

Abstract
Background and Aims
Ischemia–reperfusion injury (IRI) represents a risk factor in liver transplantation (LT). We have shown that overexpression of heme oxygenase‐1 (HO‐1) mitigates hepatic IRI in LT recipients. Here, we hypothesized that human antigen R (HuR), the stabilizer of adenylate‐uridylate (AU)‐rich mRNAs, is required for hepatoprotection in LT.
Approach and Results
In an experimental arm, HuR/HO‐1 protein expression was correlated with hepatic IRI phenotype. In an in vitro inflammation mimic model of hepatic warm IRI, induction of HuR/HO‐1 and cytoplasmic localization following cytokine preconditioning were detected in primary hepatocyte cultures, whereas HuR silencing caused negative regulation of HO‐1, followed by enhanced cytotoxicity. Using the HuR‐inhibitor, we showed that HuR likely regulates HO‐1 through its 3′ untranslated region and causes neutrophil activation (CD69+/lymphocyte antigen 6 complex locus G [Ly6‐G]). HuR silencing in bone marrow–derived macrophages decreased HO‐1 expression, leading to the induction of proinflammatory cytokines/chemokines. RNA sequencing of HuR silenced transcripts under in vitro warm IRI revealed regulation of genes thymus cell antigen 1 (THY1), aconitate decarboxylase 1 (ACOD1), and Prostaglandin E Synthase (PTGES). HuR, but not hypoxia‐inducible protein alpha, positively regulated HO‐1 in warm, but not cold, hypoxia/reoxygenation conditions. HuR modulated HO‐1 in primary hepatocytes, neutrophils, and macrophages under reperfusion. Adjunctive inhibition of HuR diminished microtubule‐associated proteins 1A/1B light chain 3B (LC3B), a marker for autophagosome, under HO‐1 regulation, suggesting a cytoprotective mechanism in hepatic IR. In a clinical arm, hepatic biopsies from 51 patients with LT were analyzed at 2 hours after reperfusion. Graft HuR expression was negatively correlated with macrophage (CD80/CD86) and neutrophil (Cathepsin G) markers. Hepatic IRI increased HuR/HO‐1 expression and inflammatory genes. High HuR–expressing liver grafts showed lower serum alanine aminotransferase/serum aspartate aminotransferase levels and improved LT survival.
Conclusions
This translational study identifies HuR as a regulator of HO‐1–mediated cytoprotection in sterile liver inflammation and a biomarker of ischemic stress resistance in LT.
Abbreviations
- ACOD1
aconitate decarboxylase 1
- AU
adenylate‐uridylate
- Bcl‐xL
B cell lymphoma extra large
- BMM
bone marrow macrophage
- BMP4
bone morphogenic protein 4
- bx
biopsy
- CCL
chemokine (C‐C motif) ligand
- CM
cytokine mix
- CXCL
chemokine (C‐X‐C motif) ligand
- DAMP
damage‐associated molecular pattern
- DMSO
dimethyl sulfoxide
- HIF‐1α
hypoxia‐inducible protein alpha
- HO‐1
heme oxygenase‐1
- HuR
human antigen R
- H/R
hypoxia/reoxygenation
- IR
Ischemia‐reperfusion
- IRI
ischemia–reperfusion injury
- IL
interleukin
- LPS
lipopolysaccharide
- LT
liver transplantation
- Ly6‐G
lymphocyte antigen 6 complex locus G
- MCP
monocyte chemoattractant protein
- PTBP2
polypyrimidine‐tract binding protein 2
- PTGES
prostaglandin E synthase‐1
- RBP
RNA‐binding protein
- RNA‐Seq
RNA‐sequencing
- sALT
serum alanine aminotransferase
- sAST
serum aspartate aminotransferase
- THY1
thymus cell antigen 1
- TLR
toll‐like receptor
- TNF
tumor necrosis factor
- UCLA
University of California, Los Angeles
- UTR
untranslated region
Liver ischemia–reperfusion injury (IRI), a common clinical condition triggered by vascular surgery, hepatic resection, or liver transplantation (LT), represents a major risk factor for acute/chronic graft rejection and is a contributing factor in organ shortage available for transplantation.1, 2 Oxidative stress, the hallmark of IRI, triggers proinflammatory cytokine/chemokine programs and creates a deleterious milieu promoting sterile inflammation, cell death, and ultimately organ failure. These adverse innate immune‐driven effects become even more significant in expanded criteria liver transplants from marginal, deceased, and non‐heart‐beating donors. However, despite obvious clinical importance, there are currently no therapeutics against or patient‐specific diagnostics of IRI‐LT. As mechanisms that account for liver IRI are not well appreciated, biomarkers of early graft function as well as strategies to improve clinical outcomes and expand the donor pool are warranted.
The University of California, Los Angeles (UCLA) group has pioneered the concept of combating liver IRI by inducing heme oxygenase‐1 (HO‐1; Hmox1, hsp32), the rate‐limiting enzyme that catalyzes the degradation of heme into biliverdin, iron, and carbon monoxide.3 In rodent models of peritransplant hepatic damage, we confirmed that pharmacological‐mediated, cell therapy‐mediated, or transgene‐mediated HO‐1 overexpression ameliorated IRI by limiting the severity of oxidative stress and promoting anti‐inflammatory, antiapoptotic, and proangiogenic function. The “healing” potential of HO‐1 was central to re‐establishing homeostasis in IR‐stressed livers, but when HO‐1 overexpression was impaired, the disease process was exacerbated, and mechanisms that ensure a return to homeostasis were blunted. This finding made us propose that HO‐1 might be considered as a denominator of donor organ “quality.”4 We then addressed a functional link between HO‐1 and toll‐like receptor (TLR) 4–driven sterile inflammation in LT by documenting that HO‐1 activation 1) promotes cytoprotection by suppressing Type‐1 interferon (IFN) downstream of TLR4 signaling5 and 2) triggers phosphoinositide 3‐kinase/Akt signaling to provide negative feedback for TLR4‐mediated sterile inflammation.6
In the liver, genome‐wide changes in RNA‐binding proteins (RBPs) regulate the half‐life of many mRNAs in response to stress, necrosis, inflammation, or immune stimuli. Human antigen R (HuR), a member of the Hu/Elav (Embryonic Lethal, Abnormal Vision) family, is an RBP ubiquitously expressed in the nucleus of unstimulated cells. When sensing cellular stress, HuR promotes rapid spatiotemporal expression of stress‐response proteins by selectively binding/stabilizing their AU‐rich mRNAs.7 Although HuR may be essential for HO‐1 transcriptional regulation under stress,8 its physiologic and pathologic functions in IR‐triggered sterile liver inflammation or LT remain unknown.
Here, we used hepatocyte‐induced hypoxia as a mimic of acute liver ischemia and compared this with preconditioned cytokine mix (CM) exposure in a murine model of warm hepatic IRI to gain further insight into the role of HuR/HO‐1 axis in LT. In parallel, we retrospectively analyzed 51 human patients with LT to decipher the impact of graft HuR expression on LT phenotype/outcomes and unpack its relationship with HO‐1. This translational study establishes HuR as a regulator of HO‐1–mediated cytoprotection in sterile liver inflammation and a biomarker of ischemic stress resistance in LT.
Experimental Procedures
Animals
C57BL/6 male mice at 6–8 weeks of age were used (Jackson Laboratory, Bar Harbor, ME). Animals were housed in the UCLA animal facility under specific pathogen‐free conditions and received humane care according criteria outlined in the “Guide for the Care and Use of Laboratory Animals” (National Institutes of Health publication 86‐23, revised 1985).
Clinical Liver Transplant Study
Fifty‐one adult primary liver transplant (LT) recipients were recruited under institutional review board protocol (13‐000143; May 10, 2013 to April 6, 2015). Patients provided informed consent before their participation in the study. The recipient and donor variables of clinical patients' cohort are shown (Supporting Table S1A,B). Study data were collected and managed using REDCap electronic data capture tools hosted at UCLA. All donor organs were perfused and stored in cold University of Wisconsin solution (ViaSpan; Bristol‐Meyers Squibb, Garden City, NY). Cold ischemia time was defined as the time from perfusion of the donor with a preservation solution to removal of the liver from cold storage. Protocol Tru‐Cut needle biopsies (bx) were obtained intraoperatively from the left lobe about 2 hours after portal reperfusion (before surgical closing of abdomen) and snap frozen. No donor organs were obtained from executed prisoners or other institutionalized persons.
Please see additional methods in the Supporting Information.
