

Abstract
The study of epigenomics has advanced in recent years to span the regulation of a single genetic locus to the structure and orientation of entire chromosomes within the nucleus. In this review, we focus on the challenges and opportunities of clinical epigenomics in cardiovascular disease. As an integrator of genetic and environmental inputs, and because of advances in measurement techniques that are highly reproducible and provide sequence information, the epigenome is a rich source of potential biosignatures of cardiovascular health and disease. Most of the studies to date have focused on the latter, and herein we discuss observations on epigenomic changes in human cardiovascular disease, examining the role of protein modifiers of chromatin, noncoding RNAs and DNA modification. We provide an overview of cardiovascular epigenomics, discussing the challenges of data sovereignty, data analysis, doctor-patient ethics and innovations necessary to implement precision health.
Keywords: Epigenetics, DNA methylation, Cardiovascular disease, Arrhythmia, Precision medicine
1. Introduction
Widespread clinical implementation of precision medicine has the potential to revolutionize clinical care as well as to unleash a myriad of privacy, cost and ethical issues. At present, we are in the early stages of integrating epigenetics into this story—where we end up will depend on appropriate research, stewardship and implementation.
Exact definitions vary but a key feature of precision medicine is the tailoring of clinical care based on an integrated patient history that includes genomic and/or epigenomic data. The power and potential of such strategy could have far reaching effects in patient care. Ideally, precision medicine would be accomplished through a multi-omics approach taking into account individual variability in the genome, epigenome, transcriptome, proteome and metabolome to optimally direct clinical care for each individual. An advantage of epigenetic measurements is that they reflect heritable factors (i.e. DNA sequence), while at the same time being dynamic and thus capable of incorporating environmental factors, which are of critical importance in the pathophysiology of disease.
Heart disease is the leading cause of death for both men and women, as well as across most racial and ethnic groups in the United States. In 2017, there were 859,125 deaths due to cardiovascular disease in the United States [1]. In addition, the total indirect and direct cost of cardiovascular disease in the United States was approximately $351.2 billion for 2014 to 2015 [1]. The implementation of clinical epigenomics in the prevention and management of cardiovascular disease has the potential for significant improvement in patient outcomes. In this review, we will outline the field of epigenomics, recent epigenomics studies in cardiovascular disease, measurement of epigenetic marks, data analysis and challenges to clinical implementation.
2. Overview of epigenetics
Epigenetic modifications can be considered anything that alters gene function without altering DNA sequence [2]. This includes modifications to the DNA itself, such as methylation [3], in addition to a cadre of proteins that directly bind DNA, such as histones and non-nucleosomal chromatin structural proteins [2].
Histone modification influences chromatin accessibility and the binding and activity of transcriptional machinery [4,5]. These modifications primarily include histone acetylation, phosphorylation and methylation, in addition to other less well-studied modifications. Histone acetylation is a dynamic process regulated by two families of enzymes: histone acetyl-transferases (HATs) and histone deacetylases (HDACs) [4,5]. HATs use acetyl CoA as a cofactor and catalyze the transfer of an acetyl group to the ε-amino group on lysine side chains [4]. The addition of an acetyl group changes lysine’s positive charge to a neutral charge which can weaken the interaction between histones and DNA. Histone phosphorylation is also a dynamic process and takes place on serines, threonines and tyrosines [4]. The phosphorylation primarily occurs in the N-terminal histone tails. The addition and removal of phosphate groups is regulated by kinases and phosphatases, respectively. The addition of a phosphate group to hydroxyl groups of the target amino acid’s side chain adds negative charge. Histone methylation can involve mono-, di- or tri-methylation and typically occurs at the side chains of lysines and arginines. In contrast to histone acetylation and phosphorylation, histone methylation does not alter the charge of the protein but is influential in chromatin accessibility [6]. While local electrostatic and other steric changes play a role in the actions of histone modifications to influence binding to proteins and DNA, it is now appreciated that histone modifications often act in concert with each other (i.e. combinations of modifications specify different transcriptional outcomes) as well as through interactions with other histone binding proteins, such as chromatin readers—these concepts have been reviewed in detail elsewhere [7–9].
DNA methylation primarily occurs at cytosines contiguous with guanine (CpGs) and plays a key role in gene expression, development and disease [3]. DNA methyltransferases catalyze the addition of a methyl group from S-adenosyl-L-methionine to the 5-carbon position of cytosine. DNA promotor methylation is associated with gene silencing though the exact mechanism is not completely understood as the methyl mark alone does not seem to be sufficient to confer silencing [3]. DNA gene body methylation, however, is associated with increased transcription. DNA methylation is one of the most studied epigenetic modifications and has been implicated in many cancers as well as cardiovascular disease processes [10].
RNA has also been implicated in epigenetic regulation of gene expression [11]. There are many classes of small RNAs and long non-coding RNAs that are regulators of chromatin structure [12]. Small RNAs alter gene expression via RNA interface pathways. In addition, some long non-coding RNAs appear to contain signals that recruit chromatin modifying complexes. A common process by which small RNAs and long non-coding RNAs modify chromatin structure and silence transcription is through the formation of RNA scaffolds. The role of RNA in epigenetic regulation has been covered extensively elsewhere [13].
Epigenetic marks are tissue specific. DNA methylation, for example, varies significantly across cell types [14]. In whole blood samples, most of the variability in DNA methylation is secondary to the cellular composition of the blood sample [15]. For this reason, it is critical that the cellular composition of the sample tissue be accounted for when studying epigenetic marks [15]. Given that the primary tissue of disease is often impractical to obtain in the clinical setting, many epigenome-wide association studies have focused on whole blood samples. Paradoxically, some epigenetic marks are conserved across tissues [16] while still exerting tissue specific effects (similar to a genetic mutation), further supporting the use of whole blood samples to study disease that primarily affects other tissues.
Epigenetic marks are dynamic and may, in some cases, be heritable [2]. These modifications can change over the course of one’s life as a result of aging, environmental exposure and disease. Smoking and adiposity, for example, can result in epigenetic changes [17]. Habitual diet quality results in differential methylation in at least 30 CpGs, 12 of which are associated with all-cause mortality [18]. Epigenetic modifications also help modulate gene expression in development and disease [19]. Due to the dynamic nature of epigenetic marks, studies analyzing the epigenetic features of disease may serve as predictors of outcomes or sequalae depending on when in the disease course the sample was obtained [20]. In addition, the dynamic nature of epigenetic marks holds the possibility to monitor disease prospectively, including surveilling response to treatment [21]. A sizable body of work in animal models has implicated epigenomic processes in cardiovascular disease and has been reviewed in detail elsewhere [7,22]. Herein we focus on studies in humans.
3. Status of epigenetics in clinical medicine
3.1. Atherosclerosis
Atherosclerosis is the causative process in peripheral artery disease, coronary artery disease and cerebrovascular disease. Epigenetic modifications have been implicated in the development and progression of atherosclerosis. Increased acetylation of histone H3 lysine 9 (H3K9ac) and histone H3 lysine 27 (H3K27ac) in smooth muscle cells are associated with advanced atherosclerotic lesions compared to healthy carotid arteries [23]. Expression of GCN5L and MYST1 which are regulated by histone acetylation is associated with plaque severity in atherosclerosis [23]. Histone deacetylase 9 (HDAC9) has been shown to regulate atherosclerotic plaque vulnerability. HDAC9 binds to inhibitory kappa B kinase (IKK) α and β which causes IKK to be deacetylated and activated which impacts inflammatory response in macrophages and endothelial cells. Inhibition of HDAC9 attenuates atherosclerotic plaque formation, disease progression and enhances plaque stability [24]. In addition, histone H3 lysine 9 (H3K9) and histone H3 lysine 27 (H3K27) hypomethylation are also associated with atherosclerotic plaques in smooth muscle cells and inflammatory cells compared to healthy carotid arteries. Methylation on histone H3 lysine 4 (H3K4) is associated with severity of atherosclerosis [23]. These results demonstrate that histone acetylation and methylation are significantly associated with atherosclerosis.
Differential DNA methylation is also associated with atherosclerotic disease. One study [25] identified 1985 CpGs associated with atherosclerosis in human aorta samples and found that the majority of these sites showed a correlation between hypermethylation and advanced atherosclerosis. These significant CpGs were associated with gene expression in the same aorta samples. In contrast, another study [26] examining DNA methylation in human femoral artery samples found that hypomethylation of differentially methylated CpGs was associated with atherosclerotic plaques and that the majority of these sites were associated with increased gene expression. In particular, the 14q32 locus was hypomethylated and associated with upregulation of several miR-NAs. In the MESA cohort, investigators [27] discovered four CpGs whose methylation at least partially mediates expression of cyan module genes ABCG1, SC4MOL and LDLR, a known network of coexpressed cholesterol metabolism genes. These studies suggest that the pathophysiology of atherosclerosis is associated with epigenetic regulation though the mechanics are inadequately understood. Epigenetic regulation, however, may be a target for therapeutics and a biomarker for disease progression.
