

Abstract
Trimethylation of histone H3 lysine 27 (H3K27me3) regulates gene repression, cell-fate determination and differentiation. We report that a conserved Bromo-Adjacent Homology (BAH) module of BAHCC1 (BAHCC1BAH) ‘recognizes’ H3K27me3 specifically and enforces silencing of H3K27me3-demarcated genes in mammalian cells. Biochemical, structural and ChIP-seq-based analyses demonstrate that direct readout of H3K27me3 by BAHCC1 is achieved through a hydrophobic trimethyl-lysine-binding ‘cage’ formed by BAHCC1BAH, mediating co-localization of BAHCC1 and H3K27me3-marked genes. BAHCC1 is highly expressed in human acute leukemias and interacts with transcriptional co-repressors. In leukemia, depletion of BAHCC1, or disruption of the BAHCC1BAH:H3K27me3 interaction, causes de-repression of H3K27me3-targeted genes that are involved in tumor suppression and cell differentiation, leading to suppression of oncogenesis. In mice, introduction of a germline mutation at Bahcc1 to disrupt its H3K27me3 engagement causes partial postnatal lethality, supporting a role in development. This study identifies an H3K27me3-directed transduction pathway in mammals that relies on a conserved BAH ‘reader’.
Post-translational modifications (PTMs) of histones represent a means for modulating gene expression and determining cellular identities during organismal development and cell lineage specification. Among various histone-modifying machineries, Polycomb Repressive Complex 2 (PRC2) catalyzes H3K27me3, a histone PTM crucial for gene repression and imprinting, epigenomic states, cell-fate determination and embryogenesis1–7. The importance of understanding H3K27me3-mediated gene regulation is further supported by a range of disease-associated gene mutations that recurrently target H3K27 or its modifying enzyme EZH2. A lysine-to-methionine missense mutation of H3K27 (H3K27M) specifies a deadly pediatric glioma subtype, which displays the altered H3K27me3 pattern8,9, and gain-of-function and loss-of-function mutations of EZH2, the catalytic subunit of PRC2, occur frequently in lymphomas and myeloid neoplasms, respectively10–14.
Histone PTMs elicit gene-regulatory effects at least partly through recruiting ‘readers’ or ‘effectors’, which subsequently control or modulate functional outputs of target chromatin such as transcription15,16. In a current H3K27me3-’readout’ model, H3K27me3 is recognized by chromodomain (CD) harbored within the chromobox (CBX) subunit of Polycomb Repressive Complex 1 (PRC1), which induces H2A mono-ubiquitination (H2Aub) and chromatin compaction3,17–21. WD40 repeats of EED, a core subunit of PRC2, also recognizes H3K27me3, allosterically activating PRC2 for propagation of H3K27me322. While CBX in PRC1 is viewed as the major downstream ‘effector’ class for transducing the H3K27me3-initiated signals in mammalian cells, interplay between H3K27me3 and PRC1 is complex. Certain PRC1 variant complexes achieve genomic targeting via an H3K27me3-independent mechanism and actually act upstream of H3K27me323–25. These observations raise a question of whether an H3K27me3 ‘reader’ other than CBX exists in mammals.
Through cellular, biochemical, biophysical and structural analyses, we report that a conserved BAH module of BAHCC1 (BAHCC1BAH) functions as a direct, specific ‘reader’ of H3K27me3 in mammalian cells. BAHCC1 is significantly overexpressed among several human acute leukemia subtypes and loss-of-function studies show BAHCC1 to be crucial for malignant growth in vitro and in vivo. Integrated chromatin immunoprecipitation-sequencing (ChIP-seq) and RNA sequencing (RNA-seq) profiling reveal that BAHCC1 directly binds H3K27me3-demarcated genes, enforcing an optimal repression state. BAHCC1 interacts with transcriptional corepressors, SAP30-binding protein (SAP30BP) and histone deacetylase (HDAC), establishing a molecular basis for BAHCC1-mediated target repression. In acute leukemias, CRISPR/Cas9-based mutagenesis of BAHCC1BAH demonstrates that BAHCC1BAH-directed H3K27me3 ‘readout’ is crucial for silencing of H3K27me3-marked genes and maintenance of an aggressive cancer phenotype; in mice, introducing a germline mutation at Bahcc1BAH to disrupt its H3K27me3 engagement leads to an incomplete postnatal lethality phenotype. Collectively, this study describes an H3K27me3 signal transduction cascade in mammalian cells, challenging the current norm that tends to regard CBX within PRC1 as the sole effector of H3K27me3.
Results
BAHCC1 is overexpressed in various human acute leukemia subtypes, sustaining tumorigenicity.
BAHCC1 (Fig. 1a), a predicted chromatin regulator of unknown cellular function, was shown to be overexpressed in acute myeloid leukemias (AML) showing poor prognosis26. Using publicly available datasets, we observed that, akin to what was seen in AML (Extended Data Fig. 1a–b), the expression of BAHCC1 is significantly elevated in acute lymphoblastic leukemia (ALL) of T-cell or B-cell lineages (T-ALL or B-ALL), compared to normal tissues (Fig. 1b and Extended Data Fig. 1c–f). BAHCC1 overexpression is associated with genetic abnormality related to worse clinical outcomes such as MLL rearrangement and E2A-PBX1 fusion (Extended Data Fig. 1f–g). The function of BAHCC1 was previously unstudied; thus, we sought to characterize BAHCC1 by inserting a 3×Flag tag to the endogenous BAHCC1 locus via a CRISPR/Cas9-based knock-in (KI) strategy27 (Extended Data Fig. 1h–j). Using independently derived leukemia lines with BAHCC1–3×Flag KI alleles, we detected an expected ~300-kD protein, which displays an exclusive nuclear localization and is readily depleted by BAHCC1-targeting shRNAs (Extended Data Fig. 1k–m); similar results were seen in HeLa cells with the same 3×Flag-KI approach (data not shown), consistent with the predicted role of BAHCC1 as a chromatin regulator.
Fig. 1. BAHCC1 sustains acute leukemia cell growth in vitro and in vivo.
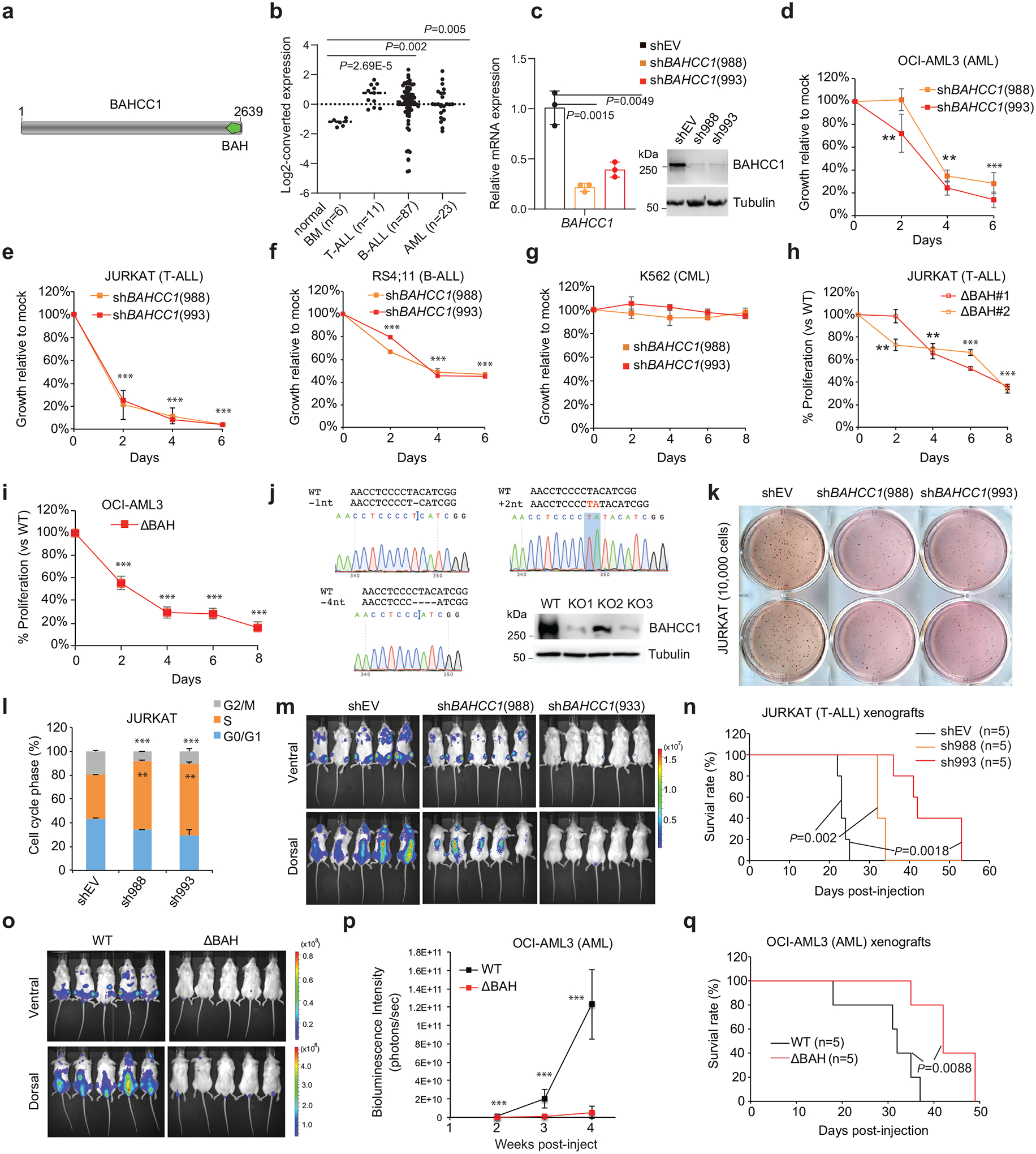
a, Domain architecture of BAHCC1.
b, Dot plot showing BAHCC1 overexpression in primary T-ALL, B-ALL and AML samples, relative to normal tissues, based on GEO dataset GSE7186.
c, RT-qPCR (left) and immunoblots (right) of BAHCC1 after its knockdown (KD) in JURKAT cells (n = 3 biologically independent samples). Data are presented as mean ± SD.
d-g, Proliferation of the indicated cells, OCI-AML3 (d), JURKAT (e), RS4;11 (f) or K562 (g), post-KD of BAHCC1 relative to mock-treated control (n = 3 biologically independent experiments). Data are presented as mean ± SD. ** P < 0.01; *** P < 0.001.
h-j, Proliferation of JURKAT T-ALL (h) and OCI-AML3 AML cells (i) following CRISPR-cas9-mediated disruption of the BAHCC1 BAH domain (BAHCC1BAH), relative to WT (n = 3 biologically independent experiments). Data are presented as mean ± SD. Panel j, Sanger sequencing for detecting homozygous frame-shift mutation of BAHCC1 and immunoblotting for BAHCC1 knockout (KO) mediated by sgRNAs that target exons encoding BAHCC1BAH. ** P < 0.01; *** P < 0.001.
k, Representative images of colonies formed by JURKAT cells, either mock-treated (shEV) or post-KD of BAHCC1 (n = 2 biologically independent experiments).
l, Quantification of cell cycle phases using JURKAT cells, either mock-treated or post-KD of BAHCC1 (n = 3 biologically independent experiments). ** P < 0.01; *** P < 0.001. Data are presented as mean ± SD.
m-n, Bioluminescence imaging (m; three weeks post-transplantation) and Kaplan-Meier survival curve (n) of NSG mice xenografted with luciferase-labeled JURKAT cells, either mock-treated (shEV) or stably transduced with a BAHCC1-targeting shRNA. P values were calculated by log-rank test; n, cohort size.
o-q, Bioluminescence imaging (o; two weeks post-transplantation), summary of bioluminescence signals (p; from dorsal imaging; n = 5 mice, with data presented as mean ± SD) and Kaplan-Meier curve (q) of NSG mice xenografted with luciferase-labeled OCI-AML3 AML cells that harbor the CRISPR-cas9-caused frameshift mutation within BAHCC1BAH, relative to WT. n, cohort size. *** P < 0.001.
Next, we assessed requirement of BAHCC1 for tumorigenesis. Using independent shRNAs (Fig. 1c), we performed BAHCC1 knockdown (KD) in a set of human acute leukemia lines that cover AML (OCI-AML3, MV4;11), T-cell ALL (JURKAT, CUTLL1, CCRF-CEM, ALL-SIL, MOLT4) and B-cell ALL (RS4;11), and found that BAHCC1 depletion significantly suppressed proliferation of these leukemia cells in vitro (Fig. 1d–f and Extended Data Fig. 2a–f). In contrast, BAHCC1 KD did not affect growth of K562 chronic myeloid leukemia cells (Fig. 1g and Extended Data Fig. 2a) or colony-forming capabilities of hematopoietic stem/progenitor cells (HSPCs) in vitro (Extended Data Fig. 2g). To identify BAHCC1 protein regions involved in malignant growth, we employed the recently described Cas9/sgRNA-based scanning approach28,29 for gene editing and mutagenesis of BAHCC1, which revealed several protein regions to be crucial for JURKAT cell proliferation, including a C-terminal BAH domain (BAHCC1BAH) (Extended Data Fig. 2h). Using individual sgRNAs, we verified that the sgRNA-induced frame-shift and disruption of BAHCC1BAH in JURKAT ALL cells and OCI-AML3 AML cells phenocopied shRNA-mediated KD of BAHCC1 resulting in significantly decreased cancer cell growth (Fig. 1h–j), while CRISPR/dCas9-mediated silencing of BAHCC1 did not affect K562 cell growth (Extended Data Fig. 2i). BAHCC1 depletion also led to defect in colony-forming capacities of acute leukemia cells (Fig. 1k and Extended Data Fig. 2j–l), a significant delay in cell cycle progression, especially the G1-S phase transition (Fig. 1l and Extended Data Fig. 2m), and a slight increase in apoptosis (Extended Data Fig. 2n). Importantly, using three different xenografted ALL (JURKAT or CUTLL1) or AML (OCI-AML3) leukemia models, we found that the shRNA-mediated KD of BAHCC1 or CRISPR/Cas9-induced disruption of BAHCC1BAH significantly suppressed the growth of xenografted leukemias in vivo (Fig. 1m–q and Extended Data Fig. 3a–c), an effect not seen with K562 cell xenografts (Extended Data Fig. 3d–e). These results highlighted a critical role of BAHCC1 and its BAHCC1BAH module for acute leukemogenesis.
BAHCC1BAH specifically ‘reads’ H3K27me3 in vitro and in cells
Sequence alignment of BAHCC1 proteins across different vertebrate species showed their BAH domains to be highly conserved (Extended Data Fig. 3f–g). To examine the putative histone-binding activity of BAHCC1BAH, we probed the BAHCC1BAH recombinant protein with a histone peptide microarray and detected its specific binding to H3K27me3/2-containing peptides (Fig. 2a and Supplementary Fig. 1a), an interaction confirmed by repeated pull-down assays with individual peptides (Fig. 2b, Extended Data Fig. 3h and Supplementary Fig. 1b). As a control, we verified a reported specific binding of histone H4 lysine 20 dimethylation (H4K20me2) by the BAH domain of Origin Recognition Complex subunit 1 (ORC1BAH)30 (Extended Data Fig. 3i and Supplementary Fig. 1c). Specific engagement of H3K27me3/2 by BAHCC1BAH was further verified by Isothermal Titration Calorimetry (ITC) measurements (Fig. 2c–e and Extended Data Fig. 3j–q). Additionally, ITC revealed a comparable H3K27me3-binding affinity of BAHCC1BAH relative to chromodomain of CBX7 (CBX7CD), a known H3K27me3 reader module31–33 (Fig. 2f and Extended Data Fig. 3q, bottom). Co-immunoprecipitation (CoIP) also demonstrated binding of endogenous BAHCC1 to H3K27me3 but not to other examined histone methylations in cells (Fig. 2g). Consistently, immunostaining showed colocalization between BAHCC1 and H3K27me3 (Fig. 2h), and not H3K9me3 (Supplementary Fig. 1d). Altogether, these observations demonstrate that BAHCC1BAH specifically ‘reads’ highly methylated H3K27.
Fig. 2. BAHCC1BAH specifically ‘reads’ H3K27me3.
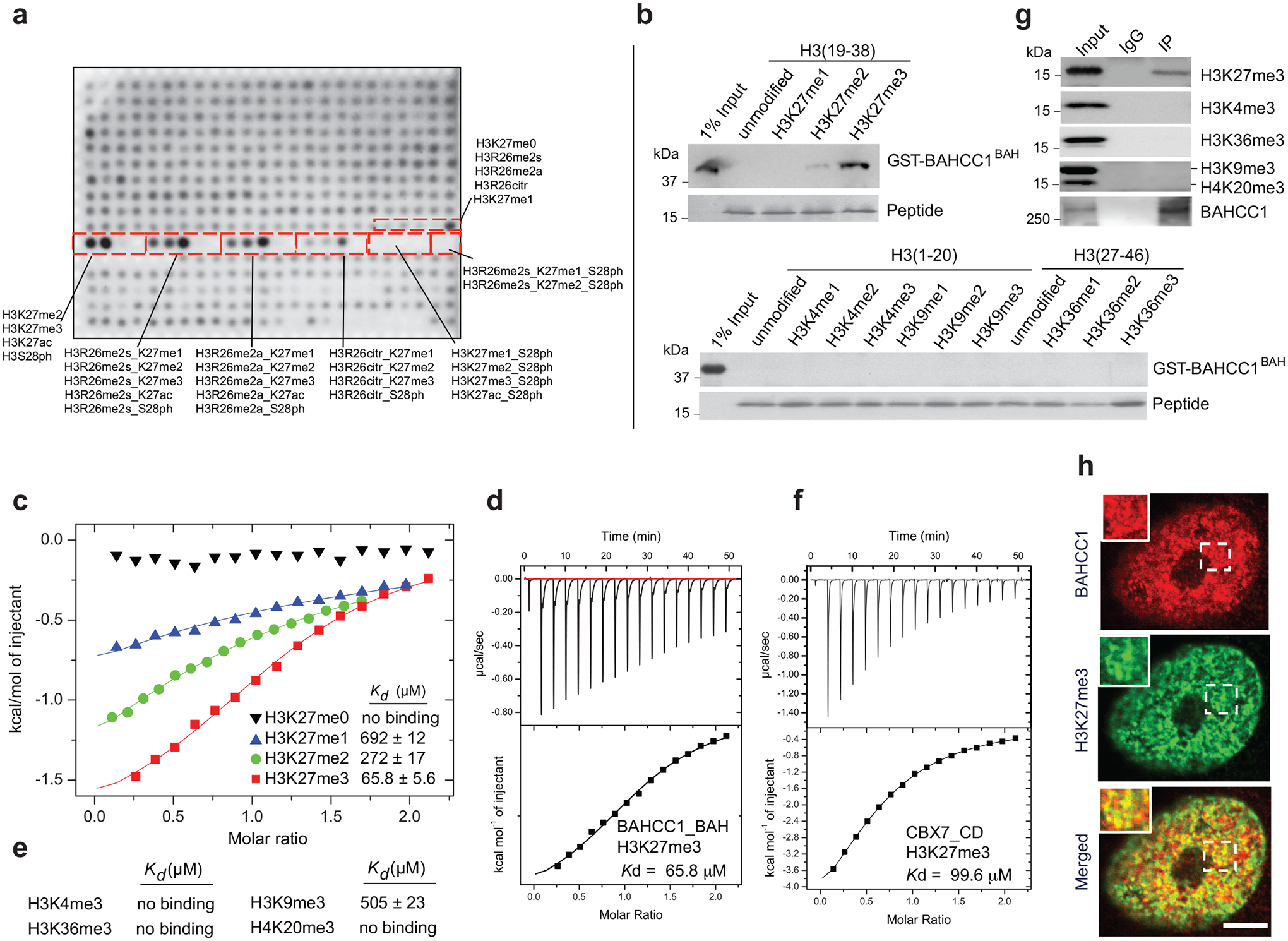
a, Histone peptide array probed with GST-BAHCC1BAH protein, followed by visualization with anti-GST antibody. Red boxes highlight peptides carrying H3K27 methylation, either singly or in combination with adjacent residue modification, with the corresponding peptide identification annotated aside.
b, Pulldown using GST-BAHCC1BAH and biotinylated histone peptide carrying H3K27 methylation (top) or the other indicated methylation (bottom).
c, ITC measuring affinity of binding between BAHCC1BAH and peptide with H3K27 methylation.
d, ITC binding curve of BAHCC1BAH recombinant protein with the H3K27me3 peptide.
e, Summary of ITC measurements revealing affinities of binding between BAHCC1BAH and the indicated histone lysine trimethylation.
f, ITC binding curve of CBX7CD protein with the H3K27me3 peptide.
g, CoIP for interaction between endogenous BAHCC1 and the indicated histone methylation in JURKAT cells.
h, Representative images of confocal immunofluorescence microscopy showing that BAHCC1 (red) colocalizes with H3K27me3 (green) in HeLa cells. Scale bar, 5 μm.
