and proteins. Moreover, it is a wise strategy to concentrate on processes that are similar between several virus families.
KEYWORDS: Enterovirus, coronavirus, main protease, 3C protease, SARS-COV-2, COVID-19
1. Introduction to enteroviruses and coronaviruses
1.1. Enteroviruses
Enteroviruses are non-enveloped RNA viruses belonging to the picornaviridae family [1]. EVs are small, icosahedral viruses, about 30 nm in size with single stranded 7–7.5 kb genome (Figure 1). They cause a huge number of acute infections, most of which are associated with mild, flu-like, symptoms [2]. Enteroviruses, however, contribute to more severe diseases when they spread from primary infection sites, i.e from gastrointestinal or respiratory tract, to secondary tissues such as pancreas, heart, liver or brain. The more severe diseases include myocarditis, acute flaccid myelitis, encephalitis, and pancreatitis, and may especially impact on immunodeficient patients and neonatal populations [2,3]. In addition to acute diseases, enteroviruses have also been associated with some chronic diseases such as type 1 diabetes (T1D), asthma, and myocardial infarction [2,4,5] which increases their clinical importance.
Figure 1.
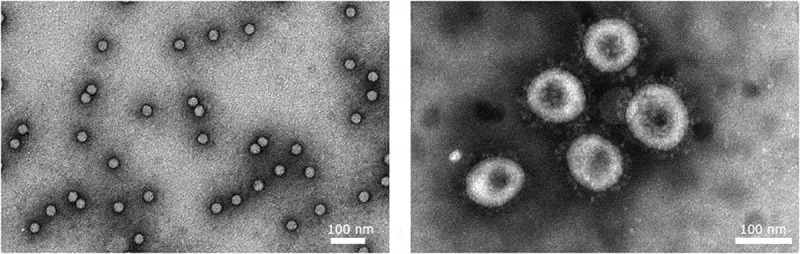
Enteroviruses (CVA9) and coronaviruses (HCoV-OC43) visualized by TEM. Purified Coxsackievirus A9 and HCoV-OC43 were negatively stained using phosphotungstic acid. Courtesy of Sailee Shroff, University of Jyväskylä, Finland
The enterovirus genus consists of 15 different species out of which four enterovirus species (EV-A, EV-B, EV-C and EV-D) and three rhinovirus (RV) species (RV-A, RV-B and RV-C) infect humans [6]. Human enterovirus species include over 100 serotypes of echoviruses, polioviruses, Coxsackieviruses and enteroviruses and over 100 serotypes of rhinovirus species, many of which have emerged during the last 10 years [7]. Although poliovirus has been almost eradicated from the world, several non-polioviruses are causing difficult epidemics all around the world, especially enterovirus-D68 belonging to EV-D group, and enterovirus-A71 belonging to EV-A group [8,9]. Enterovirus-A71 causes hand-foot-and-mouth disease and may lead to difficult neurological symptoms [9]. It has caused several difficult outbreaks especially in Asia. Infections by enterovirus-D68 have caused severe pneumonia and even deaths especially in the United States [8]. Human rhinoviruses are the primary causative agent of the common cold worldwide [10]. They cause mostly mild symptoms but can also cause muscle fatigue and headaches.
Despite their prevalence, currently there are no antivirals on the market to treat enteroviral infections. In addition, vaccines have been produced only against poliovirus and enterovirus-A71, and because of the large number of different serotypes, the production of vaccines against all enteroviruses is not feasible.
Enteroviruses may use cell surface integrins, DAF, CAR, or LDL receptor, or common cell surface molecules such as sialic acid or heparin sulfate as their receptors [11]. Some members of the enteroviruses enter the acidic clathrin endocytic pathway, such as enterovirus-A70, enterovirus-D68, echovirus 6, or rhinoviruses [12,13]. However, many enteroviruses also use a non-acidifying pathway that they can trigger from the specialized lipid microdomains, rafts, to the cytoplasm [1]. The uncoating for enteroviruses starts early in the cytoplasmic endosomes, if not already at the plasma membrane, but continues with a higher pace until around 2 h p.i [1]. For enteroviruses that are dependent on low pH for successful uncoating such as rhinoviruses and enterovirus-A71, entry to early endosomes with mildly acidic conditions are needed to promote dismantling of the virus capsid [16,14]. Many enteroviruses, such as enteroviruses belonging to the EV-B group (Coxsackie B viruses, echovirus 1, echovirus 30, and Coxsackievirus A9) do not need acidification for uncoating [1]. This is also understandable as these viruses infect humans by entering through the gastrointestinal tract and are exposed to very low pH (around 2–3) in the gut. Enteroviruses which are infecting through the upper respiratory tract do not face acidic pH upon entry and perhaps therefore, have evolved to use low pH to promote uncoating. Interestingly, some pH independent viruses, such as echovirus 7, can also enter the acidic pathway, which suggests that the virus encounters other important cues along that pathway [15]. The transfer of the RNA to the cytoplasm occurs through pores which are formed from the released VP4 from the virion capsid, while the hydrophobic N-terminus of the VP1 exposed at the later uncoating stage helps to anchor the virus on the membrane [16]. Host cell factors, such as PLA2G16 have a role in the genome release to the cytoplasm, although we still lack some mechanistic details to understand this step [17]. Rhinoviruses may also cause larger permeability through breakages in the endosomal membrane to boost their entry to the cytoplasm [18].
In the host cell cytoplasm, the enteroviral RNA is first translated as a single polyprotein via internal ribosomal entry site (IRES) in the 5ʹ non-coding region of the virus genome. Viral polyprotein processing leads to the release of several individual proteins that are needed for viral replication as well as capsid assembly [19]. Out of these proteins, the key protein is viral RNA dependent RNA polymerase 3D, which first carries out the uridylylation of viral protein 3B (VPg), which works as a primer in the replication. In addition, 3D polymerase catalyzes the replication of negative RNA strands, which then work as a template for positive strand RNA synthesis, also carried out by 3D polymerase.
The replication of enteroviruses occurs on re-modeled host cell membranes called replication organelles. Their formation requires both viral proteins and host factors. One of the important viral proteins in their formation is the viral protein 3A, which recruits the kinase phosphatidylinositol 4-kinase type IIIβ in the site of replication resulting in the increase in local phosphatidylinositol-4-phosphate lipid levels [20].
Since the enteroviral proteins are translated as a single polyprotein, the individual proteins need to be released from the polyprotein by proteolytic cleavages [21]. The structural viral proteins 0, 3, and 1 are needed for capsid assembly, while non-structural proteins 2A, 2B, 2 C, 3A, 3B, 3 C, and 3D participate in other processes such as replication. The polyprotein processing is carried out by enterovirus encoded proteases 2A and 3 C and a precursor protein 3 CD. The primary cleavage between structural and non-structural protein regions is carried out by 2A, while the 3 C (or the precursor 3 CD) protease carries out the other cleavages [22]. In addition to the polyprotein cleavage, the viral proteases have been shown to cleave different host cell factors in order to promote the infection [23]. In addition to viral proteases, a recent paper suggests that cellular proteases, the calpains, may be important in polyprotein processing [24]. It was shown in vitro that the calpains could process the polyprotein of enteroviruses and release structural viral proteins 1 and 3. Thus, inhibition of calpains may also be a potential antiviral strategy.
During enterovirus assembly in the cytoplasm, three viral structural proteins (0, 1, and 3) assemble first into 60 protomers; then, into 12 pentamers that finally assemble around the viral RNA to form a provirion. Cellular heat shock protein 90 (HSP90) helps to correctly fold the P1 precursor in order to release the individual capsid proteins [25]. In addition, glutathione has been shown to assist in the assembly of pentamers and procapsids [26]. After assembly, new progeny viruses are released from the cell either through lysis or via extracellular vesicles without breaking up the cell.
