

Abstract
Carbonic anhydrases (CAs, EC 4.2.1.1) are enzymes involved in a multitude of diseases, and their inhibitors are in clinical use as drugs for the management of glaucoma, epilepsy, obesity, and tumours. In the last decade, multitargeting approaches have been proposed by hybridisation of CA inhibitors (CAIs) of sulphonamide, coumarin, and sulphocoumarin types with NO donors, CO donors, prostaglandin analogs, β-adrenergic blockers, non-steroidal anti-inflammatory drugs, and a variety of anticancer agents (cytotoxic drugs, kinase/telomerase inhibitors, P-gp and thioredoxin inhibitors). Many of the obtained hybrids showed enhanced efficacy compared to the parent drugs, making multitargeting an effective and innovative approach for various pharmacological applications.
Keywords: Carbonic anhydrase, inhibitors, hybrid drugs, multitargeting, anticancer agents
1. Introduction
The central paradigm in drug design, at least for the last century, was the concept introduced by Paul Ehrlich of the “magic bullet”1. Considered the founder of chemotherapy, mainly due to his excellent work on the treatment of syphilis with arsphenamine (Salvarsan) and structurally related derivatives2, this scientist introduced the concept of drug target and pinpointed to the fact that an effective drug should specifically act on one target in order to manifest its therapeutic benefits. Furthermore, many of the side effects that a drug shows may be considered as being due to off-targeting, that is, interactions with diverse biomolecules than the real target, according to the magic bullet theory. As a consequence, most if not all modern drugs in clinical use were discovered considering these concepts, which continue to influence generations of medicinal chemists in the search of both new drug targets as well as compounds that should effectively, and possibly specifically interact with them for producing beneficial therapeutic effects3.
The magic bullet strategy is for sure still valid today, but started to be developed and extended in the last two decades to what is now commonly known as the multitargeting approach, which contemplates the dual/multiple (hybrid) drugs, that is, compounds that incorporate in the same molecular entity at least two different chemotypes which hit diverse drug targets4,5, as shown schematically in Figure 1. The advantages of such a hybrid drug may be multiple, among which the possible synergistic effect of the two (or more than two) chemotypes present in the hybrid, a more predictable pharmacokinetic profile in comparison to the administration of individual, single agents, as well as improved compliance for the patients who do not need to take many pills, etc. However, there are also challenges connected to this approach, such as for example attaining a balanced activity at each considered target, while simultaneously achieving the suitable pharmacokinetic and pharmacodynamic profiles as well as the necessary selectivity and efficacy4,5. For such reasons, the multiple targeting approach was initially considered mainly for multifactorial diseases such as cancer6–8, Alzheimer’s and other neurodegenerative diseases9,10, schizophrenia11, etc. However, more recently the approach was successfully extended to the discovery of antibiotics12 and antiviral drugs13, proving that it may be a general one.
Figure 1.

The multitargeting approach with a dual/multiple ligand, schematically shown as the triple arrow, which acts on multiple (in this case three) different targets, represented as A, B and C.
Here I shall review the multitargeting approach of the carbonic anhydrase (CA, EC 4.2.1.1)14,15 inhibitors (CAIs) which have been hybridised with a variety of other diverse pharmacological agents. Indeed, the CAIs per se have clinical applications as diuretics16, antiglaucoma17, antiepileptic18, antiobesity19, and antitumor20–22 agents. Furthermore, their potential in the treatment of neuropathic pain23, cerebral ischaemia24, or as anti-infectives25–29 was recently highlighted, and all these aspects make them of great interest for obtaining hybrids acting on additional targets. The main reason why the multi-targeting approach is successful with CAIs is due to the fact that these enzymes are widespread in both prokaryotes and eukaryotes14,15, organisms in which they play crucial functions connected to pH regulation, homeostasis and metabolism, and their inhibition leads to pharmacological responses30. In humans, there is a multitude of isoforms (15, of which 12 are catalytically active14,15) specifically expressed in various tissues, and the modulation of their activities has important physiological and pharmacological applications, as already mentioned above. The field of CAs and their inhibitors was reviewed recently31,32 and we will not detail here these aspects, but we will directly consider the hybrid drugs based on CAIs reported so far and their various pharmacological applications.
2. Hybrids drugs incorporating CAIs and other pharmacophores as antiglaucoma agents
2.1. Antiglaucoma hybrids incorporating CAIs and NO donors
Glaucoma is a neuropathy characterised among others by elevated intraocular pressure (IOP), the only measurable clinical abnormality, which, however, if not treated, may lead to blindness or significant vision loss17. Avoiding the death of optical nerve neurons, responsible for the vision loss which eventually will cause blindness, is nowadays the most desirable approach for the treatment of the disease (i.e. neuroprotection) together with the lowering of IOP17. Systemic or topically acting sulphonamide CAIs are in clinical use for decades for the treatment of glaucoma, being among the relevant 6 classes of such drugs available nowadays17. In the last decades, the role of nitric oxide (NO), a radical gas involved in vasodilation and produced endogenously by the enzyme nitric oxide synthase (NOS) from arginine as substrate, has been shown to be relevant for this disease33. Nitric oxide acts as a potent vasodilator that impacts numerous systems throughout the body, among which also the regulation of ocular pressure. This process is similar to the systemic pressure regulation induced by this gas-transmitter in vertebrates17,33. In fact, the endothelium within the Schlemm’s channel (SC), involved in the outflow of the aqueous humour from the eye, has a similar shear stress sensitivity as the systemic vasculature, functioning thus as a barostat17,33. The NO produced by endothelial cells from SC is thus involved in regulating IOP, which, as mentioned above, is increased in the eyes of patients suffering from glaucoma17. Thus, the idea to combine CAIs, clinically used antiglaucoma agents, as mentioned above, with NO-donating compounds, a relatively underexplored class of ocular drugs till recently, led to the development of hybrid drugs which indeed showed excellent IOP and neuroprotective action34. The increased vasodilation, coupled with potential anti-inflammatory and anti-platelet effects of NO itself (and probably also of the sulphonamides35) are only some of the various activities that a hybrid incorporating a NO-donor moiety and a CAI can offer36–39.
The first compounds to incorporate these two diverse chemotypes were based on the dorzolamide 1 saffold36. Dorzolamide 1 itself is a sulphonamide CAI (Figure 2) in clinical use since 1995 as eye drops for the topical treatment of glaucoma16,17. Hybridisation of 1 by means of various NO-donating moieties incorporated in nitrate ester functionalities, as the compounds 2–6 (Figure 2), led to hybrids with effective, low nanomolar CA inhibitory properties against the isoforms involved in aqueous humour production, such as CA II and IV, and also showed effective IOP-lowering properties in an animal model of glaucoma, that is, normotensive rabbits36. Furthermore, the X-ray crystal structure of some of these sulphonamide–NO donor hybrids, bound to hCA II, were resolved and are shown in Figure 336,37.
Figure 2.

