

PURPOSE
To analyze the prevalence of homologous recombination deficiency (HRD) in patients with pancreatic ductal adenocarcinoma (PDAC).
MATERIALS AND METHODS
We conducted a systematic review and meta-analysis of the prevalence of HRD in PDAC from PubMed, Scopus, and Cochrane Library databases, and online cancer genomic data sets. The main outcome was pooled prevalence of somatic and germline mutations in the better characterized HRD genes (BRCA1, BRCA2, PALB2, ATM, ATR, CHEK2, RAD51, and the FANC genes). The secondary outcomes were prevalence of germline mutations overall, and in sporadic and familial cases; prevalence of germline BRCA1/2 mutations in Ashkenazi Jewish (AJ); and prevalence of HRD based on other definitions (ie, alterations in other genes, genomic scars, and mutational signatures). Random-effects modeling with the Freeman-Tukey transformation was used for the analyses. PROSPERO registration number: (CRD42020190813).
RESULTS
Sixty studies with 21,842 participants were included in the systematic review and 57 in the meta-analysis. Prevalence of germline and somatic mutations was BRCA1: 0.9%, BRCA2: 3.5%, PALB2: 0.2%, ATM: 2.2%, CHEK2: 0.3%, FANC: 0.5%, RAD51: 0.0%, and ATR: 0.1%. Prevalence of germline mutations was BRCA1: 0.9% (2.4% in AJ), BRCA2: 3.8% (8.2% in AJ), PALB2: 0.2%, ATM: 2%, CHEK2: 0.3%, and FANC: 0.4%. No significant differences between sporadic and familial cases were identified. HRD prevalence ranged between 14.5%-16.5% through targeted next-generation sequencing and 24%-44% through whole-genome or whole-exome sequencing allowing complementary genomic analysis, including genomic scars and other signatures (surrogate markers of HRD).
CONCLUSION
Surrogate readouts of HRD identify a greater proportion of patients with HRD than analyses limited to gene-level approaches. There is a clear need to harmonize HRD definitions and to validate the optimal biomarker for treatment selection. Universal HRD screening including integrated somatic and germline analysis should be offered to all patients with PDAC.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is the deadliest solid malignancy, with a five-year survival rate of < 10% and an increasing public health burden considering the estimated rise of its incidence and unchanging mortality over the next 20 years.1-4 Its biologic aggressiveness is compounded by the limited availability of effective therapies and a lack of prevention strategies.5,6 Defects in DNA damage response (DDR) genes causing homologous recombination deficiency (HRD) identify a clinically relevant subgroup of patients with PDAC, with both therapeutic and preventative implications.7-10 Accumulating evidence from nonrandomized clinical trials infers HRD as a putative biomarker of therapeutic response for platinum-based chemotherapy in patients with advanced PDAC.11,12 Within HRD, germline variants in BRCA1 and BRCA2 are associated with improved progression-free survival in patients with platinum-sensitive metastatic PDAC treated with the poly (ADP-ribose) polymerase (PARP) inhibitor (PARPi) olaparib as maintenance therapy.13 Interestingly, based on preclinical evidence and phase II nonrandomized clinical trials, additional non-BRCA HRD aberrations may predict sensitivity to PARPi14-17 with other therapeutic strategies targeting DDR currently under clinical investigation (including immunotherapy, ATM, ATR, and WEE1 inhibitors).18,19 In addition, germline pathogenic variants in several HRD genes in PDAC confers cancer susceptibility, with implications for risk assessment and prevention of a broad spectrum of neoplasms in patients and healthy relatives.20
CONTEXT
Key Objective
Homologous recombination deficiency (HRD) identifies a clinically relevant subgroup of patients with pancreatic cancer (pancreatic ductal adenocarcinoma). However, clinically relevant HRD is still poorly defined and variably reported, depending on definitions and assays used. This systematic review and meta-analysis aimed to define the prevalence of HRD in pancreatic ductal adenocarcinoma.
Knowledge Generated
Surrogate readouts of HRD (ie, genomic scarring, as well as point mutational and structural variant signatures) can identify a greater proportion of patients with HRD than analyses limited to gene-level approaches. However, a clinically applicable diagnostic is yet to be developed to capture these. The rate of germline mutations in HRD genes is similar in sporadic and familial patients.
Relevance
Given the known therapeutic implications of HRD-associated pancreatic cancer (such as sensitivity to platinum analogues and possibly other targeted agents, including poly [ADP-ribose] polymerase inhibitors), identifying patients who fit into this category beyond the use of standard gene panels will be important to refine our approach to precision medicine in this disease.
Despite major efforts, HRD is still challenging to define with reported prevalence in PDAC highly variable, limiting its clinical implementation in routine practice and therapeutic development.7,11,12,18,21-29 This uncertainty is primarily because of inconsistencies in HRD measurement and definitions (gene-level tests, genomic scars, signatures, or a combination of these methods); and the difficulties in assessing the contribution of each genomic event.30,31 Specifically, a few hundred genes are proposed to be involved in homologous recombination repair, including (but not limited to) BRCA1/2, PALB2, ATR, ATM, CHEK1/2, RAD51, and FANC genes, resulting in HRD when germline or somatic inactivation occurs by mutation or epigenetic silencing.32 It is possible to analyze a broad range of these genes in a single test through next-generation sequencing (NGS) technologies. However, there is no accepted consensus on which genes and genomic regions should be included using sequencing methods that can be applied in routine care to maximize the probability of finding clinically meaningful HRD. The inconsistency of genes included in NGS-based HRD panels, and the interpretation of the functional impact of mutations, result in high variability in prevalence estimates of HRD in PDAC.11,12,18,25,28,29 Moreover, recent largescale whole-exome sequencing (WES) and whole-genome sequencing (WGS) analyses suggest that HRD likely extends beyond point mutations in core genes, implying other molecular mechanisms, which are yet to be elucidated.7,21,24,33-36
Here, we present a systematic review of the current literature on HRD in PDAC and perform a prevalence meta-analysis of the better-characterized HRD genes with known or potential clinical utility. Particular focus was given to germline variants, both in sporadic and familial cases, to assess the contribution of HRD genes to cancer susceptibility and potential intervention.
MATERIALS AND METHODS
Search Strategy, Selection, and Inclusion Criteria
The study protocol and data extraction for the systematic review and meta-analysis was designed according to Preferred Reporting Items for Systematic reviews and Meta-Analyses guidelines.37 The research protocol was registered at the International Prospective Register of Systematic Reviews (PROSPERO38 number: CRD42020190813). PubMed, Scopus, and the Cochrane Library databases, and online cancer genomic data sets were queried for articles reporting the prevalence of HRD in PDAC, published from database inception to February 28, 2020. Specific HRD genes were selected after an exhaustive review of the literature and pragmatic considerations based on which genes were studied and reported in the literature and the likely clinical utility (most frequently altered; better characterized; known role in PDAC susceptibility; used as biomarkers in clinical trials): BRCA1, BRCA2, PALB2, ATR, ATM, CHEK2, and RAD51 (including -B, C, D), and the Fanconi-Anemia (FANC) genes (at least one of the following: FANC-A, B, C, D1, D2, E, F, G, and M). Studies reporting other definitions of HRD (ie, mutations in other genes, genomic scars, signatures, and structural variation patterns) were also considered for inclusion in the systematic review but not for the pooled prevalence meta-analysis.