Results
Elevated HuR/HO‐1 Protein Expression Pattern is Correlated With Hepatic IR Stress In Vivo and In Vitro
Because we have demonstrated that HO‐1 overexpression protects livers against IRI,9 we now attempted to identify putative factors that regulate HO‐1 mRNA stability. Environmental stimuli like cytokines, hormones, and temperature shifts, as well as stresses like hypoxia and tissue injury, can control the stability of a particular mRNA. To identify RBPs that control HO‐1 mRNA stability in the acute phase of IR stress, we used a mouse (C57/BL6) model of hepatic warm IRI. When subjected to 60 minutes of portal triad blockage, gradual hepatocellular damage peaked at 6 hours of reperfusion as compared with sham‐treated controls. Indeed, hematoxylin and eosin staining of representative IR‐liver tissue samples (Fig. 1A) showed extensive sinusoidal congestion and edema/vacuolization as well as hepatocellular necrosis as measured by terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling assay and corresponding Suzuki score (mean ± SEM 5.599 ± 2.019, P < 0.05) as compared with control livers (Fig. 1B,C). The hepatocellular damage, as assessed by serum alanine aminotransferase (sALT)/serum aspartate aminotransferase (sAST) levels (Fig. 1D), as well as expression of inflammatory chemokines (chemokine [C‐C motif] ligand [CCL] 2/monocyte chemoattractant protein [MCP] 1, chemokine [C‐X‐C motif] ligand [CXCL] 10, CXCL2) (Fig. 1E) were all highly elevated in IR‐stressed mouse livers.
Figure 1.
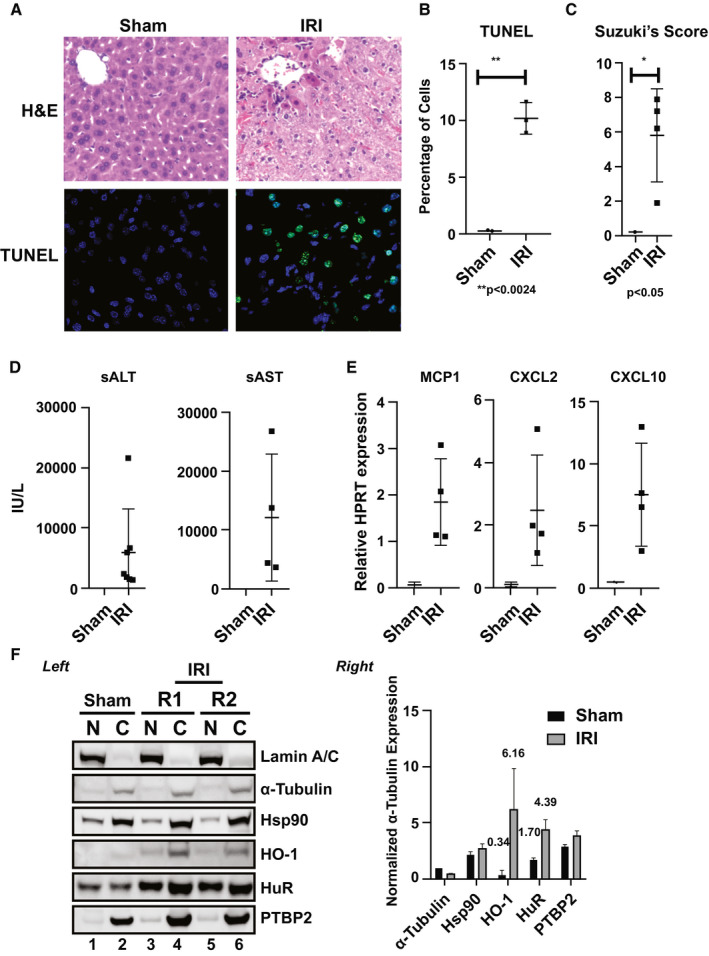
HO‐1 and HuR protein levels are up‐regulated in a mouse warm hepatic IRI model. Livers in C57/BL mice were subjected to 60 minutes of portal triad blockage and subsequent reperfusion for 6 hours. (A) Representative hematoxylin and eosin (H&E) and (B) terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling (TUNEL) staining (n = 3; original magnification, ×100; **P < 0.0024). (C) Suzuki's score (*P < 0.05; IRI vs. Sham) of the liver sections, as well as (D) sALT/sAST levels (n = 4–7/group; mean ± standard error, IU/L) and (E) mRNA levels of chemokines MCP1, CXCL2, and CXCL10 are shown. (F) Left: representative fractionation of nuclear and cytoplasmic proteins from liver tissues obtained from animals exposed to warm IRI. Lamin A/C (nuclear marker), α‐tubulin (cytosolic marker), Hsp90 (stress‐response comparison control), HO‐1 (subject of study), HuR (subject of study), and PTBP2 (downstream effector of HuR) are shown. R1 and R2 are replicates 1 and 2. Right: unpaired two‐tailed Student t test of representative samples presented were calculated relative to α‐Tubulin cytoplasmic expression. Data shown are mean ± SEM; n = 2 (repeated at least three independent times). HPRT, hypoxanthine‐guanine phosphoribosyltransferase.
We next aimed to determine whether HO‐1 was inducible under pathological conditions such as oxidative stress. As expected, HO‐1, but not Hsp90, serving as a heat‐shock cytoplasmic control nonresponsive to IRI, was induced (approximately 18‐fold; 6.16/0.34) and localized to the cytoplasm after hepatic IR (Fig. 1F). Importantly, when the induction of HuR and one of its downstream effector targets, polypyrimidine‐tract binding protein 2 (PTBP2) was tested, we observed more than 2.5‐fold (4.39/1.70) induction of HuR, both in the nucleus and cytoplasm (Fig. 1F). It was unclear, however, whether HuR induction was caused by hepatic gene expression or whether innate immune activation by infiltrating macrophages/neutrophils was causing its accumulation in IR‐stressed livers.
To test the induction of HuR and HO‐1 in distinct cell populations involved in IRI, we established an in vitro inflammation model in which primary hepatocytes isolated from livers of WT mice (C57/BL6) were preconditioned with a CM that included tumor necrosis factor (TNF)‐α, IFN‐γ, interleukin (IL)‐1β, and endotoxin lipopolysaccharide (LPS) exposure (Fig. 2A). This in vitro IRI “test‐tube” model is based on one described for human hepatocytes.10 Because IRI is associated with cellular injury, CM‐treated samples were analyzed for P62 expression, a marker for autophagy, which is an intracellular self‐digesting pathway responsible for energy homeostasis and removing long‐lived or damaged organelles/proteins.11 Not surprisingly, we observed dramatic up‐regulation of P62 protein levels (Fig. 2B) after exposure of cultured hepatocytes to the CM. Consistent with inflammatory events in IR‐stressed liver in vivo, we observed up‐regulation of HO‐1 (mean ± SEM, 0.3955 ± 0.07022, P < 0.0301 +CM vs. dimethyl sulfoxide [DMSO]) in the presence of CM‐treated samples in vitro. Importantly, HuR as well as PTBP2 showed markedly increased protein levels. These changes in gene expression profiles were paralleled by significant membrane leakage, as demonstrated by the release of lactate dehydrogenase (Fig. 2C) and western blot‐assisted detection of histone H3 alarmin (Fig. 2D) (mean ± SEM, 5.965 ± 0.2650, P < 0.0022 +CM vs. DMSO). Histones that act as damage‐associated molecular patterns (DAMPs) passively exist in free form or are bound to DNA as part of nucleosomes. Once released in the extracellular space, DAMPs activate the host immune system, causing further cytotoxicity. We also examined HO‐1 and HuR cytolocalization after cytokine stimulation by immunofluorescence staining and found they were more conspicuous in the cytoplasm after proinflammatory CM treatment (Fig. 2E), suggesting that resolution of metabolic disturbances is central to the HuR/HO‐1 axis. Thus, we have established a reliable model system to study the HuR–HO‐1 crosstalk in hepatic IRI and anti‐inflammatory functions in vivo and in vitro.
Figure 2.