3.2. Hypertension
Hypertension is a common cardiovascular disease and affects more than 1 billion people world-wide [1]. In addition, high blood pressure is a common risk factor for stroke, chronic kidney disease and heart disease [1]. Epigenetic studies of hypertension are numerous and there are clearly many epigenetic mechanisms associated with hypertension. Lower levels of 5-methylcytosine (5-mC) are present in the DNA of patients with hypertension and the 5-mC level is correlated with the stage of hypertension [28]. For example, patients with stage 2 hypertension (systolic blood pressure > 159 mmHg and/or diastolic blood pressure > 99 mmHg) have higher 5-mC levels than patients with stage 1 hypertension (systolic blood pressure 140–159 mmHg and/or diastolic blood pressure 90–99) [28].
In a genome-wide association study [29] performed in 320,251 individuals of East Asian, South Asian and European descent, 12 single nucleotide polymorphisms (SNPs) were discovered that are associated with blood pressure. These SNPs are associated with genes related to vascular smooth muscle (IGFBP3, KCNK3, PDE3A and PRDM6) and renal function (ARHGAP24, OSR1, SLC22A7 and TBX2). These genetic variants were able to predict increased left ventricular mass (secondary to concentric hypertrophy from hypertension), serum NT-proBNP levels, cardiovascular mortality and all-cause mortality. These SNPs are also enriched for association with DNA methylation at multiple nearby CpGs which suggests that DNA methylation may be part of the pathway linking sequence variation with disease at some of these loci. This study demonstrates evidence of DNA methylation’s role in blood pressure regulation.
Other epigenetic studies of hypertension focus on epigenetic determinants of blood pressure and how these relate to the efficacy of different anti-hypertensive medications. One study [30] analyzed the variable response to hydrochlorothiazide (HCTZ) related to SNPs in DOT1L, MLLT3, SIRT1 and SGK1 which encode genes in a pathway that control histone 3 lysine 79 methylation. The candidate SNPs DOT1L, MLLT3, SIRT1 and SGK1 were unable to be validated but two possible SNPs were suggested that require separate validation. The SNP rs2269879 in DOT1L could be associated with HCTZ response in Caucasians and rs12350051 in MLLT3 may be associated with untreated blood pressure in African-Americans. Studies such as this demonstrate the potential of precision medicine in hypertension management and suggest the choice of anti-hypertensive medications may be guided by tests that measure a patient’s epigenetic modification profile.
3.3. Metabolic syndrome
Metabolic syndrome refers to a cluster of disorders of metabolism that together synergistically increase the risk of heart disease more than the individual components alone. These metabolic disorders include elevated fasting blood sugar (>100 mg/dL), elevated blood pressure (Systolic blood pressure > 130 mmHg or diastolic blood pressure > 85 mmHg), increased triglyceride level (>150 mg/dL), low High Density Lipoprotein (<40 mg/dL in men or < 50 mg/dL in women) and abdominal obesity (waist circumference > 40 in. in men or > 35 in. in women) [31]. Metabolic disease affects 44% of people over the age of 50 in the United States and increases risk of myocardial infarction, stroke and diabetes.
DNA methylation has been shown to be associated with several metabolic processes related to metabolic syndrome including hypertension, diabetes and obesity. An epigenome-wide association study [32] performed in 201 people from the Metabolic Syndrome in Men (METSIM) cohort identified 13 clinical traits in 21 loci from adipocyte samples. Using expression quantitative trait loci, 18 candidate genes were identified, of which some had known associations with diabetes and obesity. This novel research helps describe the molecular effects of metabolic syndrome and increases further understanding of this disorder. Several other studies [33,34] have investigated the role of DNA methylation in features of metabolic syndrome. An epigenome-wide association study of adipose samples [35] identified 2825 genes where DNA methylation and gene expression correlated with BMI. In addition, in the same study 711 CpGs were associated with HgbA1c with 14% showing positive and 86% showing negative correlation between methylation and HgbA1c. Another epigenome-wide association study [36] identified 187 CpGs which were associated with BMI. These 187 CpGs were associated with cis expression in blood at 38 annotated genes. Altered DNA methylation identified in this study was associated with future development of diabetes, a result with significant implications for patient care.
3.4. Coronary artery disease
Cardiovascular disease is the number one cause of death globally [37] with an estimated prevalence of 121,500,000 people in the United States over the age of 20 (48% of the population over 20) with 859,125 deaths annually [1]. Coronary artery disease, a subset of cardiovascular disease, involves atherosclerotic disease of the coronary arteries that supply blood flow to the myocardium. Coronary artery disease is the most common form of heart disease in the United States and was responsible for 365,914 deaths in the United States in 2017 [1]. Several studies have investigated epigenetic risk factors for coronary artery disease.
An epigenome-wide association study [38] of blood samples in 729 individuals from northern Sweden demonstrated 211 CpGs that are associated with a history of myocardial infarction. These 211 CpGs were associated with 196 genes, 42 of which have known links to cardiac function. Another study [39] investigated the association of DNA methylation with myocardial infarction in white blood cell samples from 292 patients with a history of myocardial infarction and 292 matched controls (EPICOR study). A differentially methylated region within the ZBTB12 gene body and LINE-1 hypomethylation were discovered. Gene body hypermethylation has been shown to be associated with increased transcription [3]. The significant CpGs in the EPICOR study cohort were then replicated by mass spectrometry in 317 myocardial infarction cases and 262 controls (EPIC-NL study). In the replication cohort, DNA methylation data improved prediction of cases versus controls compared to traditional clinical myocardial infarction predictors alone. These findings are of direct clinical relevance as they can be used to screen patients for coronary artery disease and implement lifestyle changes and medications to prevent disease progression. Another epigenome-wide association study [40] of CRP levels found that CpGs associated with low grade inflammation were also associated with incident and prevalent coronary heart disease. These inflammation CpGs are another example of potential targets for medications or lifestyle interventions to mitigate the risk of coronary heart disease.
Patients with a history of coronary heart disease have different global methylation profiles in blood samples. One study [41] showed that 5-methylcytosine (5-mC) and 5-hydroxymethylcytosine (5-hmC) levels in PBMCs of patients with coronary heart disease were higher than in controls. TET proteins are responsible for oxidizing 5-mC into 5-hmC as well as 5-formylcytosine and 5-carboxylcytosine [42]. These oxidized 5-mC derivatives can then be processed by a variety of mechanisms to demethylated DNA. Interestingly, this study [41] also found that TET2 expression was significantly increased in patients with coronary heart disease.
Other studies identify epigenetic marks associated with known biomarkers for myocardial infarction. Growth-differentiation factor-15 (GDF-15) is a member of the transforming growth factor beta (TGF-β). GDF-15 levels increase secondary to pathological stress associated with inflammation or tissue damage. GDF-15 has been shown to be increased in blood from patients who have had myocardial infarction. An epigenome-wide association study [43] of GDF-15 levels in white blood cells in 717 individuals revealed 16 CpGs at 11 independent loci that were validated in a separate cohort. One of these loci is associated with MIR21 which encodes a microRNA (miR-21) that is known to be associated with the development of heart disease.
Dynamic epigenetic modification can also capture treatment effects for known cardiovascular risk factors. For example, folate and some B-vitamin (B2, B6, B12 and folic acid) supplementation reduces serum homocysteine levels, an independent risk factor for cardiovascular disease. One study [44] identified three differentially methylated regions in males and two in females, all of which had a positive correlation between hypermethylation and myocardial infarction resulting in decreased expression. They found an inverse relationship between B-vitamin intake and DNA methylation of candidate genes. Overall, they demonstrated that reduced B-vitamin intake results in OCM and Hcy gene hypermethylation, decreased gene expression and increased risk of myocardial infarction. By contrast, increased B vitamin consumption results in hypomethylation of promotors for genes such as cystathionine-beta-synthetase (CBS) which results in increased CBS activity, decreased homocysteine levels and decreased cardiovascular disease risk. Studies such as this are of significant importance in that they demonstrate response to treatment can be captured in epigenetic modification of known cardiovascular risk factors.
3.5. Heart failure
Heart failure affected 5.4 million adults in the United States in 2017 alone. The estimated total cost of heart failure in 2012 was $30.7 billion with over two thirds attributed to direct costs [1]. Several studies have investigated epigenetic signatures of heart failure subtypes such as ischemic cardiomyopathy, dilated cardiomyopathy and hypertrophic cardiomyopathy. In one study [45], targeted DNA methylation profiling identified 195 unique differentially methylated regions with 5 in hypertrophic obstructive cardiomyopathy, 151 in dilated cardiomyopathy and 55 in ischemic cardiomyopathy. These differentially methylated regions were localized to several differentially methylated genes and ncRNA—linking regulation of these loci to the distinct heart failure subtypes. Subsequent gene/ncRNA expression analysis using quantitative reverse transcription polymerase chain reaction revealed 6 genes and 2 microRNA with significantly up- or down-regulated expression consistent with the change in methylation in the corresponding heart failure group. This important study shows that gene expression related to several subtypes of heart failure are all regulated in part by differential DNA methylation. Other investigators [46] identified that in contrast to cardiac development, gene expression related to heart failure was related to alterations in active histone marks without major changes in DNA methylation and repressive histone marks. Although these studies show different specific mechanisms for regulating heart failure gene expression, both suggest the importance of epigenetic regulation of the pathophysiology of heart failure.