The search protocol was updated on May 19, 2020, after the Food and Drug Administration approval of olaparib for patients with HRD metastatic castration-resistant prostate cancer, defined according to germline or somatic mutations in the following 15 genes: BRCA1/2, ATM, BARD1, BRIP1, CDK12, CHEK1/2, FANCL, PALB2, PPP2R2A, RAD51 B/C/D, or RAD54L.39 As the data extraction was terminated at that time, only online cancer genomic data sets were queried to investigate the prevalence of these 15 genes in PDAC.
Titles and abstracts of all identified articles and publicly available data sets were independently screened by two authors (R.C. and S.P.). Articles were included if the study cohort was composed of at least 20 patients, regardless of study kind and design, sequencing methodology, DNA source, type of mutation, or ethnicity of the study cohorts. Each author worked blindly from the other, and each selected manuscript was double-checked by the other. Discrepancies were resolved through consensus by four authors (R.C., S.P., D.K.C., and V.C.). The possibility of overlapping populations was considered. The von Elm patterns of duplication were adopted.40 Further details on search strategy, selection, and data extraction are presented in the Data Supplement (online only).
Outcomes of Interest and Definitions
The main outcome measure was the pooled prevalence of germline and somatic mutations in each HRD gene listed above. Secondary outcomes included the pooled prevalence of germline mutations overall, and individually in familial and sporadic PDAC; the pooled prevalence of germline BRCA1/2 mutations in patients with Ashkenazi Jewish (AJ) ancestry; and the prevalence of HRD according to other definitions (as reported above).
The prevalence of any mutation was included regardless of whether it was germline, somatic, or founder. When reported, the details of the mutations were checked to evaluate their pathogenicity or clinical relevance, according to the current guidelines for variant interpretation.41,42 Only pathogenic or likely pathogenic or clinically relevant variants were considered for the prevalence analysis, whereas benign, likely benign, and variants of uncertain significance were excluded. If the required information was not reported in the published report, we consulted available online cancer genomic data sets or requested raw data from the authors. In case of missing data, variables were classified as not reported or unclear to avoid misinterpretation. Other definitions of HRD were reported and analyzed separately because of the high level of variability that impeded the performance of proportional meta-analysis.
For each study, data on family history were reviewed by a team member with expertise in hereditary cancer syndromes (R.C.) and every case for which the details were reported was (re-)classified as familial or sporadic according to current National Comprehensive Cancer Network and American College of Medical Genetics and Genomics criteria for genetic cancer risk assessment.20,43,44 If family history was not reported, cases were classified as unselected and excluded from the subgroup analysis of sporadic patients to avoid selection bias with potential consequent overestimation of the results.
Statistical Analysis
Meta-analysis.
A random-effect meta-analysis (DerSimonian and Laird model) was performed on the prevalence data to calculate the pooled event rate using the Freeman-Tukey transformation.45,46 A Cochran's Q test for heterogeneity was performed reporting the I2 statistic, which indicates the percentage of variation across studies because of heterogeneity rather than chance.47 Heterogeneity values of < 30%, 30%-60%, 61%-75%, and > 75% were, respectively, classified as low, moderate, substantial, and considerable.48
Publication bias and study bias.
Funnel plots of study size against log odds were used to assess publication bias.49 The funnel plot asymmetry was assessed by using the Macaskill regression test for binary data.50 A general linear (mixed-effects) meta-regression model was also computed, where observations were weighted by the inverse variance of the estimate to allow for heteroscedasticity.51
Different strategies for study quality and risk-of-bias appraisal were evaluated.52 The available options could be improved for specific application to translational cancer genomic studies and for systematic reviews that incorporate multiple study designs. Risk Of Bias In Non-randomized Studies—of Interventions (ROBINS-I) demonstrated to be the most appropriate and adaptable tool and, as a consequence, was used for these analyses.53 In addition, an internal risk-of-bias assessment at the study level was specifically developed: Translating-ROB (ie, TRANSLATIonal caNcer Genomic Risk Of Bias). According to this tool, a 25-point quality rating was applied to each study (Data Supplement).
Meta-regression.
A mixed-effects meta-regression analysis was conducted to examine the possibility of effect modification of the pooled prevalence estimates. The mixed-effects meta-regression estimates were computed accounting for the nonlinearity for risk-of-bias score using the restricted cubic spline method.48 The following variables were considered possible moderators of the dependent variable: sequencing methodology (non-NGS v NGS); stage of disease (early v metastatic); risk-of-bias score (Translating-ROB and ROBINS-I); and study sample size (Macaskill test P value was applied as indicator of the effect of the study size on outcome). The model estimates were adjusted within genes for the multiplicity of testing using the Benjamini-Hochberg correction.48
Mean and standard deviation or median with interquartile range were reported in cases of normally or non-normally distributed data, respectively. Statistical analysis was performed using R (R Foundation for Statistical Computing, Vienna, Austria, v. 4.02)54 with the metafor 2.4-055 and FactoMineR packages.56
RESULTS
General Findings
A total of 2,062 nonduplicate titles and abstracts were retrieved from PubMed, Scopus, and Cochrane databases, and hand-to-hand searches. Three publicly available cancer genomic data sets were consulted.57-59 After screening abstracts and titles, 1,918 studies were judged not relevant. After screening full texts, an additional 73 studies that did not meet the eligibility criteria were excluded, with a total of 71 meeting inclusion or exclusion criteria for the systematic review. After a more detailed analysis of the records, 12 manuscripts were further excluded (85 excluded studies are reported in the Data Supplement), and a final total of 59 were included in the systematic review (Data Supplement). Only one cancer genomic data set57 was included. The other two were excluded because of overlap with published studies, containing more detailed information.7,21,60 In addition, a further three studies included in the systematic review were excluded from the meta-analysis because of selection bias or granularity in data reporting.
Therefore, 59 studies and one cancer genomic data set with 21,842 participants from 18 countries (Data Supplement) met the systematic review's inclusion criteria, whereas 56 studies and the genomic data set were included in the meta-analysis. The flowchart of the study selection process is reported in Figure 1.
FIG 1.