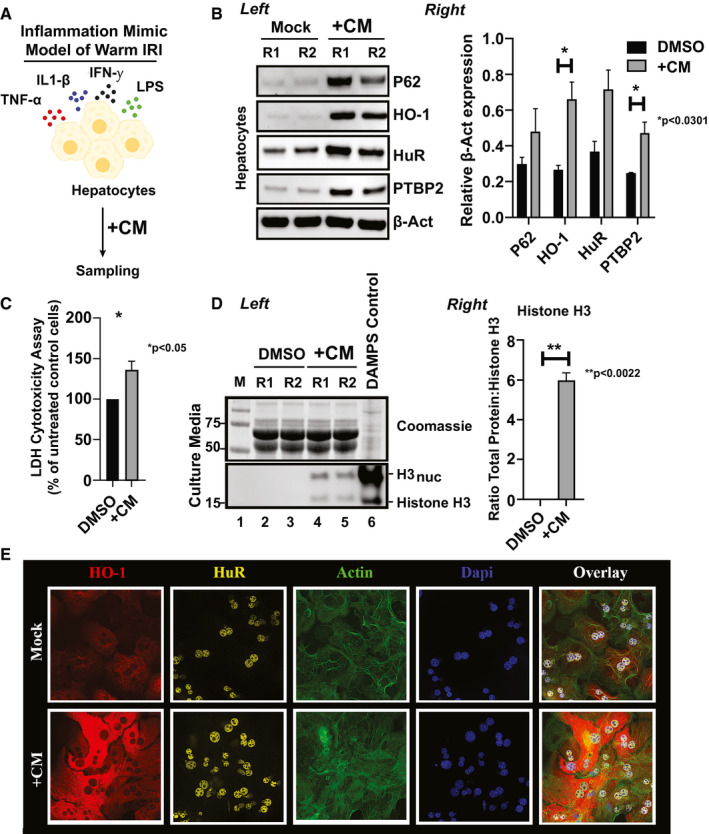
Cytokine stimulation of primary‐derived hepatocytes mimics reperfusion injury by causing the up‐regulation of HO‐1/HuR protein levels. (A) Workflow of the in vitro inflammation mimic model of warm IRI, dependent on CM)preconditioning. (B) Left: total lysates from cells treated with either DMSO or CM for 12 hours were probed by western blot for expression differences between P62 (marker for autophagy), HO‐1 (subject of study), HuR (subject of study), PTPB2 (downstream marker of HuR), or β‐Act (as loading control). Right: an unpaired two‐tailed Student t test of representative samples presented calculated relative β‐Act expression (*P < 0.0301, +CM vs. DMSO control). Data are shown as mean (n = 3/group) ± SEM. (C) Lactate dehydrogenase (LDH) release after CM hepatocytes treatment as a percentage of untreated control cells (*P < 0.05, +CM vs. DMSO control). (D) Left: hepatocellular toxicity was assessed by probing for histone H3 levels from culture media (bottom panel). Histone H3 versus H3nuc (the nucleosome‐associated form) represent minus (naked) or plus nucleosome association. Coomassie‐stained polyacrylamide gel electrophoresis samples were used as loading controls (top panel). Positive DAMPs control was obtained from hepatocytes treated 48 hours with 1× HBSS. Right: data shown are mean ± SEM of representative samples as presented. **P < 0.0022, CM versus DMSO for n = 3 pooled samples. (E) Representative immunohistochemical detection of hepatocyte HuR and HO‐1 after 12 hours CM conditioning (n = 3/group; original magnification, ×40). Act, actin; Dapi, 4′,6‐diamidino‐2‐phenylindole.
HuR Silencing Down‐Regulates HO‐1 and Associates With Increased Hepatotoxicity
We next aimed to determine the hierarchical role of HuR and HO‐1 in the mechanism of hepatocellular protection. We treated hepatocytes with siRNAs targeting HuR in the presence or absence of CM exposure and examined HO‐1 expression levels coordinately (Fig. 3A). When the HuR silencing efficiency approximated 85% [mean values (1.2445 – 0.184) – 1; Fig. 3B], we observed a positive correlation with HO‐1 silencing (r = 0.9603, P < 0.0201; Fig. 3C) and P62, although the difference for the latter failed to reach statistical significance (P = 0.2549). Importantly, interrogation of LC3B, a marker for autophagosome structure and a target of HO‐1,12 revealed a significant reduction of protein levels after HuR silencing (r = 0.9672, P < 0.0165, Fig. 3A), hinting at a putative axis through HO‐1 activity. We also show that siHuR augmented the hepatocellular damage regardless of CM preconditioning (Fig. 3D; lanes 9–10 vs. 5–6) and is quantitated in Fig. 3D (right panel) (mean ± SEM = 0.2897 ± 0.01348; P < 0.0022, siHuR DMSO vs. siControl DMSO). Interestingly, histone H3 was predominately in its naked form (lower band) in samples treated with siHuR versus CM alone (lanes 9–12 vs. 3–4), suggesting that actively excreted or passively released stressed/dying cells release DAMPs from alternative molecular pathways. Thus, our data show that HuR acts as an essential cytoprotective mediator of hepatocellular function.
Figure 3.
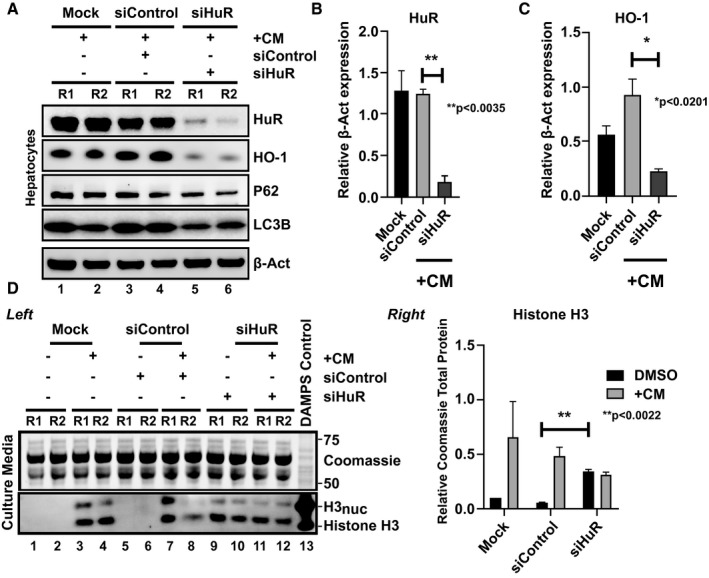
Silencing of HuR exacerbates hepatocellular toxicity during an in vitro warm IRI model. (A) Total lysates from CM‐conditioned primary hepatocyte cultures treated with siControl or siHuR siRNAs were probed by western blot for differences between HuR, HO‐1, P62, LC3B and β‐Act, as a loading control. Unpaired two‐tailed Student t test of representative samples (B) HuR and (C) HO‐1 as presented were calculated relative to β‐Act expression (for HuR expression, **P < 0.0035; for HO‐1 expression, *P < 0.0201, siHuR vs. siControl). (D) Left and right: hepatocellular toxicity was assessed similarly to Fig. 2D legend. **P < 0.0022, siHuR versus siControl in absence of CM for n = 2 (repeated at least three independent times).
HuR Inhibition Destabilizes the 3′ Untranslated Region of HO‐1 mRNA and Activates CD69+Ly6‐G+ Neutrophils
Because the regulatory function of HO‐1 depends on HuR in CM‐mediated hepatocyte cultures, we next asked whether this interaction depends on the regulatory elements encoded in the 3′ untranslated region (UTR) of HO‐1. Because HuR targets the 3′UTR of transcripts to protect against degradation, we searched for potential HuR consensus binding sites in HO‐1 mRNA. Using possible iterations of the flexible motif described elsewhere,13 the conformity to the expected nucleotide(s) at each position was scored, and 4 putative HuR‐binding motifs were identified in exon 5, 2 consisting of short uridylate (U)‐rich stretches that contained some G but no C nucleotides (Supporting Fig. S1A). To test the hypothesis that HuR stabilizes HO‐1 3′UTR, we used the coumarin‐derived HuR disrupter, CMLD‐2, which has been shown to functionally block AU‐rich interactions found mostly in the 3′UTR of protein‐coding mRNAs.14 Consistently, our data show that CMLD‐2 treatment (30 μM) significantly reduced HO‐1 protein levels in primary hepatocyte cultures, indicating that HuR likely stabilizes the 3′UTR of HO‐1 mRNA (Supporting Fig. S1B). By contrast, when the HuR nonspecific inhibitor NC‐314 was included as a negative control, no significant difference was observed in HO‐1 protein levels.
Because neutrophils (Ly6‐G+) are the early responders to hepatic IR stress and represent important components in the protracted inflammatory response/severity of IRI, we investigated the role of HuR in this cellular population. CD69, one of the neutrophil markers implicated in the pathogenesis of inflammation, can act as a costimulus during neutrophil activation in vitro.15 We hypothesized that primary cultured neutrophils treated with CMLD‐2 will exert proinflammatory functions, notably under LPS stress. Our data show that functionally blocking HuR with CMLD‐2 leads to increased neutrophil activation by evidence of CD69+Ly6‐G+ markers (quantitated in Supporting Fig. S1C as presented in Supporting Fig. S1D; mean ± SEM, 46.88 ± 3.062, P < 0.0042). Importantly, the LPS challenge significantly increased the frequency of CD69+Ly6‐G+ neutrophils at the lowest inhibitory dosage (50 μM) of CMLD‐2 (mean ± SEM, 54.05 ± 2.980, P < 0.0030), suggesting that HuR may minimize the adverse effects of neutrophil activation, a hallmark of early reperfusion mobilized through chemotaxis during IRI.