The epigenetic pathogenesis of heart failure is currently an active area of research. One study [47] focused on ischemic cardiomyopathy and used genome-wide DNA methylation analysis with RNA sequencing to understand the pathogenesis of heart failure in patients with ischemic cardiomyopathy. They discovered gene expression related to anerobic glycolysis, suppressed oxidative metabolism and altered cell remodeling. In addition, they identified KL15 as an upstream regulator of pathologic gene expression in ischemic cardiomyopathy which had epigenetic regulation by EZH2 and hypermethylation. These findings are important in identifying how coronary artery disease results in altered gene expression that can lead to heart failure. Whereas several researchers have investigated individual epigenetic mechanisms related to heart failure subtypes, one study [48] analyzed differential DNA methylation and histone modification associated with a common final pathway in end stage heart failure in patients with ischemic and idiopathic cardiomyopathy undergoing heart transplant. They found differential DNA methylation was present in promoter CpG islands, intragenic CpG islands and upregulated genes. Promoter hypermethylation has been shown to inhibit gene transcription [3,48,49], in some cases by preventing transcription factor binding. Gene body DNA methylation, however, is associated with increased transcription [3,48,49]. In end stage heart failure patients, differential histone H3 lysine 36 tri-methylation enrichment was associated with coding regions of the genome [50]. Lastly, they tested the abundance of RNA transcripts from DUX4 locus and found that expression of DUX4 was significantly reduced in idiopathic cardiomyopathy hearts compared to control. This reduced expression was also associated with hypermethylation in hearts with idiopathic cardiomyopathy. The epigenetic regulation of genes associated with end stage heart failure may reflect common targets for treating heart failure. In addition, the assessment of these differential epigenetic marks may help prognosticate patients with earlier presentation of heart failure.
3.6. Dilated cardiomyopathy
Dilated cardiomyopathy is a disease characterized by progressive dilation of the left ventricle resulting in decreased left ventricular systolic function and congestive heart failure. There is a familial component to dilated cardiomyopathy with over 40 genes causing predisposition to the phenotype. Despite known genetic risk factors, the disease course of dilated cardiomyopathy is variable, and it is thought that epigenetic modifications may play a role in pathophysiology of the disease. Differential methylation is present in left ventricular myocardium in patients with dilated cardiomyopathy [51]. Differential methylation in genes LY75, ERBB3, HOXB13 and ADORA2A are associated with dilated cardiomyopathy whereas these genes had not previously been implicated in the pathogenesis of heart failure or dilated cardiomyopathy. These findings suggest that differential methylation may result in altered gene expression in heart failure secondary to dilated cardiomyopathy.
A recent epigenome-wide association study [52] of dilated cardiomyopathy using left ventricular myocardium samples identified 27 epigenetic loci that were validated in a separate cohort. This study also identified an additional 513 genetic loci associated with dilated cardiomyopathy by performing methylation-expression quantitative trait locus analysis separately in the discovery and validation cohorts and taking the common loci to both analyses. Several key genes from other studies (LY75, PTGES, CTNNAL1, TNFSF14, MRPL16 and KIF17) were able to be replicated in this analysis. Interestingly, there were 3798 CpGs that had similar methylation in blood as left ventricular tissue which illustrates the potential for peripheral biomarkers for dilated cardiomyopathy. LY75 methylation, however, was unfortunately not conserved in blood.
In dilated cardiomyopathy, one study identified differential methylation CpGs in left ventricular myocardium compared to right ventricular myocardium [53]. Tissue samples from the same heart were used as a control as the right ventricle is not as severely affected in patients with dilated cardiomyopathy. Of the differentially methylated CpGs associated with genes, approximately 70% were hypermethylated in the left ventricle when compared to the right ventricle. These hypermethylated probes were predominantly found in promoter-proximal regions such as 200 bp upstream of the transcription start site, the first exon and the 5′ untranslated region. Promoter hypermethylation has been shown to be associated with gene silencing, in some instances by preventing transcription factor binding [3]. Differentially methylated probes were also associated with transcription factor binding sites except CCCTC-binding factor. Interestingly, the effect of DNA methylation on gene expression was found to be bi-directional with some hypermethylated genes being upregulated and some hypomethylated genes being downregulated. Differential methylation was also enriched in the cis-regulatory regions of HAND1 and TBX5 which are genes involved in left ventricular development [53].
3.7. Atrial fibrillation
Atrial fibrillation is the most common persistent cardiac arrhythmia with more than 2.7 million people affected in the United States [54]. Atrial fibrillation is associated with increased risk of stroke, heart failure, dementia, myocardial infarction and death. Several studies have investigated the contribution of genetic variation to atrial fibrillation, but the molecular mechanisms of the identified SNPs are unknown. An epigenome-wide association study [55] of patients in the Framingham cohort identified two CpGs associated with incident atrial fibrillation and five CpGs associated with prevalent atrial fibrillation. In addition, fourteen previously validated SNPs were associated with at least one of the CpGs associated with atrial fibrillation. These results demonstrate that DNA methylation is associated with the pathophysiology of atrial fibrillation.
Differential DNA methylation has also been identified in left atrial tissue of patients with permanent atrial fibrillation [56]. Previous genome-wide association studies implicated PITX2, CCDC141 and CACNA1C which were found to be differentially methylated in left atrial tissue in patients with permanent atrial fibrillation. Genes with previously described differential expression in atrial fibrillation were analyzed and 12 were found to be hypomethylated and 8 to be hypermethylated. Real time quantitative PCR of four of these 20 genes confirmed differential methylation was associated with changes in gene expression.
3.8. Epigenetic sequencing and data acquisition
Several forms of epigenetic mark have been shown to be amenable to reproducible measurement across labs. A community-wide bench-marking study involving 18 different laboratories in 7 different countries was able to generate consistent results in 21 locus-specific assays and 6 global assays [57]. For this reason, epigenetic data is well-suited for biomarker studies and clinical diagnostics.
Many platforms exist for obtaining epigenetic data from blood and tissue samples. For DNA methylation, bisulfite treated DNA can be analyzed with either methylation microarray or next generation sequencing. It must be noted, however, that analysis of bisulfite treated DNA cannot distinguish between 5-mC and 5-hmC [58], thus limiting the appreciated individual impact of 5-hmC. The most recent microarray platform is the Illumina MethylationEPIC. The MethylationEPIC microarray interrogates the methylation status of 853,307 CpGs. Microarray platforms have well established analysis, are cheaper, require little DNA and provide a uniform set of CpGs for all samples. Microarrays are limited by variation from experimental conditions and batch effects which can limit reproducibility [59].
Next generation sequencing includes whole genome bisulfite sequencing (WGBS) and reduced representation bisulfite sequencing (RRBS). Although all CpGs may be attempted to be sequenced, some sites have low coverage (<10×). In addition, WGBS though comprehensive is still too expensive to be deployed on a population scale. RRBS uses restriction enzyme digestion to cut DNA into fragments between two C’s at CCGG sites which are frequently found in CpG islands and promoters. In general, RRBS typically captures 80% of CpG islands and 60% of promoter regions. It is far more time- and cost-effective than WGBS [60]. The disadvantages of RRBS is that coverage of CpGs across multiple samples can be inconsistent, some genes lack coverage and the resulting analysis is complex. Beyond WGBS and RRBS, there is targeted bisulfite sequencing which utilizes DNA or RNA probes to target selected regions in the genome for bisulfite sequencing [61].
Tissue selection for epigenetic studies is of crucial importance. A balance must be struck between accessibility of the tissue and the association of that tissue with the primary disease process. Most epigenome-wide association studies are in blood due to the ease of obtaining this data and the extent of prior research on analysis of blood samples in epigenome-wide association studies. Epigenome-wide association studies have also been performed on adipose tissue [32,36], myocardium [19,45,50–53], brain tissue [62] and many other sources. Tissue samples contain a range of cell types that must be accounted for in the statistical analysis. Blood samples contain DNA from a variety of peripheral leukocytes including neutrophils, lymphocytes (CD8+ T cells, CD4+ T cells, CD56+ natural killer cells and CD19+ B cells) and CD14+ monocytes. Blood has also been shown to contain circulating cell-free DNA released from dying cells in organs throughout the body [63] which could be a biomarker for organ dysfunction or graft failure. Cell-free DNA has also been used to identify thousands of bacteria and viruses within the human microbiome from blood samples [64].
Single cell epigenetics allows for evaluation of epigenetic variability across a population of cells from a given tissue in a single person. Single cell epigenetics circumvents the issue of cell type heterogeneity within tissue samples which creates artificial differences in DNA methylation due to differences in cellular abundance rather than biological differences from differential CpG methylation. In humans, single cell DNA methylation has been analyzed in pre-implantation embryos [65], spermatagonial stem cells [66], colorectal cancer [67] and other populations. In mice, one study [68] analyzed transcriptome and methylome reprograming in myocyte-derived cardiac progenitor cells. Although single cell epigenetics is an emerging field of analysis, it has the potential to better refine our understanding of epigenetic regulation in distinct cell populations. In addition, this technology will prove crucial in understanding epigenetic regulation in less abundant sub-populations of cells that have disease relevance [69].