Structural analysis of BAHCC1BAH reveals a unique H3K27me3-binding pocket
To gain insight into the molecular basis underlying BAHCC1BAH-mediated recognition of H3K27me3, we solved the crystal structure of murine BAHCC1BAH in complex with an H3K27me3-containing histone peptide (Fig. 3a–b; Supplementary Table 1). The BAHCC1BAH-H3K27me3 complex belongs to the space group P31, containing eight BAHCC1BAH molecules in one asymmetric unit (Extended Data Fig. 4a). This structure reveals a twisted, five-stranded β-sheet (β6-β7-β8-β10-β11), with its inner and outer surfaces packed against the immediately upstream and downstream β-sheets (Inner: β1-β4-β5; Outer: β12-β13), respectively (Fig. 3a; arrows in Extended Data Fig. 3g). Additionally, the C-terminal ends of β7 and β8 are paired with an N-terminal two-stranded β-sheet (β2-β3) and a C-terminal three-stranded β-sheet (β9-β14-β15), respectively, together creating an elongated β-fold. The H3K27me3 peptide associates with one of the outer β-strand (β10) of the central β-sheet in an ‘L’-shape conformation (Fig. 3a–b). Notably, the side chain of H3K27me3 inserts into a deep pocket formed by residues Y2537, W2558, Y2560 and E2564 of mBAHCC1BAH (Fig. 3c and Extended Data Fig. 4b), which are conserved across species (Extended Data Fig. 3g, red stars). Next to the H3K27me3-binding pocket, the methyl group of H3A24 is embedded in a concave surface lined by mBAHCC1BAH A2530 and T2591, while the prolyl ring of H3P30 slides into a shallow groove formed by mBAHCC1BAH H2583, P2628 and T2629 (Fig. 3b and 3d), which likely contributes to the binding specificity of mBAHCC1BAH for H3K27me3 over H3K9me3. The mBAHCC1BAH-H3K27me3 association is also supported by a number of intermolecular hydrogen-bonding or electrostatic contacts (Fig. 3b and 3d): N-terminal to residue H3K27me3, H3K23 interacts with mBAHCC1BAH D2588 through electrostatic attraction, and H3A25 and H3R26 interact with mBAHCC1BAH L2528 and N2587 via main chain-main chain and main chain-side chain hydrogen bonds, respectively; C-terminal to residue H3K27me3, H3S28 interacts with mBAHCC1BAH D2585 and H2583 through main chain and/or side chain hydrogen bonds. Mutation of the caging residues of BAHCC1BAH disrupted its binding to H3K27me3-containing peptides as measured by ITC (Fig. 3e and Extended Data Fig. 4c–h) and pull-down assays (Fig. 3f). Additionally, we utilized the CRISPR/Cas9-based gene editing technology to introduce site-directed mutagenesis of BAHCC1BAH in JURKAT cells (Fig. 3g and Extended Data Fig. 4i) and found that the cellular association between BAHCC1 and H3K27me3 was disrupted by point mutation of ‘caging’ residues (Fig. 3h). These results revealed the molecular underpinnings crucial for BAHCC1BAH-directed ‘reading’ of H3K27me3.
Fig. 3. Structural analysis of BAHCC1BAH reveals a unique H3K27me3-’reading’ pocket.

a, Overall structure of BAHCC1BAH (aquamarine) bound to the H3K27me3 peptide (yellow). BAHCC1BAH and peptide are shown in ribbon and stick representation, respectively.
b, Close-up view of the BAHCC1BAH-H3K27me3 interactions. The H3K27me3-interacting residues of BAHCC1BAH and peptide are shown as pink and yellow sticks, respectively. Dashed line indicates hydrogen bond.
c, Positioning of the H3K27me3 side chain (yellow) within the aromatic cage of BAHCC1BAH. The cage residues (pink) of mouse and human BAHCC1BAH are labeled in red, with the latter shown in parentheses.
d, Schematic diagrams of the BAHCC1BAH (black)-H3K27me3 (magenta) interactions. Hydrogen bonding and electrostatic interactions are shown as black and green dashed lines, respectively. Hydrophobic interactions are colored in yellow.
e, ITC measures the binding affinity between BAHCC1BAH, either WT or mutant, and H3K27me3 peptide.
f, Pulldown using biotinylated H3K27me3 peptide and GST-BAHCC1BAH, either WT or BAH-mutated.
g, CRISPR/Cas9-mediated gene editing for introducing the Y2533A homozygous point mutation to BAHCC1 in JURKAT cells. Shown are Sanger sequencing results using cDNA as template.
h, CoIP for association between endogenous BAHCC1 and H3K27me3-containing histones in JURKAT cells that express the 3×Flag-tag knockin allele of BAHCC1, either WT or harboring a Y2533A (left, two independent lines) or W2554G (right) homozygous mutation at BAHCC1BAH.
BAHCC1 contributes to silencing of H3K27me3-demarcated genes
Next, we sought to determine the role of BAHCC1 in gene regulation first by transcriptome profiling of JURKAT leukemia cells before and after BAHCC1 depletion. RNA-seq revealed that transcriptional alterations caused by independent BAHCC1-targeting shRNAs are highly correlated (Fig. 4a, Extended Data Fig. 5a and Supplementary Table 2). Gene Ontology (GO) analysis and Ingenuity Pathway Analysis (IPA) of differentially expressed genes (DEGs), as well as Gene Set Enrichment Analysis (GSEA), consistently detected the development- and differentiation-related gene-expression programs among the most affected transcripts following BAHCC1 loss (Extended Data Fig. 5b–f). Additionally, GSEA uncovered marked correlations between BAHCC1 depletion and re-activation of genesets known to be targeted by PRC1 (Fig. 4b–c, Extended Data Fig. 5d and 5g), PRC2 (Fig. 4d–e, Extended Data Fig. 5d and 5h–k) or H3K27me3 (Extended Data Fig. 5d and 5l). Similar results were observed with Platform for Integrative Analysis of Omics (Piano)34, with BAHCC1-repressed transcripts most significantly enriched with genesets related to development, lineage specification and Polycomb- or H3K27me3-bound targets (Extended Data Fig. 5m). Notably, depletion of BAHCC1 did not affect gene expression and protein levels of the examined PRC2 or PRC1 subunits such as EZH2, EED, SUZ12, RING1A and RING1B, or global H3K27me3 (Supplementary Fig. 2a–c). Additionally, transcriptome analyses of OCI-AML3 cells, an AML line, and RS4;11 cells, a B-ALL line (Supplementary Tables 3 and 4) revealed that BAHCC1 loss was positively associated with de-repression of genes suppressed by H3K27me3, PRC2 or PRC1 in both models (Supplementary Fig. 2d–e), despite only a minor portion of BAHCC1-regulated transcripts found to be shared in these cells when compared to JURKAT cells (Supplementary Fig. 2f–g). These results thus suggest a link between BAHCC1 and the Polycomb-H3K27me3 pathway, regardless of cell background.
Fig. 4. Integrated RNA-seq and ChIP-seq analyses demonstrate a direct involvement of BAHCC1 in silencing of the H3K27me3-targeted genes.
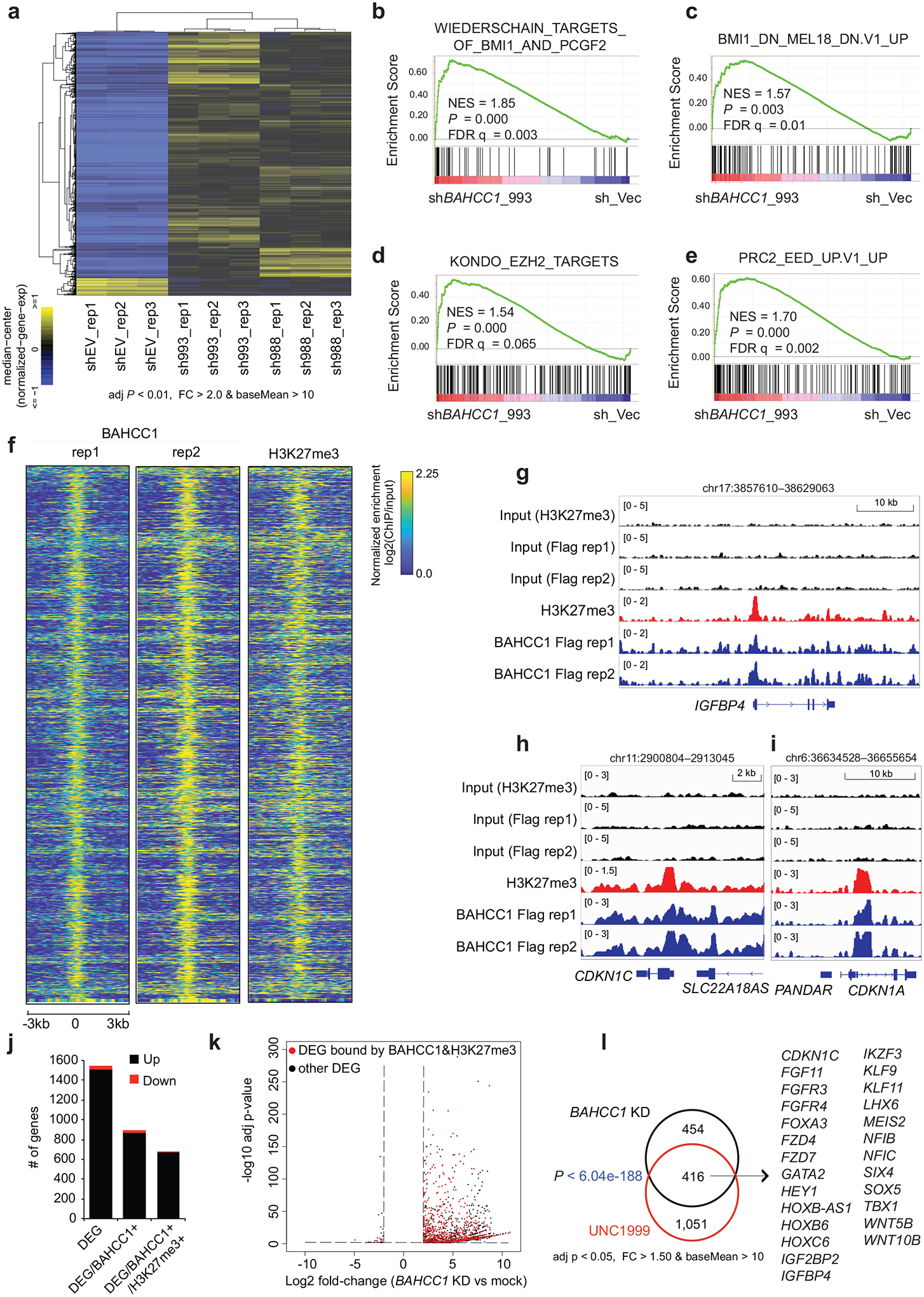
a, Heatmap showing expression of differentially expressed genes (DEGs) after BAHCC1 KD (sh993 or sh988), relative to mock (shEV), in JURKAT cells (n = 3 biologically independent experiments). The threshold of DEGs is adjusted P value (adj. p) < 0.01, fold-change (FC) > 2.0 and transcript baseMean value > 10 based on RNA-seq. Color bar, log2(FC).
b-e, GSEA revealing positive correlations between BAHCC1 depletion and reactivation of genesets related to PRC1 (b-c) or PRC2 (d-e). NES, normalized enrichment score; FDR, false discovery rate.
f, Heatmap showing ChIP-seq peaks of BAHCC1 (Flag; two replicated experiments) that overlap with H3K27me3 peaks in JURKAT cells after normalization to input and sequencing depth. Color bar, log2(ChIP/Input).
g-i, ChIP-seq profiles of BAHCC1 (blue; Flag) and H3K27me3 (red) at IGFBP4 (g), CDKN1C (h) and CDKN1A (i) in JURKAT cells.
j, Summary of the total number of DEGs (a cutoff of q < 0.01 and log2|FC| > 2), either up- (black) or down-regulated (red) after BAHCC1 depletion relative to mock, as detected by RNA-seq in JURKAT cells. DEGs are either all detected (left) or those with direct binding of BAHCC1 (middle) or both BAHCC1 and H3K27me3 (right), based on the called ChIP-seq peaks.
k, Volcano plot of DEGs, including those directly bound by both BAHCC1 and H3K27me3 (red) and the rest (black), as detected by RNA-seq in JURKAT cells after BAHCC1 KD, compared to control.
l, Venn diagram illustrating overlap for those BAHCC1 and H3K27me3 co-bound DEGs found to be upregulated after BAHCC1 depletion (black) or UNC1999 treatment (red), relative to mock, in JURKAT cells. Right panel lists the well-defined Polycomb targets commonly affected by both treatment conditions.
Next, we determined genome-wide occupancy of BAHCC1 by ChIP-seq using Flag antibody and JURKAT leukemia cells that carry the BAHCC1–3×Flag KI allele (Extended Data Fig. 1h–k). The replicated experiments produced robust and correlated ChIP-seq signals (Extended Data Fig. 6a–b). We also found a significant overlap between BAHCC1 ChIP-seq peaks and those of H3K27me3 in JURKAT cells (Fig. 4f and Extended Data Fig. 6c–d), as exemplified by those identified at the tumor suppressive loci such as IGFBP4, CDKN1A and CDKN1C (Fig. 4g–i) and development-related transcription factors (TFs) such as HOX, GATA, KLF and SMAD (Extended Data Fig. 6e–f). Integration of ChIP-seq and RNA-seq results showed that DEGs directly bound by BAHCC1 or by both BAHCC1 and H3K27me3 mainly showed up-regulation upon BAHCC1 depletion (Fig. 4j–k), lending a support for a role of BAHCC1 as transcriptional repressor. To further compare BAHCC1- and H3K27me3-controlled gene-expression programs, we treated JURKAT cells with UNC1999, a small-molecule inhibitor of EZH2 and related EZH135,36, and subsequent RNA-seq profiling (Extended Data Fig. 6g and Supplementary Table 5) revealed that UNC1999-derepressed DEGs significantly overlapped with those caused by BAHCC1 loss (Fig. 4l, P < 6.04 × 10−188 for direct target overlapping; Extended Data Fig. 6h–i). Notably, DEGs co-repressed directly by H3K27me3 and BAHCC1 included not only known PRC2-targeted lineage-specification TFs but also cell cycle inhibitors and tumor suppressors such as CDKN1A, CDKN1C and IGFBP4 (Fig. 4l), in agreement with the slowed tumor growth and defective cell cycle progression seen post-depletion of BAHCC1. Using RT-qPCR and independent EZH2-expressing acute leukemia cells (Extended Data Fig. 6j), we further verified that BAHCC1 depletion (Extended Data Fig. 6k–n) or UNC1999 treatment (Extended Data Fig. 6o–p) caused re-activation of the examined H3K27me3-marked genes. Furthermore, post-treatment of UNC1999, the overall BAHCC1 binding to chromatin was impaired, as assayed by immunoblotting after cell fractionation (Extended Data Fig. 7a), and BAHCC1 binding to its target genes significantly reduced, as assessed by ChIP-qPCR (Extended Data Fig. 7b). Collectively, these findings demonstrate an essential and direct role of BAHCC1 in silencing of H3K27me3-targeted genes.
Disruption of BAHCC1BAH:H3K27me3 interaction impacts gene repression and oncogenesis
Next, we aimed to define the involvement of BAHCC1BAH, an H3K27me3-’reading’ module, in gene silencing. First, ChIP-seq showed that, relative to WT, the BAH-mutated (Y2533A) BAHCC1 displayed the significantly reduced overall binding to target genes in JURKAT cells (Fig. 5a and Extended Data Fig. 6a, 7c). By ChIP-qPCR, we also confirmed such a BAH-dependent binding to H3K27me3-marked genes in leukemia cells harboring the W2554G-mutated BAHCC1 (Extended Data Fig. 7d), which is consistent with the overall reduced H3K27me3 interaction (Fig. 3h) and chromatin binding of W2554G-mutated BAHCC1 as observed in these cells (Extended Data Fig. 7e). Next, we performed RNA-seq profiling of JURKAT cells carrying BAHCC1BAH mutation, either Y2533A or W2554G, and found that DEGs due to these two independently introduced BAHCC1BAH mutations showed a significant overlap with DEGs caused by BAHCC1 loss (Fig. 5b–c and Supplementary Table 6). Again, DEGs include transcripts encoding cell cycle inhibitors, tumor suppressors and lineage-specification TFs, all of which are known to be repressed by H3K27me3 and PRC2 (Fig. 5c, bottom panel). GSEA further revealed that, relative to WT, the H3K27me3-engagement-defective mutation of BAHCC1BAH is positively correlated with the re-activation of genesets known to be repressed by H3K27me3 (Extended Data Fig. 7f), PRC1 (Fig. 5d and Extended Data Fig. 7f–g) or PRC2 (Fig. 5e–f and Extended Data Fig. 7f, 7h–n), which is in agreement with what was observed for BAHCC1 depletion (Extended Data Fig. 5 and Supplementary Fig. 2). Mutations of BAHCC1BAH did not change stability of the examined PRC2 proteins or global H3K27me3 (Extended Data Fig. 7p). As expected, point mutation of BAHCC1BAH also led to derepression of genesets related to cell differentiation (Extended Data Fig. 7f, 7o and Supplementary Fig. 3a). Using RT-qPCR, we further validated an essential requirement of the H3K27me3-engaging BAHCC1BAH for silencing of H3K27me3 targets in three independently derived leukemia lines— JURKAT (Extended Data Fig. 8a–b) and CUTLL1 (Fig. 5g) that harbored a Y2533A (Extended Data Fig. 8a and 8c–d) or W2554G homozygous mutation of BAHCC1BAH (Extended Data Fig. 8b); as a control, cMyc expression was unaltered in these cells (Extended Data Fig. 8e).
Fig. 5. BAHCC1BAH-mediated binding of H3K27me3 is crucial for Polycomb target gene repression and oncogenesis.
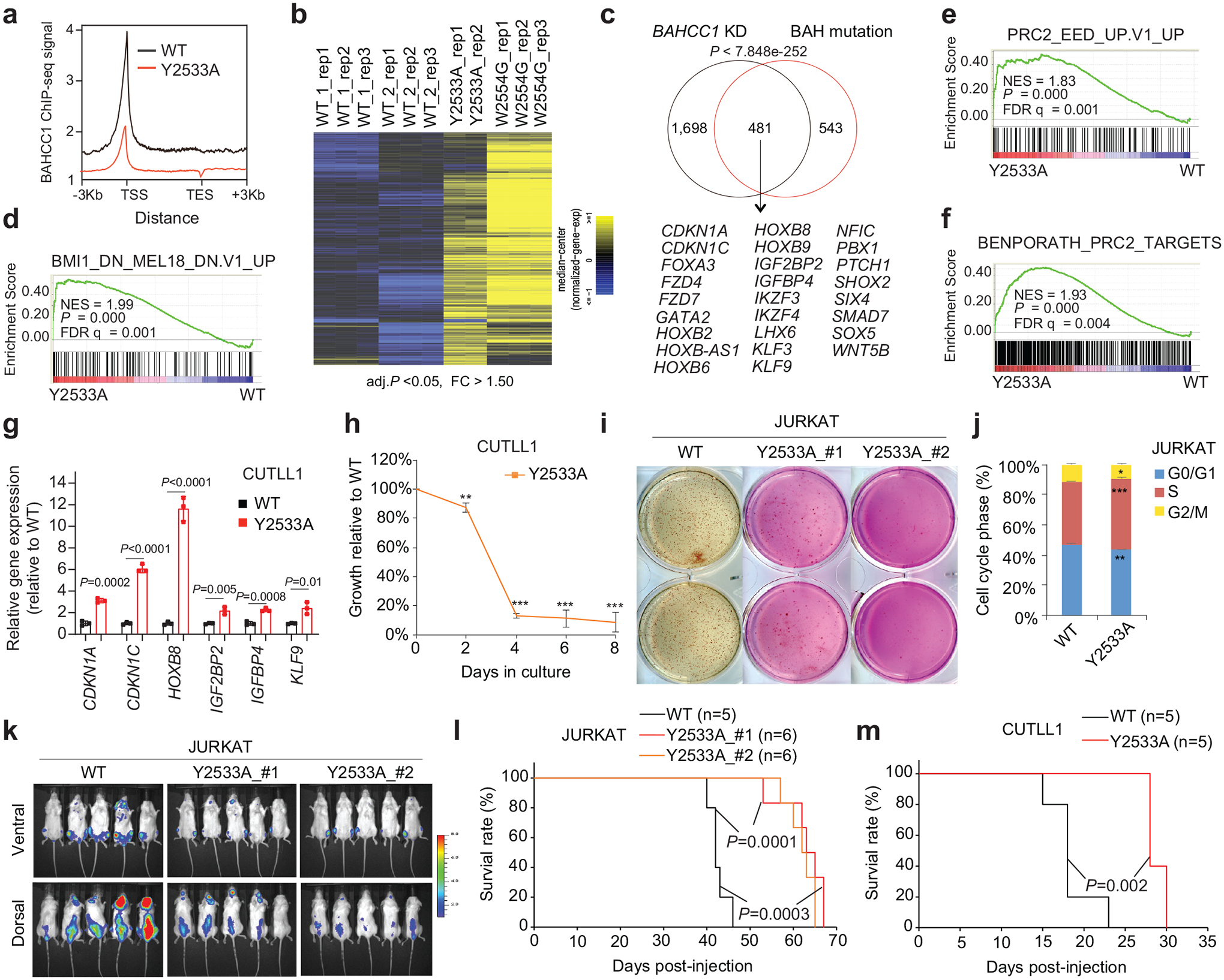
a, Averaged Flag-BAHCC1 ChIP-seq signals over ±3 kb from target genes in JURKAT cells carrying either WT or Y2533A homozygous mutation of BAHCC1BAH. ChIP-seq reads were normalized to input and sequencing depth. TSS, transcription start site; TES, transcription end site.
b, Heatmap of DEGs commonly upregulated in JURKAT cells with the Y2533A or W2554G homozygous mutation of BAHCC1BAH, relative to WT. The thresholds of DEG are adjusted P value < 0.05 and FC > 1.50.
c, Venn diagram shows overlapping of DEGs upregulated in JURKAT cells after BAHCC1 depletion (black) or BAHCC1BAH mutation (red), relative to mock.
d-f, GSEA showing positive correlations between the Y2533A homozygous mutation of BAHCC1BAH and derepression of PRC1 (panel d) or PRC2 (e-f) target genes.
g, RT-qPCR of H3K27me3-targeted genes in CUTLL1 cells carrying Y2533A homozygous mutation of BAHCC1BAH relative to WT (n = 3 independent experiments). Data were plotted as mean ± SD after normalization to GAPDH and to WT.
h, Proliferation of CUTLL1 cells with the Y2533A homozygous mutation of BAHCC1BAH, relative to WT (n = 3 independent experiments). Data are presented as mean ± SD. ** P < 0.01; *** P < 0.001.
i, Colony formation by JURKAT cells carrying WT or the Y2533A homozygous mutation of BAHCC1BAH (n = 2 biologically independent experiments).
j, Quantification of cell cycle progression using JURKAT cells carrying WT or the Y2533A homozygous mutation of BAHCC1BAH (n = 3 biologically independent experiments). Data are presented as mean ± SD. * P < 0.05; ** P < 0.01; *** P < 0.001.
k-m, Representative imaging (k; 5 weeks post-xenograft of JURKAT cells) and Kaplan-Meier curve of event-free survival (l-m) of NSG mice xenografted with luciferase-labeled JURKAT (k-l) or CUTLL1 (m) cells that carry WT or the Y2533A homozygous mutation of BAHCC1BAH. n, cohort size.