HuR Silencing Triggers Proinflammatory Cytokine Program in Bone Marrow Macrophage Cultures
Because macrophages, critical early initiators of innate immune response in the liver, orchestrate inflammation cascade subsequent to IR stress, we next aimed to determine whether HuR alters the anti‐inflammatory/proinflammatory gene expression profile in LPS‐challenged bone marrow macrophage (BMM) through regulation of HO‐1. For immunofluorescence BMM staining (Fig. 4A), cytoplasmic infiltration of HO‐1 was more conspicuous with adjunctive LPS‐challenged cells after 3 hours. In addition, this correlated with the marked increase of cytoplasmic HuR after endotoxin stimulation. Notably, HO‐1 translocated to membrane tubular structures extending from the basal BMM nuclear body, whereas HuR polarized mostly to the nucleus, suggesting that cytoplasmic overrepresentation of HuR is not required to stabilize stress‐responsive mRNAs in macrophages. Next, BMM were treated with siRNAs targeting HuR in the presence or absence of LPS exposure, and HO‐1 expression levels were examined (Fig. 4B). Indeed, siHuR BMMs showed a significant reduction of HO‐1 levels, and this correlated with increased mRNA coding for proinflammatory cytokine TNF‐α (Fig. 4C) and chemokines MCP1/CXCL10 (Fig. 4D) compared with siRNA‐treated controls. This confirms that HuR exerts an anti‐inflammatory function and likely inhibits inflammatory cytokine‐induced hepatic IRI.
Figure 4.
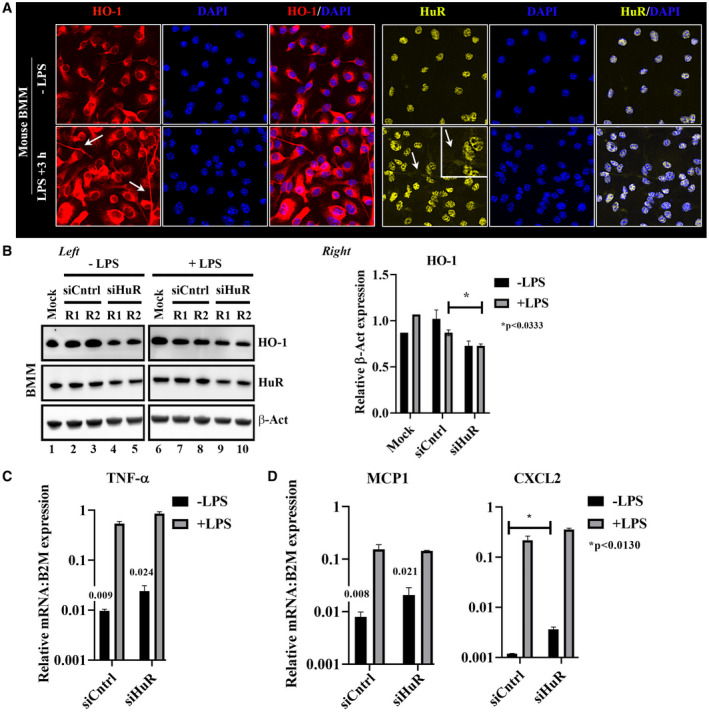
Silencing of HuR enhances proinflammatory signatures in BMMs. (A) Mouse (C57BL/6) BMM was subjected to LPS endotoxin for 3 hours followed by HO‐1 or HuR immunofluorescence staining. White arrows signify areas of cytoplasmic distribution. (B) Left: total lysates from LPS‐conditioned BMM were treated with siControl (siCntrl) or siHuR RNAs and probed by western blot for differences between HO‐1, HuR, and β‐Act as a loading control. Samples separated by a border were analyzed on the same western blot. Equal protein amounts (25 μg) from protein lysates were loaded on each lane. Right: quantitation of HO‐1, data shown are mean ± SEM, *P < 0.0333; siHuR +LPS versus siCntrl +LPS. (C) Inflammatory cytokine analyses of TNF‐α by qPCR. (D) mRNA coding for inflammatory chemokines (MCP1, CXCL2). Data were normalized to B2M gene expression, n = 3. Act, actin; DAPI, 4′,6‐diamidino‐2‐phenylindole.
HuR Indirect Targets in Cytokine‐Stimulated Hepatocytes Identified by RNA Sequencing
Because the Elav family of RBPs and HuR (Elavl1) are crucial in stabilization and translational recruitment of labile mRNA transcripts during cellular stress, we next aimed to determine HuR regulation of mRNA target genes in primary hepatocyte cultures under CM stimulation. Real‐time PCR of HuR siRNA‐mediated gene knockdown validated significant differences in the CM versus siControl CM conditions used for bioinformatic analyses (Fig. 5A). Next, these samples were used for RNA sequencing (RNA‐Seq) experiments in which 392 million total raw reads (mean 98.02 million) were obtained and 2,910 of 4,313 gene sets were up‐regulated on siHuR silencing and 1,389 were significantly enriched at nominal P value < 1%. Principal component analysis mapping showed that biological replicates associated together versus the disparate control samples (Fig. 5B). The heat map, showing the position of the hierarchically clustered mRNA reads, indicates selected transcripts that were validated by real‐time quantitative PCR (qPCR) (Fig. 5C). Gene ontology enrichment analyses were performed next for genes involved in acute inflammatory response. We validated the HuR RNA‐Seq data by performing qPCR on the potential up‐regulated siHuR‐dependent transcripts: THY1, late cornified envelope 1g, and WNT7A (Fig. 5D). Thy1 (CD90) is a cell surface glycoprotein involved in cell adhesion, migration, and signal transduction, whereas Wnt7a has been shown to significantly inhibit the expression of myeloid cell phenotype/function.16 We also analyzed a subset of transcripts that were down‐regulated under similar conditions: CCL3, ACOD1, and prostaglandin E synthase‐1 (PTGES) (Fig. 5E). CCL3 is associated with reduced stellate cell activation and liver immune cell infiltration,17 whereas ACOD1 is involved in the inhibition of the inflammatory response by acting as a negative regulator of TLR‐mediated innate responses.18 PTGES regulates liver protection/repair through EP4 signaling during hepatic IRI,19 and our data show a putative role given by HuR (Fig. 5F) (mean ± SEM, −0.4992 ± 0.09254, P < 0.0327; siHur +CM vs. mock control). Importantly, the post‐transcriptional regulation of HuR‐regulated genes represents a subset of potential targets in hepatic IRI.
Figure 5.
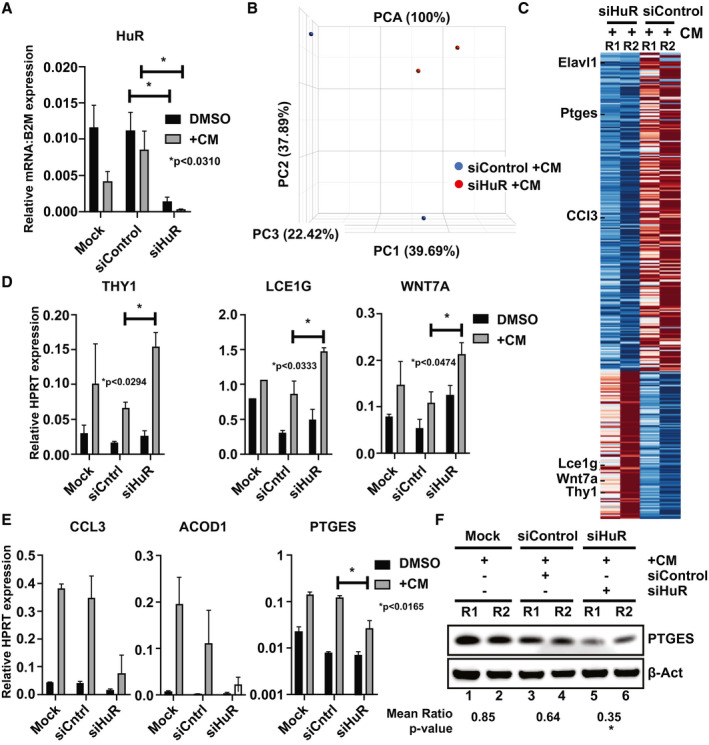
Bioinformatic and experimental validation of HuR gene targets postcytokine stimulation of primary‐derived hepatocytes. (A) Quantitative PCR was conducted to demonstrate the efficiency of HuR silencing in the presence and absence of CM treatment for next‐generation sequencing (RNA‐Seq) transcriptome profiling. Data shown are mean ± SEM, *P < 0.0310, n = 3 of representative samples siHuR +CM versus siControl +CM. (B) Principal component analysis (PCA) and (C) heatmap indicate the average relative information for log10 fragments per kilobase per million reads mapped (FPKM) values for selected genes indicated by tick marks. Color corresponds to per‐gene z‐scores that are computed from log10 FPKM (after adding 0.01). (D) Selected up‐regulated genes (THY1, Lce1g; late cornified envelope 1G, and WNT7A) and (E) down‐regulated genes CCL3, ACOD1, and PTGES after HuR silencing under in vitro warm IR conditions as probed by qPCR. (F) Total lysates from CM‐conditioned primary hepatocyte cultures treated with siControl or siHuR siRNAs (as presented in Fig. 3) were used to probe for PTGES and β‐Act by western blot. Unpaired two‐tailed Student t test of representative samples was calculated relative to β‐Act expression (*P < 0.0327, siHuR +CM vs. Mock +CM). Data are shown as mean n = 3.