Study design is of critical importance in epigenetic research. Given that epigenetic marks are dynamic, differential methylation may reflect risk factors for a disease or downstream consequence of a disease depending on the timing of sample acquisition [70]. For this reason, epigenetic case control studies cannot differentiate between risk factors for disease and sequalae of disease. Sample size requirements are also variable depending on the effect size of the epigenetic loci, mean methylation difference between cases/controls and methylation variability at loci of interest [71]. One study [72] estimated statistical power for EPIC array studies using a statistical threshold of p < 9.42 × 10–8. They estimated >80% power to detect a mean methylation difference of 5% between 100 cases and 100 controls at 85% of sites in their simulated dataset. In addition, the method of DNA methylation data acquisition also affects the requisite sample size with beta-binomial statistical models in sequencing data being more sensitive and thus requiring smaller sample size [73]. Epigenetic datasets are also large and can have possible unknown confounders such as population stratification [74] and cell type heterogeneity [15]. Ancestry data and cell type estimates should be covariates accounted for prior to data analysis [15,74]. In addition, the statistical analysis of such large datasets can lead to type I error [75]. Statistical inflation should be controlled using methods such as the Benjamini-Hochburg procedure or the Bonferroni correction [59]. Epigenetic studies should also have separate discovery and validation cohorts to further prevent spurious genetic and statistical associations [59]. Validation cohorts can involve separate samples from the same population (essentially reproducibility or internal validation) or preferably samples from a different population with either differently measured outcomes, sampling strategy or timepoint of sampling [76].
3.9. Data analysis
All epigenomic data needs to be processed using suites of often publicly available programs, as has been reviewed elsewhere [59,77,78] and in what constitutes a continually evolving aspect of this field. Our remarks here focus on DNA methylation, given its emergent role as a disease relevant biomarker in human disease. Prior to analyzing for differential methylation, the methylation data needs to be pre-processed. For Illumina microarray data, one must 1) obtain data via bead-level intensity extraction, 2) perform signal intensity adjustment, 3) calculate average beta value (methylation ratio) detection p-value, 4) perform normalization and 5) perform batch correction. These steps can be accomplished with Illumina’s GenomeStudio module or the R package ‘minfi’ [79]. For next generation bisulfite sequencing, the data must 1) have its read quality assessed and cleaned up, 2) be aligned to a reference genome and 3) CpG methylation status extraction. In RRBS, the sequencing reads require alignment and processing steps that are unique to RRBS [80].
Prior to statistical analysis of the data, it is important to consider all of the methylation covariates of the data. Age [81], smoking history [82], BMI [83] and cell type composition [14] are all known to affect DNA methylation, and these must be taken into account to analyze the biological question being investigated. Cell type heterogeneity is a common confounder of epigenome-wide association analysis and is not typically measured directly. Correcting for cell-type heterogeneity has been discussed most extensively in whole blood samples. Overall, there are two basic methodologies of correcting for cell type heterogeneity: reference-free and reference-based techniques. Reference-free methods assume that the major sources of variability in the methylation dataset are secondary to cell type heterogeneity and seek to create variables explaining this variability that serve as surrogates for cell type. These reference-free methods include RefFreeEwas [84], ReFactOR [85] and FaST-LMM-EWASher [86]. Because the first several principal components in principal component analysis (PCA) of methylation data are associated with cell type [87], principal components can be added as covariates in regression analysis to reduce statistical inflation associated with cell-type heterogeneity. ReFactOR and FaST-LMM-EWASher are in fact variants on PCA and have been shown to increase statistical power and reduce false positives in simulated and real methylation datasets [85,86]. It is notable that in all reference-free methods, true biological signals can inadvertently be removed using reference free cell type correction. Reference-free methods can also correct for systemic confounding not related to cell type heterogeneity. Referenced based methods such as the Houseman algorithm [88] estimate cell type composition from CpGs known to be associated with cell type.
Statistical methods of analyzing epigenetic data are numerous [89]. Microarray data analysis predominantly involves comparison of beta-values (methylation ratio) between two groups. Illumina Methylation Analyzer (IMA) is specifically developed for 450 K microarray data. Two group analysis with IMA involves ‘limma’ (moderated t statistics), student t statistics or Wilcoxon rank sum test. ‘Minfi’ is an R package that also compares individual CpG methylation with continuous variables by linear regression or categorical variables by F-test [60].
Next generation sequencing data can also be analyzed by ratio-based or count-based statistical methods. Ratio-based methods use the same methods described for microarray data by calculating the methylation ratio for each CpG but this ignores differences in sequencing depth at each CpG. Count-based statistical methods include contingency table tests (e.g. methylKit [90], COHCAP [91]), logistic regression (e.g. methylkit) or beta-binomial models (e.g. RADmeth [92], MethylSig [93], MOABS [94], DSS general [95], and MACAU [96]). Of these, beta-binomial models perform best in analysis, due to their ability to model overdispersion and accounting of coverage data for each CpG but are computationally intensive [73,97]. Computation time is longer using RADmeth taking approximately 1 to 2 h on a modern personal computer to analyze 39 samples with approximately 50,000 CpGs each [73]. MethylSig, MOABS, DSS general and MACAU are able to analyze the same dataset on the order of minutes [73]. Datasets with many more samples or CpGs may require a computer cluster to perform computation. DSS general, however, is more efficient and can analyze datasets with millions of CpGs on a modern personal computer in under an hour.
4. Benefits and challenges in clinical implementation
The clinical implementation of epigenetics has enormous potential, particularly in treating cardiovascular disease. Epigenetic marks can be used to monitor response to treatment of disease and predict therapeutic response. In luminal B breast cancer, DNA methylation has been shown to improve prediction of response to neoadjuvant chemotherapy [98]. DNA methylation has also been shown to be associated with response to etanercept in patients with rheumatoid arthritis [99]. Epigenetics could be used to predict disease and, subsequently, to predict the disease course and response to treatment. The clinical impact of such technology could revolutionize patient care and have far reaching effects.
These benefits, however, are not without difficulty. With the vast increase in biological information captured by state-of-the-art epigenomic technologies comes the thorny challenges of clinical implementation of these technologies. How we meet these challenges will determine the extent to which epigenomic knowledge can be deployed to influence clinical care. Privacy is of the utmost concern especially with sequencing data because individual genetic and epigenetic risk factors can be found in the dataset.
Who owns the data? The patient certainly has a right to their own epigenetic data and should be able to request their data returned later if they would like to withdraw from research. The return of epigenetic data should have a predetermined procedure to transfer the data with a genetic counselor to help explain the results, especially if the epigenetic data contains actionable information. Each patient’s preferences should also be clearly recorded during enrollment into the study including whether they would like to be contacted later about significant results or follow-up.
The cost of acquiring epigenetic data is high though as technology improves these costs will decrease. Given the dynamic nature of epigenetic data, the timing of data acquisition is of critical importance and the optimal timing of repeated sampling must be empirically determined for different disease settings. More research is required on this subject, especially as the epigenetic profiling of single patient samples become broader and are implicated in wider reaches of clinical care.
Who needs epigenetic profiling? Until this technology is affordable as standard lab tests, who needs epigenetic risk profiling will likely be determined by which patients are deemed to have high risk medical conditions based on analysis of clinical risk factors. Machine learning, a form of artificial intelligence which allows computers to learn and make predictions from data, has the advantage of being able to uncover complex associations in large datasets which is well suited to epigenetic data [100]. As such, machine learning may better help to classify disease based on epigenetic risk factors [101–103] and may be able to offer individualized treatment to improve outcomes. The long-term answer to the question of who needs epigenetic profiling will be dependent on how effectively large cohort studies demonstrate strong predictive or preventative value, thereby encouraging insurers and health systems to invest in the tools for widespread adoption. The ultimate impact of epigenomic medicine will finally rely on the empowerment of the (appropriately educated) physician to use the insights from these tools to manage health in an accountable manner over the life of the patient (Fig. 1).
Fig. 1. Integrating epigenomics into patient care.