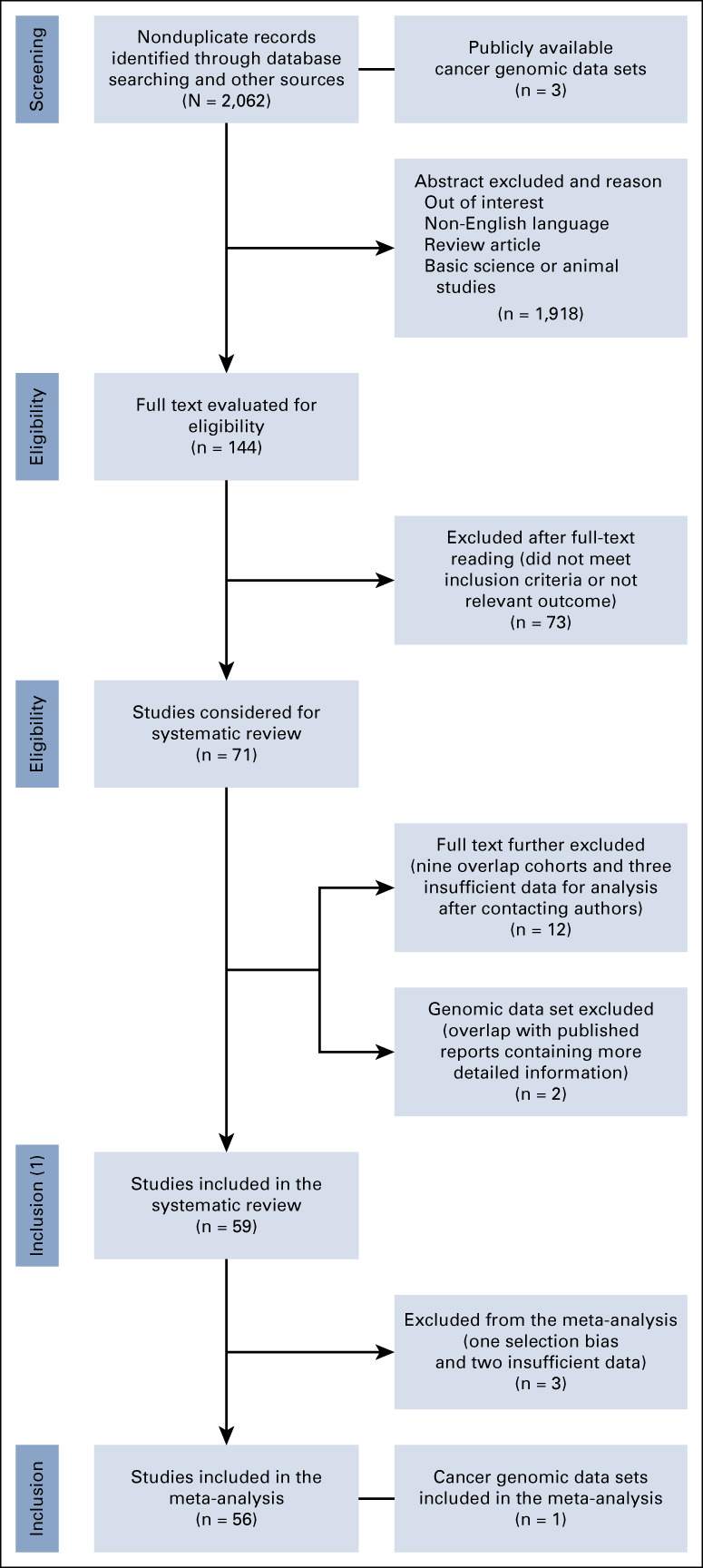
Preferred Reporting Items for Systematic Reviews and Meta-Analyses flowchart of patient selection for the meta-analysis.
The 60 studies (including the data set) included in the systematic review are summarized in the Data Supplement. The majority were from the United States (33 out of 60, 55%). The median number of patients per study was 81 (interquartile range 226). Thirteen studies were conducted in resected patients, seven in metastatic, and one in locally advanced disease, and 16 studies enrolled patients with mixed clinical stage. The remaining studies (23 out of 60, 38%) did not report this information. BRCA1/2 were the most frequently studied genes (54 out of 60 studies, of which 45 reported information on BRCA1 and 52 on BRCA2), followed by PALB2 (43 out of 60), ATM (35 out of 60), CHEK2 (30 out of 60), FANC genes (27 out of 60), RAD51 genes (26 out of 60), and ATR (21 out of 60). Germline mutations were tested in 54 studies, somatic in 19, and 13 reports included somatic and germline mutations.
Ethnicity was reported in 35 out of 60 (58%) studies (Data Supplement). When reported, Caucasian or White was the most represented ethnicity. Eleven studies (18.3%) that did not specify ethnicity were presumed to be conducted in Caucasian or White populations based on the geographic location of the participating institution. Four studies enrolled Asian patients only. A total of 17 out of 60 (28.3%) studies included patients with AJ ancestry, nine of which reported the population-specific mutation rate and were included in the subgroup analysis.
Several sequencing methodologies had been used, including targeted Sanger sequencing, targeted NGS of individual genes, targeted NGS of multiple genes, targeted capture NGS, WES, and WGS (Data Supplement).
Overall, 13 out of 60 (21.7%) studies enrolled patients with familial PDAC and 27 (45%) included unselected populations but reported details on family history. The remaining 20 (33.3%) included unselected patients and were as a consequence excluded from this specific subgroup meta-analysis (Data Supplement).
Pooled Prevalence Estimates
Detailed results of the pooled prevalence estimates of mutations in individual HRD genes are reported in Table 1. The pooled proportion of germline and somatic mutations in all included studies was BRCA1: 0.9%, BRCA2: 3.5%, PALB2: 0.2%, ATM: 2.2%, CHEK2: 0.3%, FANC: 0.5%, RAD51: 0.0%, and ATR: 0.1%. The pooled proportion of germline mutations was BRCA1: 0.9% (2.4% in AJ), BRCA2: 3.8% (8.2% in AJ), PALB2: 0.2%, ATM: 2%, CHEK2: 0.3%, and FANC: 0.4%. No significant differences in the estimates were identified between sporadic and familial cases.
TABLE 1.
Pooled Prevalence Estimates of Individual Homologous Recombination Deficiency Gene Mutations

For each main outcome, the following tests were performed and reported: the funnel plot of study size against log odds, the forest plot of the prevalence meta-analysis, the linear mixed-effect meta-regression of event rates according to bias score, and event rates according to sample size (Data Supplement). The funnel plots for the prevalence outcomes showed that globally, the event rates for the studies considered fell within the confidence bounds of the plot (low-publication bias zone), indicating an acceptable publication bias result. BRCA2 was the outcome reporting the greatest number of studies outside the 95% CI.
Meta-regression analysis identified no significant effect modifiers on mutation prevalence for the main end points including sequencing methodology, stage of disease, sample size, and risk-of-bias score at study level (Data Supplement). Detailed biostatistics is reported in the Data Supplement.
The risk of bias according to ROBINS-I and Translating-ROB as well as the validation procedure of Translating-ROB is reported in the Data Supplement. According to ROBINS-I, 45 out of 57 studies (79%) resulted at low or moderate risk of bias.
HRD Prevalence According to Other Definitions
A total of nine studies reported (1) the prevalence of mutations in additional genes beyond those selected for the meta-analysis; (2) other definitions of HRD (genomic scars, mutational signatures, and structural variation patterns); and (3) only the overall HRD prevalence, without specifying single gene alterations. HRD prevalence ranged between 14.5%-16.5% when extended NGS panels were used and 24%-44% through WGS or WES (Table 2).