Warm, But Not Cold, Hypoxia/Reoxygenation Modulates Hepatocyte HO‐1 Expression
Having demonstrated the importance of HuR/HO‐1 axis during hepatic inflammation as mimicked using various cytokine combinations, we next aimed to determine the role of HuR on HO‐1 during in vitro conditions mimicking hypoxic ischemia. This process is characterized in vivo by localized cellular metabolic disturbances resulting from glycogen consumption, lack of oxygen supply, and adenosine triphosphate (ATP) depletion. Because hypoxic ischemia is due to inadequate oxygen uptake by the centrilobular hepatocytes resulting in necrosis in vivo, we established an in vitro model to study the HuR/HO‐1 axis at various lengths of hypoxia/reoxygenation (H/R) under warm versus cold conditions in primary mouse hepatocytes.
Warm H/R caused a marked increase of HO‐1 protein expression after 1 hour of reoxygenation, with its peak at 3 hours (Fig. 6A). By contrast, after cold H/R, HO‐1's expression kinetics were slower to reach peak expression after 6 hours. When HuR was analyzed next, we observed that after warm H/R, HuR closely matched the HO‐1 time‐course, whereas after cold H/R, there was no change in HuR expression (Fig. 6B). Because hypoxia‐inducible protein (HIF‐1α) elevates the expression of HO‐1 mRNA20 and HuR elevates HIF‐1α expression,21 it was surprising to see HIF‐1α expression peak after 6 hours during warm H/R, whereas it was more responsive (after 1 hour) to cold H/R. To establish that our H/R conditions were sufficient to activate HIF‐1α, bone morphogenic protein 4 (BMP4), belonging to the TGF‐β superfamily of proteins, was included, as it has been shown to be responsive to hypoxia.22 Surprisingly, we discovered it also maintained a differential response to warm versus cold H/R stress. On extended periods of cold H/R, we observed a significant down‐regulation of BMP4 that was not seen in warm H/R conditions. Finally, because hepatocyte apoptosis during hepatic IRI is a vital promoter as well as a marker for hepatocellular damage, we analyzed antiapoptotic B cell lymphoma extra large (Bcl‐xL) and observed its increased expression in warm H/R but not during cold H/R settings over time. These data indicate that the HuR–HO‐1 pathway is required for warm, but not cold, hypoxic ischemia hepatic insult.
Figure 6.
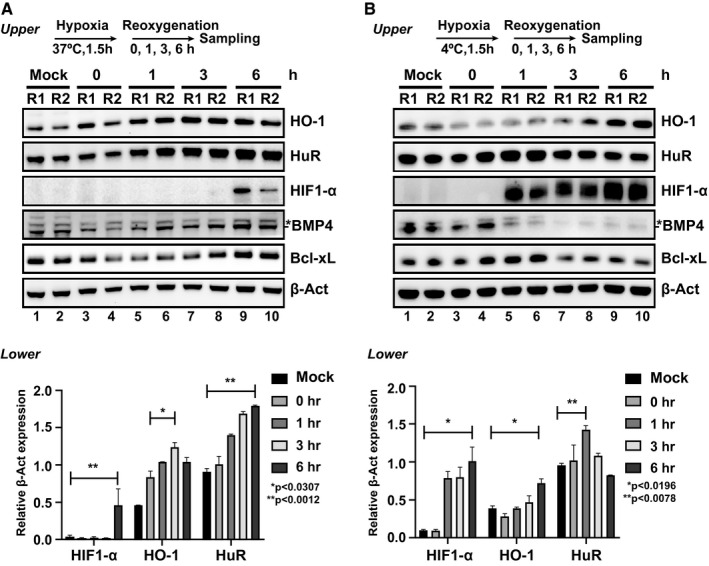
HuR regulation of HO‐1 differs under warm versus cold hypoxia and reoxygenation. Primary‐derived hepatocytes were cultured in serum‐free medium incubated under acute (1.5 hours) (A) warm or (B) cold hypoxia conditions followed by standard normoxia incubation for the indicated times. Total lysates were probed by western blot for differences between HO‐1, HuR, HIF‐1α, BMP4, Bcl‐xL, and β‐Act, as a loading control. Bottom: data below figures show unpaired two‐tailed Student t test of representative samples showing HIF‐1α, HO‐1, and HuR calculated relative to β‐Act expression. Data shown are mean ± SEM, n = 2 (repeated at least three times). Act, actin.
HO‐1 Graft Levels Positively Correlate With HuR Expression in Human Patients with LT
Having demonstrated that the HuR/HO‐1 axis facilitated hepatoprotection in murine in vivo/in vitro models, we next aimed to validate the relevance of these findings in human LT. Postreperfusion hepatic bx from 51 human patients with LT obtained 2 hours after portal reperfusion (before the abdominal closure) (Fig. 7A) were screened by western blots for HO‐1/HuR expression. We found no significant correlation between high post‐transplant HO‐1 protein levels compared with HO‐1 mRNA levels (r = 0.1191, P = 0.4305, data not shown). In contrast, HuR and HO‐1 protein levels showed a significant positive correlation among our human LT cohort (r = 0.4018, P = 0.0035, Fig. 7B). To rule out the possibility that this correlation suggested negative feedback on HuR expression by HO‐1 signaling, we went back to our experimental settings and treated murine hepatocytes with siRNAs targeting HO‐1 in the presence or absence of CM exposure and examined HuR expression levels coordinately (Supporting Fig. S2A). Indeed, although siHO‐1 treatment augmented hepatocellular damage as expected (Supporting Fig. S2B), we did not see any measurable effect on HuR regulation.
Figure 7.
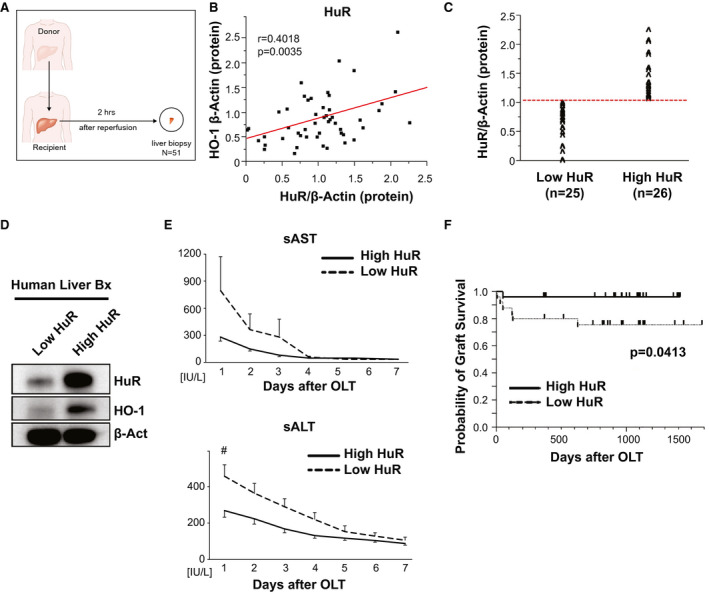
High HuR expression at the graft site correlates with improved hepatocellular function and overall survival in patients with LT. (A) Hepatic bx samples were collected from 51 human LT cases approximately 2 hours after portal reperfusion. (B) Western blot–assisted protein levels of HO‐1 were positively correlated with HuR. (C) LT cases were classified into low (n = 25) and high (n = 26) expression groups based on HuR protein levels with β‐actin normalization. (D) Representative example of human liver bx with high versus low HuR expression (E) sAST and sALT levels up to 7 days post‐LT. (F) The cumulative probability of graft survival (Kaplan–Meier method). Dotted line indicates low HuR, whereas the solid line indicates high HuR groups. Act, actin; OLT, orthotopic liver transplantation.