Structure of the nitrate ester derivatives of dorzolamide 1 (R = H) of types 2–6.
Figure 3.

Binding of sulphonamide–NO donors to hCA II as determined by X-ray crystallographic experiments. (A) Compound 6 (PDB 3K2F) in magenta, and (B) sulphonamide 8 (PDB 3NI5) is shown in green. The zinc ion from the CA active site is shown as a gold sphere, being coordinated by three His residues (His 94, 96 and 119 – not numbered in the figure for the sake of simplicity) and the fourth metal ion ligand being the deprotonated sulphonamide from the inhibitor. The other amino acid residues involved in contacts with the inhibitors are also highlighted.
In subsequent studies, various other CAI–NO donating compounds were synthesised and investigated as CAIs and IOP lowering agents, among which 4-carboxy-benzensulphonamide, 1,3,4-thiadiazole-2-sulphonamide, 4–(2-carboxyethyl)-benzensulphonamide and 4-hydroxybenzensulphonamides (Figure 4)37,38. Some of these derivatives, such as compounds 7–9 were low nanomolar in vitro CA inhibitors of isoforms involved in aqueous humour formation, such as CA II, CA IV and CA XII37,38. By means of X-ray crystallographic studies, the factors associated with their significant inhibitory activity were also revealed, as shown in Figure 3(B) for the adduct of 8 with hCA II (h = human isoform)37.
Figure 4.

Chemical structure of aromatic/heterocyclic sulphonamides incorporating NO-donating moieties of the aliphatic nitrate ester type, 7–9.
In fact, for both hybrids incorporating sulphonamides and NO donating moieties which have been characterised by crystallography when bound to hCA II (compounds 6 and 8, see Figure 3), the deprotonated sulphonamide group of the hybrid was observed bound to the zinc ion, the organic scaffold was participating in a large number of favourable interactions with amino acid residues from the active site (shown in detail in Figure 3), and the tail NO-donating fragment of the hybrid was also observed intact, demonstrating that the hybrids were stable enough for exerting their dual effect36,37.
Compound 7, also known as NCX-25038, was investigated in detail as an antiglaucoma agent from the pharmacological viewpoint. In a rabbit model of transient ocular hypertension NCX-250 was 2 times a more effective IOP lowering agent compared to dorzolamide, a clinically used drug, as mentioned above17,38. It also reduced short-term elevated IOP in another animal model of the disease, that is, the rabbit with carbomer-induced glaucoma38. The NO-donating properties of the compound were also assessed using isosorbide mononitrate as standard NO-donor, being observed a slow but steadfast release of the gaso transmitter. It was thus concluded that the effective IOP lowering caused by NCX-250 treatment is due both to its CA inhibitory effects and to the vasodilation induced by the liberated NO, as expected for this type of hybrid drug38. In the same study was then reported that the chronic administration of NCX-250 as 2% eye drops in carbomer-induced glaucomatous New Zealand albino rabbits led to a significant and durable IOP lowering, which was superior to that observed with the single, standard drugs, dorzolamide or isosorbide mononitrate38. By using echo-doppler measurements of the ophthalmic artery in animals treated with NCX-250, reduced systolic and diastolic velocities were observed, suggesting a beneficial effect of this class of hybrids incorporating sulphonamide CAIs and NO-donating nitrate esters for supplying blood to the optic nerve, an effect which has not been observed when dorzolamide alone was administered to the experimental animals.
Another study reported thereafter sulphonamide CAIs incorporating furazan and furoxan derivatives as NO-donating moieties, of types 10 (a–p) (Figure 5)39. Although effective CA inhibitory effects (in the low nanomolar range) and IOP lowering (slightly more effective than dorzolamide) were observed with hybrids of type 1039, they were not superior to the nitrate esters mentioned above, which are easier to be prepared and more stable compared to the furazans38,39.
Figure 5.

Sulphonamides 10 (a–p) incorporating furazan and furoxan as NO-donating moieties.
2.2. Sulphonamides conjugated with prostaglandin F receptor agonists
The prostaglandin F (PGF) receptor agonists (PGFR) constitute a well-known class of antiglaucoma drugs, with several compounds in clinical use for more than two decades, the best known one being latanoprost40. They decrease IOP by increasing the uveoscleral ocular aqueous outflow17. Long et al.41 proposed a hybridisation of sulphonamide CAIs with PGFR agonists as a new approach for the treatment of glaucoma. The heterocyclic (1,3,4-thiadiazole-sulphonamide) and aromatic (benezenesulphonamide) hybrids of types 11 and 12, Figure 6, have been obtained, which possessed good ocular permeability, but no in vivo antiglaucoma data and detailed CA inhibition experiments have been reported to date for these presumably dual-acting compounds41. However, this strategy represents and interesting and innovative approach for the management of glaucoma.
Figure 6.
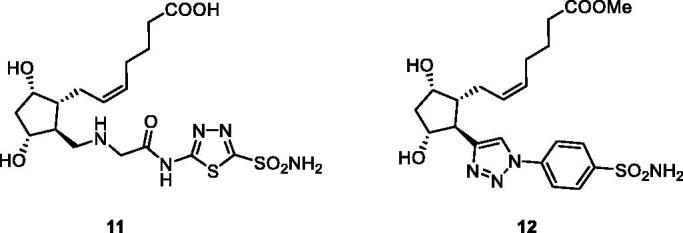
Prostaglandin receptor agonists–sulphonamide CAIs hybrids of types 11 and 12.
2.3. CAI–β-adrenergic receptors antagonists hybrids
One of the mostly used antiglaucoma drug eye drops formulations is COSOPT®, which combines two drugs, the β-adrenergic receptor (AR) antagonist timolol and the CAI dorzolamide17. β-Blockers such as timolol and many other derivatives structurally related to it reduce IOP through the blockade of sympathetic nerve endings within the ciliary epithelium, whereas the CAIs reduce the secretion of the aqueous humour due to the inhibition of the ciliary process CA isoforms CA II, IV and XII involved in the production of bicarbonate present in the aqueous humour17. Thus, a multi-targeted drug design strategy of hybrid drugs incorporating CAIs of the sulphonamide type and β-ARs antagonists was proposed by Nocentini et al. (Figure 7(A))42. Dual derivatives of types 13–16, showed a remarkable CA inhibitory potency, in the nanomolar range, against isoforms hCA II and XII (involved in glaucoma), also possessing an acceptable affinity to β-ARs (of type β1 and β2) in the low micromolar range, comparable to that of the racemic, clinically used β-blocker drug atenolol17,42. The X-ray crystal structure of hCA II in complex with the dual compound 14 was determined and explained why the compound is a highly effective CAI against hCA II (Figure 7(B))42. These derivatives showed an IOP lowering effect comparable or better than those of dorzolamide, timolol and their combination (COSOPT), 60 min post-treatment. On the other hand, for some of these dual-acting compounds, the IOP lowering efficacy was two-fold higher compared to the parent molecules after 1 and 2 h post-administration, demonstrating thus the great efficacy and longer duration of action of the multi-targeted strategy with this type of hybrids42.
Figure 7.

(A) Sulphonamide CAI–β-blocker hybrids of types 13–16. (B) X-ray crystal structure for the adduct of 14 with hCA II: the zinc ion (gray sphere) with its three protein ligands (His94, 96, 119) are evidenced. The inhibitor 14 (blue sky) is coordinated with the deprotonated sulphonamide moiety to the metal ion, and its scaffold participates in a range of favourable interactions with active site residues, such as Thr199, Leu198, Phe131, Val135. Hydrogen bonds are shown as dashed lines.
3. Hybrids of CAIs with anti-inflammatory agents
3.1. CAI–CO releasing molecules (CORMs) hybrids
Similar to NO discussed in the preceding paragraph, CO is another gaso-transmitter involved in a multitude of physiological processes, although being a highly toxic molecule43. CO, unlike NO, is a rather stable gas and it is endogenously produced through the degradation haem under the action of the enzyme haem oxygenases (HO; EC 1.14.99.3), more precisely its isoforms HO1 and HO244. These enzymes and the CO produced through their activity, play a protective role against intracellular oxidative stress, and an overexpression of HO1 was reported in several inflammatory and autoimmune diseases45. Thus, although CO is highly poisonous compound due to its strong affinity for haemoglobin (Hb), this gas also binds to other haem-containing proteins, such as the soluble guanylyl cyclase (sGC), the NO synthase (NOS), the NADPH oxidase as well as mitochondrial cytochrome C oxidase, and this probably endows CO with cytoprotective and homeostatic antiiflammatory properties46. Indeed, in the last decades, CO releasing molecules (CORMs) which allow a controlled release of small amounts of this toxic compound started to be considered for therapeutic applications46,47. The alkyne hexacarbonyl dicobalt(II) complexes are the most well-known CORMs, although many other such chemotypes have been developed46,47.
Thus, a strategy to obtain CAI–CORM hybrids, which may exploit the pharmacological properties of both chemotypes, was recently proposed by Berrino et al.48 and it is shown schematically in Figure 8. The idea is based on the fact that the dicobalt(II)octacarbonyl ha a high affinity to form complexes with terminal alkynes, such as 17 (losing 2 CO molecules) leading thus to derivatives of type 18 (Figure 8), which are stable but may release CO depending on the substitution pattern at the alkyne moiety46–48.
Figure 8.

General strategy for obtaining CAI–CORM hybrids 18 based on the hexacarbonyl dicobalt(II) complexes.
All common CAIs, such as the aromatic sulphonamides (compound 19), hydroxycoumarins (compound 20), 7-aminocoumarins (derivative 21) and 6-hydroxysulphocoumarins (derivative 22) have been hybridised in this way with hexacarbonyl dicobalt(II) moieties, as shown in Figure 9. The isoforms hCA I, II, IV, IX, XII were effectively and sometimes selectively inhibited by such derivatives, with efficacies in the nanomolar range, whereas the pain-relieving properties of several hybrids were also assessed in an animal model of rheumatoid arthritis (RA), by using the paw-pressure and incapacitance tests, which demonstrated an efficacy similar or slightly better than that of the clinically used drug ibuprofen, belonging to the cyclooxygenase (COX) inhibitors48. In subsequent papers from the same group49,50, it has been demonstrated that the CAI–CORM hybrids modulate lipopolysaccharide (LPS) stimulation in macrophages, exerting beneficial anti-inflammatory effects and counteracting inflammatory stimuli by reducing the release of tumour necrosis factor alpha (TNF-α), a pro-inflammatory protein49. Tendon recovery after injury is another inflammation model in which the utility of the CAI–CORM compounds was recently demonstrated50. Tenocyes (tendon cells) were shown to be protected by CAI–CORM hybrids from oxidative stress induced by inflammation and to have an enhanced viability after treatment with the hybrid drug, which may be another useful application of these compounds.
Figure 9.

CAI–CORM hybrids of types 19–22 incorporating sulphonamide, 7-hydroxycoumarin, 7-aminocoumarin and 6-hydroxysulphocoumarin as scaffolds with CA inhibitory properties and hexacarbonyl dicobalt(II) as CO releasing fragment.
3.2. CAI–cyclooxygenase inhibitor (non steroidal anti-inflammatory drugs, NSAIDs) hybrids
Cyclooxygenases (COXs) are involved in the biosynthesis of prostaglandins, essential inflammation modulators, and their inhibition by COX inhibitors, also known as non steroidal anti-inflammatory drugs (NSAIDs) is one of the main therapeutic approach for controlling various inflammatory processes, including pain, fever, arthritis, etc.51–56. Several CA isoforms, among which CA II, IV, IX and XII were demonstrated to be involved in arthritis-like diseases57, being overexpressed in the synovial fluid and synoviocytes from patients suffering of rheumatoid arthritis (RA). Thus, hybrids were designed to target such CA isoforms involved in this condition, incorporating both a CA-binding moiety of the coumarins or sulphonamide type, and a COX inhibitor of the NSAID type such as the carboxylic acid derivatives indomethacin, sulindac, ibuprofen, flurbiprofen, ketoprofen, diclofenac, ketorolac, naproxen, etc. (Figure 10)51–56.
Figure 10.

General structures of the COX inhibitor-CAI hybrids A and B, and several coumarin–NSAIDs hybrids of type 23–27.
Both 6- and 7-hydroxy-coumarins, such as those shown in Figure 1051,53 as well as the corresponding derivatives incorporating esters instead of amides52, but also the sulphonamide–COX inhibitor hybrids (of type B in Figure 10)54 were obtained and investigated for their stability in plasma and for inhibition of the CA isoforms involved in inflammatory processes (hCA II, IV, IX and XII), and in some cases also for the inhibition of COX1/2 isoforms56,57. The hybrids were shown to be quite stable to hydrolysis (except for some coumarin esters) and to potently inhibit both CAs and COX isoforms involved in inflammation, with high efficacy being thus rather equilibrated hybrids. But more importantly, in several inflammation models, they showed excellent in vivo activity, superior to the parent compounds (CAI alone, COX inhibitor alone and the combination of the two drugs), proving the efficacy of the multitargeting approach51–56. Indeed, some of the obtained coumarin and sulphonamide hybrids, were assessed by means of the paw-pressure and incapacitance tests in a rat in vivo model of RA47. Potent antihyperalgesic effects were observed for all of them, in a dose-dependent manner, starting already at very low concentrations of hybrids, as low as 1 mg kg−1. For example, the 7-coumarin substituted ibuprofen derivative 25 showed long-lasting antihyperalgesic effects, in the two in vivo models mentioned above, superior to both ibuprofen or the 7-hydroxycoumarin (and their combination)51,55. Some of these derivatives as well as the corresponding sulphonamide hybrids also showed potent anti-inflammatory activity in a lung fibrosis animal model56, making this type of hybrids of great interest as novel anti-inflammatory agents.
4. Antitumor hybrid drugs incorporating CAIs conjugated with other cheotypes
4.1. CAI–cytotoxic agents hybrids
At least two CA isoforms, hCA IX and XII, are overexpressed in solid tumours as a consequence of the hypoxic environment and activation of the hypoxia inducible factor (HIF) signalling cascade20–22. These two tumour-associated CAs were validated as antitumor/antimetastatic drug targets, with at least one small molecule compound, the sulphonamide SLC-0111 in Phase Ib/II clinical trials for the management of hypoxic, metastatic solid tumours58. Since cancer is a multifactorial disease, and as CA IX/XII were only recently validated as drug targets, a multitude of hybridisation approaches involving CA IX/XII inhibitors and other antitumor chemotypes were proposed in the last period.
The high affinity of aromatic/heterocyclic sulphonamides acting as CAIs for CA IX and XII, which are found in rather low concentration in most normal tissues but are overexpressed in many hypoxic tumours20–22 led Neri’s and other groups to propose them for hybridisation with cytotoxic payloads possessing a variety of chemical functionalities in order to specifically target tumours without affecting the healthy tissues Figure 1159–63. The cytotoxins included in these studies were monomethyl auristatin (compound 28 in Figure 11)60,61, tubulysin B (scaffold 29 in Figure 11)62, the maytansinoids 30 and 3159, as well as mertansine 3259 (Figure 11). All these toxins are highly effective in killing tumour cells but they are equally toxic to normal cells, and for this reason are impossible to be used in therapy. However, the conjugation of these toxins with CAIs of the sulphonamide type as “warheads,” through oligopeptide linkers as shown in Figure 11, led to dual agents which showed effective anticancer effects due to the action of the delivered cytotoxin only within tumour cells, as well as due to the inhibition CA IX and XII, which, as mentioned above, are tumour-associated and are present in low amounts in normal tissues, thus interfering with the metabolism and pH regulation in tumours20–22,58–63.
Figure 11.

General strategy for hybridisation of CAIs with cytotoxins in compounds of type C. CA IX/XII–cytotoxic agent hybrids, incorporate toxic payloads 28–32, whereas the CA inhibitory warheads may be benzenesulphonamide or 1,3,4-thiadiazole-2-sulphonamide derivatives (although coumarins and other CAI chemotypes can be used). The linker is usually a tetrapeptide incorporating water-solubilizing residues (Asp, Arg, Cys).
Hybrids as those shown in Figure 11, incorporating aromatic/heterocyclic sulphonamides as CA inhibitory chemotype, possessed low nanomolar affinity for CA IX as well as in vivo activity, in SK-RC-52 renal cell carcinoma cancer xenograft models of cancer in experimental animals59–61,63.
4.2. Dihydroartemisinin–CAI hybrids
Artemisinin and dihydroartemisinin (DHA), which is more hydrophobic than the parent compound, are well-known antimalarial agents, which have been also shown to possess cytotoxic activity against several cancer cell lines64. Guo’s group reported interesting hybrids incorporating DHA and CAIs of the type shown in Figure 1264–66.
Figure 12.

DHA–CAI hybrids incorporating sulphonamide (33 and 35) and coumarin moieties (34) as CA inhibitory fragment. In compounds 35R = sulfamoyl, in meta or para positions. The linkers may be 1,2,3-triazole based moieties obtained by click chemistry, –O–(CH2)n–O–, n = 2–4, etc.
The sulphonamides of type 33 and 35 have been tested as hCA I, II, IX and XII inhibitors and showed generally low micromolar inhibitory action, and anti-proliferative activity against several cancer cell lines, such as MDA-MD-231 (breast), HT-29 (colon) and no toxicity against the normal MCF-10A epithelial cells64. The coumarins 34 were not tested as CAIs, although these compounds are known to possess remarkable such activity14,15,67,68, coupled with selectivity for the inhibition of the tumour-associated versus the cytosolic isoforms. Thus, the coumarins were only tested for their antiproliferative activity against the same cancer cell lines mentioned for 35, showing interesting activities65,66. Some of the coumarins reported in these interesting studies were docked within the CA IX active site as hydrolysed derivatives, as indeed, the inhibition mechanism by this class of compounds involves their hydrolysis at the lactone ring and occlusion of the CA active site entrance by the obtained hydroxycinnamic acids67,68. However, the docking performed in the mentioned studies65,66 reported the carboxylate moieties of the hydrolysed coumarin as coordinating the active site zinc ion, which is not in agreement with the X-ray crystallographic data mentioned above67,68.
4.3. Other CAI–anticancer drug hybridisation approaches
Some of the most widely used anticancer drugs nowadays are the tyrosine kinase inhibitors and the tubulin polymerisation inhibitors, with many new representatives of these classes being launched each year69. Thus, in this paragraph I will examine a set of hybridisation approaches which involve CAIs and several other, rather heterogeneous classes of antitumor agents, among which the epidermal growth factor receptor (EGFR) antagonists (this protein has kinase activity)70, the 15-lipoxygenase (15-LOX)/COX-2 (multitargeting of tree different proteins) inhibitors71, the telomerase inhibitors72, the P-glycoprotein (P-gp) inhibitors73, and the thioredoxin inhibitors74,75.
Zhang et al.70 reported hybrids incorporating the tyrosine kinase inhibitory fragments found in erlotinib and gefitinib, clinically used anticancer agents69 and benzenesulphonamides with CA inhibitory activity, of types 36 (Figure 13) which were shown to possess antiproliferative activity against several cell lines, such as A549, A431 and H1975. Few of the compounds were also tested for the inhibition of EGFR (wild type and mutant) and hCA II and IX, showing nanomolar activity for the first protein, and micromolar ones for the CAs. However, the docking of some of the primary sulphonamides within the hCA IX active site was erroneously performed by these authors70. The sulphonamide was docked as neutral molecule and not as anion (although sulphonamides bind to these enzymes in the anionic, sulphonamidate form14–16) and as a consequence they were reported to inhibit CAs due to the hydrogen bonds formed with Thr199, Thr200 and several other amino acid residues, which is completely erroneous.
Figure 13.

Multitargeting compounds incorporating the kinase inhibitory fragment from erlotinib and sulphonamide CAIs of type 36 and compounds 37 and 38 targeting CAs, COX-2, and 15-LOX.
Elzahhar et al.71 reported a multitargeting strategy involving three different targets: CAs, COX-2 and 15-LOX, all enzymes found to be overexpressed in some types of tumours. Some of the reported derivatives, such as 37 and 38 (Figure 13) showed nanomolar CA IX/XII and COX-2 inhibitory action and micromolar inhibition of 15-LOX, and possessed effective antiproliferative action against hepatic (HepG2), breast (MCF7) and lung (A549) cancer cell lines71. These compounds were also docked within the active sites of the three enzymes, showing favourable interaction, but as for the preceding study, the docking with the CA IX active site was performed with the neutral and not the deprotonated sulphonamide. However, the interaction with the zinc ion was in this case observed, being in fact one of the main factors responsible for the effective inhibition of these enzymes71.
Berrino et al.72 reported a highly innovative hybridisation approach of CAIs with telomerase inhibitors based on the azidothimidine (AZT) scaffold (Figures 14 and 15) by using click chemistry procedures.
Figure 14.

Hybridisation of CAIs and telomerase inhibitors of the AZT type.
Figure 15.

Telomerase-CAI hybrids 39 (a–u) incorporating sulphonamide, coumarin, sulphocoumarin fragments with CA inhibitory action, AZT as telomerase inhibitor and various linkers (ether, thioether, amine, amide, ester, selenoether, carbamate, etc.).
As seen from Figure 15, a large number of telomerase-CAI hybrids of type 39 (a–u) were reported, which incorporate the main CA inhibitory chemotypes, of the primary sulphonamide, coumarin, and sulphocoumarin type72. The telomerase inhibitor was on the other hand always based on the AZT fragment, but the linkers between the two chemotypes were highly variable, as shown in Figure 15. Many such derivatives showed low nanomolar CA IX/XII inhibition and potent antitelomerase activity in PC-3 and HT-29 cell lines, with IC50 values ranging from 5.2 to 9.1 μM against this target72. Some of the sulphonamides were also crystallised in complex with some CA isoforms, such as hCA II, demonstrating the structural reasons of their strong affinity for this target72, as shown in Figure 16.
Figure 16.

The adduct of hCA II with 39a as determined by X-ray crystallography. Left: hCA II active site, with the zinc ion (gray sphere) and its histidine ligands (His94, 96 and 119) and the inhibitor showed with its electronic density. Right: detailed interactions in which the sulphonamide 39a participates with amino acid residues from the enzyme active site. Red spheres are water molecules, dashed lines represent hydrogen bonds. Residues involved in the binding are evidenced.
Teodori et al.73 proposed hybrids incorporating P-gp inhibitors of the amine (40) type and CAIs of the coumarin (41) and sulphonamide (42) types, showed in Figure 17.
Figure 17.

CAI–P-gp inhibitor hybrids of the coumarin 41 and sulphonamide 42 types.
The P-glycoprotein (P-gp) is involved in the resistance problems observed with many antitumor agents, as this transporter extrudes the anticancer drugs from the cells73. Tertiary amines of the verapamil type (general structure 40) act as effective inhibitors of P-gp, and this was the rationale for obtaining hybrids of P-gp inhibitors and CAIs targeting the tumour-associated enzymes CA IX and XII. Indeed, compounds of types 41 and 42 incorporate the N,N-bis(alkanol)amine diester scaffold to which the CA inhibitory fragment has been appended. They showed effective CA IX/XII inhibitory action, some of them in the low nanomolar range, as well as inhibitory activity against P-gp. In a rhodamine-based test for evaluating multidrug resistance to antitumor agents in LoVo/DOX cells, that overexpress both P-gp and CA XII, some of the coumarin derivatives 41 were able to revert the phenomenon, leading to an increased uptake of doxorubicin within the tumour cell, demonstrating thus the efficacy of the hybridisation approach73.
Recently, Krasavin et al.74,75 proposed another interesting multitargeting approach of two enzymes involved in cancerogenesis: thioredoxin (TrxR, which is overexpressed in cancers as a defence against oxidative stress) and CA IX/XII, involved in metabolism and pH regulation in tumours (Figure 18). Indeed, variously substituted sulphocoumarins acted as low nanomolar hCA IX/XII inhibitors and showed micromolar activity against TrxR, a selenium-containing enzyme (Figure 18). The effiocacy of the dual targeting was also tested ex vivo, in the pancreatic cancer cell line PANC-1, showing superior efficacy to the single agents75.
Figure 18.

Sulphocoumarins targeting tumour-associated CAs and thioredoxin.
5. Conclusions
Due to the fact that the CAs are enzymes involved in a variety of physiologic and pathologic processes, and since highly effective CAI inhibitors are available belonging to many chemical classes76–78, their hybridisation with other pharmacologic agents, in a multitargeting approach, saw important developments over the last 15 years. The most advanced applications are those in the field of glaucoma, since the sulphonamide CAIs were combined with NO-donating moieties, PG receptor agonists, and β-blockers. Many of the obtained hybrids showed excellent affinity for both targets and an enhanced biological activity, in this case IOP lowering, in animal models of glaucoma. However, there are still several classes of antiglaucoma agents which have not yet been hybridised with CAIs, such as for example the α-adrenergic agonists, the rock-kinase inhibitors, etc., and they should offer interesting possibilities to obtain novel antiglaucoma drugs. Interesting developments were also registered for the hybrids of CAIs with anti-inflammatory agents such as CORMs, for obtaining compounds which release in a controlled and slow manner CO, as well as with classical NSAIDs belonging to the aryl-acetic or aryl-propionic acid drugs. Sulphonamides, coumarins and sulphocoumarin CAIs were hybridised with such derivatives by using different derivatization techniques, which showed increased anti-inflammatory action in various disease models in experimental animals.
The anticancer field also saw interesting multitargeting approaches, as sulphonamide/coumarin CAIs have been hybridised with cytotoxic agents, kinase inhibitors, telomerase inhibitors, P-gp inhibitors or thioredoxin inhibitors. Many such derivatives showed enhanced anti-proliferative activity compared to the single individual agents or their combination, proving the efficacy of the multitargeting. However, there are many other classes of pharmacological agents which would be amenable to obtaining hybrids with CAIs which were not yet investigated, and presumably in the future some of them will be explored.
Funding Statement
Research from the author’s laboratory was financed by the Italian Ministry for Education and Science (MIUR), grant PRIN: rot. [2017XYBP2R] and by Ente Cassa di Risparmio di Firenze (ECRF), grant [CRF2020.1395].
Disclosure statement
CT Supuran is Editor-in-Chief of Journal of Enzyme Inhibition and Medicinal Chemistry and he was not involved in the assessment, peer review or decision making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- 1.Strebhardt K, Ullrich A.. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer 2008;8:473–80. [DOI] [PubMed] [Google Scholar]
- 2.Gelpi A, Gilbertson A, Tucker JD.. Magic bullet: Paul Ehrlich, Salvarsan and the birth of venereology. Sex Transm Infect 2015;91:68–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atanasov AG, Zotchev SB, Dirsch VM, Supuran CT.. International Natural Product Sciences Taskforce, Supuran CT. Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov 2021;20:200–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morphy R, Kay C, Rankovic Z.. From magic bullets to designed multiple ligands. Drug Discov Today 2004;9:641–51. [DOI] [PubMed] [Google Scholar]
- 5.Meunier B. Hybrid molecules with a dual mode of action: dream or reality? Acc Chem Res 2008;41:69–77. [DOI] [PubMed] [Google Scholar]
- 6.Gediya LK, Njar VCO.. Promise and challenges in drug discovery and development of hybrid anticancer drugs. Expert Opin Drug Discov 2009;4:1099–111. [DOI] [PubMed] [Google Scholar]
- 7.Pedrosa MD, da Cruz RMD, Viana JD.. Hybrid compounds as direct multitarget ligands: a review. Curr Top Med Chem 2017;17:1044–79. [DOI] [PubMed] [Google Scholar]
- 8.Szumilak M, Wiktorowska-Owczarek A, Stanczak A.. Hybrid drugs-a strategy for overcoming anticancer drug resistance? Molecules 2021;26:2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pirolla NFF, Batista VS, Dias Viegas FP, et al. Alzheimer’s disease: related targets, synthesis of available drugs, bioactive compounds under development and promising results obtained from multi-target approaches. Curr Drug Targets 2021;22:505–38. [DOI] [PubMed] [Google Scholar]
- 10.Malafaia D, Albuquerque HMT, Silva AMS.. Amyloid-β and tau aggregation dual-inhibitors: a synthetic and structure-activity relationship focused review. Eur J Med Chem 2021;214:113209. [DOI] [PubMed] [Google Scholar]
- 11.Stępnicki P, Kondej M, Koszła O, et al. Multi-targeted drug design strategies for the treatment of schizophrenia. Expert Opin Drug Discov 2021;16:101–14. [DOI] [PubMed] [Google Scholar]
- 12.Wetzel C, Lonneman M, Wu C.. Polypharmacological drug actions of recently FDA approved antibiotics. Eur J Med Chem 2021;209:112931. [DOI] [PubMed] [Google Scholar]
- 13.Ma Y, Frutos-Beltrán E, Kang D, et al. Medicinal chemistry strategies for discovering antivirals effective against drug-resistant viruses. Chem Soc Rev 2021;50:4514–40. [DOI] [PubMed] [Google Scholar]
- 14.(a) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. [DOI] [PubMed] [Google Scholar]; (b) Supuran CT. Emerging role of carbonic anhydrase inhibitors. Clin Sci 2021;35:1233–49. [DOI] [PubMed] [Google Scholar]
- 15.Mishra CB, Tiwari M, Supuran CT.. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev 2020;40:2485–565. [DOI] [PubMed] [Google Scholar]
- 16.Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. [DOI] [PubMed] [Google Scholar]
- 17.Mincione F, Nocentini A, Supuran CT.. Advances in the discovery of novel agents for the treatment of glaucoma. Expert Opin Drug Discov 2021. [DOI] [PubMed] [Google Scholar]
- 18.Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21. [DOI] [PubMed] [Google Scholar]
- 19.Scozzafava A, Supuran CT, Carta F.. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35. [DOI] [PubMed] [Google Scholar]
- 20.Neri D, Supuran CT.. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. [DOI] [PubMed] [Google Scholar]
- 21.Supuran CT. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs 2018;27:963–70. [DOI] [PubMed] [Google Scholar]
- 22.Supuran CT. Experimental carbonic anhydrase inhibitors for the treatment of hypoxic tumors. J Exp Pharmacol 2020;12:603–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother 2016;16:961–8. [DOI] [PubMed] [Google Scholar]
- 24.Bulli I, Dettori I, Coppi E, et al. Role of carbonic anhydrase in cerebral ischemia and carbonic anhydrase inhibitors as putative protective agents. Int J Mol Sci 2021;22:5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Supuran CT, Capasso C.. Antibacterial carbonic anhydrase inhibitors: an update on the recent literature. Expert Opin Ther Pat 2020;30:963–82. [DOI] [PubMed] [Google Scholar]
- 26.Vermelho AB, Rodrigues GC, Supuran CT.. Why hasn’t there been more progress in new Chagas disease drug discovery? Expert Opin Drug Discov 2020;15:145–58. [DOI] [PubMed] [Google Scholar]
- 27.Kaur J, Cao X, Abutaleb NS, et al. Optimization of acetazolamide-based scaffold as potent inhibitors of vancomycin-resistant Enterococcus. J Med Chem 2020;63:9540–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hewitt CS, Abutaleb NS, Elhassanny AEM, et al. Structure-activity relationship studies of acetazolamide-based carbonic anhydrase inhibitors with activity against Neisseria gonorrhoeae. ACS Infect Dis. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Supuran CT, Capasso C.. A highlight on the inhibition of fungal carbonic anhydrases as drug targets for the antifungal armamentarium. Int J Mol Sci 2021;22:4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Supuran CT. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin Drug Discov 2020;15:671–86. [DOI] [PubMed] [Google Scholar]
- 32.Nocentini A, Angeli A, Carta F, et al. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J Enzyme Inhib Med Chem 2021;36:561–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Supuran CT, Carbonic anhydrase inhibitor – NO donor hybrids and their pharmacologic applications. In Bonavida B, Morbidelli L, eds. Therapeutic applications of nitric oxide in cancer and inflammatory-related disorders. Amsterdam (The Netherlands): Elsevier; 2019: 229–242. [Google Scholar]
- 34.Weinreb RN, Liebmann JM, Martin KR, et al. Latanoprostene Bunod 0.024% in subjects with open-angle glaucoma or ocular hypertension: pooled phase 3 study findings. J Glaucoma 2018;27:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eysteinsson T, Gudmundsdottir H, Hardarson AO, et al. Carbonic anhydrase inhibitors of different structures dilate pre-contracted porcine retinal arteries. Int J Mol Sci 2019;20:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steele RM, Benedini F, Biondi S, et al. Nitric oxide-donating carbonic anhydrase inhibitors for the treatment of open-angle glaucoma. Bioorg Med Chem Lett 2009;19:6565–70. [DOI] [PubMed] [Google Scholar]
- 37.Mincione F, Benedini F, Biondi S, et al. Synthesis and crystallographic analysis of new sulphonamides incorporating NO-donating moieties with potent antiglaucoma action. Bioorg Med Chem Lett 2011;21:3216–21. [DOI] [PubMed] [Google Scholar]
- 38.Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47. [DOI] [PubMed] [Google Scholar]
- 39.Chegaev K, Lazzarato L, Tamboli Y, et al. Furazan and furoxan sulphonamides are strong α-carbonic anhydrase inhibitors and potential antiglaucoma agents. Bioorg Med Chem 2014;22:3913–21. [DOI] [PubMed] [Google Scholar]
- 40.Angeli A, Supuran CT.. Prostaglandin receptor agonists as antiglaucoma agents (a patent review 2013 - 2018). Expert Opin Ther Pat 2019;29:793–803. [DOI] [PubMed] [Google Scholar]
- 41.Long DD, Frieman B, Hegde SS, et al. A multivalent approach towards linked dual-pharmacology prostaglandin F receptor agonist/carbonic anhydrase-II inhibitors for the treatment of glaucoma. Bioorg Med Chem Lett 2013;23:939–43. [DOI] [PubMed] [Google Scholar]
- 42.Nocentini A, Ceruso M, Bua S, et al. Discovery of β-adrenergic receptors blocker-carbonic anhydrase inhibitor hybrids for multitargeted antiglaucoma therapy. J Med Chem 2018;61:5380–94. [DOI] [PubMed] [Google Scholar]
- 43.Papapetropoulos A, Foresti R, Ferdinandy P.. Pharmacology of the ‘gasotransmitters’ NO, CO and H2S: translational opportunities. Br J Pharmacol 2015;172:1395–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tenhunen R, Marver HS, Schmid R.. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA 1968;61:748–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willis D, Moore AR, Willoughby DA.. Heme oxygenase isoform expression in cellular and antibody-mediated models of acute inflammation in the rat. J Pathol 2000;190:627–34. [DOI] [PubMed] [Google Scholar]
- 46.Motterlini R, Otterbein LE.. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov 2010;9:728–43. [DOI] [PubMed] [Google Scholar]
- 47.Ott I, Kircher B, Dembinski R, Gust R.. Alkyne hexacarbonyl dicobalt complexes in medicinal chemistry and drug development. Expert Opin Ther Pat 2008;18:327–37. [Google Scholar]
- 48.Berrino E, Milazzo L, Micheli L, et al. Synthesis and evaluation of carbonic anhydrase inhibitors with carbon monoxide releasing properties for the management of rheumatoid arthritis. J Med Chem 2019;62:7233–49. [DOI] [PubMed] [Google Scholar]
- 49.Berrino E, Carradori S, Angeli A, et al. Dual carbonic anhydrase IX/XII inhibitors and carbon monoxide releasing molecules modulate LPS-mediated inflammation in mouse macrophages. Antioxidants 2021;10:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gallorini M, Berardi AC, Ricci A, et al. Dual acting carbon monoxide releasing molecules and carbonic anhydrase inhibitors differentially modulate inflammation in human tenocytes. Biomedicines 2021;9:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bua S, Di Cesare Mannelli L, Vullo D, et al. Design and synthesis of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the treatment of rheumatoid arthritis. J Med Chem 2017;60:1159–70. [DOI] [PubMed] [Google Scholar]
- 52.Bua S, Lucarini L, Micheli L, et al. Bioisosteric development of multitarget nonsteroidal anti-inflammatory drug-carbonic anhydrases inhibitor hybrids for the management of rheumatoid arthritis. J Med Chem 2020;63:2325–42. [DOI] [PubMed] [Google Scholar]
- 53.Menicatti M, Pallecchi M, Bua S, et al. Resolution of co-eluting isomers of anti-inflammatory drugs conjugated to carbonic anhydrase inhibitors from plasma in liquid chromatography by energy-resolved tandem mass spectrometry. J Enzyme Inhib Med Chem 2018;33:671–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akgul O, Di Cesare Mannelli L, Vullo D, et al. Discovery of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the management of rheumatoid arthritis. J Med Chem 2018;61:4961–77. [DOI] [PubMed] [Google Scholar]
- 55.Micheli L, Bozdag M, Akgul O, et al. Pain relieving effect of-NSAIDs-CAIs hybrid molecules: systemic and intra-articular treatments against rheumatoid arthritis. Int J Mol Sci 2019;20:1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lucarini L, Durante M, Sgambellone S, et al. Effects of new NSAID-CAI hybrid compounds in inflammation and lung fibrosis. Biomolecules 2020;10:1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Margheri F, Ceruso M, Carta F, et al. Overexpression of the transmembrane carbonic anhydrase isoforms IX and XII in the inflamed synovium. J Enzyme Inhib Med Chem 2016;31:60–3. [DOI] [PubMed] [Google Scholar]
- 58.McDonald PC, Chia S, Bedard PL, et al. A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am J Clin Oncol 2020;43:484–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krall N, Pretto F, Decurtins W, et al. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014; 53:4231–5. [DOI] [PubMed] [Google Scholar]
- 60.Cazzamalli S, Ziffels B, Widmayer F, et al. Enhanced therapeutic activity of non-internalizing small-molecule-drug conjugates targeting carbonic anhydrase IX in combination with targeted interleukin-2. Clin Cancer Res 2018; 24:3656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cazzamalli S, Dal Corso A, Widmayer F, Neri D.. Chemically defined antibody- and small molecule-drug conjugates for in vivo tumor targeting applications: a comparative analysis. J Am Chem Soc 2018;140:1617–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marks IS, Gardeen SS, Kurdziel SJ, et al. Development of a small molecule tubulysin B conjugate for treatment of carbonic anhydrase IX receptor expressing cancers. Mol Pharm 2018;15:2289–96. [DOI] [PubMed] [Google Scholar]
- 63.Buller F, Steiner M, Frey K, et al. Selection of carbonic anhydrase IX inhibitors from one million DNA-encoded compounds. ACS Chem Biol 2011;6:336–44. [DOI] [PubMed] [Google Scholar]
- 64.An R, Lin B, Zhao S, et al. Discovery of novel artemisinin-sulphonamidehybrids as potential carbonicanhydrase IX inhibitors with improved antiproliferative activities. Bioorg Chem 2020;104:104347. [DOI] [PubMed] [Google Scholar]
- 65.Yu H, Hou Z, Tian Y, et al. Design, synthesis, cytotoxicity and mechanism of novel dihydroartemisinin-coumarin hybrids as potential anti-cancer agents. Eur J Med Chem 2018;151:434–49. [DOI] [PubMed] [Google Scholar]
- 66.Tian Y, Liang Z, Xu H, et al. Design, synthesis and cytotoxicity of novel dihydroartemisinin-coumarin hybrids via click chemistry. Molecules 2016;21:758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62. [DOI] [PubMed] [Google Scholar]
- 68.Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44. [DOI] [PubMed] [Google Scholar]
- 69.Peerzada MN, Hamel E, Bai R, et al. Deciphering the key heterocyclic scaffolds in targeting microtubules, kinases and carbonic anhydrases for cancer drug development. Pharmacol Ther 2021;225:107860. [DOI] [PubMed] [Google Scholar]
- 70.Zhang B, Liu Z, Xia S, et al. Design, synthesis and biological evaluation of sulfamoylphenyl-quinazoline derivatives as potential EGFR/CAIX dual inhibitors. Eur J Med Chem 2021;216:113300. [DOI] [PubMed] [Google Scholar]
- 71.Elzahhar PA, Abd El Wahab SM, Elagawany M, et al. Expanding the anticancer potential of 1,2,3-triazoles via simultaneously targeting cyclooxygenase-2,15-lipoxygenase and tumor-associated carbonic anhydrases. Eur J Med Chem 2020;200:112439. [DOI] [PubMed] [Google Scholar]
- 72.Berrino E, Angeli A, Zhdanov DD, et al. Azidothymidine “clicked” into 1,2,3-triazoles: first report on carbonic anhydrase-telomerase dual-hybrid inhibitors. J Med Chem 2020;63:7392–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Teodori E, Braconi L, Bua S, et al. Dual P-glycoprotein and CA XII inhibitors: a new strategy to reverse the P-gp mediated multidrug resistance (MDR) in cancer cells. Molecules 2020;25:1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krasavin M, Sharonova T, Sharoyko V, et al. Combining carbonic anhydrase and thioredoxin reductase inhibitory motifs within a single molecule dramatically increases its cytotoxicity. J Enzyme Inhib Med Chem 2020;35:665–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Krasavin M, Žalubovskis R, Grandāne A, et al. Sulphocoumarins as dual inhibitors of human carbonic anhydrase isoforms IX/XII and of human thioredoxin reductase. J Enzyme Inhib Med Chem 2020;35:506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Innocenti A, Gülçin I, Scozzafava A, Supuran CT.. Carbonic anhydrase inhibitors. Antioxidant polyphenols effectively inhibit mammalian isoforms I-XV. Bioorg Med Chem Lett 2010;20:5050–3. [DOI] [PubMed] [Google Scholar]
- 77.Briganti F, Pierattelli R, Scozzafava A, Supuran CT.. Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: kinetic and spectroscopic investigations. Eur J Med Chem 1996;31:1001–10. [Google Scholar]
- 78.Berrino E, Michelet B, Martin-Mingot A, et al. Modulating the efficacy of carbonic anhydrase inhibitors through fluorine substitution. Angew Chem Int Ed Engl 2021. [DOI] [PubMed] [Google Scholar]