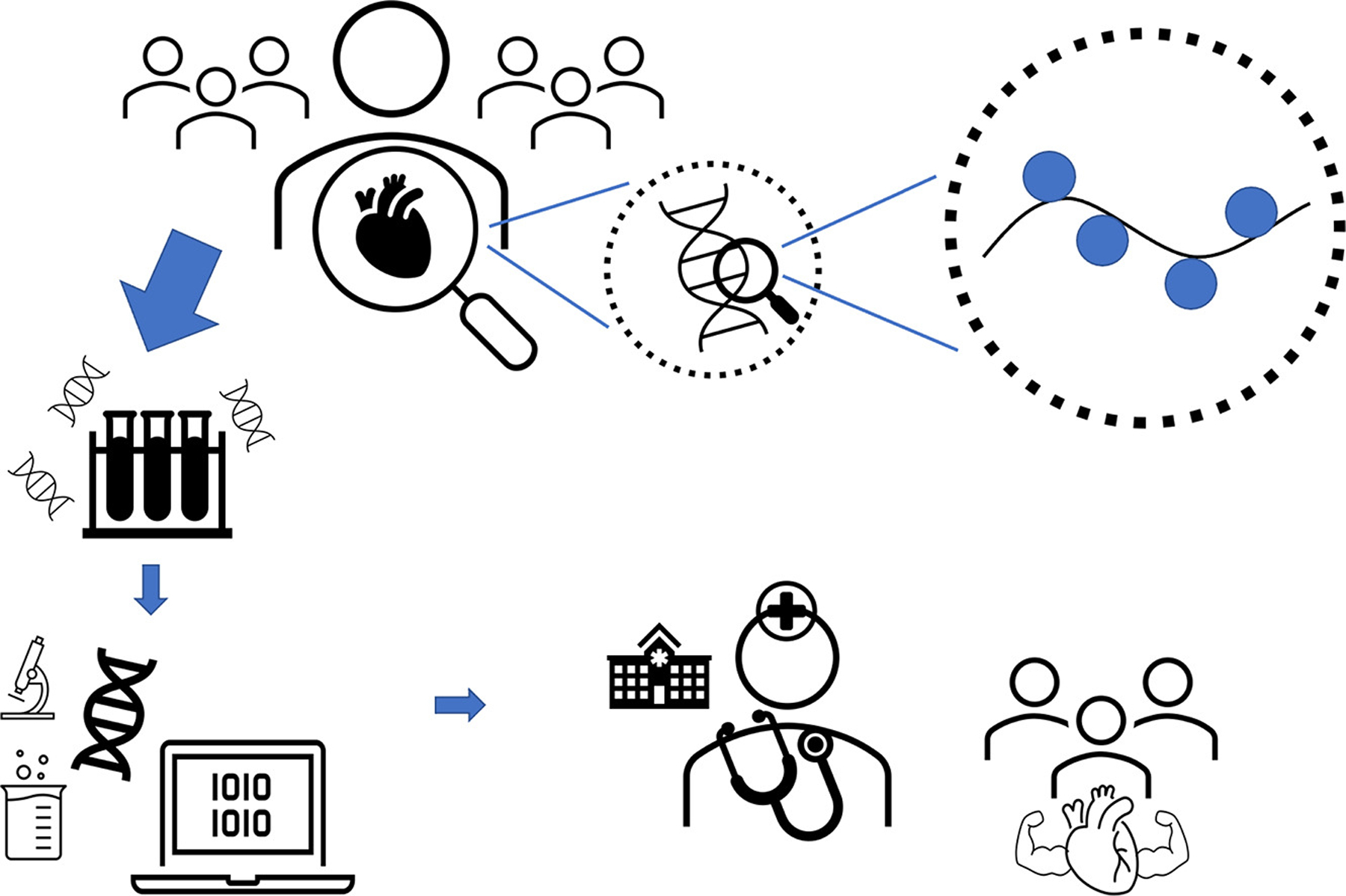
Epigenetic marks incorporate environmental and genetic factors into the pathophysiology of many aspects of cardiovascular disease. These epigenetic biosignatures can be used by clinicians to treat disease and monitor individual response to treatment. Integrating epigenetic data, analysis and clinician education into patient care are crucial next steps in implementation to improve clinical care at a population level.
Acknowledgements
The authors thank Elizabeth Soehalim for assistance with the figure. The author as also thankful for support from the Department of Anesthesiology & Perioperative Medicine, the David Geffen School of Medicine and the Clinical and Translational Science Institute (NIH UL1TR000124), all at UCLA.
References
- [1].Virani Salim S., Benjamin Emelia J., Bittencourt Marcio S., Callaway Clifton W., Carson April P., Chamberlain Alanna M., Chang Alexander R., Cheng Susan, Delling Francesca N., Djousse Luc, Elkind Mitchell S.V., Ferguson Jane F., Fornage Myriam, Khan Sadiya S., Kissela Brett M., Knutson Kristen L., Kwan Tak W., Lackland Daniel T., Lewis Teńe T., Lichtman Judith H., Longenecker Chris T., Loop Matthew Shane, Lutsey Pamela L., Martin Seth S., Matsushita Kunihiro, Moran Andrew E., Mussolino Michael E., Perak Amanda Marma, Rosamond Wayne D., Roth Gregory A., Sampson Uchechukwu K. A., Satou Gary M., Schroeder Emily B., Shah Svati H., Shay Christina M., Spartano Nicole L., Stokes Andrew, Tirschwell David L., VanWagner Lisa B., Tsao Connie W., American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee, Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association, Circulation 141 (9) (2020) e139–e596. [DOI] [PubMed] [Google Scholar]
- [2].Allis CD, The molecular hallmarks of epigenetic control, Nat Rev Genet 17 (8) (2016) 487–500. [DOI] [PubMed] [Google Scholar]
- [3].Greenberg MVC, The diverse roles of DNA methylation in mammalian development and disease, Nat Rev Mol Cell Biol. 20 (10) (2019) 590–607. [DOI] [PubMed] [Google Scholar]
- [4].Bannister Andrew J., Regulation of chromatin by histone modifications, Cell Res 21 (3) (2011) 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cedar H, Linking DNA methylation and histone modification: patterns and paradigms, Nat Rev Genet 10 (5) (2009) 295–304. [DOI] [PubMed] [Google Scholar]
- [6].Jambhekar A, Shi Y, Roles and regulation of histone methylation in animal development, Nat Rev Mol Cell Biol. 20 (10) (2019) 625–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Keating Samuel T., Epigenetics and metabolism, Circulation Research 116 (4) (2015) 715–736. [DOI] [PubMed] [Google Scholar]
- [8].Zhang W, Qu J, Liu GH, Epigenetic Modifications in Cardiovascular Aging and Diseases, Circ Res. 123 (7) (2018). [DOI] [PubMed] [Google Scholar]
- [9].Gillette TG, Readers, writers, and erasers: chromatin as the whiteboard of heart disease, Circ Res. 116 (7) (2015) 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jones PA, Functions of DNA methylation: islands, start sites, gene bodies and beyond, Nat Rev Genet 13 (2012) 484–492. [DOI] [PubMed] [Google Scholar]
- [11].Mattick JS, Dinger ME, Mercer TR, Mehler MF, RNA regulation of epigenetic processes, Bioessays. 31 (1) (2009) 51–59. [DOI] [PubMed] [Google Scholar]
- [12].Wei JW, Yang C, Kang CS, Non-coding RNAs as regulators in epigenetics, Oncol Rep. 37 (1) (2017) 3–9. [DOI] [PubMed] [Google Scholar]
- [13].Holoch Daniel, RNA-mediated epigenetic regulation of gene expression, Nat Rev Genet 16 (2) (2015) 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Udo Baron IT, Hellwag Alexander, Eckhardt Florian, Berlin Kurt, Hoffmuller Ulrich, Gardina Paul, Olek Sven, DNA methylation analysis as a tool for cell typing, Epigenetics 1 (1) (2006) 55–60. [DOI] [PubMed] [Google Scholar]
- [15].Jaffe AE, Irizarry RA, Accounting for cellular heterogeneity is critical in epigenome-wide association studies, Genome Biol. 15 (2) (2014) R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hyang-Min Byun KDS, Pan Fei, Weisenberger Daniel J., Kanel Gary, Laird Peter W., Yang Allen S., Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual- specific DNA methylation patterns, Human Molecular Genetics 18 (24) (2009) 4808–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].van Jenny Dongen MGN, Willemsen Gonneke, Hottenga Jouke-Jan, Helmer Quinta, Dolan Conor V., Ehli Erik A., Davies Gareth E., van Iterson Maarten, Breeze Charles E., Beck Stephan, H. Eka Suchiman BIOS Consortium, Jansen Rick, van Meurs Joyce B., Heijmans Bastiaan T., Slagboom P. Eline, Boomsma Dorret I., Genetic and environmental influences interact with age and sex in shaping the human methylome, Nature Commun 7 (2016) 11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jiantao Ma CMR, Braun Kim V.E., Reynolds Lindsay M., Aslibekyan Stella, Xia Rui, Biligowda Niranjan G., Huan Tianxiao, Liu Chunyu, Mendelson Michael M., Joehanes Roby, Hu Emily A., Vitolins Mara Z., Wood Alexis C., Lohman Kurt, Ochoa-Rosales Carolina, van Meurs Joyce, Uitterlinden Andre, Liu Yongmei, Elhadad Mohamed A., Heier Margit, Waldenberger Melanie, Peters Annette, Colicino Elena, Whitsel Eric A., Baldassari Antoine, Gharib Sina A., Sotoodehnia Nona, Brody Jennifer A., Sitlani Colleen M., Tanaka Toshiko, Hill W. David, Corley Janie, Deary Ian J., Zhang Yan, Schöttker Ben, Brenner Hermann, Walker Maura E., Ye Shumao, Nguyen Steve, Pankow Jim, Demerath Ellen W., Zheng Yinan, Hou Lifang, Liang Liming, Lichtenstein Alice H., Hu Frank B., Fornage Myriam, Voortman Trudy, Daniel Levy Whole Blood DNA Methylation Signatures of Diet Are Associated With Cardiovascular Disease Risk Factors and All-Cause Mortality, Circ Genom Precis Med 13 (4) (2020) e002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ralf Gilsbach SP, Grüning Björn A., Schnick Tilman, Burger Lukas, Benes Vladimir, Würch Andreas, Bönisch Ulrike, Günther Stefan, Backofen Rolf, Fleischmann Bernd K., Schübeler Dirk, Hein Lutz, Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease, Nat Commun 5 (2014) 5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ng JW, Wong A, Kuh D, Smith GD, Relton CL, The role of longitudinal cohort studies in epigenetic epidemiology: challenges and opportunities, Genome Biol. 13(6) (2012) 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Goud Alladi C, Bellivier F, Marie-Claire C, DNA Methylation as a Biomarker of Treatment Response Variability in Serious Mental Illnesses: A Systematic Review Focused on Bipolar Disorder, Schizophrenia, and Major Depressive Disorder, Int J Mol Sci. 19 (10) (2018) 3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rosa-Garrido Manuel, Vondriska Thomas M., Epigenomes in Cardiovascular Disease, Circulation Research 122 (11) (2018) 1586–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Greißel Anna, Burgkart Rainer, Zimmermann Alexander, Eckstein Hans-Henning, Zernecke Alma, Jaroslav Pelisek Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques, Cardiovascular Pathology 25 (2) (2016) 79–86. [DOI] [PubMed] [Google Scholar]
- [24].Asare Y, Bokov Y, et al. , Histone Deacetylase 9 Activates IKK to Regulate Atherosclerotic Plaque Vulnerability, Circ Res. 127 (6) (2020) 811–823. [DOI] [PubMed] [Google Scholar]
- [25].Valencia-Morales MDP, Zaina S, Heyn H, et al. , The DNA methylation drift of the atherosclerotic aorta increases with lesion progression, BMC Med Genomics 8 (7) (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Einari Aavik HL, Leppänen Olli, Wirth Thomas, Häkkinen Sanna-Kaisa, Bräsen Jan-Hinrich, Beschorner Ulrich, Zeller Thomas, Braspenning Maarten, van Criekinge Wim, Mäkinen Kimmo, Ylä-Herttuala Seppo, Global DNA methylation analysis of human atherosclerotic plaques reveals extensive genomic hypomethylation and reactivation at imprinted locus 14q32 involving induction of a miRNA cluster, Eur Heart J. 36 (16) (2015) 993–1000. [DOI] [PubMed] [Google Scholar]