HuR Graft Levels Negatively Correlate With Adaptive and Innate Immune Activation in Human LT
We next aimed to evaluate putative association between graft HuR levels and innate/adaptive immune gene signatures in patients with LT. HuR expression correlated with mRNA coding for cathepsin G: r = −0.3907, P = 0.0073 (Supporting Fig. S3A), CD4: r = −0.2957; P = 0.0460, CD8: r = −0.3938; P = 0.0061 (Supporting Fig. S3B), CD28: r = −0.2559; P = 0.0861 (Supporting Fig. S3C), CD80: r = −0.3555; P = 0.0153, and CD86: r = −0.3117; P = 0.0349 (Supporting Fig. S3D), implying that increased HuR induction coincided with decreased neutrophil, T‐cell, and macrophage infiltration/activation, respectively, at the graft site. It is notable that IL‐17, one of the prominent T‐lymphocyte cytokines to switch from an innate to adaptive immune activation in human LT,23 showed down‐regulation postreperfusion with respect to graft HuR levels (r = −0.3138, P = 0.0337; Supporting Fig. S3E), indicating increased HuR was associated with a suppressed adaptive immune signature in human LT.
Increased Graft HuR Levels Correlate With Improved Hepatocellular Function and Survival in Patients with LT
To study whether graft HuR expression may have affected clinical outcomes, LT recipients were divided into low HuR (n = 25) and high HuR (n = 26) expression groups (Fig. 7C) based on the western blot‐assisted HuR protein levels (Fig. 7D). There was no correlation between HuR grouping and donor age, sex, weight, body mass index, cold ischemia time, and donation status (donation after cardiac death or donation after brain death), disease etiology, prevalence of hepatocellular carcinoma, ABO compatibility, or Model for End‐Stage Liver Disease score (Supporting Table S1A,B). There was also no correlation between HuR grouping and surgical factors, including intraoperative blood loss and operation procedure time. Interestingly, and consistently with Supporting Fig. S3, patients with LT characterized by increased hepatic HuR expression postreperfusion showed improved early liver graft function, as measured by sAST/sALT levels (Fig. 7E). Strikingly, patients with increased HuR levels at the graft site trended toward better survival at 2 years compared with the low HuR group (P = 0.0413, log‐rank test, Fig. 7F). These data indicate that the HuR pathway is essential for HO‐1–mediated hepatoprotection against IR stress in human patients with LT.
Discussion
This study explores the cytoprotective phenotype of the HuR/HO‐1 axis in hepatic IRI‐LT in mice and humans. Although anti‐inflammatory signaling is critical for HO‐1 cytoprotection, our findings document the RBP HuR as a mechanistic component of its biological activity. During the acute phase of inflammatory response to LT, there is a well‐orchestrated response to IR stress and tissue injury that involves the up‐regulation of HO‐1 protein. Indeed, the short‐term adaptation of HO‐1 mRNA to local inflammatory conditions in the current clinical arm did not correlate with post‐transplant HO‐1 protein levels. Because the availability of resources for protein biosynthesis strongly influences the relationship between protein levels and their coding transcripts, we aimed to investigate the mechanisms controlling HO‐1 mRNA fluctuations. A literature search of RBPs that regulate steady‐state mRNAs led to potential candidates (AUF1, KSRP, BRF1, CUG‐BP1, and tristetraprolin) all decay‐promoting RBPs versus stability‐promoting RBP candidates (NF90, CP1 nucleolin, RNPC1, CUG‐BP2, PAIP2, and members of the Hu/Elav family). On the other hand, HuR was shown to elevate the mRNA stability/translation of HO‐1 in several in vitro studies using fibroblasts,24 human neuroblastoma cells,25 and human pancreatic cancer cells.26 Although the transcriptional role of HO‐1 has been explored, we have limited knowledge of the post‐transcriptional mechanisms that regulate its expression.
HuR cellular activity was significantly increased in liver tissue under warm IR stress (Fig. 1). In accordance with another recent study,27 we found that HuR plays a key role in the post‐transcriptional regulation of HO‐1 through the AU‐rich or U‐rich elements in its 3′UTR to control mRNA stability/translation (Supporting Fig. S1). Because we show that modulators of inflammation (e.g., LPS, chemokines, and cytokines) can influence the cytoplasmic localization of both HuR and HO‐1 (Figs. 2E and 4A), it will be important to determine how their influence acts as a post‐transcriptional mechanism that modulates HO‐1 expression. For example, certain proinflammatory mediators reduce HuR occupancy on the carboxypeptidase B2 3'UTR,28 whereas IL‐6 enhances HuR association with C‐reactive protein, a major component of inflammatory reaction that augments innate immunity.29
Because data on the role of HuR in animal models are limited and often conflicting, the in vivo function of this RBP remains poorly understood. For example, whereas changes in HuR levels or localization in clinical samples from patients with inflammatory disease or cancer have suggested that HuR functions in a proinflammatory and protumorigenic manner,30 the overexpression of HuR was shown to impair tumor growth in a mouse model of estrogen receptor–negative breast cancer31 and control adipose triglyceride lipase and lipolysis in an obesity model.32 Because IR‐triggered liver injury involves the activation of Kupffer cells and neutrophils and the production of cytokines/chemokines and infiltration by lymphocytes/monocytes, we investigated HuR in the context of these immune cascades.
The consequences of the amplified proinflammatory function of HuR in hepatocytes were revealed by the passive release of histone H3 following RNA interference, indicating increased hepatotoxicity (Fig. 3). DAMPs, when released from damaged and/or necrotic tissues, stimulate innate immune cell activation through TLR4/TLR9 signaling. Nuclear histone proteins have been identified as the potential endogenous ligand of TLR9 in the liver, and the neutralization of histones has been shown to be cytoprotective.33 TLR9 functions in cells derived from bone marrow, particularly neutrophils, during liver IR stress, to boost production of proinflammatory mediators, and HuR was shown to contribute to TLR9 signaling by enhancing the growth/metastatic potential of human lung cancer cells.34 It is notable that our studies using CMLD‐2 on neutrophils showed significant CD69 expression on the surface of activated Ly6‐G+ cells, connecting an insofar largely unexplored role for TLR9 signaling in liver IRI to HuR regulation (Supporting Fig. S1). By contrast, the functions of inflammatory macrophages in LT are regulated by chemokine/chemokine receptor interactions to differentiate between their distinctive migration, invasion, and retention patterns. With the loss of HuR function in myeloid cells, the pivotal regulators of innate‐driven inflammation, we discovered HO‐1 was more susceptible to LPS‐induced septic shock than controls (Fig. 4). As shown herein, HuR macrophages displayed alterations in specific chemokines guiding inflammatory recruitment, such as MCP1. Unlike in hepatocytes, however, the loss of myeloid‐derived HuR not only limited HO‐1 expression, but it also rendered it unresponsive to LPS. It is logical to conceptualize a mechanism in which HuR regulates macrophage polarization to protect against liver IRI. To that end, we have recently shown that HO‐1 expression modulates hepatic macrophage polarization and protects against IRI at least in part by favoring an M2 phenotype.35
To strengthen and extend our findings, we sought to investigate how the hypoxic environment (cold or warm) modulates HO‐1 signaling in hepatic IRI. It has been shown that HIF1‐α promotes HO‐1 mRNA/protein expression in response to hypoxia in rat lung, liver, heart, or aortic vascular smooth muscle cells.20 Although the significance of increased HO‐1 expression may relate to the production of intracellular gases (carbon monoxide/nitric oxide), which can lead to elevated cGMP levels, causing changes in vascular tone, endothelial permeability, and/or coagulant function, HIF‐1α is thought to coordinate homeostatic transcriptional responses to cellular hypoxia, thereby leading to the induction of hypoxia‐inducible factor target genes, which facilitates adaptive responses to low oxygen stress. In our study, HO‐1 protein levels correlated with HIF‐1α expression under cold, but not warm, H/R (Fig. 6). By contrast, the regulation of HO‐1 signaling appears coordinated by HuR after warm, but not cold, H/R. To validate the relevance of these findings in humans, we observed a significant positive correlation (r = 0.2956, P = 0.0316, Supporting Fig. S4A) between HIF‐1α and HO‐1 levels in pretransplant donor liver bx. This is consistent with findings by Yang et al. that identified HO‐1 as a HIF‐1α target gene in renal medullary interstitial cells.36 In contrast, there was no correlation between HIF‐1α and HO‐1 (Supporting Fig. S4B) or HuR (Supporting Fig. S4C) post‐transplant after reperfusion. Because this HO‐1 discrepancy between warm and cold hypoxia has not been well documented and remains poorly understood, we suggest that future studies focus on the relevance of mitogen‐activated protein kinase family members, including stress‐activated protein kinases (SAPKs), which are targets of environmental stress and associated with cell apoptosis.37 In support of this supposition, c‐Jun N‐terminal kinase 1/SAPK1 is known to be stimulated in vivo after rat LT38 and to mediate hepatic IRI.