Next, we sought to determine the requirement of BAHCC1BAH-mediated H3K27me3 ‘readout’ for tumorigenesis. Here, we found that, despite a lack of apparent morphological changes (Extended Data Fig. 8f), ALL (JURKAT or CUTLL1) and AML (OCI-AML3) cells with the Y2533A or W2554G homozygous mutation of BAHCC1BAH all exhibited significantly decelerated proliferation (Fig. 5h and Extended Data Fig. 8g–h), decreased colony-forming capabilities (Fig. 5i and Extended Data Fig. 8i) and a dysregulated cell cycle progression (Fig. 5j and Extended Data Fig. 8j), when compared to their WT counterpart cells. Strikingly, in three independent tumor xenografted models, BAHCC1BAH mutation caused a significant delay in tumor progression as evaluated by bioluminescence imaging of tumor growth in live animals (Fig. 5k and Extended Data Fig. 8k–m) and event-free survival (Fig. 5l–m and Extended Data Fig. 8n). Collectively, we show that engagement of H3K27me3 by BAHCC1BAH ensures an optimal silencing state of genes bound by H3K27me3 in acute leukemias, a process involved in the maintenance of tumorigenicity.
BAHCC1 associates with transcriptional co-repressors, maintaining hypoacetylation at target genes
Next, we aimed to gain insight into molecular mechanisms underlying the BAHCC1-mediated gene silencing. Mass spectrometry-based analysis of BAHCC1 pulldowns identified SAP30-binding protein (SAP30BP, also known as HTRP37) among the top hits of BAHCC1-associated proteins in HeLa cells carrying the engineered BAHCC1–3×Flag allele (Extended Data Fig. 8o). SAP30BP interacts with SAP3037, a transcriptional corepressor that binds histone deacetylase (HDAC) complexes38,39. Using CoIP in JURKAT cells, we further validated the physical interactions of BAHCC1 with SAP30BP, HDAC1 and HDAC-associated Inhibitor of Growth (ING) proteins40 (Fig. 6a–b) whereas there is no detectable interaction between BAHCC1 and the examined components of PRC2 or PRC1 (Fig. 6a, Extended Data Fig. 8p). These results suggested an involvement of HDAC1 complex in the BAHCC1-mediated gene silencing. Consistent with this notion, GSEA and Piano analyses of RNA-seq datasets found a strong correlation of re-activation of HDAC target genes with either depletion of BAHCC1 (Supplementary Fig. 3b) or introduction of an H3K27me3-binding-defective mutation to BAHCC1BAH (Supplementary Fig. 3b–c). ChIP-seq indeed detected the significantly elevated levels of histone acetylation at the BAHCC1 target genes in cells carrying the BAHCC1BAH mutation (W2554G), relative to WT controls (Fig. 6c–d), as exemplified by those seen at tumor suppressive loci (CDKN1C, IGF2BP2, IGFBP4) and the HOX cluster genes (Fig. 6e and Extended Data Fig. 9a). ChIP-qPCR verified similar effects of an independent BAHCC1BAH mutation, Y2533A, on histone acetylation at the examined promoters of H3K27me3 targets (Fig. 6f), but not a negative control locus (Extended Data Fig. 9b). Depletion of HDAC1 or SAP30BP (Extended Data Fig. 9c–d), as well as treatment with HDAC inhibitors, all led to derepression of the tested BAHCC1 target genes (Fig. 6g–h and Extended Data Fig. 9e–f) and the slowed leukemia cell growth (Fig. 6i–j and Extended Data Fig. 9g). Thus, engagement of H3K27me3 by BAHCC1 maintains a histone deacetylated state at H3K27me3-targeted genes.
Fig. 6. BAHCC1 interacts with corepressors, maintaining a hypoacetylated chromatin state at target genes.
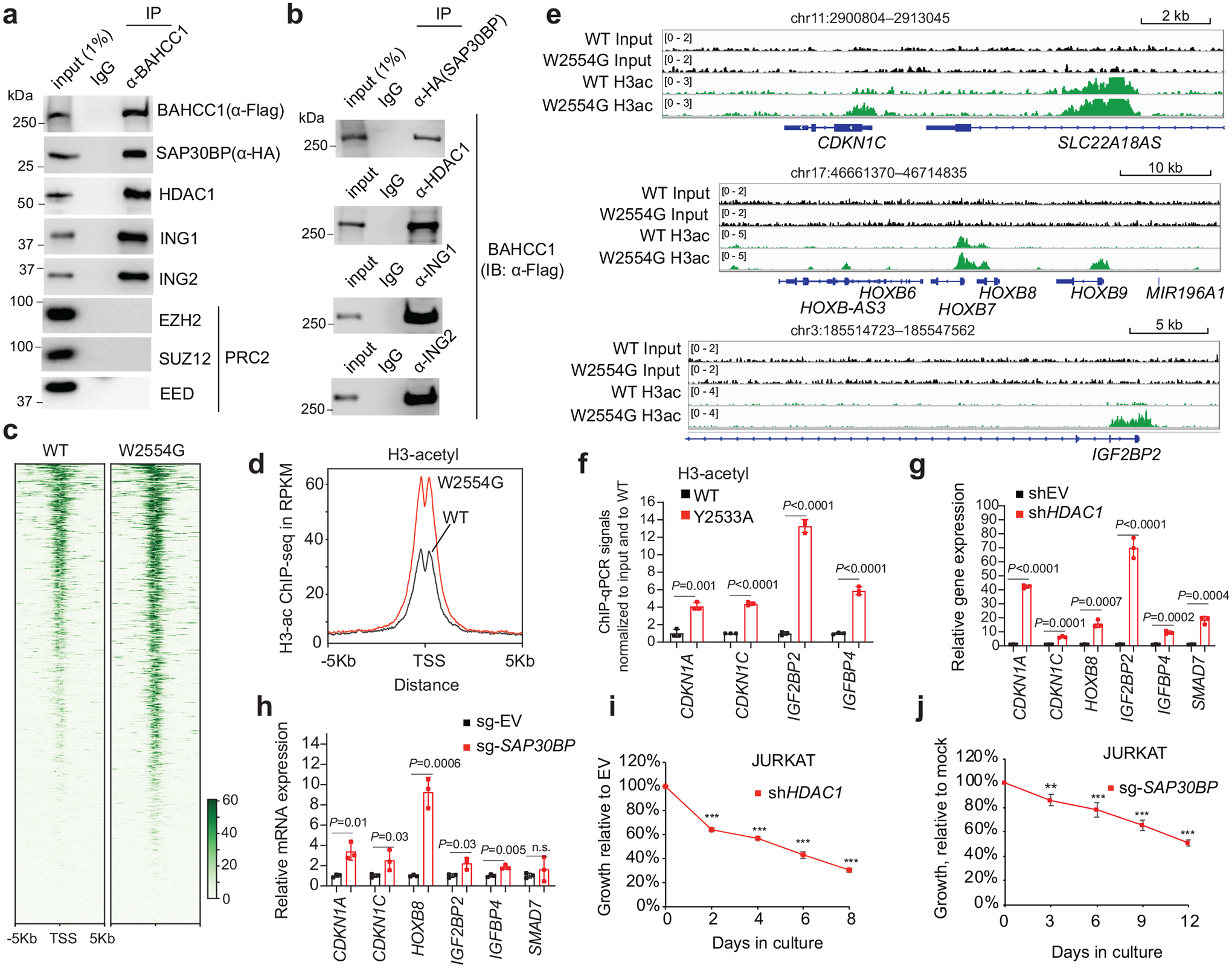
a-b, CoIP for interaction between endogenous Flag-BAHCC1 and the indicated HDAC1 or PRC2 complex component in JURKAT cells.
c-d, Heatmap (c) and averaged ChIP-seq signals (d) of histone acetylation across ±5 kb from TSS in JURKAT cells carrying either WT or the W2554G homozygous mutation of BAHCC1BAH.
e, ChIP-seq profiles of histone acetylation at the indicated gene in JURKAT cells harboring WT or the W2554G homozygous mutation of BAHCC1BAH.
f, ChIP-qPCR of histone acetylation at the indicated gene promoter in JURKAT cells that carry WT or the Y2533A homozygous mutation of BAHCC1BAH (n = 3 biologically independent samples). Data are presented as mean ± SD. ** P < 0.01; *** P < 0.001.
g-h, RT-qPCR of H3K27me3-marked genes post-depletion of HDAC1 (g) or SAP30BP (h) in JURKAT cells, compared to mock (n = 3 biologically independent samples). Data are presented as mean ± SD. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
i-j, Proliferation of JURKAT cells post-depletion of HDAC1 (i) or SAP30BP (j), compared to mock (n = 3 biologically independent experiments). Data are presented as mean ± SD. ** P < 0.01; *** P < 0.001.
An H3K27me3-engagement-defective mutant of Bahcc1 increases neonatal lethality
Next, we queried whether or not BAHCC1BAH functions in a biological system other than human leukemia. Towards this end, we generated genetically modified mice that harbor a germline mutation of Bahcc1Y2537A (Extended Data Fig. 9h), which is equivalent to human BAHCC1Y2533A (Fig. 3c). Breeding of heterozygous mice gave rise to all expected genotypes; however, about 23% of pups with the homozygous mutation of Bahcc1Y2537A died at birth, whereas lethality was not seen in their WT or heterozygous littermates (Fig. 7a). At birth, the average body weight of Bahcc1Y2537A homozygous mice was found to be significantly lower, compared to control (Extended Data Fig. 9i). These observed phenotypes of partially penetrant neonatal lethality and mild dwarfism indicate that BAHCC1BAH is involved in embryonic development. Furthermore, we assessed whether the introduced Bahcc1Y2537A mutation affects hematopoiesis in adult mice and found a lack of apparent alterations in all examined hematopoietic cell compartments in the bone marrow and spleen, including various examined committed lineages (Supplementary Fig. 4a–b) or HSPCs (Supplementary Fig. 4c). This finding indicates that BAHCC1 is largely dispensable for adult hematopoiesis.
Fig. 7. BAHCC1 and PRC1 corepress the H3K27me3-marked genes in cells of different lineage origin.
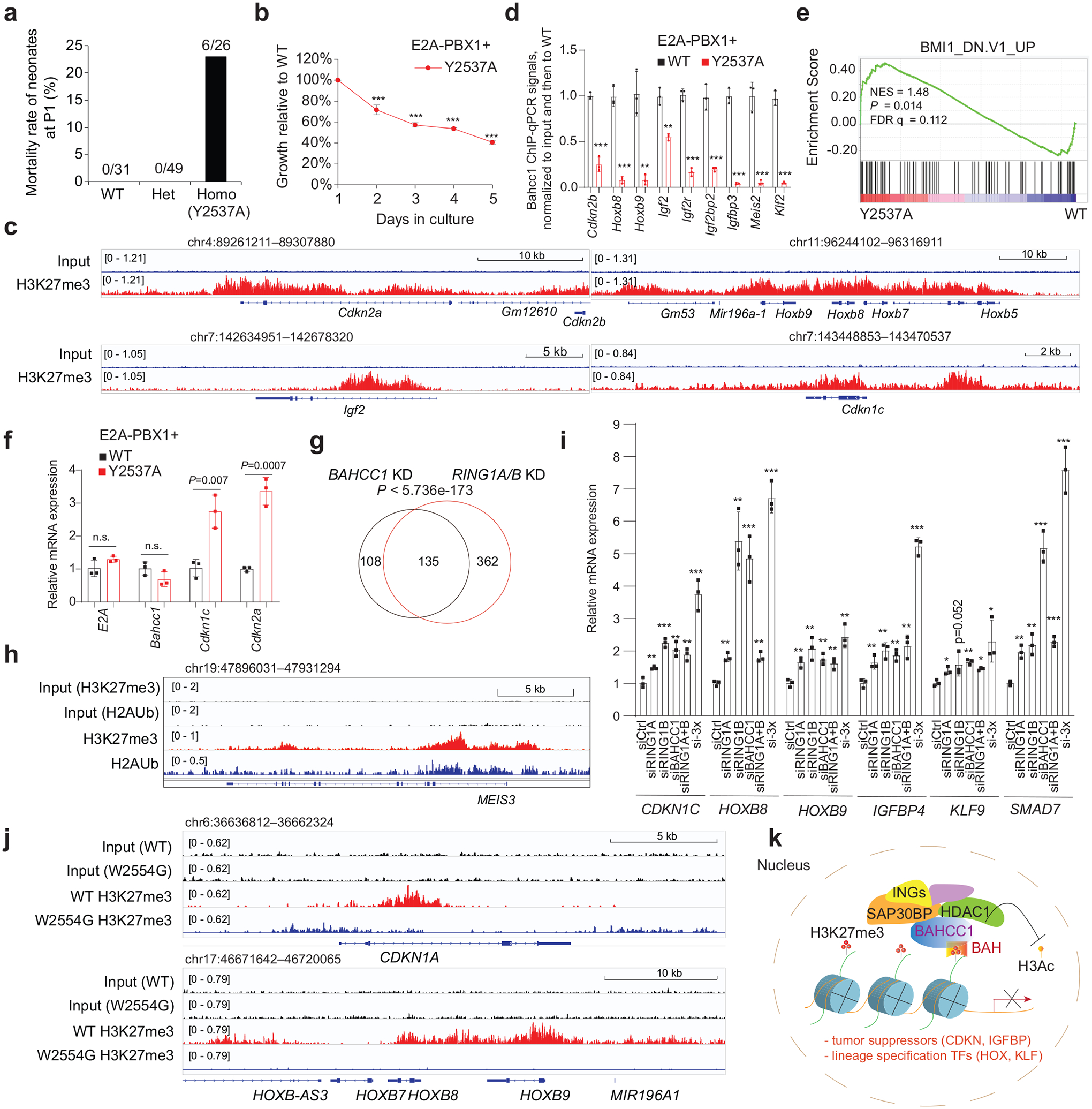
a, Mortality of mouse pups carrying either WT or the Y2537A heterozygous or homozygous mutation of Bahcc1, at day 1 post-birth (P1).
b, Proliferation of E2A-PBX1-transformed murine leukemia with the Y2537A homozygous mutation of Bahcc1BAH, relative to WT (n = 3 biologically independent experiments). Data are presented as mean ± SD. *** P < 0.001
c, H3K27me3 ChIP-seq profiles at classic Polycomb targets in E2A-PBX1-transformed murine leukemia cells.
d, ChIP-qPCR for Bahcc1 binding to TSS of the indicated gene in E2A-PBX1-transformed murine leukemia cells with WT or the Y2537A homozygous mutation of Bahcc1BAH (n = 3 independent experiments, with data plotted as mean ± SD after normalization to input and to WT). ** P < 0.01; *** P < 0.001.
e, GSEA reveals that, relative to WT, the H3K27me3-binding-defective mutation (Y2537A) of Bahcc1BAH is positively correlated to derepression of Polycomb genes.
f, RT-qPCR of E2A-PBX1, Bahcc1 and H3K27me3-targeted genes in E2A-PBX1-transformed murine leukemia cells with WT or the Y2537A homozygous mutations of Bahcc1BAH (n = 3 biologically independent samples). Data are presented as mean ± SD.
g, Venn diagram shows overlapping of DEGs upregulated post-depletion of BAHCC1 (black) or PRC1 (RING1A/1B; red), relative to mock, in 293 cells.
h, H3K27me3 and H2Aub ChIP-seq profiles at MEIS3, a classic Polycomb target, in 293 cells.
i, RT-qPCR of H3K27me3-targeted genes in 293 cells post-KD of RING1A, RING1B, BAHCC1, RING1A plus RING1B, or all three genes (si-3×), relative to mock (siCtrl). n = 3 biologically independent samples. Data are presented as mean ± SD. * P < 0.05; ** P < 0.01; *** P < 0.001.
j, H3K27me3 ChIP-seq profiles at Polycomb targets in JURKAT cells with WT or the W2554G homozygous mutation of BAHCC1BAH.
k, Scheme showing that a previously unappreciated H3K27me3-transduction pathway, which functions through an H3K27me3-specific ‘reader’ module of BAHCC1BAH and BAHCC1-associated corepressors, exists in mammals for silencing the H3K27me3-bound targets, implicative of a new mechanism underlying the H3K27me3 readout. Of equal importance, such a process is crucially involved in oncogenesis.
Given that BAHCC1 overexpression is associated with acute leukemias with genetic abnormalities such as E2A-PBX1 fusion (Extended Data Fig. 1f), we retrovirally transduced E2A-PBX1 into murine HSPCs isolated from WT or Bahcc1Y2537A-mutated mice and established acute leukemia cells as described before41 (Extended Data Fig. 9j). We found that the Bahcc1Y2537A mutation interfered with proliferation and colony-forming capabilities of the E2A-PBX1-transformed murine leukemias (Fig. 7b and Extended Data Fig. 9k–l) without affecting differentiation (Extended Data Fig. 9j). ChIP-seq with E2A-PBX1-transformed leukemia cells revealed the H3K27me3-demarcated genes, which included transcription factors such as Hox and tumor suppressors such as Cdkn2a/b (Fig. 7c and Extended Data Fig. 9m), and the Bahcc1 occupancy at these genes was severely impaired by the Y2537A mutation (Fig. 7d and Extended Data Fig. 9n). GO and GSEA analyses of the RNA-seq profiling of these leukemia cells further revealed that, relative to WT, the Bahcc1Y2537A mutation is positively correlated with the reactivation of genesets associated with development (Supplementary Fig. 5a) and Polycomb or HDAC corepressors (Fig. 7e, Extended Data Fig. 9o and Supplementary Table 7). RT-qPCR further confirmed that, relative to WT, the Bahcc1Y2537A mutation led to de-repression of Cdkn1c and Cdkn2a, two classic Polycomb targets, in murine leukemia cells (Fig. 7f).