TABLE 2.
Overall HRD Frequency

The estimated prevalence of the HRD 15-gene list used clinically for metastatic castration-resistant prostate cancer was calculated using an integrated analysis of data from this study and the three online publicly available data sets mentioned in the search strategy. Online data sets provide data only on somatic mutations. Therefore, the latter information was combined with the pooled prevalence estimated of germline mutations in 8 out of 15 overlapping genes from this meta-analysis. The prevalence of the HRD 15-gene list in PDAC was approximately 12% (likely underestimated because of the methodologic limitations in the computation) (Data Supplement).
LOH (Loss of the Wild-Type Allele) and Somatic Versus Germline Status
Only 10 studies evaluated the somatic event in the second allele. Significant variation in the somatic event (from 0% to 100%) was observed. When only studies with larger cohorts were considered, the overall rate of LOH for all genes tested was approximately 50% (Data Supplement).24,33,61,62
Considering the low numbers, the frequency of germline versus somatic mutations was computed for BRCA1 and BRCA2 only, with 67% of BRCA mutations germline and 33% somatic (Data Supplement).
DISCUSSION
To our knowledge, this is the first systematic review and meta-analysis assessing the prevalence of HRD in PDAC. There was significant variation in the prevalence of HRD estimates based on current and variable HRD definitions and methods of assessment, with a prevalence of 7.7% using mutation testing of the better characterized HRD genes, 14.5%-16.5% through extended NGS panels, and 24%-44% through WES or WGS. The main contribution to HRD was through BRCA2, BRCA1, and ATM, followed by FANC genes, CHEK2, and PALB2. The prevalence of aberrations in RAD51, ATR, BRIP1, BARD1, CDK12, and CHEK1 was markedly less, whereas evidence for rarer genes, such as RAD54L and PPP2R2A, is lacking. These findings support the increasing interest in ATM as it constitutes one of the more common and potentially actionable HRD genes in PDAC.63-67
We estimated that the 15-gene list used in prostate cancer may capture approximately 12% of patients with PDAC with HRD tumors. Nonetheless, it is important to underline that although the extension of the genes included in NGS panel testing increases the probability of HRD identification, the clinical relevance of HRD candidates, beyond the core BRCA1, BRCA2, and PALB2 genes, has yet to be established.68,69 Many other outstanding questions still need to be addressed, including the clinical differences between somatic versus germline mutations, monoallelic versus biallelic inactivation (including epigenetic silencing), the specific functional consequence of each molecular alteration in predicting therapeutic sensitivity to therapy with platinum, PARPi, and other novel agents that target DDR, and the mechanisms of primary or secondary resistance (including the role of secondary mutations in DDR genes). A possible solution to assessing the functional contribution of each specific alteration, or potentially biomarkers in their own right, is to assess various patterns in genomic aberrations across the genome that represent defects in DDR mechanisms. These surrogate genomic readouts of HRD such as genomic scarring as well as point mutational and structural variant signatures70-72 have the potential to deliver clinically relevant genomic information and to detect HRD beyond point mutations in known HR genes in an additional 10%-15% of tumors in cohort-based and preclinical studies.7,36,70,73 Our results showed that when these surrogate measures are used, like in studies based on WGS technologies, the probability of capturing HRD rises significantly (up to 44%).7,33 The central challenge is that although WGS can define putative biomarkers of therapeutic response in cohort studies of breast73 and pancreatic cancer,19 these methodologies are not currently translatable to the clinic. Routine formalin-fixed clinical biopsies used for sequencing are often small, and current diagnostic assays are mostly focused on the coding regions. Technology continues to advance, and one day, WGS may integrate seamlessly into the health system and deliver routine results; however, in the meantime, we require a feasible diagnostic that can capture surrogate readouts of HRD that can be tested in clinical trials. An additional current challenge is the probability of loss of the second allele in a given HRD gene. Although this occurs in 90% or more in the case of germline BRCA1 and BRCA2,74,75 allowing the assumption that if a mutation in one allele is detected, the second is inactivated in 90%, the rate of loss of the second allele for somatic BRCA mutations is not well characterized. In particular, germline and somatic events in other HRD genes are largely undefined. The second allele is often inactivated through copy-number alterations and structural variants and can be difficult to detect and interpret, especially if the epithelial cellular content falls below 30%, with rates of epigenetic inactivation largely unknown beyond BRCA. This different rate of second allelic loss impacts on penetrance for predisposition assessments of novel candidate genes and substantially affects therapeutic development for non-BRCA HRD genes. HRD driven by gene-level events beyond the core HRD genes may potentially be the consequence of a large diverse group of genes with low rates of second allelic inactivation, making surrogate measures more attractive. Setting a threshold to define HRD and the predictive therapeutic value of HRD defined in this way as with noncore HRD genes will require clinical testing and validation.7,26,33,76,77 What is needed is a feasible diagnostic that can assess gene-level events and signatures and can use formalin-exposed material from small biopsies (Fig 2).78,79 Park et al80 recently showed that pathogenic somatic or germline mutations in core HRD genes (BRCA1/2, PALB2) and biallelic loss of other rarer HRD genes determined through targeted-capture NGS are associated with improved survival in patients with advanced HRD PDAC treated with platinum-based chemotherapy. Loss of the second allele was more prevalent in BRCA1 and BRCA2 versus other HR genes. They were also able to determine surrogate measures of HRD in a subgroup of patients (large-scale transition, point mutational Signature 3,70 and genomic instability) from the same assay, which predicted platinum response and improved survival. Notably, HRD PDAC is associated with increased tumor mutation burden,81 offering opportunities for combining immunotherapy with PARP inhibition in this subgroup.80,82
FIG 2.

Overview of HRD identification and clinical implications. Although WGS represents the most comprehensive method for HRD identification as it delivers integrated analyses of all genomic events, many barriers limit its utilization in the clinic, feasibility of accessing fresh biopsy material of sufficient size, cost, and analytic complexity. WES is a more accessible strategy and is often proposed as the second choice. However, it seems not to be the optimal method for cancer profiling as many driver events occur outside the coding exome may be missed, on one hand, and the majority of included genes are not cancer genes, on the other. Despite some technical limitations, targeted-capture sequencing delivering comprehensive genomic information, including individual gene mutations, signatures, and structural variation patterns, may represent a reasonable option for real-world applicability (practical and financial advantages compared with WES and WGS). Rating level of sequencing technologies: ++, optimal; +, good; ±, low; –, poor. aFunctional assays for real-time HRD status require in vivo or in vitro experiments. CNA, copy-number alterations; HRD, homologous recombination deficiency; LOH, loss of heterozygosity; LST, large-scale transitions; NGS, next-generation sequencing; PDCL, patient-derived cell lines; PDO, patient-derived organoids; PDX, patient-derived xenograft; TAI, telomeric allelic imbalance; WES, whole-exome sequencing; WGS, whole-genome sequencing.
Concerning germline alterations, our results further support the current National Comprehensive Cancer Network guidelines recommending routine screening for germline variants in patients with PDAC at diagnosis, regardless of age, ancestry, and family or personal history of cancer, including not only BRCA1/2 but also ATM, CDKN2A, PALB2, STK11, TP53, MLH1, MSH2, MSH6, and PMS2.20 Based on our findings, FANC genes and CHEK2 should be added to that list. Given the relevant clinical implications of identifying mutation carriers without a family history, current guidelines for germline testing should be reassessed.62,83,84 Broader testing would maximize the identification of patients suitable for approved and investigational therapies and would positively impact on prevention strategies of a broad spectrum of tumors in healthy family members through cascade testing.13,85,86
The result of this systematic review and meta-analysis should be interpreted with caution, given the following limitations: (1) ascertainment bias because of the exclusion of manuscripts that were not considered of interest because of vague methods or reporting of results; (2) limited data on baseline patient characteristics in the majority of studies; (3) heterogeneity among studies and included patient populations; (4) variability in sequencing methodologies; (5) considerable heterogeneity in some meta-analysis results; (6) inherent publication bias, where research on the topic may be skewed toward the publication of significant results only47,87; and (7) risk of bias at study level. In this regard, there was significant inaccuracy and inconsistency in reporting methods and results, thus making conclusions partially comparable only. This, together with the aforementioned limitations, likely hampers analysis and interpretation. To improve study quality and ensure transparency and standardization of reporting results, we propose a modified (m-) REMARK criteria88 as a checklist for future translational cancer genomic studies (Table 3). m-REMARK has also been used to derive an internal risk-of-bias assessment tool at study level, specifically developed for studies focused on germline or somatic mutation analysis. After this initial validation, further investigation will be essential to more accurately validate Translating-ROB, as well as m-REMARK.
TABLE 3.
Modified REMARK

Because of general poor reporting of information, it was not possible to systematically assess prevalence variations according to disease stage or patient age. However, individual studies did not report substantial discrepancies in HRD frequency between early-stage and late-stage patients.8,28,94 Similarly, the majority of studies did not identify statistically significant age variations between germline mutation carriers and wild-type patients,8,61,95-98 and three studies evaluating the mutation rate in young-onset patients (< 50 years) did not find a significant difference compared with older patients.25,83,99 Further large studies are necessary to clarify these important aspects.
Last, general poor reporting of ethnicity and focus on Caucasian or White populations of the majority of studies not only represents a limitation that hampers the wide generalizability of research findings to underrepresented populations, but also highlights major disparities in access to cancer research programs. Interestingly, geographic and ethnic heterogeneity of BRCA mutation prevalence among patients with PDAC has recently been described,100 but further studies are needed to understand ethnic variations of genomic events and related clinical implications.
In conclusion, HRD constitutes a prevalent and clinically relevant pathway in PDAC. Preclinical and clinical data support that every patient with newly diagnosed PDAC should be tested for HRD and ideally enrolled in biomarker-enriched clinical trials (Table 4). Based on our study and available literature, integrated HRD assessment, including germline and somatic analysis, represents the current ideal approach, with the highest potential to drive therapeutic choices not only in metastatic but also in early-stage disease.13,80,85,86,101 Nevertheless, major efforts are necessary to harmonize HRD definition and to find the optimal biomarker for treatment selection. Although surrogate readouts of HRD can identify a greater proportion of patients with HRD than analyses limited to gene-level approaches, they need to be assessed in clinical trials, and before widespread adoption would require a diagnostic capable of feasibly detecting genomic signatures in the majority of patients. Expanding research on integrated WGS or WES and transcriptomic profiling, together with functional analyses, is also necessary to unravel the complex biology of HRD in PDAC, to elucidate the predictive value of HRD aberrations beyond core genes, and to understand the real-time HRD status.
TABLE 4.
Clinical Recommendations on HRD Testing (including HR-related cancer susceptibility genes)

ACKNOWLEDGMENT
The authors gratefully thank the Italian BIostatistics Group (IBIG) for their contribution to this research.
Philip A. Beer
Employment: Tessellex Ltd
Stock and Other Ownership Interests: Karus Therapeutics
Talia Golan
Honoraria: MSD, Rafael Pharmaceuticals
Consulting or Advisory Role: AbbVie, AstraZeneca, Bayer, MSD, Teva
Speakers' Bureau: AbbVie, AstraZeneca
Research Funding: AstraZeneca, MSD
Travel, Accommodations, Expenses: AstraZeneca, MSD
Chiara Braconi
Honoraria: Bayer, Menarini Silicon Biosystems, Pfizer, Merck Serono, Lilly
Consulting or Advisory Role: Incucyte
Michele Milella
Honoraria: Pfizer, MSD, AstraZeneca, Roche, EUSA Pharma, Boehringer Ingelheim, Ipsen
Aldo Scarpa
Consulting or Advisory Role: Incyte
Speakers' Bureau: MSD, Amgen, GlaxoSmithKline/Tesaro
Giuseppe Malleo
Research Funding: FibroGen
David K. Chang
Speakers' Bureau: Celgene, Viatris
Research Funding: Celgene, AstraZeneca, MSD Oncology
Travel, Accommodations, Expenses: Celgene
Andrew V. Biankin
Employment: AstraZeneca/MedImmune, BMSi
Leadership: Cambridge Cancer Genomics, Concr, Wollemia Oncology, Gabriel Precision Oncology, Cumulus Oncology
Stock and Other Ownership Interests: Cumulus Oncology, Modulus Oncology, Wollemia Oncology, Concur, Cambridge Cancer Genomics, Gabriel Precision Oncology, human.ai
Honoraria: Havas Lynx Group
Consulting or Advisory Role: AstraZeneca/MedImmune
Speakers' Bureau: Celgene
Research Funding: Celgene, AstraZeneca/MedImmune
Patents, Royalties, Other Intellectual Property: Agilent Technologies—Royalty payments to Institute (University of Glasgow)
No other potential conflicts of interest were reported.
SUPPORT
Associazione Italiana Ricerca Cancro (AIRC 5x1000 no. 18718). Associazione Italiana Ricerca Cancro (AIRC 5x1000 no. 12182). Fondazione Italiana Malattie Pancreas—Italian Ministry of Health (CUP_J38D19000690001). Fondazione Cariverona: Oncology Biobank Project Antonio Schiavi (prot. 203885/2017). Cancer Research UK C29717/A17263, C29717/A18484, C596/A18076, C596/A20921, C29717/A23526. Wellcome Trust Senior Investigator Award: 103721/Z/14/Z. Pancreatic Cancer UK Future Research Leaders Fund FLF2015_04_Glasgow. Scottish Genomes Partnership SEHHD-CSO 1175759/2158447. MRC/EPSRC Glasgow Molecular Pathology Node MR/N005813/1. The Howat Foundation.
AUTHOR CONTRIBUTIONS
Conception and design: Raffaella Casolino, Salvatore Paiella, Talia Golan, Aldo Scarpa, Giuseppe Malleo, Roberto Salvia, Claudio Bassi, David K. Chang, Andrew V. Biankin
Financial support: Vincenzo Corbo, Aldo Scarpa, Andrew V. Biankin
Administrative support: David K. Chang, Andrew V. Biankin
Provision of study materials or patients: Raffaella Casolino, David K. Chang
Collection and assembly of data: Raffaella Casolino, Salvatore Paiella, Giuseppe Malleo, Roberto Salvia, Claudio Bassi, David K. Chang
Data analysis and interpretation: Raffaella Casolino, Salvatore Paiella, Danila Azzolina, Philip A. Beer, Vincenzo Corbo, Giulia Lorenzoni, Dario Gregori, Talia Golan, Chiara Braconi, Fieke E. M. Froeling, Michele Milella, Aldo Scarpa, Antonio Pea, Giuseppe Malleo, Roberto Salvia, Claudio Bassi, David K. Chang
Manuscript writing: Raffaella Casolino, Andrew V. Biankin
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Homologous Recombination Deficiency in Pancreatic Cancer: A Systematic Review and Prevalence Meta-Analysis
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Philip A. Beer
Employment: Tessellex Ltd
Stock and Other Ownership Interests: Karus Therapeutics
Talia Golan
Honoraria: MSD, Rafael Pharmaceuticals
Consulting or Advisory Role: AbbVie, AstraZeneca, Bayer, MSD, Teva
Speakers' Bureau: AbbVie, AstraZeneca
Research Funding: AstraZeneca, MSD
Travel, Accommodations, Expenses: AstraZeneca, MSD
Chiara Braconi
Honoraria: Bayer, Menarini Silicon Biosystems, Pfizer, Merck Serono, Lilly
Consulting or Advisory Role: Incucyte
Michele Milella
Honoraria: Pfizer, MSD, AstraZeneca, Roche, EUSA Pharma, Boehringer Ingelheim, Ipsen
Aldo Scarpa
Consulting or Advisory Role: Incyte
Speakers' Bureau: MSD, Amgen, GlaxoSmithKline/Tesaro
Giuseppe Malleo
Research Funding: FibroGen
David K. Chang
Speakers' Bureau: Celgene, Viatris
Research Funding: Celgene, AstraZeneca, MSD Oncology
Travel, Accommodations, Expenses: Celgene
Andrew V. Biankin
Employment: AstraZeneca/MedImmune, BMSi
Leadership: Cambridge Cancer Genomics, Concr, Wollemia Oncology, Gabriel Precision Oncology, Cumulus Oncology
Stock and Other Ownership Interests: Cumulus Oncology, Modulus Oncology, Wollemia Oncology, Concur, Cambridge Cancer Genomics, Gabriel Precision Oncology, human.ai
Honoraria: Havas Lynx Group
Consulting or Advisory Role: AstraZeneca/MedImmune
Speakers' Bureau: Celgene
Research Funding: Celgene, AstraZeneca/MedImmune
Patents, Royalties, Other Intellectual Property: Agilent Technologies—Royalty payments to Institute (University of Glasgow)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Kamisawa T Wood LD Itoi T, et al. : Pancreatic cancer. Lancet 388:73-85, 2016 [DOI] [PubMed] [Google Scholar]
- 2.Bray F Ferlay J Soerjomataram I, et al. : Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394-424, 2018 [DOI] [PubMed] [Google Scholar]
- 3.Rahib L Wehner MR Matrisian LM, et al. : Estimated projection of US cancer incidence and death to 2040. JAMA Netw Open 4:e214708, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carioli G Malvezzi M Bertuccio P, et al. : European cancer mortality predictions for the year 2021 with focus on pancreatic and female lung cancer. Ann Oncol 32:478-487, 2021 [DOI] [PubMed] [Google Scholar]
- 5.Pereira SP Oldfield L Ney A, et al. : Early detection of pancreatic cancer. Lancet Gastroenterol Hepatol 5:698-710, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christenson ES, Jaffee E, Azad NS: Current and emerging therapies for patients with advanced pancreatic ductal adenocarcinoma: A bright future. Lancet Oncol 21:e135-e145, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waddell N Pajic M Patch AM, et al. : Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518:495-501, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holter S Borgida A Dodd A, et al. : Germline BRCA mutations in a large clinic-based cohort of patients with pancreatic adenocarcinoma. J Clin Oncol 33:3124-3129, 2015 [DOI] [PubMed] [Google Scholar]
- 9.Slavin TP Neuhausen SL Nehoray B, et al. : The spectrum of genetic variants in hereditary pancreatic cancer includes Fanconi anemia genes. Fam Cancer 17:235-245, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wattenberg MM Asch D Yu S, et al. : Platinum response characteristics of patients with pancreatic ductal adenocarcinoma and a germline BRCA1, BRCA2 or PALB2 mutation. Br J Cancer 122:333-339, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sehdev A Gbolahan O Hancock BA, et al. : Germline and somatic DNA damage repair gene mutations and overall survival in metastatic pancreatic adenocarcinoma patients treated with FOLFIRINOX. Clin Cancer Res 24:6204-6211, 2018 [DOI] [PubMed] [Google Scholar]
- 12.Park W Chen J Chou JF, et al. : Genomic methods identify homologous recombination deficiency in pancreas adenocarcinoma and optimize treatment selection. Clin Cancer Res 26:3239-3247, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Golan T Hammel P Reni M, et al. : Olaparib as maintenance treatment following first-line platinumbased chemotherapy in patients with a germline BRCA mutation and metastatic pancreatic cancer: Phase III POLO trial. Ann Oncol 30, 2019. (abstr IV152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Golan T Stossel C Atias D, et al. : Recapitulating the clinical scenario of BRCA-associated pancreatic cancer in pre-clinical models. Int J Cancer 143:179-183, 2018 [DOI] [PubMed] [Google Scholar]
- 15.Zhu H Wei M Xu J, et al. : PARP inhibitors in pancreatic cancer: Molecular mechanisms and clinical applications. Mol Cancer 19:49, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Golan T Varadhachary GR Sela T, et al. : Phase II study of olaparib for BRCAness phenotype in pancreatic cancer. J Clin Oncol 36:297, 2018 [Google Scholar]
- 17.Javle M Shacham-Shmueli E Xiao L, et al. : Olaparib monotherapy for previously treated pancreatic cancer with DNA damage repair genetic alterations other than germline BRCA variants: Findings from 2 phase 2 nonrandomized clinical trials. JAMA Oncol 7:693-699, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perkhofer L Gout J Roger E, et al. : DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 70:606-617, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dreyer SB Upstill-Goddard R Paulus-Hock V, et al. : Targeting DNA damage response and replication stress in pancreatic cancer. Gastroenterology 160:362-377.e13, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daly MB Pilarski R Yurgelun MB, et al. : NCCN guidelines insights: Genetic/familial high-risk assessment: Breast, ovarian, and pancreatic, version 1.2020. J Natl Compr Canc Netw 18:380-391, 2020 [DOI] [PubMed] [Google Scholar]
- 21.Bailey P Chang DK Nones K, et al. : Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531:47-52, 2016 [DOI] [PubMed] [Google Scholar]
- 22.Heeke AL Pishvaian MJ Lynce F, et al. : Prevalence of homologous recombination-related gene mutations across multiple cancer types. JCO Precis Oncol 10.1200/PO.17.00286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Witkiewicz AK McMillan EA Balaji U, et al. : Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6:6744, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Connor AA Denroche RE Jang GH, et al. : Association of distinct mutational signatures with correlates of increased immune activity in pancreatic ductal adenocarcinoma. JAMA Oncol 3:774-783, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singhi AD George B Greenbowe JR, et al. : Real-time targeted genome profile analysis of pancreatic ductal adenocarcinomas identifies genetic alterations that might be targeted with existing drugs or used as biomarkers. Gastroenterology 156:2242-2253.e4, 2019 [DOI] [PubMed] [Google Scholar]
- 26.Shahda S Timms KM Ibrahim AA, et al. : Homologous recombination deficiency in patients with pancreatic ductal adenocarcinoma and response to chemotherapy. JCO Precis Oncol 10.1200/PO.17.00087 [DOI] [PubMed] [Google Scholar]
- 27.Golan T, Javle M: DNA repair dysfunction in pancreatic cancer: A clinically relevant subtype for drug development. J Natl Compr Canc Netw 15:1063-1069, 2017 [DOI] [PubMed] [Google Scholar]
- 28.Pishvaian MJ Blais EM Brody JR, et al. : Outcomes in patients with pancreatic adenocarcinoma with genetic mutations in DNA damage response pathways: Results from the know your tumor program. JCO Precis Oncol 10.1200/PO.19.00115 [DOI] [PubMed] [Google Scholar]
- 29.Kondo T Kanai M Kou T, et al. : Association between homologous recombination repair gene mutations and response to oxaliplatin in pancreatic cancer. Oncotarget 9:19817-19825, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golan T, Brody JR: Targeting homologous recombination addicted tumors: Challenges and opportunities. Ann Pancreat Cancer 3:6, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller RE Leary A Scott CL, et al. : ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann Oncol 31:1606-1622, 2020 [DOI] [PubMed] [Google Scholar]
- 32.O'Connor MJ: Targeting the DNA damage response in cancer. Mol Cell 60:547-560, 2015 [DOI] [PubMed] [Google Scholar]
- 33.Aguirre AJ Nowak JA Camarda ND, et al. : Real-time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discov 8:1096-1111, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biankin AV Waddell N Kassahn KS, et al. : Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491:399-405, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Witkiewicz AK McMillan EA Balaji U, et al. : Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6:6744, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golan T O'Kane GM Denroche RE, et al. : Genomic features and classification of homologous recombination deficient pancreatic ductal adenocarcinoma. Gastroenterology 160:2119-2132.e9, 2021 [DOI] [PubMed] [Google Scholar]
- 37.Moher D Liberati A Tetzlaff J, et al. : Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med 6:e1000097, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.National Institute for Health Research: International prospective register of systematic reviews. https://www.crd.york.ac.uk/prospero/
- 39.de Bono J Mateo J Fizazi K, et al. : Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med 382:2091-2102, 2020 [DOI] [PubMed] [Google Scholar]
- 40.von Elm E Poglia G Walder B, et al. : Different patterns of duplicate publication: An analysis of articles used in systematic reviews. JAMA 291:974-980, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Li MM Datto M Duncavage EJ, et al. : Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 19:4-23, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richards S Aziz N Bale S, et al. : Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405-424, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hampel H Bennett RL Buchanan A, et al. : A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet Med 17:70-87, 2015 [DOI] [PubMed] [Google Scholar]
- 44.Bashford MT Kohlman W Everett J, et al. : Addendum: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet Med 21:2844, 2019 [DOI] [PubMed] [Google Scholar]
- 45.Freeman MF, Tukey JW: Transformation related to the angular and the square root. Ann Math Statist 21:607-611, 1950 [Google Scholar]
- 46.DerSimonian R, Laird N: Meta-analysis in clinical trials. Control Clin Trials 7:177-188, 1986 [DOI] [PubMed] [Google Scholar]
- 47.Higgins JP Thompson SG Deeks JJ, et al. : Measuring inconsistency in meta-analyses. BMJ 327:557-560, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guyatt GH Oxman AD Kunz R, et al. : GRADE guidelines: 7. Rating the quality of evidence—Inconsistency. J Clin Epidemiol 64:1294-1302, 2011 [DOI] [PubMed] [Google Scholar]
- 49.Hunter JP Saratzis A Sutton AJ, et al. : In meta-analyses of proportion studies, funnel plots were found to be an inaccurate method of assessing publication bias. J Clin Epidemiol 67:897-903, 2014 [DOI] [PubMed] [Google Scholar]
- 50.Macaskill P, Walter SD, Irwig L: A comparison of methods to detect publication bias in meta-analysis. Stat Med 20:641-654, 2001 [DOI] [PubMed] [Google Scholar]
- 51.Begg CB: A comparison of methods to detect publication bias in meta-analysis by P. Macaskill, S. D. Walter and L. Irwig, Statistics in Medicine, 2001; 20:641-654. Stat Med 21:1803, 2002; author reply 1804 [DOI] [PubMed] [Google Scholar]
- 52.Farrah K Young K Tunis MC, et al. : Risk of bias tools in systematic reviews of health interventions: An analysis of PROSPERO-registered protocols. Syst Rev 8:280, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sterne JA Hernan MA Reeves BC, et al. : ROBINS-I: A tool for assessing risk of bias in non-randomised studies of interventions. BMJ 355:i4919, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toma S Emionite L Scaramuccia A, et al. : Retinoids and human breast cancer: In vivo effects of an antagonist for RAR-alpha. Cancer Lett 219:27-31, 2005 [DOI] [PubMed] [Google Scholar]
- 55.Viechtbauer W: Conducting meta-analyses in R with the metafor package. J Stat Softw 36:1-48, 2010 [Google Scholar]
- 56.Lê S, Josse J, Husson F: FactoMineR: An R package for multivariate analysis. J Stat Softw 25:1-18, 2008 [Google Scholar]
- 57.cBioPortal for Cancer Genomics. https://www.cbioportal.org/
- 58.Genomic Data Commons Data Portal. https://portal.gdc.cancer.gov/
- 59.International Cancer Genome Consortium Data Portal. https://dcc.icgc.org/
- 60.Cancer Genome Atlas Research Network : Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32:185-203.e13, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yurgelun MB Chittenden AB Morales-Oyarvide V, et al. : Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet Med 21:213-223, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lowery MA Wong W Jordan EJ, et al. : Prospective evaluation of germline alterations in patients with exocrine pancreatic neoplasms. J Natl Cancer Inst 110:1067-1074, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Armstrong SA Schultz CW Azimi-Sadjadi A, et al. : ATM dysfunction in pancreatic adenocarcinoma and associated therapeutic implications. Mol Cancer Ther 18:1899-1908, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cavaciuti E Lauge A Janin N, et al. : Cancer risk according to type and location of ATM mutation in ataxia-telangiectasia families. Genes Chromosomes Cancer 42:1-9, 2005 [DOI] [PubMed] [Google Scholar]
- 65.Roberts NJ Jiao Y Yu J, et al. : ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov 2:41-46, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pritchard CC Mateo J Walsh MF, et al. : Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med 375:443-453, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marabelli M, Cheng SC, Parmigiani G: Penetrance of ATM gene mutations in breast cancer: A meta-analysis of different measures of risk. Genet Epidemiol 40:425-431, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tung NM Robson ME Ventz S, et al. : TBCRC 048: Phase II study of olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J Clin Oncol 38:4274-4282, 2020 [DOI] [PubMed] [Google Scholar]
- 69.Hussain M Mateo J Fizazi K, et al. : Survival with olaparib in metastatic castration-resistant prostate cancer. N Engl J Med 383:2345-2357, 2020 [DOI] [PubMed] [Google Scholar]
- 70.Alexandrov LB Kim J Haradhvala NJ, et al. : The repertoire of mutational signatures in human cancer. Nature 578:94-101, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McBride DJ Etemadmoghadam D Cooke SL, et al. : Tandem duplication of chromosomal segments is common in ovarian and breast cancer genomes. J Pathol 227:446-455, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ng CK Cooke SL Howe K, et al. : The role of tandem duplicator phenotype in tumour evolution in high-grade serous ovarian cancer. J Pathol 226:703-712, 2012 [DOI] [PubMed] [Google Scholar]
- 73.Davies H Glodzik D Morganella S, et al. : HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 23:517-525, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nguyen L Martens JWM Van Hoeck A, et al. : Pan-cancer landscape of homologous recombination deficiency. Nat Commun 11:5584, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sokol ES Pavlick D Khiabanian H, et al. : Pan-cancer analysis of BRCA1 and BRCA2 genomic alterations and their association with genomic instability as measured by genome-wide loss of heterozygosity. JCO Precis Oncol 10.1200/PO.19.00345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.O'Kane GM Denroche R Picardo SL, et al. : Homologous recombination deficiency (HRD) scoring in pancreatic ductal adenocarcinoma (PDAC) and response to chemotherapy. J Clin Oncol 38:741, 2020 [Google Scholar]
- 77.Swanton C Soria JC Bardelli A, et al. : Consensus on precision medicine for metastatic cancers: A report from the MAP conference. Ann Oncol 27:1443-1448, 2016 [DOI] [PubMed] [Google Scholar]
- 78.Beer PA Cooke SL Chang DK, et al. : Reasons to be testing: The dawn of complex molecular profiling in routine oncology practice. Ann Oncol 30:1691-1694, 2019 [DOI] [PubMed] [Google Scholar]
- 79.Beer PA Cooke SL Chang DK, et al. : Defining the clinical genomic landscape for real-world precision oncology. Genomics 112:5324-5330, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Park W Chen J Chou JF, et al. : Genomic methods identify homologous recombination deficiency in pancreas adenocarcinoma and optimize treatment selection. Clin Cancer Res 26:3239-3247, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Humphris JL Patch AM Nones K, et al. : Hypermutation in pancreatic cancer. Gastroenterology 152:68-74.e2, 2017 [DOI] [PubMed] [Google Scholar]
- 82.Seeber A Zimmer K Kocher F, et al. : Molecular characteristics of BRCA1/2 and PALB2 mutations in pancreatic ductal adenocarcinoma. ESMO Open 5:e000942, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salo-Mullen EE O'Reilly EM Kelsen DP, et al. : Identification of germline genetic mutations in patients with pancreatic cancer. Cancer 121:4382-4388, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shindo K Yu J Suenaga M, et al. : Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol 35:3382-3390, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Golan T Barenboim A Lahat G, et al. : Increased rate of complete pathologic response after neoadjuvant FOLFIRINOX for BRCA mutation carriers with borderline resectable pancreatic cancer. Ann Surg Oncol 27:3963-3970, 2020 [DOI] [PubMed] [Google Scholar]
- 86.Yu S Agarwal P Mamtani R, et al. : Retrospective survival analysis of patients with resected pancreatic ductal adenocarcinoma and a germline BRCA or PALB2 mutation. JCO Precis Oncol 10.1200/PO.18.00271 [DOI] [PubMed] [Google Scholar]
- 87.Kicinski M, Springate DA, Kontopantelis E: Publication bias in meta-analyses from the cochrane database of systematic reviews. Stat Med 34:2781-2793, 2015 [DOI] [PubMed] [Google Scholar]
- 88.McShane LM Altman DG Sauerbrei W, et al. : REporting recommendations for tumour MARKer prognostic studies (REMARK). Br J Cancer 93:387-391, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jennings LJ Arcila ME Corless C, et al. : Guidelines for validation of next-generation sequencing-based oncology panels: A joint consensus recommendation of the Association for Molecular Pathology and College of American Pathologists. J Mol Diagn 19:341-365, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Human Gene Mutation Database. http://www.hgmd.org
- 91.COSMIC Catalog of Somatic Mutations in Cancer. http://cancer.sanger.ac.uk/cosmic
- 92.Ensembl Variant Effect Predictor (VEP). https://www.ensembl.org/info/docs/tools/vep/index.html
- 93.OMIM: National Center for Biotechnology Information. http://www.ncbi.nlm.nih.gov/omim
- 94.Smith AL Alirezaie N Connor A, et al. : Candidate DNA repair susceptibility genes identified by exome sequencing in high-risk pancreatic cancer. Cancer Lett 370:302-312, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Takeuchi S Doi M Ikari N, et al. : Mutations in BRCA1, BRCA2, and PALB2, and a panel of 50 cancer-associated genes in pancreatic ductal adenocarcinoma. Sci Rep 8:8105, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brand R Borazanci E Speare V, et al. : Prospective study of germline genetic testing in incident cases of pancreatic adenocarcinoma. Cancer 124:3520-3527, 2018 [DOI] [PubMed] [Google Scholar]
- 97.Lucas AL Frado LE Hwang C, et al. : BRCA1 and BRCA2 germline mutations are frequently demonstrated in both high-risk pancreatic cancer screening and pancreatic cancer cohorts. Cancer 120:1960-1967, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ferrone CR Levine DA Tang LH, et al. : BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J Clin Oncol 27:433-438, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smith AL Wong C Cuggia A, et al. : Reflex testing for germline BRCA1, BRCA2, PALB2, and ATM mutations in pancreatic cancer: Mutation prevalence and clinical outcomes from two Canadian research registries. JCO Precis Oncol 10.1200/PO.17.00098 [DOI] [PubMed] [Google Scholar]
- 100.Golan T Kindler HL Park JO, et al. : Geographic and ethnic heterogeneity of germline BRCA1 or BRCA2 mutation prevalence among patients with metastatic pancreatic cancer screened for entry into the POLO trial. J Clin Oncol 38:1442-1454, 2020 [DOI] [PubMed] [Google Scholar]
- 101.Casolino R Braconi C Malleo G, et al. : Reshaping preoperative treatment of pancreatic cancer in the era of precision medicine. Ann Oncol 32:183-196, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]