1.2. Coronaviruses
Coronaviruses, belonging to the order of Nidovirales, circulate among humans causing common cold, reminiscent of rhinovirus infections. HCoV-229E and HCoV-OC43 cause typically seasonal infections [27]. However, from time to time, coronaviruses cause epidemic outbreaks with severe respiratory symptoms and high mortality [16,28]. The first two regional epidemics were caused by SARS-CoV (Severe acute respiratory syndrome) in China and 28 other countries, e.g., Canada, Singapore, and Vietnam between 2002 and 2003, and by MERS-CoV (Middle east respiratory syndrome), mostly in the Arab Peninsula and South Korea between 2012 and 20, but the epidemic still continues at a low pace. In 2019, in Wuhan, China, a novel COVID-19 coronavirus, also called SARS-CoV-2, eventually caused a pandemic which, by spring 2021 has already infected 124.2 million people and killed over 2.73 million people [29]. While SARS-CoV in 2003 was causing infection in the lower respiratory system, SARS-CoV-2 infection is also causing virus spread also from the upper respiratory, nasopharyngeal area, possibly explaining the more efficient spread and worldwide pandemic.
Coronaviruses are also RNA viruses containing a single-strand genome of positive polarity [30]. The size of the genome is huge, one of the largest among RNA viruses, approximately 30 kb. The size of the virus is around 120–160 nm in diameter and, being an enveloped virus, the shape is somewhat irregular, rounded, or oval (Figure 1). The spikes on the envelope, formed by S protein, bring the peculiar corona outlook for the virus. The coronavirus also encodes three other structural proteins, membrane (M) protein, nucleocapsid (N) protein, and envelope (E) protein. The N protein binds to the viral RNA and is involved in viral replication and translation. The M protein, located in the envelope is responsible for the coronavirus assembly. The S-protein is important for binding to the receptor(s) on the cell surface and in fusion with the endosomal membrane to release the viral RNA to the cytoplasm for efficient translation and replication. The small E protein has roles in virus binding to cells, in virus assembly and membrane permeability. The genome altogether encodes for 16 non-structural proteins (Nsp1-16), including the proteases Nsp3, a multi-domain protein containing a papain-like protease ([31] Lei et al., Antiviral Res. 2018) and Nsp5, the main protease, or 3 C-like protease [28].
Coronavirus spike protein binds to the host cell receptors on the plasma membrane. Those receptors include human aminopeptidase N for HCoV-229E and angiotensin-converting enzyme2 (ACE2) for SARS-Cov and SARS CoV-2 [12]. The Spike-ACE2 complex has been solved by Cryo-EM and shows that 16 residues from the S and 20 residues from ACE-2 are involved in the binding [32,33]. The binding residues are conserved between SARS viruses,but due to having more interactions with ACE-2, SARS-CoV-2 was suggested to bind ACE-2 with higher affinity than SARS-CoV [32,33]. Recently, a new player was shown to be involved in binding to the host cells, namely neuropilin receptor (NRP1 and NRP2) [34]. Specific to SARS-CoV-2 and some other highly virulent pathogenic viruses such as Ebola and avian influenza, their spike proteins have a polybasic cleavage site, which, after proteolytic cleavage, exposes a conserved C-terminal motif (RXXROH) which is able to bind and activate neuropilin receptors. Neuropilin receptors are known to be abundantly expressed in the olfactory system, including the neuronal cells, suggesting that neuropilins may truly promote successful infection in the upper respiratory tract and may be the basis of the neurological symptoms in several infected subjects.
Coronaviruses rely on acidic conditions inside the endosomes to gain entry to the host cell cytoplasm [28]. The spike protein contains two domains, S1 responsible for receptor binding and S2 for the fusion. During virus assembly in the host cells, the spike protein is cut by furin in the secretory pathway so that the spike is already primed by one proteolytic cleavage. During virus entry, after receptor binding the spike is primed the second time for fusion by proteolytic cleavage at S2´site to expose the fusion peptide. This second cleavage may occur at neutral pH on the plasma membrane by the host cell TMPRSS2, or after entry to the acidic endosomes by the host cell cathepsin L [35,36]. Exposure of the fusion peptide promotes fusion between the coronavirus envelope with the limiting membrane of the endosomes thus releasing the nucleocapsid to the cytoplasm. The nucleocapsid will then need a second uncoating step to release the nucleocapsid protein from the genome. This happens in cytoplasmic proteasomes by degrading the N protein from around the genome, thus allowing the single strand genome to act as a messenger RNA to start translation in the cytoplasm.
The coronaviruses have a polycistronic genome organization [28]. The structural proteins are encoded in the 3´end of the genome and expressed from a nested set of subgenomic mRNAs after a discontinuous transcription. The replicase proteins are encoded by two 5´end open reading frames 1a and 1b (ORF1a and ORF1b), connected by a ribosomal frameshift site. The translation products of ORF1a and ORF1b are cleaved by viral proteases at 14 sites. The envelope-located spike protein will undergo co-translational processing of the glycoprotein and maturation cleavages along the exocytic pathway on its way to the plasma membrane. The varying activities and kinetics of the viral proteases to release the replicase components and accessory proteins have a strong regulatory effect on the replication and pose a valid target for antiviral action. The synthesis of both negative and positive RNA occurs in double membrane vesicles [37]. The positive strand RNA copies are transferred through a pore through the double membrane vesicles, after which N proteins start encapsidation of the genomic RNA to the assembling virions [38].
The genome is translated into polyproteins pp1a and pp1ab containing the non-structural proteins important for replication and translation. These 16 non-structural proteins are proteolytically processed from the polyproteins by the main protease Mpro [23]. The main protease was originally called 3 C-like proteinase, 3 CLpro due to the fact that it closely resembles the enterovirus 3 C protease. Mpro resides upstream of the ribosomal frameshift side, in the most conserved area of the virus genome. This Mpro is autocatalytically processed from the polyprotein and it further processes all downstream proteins of the replicase polyprotein. The amino-terminal proteins of the replicase polyprotein are released by the action of the viral papain-like cysteine proteinase.
Coronaviruses are pleiomorphic viruses leading to variability in size and shape of the newly assembled virions. In the assembly process, the integral membrane protein M first attaches to the membrane and orchestrates the other proteins to gather in the budding area: N protein, which makes helical arrangements with the viral RNA, spike protein S and E protein [39]. M protein dimers and oligomers induce curvature to the membrane with the help of N, S, and E. It seems that all these proteins together keep the virion size more compact leading to exit of round or oval coronaviruses of a rather compact size range.
2. Similarities between coronaviruses and enteroviruses
From first sight, there is very little in common between coronaviruses and enteroviruses (Table 1). Despite both being RNA viruses and having a single strand RNA genome of positive polarity, most other details differentiate the viruses far apart from each other. First of all, the size difference is huge, enteroviruses being one of the smallest viruses with a diameter of 30 nm and small genome of 7 kb while coronaviruses are one of the largest viruses with genome encoding 30 kb and diameter of 100–140 nm. Furthermore, coronaviruses are enveloped and enteroviruses non-enveloped indicating many differences in their uncoating, entry, and assembly. The roles of non-structural proteins of enteroviruses are rather well known but, for coronaviruses, several non-structural and accessory proteins still lack the information of their function. One major similarity is already known between these virus groups: namely, the main protease and 3 C protease in enteroviruses. In addition, very recently, it was discovered that there may be similarities in the mechanisms to initiate viral RNA synthesis by covalently modifying selected non-structural proteins [40,41]. We thus focus in the next chapter on these similarities, and what is known from wet lab studies with potential drugs targeting both groups. Publications of both virus groups and deposited structures for viral proteases have been thoroughly gone through for this study.
Table 1.