BAHCC1 and PRC1 co-repress H3K27me3-marked genes in different cell lineages
Given that PRC1 is known to mediate repression of H3K27me3 target genes, we have also carried out KD of RING1B, a catalytic subunit of PRC1, in JURKAT cells and observed significant reactivation of targets repressed by BAHCC1 and H3K27me3 such as CDKN1C, IGFBP4 and HOXB8 (Extended Data Fig. 10a). Furthermore, we turned to 293 cells, a common cell line of embryonic kidney origin, and performed RNA-seq profiling post-KD of BAHCC1 or the essential subunit of PRC1 (Supplementary Table 8). Genes repressed by BAHCC1 and those by PRC1 displayed a significant overlap (Fig. 7g), among which include classical Polycomb targets demarcated by both H3K27me3 and H2Aub (Fig. 7h; Extended Data Fig. 10b and Supplementary Fig. 5b). Furthermore, we performed KD of either BAHCC1 alone, RING1A plus RING1B, or all three in 293 cells, and subsequent RT-qPCR of several H3K27me3-targeted genes revealed that BAHCC1 and PRC1 likely act cooperatively in repressing their target genes (Extended Data Fig. 10c and Fig. 7i). To examine chromatin alteration post-KD of BAHCC1 in 293 cells, we performed a time-course study of gene expression and histone modification changes and found that, concurrent with elevated expression of H3K27me3-targeted genes (Extended Data Fig. 10d–e), local levels of acetylated histone H3 were increased and H3K27me3 reduced simultaneously following KD of BAHCC1 (Extended Data Fig. 10f–g). Consistently, H3K27me3 ChIP-seq in JURKAT cells with the W2554G-mutated BAHCC1 showed similarly decreased levels of H3K27me3 at genes de-repressed due to this mutation of BAHCC1BAH (Fig. 7j and Extended Data Fig. 10h). Altogether, we show that BAHCC1 and PRC1 can co-repress H3K27me3-targeted genes in cell types of different developmental lineages (JURKAT and 293 cells), further supporting a generalized functionality of BAHCC1 in mammalian cells.
Discussion
H3K27me3 strongly associates with a repressed state of gene transcription and plays a critical role in many biological processes, including lineage specification, cell-fate determination, cell-cycle control and senescence. Functional transduction of H3K27me3 in mammals has been believed to be achieved largely through the CBX subunit of PRC1, a class of H3K27me3 ‘readers’ extensively investigated by numerous studies31–33,42–45. Our biochemical and structural studies identified BAHCC1BAH as a specific and direct ‘reader’ of H3K27me3, with a binding affinity comparable to CBX7 (Fig. 2d–f). Comparison of the BAHCC1BAH-H3K27me3 complex with the previously determined CBX7CD-H3K27me3 complex31 reveals similar, but distinct patterns of intermolecular interactions. Notably, CBX7CD presents a shallow surface groove, formed by four aromatic residues (F11, W32, W35 and Y39), to harbor the side chain of H3K27me3 (Extended Data Fig. 10i versus Fig. 3b–c). In contrast, BAHCC1BAH forms a deep pocket with mixed aromatic and acidic residues, recognizing H3K27me3 via a cavity insertion mechanism46. Furthermore, CBX7CD engages sequence-specific interactions mainly with the residues upstream of H3K27me3 whereas BAHCC1BAH recognizes the residues from both N- and C-terminal to H3K27me3. These differences highlight a divergent H3K27me3-recognition mechanism between CBXCD and BAHCC1BAH. It is worth noting that the H3K27me3-binding interface of BAHCC1BAH resembles the H4K20me2-binding site of ORC1BAH (Extended Data Fig. 10j), although the two complexes involve distinct sequence-specific interactions for histone recognition.
Using the integrated genomic profiling and CRISPR/Cas9-based editing approaches, we demonstrated that BAHCC1BAH functions to facilitate binding of BAHCC1 to H3K27me3-marked loci to enforce gene silencing. BAHCC1 interacts with transcriptional corepressors such as HDACs and SAP30BP, providing a mechanism for ensuring a hypoacetylated chromatin state seen at the H3K27me3-targeted genes (see Fig. 7k for a model). Previously, Zhao et al. reported that the BAH domain of BAHD1 binds H3K27me3 in an in vitro assay47 but the gene-regulatory role of this BAH domain remains undetermined and warrants further investigation. Additionally, two plant-specific proteins SHL48 and EBS49,50 were shown to engage H3K27me3 via an embedded BAH ‘reader’; thus, the BAH-directed H3K27me3 ‘readout’ mechanism appears conserved across plant and animal species. Altogether, this study challenges the current norm that tends to emphasize CBX within PRC1 as the sole/main effector of H3K27me3 in mammals. It is conceivable that, among different cell types, BAHCC1BAH and CBXCD may have both common and differential effects for mediating repression of the H3K27me3-targeted genes, the details of which requires more investigation in future.
Molecular players related to H3K27me3 are frequently found to be associated with pathogenesis, notably cancer13. Hyperactivation of PRC2 due to gain-of-function mutation or overexpression of EZH2 is a recurrent event in B-cell lymphoma and solid cancer, respectively. Our initial interest towards BAHCC1 stems from its overexpression pattern across several subtypes of acute leukemias showing poor outcomes. Here, we further demonstrated the tumor-promoting function of BAHCC1 in acute leukemia by using a set of in vitro and in vivo models. In particular, the BAHCC1BAH-mediated recognition of H3K27me3 is crucial for optimal silencing of gene-expression programs intimately associated with tumor growth and aggressiveness, such as cell cycle inhibitors (CDKNs), and transcripts related to terminal differentiation. Targeting excessive H3K27me3-associated signaling represents an attractive strategy for treatment of the affected cancers, as exemplified by the EZH2 catalytic inhibitors currently under clinical evaluation13. BAHCC1BAH-directed ‘readout’ of H3K27me3 is crucially involved in sustaining leukemogenesis, implicating BAHCC1BAH to be a valuable drug target site. Further investigation is warranted to develop inhibitors of BAHCC1BAH as a potential strategy for suppressing H3K27me3-enforced oncogenic programs in cancer. It is noteworthy that PRC2 and EZH2 can act as oncogene or tumor suppressor under different biological contexts10–13,35,51–57; additionally, EZH2 can interact with non-PRC2 partners such as the androgen receptor in advanced prostate cancer58. A detailed understanding of the functional cooperation between BAHCC1 and various Polycomb complexes requires careful and systematic studies in future.
Methods
Analysis of public transcriptome datasets of cancer samples.
The publicly available gene expression profiling datasets used in the study for examination of BAHCC1 (also known as KIAA1447 or BAHD2) included the Cancer Cell Line Encyclopedia (CCLE) data59, the NCBI Gene Expression Omnibus (GEO) datasets of acute leukemia patients under accession numbers GSE1159, GSE24505, GSE13204, GSE33315, GSE7186, GSE28460 and GSE34861, and dataset of the St Jude Hospital “Pediatric Acute Lymphoblastic Leukemia (ALL)” cohort (http://www.stjuderesearch.org/data/ALL3).
Cell lines and tissue culture.
Cell lines used in the study included HEK293 (ATCC #CRL-1573), HEK293T (ATCC #CRL-3216), HeLa (ATCC #CCL-2) and a panel of human leukemia cell lines, which cover AML (MV4;11, ATCC #CRL-9591; OCI-AML3, a gift of M. Minden [Princess Margaret Hospital, Canada]), B-ALL (RS4;11, ATCC #CRL-1873), T-ALL (JURKAT, ATCC #TIB-152; CCRF-CEM, ATCC #CCL-119; MOLT4, ATCC #CRL-1582; ALL-SIL, DSMZ # ACC 511; CUTLL1, a kind gift of P. Ntziachristos [Northwestern University]) and CML (K562, ATCC #CCL-243). All cell lines were maintained using vendor-recommended culture conditions. A luciferase-expressing line of JURKAT cells (a gift of M. Brenner, Baylor College of Medicine) was maintained as described before60. Authentication of cell line identities, including parental and derived lines, was ensured by the Tissue Culture Facility affiliated to UNC Lineberger Comprehensive Cancer Center with the genetic signature profiling and fingerprinting analysis61. A routine examination of cell lines in culture for any possible mycoplasma contamination was performed every month with detection kits (Lonza). Cells with less than of 10 times of passages were used in this study.
Plasmids.
The pLKO.1-Puro based shRNA plasmids for knockdown (KD) of BAHCC1 (human or mouse) and HDAC1 were purchased from Sigma, with the detailed information and shRNA stem sequence listed in Supplementary Table 9. A BAHCC1 cDNA clone (NCBI Reference Sequence: NM_001291324.1) was obtained from Open Biosystems. The BAH domain of BAHCC1 was cloned into pGEX-4T1 (GE Healthcare Life Science) by PCR. Point mutation was generated using a site-directed mutagenesis kit (Stratagene). A pGEX-6P1-based GST-fusion ORC1 BAH domain construct was previously descibed30. A cDNA clone of SAP30BP (NM_013260) was obtained from the human cDNA library ORFeome version 8.1, followed by in-frame fusion with 3×HA tag by PCR and subsequent cloning into an MSCV-based retroviral expression vector (Clontech). Plasmids used for CRISPR/Cas9-based genome editing include a Flag-epitope-tag-ChIP (pFETCh)27-based donor vector (Addgene, cat#63934), a Cas9-containing PX458 plasmid (pSpCas9_BB_2A-GFP; Addgene, cat#48138), a dCas9-KRAB plasmid (pLV_hU6-sgRNA_hUbC-dCas9-KRAB-T2a-Puro; Addgene, cat#71236), a pcDNA-dCas9-p300 Core plasmid (Addgene, cat #61357), pcDNA-dCas9-p300 Core mutant (D1399Y) (Addgene, cat#61358) and lentiGuide-Puro (Addgene, cat#52963). All plasmids were confirmed by Sanger sequencing before use. Primer sequences information used for cloning are listed in Supplementary Table 9.
Viral production, stable cell line generation and siRNA transfection.
Packaging of virus and generation of stable cell lines were performed as previously described35,62. The cells were transfected with siRNA using lipofectamine RNAiMAX according to manufacturer instructions. ON-TARGETplus Human BAHCC1 (57597) siRNA SMARTpool (#L-023331-02-0005) was ordered from Dharmacon. Human Silencer™ Select Pre-Designed siRNA RING1A (siRNA ID: s12036) and RING1B (siRNA ID: s12068) were ordered from Thermo Fisher Scientific.
Purification, Retroviral Transduction, and Cultivation of Primary Murine Hematopoietic Stem/Progenitor Cells (HSPCs)
Primary mouse HSPC cells were enriched by using a previously described lineage-negative enrichment protocol (Miltenyi Biotech), followed by cultivation and viral transduction as previously described62–64.
Cell proliferation and colony-forming unit (CFU) assays.
Measurements of proliferation by direct cell number counting were previously described35,62. CFU assays with mouse HSPC cells were carried out by using a semi-solid cultivation medium system according to vendor’s specifications (StemCell Tech., MethoCult cat #M3434). For CFU assay with human leukemia cell lines, 5,000 or 10,000 cells per well were plated in triplicate in 6-well plates that contain the complete growth medium supplemented with 0.3% and 0.5% of melted soft agar, respectively, in the upper and bottom layers. To prevent desiccation, 1 ml of complete growth medium was added on top of cultures. After cultivation for 2 weeks, colonies were stained with iodonitrotetrazolium chloride (Sigma), followed by direct quantification of colony numbers in each well.
Cell cycle progression analysis.
Staining of cells with Propidium Iodide (PI; Sigma) was used for quantifying the DNA content and analyzing cell cycle progression. In brief, cells were fixed in 80% of ice-cold methanol and incubated at −20°C for overnight. Methanol was decanted completely and cells were resuspended in 2–3 ml of PBS with 0.5% of BSA for 5 minutes. After centrifugation, cell pellets were resuspended in 0.4–0.5 ml of freshly prepared staining solution (0.1% Triton X-100, 200 μg/ml RNase A and 20 μg/ml PI in PBS). Cells, covered with foil, were kept at room temperature for 30 min, followed by flow cytometry analysis. DNA content was examined using a Cyan flow cytometer. Data were collected and analyzed using FlowJo and ModFit software.
In vivo growth of tumor cell xenografted models.
All animal experiments were approved by and performed in accordance with the guidelines of the University of North Carolina (UNC) Institutional Animal Care and Use Committee (IACUC). NOD/SCID/IL2Rgamma-null (NSG) mice were purchased from Jax Lab and maintained by the Animal Studies Core affiliated to the UNC Lineberger Comprehensive Cancer Center. Mice were housed in a germ-free environment with food and tap water ad libitum. Room temperature and relative humidity were held at 22° ± 2° C and 30–70% respectively. Automatic light control guaranteed a 12-hour light and 12-hour darkness cycle (7am-7pm/7pm-7am). Three million of JURKAT T-ALL cells with BAHCC1 KD, or one million of JURKAT cells with mutation of BAHCC1, were transplanted via tail-vein injection into each of the sub-lethally irradiated (200 rads) NSG recipient mice (age of 8 weeks). K562 cell xenografts were established by subcutaneously injecting three million of cells, which were resuspended in 200 μl of high concentration matrigel matrix (50 million cells/ml, BD cat #354248), into both flanks of the NSG mice. Mice were monitored and sizes of subcutaneously xenografted tumors measured every other day. Bioluminescence imaging of live mice xenografted with tumor cells stably expressing a luciferase reporter was performed as described before65. The investigators were blinded to allocation of mice and their outcome assessment (carried out by Animal Studies Core, UNC Cancer Center).
Wright-Giemsa Staining.
Wright-Giemsa staining of leukemia cells, prepared on glass slides, was carried out as described before35,63. Images of cell staining were captured with an EVOS-XL Cell Imaging System (Life Technologies).
CRISPR/Cas9-based editing of BAHCC1.
A previously described approach termed as pFETCh27 was utilized to introduce a 3×Flag tag in-frame to the C-terminus of BAHCC1 in cells. Briefly, two homology arms located before and after the stop codon of BAHCC1 were synthesized as dsDNA genomic blocks (gBlocks; ordered from Integrated DNA Technologies Inc., with the sequence information provided in Supplementary Table 9), followed by cloning into a 3×Flag-P2A-Neomycin (Neo) cassette-containing pFETCh donor vector (Addgene, #63934) by the Gibson Assembly method (New England BioLabs), which produced the pFETCh-BAHCC1-×Flag targeting vector. CRISPR guide RNAs (gRNAs) were designed using an online tool (http://chopchop.cbu.uib.no/) to target genomic sites in close proximity to the stop codon of BAHCC1 (the sequence information of sgRNAs provided in Supplementary Table 9) and subsequently cloned into a Cas9-containing pSpCas9_BB_2A-GFP plasmid (Addgene, #48138). After co-transfection of both pFETCh-BAHCC1–3×Flag and gRNA-containing PX458 plasmids by lipofectamine 2000 (Invitrogen), cells were subjected to drug selection with 300 μg/ml of G418 for two weeks. The produced drug-resistant cells were split into 96-well plates to establish clonal lines for genotyping. As previously recommended27, different sets of genotyping primers that span either 5’- or 3’-homologous recombination site within the BAHCC1 genomic regions were designed for detection of the desired homologous recombination events; as well, the homology arm-specific primer sets that span the 3×Flag-P2A-Neo cassette were used to assess homozygous versus heterozygous insertion (the sequence information of genotyping primers provided in Supplementary Table 9). As shown in Extended Data Figure 1i, correct events of homologous recombination give rise to the amplicon with a size of 1,451 and 943 bp by using 5’-end (red) and 3’-end (blue) genotyping primer sets, respectively; two additional genotyping primer sets spanning over the knockin (KI) cassette, i.e. Middle-1 (purple) and Middle-2 (green), produce the 1,367-bp and 1,380-bp amplicons, respectively, and are used to differentiate heterozygous versus homozygous KI. Direct Sanger sequencing of all genotyping amplicons, with gDNA or cDNA as PCR template, and Flag immunoblotting were carried out to ensure accuracy of homologous recombination for the used BAHCC1–3×Flag KI lines. Independently derived lines were used for subsequent analysis.
To carry out the CRISPR/Cas9-based protein domain scanning of BAHCC1 in leukemia cells, we first established clonal JURKAT cell lines that stably expressed Cas9 with the previously described system28, and cell clones showing optimal efficiency of Cas9-mediated gene editing identified by using sgRNAs that target essential genes (i.e., BRD4 and RPA3), compared to those targeting the non-essential ROSA26 locus, as described before28. To generate the BAHCC1-targeted sgRNA library, we used a previously described protocol with the BAHCC1 cDNA fragments isolated from JURKAT cells by PCR cloning29. JURKAT cells post-transduction of the BAHCC1-targeted sgRNA viral library were collected at day 1 and day 10, followed by PCR-based amplification of the sgRNA-containing genomic regions and subsequent deep sequencing as described29. Quantification of sgRNA distribution and plotting of sgRNA enrichment, as well as individual sgRNA-based editing and verification, were carried out as described before28,29. In brief, we split the BAHCC1 coding region into 20-bp bins, followed by maximum-likelihood analysis of essentiality of each bin as assessed by the beta-value and P value using by the “Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout” (MAGeCK) module66.
Quantitative RT-PCR (RT-qPCR).
The real-time PCR following reverse transcription (RT) of total RNA was carried out as previously described67. In brief, 1 μg of total RNA was extracted with the RNeasy plus kit (Qiagen) and reverse transcription carried out by using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to provider’s protocols. Quantitative PCR was run in triplicate using the SYBR green master mix reagent (BioRad) on an ABI 7900HT fast real-time PCR system (ABI). Data from at least three independent experiments are presented as mean ± standard deviation (SD) after normalization. The information of primers used for RT-qPCR was provided in Supplementary Table 9.
Recombinant Glutathione S-Transferase (GST) Protein Production.
GST-fusion proteins were generated and purified as previously described64,67.
Antibodies.
The information of antibodies used for immunoblotting, immunofluoresence, CoIP and ChIP was listed in Supplementary Table 9. To generate antibodies against human BAHCC1, we first chose the amino acids 1200–1350 of BAHCC1 (NCBI accession # NP_001278253.1; the sequence information listed in Supplementary Table 9) as immunogen due to a predicted specificity and surface presentation of this segment. PCR-based amplification of this segment was carried out using total cDNAs of OCI-AML3 leukemia cells, following by cloning into a bacterial expression vector pGEX-6P2 (GE Healthcare Life Science). The GST-BAHCC1 fusion recombinant protein was subsequently expressed in the BL21 Escherichia coli (Novagen) and purified with the glutathione beads as described before64,67. The purified protein was used as immunogen in rabbits (1 mg of purified protein per rabbit) followed by the ELISA-based examination of bleeds and affinity purification (carried out by Pocono Laboratory Inc.).
Immunoblotting.
Total protein samples were prepared with the NP40 lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% of IGEPAL® CA-630) followed by brief sonication of cells and centrifugation. Same amounts of extracted samples were loaded into SDS-PAGE gels for immunoblotting analysis.
Immunofluoresence.
For immunofluoresence, cells were fixed in 4% of paraformaldehyde for 15 min at room temperature, followed by incubation in PBS containing 0.25% of Triton X-100 for 10 min to permeabilize the cells. Fixed cells were first stained with primary antibodies and then with Alexa-488/594 conjugated secondary antibodies (Life Technologies) as described before67. Nuclei were stained with 4, 6-diamino-2-phenylindole (DAPI, 0.1 μg/ml). Fluorescence signal was detected by using the FV1000 confocal microscope (Olympus, UNC Imaging Core).
Cell fractionation
Whole cell lysates were fractioned into soluble nucleoplasmic and chromatin-bound fractions as previously described68 with the following modifications. In brief, cells were lysed on ice for 30 min in the lysis buffer containing 10 mM PIPES (pH 7.0), 300 mM sucrose, 200 mM NaCl, 3 mM MgCl2, 0.5% Triton X-100 and 1× EDTA-free protease inhibitor (Roche), followed by centrifugation at 1,300 × g for 5 minutes at 4 °C to separate the supernatant and the pellet, which represents soluble and chromatin-bound fractions, respectively68. Lysates prepared from the equal numbers of cells were loaded on SDS-PAGE for western blot analysis. Histone H3 and β-tubulin antibodies were used as immunoblotting controls for monitoring the purity of fractionation.
Protein expression and purification for crystallization.
A cDNA fragment encoding the C-terminal residues 2,485–2,643 or 2,460–2,643 of murine BAHCC1, encompassing the BAH domain (mBAHCC1BAH), was inserted into a modified pRSF-Duet vector preceded by an N-terminal hexahistidine (His6)-SUMO tag and an ubiquitin-like protease 1 (ULP1) cleavage site. BL21(DE3) RIL cells harboring the expression plasmids were induced by addition of 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) when the cell density reached optical density at 600 nm (A600) of 0.8 and continued to grow at 16 °C overnight. The cells were harvested and lysed in buffer containing 50 mM Tris-HCl (pH 8.0), 1 M NaCl, 25 mM Imidazole, 10% glycerol and 1 mM PMSF. Subsequently, the fusion proteins were purified through a nickel column, followed by removal of His6-SUMO tag by ULP1 cleavage, ion-exchange chromatography and size-exclusion chromatography. The purified protein samples were concentrated in 25 mM Tris-HCl (pH 7.5), 100 mM NaCl, 5% glycerol and 5 mM DTT, and stored at −80 °C freezer. The mBAHCC1BAH domain mutants were constructed by site-directed mutagenesis and purified in the same way as described above.
Histone peptide arrays.
Probing protein binding with a histone peptide array platform (Active Motif # 13005) was carried out according to the manufacturer’s specifications with slight modifications described before64,67. Briefly, after a short blocking with 5% of milk in TBST (150 mM NaCl, 2 mM KCl, 25 mM Tris pH 7.4) for 1hr at RT, the peptide array slides were incubated with GST-BAHCC1BAH proteins in the binding buffer (TBST, 1% milk, 0.05% NP40, 0.1 mM DTT, 0.1 mM PMSF and protease inhibitor cocktail (Roche)) overnight at 4°C with gentle agitation. After extensive washing with TBST, the slides were probed with HRP-conjugated anti-GST antibodies (GeneTex) and visualized with ImageQuant LAS 4000 Luminescent Image Analyzer (GE Healthcare).