Our study documents the beneficial impact of HuR induction and overexpression in patients with liver transplants. In support of this finding, we analyzed HuR/Bcl‐xL (Supporting Fig. S5A) and HuR/Bcl‐2 (Supporting Fig. S5B) levels in post‐transplant samples and observed a significant positive correlation among the latter in our patients cohorts that were high HuR versus low HuR (r = 0.3748, P = 0.0067). Indeed, high HuR levels in human postreperfusion hepatic bx correlated with improved hepatocellular graft function (sALT/sAST) at postoperative day 1–7 and better overall survival at 2 years as compared with the patient group that was low HuR (Fig. 7). This finding is consistent with our recent study in which post‐transplant low HO‐1 levels was a reliable predictor of exacerbated hepatic IR damage in patients with LT.39 Although the link between LT and recipient long‐term outcomes and hepatic IRI severity remain controversial,9 future studies with a larger cohort of patients with LT are warranted. Clearly, many questions remain, and among the most clinically relevant are the following: How is HuR overexpression protective in the ischemia versus reperfusion stages of hepatic IRI, and is the protection dependent on the presence of a strong inflammatory component? How does the interplay of molecular factors (e.g., HuR, HIF‐1α) among others help to promote the anti‐inflammatory phenotype of HO‐1 signaling? One clue comes from studies of adenosine metabolism and signaling and the group of inducible transcription factors (HIFs) that mediate a plethora of cellular adaptations in response to hypoxia.40 Extracellular adenosine, when induced during limited oxygen availability or acute injury, can signal through distinct adenosine receptors to facilitate cytoprotective activation. For example, up‐regulation of CD39/CD73 by inflammatory stimulation can convert ATP into adenosine, which then binds to P1 receptors to facilitate M2 macrophage activation.41 Adenosine signaling can also down‐regulate intracellular ROS levels by inducing HO‐1 expression in microglia.42
Our data, suggesting a connection between HuR and autophagy‐related P62 and LC3B (Fig. 3), may provide another mechanistic clue (Fig. 8). For example, recently, HuR has been shown to play a central role in autophagosome formation,43 and P62 has been linked to the ubiquitin–proteasome pathway. Moreover, we have recently shown that ectopic expression of adenovirus delivered HO‐1 in local BMM enhanced SIRT1/LC3B expression and alleviated IRI in a mouse LT.12 Because warm/cold hypoxia and reperfusion represent the two aspects of sterile inflammation during liver surgery that may cause severe tissue damage, better understanding of the pharmacological induction of the HO‐1 pathway may have strong therapeutic potential in various clinical liver inflammation settings.
Figure 8.
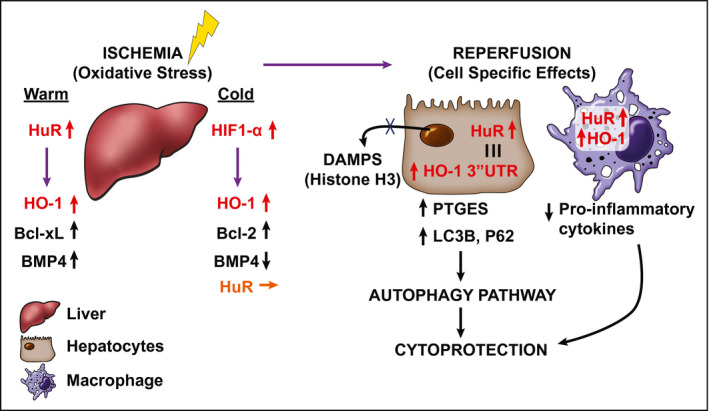
A mechanistic scheme of HuR/HO‐1 molecular axis promoting cytoprotection through autophagy signaling in IR‐stressed mouse and human LT. In primary mouse hepatocytes exposed to warm oxidative stress, HO‐1 is regulated by HuR and not HIF‐1α. Protein expression of antiapoptotic Bcl‐xL and BMP4 increase correspondingly. By contrast, HIF‐1α but not HuR is positively correlated with HO‐1 in cold‐stored human livers and may serve as a regulator in mouse hepatocytes cultured under cold hypoxic conditions. Cold hypoxia also causes a significant decline of BMP4 but not Bcl‐2, which is correlated with HuR levels in human LT. The reperfusion triggers hepatocyte DAMPs to stimulate the release of proinflammatory cytokines/chemokines. Cytoplasmic HuR stabilization of HO‐1 3′UTR mRNA in hepatocytes induces cytoprotection by PTGES/LC3B activation of autophagy signaling. Similarly, cytotoxic immunological cascades are mitigated/prevented in HuR‐expressing macrophages when HO‐1 protein levels are stabilized.
In conclusion, our study uncovered what we believe to be a HuR/HO‐1 regulatory axis of cytoprotection in sterile liver inflammation in mouse and a biomarker of ischemic stress resistance in human LT.
Author Contributions
K.J.D., K.N., K.K., H.H. and J.W.K.W. were responsible for the study concept and design. K.J.D., K.N., K.K., H.H. were responsible for experimental data acquisition. S.K., F.M.K. and K.N. performed surgical procedures. T.I., K.N. and K.K. conducted clinical data analyses. K.N., H.K. and R.W.B. discussed and reviewed the manuscript. K.D. and J.W.K.W. drafted the manuscript. R.W.B. and J.W.K.W. obtained funding. All authors read and edited the manuscript.
Supporting information
Acknowledgment
We thank Justine Aziz and Stephanie Younan for helping with clinical data collection and Jeff Aubé, Ph.D., for the gift of NC‐3.
Supported by National Institutes of Health Grants P01 AI120944 and R01 DK062357, DK107533, and DK102110 (to J. W. K. W.).
Potential conflict of interest: All authors have no relevant conflicts of interest to disclose.
[The copyright line for this article was changed on June 23, 2021 after original online publication.]
References
Author names in bold designate shared co‐first authorship.