Differences (red) and similarities (green) between enteroviruses and coronaviruses
| Attribute | Enteroviruses | Coronaviruses |
|---|---|---|
| Virion size | small, 30 nm | large, 120–160 nm |
| Genome size | 7–7,5 kb | 30 kb |
| Structure | Non-enveloped | Enveloped |
| Infection route | Fecal-oral and respiratory route | Respiratory route |
| Uncoating/release from vesicles | Largely independent of low pH | Dependent on low pH |
| Genome release | In endosomes, through endosomal membrane | In the cytoplasm, with assistance from proteasomes |
| Genome | ssRNA+ polarity | ssRNA + polarity |
| Polyprotein processing | By viral proteases | By viral proteases |
| Major protease | 3 C protease (3 Cpro) | Main protease (Mpro) |
| Protein-primed RNA synthesis | yridylylation of Nsp9 and Nsp8 | yridylylation of VPg |
| Severe epidemic causing serotypes | EV-D68, EV-A71, EV30 | SARS Cov-1, SARS CoV-2, MERS |
3. Main and 3 C protease of coronaviruses and enteroviruses
3.1. Protease structures
The picornavirus 3 C protease (Enzyme Classification, EC number 3.4.22.28) and SARS-CoV-2 main protease (EC 3.4.22.69) both belong into an enzyme subclass of cysteine endopeptidases (EC 3.4.22). Their three-dimensional structure has resemblance to the famous chymotrypsin-like fold with two beta-barrels (Figure 2). However, active sites in enteroviral 3 C proteases and in coronavirus Mpro differ. In the former, the active site harbors canonical Cys-His-Glu(Asp) catalytic triad where the reactive cysteine acts as a nucleophile triggering the scission of a peptide bond in the substrate. On the contrary, the SARS-CoV-2 main protease is evolutionarily diverged, its active site is composed of Cys-His catalytic dyad. In the free enzyme, the catalytic Cys145 is protonated whereas the proximal His41 is neutral. Upon activation, the His41 abstracts a proton from Cys145 rendering it nucleophilic, followed by the attack of sulfur to carbonyl carbon of the scissile peptide bond between P1-P1ʹ [42].
Figure 2.
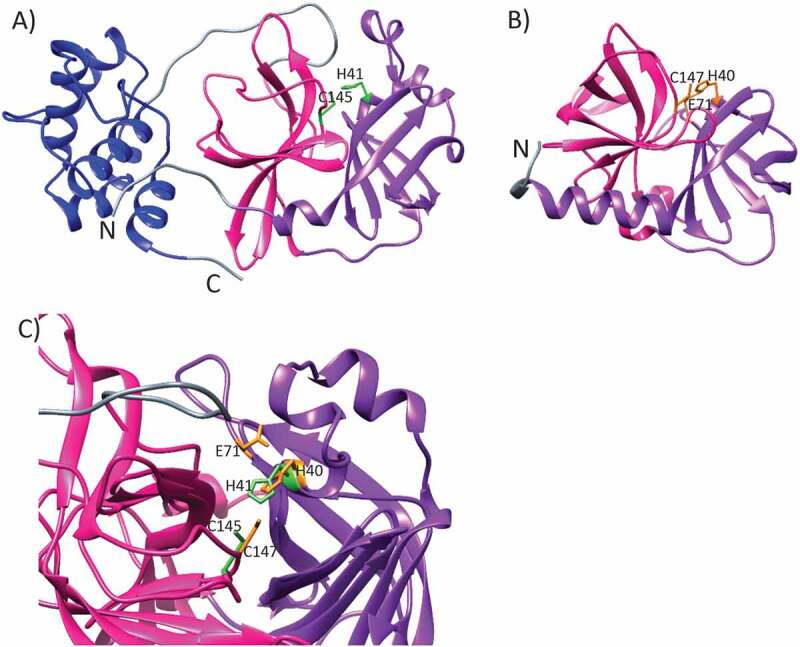
The ribbon representations of (a) SARS-CoV-2 main protease (PDB id 7K3T, DOI: 10.2210/pdb7K3T/pdb) and (b) Human Enterovirus 71 3 C protease (PDB id 3OSY [43],). The two β-barrel domains I and II, participating in the catalytic activity, are shown with purple and pink, respectively. The dimerization domain, found in SARS-CoV-2 main protease only, is colored with blue. The linker regions are colored with gray. The side chains of residues critical for the catalytic activity are marked with light green and orange for SARS-CoV-2 Mpro and enterovirus 71 3 C protease, respectively. (c) SARS-CoV-2 Mpro superimposed with the enterovirus 71 3 C protease. The catalytic sites are denoted as in A) and B). It should be noted that for the sake of clarity, only one protomer of SARS-CoV-2 dimer is shown in A) and C)
Quite strikingly, at the time of writing this review article, there exist almost 300 (297) high resolution 3D structures of SARS-CoV-2 main protease in the protein data bank, PDB, either determined as a free enzyme or as complexes with non-inhibitory fragments or together with numerous inhibitors developed by the scientific community. Recent crystal structures of SARS-CoV-2 Mpro highlight a homodimeric structure, yet, the homodimerization is elemental for the catalytic activity of coronavirus Mpro [42,44,45]. Each monomer comprises three domains. The domains I (residues 8–101) and II (residues 102–184) participate in the catalytic activity with a typical beta-barrel chymotrypsin fold. The domain III (res. 201–303) is composed of five α-helices and is essential for the dimerization. The domain III interacts mostly with the domain II as well as with the so-called N-finger from the other protomer. The dimerization of SARS-CoV-2 Mpro is mainly driven by the salt-bridge established between the negatively charged Glu290 from one protomer and positively charged Arg4 from the other [46]. Dimerization is a prerequisite for the catalytic activity of SARS-CoV-2 Mpro. Indeed, the so-called N-finger, composed of the most N-terminal residues of one protomer, makes contacts with the Glu166 of the other protomer to complete the active site.
The structure is highly similar to SARS-CoV-1 Mpro with root-mean-square-deviation of 0.53 Å between Cα atoms, as expected owing to 96% amino acid sequence identity between the two enzymes. The active site is 100% conserved between SARS-CoV-1 and SARS-CoV-2 Mpro [45].
3.2. Molecules targeting the active site of the protease
Proteases are important players in the virus replication of most viruses. The 3 C protease and Mpro are crucial for the replication of enteroviruses and coronaviruses, respectively. When designing efficient drugs against virus infection, these evolutionarily conserved proteases have been considered a valid target.
For Rhinoviruses, peptidomimetics drugs were developed against the 3 C protease, based on the active site structure and cleavage specificity. One such peptidomimetic drug, rupintrivir (AG7088) showed great in vitro efficacy against replication of all rhinovirus serotypes, and wider activity also in picornaviruses [47] and it advanced to phase II trial [48]. Rupintrivir has a lactam ring to mimic Gln at the P1 position and an a,b-unsaturated ester at P1´as a Michael acceptor to form a covalent bond with the Cys in the active site. The P1 Gln site in the substrate is shared between enteroviruses, a hydrophobic residue at the P2 position, and a small amino acid residue at the P1´position.
However, there is no sequence homology between coronaviruses and enteroviruses at the active site. Therefore, antivirals fitting the active site demonstrate varying efficacies for coronavirus or enterovirus infections. AG7088 was not functional for coronaviruses, while rupintrivir triggered further development of coronavirus protease inhibitors and some analogues developed since then have exhibited greater potency [49]. Lee et al. [46] presented peptidomimetic inhibitors that had shown earlier potency for Coxsackievirus B3 3 Cpro but now showed activity against CoV 229E and SARS-CoV Mpro as well. They solved crystal structures of 3 C and Mpro proteases with similar inhibitors to understand the structural basis of inhibitor action. The four peptidomimetic compounds tested showed lower activity for Coxsackievirus B3 3 Cpro in vitro than for SARS-CoV Mpro o due to a more open active site of 3 Cpro but also due to other differences in the binding site. These inhibitors showed 10–20 times higher IC50 for Coxsackievirus B3 in comparison to SARS-CoV-2. In these inhibitors the lactam ring mimicking Gln provides-fold better inhibitory action than Gln owing to rigidification of the flexible Gln side-chain through the ring closure that reduces the loss of conformational entropy upon binding to the protease. However, the isobutyl (leucine) in Mpro P2 position was reasoned later in the paper by Zhang et al. [50] to be too small to fill the S2 pocket of enterovirus-A71 3 Cpro and Coxsackievirus B3 3 Cpro.