- [27].Ding J, Zeller T, Müller C, Lohman K, Nicklas BJ, Kritchevsky SB, Huang Z, de la Fuente A, Soranzo N, Settlage RE, Chuang CC, Howard T, Xu N, Goodarzi MO, Chen YD, Rotter JI, Siscovick DS, Parks JS, Murphy S, Jacobs DR Jr., Post W, Tracy RP, Wild PS, Blankenberg S, Hoeschele I, Herrington D, McCall CE, Liu Y, Alterations of a Cellular Cholesterol Metabolism Network Are a Molecular Feature of Obesity-Related Type 2 Diabetes and Cardiovascular Disease, Diabetes 64 (10) (2015) 3464–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Iwona Smolarek EW, Barciszewska Anna M., Nowak Stanislaw, Gawronska Iwona, Jablecka Anna, Barciszewska Miroslawa Z., Global DNA methylation changes in blood of patients with essential hypertension, Med Sci Monit 16 (3) (2010) CR149–155. [PubMed] [Google Scholar]
- [29].Norihiro Kato ML, Takeuchi Fumihiko, et al. , Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation, Nat Genet. 47 (11) (2015) 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Duarte Julio D., Burkley Ben, Gong Yan, Langaee Taimour Y., Turner Stephen T., Chapman Arlene B., Boerwinkle Eric, Gums John G., Cooper-Dehoff Rhonda M, Beitelshees Amber L, Bailey Kent R, Fillingim Roger B, Kone Bruce C, Johnson Julie A, Effects of genetic variation in H3K79 methylation regulatory genes on clinical blood pressure and blood pressure response to hydrochlorothiazide, J Transl Med. 10 (2012) 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].AH Association, “About Metabolic Syndrome”. https://www.heart.org/en/health-topics/metabolic-syndrome/about-metabolic-syndrome. (Accessed September 16th, 2020).
- [32].Luz D Orozco CF, Hale Christopher, Rubbi Liudmilla, Rinaldi Arturo, Civelek Mete, Pan Calvin, Lam Larry, Montoya Dennis, Edillor Chantle, Seldin Marcus, Boehnke Michael, Mohlke Karen L., Jacobsen Steve, Kuusisto Johanna, Laakso Markku, Lusis Aldons J., Pellegrini Matteo, Epigenome-wide association in adipose tissue from the METSIM cohort, Human Molecular Genetics 27 (10) (2018) 1830–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Al Muftah WA, Zaghlool SB, Visconti A, Tsai PC, Kumar P, Spector T, Bell J, Falchi M, Suhre K, Epigenetic associations of type 2 diabetes and BMI in an Arab population, Clin Epigenetics 8 (2016) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chambers JC, Lehne B, Drong A, Kriebel J, Motta V, Wahl S, Elliott HR, Rota F, Scott WR, Zhang W, Tan ST, Campanella G, Chadeau-Hyam M, Yengo L, Richmond RC, Adamowicz-Brice M, Afzal U, Bozaoglu K, Mok ZY, Ng HK, Pattou F, Prokisch H, Rozario MA, Tarantini L, Abbott J, Ala-Korpela M, Albetti B, Ammerpohl O, Bertazzi PA, Blancher C, Caiazzo R, Danesh J, Gaunt TR, de Lusignan S, Gieger C, Illig T, Jha S, Jones S, Jowett J, Kangas AJ, Kasturiratne A, Kato N, Kotea N, Kowlessur S, Pitkäniemi J, Punjabi P, Saleheen D, Schafmayer C, Soininen P, Tai ES, Thorand B, Tuomilehto J, Wickremasinghe AR, Kyrtopoulos SA, Aitman TJ, Herder C, Hampe J, Cauchi S, Relton CL, Froguel P, Soong R, Vineis P, Jarvelin MR, Scott J, Grallert H, Bollati V, Elliott P, McCarthy MI, Kooner JS, Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study, Lancet Diabetes Endocrinol. 3 (7) (2015) 526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rönn T, Gillberg L, Kokosar M, Perfilyev A, Jacobsen AL, Jørgensen SW, Brøns C, Jansson PA, Eriksson KF, Pedersen O, Hansen T, Groop L, Stener-Victorin E, Vaag A, Nilsson E, Ling C, Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood, Hum Mol Genet. 24 (13) (2015) 3792–3813. [DOI] [PubMed] [Google Scholar]
- [36].Wahl S, Drong A, Lehne B, et al. , Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity, Nature 541 (2017) 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].W.H. Organization, Cardiovascular Diseases. https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds). (Accessed September 16th, 2020.
- [38].Rask-Andersen D.M. Mathias, Ahsan Muhammad, Enroth Stefan, Ek Weronica E., Gyllensten Ulf, Johansson Åsa, Epigenome-wide association study reveals differential DNA methylation in individuals with a history of myocardial infarction, Human Molecular Genetics 25 (21) (2016) 4739–4748. [DOI] [PubMed] [Google Scholar]
- [39].Guarrera G.F. Simonetta, Onland-Moret N. Charlotte, Russo Alessia, Agnoli Claudia, Allione Alessandra, Di Gaetano Cornelia, Mattiello Amalia, Ricceri Fulvio, Chiodini Paolo, Polidoro Silvia, Frasca Graziella, Verschuren Monique W. M., Boer Jolanda M.A., Iacoviello Licia, van der Schouw Yvonne T., Tumino Rosario, Vineis Paolo, Krogh Vittorio, Panico Salvatore, Sacerdote Carlotta, Matullo Giuseppe, Gene-specific DNA methylation profiles and LINE-1 hypomethylation are associated with myocardial infarction risk, Clin Epigenetics 7 (2015) 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ligthart C.M. Symen, Aslibekyan Stella, Mendelson Michael M., Conneely Karen N., Tanaka Toshiko, Colicino Elena, Waite Lindsay L., Joehanes Roby, Guan Weihua, Brody Jennifer A., Elks Cathy, Marioni Riccardo, Jhun Min A., Agha Golareh, Bressler Jan, Ward-Caviness Cavin K., Chen Brian H., Huan Tianxiao, Bakulski Kelly, Salfati Elias L., WHI-EMPC Investigators; Fiorito Giovanni, CHARGE epigenetics of Coronary Heart Disease; Wahl Simone, Schramm Katharina, Sha Jin, Hernandez Dena G, Just Allan C, Smith Jennifer A, Sotoodehnia Nona, Pilling Luke C, Pankow James S, Tsao Phil S, Liu Chunyu, Zhao Wei, Guarrera Simonetta, Michopoulos Vasiliki J, Smith Alicia K, Peters Marjolein J, Melzer David, Vokonas Pantel, Fornage Myriam, Prokisch Holger, Bis Joshua C, Chu Audrey Y, Herder Christian, Grallert Harald, Yao Chen, Shah Sonia, McRae Allan F, Lin Honghuang, Horvath Steve, Fallin Daniele, Hofman Albert, Wareham Nicholas J, Wiggins Kerri L, Feinberg Andrew P, Starr John M, Visscher Peter M, Murabito Joanne M, Kardia Sharon L R, Absher Devin M, Binder Elisabeth B, Singleton Andrew B, Bandinelli Stefania, Peters Annette, Waldenberger Melanie, Matullo Giuseppe, Schwartz Joel D, Demerath Ellen W, Uitterlinden Andŕe G, van Meurs Joyce B J, Franco Oscar H, Ida Chen Yii-Der, Levy Daniel, Turner Stephen T, Deary Ian J, Ressler Kerry J, Dupuis Jośee, Ferrucci Luigi, Ong Ken K, Assimes Themistocles L, Boerwinkle Eric, Koenig Wolfgang, Arnett Donna K, Baccarelli Andrea A, Benjamin Emelia J, Dehghan Abbas, DNA methylation signatures of chronic low-grade inflammation are associated with complex diseases, Genome Biol. 17 (1) (2016) 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dan Jiang MS, You Linna, Lu Kai, Gao Lei, Hu Chunxiao, Wu Shiyong, Chang Guanglei, Tao Hongmei, Zhang Dongying, DNA methylation and hydroxymethylation are associated with the degree of coronary atherosclerosis in elderly patients with coronary heart disease, Life Sci. 224 (2019) 241–248. [DOI] [PubMed] [Google Scholar]
- [42].Shen L, He C, Zhang Y, Mechanism and function of oxidative reversal of DNA and RNA methylation, Annu Rev Biochem. 83 (2014) 585–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ek Weronica E., Enroth Stefan, Morris Andrew P., Lindgren Cecilia M., Mahajan Anubha, Gustafsson Stefan, Gyllensten Ulf, Lind Lars, Johansson Åsa, Genome-wide DNA methylation study identifies genes associated with the cardiovascular biomarker GDF-15, Human Molecular Genetics 25 (4) (2016) 817–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fiorito SG, Valle C, Ricceri F, Russo A, Grioni S, Mattiello A, Di Gaetano C, Rosa F, Modica F, Iacoviello L, Frasca G, Tumino R, Krogh V, Panico S, Vineis P, Sacerdote C, Matullo G, B-vitamins intake, DNA-methylation of One Carbon Metabolism and homocysteine pathway genes and myocardial infarction risk: the EPICOR study, Nutr Metab Cardiovasc Dis. 24 (2014) 483–488. [DOI] [PubMed] [Google Scholar]