Biotinylated peptide pull-down.
Pull-down assays using the biotinylated histone peptide were conducted as previously described with the following modifications64,67. Biotin-conjugated histone peptides were incubated for 4 hrs at room temperature with high-capacity neutral Avidin resin (Pierce), followed by extensive washing to eliminate unbound peptide. 20 μl of such prepared peptide-Avidin resin complexes were incubated with 20 μg of the purified GST-BAHCC1BAH protein, either wildtype (WT) or BAH-mutated, in a buffer containing 20 mM Hepes (pH 7.9), 200 mM KCl, 0.05% IGEPAL® CA-630, 5% glycerol, 1 mM DTT, 0.1 mM PMSF and protease inhibitor cocktail (Roche). Similarly, the same amount of peptide-Avidin resin was typically used to pulldown with total cell extracts that contain the candidate proteins. After extensive washing, proteins bound to resin were separated by SDS-PAGE, followed by silver staining or western blotting, as described before64,67.
Crystallization and data collection.
For crystallization, 2 mM mBAHCC1BAH (residues 2,485–2,643) was mixed with the H321–33K27me3 peptide, with a C-terminal tyrosine, in a molar ratio of 1:3, before incubated with 1.6–1.8 M NaH2PO4/K2HPO4 (pH 5.4) using the hanging drop vapor-diffusion method at 4 °C. The crystals were soaked in the well solution supplemented with 25% (v/v) glycerol before flash frozen in liquid nitrogen. X-ray diffraction data were collected on beamlines 5.0.1 at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory. The datasets were processed with the HKL2000 program69. The structure of the mBAHCC1BAH-H3K27me3 complex was solved by molecular replacement with PHENIX PHASER module70 using the mouse ORC1BAH structure (PDB:4DOV) as a search model. Iterative cycles of model rebuilding and refinement were carried out using COOT (v0.8.9.2)71 and PHENIX (1.52.2_3472)72, respectively. The statistics for data processing and structure refinements are summarized in Supplementary Table 1.
ITC measurements.
Murine BAHCC1BAH (residues 2,460–2,643) and peptides were dialyzed at 4°C overnight against a buffer containing 25 mM Tris-HCl (pH 7.5) and 100 mM NaCl. The final concentrations, determined based on ultraviolet absorption at 280 nm, were 0.2 mM for mBAHCC1BAH and 2 mM for peptides. A MicroCal iTC200 system (GE Healthcare) was used to conduct the ITC measurements. A total of 15~17 injections with a spacing of 180 second and a reference power of 5 μcal/s were performed after the temperature was equilibrated to 7°C. The ITC curves were processed with software ORIGEN (MicroCal) using one-site fitting model as described before64,67.
CRISPR/Cas9-based genome editing for site-specifically mutating BAHCC1BAH.
The pFETCh-based approach27 was used to introduce point mutation into the C-terminus BAH domain of BAHCC1 in cells. Here, the pFETCh-BAHCC1–3×Flag plasmid was generated using the same strategy as described above, except that the synthesized homology arm-1 gBlock (IDT Inc.) carries a desired mutation (such as W2554G). Plasmid transfection, drug selection and genotyping were performed as described above. Homozygous mutation was verified by direct Sanger sequencing of genotyping PCR products. In addition, an independent method using the Alt-R® CRISPR-Cas9 System (IDT) in combination with a single-stranded oligodeoxynucleotide donor (ssODN) was used to introduce point mutation into the BAH domain of BAHCC1. For Alt-R® CRISPR-Cas9 System, the trans-activating CRISPR RNA (tracrRNA) with ATTO™ 550, crRNA targeting the site to be mutated (such as Y2533; the sequence information of crRNA listed in Supplementary Table 9, the S.p. HiFi Cas9 Nuclease V3 and Electroporation Enhancer were all ordered through IDT Inc.. The ssODN was custom designed (the sequence information of ssODN listed in Supplementary Table 9; using https://benchling.com/crispr) and then ordered as the PAGE-purified Ultramer® DNA oligonucleotides from commercial sources (IDT). Three phosphorothioate bonds were added at 5’ and 3’ ends of ssODN to both stabilize the donor oligo and make homology-directed repair (HDR) more efficient. To prevent re-cutting the target sequence after the desired point mutation is introduced, crRNA seed sequence is mutated in the position 17 nt upstream of the PAM. For Y2533A mutation, this position was mutated from TAC to GCG, generating a PvuI cut site to assist with genotyping. Preparation and delivery of a CRISPR-Cas9 ribonucleoprotein (RNP) complex (Alt-R® CRISPR-Cas9 crRNA:tracrRNA and S.p. HiFi Cas9 Nuclease), Electroporation Enhancer and ssODN into cells were performed according to the manufacturer’s user guide. In brief, crRNA and tracrRNA were mixed in equimolar concentration, followed by heating at 95°C for 5 min. Then the mixture was cooled down to room temperature (15–25°C) on the bench top, allowing to form the crRNA:tracrRNA duplex. RNP complex was made by diluting the crRNA:tracrRNA duplex and Cas9 enzyme components in PBS, followed by incubation at room temperature for 10–20 minutes. The RNP complex mixed with Electroporation Enhancer and ssODN was delivered to cells using electroporation with Amaxa Cell Line Nucleofector Kit V (Lonza). Thirty-six hours after electroporation, ATTO™ 550 positive cells were sorted out by FACS (UNC flow core) and split into 96-well plates to establish clonal lines for PCR and sequencing-based genotyping. After genotyping, lines with homozygous mutation were further validated at both DNA and RNA levels by direct sequencing of PCR products that cover the mutation site.
Chemical compound.
The enzymatic inhibitor of EZH2 and EZH1, UNC1999, was reported previously35,36. UNC1999 was dissolved in dimethylsulfoxide (DMSO) as 5 mM of stocks and a three-day treatment with 5 μM of UNC1999 was used in this study. Two HDAC inhibitors, Cl-994 (Tacedinaline, Sigma) and Entinostat (Selleckchem), were used according to vendor’s specifications, with a final concentration of 5 μM and 0.25 μM utilized for cell treatment studies, respectively.
Mass spectrometry–based analysis of protein complexes.
As previously described67, nuclear extracts were prepared using the Dignam protocol from the nuclei of HeLa stable cells carrying the BAHCC1–3×Flag-KI alleles, with non-tagged parental HeLa cells used as negative control. Following dialysis against low salt buffer (150 mM NaCl, 20 mM Hepes, pH 7.90, 0.025% IGEPAL® CA-630, 0.2 mM EDTA, 10% glycerol, 1 mM DTT, 0.2 mM PMSF), nuclear extracts were centrifuged at high speed to remove precipitation and supernatants incubated with Flag beads (Sigma; M2). After extensive washing, resin and bound proteins from both experimental and negative control cells were submitted to the Proteomics Core facility, UNC School of Medicine for mass spectrometry-based protein identification. Peptides identified are listed in Supplementary Table 10.
Co-immunoprecipitation (CoIP).
Total protein samples were prepared by NP40 lysis buffer (50mM Tris-HCl pH 8.0, 150mM NaCl, 1% IGEPAL® CA-630) followed by brief sonication and high-speed centrifugation. Extracted samples were incubated with antibodies on a rotator overnight at 4 degree. Dynabeads (Invitrogen) were added for additional 2 hrs. Beads were washed three times in NP40 lysis buffer, resuspended in 50 μl of 2× sample loading buffer, and boiled at 90 degree for 5 min before loading onto SDS-PAGE gel. Western blotting was performed with standard protocols using the PVDF membrane, and signals were visualized with ImageQuant LAS 4000 Luminescent Image Analyzer (GE Healthcare).
RNA sequencing (RNA-seq) and Data Analysis.
RNA-seq was performed with a previously described protocol65,73. In brief, total RNA was extracted using the RNeasy Mini Kit (QIAGEN) and then prepared using the TruSeq RNA Sample Preparation Kit v2 (Illumina) as previously described73. The multiplexed RNA-seq libraries were subjected for deep sequencing using the Illumina Hi-Seq 2500 or 4000 platform (UNC High-throughput Sequencing Facility). FastQC was used for quality control of high-throughput raw sequence data. For data analysis, RNA-seq reads were mapped using MapSplice73 and quantified using RSEM73. Read counts were upper-quantile normalized and log2 transformed. Raw read counts were used for differential gene expression analysis by DESeq2 (v1.22)73.
Gene Ontology (GO), Ingenuity pathway analysis (IPA), Gene Set Enrichment Analysis (GSEA) and Platform for Integrative Analysis of Omics (Piano) analysis.
GO, IPA and GSEA analyses were conducted as previously described62,73,74. In brief, Panther Classification System75 (http://www.pantherdb.org) and IPA software (http://www.ingenuity.com) were utilized for GO and IPA analysis, respectively, with the determined DEGs based on RNA-seq profiles. GSEA was carried out using the GSEA 2–2.2.0 software76 for testing enrichment of curated gene sets (C2). Gene sets used were obtained from the Molecular Signatures Database v6.2 (MSigDB, http://www.broadinstitute.org/gsea/msigdb/index.jsp, C2 curated gene sets or C6 oncogenic signatures). Platform for Integrative Analysis of Omics (Piano) was performed as described before34.
Chromatin immunoprecipitation (ChIP) followed by qPCR (ChIP-qPCR).
ChIP-qPCR was performed as previously described63,64. Briefly, cells were cross-linked with 1% of formaldehyde at room temperature for 10 minutes, followed by adding glycine to terminate crosslinking. After washing with cold PBS, cell sonication was conducted by Bioruptor (Diagenode) and the cleared chromatin samples incubated with antibody-conjugated Dynabeads (Invitrogen) overnight at 4 °C. Beads bound with chromatin were then extensively washed and eluted. Eluted chromatin was de-crosslinked overnight at 65 °C, followed by proteinase K digestion and DNA purification with MinElute PCR Purification Kit (QIAGEN). The purified ChIP DNA samples and their input DNA were diluted and used for ChIP-qPCR. ChIP signals were generated from 3 independent experiments followed by normalization to input signals and presented as mean ± SD. The primer sequences used for ChIP-qPCR are available in Supplementary Table 9.
ChIP followed by deep sequencing (ChIP-seq) and data analysis.
ChIP-seq was carried out with a previously described protocol62,65,73. After cell fixation and sonication, antibody enrichment and ChIP DNA recovery, library generation and parallel sequencing were conducted using an Illumina HiSeq-2500/4000 Sequencer (UNC High-throughput Sequencing Facility). FastQC was used for quality control of high throughput raw sequence data. ChIP-Seq data alignment, filtration, peak calling and assignment, and cross-sample comparison were performed as previously described35,62,73 with slight modifications. In brief, Trim galore version 0.6.1 was used for adapter trimming and quality filter for all reads. ChIP-seq reads were aligned to human genome build GRCh37/hg19 using STAR version 2.7.1a77. Samtools (v1.9), Picard MarkDuplicates funtion (ver 2.20.4), and bedtools (v2.28.0) were used to remove the non-primary alignment, PCR duplicates, or blacklist regions from aligned data. MACS2(v2.1.1) was used for peak calling. Deeptools (v3.3.0) was used to make bigwig files, heatmaps, and averaged plottings of ChIP-seq signal78. Genomic binding profiles were generated using the deepTools “bamCompare” functions with options [--operation ratio ---pseudocount 1 -binSize 10 --extendReads 250] and normalized to the matched input. Custom scripts of R(v3.5.0) or Python(v3.6) were used for statistical analysis. For the H3K27me3 profiles of parental JURKAT cells, ChIP-seq data (fastq file) were downloaded from NCBI GEO GSE85601 (Sample Accession # GSM2279072 and run #SRR4031400) and its matched input chromatin was kindly provided by Dr. P. Schaer (University of Basel, Switzerland) and sequenced in this study, followed by alignment and normalization of coverage as described above. The resulting bigWig files were visualized in the Integrative Genome Viewer (IGV). Heatmaps were generated using the deepTools “computeMatrix” and “plotHeatmap” functions. As the regions used for heatmap and averaged signal plotting, the genomic regions covering the transcription start site (TSS) to transcription end site (TES) for all genes annotated in the human genome (NCBI Reference Sequences [RefSeq]; GRCh37/hg19) were obtained from UCSC table browser.
CRISPR/Cas9-based knockin in mice and phenotypic characterization.
Bahcc1Y2537A knock-in mice were generated in the Animal Models Core facility affiliated to UNC School of Medicine by pronuclear microinjection of CRISPR/Cas9 reagents in mouse embryos with a previously described protocol79.Cas9 guide RNAs targeting the mouse Bahcc1 Y2537 codon were identified using Benchling software. Three guide RNAs overlapping the Y2537 codon were selected for activity testing. Selected guide RNAs were cloned into a T7 promoter vector followed by in vitro transcription and spin column purification. Functional testing was performed by transfecting a mouse embryonic fibroblast cell line with guide RNA and recombinant Cas9 protein (UNC Protein Expression and Purification Core Facility). Following transfection, the guide RNA target site in Bahcc1 was PCR amplified from transfected cells and analyzed by T7endo1 assay to detect Cas9-mediated mutation. Guide RNAs Bahcc1-esg85B (protospacer sequence 5’- gCTCGATGCGGCCTATGT −3’; lower case g represents a heterologous guanine added for T7 in vitro transcription) and Bahcc1-esg88B (protospacer sequence 5’- gTCGATGCGGCCTATGTA −3) were selected for micro-injection to produce founder animals. An oligonucleotide donor was designed for precise insertion of the Y2537A (TAC to GCT) mutation (with oligo sequence listed in Supplementary Table 9).
Zygotes of mice were produced by mating superovulated females to stud males. Embryos were collected the morning after mating and microinjected with 400 nM Cas9 protein, 50 ng/μl guide RNA and 50 ng/μl donor oligonucleotide in microinjection buffer (5 mM Tris-HCl pH 7.5, 0.1 mM EDTA). Injected embryos were implanted in recipient pseudopregnant females. Resulting pups were screened by sequencing PCR amplicons for the presence of the knock-in allele. PCR was performed with primers Bahcc1-ScF1and Bahcc1-ScR1 and amplicons were sequenced with Bahcc1-SqF1. Primers sequence are listed in Supplementary Table 9. Microinjection of esg85B yielded 15 animals, with 2 harboring the Y2537A mutation. Microinjection of esg88B yielded 6 animals, with 2 harboring the Y2537A mutation. One positive male founder from each guide RNA microinjection was mated to females for germline transmission of the Y2537A mutation. F1 animals heterozygous for the correct knock-in allele were used for subsequent breeding to establish the knock-in colonies. Colony maintenance, daily care and monitoring of the lines were carried out by the Animal Studies core of UNC Cancer Center. Analysis of various mouse hematopoietic cell compartments in adult mice by flow cytometry was performed as previously described80.
Statistics and reproducibility.
Experimental data are presented as the mean ± SD of three independent experiments unless otherwise noted. Statistical analysis was performed using unpaired two-sided Student’s t-test for comparing two sets of data with assumed normal distribution. We used a log-rank test for Kaplan-Meier survival curves to determine statistical significance; and for in vivo tumor progression studies, a two-sided Student’s t-test was performed to determine the statistical difference of size or imaging scores of tumor xenografts. The results for immunoblotting are representative of at least three biologically independent experiments. All statistical analyses and visualization were performed by using GraphPad (Prism v8.4.2). P < 0.05 was considered significant. Statistical significance levels are denoted as follows: * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. No statistical methods were used to predetermine sample size.
Extended Data
Extended Data Fig. 1|. BAHCC1, a nuclear factor of unknown function, shows overexpression among acute leukemias.
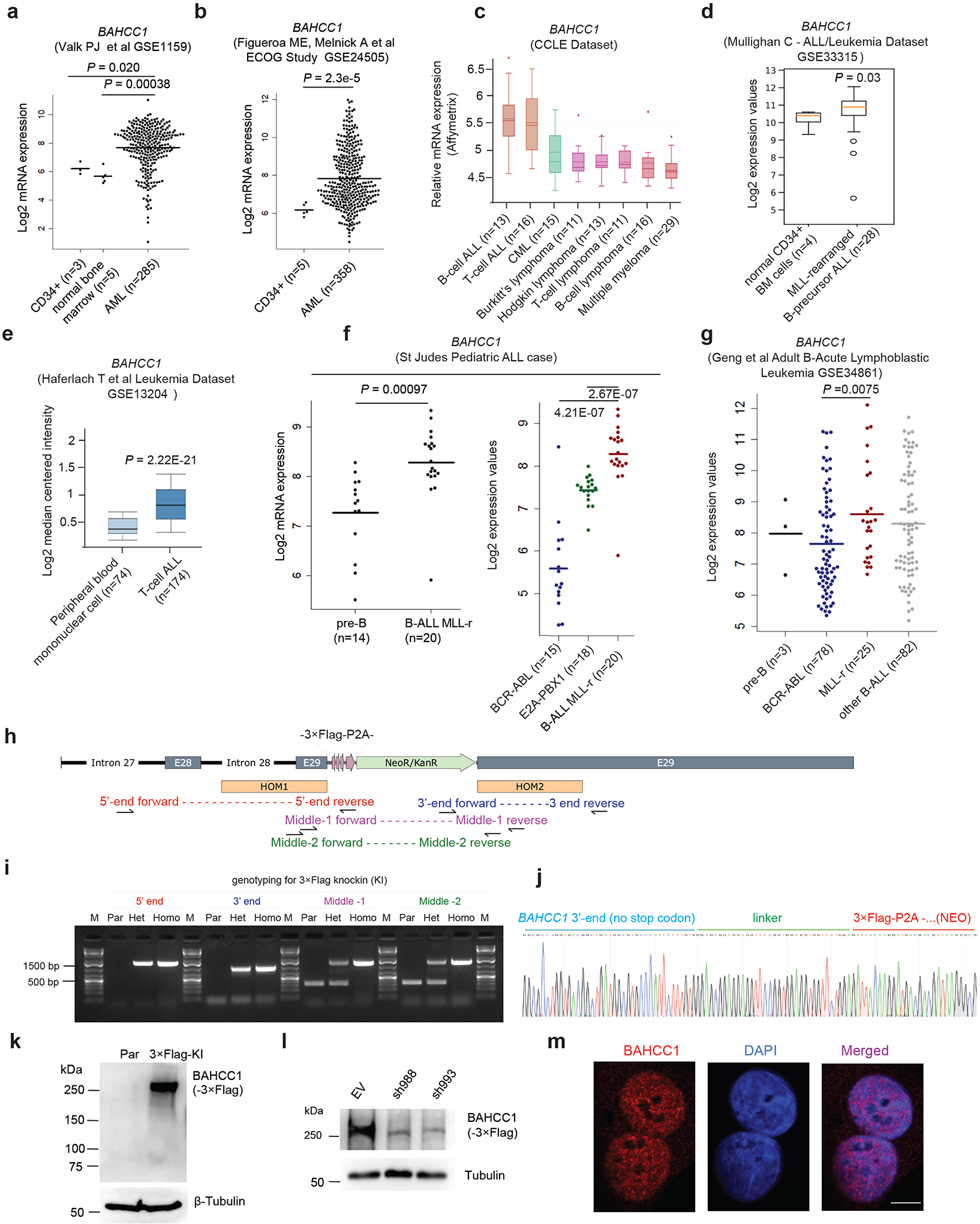
a-g, Boxplots showing BAHCC1 expression among primary AMLs (based on GEO dataset GSE115959 in panel a and GSE24505 in b), hematological cancer lines according to Cancer Cell Line Encyclopedia dataset (c), and primary ALLs carrying genetic abnormality such as MLL rearrangement (MLL-r), BCR-ABL or E2A-PBX1 according to GEO datasets GSE33315 (d) and GSE13204 (e), the St Jude Hospital “Pediatric Acute Lymphoblastic Leukemia” cohort (f) or GSE34861 (g), relative to the indicated normal controls. The line indicates the mean and two-sided Wilcoxon test was used for calculating P value denoted on top of the panel. n, sample size.
h-i, Schematic diagram (h) and PCR genotyping (i) of Flag Epitope Tag ChIP (pFETCh)-based strategy utilized to introduce a 3×Flag-P2A-NeoR+ cassette in-frame into the C-terminus of endogenous BAHCC1 gene. HOM1/2 (h), homology arm 1/2; E28/29, exons 28/29 of BAHCC1. Genotyping primers used for validating the cassette knockin are denoted in different colors. Agarose gel image (i) shows DNA ladder (M) and genotyping products using the indicated primers and genomic DNA of parental (Par) cells or those carrying heterozygous (Het) or homozygous (Homo) BAHCC1-3×Flag knockin (KI) alleles.
j, Representative Sanger sequencing results showing correct recombination and cassette knockin in the produced JURKAT cells carrying homozygous BAHCC1-3×Flag KI alleles.
k-l, Anti-Flag immunoblotting for endogenous BAHCC1–3×Flag protein, which was detected as ∼300kD in size (k) and readily depleted by BAHCC1-targeting shRNAs (sh988 or sh993; l), in JURKAT cells carrying the BAHCC1-3×Flag KI alleles. Parental (par) cells and those transduced with empty vector (EV) were used as control.
m, Representative images of confocal immunofluorescence reveal the exclusive nuclear localization of endogenous BAHCC1 (left), relative to DNA staining (middle), in HeLa cells. Scale bar, 5 μm.
Extended Data Fig. 2|. BAHCC1 promotes the in vitro growth, colony formation and cell cycle progression of acute leukemia cells.
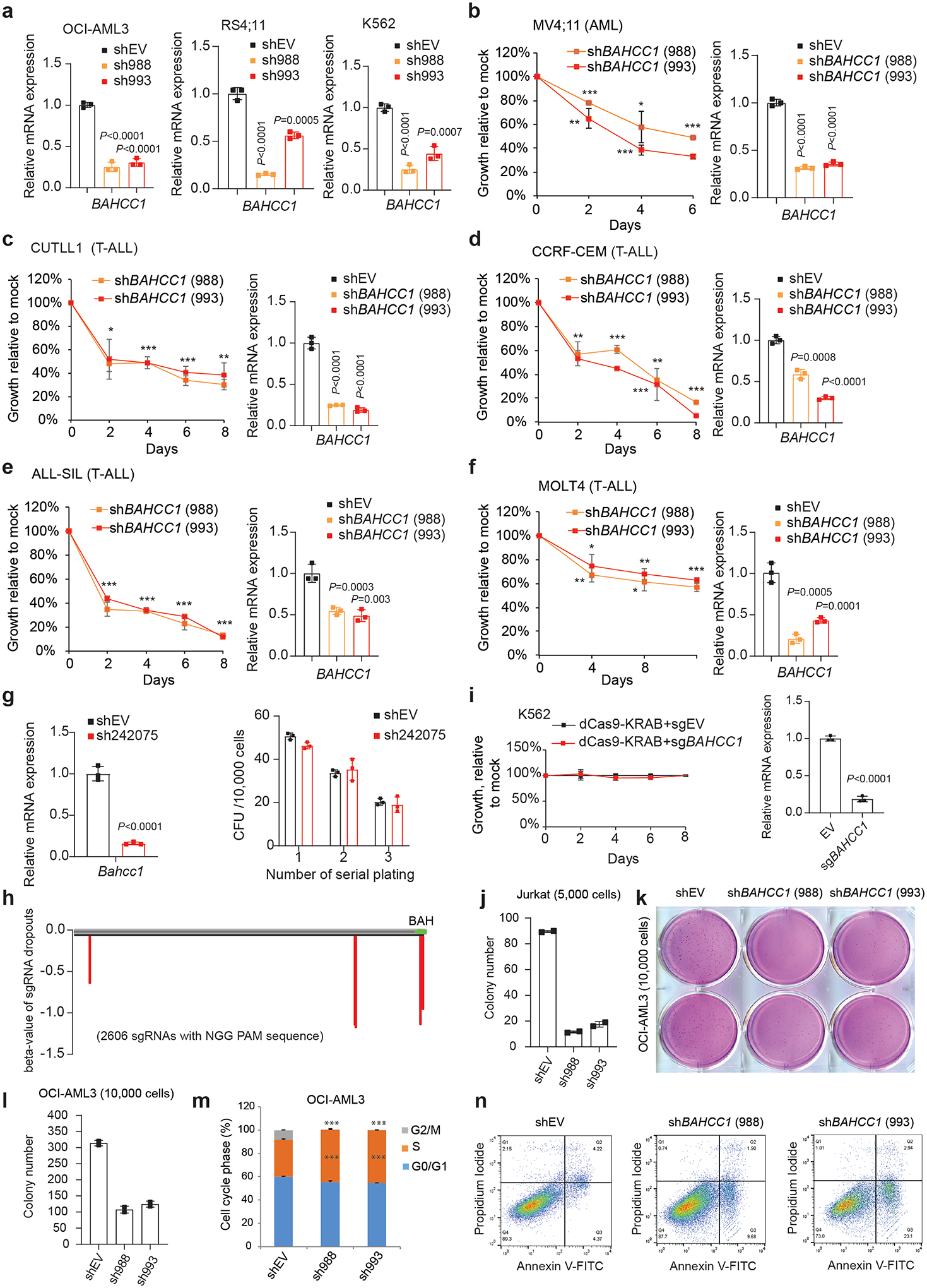
a, RT-qPCR validating shRNA-mediated BAHCC1 knockdown (KD; with independent hairpins sh988 and sh993) in OCI-AML3, RS4;11 and K562 cells (n=3 biologically independent samples). Data are presented as mean ± SD.
b-f, Proliferation (left) after BAHCC1 KD (RT-qPCR shown in the right panel), relative to empty vector (shEV)-treated control, in acute leukemia lines, including AML (MV4;11, b) and T-ALL (CUTLL1, CCRF-CEM, ALL-SIL and MOLT4; c-f). n=3 biologically independent experiments. Data are presented as mean ± SD.
g, RT-qPCR of Bahcc1 (left) and summary of colony-forming unit (CFU) assay (right) post-KD of Bahcc1 (sh242075), relative to shEV, in mouse primary HSPCs (n=3 biologically independent experiments). Data are presented as mean ± SD.
h, Dropout plot suggests that BAHCC1 regions targeted by the indicated sgRNAs are potentially crucial for JURKAT cell growth. Plotted at the bottom are sgRNAs showing significant dropout 10 days post-transduction of a viral BAHCC1 sgRNA pool, compared to day 1, among the same infected cells. Y-axis represents dropout beta-value, produced by the “Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout” module, with a cutoff of P value < 0.08.
i, K562 cell proliferation (left) after dCas9-KRAB-mediated repression of BAHCC1 (RT-qPCR shown on the right; n=3 biologically independent experiments).
j, Quantification of colonies formed by JURKAT cells post-KD of BAHCC1 compared to mock (n=2 biologically independent experiments). Data are presented as mean ± SD.
k-l, Representative plate scan images (k) and quantification of colony formation (l) using OCI-AML3 cells post-KD of BAHCC1 relative to mock (n=2 biologically independent experiments).
m, Cell cycle phase quantification using OCI-AML3 cells post-KD of BAHCC1 relative to mock (n=3 biologically independent experiments). ***, P < 0.001. Data are presented as mean ± SD.
n, Measurement of apoptosis in JURKAT cells post-KD of BAHCC1 relative to mock.
Extended Data Fig. 3|. BAHCC1 potentiates acute leukemia growth in vivo, a function that relies on an evolutionarily conserved BAH domain (BAHCC1BAH) as an H3K27me3/2-specific ‘reader’.
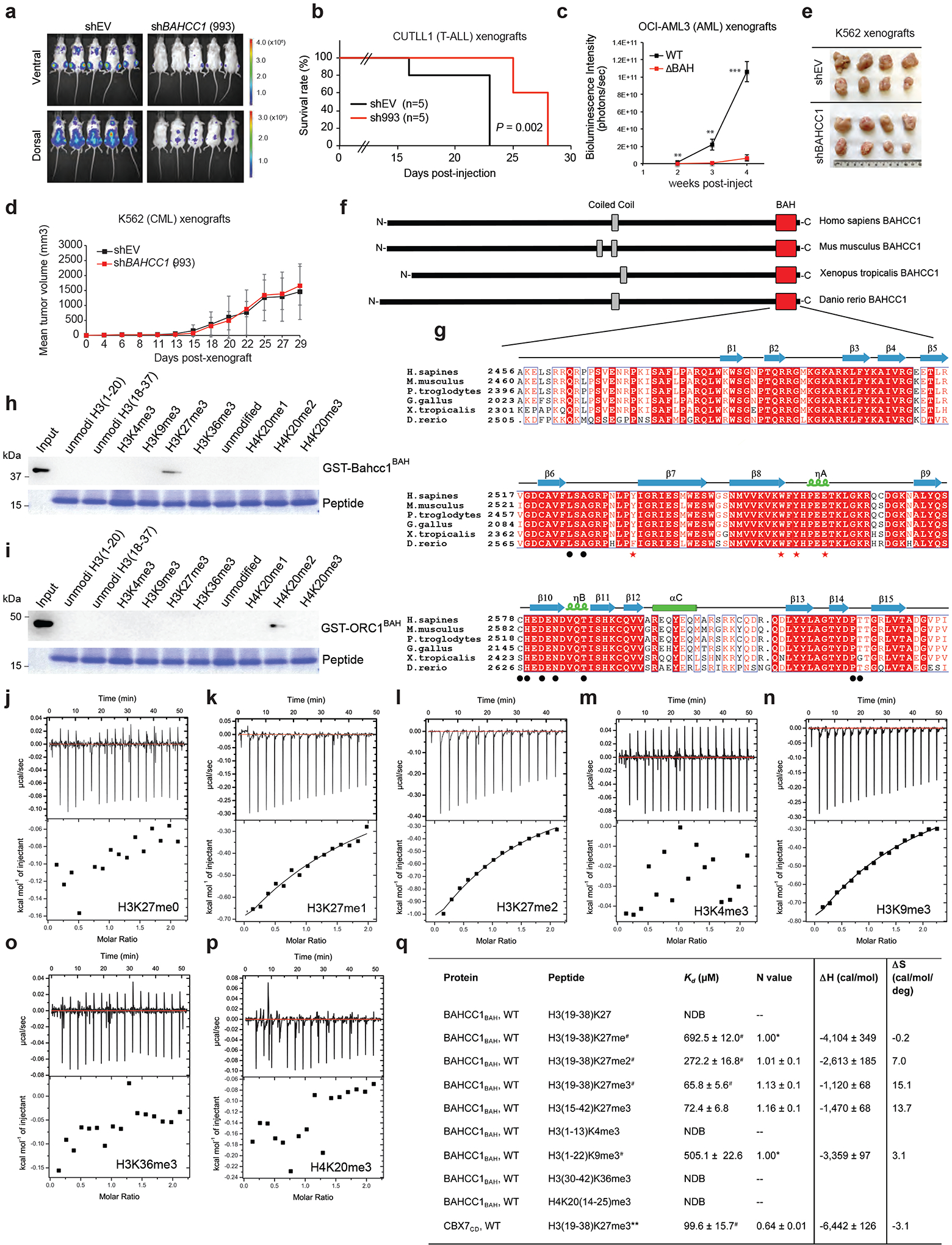
a-b, Bioluminescence imaging (a, two weeks post-transplantation) and Kaplan-Meier survival curve (b) of NSG mice xenografted with luciferase-labeled CUTLL1 cells post-transduction of empty vector or a BAHCC1-targeting shRNA.
c, Averaged bioluminescence signals (collected from ventral view) of NSG mice xenografted with luciferase-labeled OCI-AML3 cells that harbor the CRISPR-Cas9-introduced disruption of BAHCC1BAH (ΔBAH), relative to WT (n=5 mice). Data are presented as mean ± SD.
d, Plot showing the in vivo growth of K562 cell subcutaneous xenografts, after BAHCC1 KD or mock treatment. Data of tumor volume was shown as mean ± SD for each group (n=8).
e, Images of K562 subcutaneous xenografts excised from the indicated cohort at the end of experiments.
f, Domain architecture of the Homo sapiens, Mus musculus, Xenopus tropicalis and Danio rerio BAHCC1 proteins, revealing a conserved BAH domain (BAHCC1BAH).
g, Alignment using the amino acid sequences of BAHCC1BAH from different species. Conserved residues are highlighted in red. The secondary structure is drawn based on the BAHCC1:H3K27me3 co-crystal structure, with the H3K27me3-caging and other histone-binding residues designated by red stars and black dots, respectively.
h-i, Peptide pulldown using GST-BAHCC1BAH (h) or GST-ORC1BAH (i) protein and biotinylated histone H3.1 or H4 peptide carrying the indicated methylation (bottom). Unmodi, un-modified.
j-p, ITC fitting curves of BAHCC1BAH with the indicated histone peptide.
q, Summary of thermodynamic and curve fitting parameters for ITC assays using murine BAHCC1BAH recombinant protein and histone peptide. NDB, no detectable binding. *, N value set manually. #, mean and SD values derived from two independent measurements. For all the other titrations, ITC parameters were determined by curve fitting of single titration. **, due to behavior of CBX7CD and for a comparable measurement, ITC was performed at 25°C instead of 7°C for other measurements.
Extended Data Fig. 4|. Characterization of BAHCC1BAH binding to H3K27me3 through their co-crystal structure and structure-based mutagenesis.
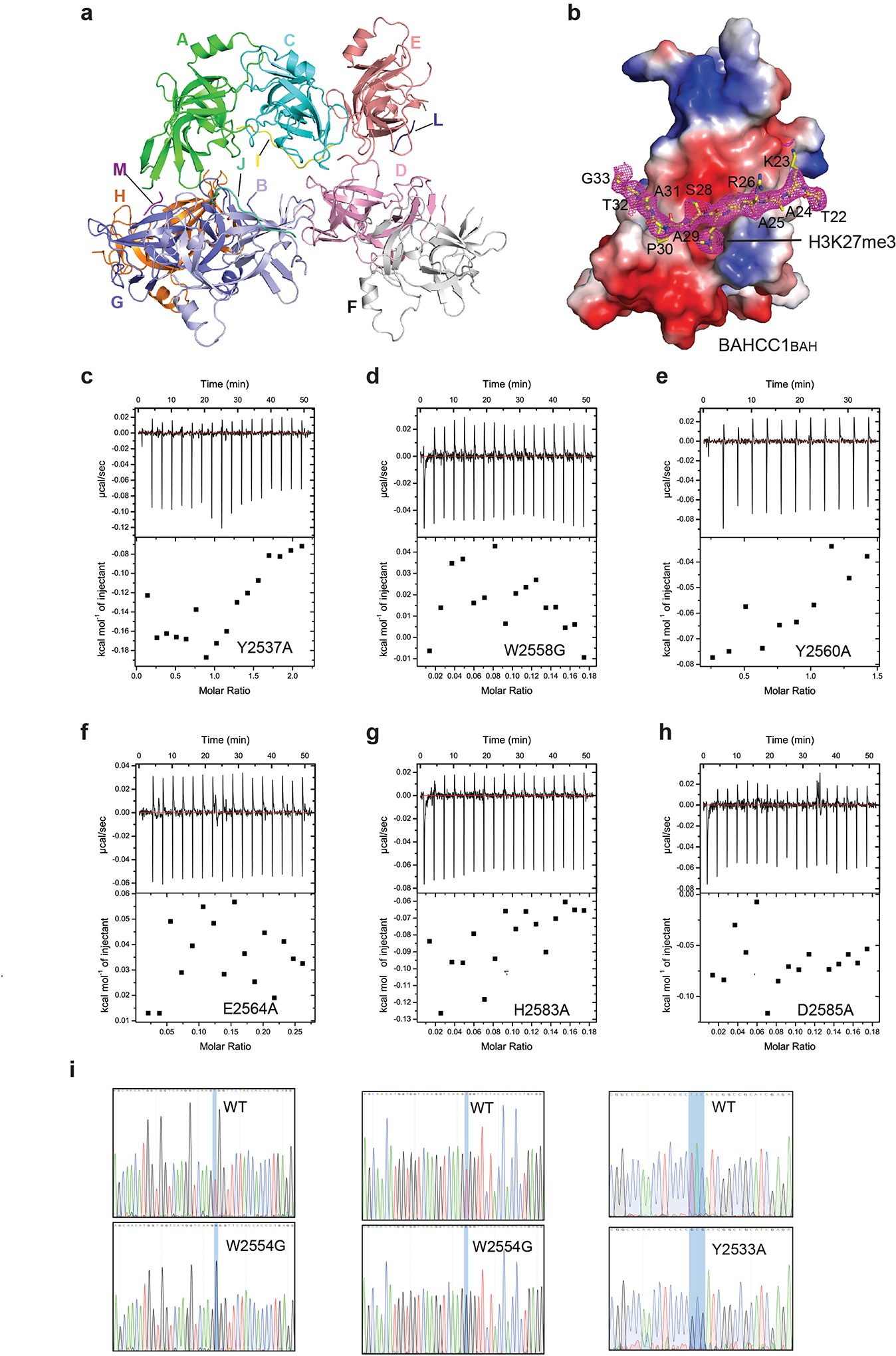
a, Crystal structures of the eight color-coded murine BAHCC1BAH molecules in one asymmetric unit, with chain ID labeled. Four of the BAHCC1BAH molecules (chain B, C, E and G) are complexed with H3K27me3 peptides (chain J, I, L and M, respectively.). The BAHCC1BAH:H3K27me3 complex with the best model-to-map fit (Chain C and I) was selected for structural analysis.
b, Electrostatic surface view of murine BAHCC1BAH bound to the H3K27me3 peptide (yellow sticks). The Fo-Fc omit map for the H3K27me3 peptide, contoured at 1.5 sigma level, was shown as mesh in magenta.
c-h, ITC binding curves of the H3K27me3 peptide with murine BAHCC1BAH recombinant protein carrying an indicated mutation, either Y2537A (c), W2558G (d), Y2560A (e), E2564A (f), H2583A (g) or D2585A (h).
i, CRISPR-cas9-mediated genomic editing to introduce homozygous point mutation of W2554G (left two panels) or Y2533A (right) into the BAHCC1 gene in JURKAT cells. Shown are Sanger sequencing results using cDNA (middle) or genomic DNA (left and right) as template. See also Extended Data Fig. 3g for murine and human BAHCC1 amino acid numerations.
Extended Data Fig. 5|. Transcriptome profiling reveals the gene-expression programs regulated by BAHCC1 in JURKAT cells.
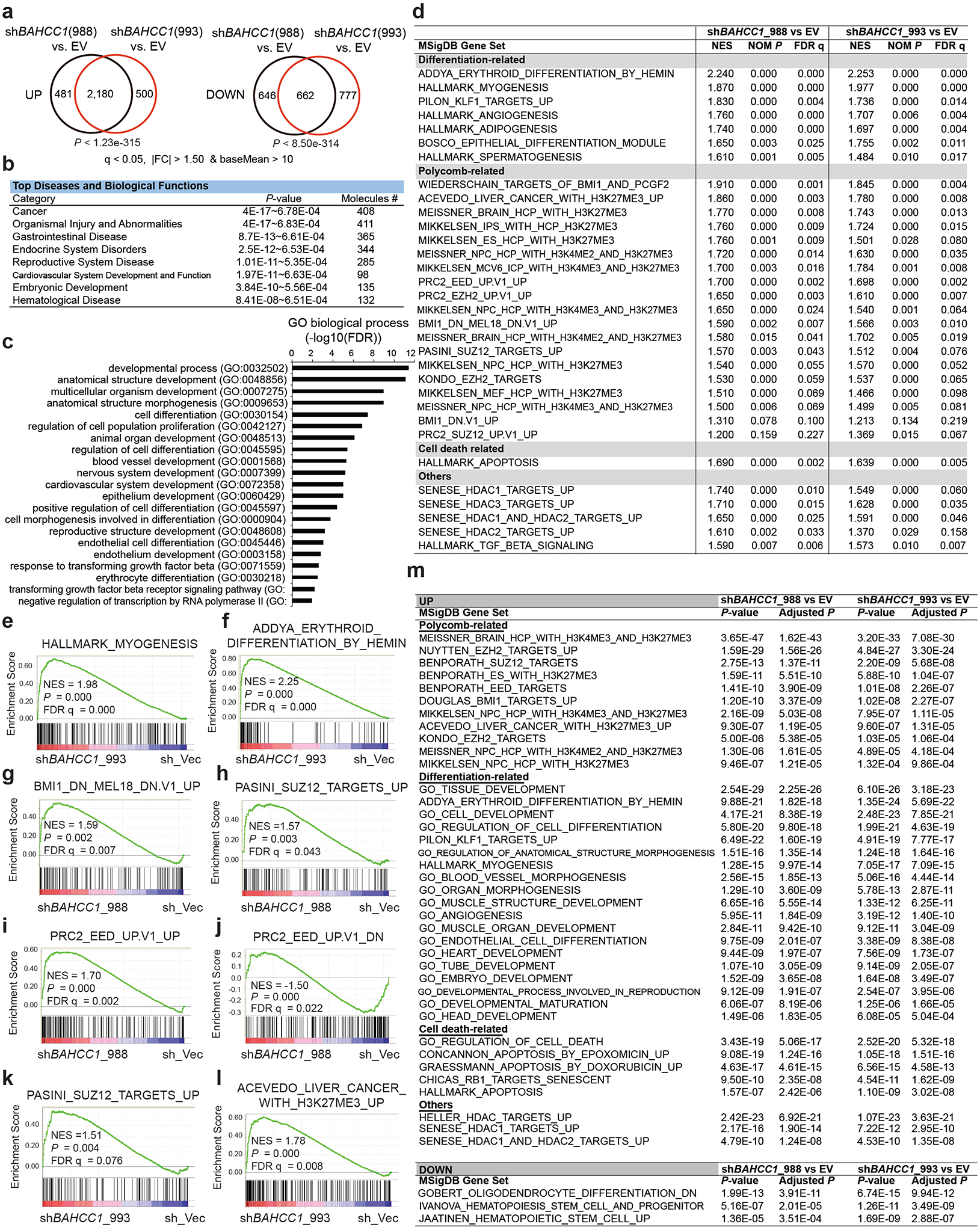
a, Venn diagrams show overlapping for upregulated (left) or downregulated (right) transcripts in JURKAT cells post-transduction of a BAHCC1-targeting shRNA, relative to empty vector-treated control (EV), as revealed by RNA-seq (n=3 biologically independent samples). The threshold for differentially expressed genes (DEGs) is an adjusted p (q) value of less than 0.05 and fold-change (FC) of over 1.50 for transcripts with baseMean value of more than 10.
b-c, Ingenuity Pathway Analysis (IPA; b) and Gene Ontology (GO; c) analyses with the genes found to be commonly upregulated post-KD of BAHCC1 by the two independent shRNAs, relative to mock, in JURKAT cells.
d, Summary of GSEA using RNA-seq profiles of JURKAT cells post-KD of BAHCC1 by sh988 (left) or sh993 (right), relative to EV. Representative genesets are categorized into differentiation, Polycomb (PRC2 and PRC1) or cell death-related or others.
e-f, GSEA demonstrates that BAHCC1 KD is correlated with derepression of genesets associated with myogenesis (e) and erythroid differentiation (f).
g-l, GSEA showing BAHCC1 KD correlated with reactivation of genesets suppressed by PRC1 (g) or PRC2 (h-k) and the H3K27me3-bound genes (l).
m, Platform for Integrative Analysis of Omics data (Piano) analysis results using RNA-seq profiles of JURKAT cells post-KD of BAHCC1 by sh988 (left) or sh993 (right), relative to EV.
Extended Data Fig. 6|. BAHCC1 co-localizes with H3K27me3, mediating optimal repression of Polycomb target genes in multiple examined leukemia cells.
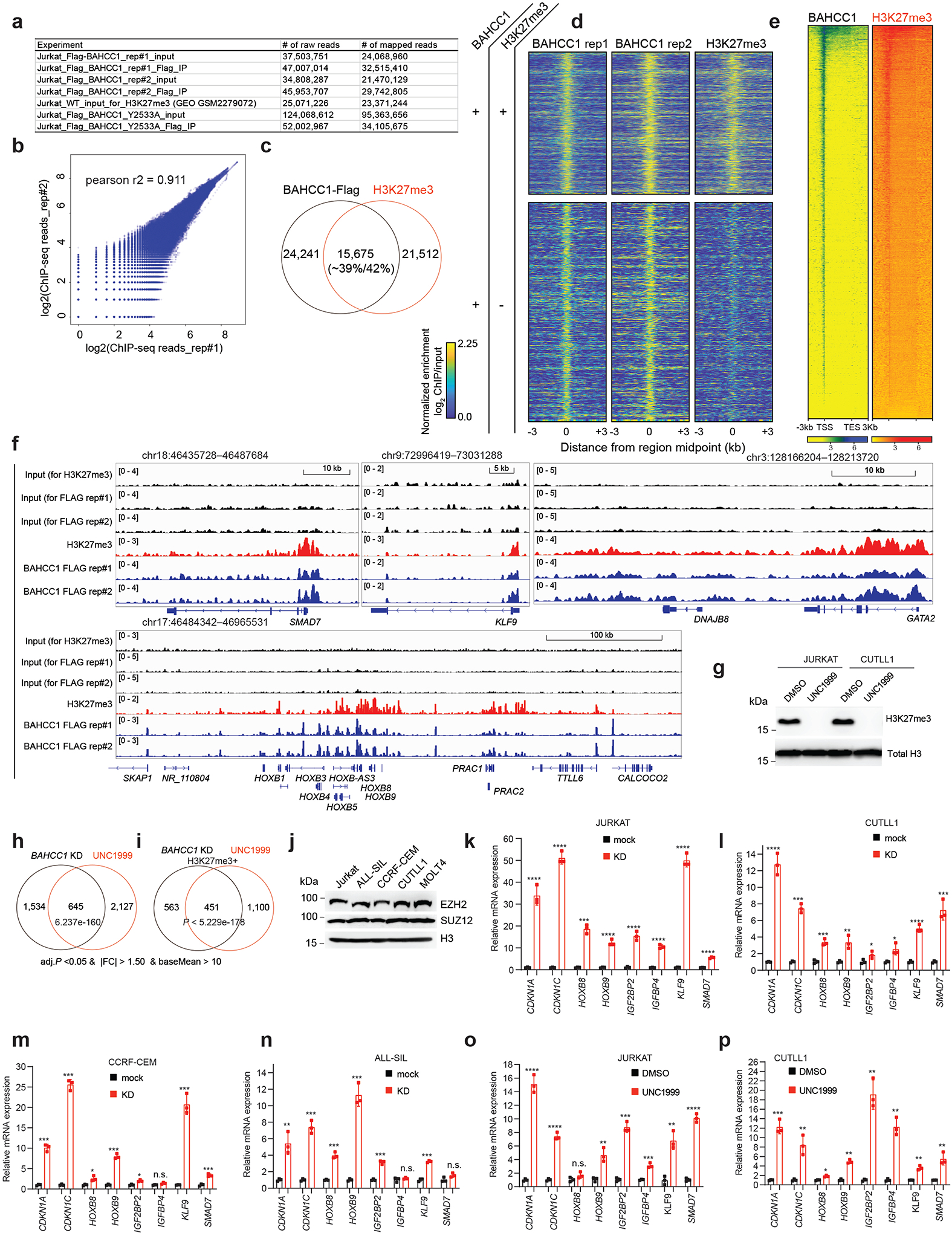
a, Summary of read counts of Flag ChIP-seq using JURKAT cells carrying 3×Flag-KI-alleles of BAHCC1, either wildtype or Y2533A-mutated.
b, Correlation analysis of replicated Flag-BAHCC1 ChIP-seq data in JURKAT cells.
c, Venn diagram showing overlapping of called BAHCC1 and H3K27me3 peaks in JURKAT cells.
d, Heatmap showing the called BAHCC1 peaks (Flag; two replicated experiments) that overlap (top) or does not overlap (bottom) with H3K27me3 in JURKAT cells after normalization to input and sequencing depth. Color bar, log2(ChIP/Input).
e, Heatmap showing BAHCC1 and H3K27me3 ChIP-seq peaks across ±3 kb from all genes in JURKAT cells. TSS, transcription start site; TES, transcription end site.
f, Flag-tagged BAHCC1 (blue) and H3K27me3 (red) ChIP-seq profile at classic Polycomb/H3K27me3-targeted loci (SMAD7, KLF9, GATA2 and the HOXB cluster) in JURKAT cells.
g, H3K27me3 and H3 immunoblots using JURKAT and CUTLL1 cells post-treatment with UNC1999.
h-i, Venn diagrams show significant overlapping for DEGs, either all DEGs (h) or those directly bound by H3K27me3 (i), that were found upregulated post-KD of BAHCC1 relative to mock (black) or post-treatment with UNC1999 relative to DMSO (red) in JURKAT cells.
j, EZH2, SUZ12 and H3 immunoblotting in various used T-ALL cell lines.
k-n, RT-qPCR of the indicated H3K27me3-targeted gene post-KD of BAHCC1 (red), relative to mock (black), in T-ALL cells including JURKAT (k), CUTLL1 (l), CCRF-CEM (m) and ALL-SIL (n). Data of three independent experiments are plotted as mean ± SD after normalization to GAPDH and to mock-treated cells. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n.s., not significant.
o-p, RT-qPCR of the indicated H3K27me3-targeted gene after UNC1999 treatment, compared to mock, in JURKAT (o) and CUTLL1 (p) cells. Data of three independent experiments are plotted as mean ± SD after normalization to GAPDH and to mock-treated cells. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n.s., not significant.
Extended Data Fig. 7|. The H3K27me3-binding-defective mutation of BAHCC1BAH decreases overall occupancy of BAHCC1 at H3K27me3-bound genes, leading to their concurrent derepression.
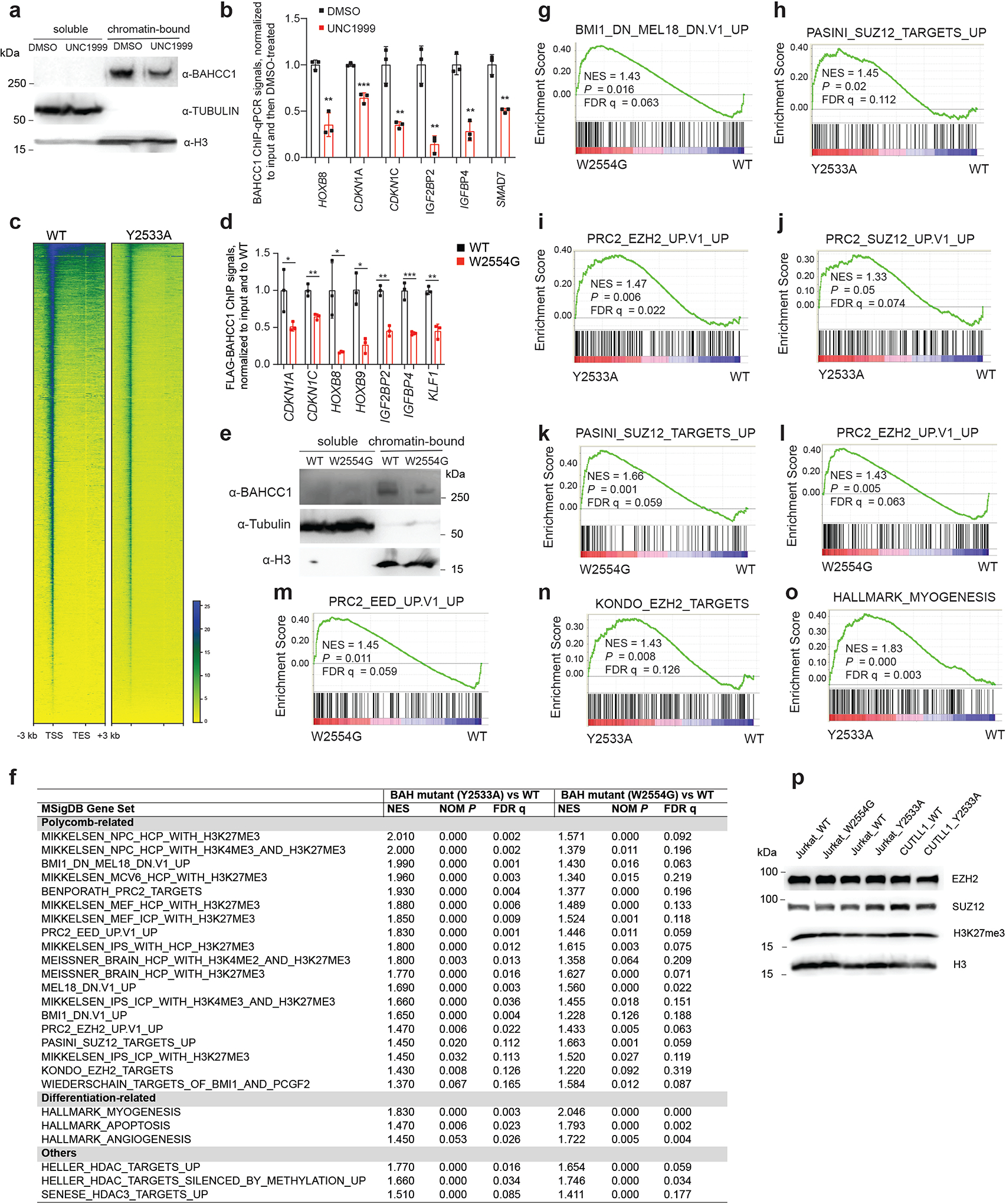
a, Immunoblots of BAHCC1, Tubulin and histone H3 using chromatin-bound and soluble fractions of 293 cells post-treatment with DMSO or UNC1999.
b, Flag ChIP-qPCR to examine binding of endogenous 3×Flag-BAHCC1 to TSS of the indicated gene in JURKAT cells post-treatment with UNC1999 versus DMSO. Data of three independent experiments are plotted as mean ± SD after normalization to those of input and to DMSO-treated. **P < 0.01; ***P < 0.001.
c, Heatmap of Flag-BAHCC1 ChIP-seq signals across all genes in JURKAT cells carrying either WT or the Y2533A homozygous mutation of 3×Flag-BAHCC1 (endogenous).
d, Flag ChIP-qPCR to examine the endogenous Flag-BAHCC1 binding to TSS of the indicated gene in JURKAT cells that carry either WT or the W2554G homozygous mutation of BAHCC1. Data of three independent experiments are plotted as mean ± SD after normalization to those of input and to WT. *P < 0.05; **P < 0.01; ***P < 0.001.
e, Immunoblots of BAHCC1, Tubulin and H3 using chromatin-bound and soluble fractions of JURKAT cells carrying WT or the W2554G homozygous mutation of BAHCC1BAH.
f, Summary of GSEA using RNA-seq profiles of JURKAT cells carrying an indicated H3K27me3-binding-defective mutation of BAHCC1BAH and their WT counterpart cells. Representative genesets showing the most significant correlations to Y2533A (left) or W2554G (right) homozygous mutation of BAHCC1BAH, relative to WT, are categorized into Polycomb-related, differentiation-related or others.
g-o, GSEA reveals that, relative to WT, the H3K27me3-binding-defective mutation of BAHCC1BAH is positively correlated to derepression of gene signatures related to PRC1 (g), SUZ12 (h, j, k), EZH2 (i, l, n), EED (m) or myogenesis (o).
p, Immunoblotting of EZH2, SUZ12, H3K27me3 and H3 in the indicated cell lines with WT or the H3K27me3-binding-defective mutation of BAHCC1BAH.
Extended Data Fig. 8|. The H3K27me3-binding-defective mutation of BAHCC1BAH slowed growth of acute leukemia in vitro and in vivo.
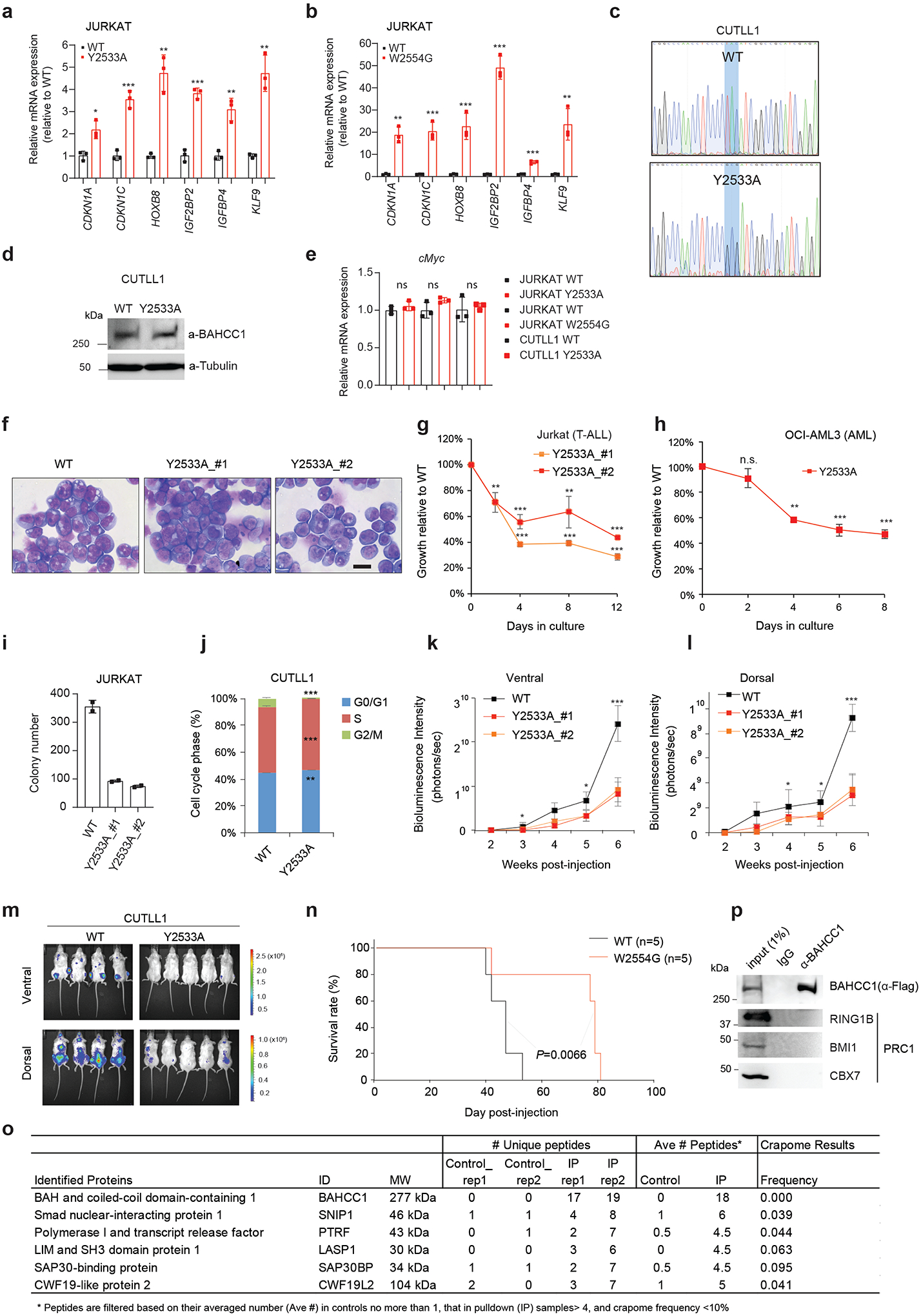
a-b, RT-qPCR of H3K27me3-marked genes in JURKAT cells harboring Y2533A (a) or W2554G (b) homozygous mutation of BAHCC1BAH, relative to WT (n=3 independent experiments). Data are plotted as mean ± SD after normalization to GAPDH and to WT. *P < 0.05; **P < 0.01; ***P < 0.001.
c-d, Sanger sequencing (c) and Flag immunoblotting (d) using CUTLL1 cells with either WT or Y2533A homozygous mutation of endogenous 3×Flag-BAHCC1.
e, RT-qPCR of cMyc in JURKAT and CUTLL1 cells with the indicated BAHCC1BAH homozygous mutation relative to WT (n=3 independent experiments). Data are plotted as mean ± SD after normalization to GAPDH and to WT. n.s., not significant.
f, Wright-Giemsa staining of JURKAT cells with WT or the Y2533A homozygous mutation of BAHCC1BAH (n=2 independent lines). Scale bar, 10 μm.
g-h, Proliferation of JURKAT (g) or OCI-AML3 cells (h) carrying homozygous mutation of BAHCC1BAH relative to WT (n=3 biologically independent experiments). Data are presented as mean ± SD.
i, Quantification of colonies formed by JURKAT cells carrying WT or Y2533A homozygous mutation of BAHCC1 (n=2 biologically independent experiments). Data are presented as mean values ± SD.
j, Quantification of cell cycle progression of CUTLL1 cells carrying WT or Y2533A homozygous mutation of BAHCC1 (n=3 biologically independent experiments). Data are presented as mean ± SD. **P < 0.01; ***P < 0.001.
k-l, Averaged bioluminescence signals collected from ventral (k) or dorsal view (l) of NSG mice (n=5) xenografted with luciferase-labeled JURKAT cells that carry WT or the Y2533A homozygous mutation (two independent lines) of BAHCC1BAH. Data are presented as mean ± SD. *P < 0.05; ***P < 0.001.
m, Bioluminescence images (two weeks post-xenograft) of NSG mice xenografted with luciferase-labeled CUTLL1 cells that carry WT or the Y2533A homozygous mutation of BAHCC1BAH.
n, Kaplan-Meier curve showing event-free survival of NSG mice xenografted with luciferase-labeled JURKAT cells that carry WT or the W2554G homozygous mutation of BAHCC1BAH. n, cohort size.
o, Summary for top hits of the BAHCC1-interacting proteins identified by Flag pulldown and mass spectrometry analyses with HeLa cells carrying BAHCC1–3×Flag KI alleles. Parental HeLa cells were used as control for Flag pulldown (n=2 biologically independent experiments). Rep, replicate.
p, CoIP for endogenous Flag-BAHCC1 and PRC1 in JURKAT cells.
Extended Data Fig. 9|. BAHCC1 interacts with corepressors SAP30BP and HDAC1, maintaining a hypoacetylated state at target genes.
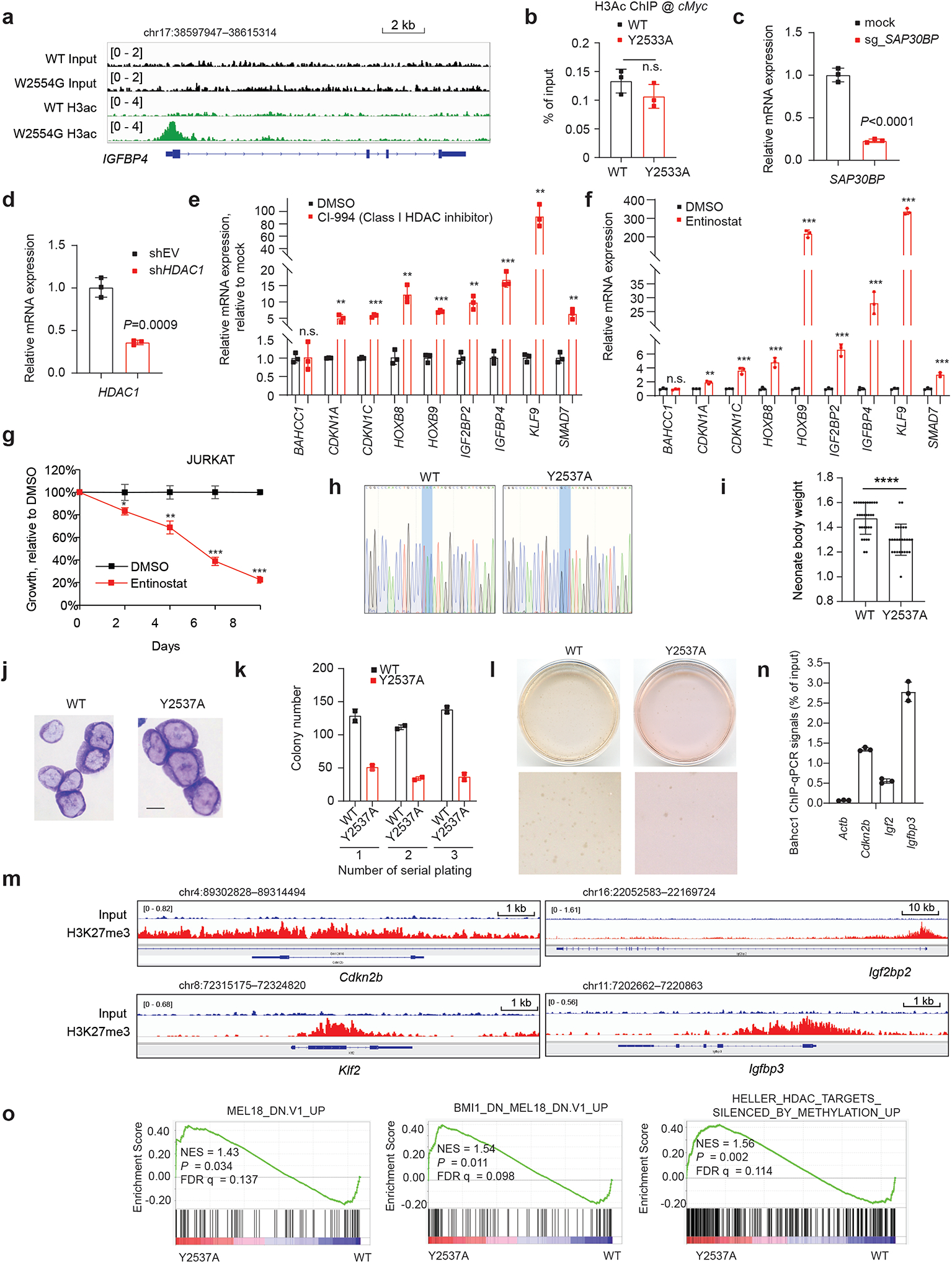
a, ChIP-seq profile of histone acetylation at the BAHCC1 target gene IGFBP4 in JURKAT cells carrying WT or the W2554G homozygous mutation of BAHCC1BAH.
b, ChIP-qPCR of histone acetylation at the cMyc promoter in JURKAT cells that carry WT or the Y2533A homozygous mutation of BAHCC1 (n=3 biologically independent samples). Data are presented as mean ± SD.
c-d, RT-qPCR of SAP30BP or HDAC1 after the dCAS9-KRAB/sgRNA-mediated repression of SAP30BP (c) or transduction of HDAC1-targeting shRNAs (d), relative to mock, in JURKAT cells (n=3 biologically independent samples). Data are presented as mean ± SD.
e-f, RT-qPCR of BAHCC1-targeted genes post-treatment of JURKAT cells with 5 μM of CI-994 (e) or 0.25 μM of Entinostat (f) for 2 days (n=3 biologically independent samples). Data are presented as mean values ± SD. **P < 0.01; ***P < 0.001; n.s., not significant.
g, JURKAT cell proliferation post-treatment with 0.25 μM of Entinostat relative to mock (n=3 biologically independent experiments). Data are presented as mean ± SD. * P < 0.05; **P < 0.01; ***P < 0.001.
h, Sanger sequencing to confirm the mouse genotype, either WT (left) or Y2537A-mutated (right) Bahcc1 allele.
i, Averaged body weight of mouse pups with either WT (left, n=31) or the Y2537A homozygous alleles (right, n=26) of Bahcc1, at day 1 post-birth (P1). Data are presented as mean ± SD. ****P < 0.0001.
j, Wright-Giemsa staining of E2A-PBX1-transformed murine leukemia cells, established with HSPCs from mice carrying either WT or the Y2537A homozygous mutation of BAHCC1BAH. Scale bar, 10 μM.
k-l, CFU quantification (k) and representative plate images (l) using the indicated E2A-PBX1-transformed murine leukemia cells (duplicate per group; data presented as mean ± SD).
m, ChIP-qPCR detects Bahcc1 binding to TSS of H3K27me3-marked genes relative to a negative locus, beta-actin (left), in the WT E2A-PBX1-transformed murine leukemia cells (n=3 independent experiments; mean ± SD after normalization to input).
n, H3K27me3 ChIP-seq profile of the indicated gene in the WT E2A-PBX1-transformed leukemia cells.
o, GSEA reveals that, relative to WT, the Y2537A mutation of Bahcc1BAH is positively correlated to derepression of Polycomb- or HDAC-related gene signature.
Extended Data Fig. 10|. BAHCC1 and PRC1 corepress H3K27me3-targeted genes in different cell lineages, suggesting a generalized functionality of BAHCC1.
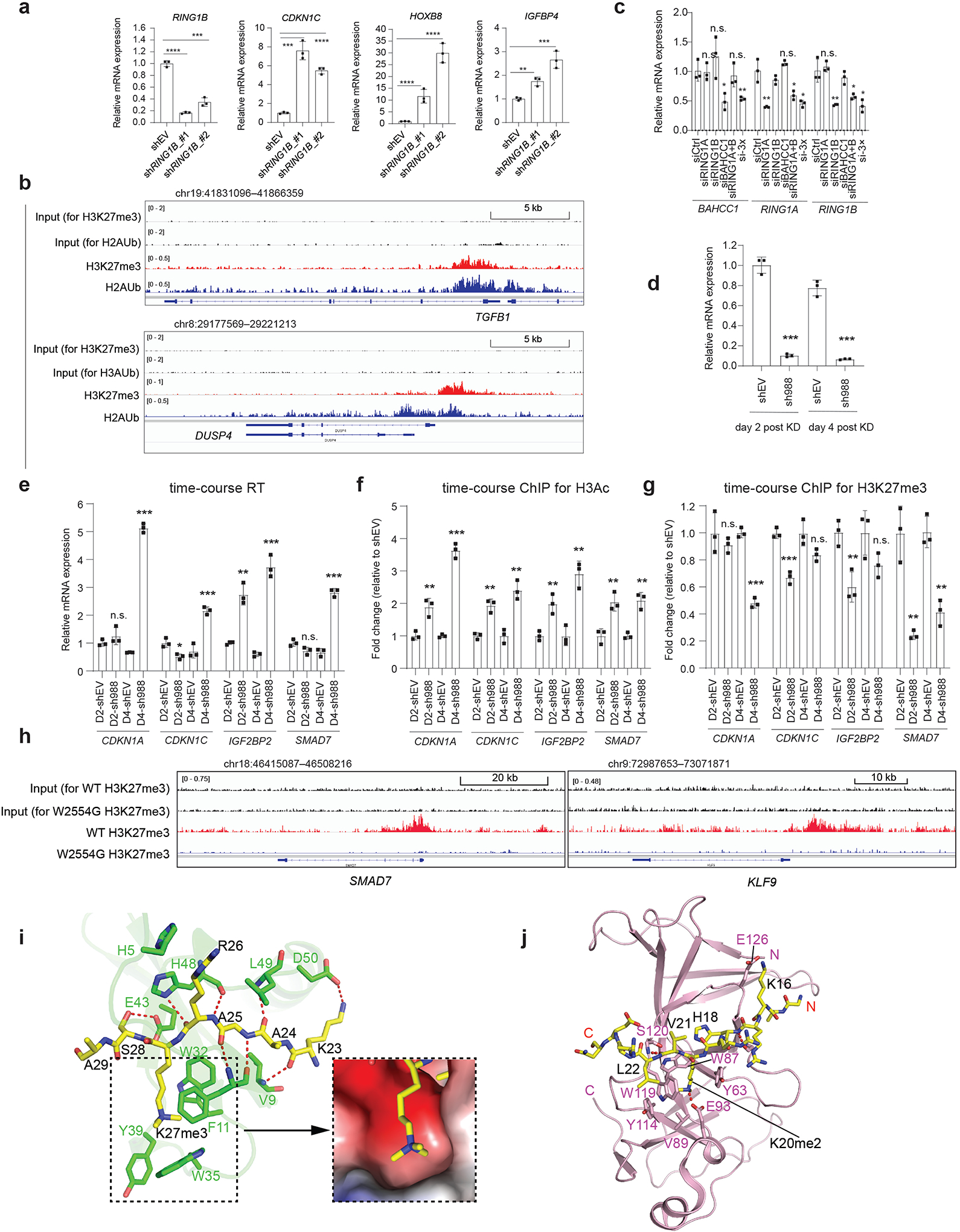
a, RT-qPCR of the indicated gene post-depletion of RING1B by independent shRNAs, relative to mock, in JURKAT cells (n=3 biologically independent samples). Data are presented as mean ± SD. **P < 0.01; ***P < 0.001; ****P < 0.0001.
b, ChIP-seq profiles of H3K27me3 and H2Aub at BAHCC1- and PRC1-corepressed genes in 293 cells.
c, RT-qPCR of BAHCC1, RING1A and RING1B in 293 cells transfected with siRNA of control (siCtrl), RING1A alone (siRING1A), RING1B alone (siRING1B), BAHCC1 alone (siBAHCC1), RING1A plus RING1B (siRING1A+B), or all three genes (si-3×). Data of three independent experiments are plotted as mean ± SD after normalization to GAPDH and to siCtrl-treated. *P < 0.05; **P < 0.01; n.s., not significant.
d-e, RT-qPCR of BAHCC1 (d) and the indicated target gene (e) two and four days (D2 and D4) after BAHCC1 KD (sh988), compared to mock, in 293 cells (n=3 independent experiments). Data are plotted as mean ± SD after normalization to GAPDH and to shEV. *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant.
f-g, H3 acetylation (f) and H3K27me3 (g) ChIP-qPCR for the indicated gene promoter at different timepoints after shRNA-mediated BAHCC1 KD in 293 cells (n=3 independent experiments). Data are plotted as mean ± SD after normalization to shEV cells. **P < 0.01; ***P < 0.001; n.s., not significant.
h, H3K27me3 ChIP-seq profile at BAHCC1 targets in JURKAT cells carrying WT or the W2554G homozygous mutation of BAHCC1.
i, Intermolecular interactions between CBX7CD (PDB 4X3K) and H3K27me3 peptide (yellow; left), along with the surface view of the H3K27me3-binding groove (right). The side chains of interacting residues are shown in stick representation and hydrogen bonds in dashed lines.
j, Ribbon representation of ORC1BAH (pink) bound to H4K20me2 peptide (yellow sticks) (PDB 4DOV). The H4K20me2-binding residues of ORC1BAH are labeled. The hydrogen bonding interactions are shown as dashed lines.
Supplementary Material
Acknowledgments
We thank Drs. P. Ntziachristos, M. Brenner and M. Minden for graciously providing reagents and cell lines used in the study, and the Wang laboratory members, T. Ptacek, H. Uryu and J. Simon for helpful discussions and technical support. We thank UNC’s facilities, including Imaging Core, High-throughput Sequencing Facility (HTSF), Bioinformatics Core, Flow Cytometry Core, Tissue Culture Facility, Proteomics Core and Animal Studies Core, for their professional assistance of this work. The core facilities affiliated with the UNC Cancer Center are supported in part by the UNC Lineberger Comprehensive Cancer Center Core Support Grant P30-CA016086. We would like to thank staff members at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory for access to X-ray beamlines. This work was supported by NIH grants (R01-CA215284 and R01-CA218600 to G.G.W.; R35GM119721 to J.S.; R35GM126900 to B.D.S.), a V Scholar Award (to G.G.W.), Kimmel Scholar Awards (to J.S. and G.G.W.) and University of California Cancer Research Coordinating Committee (UC CRCC) grant (CRR-20-634140 to J.S.), a Concern Foundation for Cancer Research grant (to G.G.W.), Gabrielle’s Angel Foundation for Cancer Research (to G.G.W.), Gilead Sciences Research Scholars Program in hematology/oncology (to G.G.W.) and When Everyone Survives (WES) Leukemia Research Foundation (to G.G.W.). G.G.W. is an American Cancer Society (ACS) Research Scholar, an American Society of Hematology (ASH) Scholar in basic science and a Leukemia and Lymphoma Society (LLS) Scholar.
Footnotes
The authors declare no competing financial interests.
Declaration of interests
B.D.S. is a co-founder and SAB member of EpiCypher. The Wang laboratory received research funds from Deerfield Management/Pinnacle Hill Company.
Data availability
Next-generation sequencing data have been deposited with the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) under accession number GSE151578. Coordinates and structure factors for the BAHCC1 BAH-H3K27me3 complex have been deposited in the Protein Data Bank under accession code 6VIL. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD021679. The publicly available gene expression profiling datasets used in the study include the Cancer Cell Line Encyclopedia (CCLE) data, the NCBI GEO datasets under accession numbers GSE1159, GSE24505, GSE13204, GSE33315, GSE7186, GSE28460 and GSE34861, and dataset of the St Jude Hospital “Pediatric Acute Lymphoblastic Leukemia (ALL)” cohort (http://www.stjuderesearch.org/data/ALL3).
References
- 1.Cao R et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–43 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Margueron R & Reinberg D The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Croce L & Helin K Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol 20, 1147–55 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Inoue A, Jiang L, Lu F, Suzuki T & Zhang Y Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 547, 419–424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller J et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111, 197–208 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Xie H et al. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell 14, 68–80 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bracken AP, Dietrich N, Pasini D, Hansen KH & Helin K Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 20, 1123–36 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartzentruber J et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–31 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Lewis PW et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–61 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nikoloski G et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 42, 665–7 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Morin RD et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42, 181–5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sneeringer CJ et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A 107, 20980–5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu B, Konze KD, Jin J & Wang GG Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp Hematol 43, 698–712 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gu Z et al. Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discov 9, 1228–1247 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chi P, Allis CD & Wang GG Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 10, 457–69 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strahl BD & Allis CD The language of covalent histone modifications. Nature 403, 41–5 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Wang H et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature 431, 873–8 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Schwartz YB & Pirrotta V A new world of Polycombs: unexpected partnerships and emerging functions. Nature reviews. Genetics 14, 853–64 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Min J, Zhang Y & Xu RM Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev 17, 1823–8 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leeb M et al. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev 24, 265–76 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grau DJ et al. Compaction of chromatin by diverse Polycomb group proteins requires localized regions of high charge. Genes Dev 25, 2210–21 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Margueron R et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tavares L et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 148, 664–78 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blackledge NP et al. Variant PRC1 Complex-Dependent H2A Ubiquitylation Drives PRC2 Recruitment and Polycomb Domain Formation. Cell (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Almeida M et al. PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 356, 1081–1084 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gough SM et al. NUP98-PHF23 is a chromatin-modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD histone reader function. Cancer discovery 4, 564–77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savic D et al. CETCh-seq: CRISPR epitope tagging ChIP-seq of DNA-binding proteins. Genome Res 25, 1581–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi J et al. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol 33, 661–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng J et al. A Molecular Chipper technology for CRISPR sgRNA library generation and functional mapping of noncoding regions. Nat Commun 7, 11178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuo AJ et al. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature 484, 115–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren C et al. Small-molecule modulators of methyl-lysine binding for the CBX7 chromodomain. Chem Biol 22, 161–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaustov L et al. Recognition and specificity determinants of the human cbx chromodomains. J Biol Chem 286, 521–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernstein E et al. Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol Cell Biol 26, 2560–9 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Varemo L, Nielsen J & Nookaew I Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res 41, 4378–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu B et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 125, 346–57 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konze KD et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS chemical biology 8, 1324–34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li JF et al. HTRP--an immediate-early gene product induced by HSV1 infection in human embryo fibroblasts, is involved in cellular co-repressors. J Biochem 136, 169–76 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Sardiu ME et al. Suberoylanilide hydroxamic acid (SAHA)-induced dynamics of a human histone deacetylase protein interaction network. Mol Cell Proteomics 13, 3114–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banks CAS et al. A Structured Workflow for Mapping Human Sin3 Histone Deacetylase Complex Interactions Using Halo-MudPIT Affinity-Purification Mass Spectrometry. Mol Cell Proteomics 17, 1432–1447 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi X et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–9 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pi WC et al. E2A-PBX1 functions as a coactivator for RUNX1 in acute lymphoblastic leukemia. Blood 136, 11–23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plys AJ et al. Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev 33, 799–813 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fursova NA et al. Synergy between Variant PRC1 Complexes Defines Polycomb-Mediated Gene Repression. Mol Cell 74, 1020–1036 e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tamburri S et al. Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol Cell 77, 840–856 e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beguelin W et al. EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell 30, 197–213 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taverna SD, Li H, Ruthenburg AJ, Allis CD & Patel DJ How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol 14, 1025–40 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao D et al. The BAH domain of BAHD1 is a histone H3K27me3 reader. Protein Cell 7, 222–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qian S et al. Dual recognition of H3K4me3 and H3K27me3 by a plant histone reader SHL. Nat Commun 9, 2425 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Z et al. EBS is a bivalent histone reader that regulates floral phase transition in Arabidopsis. Nat Genet 50, 1247–1253 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Z, Fu X, Wang Y, Liu R & He Y Polycomb-mediated gene silencing by the BAH-EMF1 complex in plants. Nat Genet 50, 1254–1261 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Ntziachristos P et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 18, 298–301 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neff T et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci U S A 109, 5028–33 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim W et al. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol 9, 643–50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka S et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood 120, 1107–17 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Beguelin W et al. Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell 37, 655–673 e11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.LaFave LM et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med 21, 1344–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mohammad F et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 23, 483–492 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Xu K et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 338, 1465–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dignam JD, Lebovitz RM & Roeder RG Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11, 1475–89 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu M et al. A resource for cell line authentication, annotation and quality control. Nature 520, 307–11 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Lu R et al. Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer Cell 30, 92–107 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang GG, Cai L, Pasillas MP & Kamps MP NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol 9, 804–12 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Wang GG et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 459, 847–51 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ren Z et al. PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation. Blood 134, 1176–1189 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cai L et al. An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol Cell 49, 571–82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rothbart SB et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat Struct Mol Biol 19, 1155–60 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Otwinowski Z & Minor W Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 70.McCoy AJ et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–32 (2004). [DOI] [PubMed] [Google Scholar]
- 72.Adams PD et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58, 1948–54 (2002). [DOI] [PubMed] [Google Scholar]
- 73.Cai L et al. ZFX Mediates Non-canonical Oncogenic Functions of the Androgen Receptor Splice Variant 7 in Castrate-Resistant Prostate Cancer. Mol Cell 72, 341–354 e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu B et al. The Chromatin Remodeler BPTF Activates a Stemness Gene-Expression Program Essential for the Maintenance of Adult Hematopoietic Stem Cells. Stem Cell Reports 10, 675–683 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mi H, Poudel S, Muruganujan A, Casagrande JT & Thomas PD PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res 44, D336–42 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–50 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ramirez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li F et al. Efficient genetic manipulation of the NOD-Rag1−/−IL2RgammaC-null mouse by combining in vitro fertilization and CRISPR/Cas9 technology. Sci Rep 4, 5290 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.He S, Kim I, Lim MS & Morrison SJ Sox17 expression confers self-renewal potential and fetal stem cell characteristics upon adult hematopoietic progenitors. Genes Dev 25, 1613–27 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Next-generation sequencing data have been deposited with the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) under accession number GSE151578. Coordinates and structure factors for the BAHCC1 BAH-H3K27me3 complex have been deposited in the Protein Data Bank under accession code 6VIL. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD021679. The publicly available gene expression profiling datasets used in the study include the Cancer Cell Line Encyclopedia (CCLE) data, the NCBI GEO datasets under accession numbers GSE1159, GSE24505, GSE13204, GSE33315, GSE7186, GSE28460 and GSE34861, and dataset of the St Jude Hospital “Pediatric Acute Lymphoblastic Leukemia (ALL)” cohort (http://www.stjuderesearch.org/data/ALL3).