Further improvement for peptidomimetics came from Zhang et al. [50] who introduced capped dipeptide α-ketoamides as broad-spectrum inhibitors for the main protease of SARS-CoV and 3 C proteases of Coxsackievirus B3 and enterovirus-A71. Six crystal structures of protease-inhibitor complexes were determined in this study. Recombinant proteases were tested against inhibitors and for viral infections of several enteroviruses in cell culture using luciferase-based reporter assays. The α-ketoamide warhead (-CO-CO-NH-) proved to be better than Michael acceptor or aldehydes because of containing two acceptors for hydrogen bonding. Furthermore, they found that the S2 pocket showed variations between enzymes: The S2 pocket is large and covered in the SARS-CoV Mpro but large and open in enteroviruses. In SARS-CoV-2 Mpro S2 is a deep hydrophobic pocket, while enteroviruses lacks a lid, i.e., it is open to one side and thus has fewer interaction points. Therefore, large aromatic substituents such as benzyl are favored by enteroviruses. The cyclohexylmethyl proved to be best for P2 and was most broadly active. A cyclopentylmethyl substitutent in P2 showed near-equipotency against coronaviruses and enteroviruses. The chosen lead compound showed submicromolar IC50 values for Coxsackievirus B3 3 Cpro and SARS-CoV Mpro. Also, in infected cell cultures, it showed values 0.7 µM or below for human rhinovirus 2, human rhinovirus 14, and enterovirus-D68 with the selectivity values over 27.
Covalently bound irreversible inhibitors were also introduced, including chloromethylketones [51]. However, they proved to be potentially cytotoxic. Already during the first SARS epidemic, a compound PF-00835231, a hydroxymethylketone was developed and showed potency, but after the 2003 epidemic, the clinical testing was stopped. This drug is an irreversible, ketone-based inhibitor, showing good activity against SARS-CoV-1 infection and good activity toward SARS-CoV-2 protease. The drug possesses acceptable solubility, stability in plasma, and low in vitro and in vivo clearances.
Several high-throughput screens have provided novel drugs to inhibit 3 C proteases. Kuo et al. [52] performed a high-throughput screening containing 6800 small-molecule-compounds. One candidate inhibited picornaviruses and CoV equally: a compound containing dihydropyrazole ring with three substituents, two phenyl groups and N-butyl-benzimidazolylamino-toluene. Also, analogues of this showed good potency against picornaviruses and coronaviruses. These are competitive inhibitors indicating that they bind to the active site. Several other molecules based on this hit were searched from other libraries and tested. This testing revealed several drugs inhibiting both SARS-CoV, HCov-229, Coxsackievirus B3, enterovirus-A71, and human rhinovirus 14 with low µM concentration.
Blanchard et al. [53] screened 50,000 drug-like small molecules with a FRET-based assay using purified SARS-CoV main protease. Five novel small molecules were chosen for further studies from this High-throughput screening. One drug, pyridinyl thiophene ester showed equal activity against both HAV (Hepatitis A virus) belonging to picornaviruses and SARS-CoV with IC50 0.5 µM.
Ma et al. [54] screened through a focused Selleckchem bioactive compound library of protease inhibitors using a FRET-based enzymatic assay for SARS-CoV-2. The screen produced proteasome inhibitors, HIV protease inhibitors, y-secretase inhibitors, hepatitis C virus protease, and DPP-4 inhibitors, some serine protease inhibitors, cathepsin and calpain protease inhibitors. The hits included simpeprevir, boceprevir, narlaprevir, and MG-132. However, as MG132 was not active against enterovirus-A71 2A or 3 C proteases and as it showed high cytotoxicity, they widened the screen for more specific calpain inhibitors, which led to very interesting finding that several calpain inhibitors showed potency against proteases for both virus groups: calpain inhibitors II and XII showed good inhibition (0.97 µM and 0.45 µM, respectively,). As calpain protease inhibitors have been shown to reduce coronavirus and enterovirus infections in earlier studies, it was not surprising though that some calpain inhibitors were active against the 3 C and main proteases [55–58]. Ma et al. [54] also included in their studies the already discovered main protease inhibitor, GC-376, which was designed earlier to target several viral proteases including MERS-CoV, norovirus, and feline infectious peritonitis virus. GC-376 showed to be very potent for SARS-CoV-2 in 0.03 µM concentration and shifted the melting curve by 18.3°C upon binding in a thermal shift assay. GC-376 proved to be active also for enterovirus-A71 3 C but not for 2A.
3.3. Structural aspects of active site targeting
The SARS-CoV-2 Mpro prefers substrates Leu(P2)-Gln(P1) ↮ Ser/Ala/Gly(P1ʹ), where ↮ indicates the scissile peptide bond. Indeed, the autocleavage of SARS-CoV-2 Mpro o takes place when C-terminal substrate Ser301-Gln306 occupies the active site, that is Ser301(P6)-Gly302(P5)-Val303(P4)-Thr304(P3)-Phe305(P2)-Gln306(P1). It is notable that only the C-terminal autocleavage substrate contains phenylalanine in the position P2, whereas this site is otherwise occupied with leucine or valine [59]. The active site is depicted in Figure 3(a), where the structure of SARS-CoV-2 Mpro is shown in complex with a potential SARS-CoV-2 Mpro o antiviral, a Michael acceptor inhibitor aka N3 that was originally developed using computer-aided drug design toward SARS-CoV-1 as well as MERS-CoV Mpro’s [60] (PDB id 7BQY). More specifically, the hydrogen bond is established between the N-terminus (Ser1) of protomer B and the Oε1 atom of Glu166 of protomer A upon dimerization. This specific interaction between two protomers complements the substrate binding site, S1, which accommodates the substrate peptide P1 (Figure 3(a)).
Figure 3.
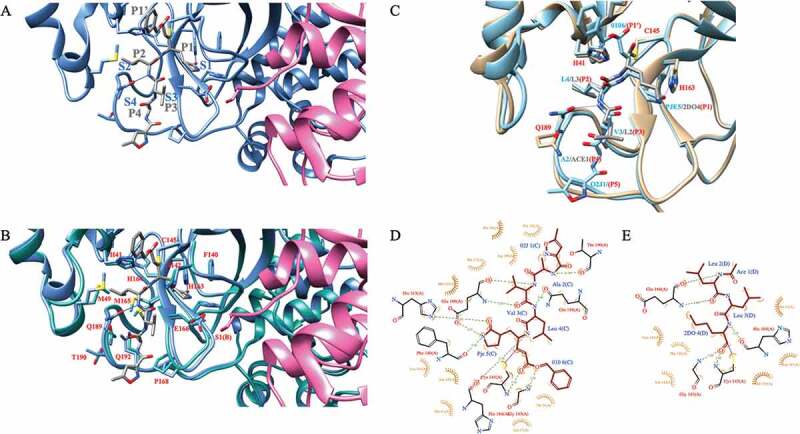
A) SARS-CoV-2 Mpro in complex with a Michael acceptor inhibitor, N3 (PDB id 7BQY). The protomer A of Mpro is shown in cornflower blue whereas the protomer B is shown in pink. The backbone of N3 is shown in gray. Subsite nomenclature, from the scissile bond in the substrate, toward the N-terminus, is P1-P4 and toward C-terminus P1ʹ (gray). Complementary sites at the protease active site are S1-S4 and S1ʹ, respectively (blue). B) SARS-CoV-2 Mpro (cornflower blue) in complex with N3 (gray) overlaid with free SARS-CoV-2 main protease (light sea green; PDB id 6YB7). The protomer B is shown in pink. Key residues are highlighted and marked in red using single amino acid codes and numbering. Hydrogen bonds from Gln189 and His163 side chains to the ligand are shown in blue line. C) SARS-CoV-2 Mpro (light blue) in complex with N3 (main chain shown in light blue) overlaid with the structure of Mpro (light brown) in complex with the calpain I inhibitor (main chain shown in gray). D) 2D ligand–protein interaction diagram for SARS-CoV-2 Mpro: N3 complex. The diagram was generated with the LigPlot+ software [70]. E) 2D ligand–protein interaction diagram for SARS-CoV-2 M protease in complex with the calpain I inhibitor. 2DO is equal to (2S)-2-aminohexane-1,1-diol; ACE is acetyl group; PJE is (E,4S)-4-azanyl-5-[(3S)-2-oxidanylidenepyrrolidin-3-yl]pent-2-enoic acid; 010 is phenylmethanol; 02 J is 5-methyl-1,2-oxazole-3-carboxyclic acid. All SARS-CoV-2 Mpro figures were generated with the Chimera software package (DOI: 10.1002/jcc.20084)
N3 is a covalent inhibitor, which establishes a covalent bond from its vinyl group with the C145 ◸-sulfur atom of protomer A in SARS-CoV-2 Mpro [44]. The structure with N3 was the first complex structure of the main protease of the new SARS-CoV-2 virus with the potential inhibitor. Since then, numerous complex structures with different and often more potent inhibitors have been published (see Table 1). Upon the binding of inhibitor N3 to the active site of the free enzyme (PDB id 6YB7), several residues, most notably, Met49, Met165 as well as Gln189 undergo a dramatic conformational change to avoid clashing with the leucine at P2 and to accommodate the ligand (Figure 3(b)). Gln189 creates a hydrogen bond with the leucine backbone amide group.
Owing to the similarity between 3 C proteases of enteroviruses and SARS-CoV-2 Nsp5, it is likely that antivirals, most notably the calpain inhibitors, are shown to be effective toward enteroviral infections and carry potential also toward COVID-19 infections. Indeed, a recent, yet unpublished SARS-CoV-2 Mpro structure in a complex with the Calpain I inhibitor is shown in Figure 3(c) (PDB id 7LBN). The complex structure is overlaid with the structure of SARS-CoV-2 in complex with N3 (PDB id 7BQY). It is evident that both ligands establish a covalent linkage between the catalytic Cys145 ◸-sulfur and carbon atom of the ligand. However, due to the long hydrophobic alkyl chain at P1 position, its accommodation to the S1 site is driven by hydrophobic interactions. Also, the orientation of Gln189 side-chain is different, preventing the formation of an hydrogen bond between the Gln189 side-chain carbonyl group and the amide group of leucine (Figure 3(c-e)).
4. Similar start of viral RNA synthesis for enteroviruses and coronaviruses?
Enteroviruses are known to trigger their RNA synthesis by covalently modifying one of their non-structural proteins, the VPg (Viral protein genome-linked) protein [61]. First, VPg is cleaved from the large polyprotein by the viral protease precursor 3 CD. Then, viral 3D polymerase will covalently link the VPg peptide with uridine monophosphate resulting in a VPg-pU. This peptide is then linked with another monophosphate to create a dinucleotide that will then bind to the viral poly(A)template to start the viral synthesis [62]. VPg will remain covalently attached to the forming genomic RNA. Now, very recently, it was discovered that an analogous transfer of a nucleoside monophosphate to a nonstructural protein 9 is essential for coronaviruses replication [40] The non-structural protein 9 is an RNA-binding protein and known to be involved in coronavirus replication [63]. It was discovered that a Nidovirus-specific RdRp-associated Nucleotidyl transferase domain (NiRAN) in a nonstructural protein 12 has catalytic activity to transfer a nucleoside monophosphate to the amino terminus of the non-structural protein 9 [40]. This transfer preferentially uses uridine nucleotides, although the NiRAN domain is also ableto catalyze other nucleotides [40]. Before the transfer, similar to enteroviruses, the main protease releases both the nonstructural proteins 12 and 9 from the polyprotein. In addition to Nsp9, similar catalytic activity of NiRAN in the non-structural protein 12 was suggested to also involve the non-structural protein 8 (Nsp8; 41). In this recent article by Shannon and coworkers (2021), Nsp12 was shown to uridylate a viral cofactor of non-structural protein 8, thus forming a UMP-Nsp8 covalent intermediate that can prime RNA synthesis from a poly(A) template. Importantly, a 5´-triphosphate analogue, AT-9010 was shown to bind tightly to NiRAN and inhibit uridylylation of Nsp8 and RNA synthesis. AT-9010 is presently in phase II clinical trials and thus is a promising antiviral candidate for coronaviruses. It will be interesting to find out in future studies, whether the functions of the primed nonstructural proteins 8 and 9 are similar, and if the priming of the non-structural protein 9 can be also halted with AT-9010.
5. Conclusions
It is important to develop antiviral drugs to combat present and future epidemics and pandemics. Vaccines may need constant development because of emerging mutations that potentially avoid available options. This is especially true with influenza and SARS-CoV-2, although the latter shows a lower rate of mutagenesis in comparison to influenza. There are available vaccines for enteroviruses such as polio and some vaccines are available for enterovirus-A71. A multivalent vaccine against all Coxsackie B viruses is being developed [65] but it is not a solution to newly emerging enteroviruses because vaccines are strictly specific to the epitopes presented. It is important to develop drugs that target more evolutionarily conserved processes and proteins. In addition, it is a wise strategy to concentrate on processes that would be similar between several virus families. Here we have concentrated on the similarities between otherwise quite distant viruses, coronaviruses and enteroviruses. Although some molecules may turn up in the future to inhibit both virus groups directly or to inhibit cell binding, there is present proof that both virus groups may be targeted with similar drugs inhibiting the proteases that proteolytically process the viral polyprotein. The main protease in coronaviruses and 3 C protease in enteroviruses are similar enough to be targeted with some selected molecules. Here, we have gone through the most promising present molecules that show activity toward both proteases. Clearly, it is challenging to have equimolar effects on proteases in both virus groups as the active sites between viruses are not identical. However, some molecules do show good inhibitory efficacy against proteases from members of both virus groups [50, Table 1].
An exciting development to inhibit protease activity has emerged from inhibiting the host cell calpain proteases. Calpain proteases are capable of proteolytically processing several target molecules, also including viral polyprotein that has been shown for enterovirus polyprotein [24]. Importantly, inhibitors against calpain proteases also efficiently inhibit the viral 3 C and M proteases [54]. If it turns out that calpain proteases can also contribute to processing of coronavirus polyproteins, inhibiting calpain proteases may prove to be a significant and more efficient strategy to cut down polyprotein processing and virus infection in cells and tissues.
6. Expert opinion
We clearly need efficient antivirals for SARS-COV-2. Present efforts to make vaccines widely available and increase their uptake are not rapid enough. We also need drugs that alleviate symptoms and complications for patients in intensive care units. In addition to coronaviruses, enteroviruses lack clinically approved antiviral drugs. Enteroviruses also cause difficult acute and chronic states, which sometimes need hospitalization. It would be great if we could find broadly acting drugs that can shorten the virus infection for coronaviruses and enteroviruses and possibly other virus groups and thus decrease the symptoms and hospitalization periods, even mortality. Despite possible future vaccines, several people could still get infected and need other means to stop the infection.
While there are mostly differences between the groups of enteroviruses and coronaviruses, there are also striking similarities between one of the proteases, which cut the viral polyprotein, namely the main protease in coronaviruses and 3 C protease in enteroviruses (Table 1). Both virus groups also have other proteases, like the papain-like protease in coronaviruses, and 2A protease in enteroviruses, but those proteases have not been declared to share similarities with each other. The main/3 C proteases share a rather similar active site. Also, the unique substrate preference for glutamine at the P1 site (Leu-Gln/Ser, Ala, and Gly) makes the viral proteases different from host cell proteases. This has been considered to offer a unique possibility to create inhibitors that show less or no inhibitory activity against cellular proteases. Several molecules have already been discovered to exert inhibitory activity on both groups, although the differences in the binding pockets between enteroviruses and coronaviruses lead to medium to large differences in the efficiencies against both groups (Table 2). Some molecules, like alpha-ketoamides, however, have been found to show equimolar efficiency against both protease types [50]. The active site of the protease is wider in enteroviruses and without a lid over the active site, indicating that for a good fit for enteroviruses, the inhibitor needs to have bulkier groups at specific sites. This again may lead to lower binding activity and thus inhibitory activity for coronaviruses. Thus, it seems compromises need to be made in order to achieve great dual efficacy for the optimal inhibitors.
Table 2.
Examples of efficacies of drugs against the enterovirus 3 C protease (3 Cpro) and coronavirus main protease (Mpro)
| Drug | 3 Cpro | Mpro |
|---|---|---|
| alpha-ketoamide (11a) [50] | 6.56 ± 3.10 (CVB3) | 1.95 ± 0.24 |
| alpha-ketoamide (11 r) [50] | 0.95 ± 0.15 (CVB3) | 0.71 ± 0.36 |
| alpha-ketoamide (11 u) [50] | 1.93 ± 0.43 (CVB3) | 1.27 ± 0.34 |
| PF-00835231 [51] | 1.70 ± 1.00 (HRV) | 0.004 ± 0.0003 |
| 43,146 [52] | 5.40 ±- 0.20 (CVB3) | 8.10 ± 0.90 |
| PSI [54] | 13.74 ± 3.86 (EV-A71) | 10.38 ± 2.90* |
| GC-376 [54] | 0.14 ± 0.03 (EV-A71) | 0.03 ± 0.01* |
| Calpain inhibitor II [54] | >20 (EV-A71) | 0.97 ± 0.27* |
| Calpain inhibitor XII [54] | >20 (EV-A71) | 0.45 ± 0.06* |
| MAC-5576 [53] | 0.54 ± 0.09 (HAV) | 0.50 ± 0,.0 |
| MAC-22,272 [53] | 0.90 ± 0.10 (HAV) | 2.60 ± 0,.0 |
IC50 valuesIC50 values
IC50 values (µM) of drugs tested against marked enterovirus 3 Cpro and SARS-CoV-1 Mpro. *Tested against SARS-CoV-2
Another approach is suggested by the recent findings that inhibitors against cellular calpain proteases also inhibit replication of coronaviruses and enteroviruses [55–58]. While there were very few details available for a long time, what may be the target of calpain inhibitors for both virus groups, some information has been recently discovered.
Calpains are papain-like cysteine proteinases, which are ubiquitous in several cell types [65,64]. There are altogether 15 calpains expressed in humans, the calpains 1 and 2 being the canonical types that have mostly been studied in detail. Calpains are expressed as proenzymes in the cytoplasm and are activated by elevated Ca2+ concentration and presumably also due to membrane association locally. Calpains have several cellular targets belonging to cytoskeletal proteins, transcription factors, and signaling proteins to name but a few. Instead of totally degrading their target proteins, calpains undergo limited proteolytic processing of the target sites, e.g., inter-domain unstructured sites in their targets. Although some prediction can be made about the substrates to be processed, there is no clear consensus sequence, but rather the 3D structure of the target substrate plays a more important role.
Inhibition of calpains have been shown to lead to marked inhibition of enterovirus infection [55]. The inhibitory action was earlier suggested targeting early replication/translation of echovirus 1 as well as vesicle trafficking, necrosis, and apoptosis during Coxsackievirus B3 and B4 infection [55,56,65]. The molecular mechanisms of action were not detailed until recently, Laajala et al. showed that calpain proteases can participate in the proteolytic processing of the viral polyprotein [24]. Thus, calpains could contribute to the same action as what is performed by the 3 C viral protease. Indeed, in the study by Laajala et al. [24], the polyprotein was processed by calpain proteases in vitro to deliver capsid proteins with very similar but not identical cutting site between the capsid proteins. The study also showed that the used calpain inhibitors I and II totally inhibited the action of expressed and purified viral 3 C protease. This indicates that calpain inhibitors could knock down the activity of both viral and cellular proteases. If calpain proteases could participate in real infection also in the polyprotein processing, inhibiting calpain proteases would be important and efficient as it would then knock down the total polyprotein processing to provide structural proteins in enteroviruses.
For coronaviruses, it has not yet been shown if calpain inhibitors can proteolytically process the parts of the polyprotein that are executed by the main protease or papain-like protease. If parts of the coronavirus polyprotein could be processed by host cell calpains, then inhibiting calpain proteases could more efficiently stop the polyprotein processing altogether. Recent finding that calpain inhibitors II and XII can inhibit the action of the main protease [54] suggests further that, as for enteroviruses, it is quite likely that calpain proteases can help the coronaviruses to process the polyprotein. That could explain, at least partially, the inhibitory action of calpain inhibitors for coronavirus infection.
Inhibition of calpain proteases has also shown positive effects at alleviating inflammation: in a pre-clinical experiment on mice, BLD-2660, a new, synthetic, orally administered calpain 1 inhibitor effectively lowered the pro-inflammatory IL-6 levels, both at protein and transcriptional level (https://ichgcp.net/fi/clinical-trials-registry/NCT04334460). The efficacy was demonstrated in a bleomycin-induced lung injury model. After this preclinical proof, a phase 2, randomized, double blinded, Placebo-controlled human study is under way to see if BLD-2660 as an add-on therapy will alleviate the difficult symptoms among hospitalized COVID-19 patients.
In addition to inhibiting proteases through broadly acting novel drugs or calpain inhibitors targeting both viral and cellular proteases, there are also other possibilities to find common targets between coronaviruses and enteroviruses. The recently discovered similarity at the start of the viral RNA synthesis between coronaviruses and enteroviruses may provide future possibilities to target both virus groups (Table 1). The present drug targeting the NiRAN domain in the coronavirus structural protein 12, AT-9010, will probably not work as the NiRAN domain is specific to the Nidovirales group where coronaviruses belong. However, perhaps other targets in the RNA synthesis startup may offer future possibilities for broadly acting drugs.
The first step during infection involves specific binding of the virions to the respective receptors on the plasma membrane. By screening various molecular libraries containing novel or FDA-approved drugs, it is possible to find molecules that bind to the virion surface, either the spike protein in the coronavirus or various binding pockets on the enterovirus surface. If the molecules bind to the site that blocks receptor binding, the efficiency of gaining entry to cells will be dramatically dropped. The binding may also interfere with the proteolytic processing of the furin cleavage site in the spike protein.
-Enteroviruses have been shown to possess more than one binding pocket that are associated with stability and receptor binding of the virions. The canonical hydrophobic pocket of enteroviruses has long been considered to be involved with the stability of the viruses [66]. The aliphatic fatty acids inside the pockets are typically partially or totally lost during the opening of the virion structure and RNA release. Therefore, various drugs have been developed to replace the fatty acid with a stronger binder to confer stability and possibly to interfere with receptor binding and cell entry. After development of Pleconaril, several other molecules have also been developed with great efficacy as inhibitors of infection [67]. In addition to the hydrophobic pocket found underneath the structural protein 1 in every protomer, 60 sites in total for 1 enterovirus, another druggable pocket was also recently discovered involving another area on the virus surface and found in both rhinoviruses and other and enteroviruses [68]. There will probably be more discoveries of new druggable areas in the enteroviruses as well as coronaviruses. It will thus be important to screen molecular libraries and search for drugs that show the potential of inhibiting several viruses. In addition to finding such molecules, it will be important to understand the mechanism of action behind the inhibitory effect. This will then help to develop even more effective future drugs with suitable pharmacokinetics and real use as future drugs.
Funding Statement
This work was supported by the grant from the Academy of Finland [#336487].
Article highlights
There are no clinically approved, widely available antivirals for enteroviruses, but Remdesivir is approved for coronavirus treatment.
Despite differences in size and shape and being enveloped and non-enveloped respectively, coronaviruses and enteroviruses share a very similar protease [the main protease/3C-like protease in coronaviruses, and the 3C protease in enteroviruses].
The 3C/M proteases share a similar but not identical active site, which serves as a good therapeutic target for both virus groups.
The proteases process viral polyproteins to release viral capsid proteins or viral proteins assisting virus replication/translation.
Some molecules can efficiently inhibit both proteases; although, depending on the choice of the critical amino acids in the binding pocket, the drugs may be more efficient on a particular virus group protease.
Calpain proteases share similarities in proteolytic processing, hence drugs acting on calpains show activity toward enterovirus and coronavirus proteases.
Calpain inhibition may prove to be a method of choice to silence both viral proteases and host cell proteases that may participate in viral polyprotein processing.
It is important to develop drugs that target more evolutionarily conserved processes and proteins.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Article highlights
There are no clinically approved, widely available antivirals for enteroviruses, but Remdesivir is approved for coronavirus treatment.
Despite differences in size and shape and being enveloped and non-enveloped respectively, coronaviruses and enteroviruses share a very similar protease [the main protease/3C-like protease in coronaviruses, and the 3C protease in enteroviruses].
The 3C/M proteases share a similar but not identical active site, which serves as a good therapeutic target for both virus groups.
The proteases process viral polyproteins to release viral capsid proteins or viral proteins assisting virus replication/translation.
Some molecules can efficiently inhibit both proteases; although, depending on the choice of the critical amino acids in the binding pocket, the drugs may be more efficient on a particular virus group protease.
Calpain proteases share similarities in proteolytic processing, hence drugs acting on calpains show activity toward enterovirus and coronavirus proteases. Calpain inhibition may prove to be a method of choice to silence both viral proteases and host cell proteases that may participate in viral polyprotein processing.
It is important to develop drugs that target more evolutionarily conserved processes and proteins.
Reviewer disclosures
Peer reviewers in this manuscript have no relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Marjomaki V, Turkki P, Huttunen M.. Infectious entry pathway of enterovirus B species. Viruses. 2015;7(12):6387–6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tapparel C, Siegrist F, Petty TJ, et al. Picornavirus and enterovirus diversity with associated human diseases. Infect Genet Evol. 2013;14:282–293. [DOI] [PubMed] [Google Scholar]
- 3.Huber S, Ramsingh AI. Coxsackievirus-Induced Pancreatitis. Viral Immunol. 2004;17:358–369. [DOI] [PubMed] [Google Scholar]
- 4.Laitinen OH, Honkanen H, Pakkanen O, et al. Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes. 2014;63:446–455. [DOI] [PubMed] [Google Scholar]
- 5.Roivainen M, Alfthan G, Jousilahti P, et al. Enterovirus infections as a possible risk factor for myocardial infarction. Circulation. 1998;98:2534–2537. [DOI] [PubMed] [Google Scholar]
- 6.International Committee on Taxonomy of Viruses . ICTV master species list 2019 v2. Checklist dataset; 2019. doi: 10.15468/i4jnfv. Available from: https://www.gbif.org/dataset/e01b0cbb-a10a-420c-b5f3-a3b20cc266ad [DOI] [Google Scholar]
- 7.Harvala H, Jasir A, Penttinen P, et al. Surveillance and laboratory detection for non-polio enteroviruses in the European Union/European Economic Area, 2016. Euro Surveill. 2017;22. DOI: 10.2807/1560-00807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention . Clusters of acute respiratory illness associated with human enterovirus 68 —Asia, Europe, and United States, 2008 –2010. MMWR Morb Mortal Wkly Rep. 2011;60:1301–1304. [PubMed] [Google Scholar]
- 9.Hu Y, Jiang L, Peng HL. Clinical analysis of 134 children with nervous system damage caused by enterovirus 71 infection. Pediatr Infect Dis J. 2015;34:718–723. pmid:25860536 [DOI] [PubMed] [Google Scholar]
- 10.Royston L, Tapparel C. Rhinoviruses and respiratory enteroviruses: not as simple as ABC. Viruses. 2016;8(1). DOI: 10.3390/v8010016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossmann MG, He Y, Kuhn RJ. Picornavirus-receptor interactions. Trends Microbiol. 2002;10(7):324–331. [DOI] [PubMed] [Google Scholar]
- 12.Baggen J, Thibaut HJ, Strating JRPM, et al. The life cycle of non-polio enteroviruses and how to target it. Nat Rev Microbiol. 2018;16(6):368–381. [DOI] [PubMed] [Google Scholar]
- 13.Fuchs R, Blaas D. Uncoating of human rhinoviruses. Rev Med Virol. 2010;20(5):281–297. [DOI] [PubMed] [Google Scholar]
- 14.Tuthill TJ, Groppelli E, Hogle JM, et al. Picornaviruses. Curr Top Microbiol Immunol. 2010;343:43–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim C, Bergelson JM. Echovirus 7 entry into polarized intestinal epithelial cells requires clathrin and Rab7. mBio. 2012;3(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panjwani A, Strauss M, Gold S, et al. Capsid protein VP4 of human rhinovirus induces membrane permeability by the formation of a size-selective multimeric pore. PLoS Pathog. 2014;10(8):e1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staring J, von Castelmur E, Blomen VA, et al. PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nature. 2017;541(7637):412–416. [DOI] [PubMed] [Google Scholar]
- 18.Brabec M, Schober D, Wagner E, et al. Opening of size-selective pores in endosomes during human rhinovirus serotype 2 in vivo uncoating monitored by single-organelle flow analysis. J Virol. 2005;79(2):1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Der Linden L, Wolthers KC, Van Kuppeveld FJ. Replication and inhibitors of enteroviruses and parechoviruses. Viruses. 2015;7(8):4529–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsu NY, Ilnytska O, Belov G, et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141(5):799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palmenberg AC. Proteolytic processing of picornaviral polyprotein. Annu Rev Microbiol. 1990;44:603–623. [DOI] [PubMed] [Google Scholar]
- 22.Anand K, Ziebuhr J, Wadhwani P, et al. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–1767. [DOI] [PubMed] [Google Scholar]
- 23.Laitinen OH, Svedin E, Kapell S, et al. Enteroviral proteases: structure, host interactions and pathogenicity. Rev Med Virol. 2016;26:251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laajala M, Hankaniemi MM, Maatta JAE, et al. Calpains can cleave structural proteins from the enterovirus polyprotein. Viruses. 2019;11. DOI: 10.3390/v11121106 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study was the first to show that calpains can proteolytically process enterovirus polyprotein. They also showed that calpain inhibitors also inhibit 3C and 2A viral proteases.
- 25.Geller R, Vignuzzi M, Andino R, et al. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. 2007;21:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma HC, Liu Y, Wang C, et al. An Interaction between glutathione and the capsid is required for the morphogenesis of C-cluster enteroviruses. PLoS Pathog. 2014;10:e1004052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.V’kovski P, Kratzel A, Steiner S, et al. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021;19:155–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.WHO Coronavirus (COVID-19) dashboard. [cited 2021 Apr 30]. Available from: https://covid19.who.int/
- 30.Boopathi S, Poma AB, Kolandaivel P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J Biomol Struct Dyn. 2020;30:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lei J, Kusov Y, Hilgenfeld R. Nsp3 of coronaviruses: structures and functions of a large multi-domain protein. Antiviral Res. 2018. Jan;149:58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. [DOI] [PubMed] [Google Scholar]
- 33.Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cantuti-Castelvetri L, Ojha R, Pedro LD, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370(6518):856–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuyama S, Nagata N, Shirato K, et al. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol. 2010;84(24):12658–12664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simmons G, Gosalia DN, Rennekamp AJ, et al. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A. 2005;102(33):11876–11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snijder EJ, Limpens RWAL, De Wilde AH, et al. A unifying structural and functional model of the coronavirus replication organelle: tracking down RNA synthesis. PLoS Biol. 2020;18(6):e3000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolff G, Limpens RWAL, Zevenhoven-Dobbe JC, et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science. 2020;369(6509):1395–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neuman BW, Kiss G, Kunding AH, et al. A structural analysis of M protein in coronavirus assembly and morphology. J Struct Biol. 2011;174(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Slanina H, Madhugiri R, Bylapudi G, et al. Coronavirus replication-transcription complex: vital and selective NMPylation of a conserved site in nsp9 by the NiRAN-RdRp subunit. Proc Natl Acad Sci USA. 2021;118(6):e2022310118. . [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study was the first to show that a coronavirus non-structural protein involved in replication may be primed by NMPylation by a viral protein, similar to picornaviruses.
- 41.Shannon A, Fattorini V, Sama B, et al. Protein-primed RNA synthesis in SARS-CoVs and structural basis for inhibition by AT-527. bioRxiv. 2021;03:23.436564. . [Google Scholar]; •• This study showed that the coronavirus nsp8 can be primed for RNA synthesis, and the process can be targeted by a drug to inhibit viral infection
- 42.Kneller DW, Phillips G, O’Neill HM, et al. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat Commun. 2020;11:3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cui S, Wang J, Fan T, et al . Crystal structure of human enterovirus 71 3C protease. J Mol Biol. 2011;408(3): 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]; • In this paper, the first 3D structure of SARS-CoV-2 3CLpro was deposited in PDB.
- 44.Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. [DOI] [PubMed] [Google Scholar]; • In this paper, the first 3D structure of SARS-CoV-2 3CLpro was deposited in PDB.
- 45.Zhang L, Lin D, Sun X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020. Apr 24;368:409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]; • In this paper, the first 3D structure of SARS-CoV-2 3CLpro was published.
- 46.Lee CC, Kuo CJ, Ko TP, et al. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J Biol Chem. 2009;284(12):7646–7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patick AK, Binford SL, Brothers MA, et al. In vitro antiviral activity of AG7088, a potent inhibitor of human rhinovirus 3C protease. Antimicrob Agents Chemother. 1999;43(10):2444–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hayden FG, Turner RB, Gwaltney JM, et al. Phase II, randomized, double-blind, placebo-controlled studies of ruprintrivir nasal spray 2-percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob Agents Chemother. 2003;47(12):3907–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang PH. Characterization and inhibition of SARS-coronavirus main protease. Curr Top Med Chem. 2006;6(4):361–376. [DOI] [PubMed] [Google Scholar]
- 50.Zhang L, Lin D, Kusov Y, et al. α-ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J Med Chem. 2020;63(9):4562–4578. [DOI] [PubMed] [Google Scholar]; •• This study found α-ketoamides showing near-equipotent antiviral activity against the main and 3C proteases.
- 51.Hoffman RL, Kania RS, Brothers MA, et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J Med Chem. 2020;63(21):12725–12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuo CJ, Liu HG, Lo YK, et al. Individual and common inhibitors of coronavirus and picornavirus main proteases. FEBS Lett. 2009;583(3):549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blanchard JE, Elowe NH, Huitema C, et al. High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem Biol. 2004;11(10):1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma C, Sacco MD, Hurst B, et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study was first to show inhibitory action on 3CL protease of coronaviruses by calpain inhibitors.
- 55.Upla P, Marjomaki V, Nissinen L, et al. Calpain 1 and 2 are required for RNA replication of echovirus 1. J Virol. 2008;82:1581–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study was first to show antiviral action of calpain inhibitors on enterovirus infection. Dependence on calpains was shown by inhibitory drugs on calpains as well as knockdown of calpains 1 and 2.
- 56.Bozym RA, Patel K, White C, et al. Calcium signals and calpain-dependent necrosis are essential for release of coxsackievirus B from polarized intestinal epithelial cells. Mol Biol Cell. 2011;22:3010–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barnard DL, Hubbard VD, Burton J, et al. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARSCoV) by calpain inhibitors and beta-D-N4-hydroxycytidine. Antivir Chem Chemother. 2004;15(1):15–22. [DOI] [PubMed] [Google Scholar]; •• This study was first to show antiviral action of calpain inhibitors on coronavirus infection.
- 58.Schneider M, Ackermann K, Stuart M, et al. Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome-independent inhibition of M-calpain. J Virol. 2012;86(18):10112–10122. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study showed inhibition of coronavirus infection by calpain inhibitors, also by using m-calpain knockdown experiments further confirming that m-calpain is needed for coronavirus infection.
- 59.Lee J, Worrall LJ, Vuckovic M, et al. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat Commun. 2020;11:5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang H, Xie W, Xue X, et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3(10):e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee YF, Nomoto A, Detjen BM, et al. A protein covalently linked to poliovirus genome RNA. Proc Natl Acad Sci U SA. 1977;74:59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paul AV, Peters J, Mugavero J, et al. Biochemical and genetic studies of the VPg uridylylation reaction catalyzed by the polymerase of poliovirus. J Virol. 2002;88(2):891–904. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study was the first to show that the enterovirus VPg can be uridylylated by RNA polymerase.
- 63.Egloff MP, Ferron F, Campanacci V, et al. Canard . The severe acute respiratory syndrome-coronavirus replicative protein nsp9 is a single-stranded RNA-binding subunit unique in the RNA virus world. Proc Natl Acad Sci U S A. 2004;101:3792–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stone VM, Hankaniemi MM, Laitinen OH, et al. A hexavalent Coxsackievirus B vaccine is highly immunogenic and has a strong protective capacity in mice and nonhuman primates. Sci Adv. 2020;6(19):eaaz2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goll DE, Thompson VF, Li H, et al. The calpain system. Physiol Rev. 2003. Jul;83(3):731–801. [DOI] [PubMed] [Google Scholar]
- 66.Yoon SY, Ha YE, Choi JE, et al. Coxsackievirus B4 uses autophagy for replication after calpain activation in rat primary neurons. J Virol. 2008. Dec;82(23):11976–11978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smyth M, Pettitt T, Symonds A, et al. Identification of the Pocket Factors in a Picornavirus. Arch Virol. 2003;148:1225–1233. [DOI] [PubMed] [Google Scholar]
- 68.Pevear DC, Tull TM, Seipel ME, et al. Activity of Pleconaril Against Enteroviruses. Antimicrob Agents Chemother. 1999;43:2109–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abdelnabi R, Geraets JA, Ma Y, et al. A novel druggable interprotomer pocket in the capsid of rhino- and enteroviruses. PLoS Biol. 2019;17:e3000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laskowski RA, Swindells MB. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery.J Chem Inform Model. 2011;51(10):2778–2786. [DOI] [PubMed] [Google Scholar]