- [45].Nadezhda Glezeva BM, Collier Patrick, Moravec Christine S., Phelan Dermot, Donnellan Eoin, Adam Russell-Hallinan Darran P. O Connor, Gallagher William M., Gallagher Joe, Kenneth McDonald Mark Ledwidge, Baugh John, Das Sudipto, Watson Chris J., Targeted DNA Methylation Profiling of Human Cardiac Tissue Reveals Novel Epigenetic Traits and Gene Deregulation Across Different Heart Failure Patient Subtypes 12, 2019, p. e005765, 3. [DOI] [PubMed] [Google Scholar]
- [46].Gilsbach R, Preissl S, et al. , Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo, Nat Commun 9(1) (2018) 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Pepin ME, Crossman DK, et al. , Genome-wide DNA methylation encodes cardiac transcriptional reprogramming in human ischemic heart failure, Lab Invest. 99 (3) (2019) 371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gilsbach R, Schwaderer M, Preissl S, Gruning BA, Kranzhofer D, Schneider P, Nuhrenberg TG, Mulero-Navarro S, Weichenhan D, Braun C, Dressen M, Jacobs AR, Lahm H, Doenst T, Backofen R, Krane M, Gelb BD, Hein L, Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo, Nat Commun 9(1) (2018) 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chen H, Orozco L, Wang J, Rau CD, Rubbi L, Ren S, Wang Y, Pellegrini M, Lusis AJ, Vondriska TM, DNA methylation indicates susceptibility to isoproterenol-inducd cardiac pathology and is associated with chromatin states, Circ Res 118 (2016) 786–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mehregan Movassagh M-KC, Knowles David A., Cordeddu Lina, Haider Syed, Down Thomas, Siggens Lee, Vujic Ana, Simeoni Ilenia, Penkett Chris, Goddard Martin, Lio Pietro, Bennett Martin R., Foo Roger S.-Y., Distinct epigenomic features in end-stage failing human hearts, Circulation 124 (22) (2011) 2411–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jan Haas KSF, Park Yoon Jung, Keller Andreas, Vogel Britta, Lindroth Anders M., Weichenhan Dieter, Franke Jennifer, Fischer Simon, Bauer Andrea, Marquart Sabine, Sedaghat-Hamedani Farbod, Kayvanpour Elham, Köhler Doreen, Wolf Nadine M., Hassel Sarah, Nietsch Rouven, Wieland Thomas, Ehlermann Philipp, Schultz Jobst-Hendrik, Andreas Dösch Derliz Mereles, Hardt Stefan, Backs Johannes, Hoheisel Jörg D., Plass Christoph, Katus Hugo A., Meder Benjamin, Alterations in cardiac DNA methylation in human dilated cardiomyopathy, EMBO Mol Med 5 (3) (2013) 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Benjamin Meder JH, Sedaghat-Hamedani Farbod, Kayvanpour Elham, Frese Karen, Lai Alan, Nietsch Rouven, Scheiner Christina, Mester Stefan, Bordalo Diana Martins, Amr Ali, Dietrich Carsten, Pils Dietmar, Siede Dominik, Hund Hauke, Bauer Andrea, Holzer Daniel Benjamin, Ruhparwar Arjang, Mueller-Hennessen Matthias, Weichenhan Dieter, Plass Christoph, Weis Tanja, Backs Johannes, Wuerstle Maximilian, Keller Andreas, Katus Hugo A., Posch Andreas E., Epigenome-Wide Association Study Identifies Cardiac Gene Patterning and a Novel Class of Biomarkers for Heart Failure, Circulation 136 (16) (2017). [DOI] [PubMed] [Google Scholar]
- [53].Bong-Seok I-UK, Bae Jae-Bum, Yu Ho-Yeong, Jeon Eun-Seok, Lee Hae-Young, Kim Jae-Joong, Choi Murim, Choi Sun Shim, Methylome analysis reveals alterations in DNA methylation in the regulatory regions of left ventricle development genes in human dilated cardiomyopathy, Genomics 108 (2) (2016). [DOI] [PubMed] [Google Scholar]
- [54].Du X, Ma C, Is Atrial Fibrillation a Preventable Disease? J Am Coll Cardiol. 69 (15) (2017) 1968–1982. [DOI] [PubMed] [Google Scholar]
- [55].Lin X.Y. Honghuang, Xie Zhijun, Lunetta Kathryn L., Lubitz Steven A., Larson Martin G., Ko Darae, Magnani Jared W., Mendelson Michael M., Liu Chunyu, McManus David D., Levy Daniel, Ellinor Patrick T., Benjamin Emelia J., Methylome-wide Association Study of Atrial Fibrillation in Framingham Heart Study, Sci Rep 7 (2017) 40377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Guochang Zhao JZ, Gao Jie, Liu Yan, Gu Song, Zhang Xitao, Su Pixiong, Genome-wide DNA methylation analysis in permanent atrial fibrillation, Mol Med Rep. 16 (4) (2017) 5505–5514. [DOI] [PubMed] [Google Scholar]
- [57].Bock F.H. Christoph, Carmona Francisco J., Tierling Sascha, Datlinger Paul, Assenov Yassen, Berdasco María, Bergmann Anke K., Booher Keith, Busato Florence, Campan Mihaela, Dahl Christina, Dahmcke Christina M., Diep Dinh, Fernández Agustín F., Gerhauser Clarissa, Haake Andrea, Heilmann Katharina, Holcomb Thomas, Hussmann Dianna, Ito Mitsuteru, Kläver Ruth, Kreutz Martin, Kulis Marta, Lopez Virginia, Nair Shalima S., Paul Dirk S., Plongthongkum Nongluk, Qu Wenjia, Queirós Ana C., Reinicke Frank, Sauter Guido, Schlomm Thorsten, Statham Aaron, Stirzaker Clare, Strogantsev Ruslan, Urdinguio Rocío G., Walter Kimberly, Weichenhan Dieter, Weisenberger Daniel J., Beck Stephan, Clark Susan J., Esteller Manel, Ferguson-Smith Anne C., Fraga Mario F., Guldberg Per, Hansen Lise Lotte, Laird Peter W., Martín-Subero Jośe I., Nygren Anders O.H., Peist Ralf, Plass Christoph, Shames David S., Siebert Reiner, Sun Xueguang, Tost Jörg, Walter Jörn, Zhang Kun, Quantitative comparison of DNA methylation assays for biomarker development and clinical applications, Nat Biotechnol. 34 (7) (2016) 726–737. [DOI] [PubMed] [Google Scholar]
- [58].Nestor C, Meehan R, Dunican D, Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA, Biotechniques. 48 (4) (2010) 317–319. [DOI] [PubMed] [Google Scholar]
- [59].Wilhelm-Benartzi CS, Karagas MR, Flanagan JM, Christensen BC, Kelsey KT, Marsit CJ, Houseman EA, Brown R, Review of processing and analysis methods for DNA methylation array data, Br J Cancer 109 (6) (2013) 1394–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sun J.C. Zhifu, Slager Susan, Kocher Jean-Pierre, Base resolution methylome profiling: considerations in platform selection, data preprocessing and analysis, Epigenomics 7 (5) (2015) 813–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ziller Michael J., Gu Hongcang, Gnirke Andreas, Meissner Alexander, Targeted bisulfite sequencing of the dynamic DNA methylome, Epigenetics and Chromatin 9 (2016) 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lohoff FW, Roy A, Jung J, et al. , Epigenome-wide association study and multi-tissue replication of individuals with alcohol use disorder: evidence for abnormal glucocorticoid signaling pathway gene regulation, Mol Psychiatry (2020), 10.1038/s41380-020-0734-4. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Roni Lehmann-Werman DN, Zemmour Hai, Moss Joshua, Magenheim Judith, Vaknin-Dembinsky Adi, Rubertsson Sten, Bengt Nellgård Kaj Blennow, Zetterberg Henrik, Spalding Kirsty, Haller Michael J., Wasserfall Clive H., Schatz Desmond A., Greenbaum Carla J., Dorrell Craig, Grompe Markus, Zick Aviad, Hubert Ayala, Maoz Myriam, Fendrich Volker, Bartsch Detlef K., Golan Talia, Ben Sasson Shmuel A., Zamir Gideon, Razin Aharon, Cedar Howard, Shapiro A.M. James, Glaser Benjamin, Shemer Ruth, Dor Yuval, Identification of tissue-specific cell death using methylation patterns of circulating DNA, Proc Natl Acad Sci 113 (13) (2016) E1826–E1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kowarsky J.C.-S. Mark, Kertesz Michael, De Vlaminck Iwijn, Koh Winston, Pan Wenying, Martin Lance, Neff Norma F., Okamoto Jennifer, Wong Ronald J., Kharbanda Sandhya, El-Sayed Yasser, Blumenfeld Yair, Stevenson David K., Shaw Gary M., Wolfe Nathan D., Quake Stephen R., Numerous uncharacterized and highly divergent microbes which colonize humans are revealed by circulating cell-free DNA, Proc Natl Acad Sci 114 (36) (2017) 9623–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhu P, Guo H, Ren Y, et al. , Single-cell DNA methylome sequencing of human preimplantation embryos, Nat Genet. 50 (2018) 12–19. [DOI] [PubMed] [Google Scholar]
- [66].Guo J, Yi C, et al. , Chromatin and Single-Cell RNA-Seq Profiling Reveal Dynamic Signaling and Metabolic Transitions during Human Spermatogonial Stem Cell Development, Cell Stem Cell 21 (4) (2017) 533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bian S, Zhou X, et al. , Single-cell multiomics sequencing and analyses of human colorectal cancer, Science 362 (6418) (2018) 1060–1063. [DOI] [PubMed] [Google Scholar]
- [68].Chen X, Zhang Y, Li X, Zhong JF, Wang C, Single-cell transcriptome and epigenomic reprogramming of cardiomyocyte-derived cardiac progenitor cells, Sci Data 3 (2016) 160079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ino MV, Karemaker D, Single-Cell DNA Methylation Profiling: Technologies and Biological Applications, Trends Biotechnol 36 (9) (2016) 952–965. [DOI] [PubMed] [Google Scholar]
- [70].Birney E, Greally JM, Epigenome-wide Association Studies and the Interpretation of Disease -Omics, PLoS Genet 12 (6) (2016), e1006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Tsai PC, Power and sample size estimation for epigenome-wide association scans to detect differential DNA methylation, Int J Epidemiol 44 (4) (2015) 1429–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Mansell G, Bao Y, Kumari M, Schalkwyk LS, Mill J, Hannon E, Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array, BMC Genomics 20(1) (2019) 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhang S.B. Yun, Sun Zhifu, Statistical method evaluation for differentially methylated CpGs in base resolution next-generation DNA sequencing data, Brief Bioinform 19 (3) (2018) 374–386. [DOI] [PubMed] [Google Scholar]
- [74].Barfield RT, Kilaru V, Smith AK, Mercer KB, Duncan R, Klengel T, Mehta D, Binder EB, Epstein MP, Ressler KJ, Conneely KN, Accounting for population stratification in DNA methylation studies, Genet Epidemiol. 38 (3) (2014) 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Rao TJ, A Framework for Interpreting Type I Error Rates from a Product-Term Model of Interaction Applied to Quantitative Traits, Genet Epidemiol. 40 (2) (2016) 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].König IR, Validation in genetic association studies, Brief Bioinform 12 (3) (2011) 253–258. [DOI] [PubMed] [Google Scholar]
- [77].Fortin JP, Hansen KD, Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi, Bioinformatics 33 (4) (2017) 558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wreczycka K, Yusuf D, Grüning B, Assenov Y, Akalin A, Strategies for analyzing bisulfite sequencing data, J Biotechnol. 261 (2017) 105–115. [DOI] [PubMed] [Google Scholar]
- [79].Aryee MJ, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA, Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays, Bioinformatics 30 (10) (2014) 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gu H, Bock C, Boyle P, Gnirke A, Meissner A, Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling, Nat Protoc. 6 (4) (2011) 468–481. [DOI] [PubMed] [Google Scholar]
- [81].Field AE, Wang T, Havas A, Ideker T, Adams PD, DNA Methylation Clocks in Aging: Categories, Causes, and Consequences, Mol Cell. 71 (6) (2018) 882–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lee KW, Cigarette smoking and DNA methylation, Front Genet. 4 (2013) 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Sun D, Su S, et al. , Body Mass Index Drives Changes in DNA Methylation: A Longitudinal Study, Circ Res. 125 (9) (2019) 824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Houseman EA, Molitor J, Marsit CJ, Reference-free cell mixture adjustments in analysis of DNA methylation data, Bioinformatics 30 (10) (2014) 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Rahmani N.Z. Elior, Baran Yael, Eng Celeste, Hu Donglei, Galanter Joshua, Sam Oh, Burchard Esteban G., Eskin Eleazar, Zou James, Halperin Eran, Sparse PCA Corrects for Cell-Type Heterogeneity in Epigenome-Wide Association Studies, Nat Methods 13 (5) (2017) 443–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].James Zou CL, Heckerman David, Aryee Martin, Listgarten Jennifer, Epigenome-wide association studies without the need for cell-type composition, Nat Methods 11 (3) (2014) 309–311. [DOI] [PubMed] [Google Scholar]
- [87].Koestler DC, Karagas MR, Marsit CJ, Langevin SM, Kelsey KT, Wiencke JK, Houseman EA, Blood-based profiles of DNA methylation predict the underlying distribution of cell types: a validation analysis, Epigenetics 8 (8) (2013) 816–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Eugene Andres Houseman WPA, Koestler Devin C., Christensen Brock C., Marsit Carmen J., Nelson Heather H., Wiencke John K., Kelsey Karl T., DNA methylation arrays as surrogate measures of cell mixture distribution, BMC Bioinformatics 13 (2012) 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Angarica VE, Bioinformatics Tools for Genome-Wide Epigenetic Research, Adv Exp Med Biol 978 (489–512) (2017). [DOI] [PubMed] [Google Scholar]
- [90].Akalin A, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE, MethylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles, Genome Biol. 13 (10) (2012) R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Warden CD, Tompkins JD, Li X, Wang C, Riggs AD, Yu H, Jove R, Yuan YC, COHCAP: an integrative genomic pipeline for single-nucleotide resolution DNA methylation analysis, Nucleic Acids Res. 41 (11) (2013) e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Dolzhenko E, Using beta-binomial regression for high-precision differential methylation analysis in multifactor whole-genome bisulfite sequencing experiments, BMC Bioinformatics 15 (2014) 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Park Y, Rozek LS, Sartor MA, MethylSig: a whole genome DNA methylation analysis pipeline, Bioinformatics 30 (17) (2014) 2414–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Sun D, Rodriguez B, Park HJ, Tong P, Meong M, Goodell MA, Li W, MOABS: model based analysis of bisulfite sequencing data, Genome Biol. 15 (2) (2014) R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Differential methylation analysis for BS-seq data under general experimental design, Bioinformatics 32 (10) (2016) 1446–1453. [DOI] [PubMed] [Google Scholar]
- [96].Lea AJ, Zhou X, A Flexible, Efficient Binomial Mixed Model for Identifying Differential DNA Methylation in Bisulfite Sequencing Data, PLoS Genet. 11 (11) (2015), e1005650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Adib Shafi CM, Nguyen Tin, Draghici Sorin, A survey of the approaches for identifying differential methylation using bisulfite sequencing data, Brief Bioinform 19 (5) (2018) 737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Sigin VO, Kuznetsova EB, et al. , DNA methylation markers panel can improve prediction of response to neoadjuvant chemotherapy in luminal B breast cancer., Sci Rep. 10(1) (2020) 9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Plant D, Nair N, et al. , Differential Methylation as a Biomarker of Response to Etanercept in Patients With Rheumatoid Arthritis, Arthritis Rheumatol. 68 (6) (2016) 1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Rauschert S, Melton PE, Huang RC, Machine learning and clinical epigenetics: a review of challenges for diagnosis and classification, Clin Epigenetics 12(1) (2020) 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Tian Q, Tang J, Fang Y, Yu Z, Fan S, MRCNN: a deep learning model for regression of genome-wide DNA methylation, BMC Genomics 20 (2019) (2019) 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Crowgey EL, Robinson KG, Yeager SK, Akins RE, Epigenetic machine learning: utilizing DNA methylation patterns to predict spastic cerebral palsy, BMC Bioinformatics 19(1) (2018) 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Jurmeister P, Seegerer P, Bockmayr T, Treue D, Montavon G, Vollbrecht C, Arnold A, Teichmann D, Bressem K, Schüller U, von Laffert M, Müller KR, Capper D, Klauschen F, Machine learning analysis of DNA methylation profiles distinguishes primary lung squamous cell carcinomas from head and neck metastases, Sci Transl Med. 11 (509) (2019) eaaw8513. [DOI] [PubMed] [Google Scholar]