- 1. Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec‐Weglinski JW. Ischaemia‐reperfusion injury in liver transplantation–from bench to bedside. Nat Rev Gastroenterol Hepatol 2013;10:79‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dar WA, Sullivan E, Bynon JS, Eltzschig H, Ju C. Ischaemia reperfusion injury in liver transplantation: cellular and molecular mechanisms. Liver Int 2019;39:788‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Amersi F, Buelow R, Kato H, Ke B, Coito AJ, Shen XD, et al. Upregulation of heme oxygenase‐1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J Clin Invest 1999;104:1631‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsuchihashi S, Livhits M, Zhai Y, Busuttil RW, Araujo JA, Kupiec‐Weglinski JW. Basal rather than induced heme oxygenase‐1 levels are crucial in the antioxidant cytoprotection. J Immunol 2006;177:4749‐4757. [DOI] [PubMed] [Google Scholar]
- 5. Tsuchihashi S, Zhai Y, Bo Q, Busuttil RW, Kupiec‐Weglinski JW. Heme oxygenase‐1 mediated cytoprotection against liver ischemia and reperfusion injury: inhibition of type‐1 interferon signaling. Transplantation 2007;83:1628‐1634. [DOI] [PubMed] [Google Scholar]
- 6. Ke B, Shen XD, Ji H, Kamo N, Gao F, Freitas MC, et al. HO‐1‐STAT3 axis in mouse liver ischemia/reperfusion injury: regulation of TLR4 innate responses through PI3K/PTEN signaling. J Hepatol 2012;56:359‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ghosh M, Aguila HL, Michaud J, Ai Y, Wu MT, Hemmes A, et al. Essential role of the RNA‐binding protein HuR in progenitor cell survival in mice. J Clin Invest 2009;119:3530‐3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Papadopoulou C, Ganou V, Patrinou‐Georgoula M, Guialis A. HuR‐hnRNP interactions and the effect of cellular stress. Mol Cell Biochem 2013;372:137‐147. [DOI] [PubMed] [Google Scholar]
- 9. Nakamura K, Zhang M, Kageyama S, Ke B, Fujii T, Sosa RA, et al. Macrophage heme oxygenase‐1‐SIRT1‐p53 axis regulates sterile inflammation in liver ischemia‐reperfusion injury. J Hepatol 2017;67:1232‐1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tuzuner E, Liu L, Shimada M, Yilmaz E, Glanemann M, Settmacher U, et al. Heme oxygenase‐1 protects human hepatocytes in vitro against warm and cold hypoxia. J Hepatol 2004;41:764‐772. [DOI] [PubMed] [Google Scholar]
- 11. Rusten TE, Stenmark H. p62, an autophagy hero or culprit? Nat Cell Biol 2010;12:207‐209. [DOI] [PubMed] [Google Scholar]
- 12. Nakamura K, Kageyama S, Yue S, Huang J, Fujii T, Ke B, et al. Heme oxygenase‐1 regulates sirtuin‐1‐autophagy pathway in liver transplantation: from mouse to human. Am J Transplant 2018;18:1110‐1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lopez de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Identification of a target RNA motif for RNA‐binding protein HuR. Proc Natl Acad Sci U S A 2004;101:2987‐2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu X, Lan L, Wilson DM, Marquez RT, Tsao WC, Gao P, et al. Identification and validation of novel small molecule disruptors of HuR‐mRNA interaction. ACS Chem Biol 2015;10:1476‐1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Atzeni F, Schena M, Ongari AM, Carrabba M, Bonara P, Minonzio F, et al. Induction of CD69 activation molecule on human neutrophils by GM‐CSF, IFN‐gamma, and IFN‐alpha. Cell Immunol 2002;220:20‐29. [DOI] [PubMed] [Google Scholar]
- 16. Wallace J, Lutgen V, Avasarala S, St Croix B, Winn RA, Al‐Harthi L. Wnt7a induces a unique phenotype of monocyte‐derived macrophages with lower phagocytic capacity and differential expression of pro‐ and anti‐inflammatory cytokines. Immunology 2018;153:203‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heinrichs D, Berres ML, Nellen A, Fischer P, Scholten D, Trautwein C, et al. The chemokine CCL3 promotes experimental liver fibrosis in mice. PLoS ONE 2013;8:e66106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nair S, Huynh JP, Lampropoulou V, Loginicheva E, Esaulova E, Gounder AP, et al. Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. J Exp Med 2018;215:1035‐1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nishizawa N, Ito Y, Eshima K, Ohkubo H, Kojo K, Inoue T, et al. Inhibition of microsomal prostaglandin E synthase‐1 facilitates liver repair after hepatic injury in mice. J Hepatol 2018;69:110‐120. [DOI] [PubMed] [Google Scholar]
- 20. Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, et al. Hypoxia‐inducible factor‐1 mediates transcriptional activation of the heme oxygenase‐1 gene in response to hypoxia. J Biol Chem 1997;272:5375‐5381. [PubMed] [Google Scholar]
- 21. Galban S, Kuwano Y, Pullmann R Jr, Martindale JL, Kim HH, Lal A, et al. RNA‐binding proteins HuR and PTB promote the translation of hypoxia‐inducible factor 1alpha. Mol Cell Biol 2008;28:93‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu DC, Paulson RF. Hypoxia regulates BMP4 expression in the murine spleen during the recovery from acute anemia. PLoS ONE 2010;5:e11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sosa RA, Zarrinpar A, Rossetti M, Lassman CR, Naini BV, Datta N, et al. Early cytokine signatures of ischemia/reperfusion injury in human orthotopic liver transplantation. JCI Insight 2016;1:e89679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuwano Y, Rabinovic A, Srikantan S, Gorospe M, Demple B. Analysis of nitric oxide‐stabilized mRNAs in human fibroblasts reveals HuR‐dependent heme oxygenase 1 upregulation. Mol Cell Biol 2009;29:2622‐2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Amadio M, Scapagnini G, Davinelli S, Calabrese V, Govoni S, Pascale A. Involvement of ELAV RNA‐binding proteins in the post‐transcriptional regulation of HO‐1. Front Cell Neurosci 2014;8:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jakstaite A, Maziukiene A, Silkuniene G, Kmieliute K, Gulbinas A, Dambrauskas Z. HuR mediated post‐transcriptional regulation as a new potential adjuvant therapeutic target in chemotherapy for pancreatic cancer. World J Gastroenterol 2015;21:13004‐13019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li S, Qiu B, Lu H, Lai Y, Liu J, Luo J, et al. Hyperhomocysteinemia accelerates acute kidney injury to chronic kidney disease progression by downregulating heme oxygenase‐1 expression. Antioxid Redox Signal 2019;30:1635‐1650. [DOI] [PubMed] [Google Scholar]
- 28. Komnenov D, Scipione CA, Bazzi ZA, Garabon JJ, Koschinsky ML, Boffa MB. Pro‐inflammatory cytokines reduce human TAFI expression via tristetraprolin‐mediated mRNA destabilisation and decreased binding of HuR. Thromb Haemost 2015;114:337‐349. [DOI] [PubMed] [Google Scholar]
- 29. Kim Y, Noren Hooten N, Dluzen DF, Martindale JL, Gorospe M, Evans MK. Posttranscriptional regulation of the inflammatory marker C‐reactive protein by the RNA‐binding protein HuR and microRNA 637. Mol Cell Biol 2015;35:4212‐4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lopez de Silanes I, Lal A, Gorospe M. HuR: post‐transcriptional paths to malignancy. RNA Biol 2005;2:11‐13. [DOI] [PubMed] [Google Scholar]
- 31. Gubin MM, Calaluce R, Davis JW, Magee JD, Strouse CS, Shaw DP, et al. Overexpression of the RNA binding protein HuR impairs tumor growth in triple negative breast cancer associated with deficient angiogenesis. Cell Cycle 2010;9:3337‐3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li J, Gong L, Liu S, Zhang Y, Zhang C, Tian M, et al. Adipose HuR protects against diet‐induced obesity and insulin resistance. Nat Commun 2019;10:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through toll‐like receptor 9 in mice. Hepatology 2011;54:999‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li YJ, Wang CH, Zhou Y, Liao ZY, Zhu SF, Hu Y, et al. TLR9 signaling repressed tumor suppressor miR‐7 expression through up‐regulation of HuR in human lung cancer cells. Cancer Cell Int 2013;13:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang M, Nakamura K, Kageyama S, Lawal AO, Gong KW, Bhetraratana M, et al. Myeloid HO‐1 modulates macrophage polarization and protects against ischemia‐reperfusion injury. JCI Insight 2018;3:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang ZZ, Zou AP. Transcriptional regulation of heme oxygenases by HIF‐1alpha in renal medullary interstitial cells. Am J Physiol Renal Physiol 2001;281:F900‐F908. [DOI] [PubMed] [Google Scholar]
- 37. Crenesse D, Gugenheim J, Hornoy J, Tornieri K, Laurens M, Cambien B, et al. Protein kinase activation by warm and cold hypoxia‐ reoxygenation in primary‐cultured rat hepatocytes‐JNK(1)/SAPK(1) involvement in apoptosis. Hepatology 2000;32:1029‐1036. [DOI] [PubMed] [Google Scholar]
- 38. Bradham CA, Stachlewitz RF, Gao W, Qian T, Jayadev S, Jenkins G, et al. Reperfusion after liver transplantation in rats differentially activates the mitogen‐activated protein kinases. Hepatology 1997;25:1128‐1135. [DOI] [PubMed] [Google Scholar]
- 39. Kageyama S, Hirao H, Nakamura K, Ke B, Zhang M, Ito T, et al. Recipient HO‐1 inducibility is essential for posttransplant hepatic HO‐1 expression and graft protection: from bench‐to‐bedside. Am J Transplant 2019;19:356‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bowser JL, Lee JW, Yuan X, Eltzschig HK. The hypoxia‐adenosine link during inflammation. J Appl Physiol 1985;2017:1303‐1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lu L, Zhou H, Ni M, Wang X, Busuttil R, Kupiec‐Weglinski J, et al. Innate immune regulations and liver ischemia‐reperfusion injury. Transplantation 2016;100:2601‐2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Min KJ, Kim JH, Jou I, Joe EH. Adenosine induces hemeoxygenase‐1 expression in microglia through the activation of phosphatidylinositol 3‐kinase and nuclear factor E2‐related factor 2. Glia 2008;56:1028‐1037. [DOI] [PubMed] [Google Scholar]
- 43. Ji E, Kim C, Kang H, Ahn S, Jung M, Hong Y, et al. RNA binding protein HuR promotes autophagosome formation by regulating expression of autophagy‐related proteins 5, 12, and 16 in human hepatocellular carcinoma cells. Mol Cell Biol 2019;39:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials