

Abstract
The vascular interface of the brain, known as the blood–brain barrier (BBB), is understood to maintain brain function in part via its low transcellular permeability1–3. Yet, recent studies have demonstrated that brain ageing is sensitive to circulatory proteins4,5. Thus, it is unclear whether permeability to individually injected exogenous tracers–as is standard in BBB studies–fully represents blood-to-brain transport. Here we label hundreds of proteins constituting the mouse blood plasma proteome, and upon their systemic administration, study the BBB with its physiological ligand. We find that plasma proteins readily permeate the healthy brain parenchyma, with transport maintained by BBB-specific transcriptional programmes. Unlike IgG antibody, plasma protein uptake diminishes in the aged brain, driven by an age-related shift in transport from ligand-specific receptor-mediated to non-specific caveolar transcytosis. This age-related shift occurs alongside a specific loss of pericyte coverage. Pharmacological inhibition of the age-upregulated phosphatase ALPL, a predicted negative regulator of transport, enhances brain uptake of therapeutically relevant transferrin, transferrin receptor antibody and plasma. These findings reveal the extent of physiological protein transcytosis to the healthy brain, a mechanism of widespread BBB dysfunction with age and a strategy for enhanced drug delivery.
To support brain function, numerous transporters selectively traffic solutes across the brain’s vascular and cerebrospinal fluid (CSF) barriers1–3,6. In parallel, these barriers are understood to exhibit low permeability to most macromolecules, as measured with various exogenous tracers (Supplementary Table 1) since around the year 19007. But this leaves the brain’s physiological interaction with and permeability to thousands of endogenous blood proteins largely unexplored. After all, with each heartbeat, brain endothelial and ependymal epithelial cells are met by a new wave of diverse blood plasma proteins, probably forming a dynamic interface teeming with binding, signaling and trafficking events (Fig. 1a, Extended Data Fig. 1a–c).
Fig. 1|. Circulatory proteins permeate the healthy adult brain.
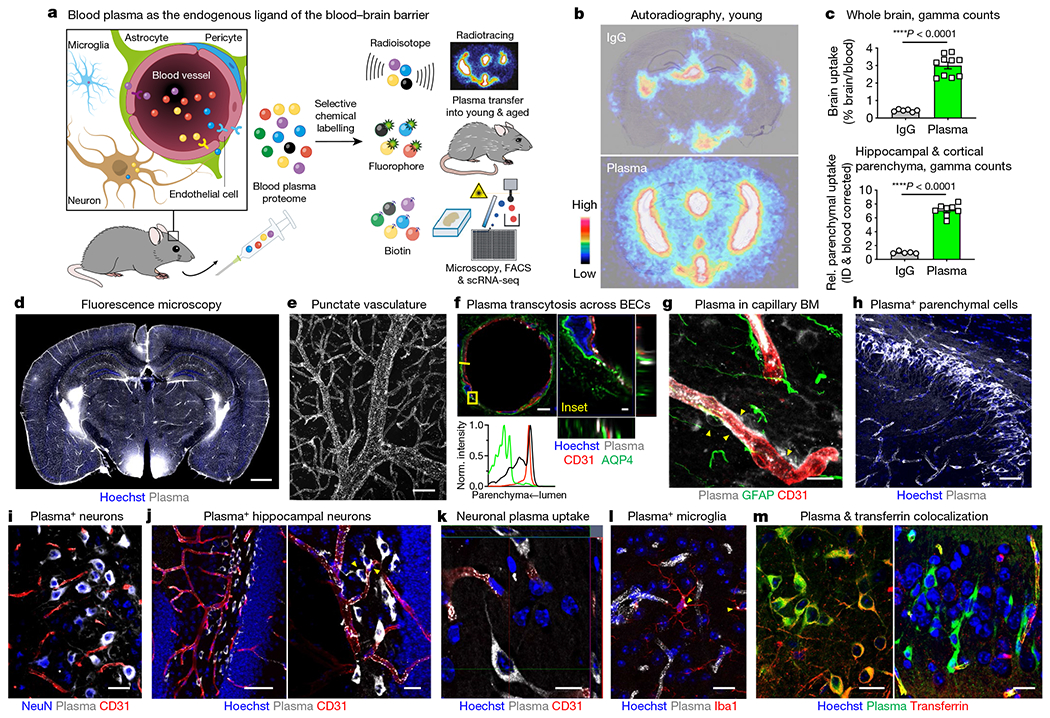
a, Study rationale. b, Brain autoradiography of 64Cu-labelled IgG and albumin (Alb)/IgG-depleted plasma (7.7 MBq matched dose, about 20 μg) injected intravenously and assessed 20 h later. Images are overlaid with Nissl stain. c, Gamma counting of whole brains (top) and devascularized hippocampal and cortical parenchyma (bottom) following the paradigm shown in a (n = 6 mice IgG and n = 10 plasma whole brain; n = 5 IgG and n = 8 plasma parenchyma; two-sided t-test; mean ± s.e.m.). ID, injected dose. d, Fluorescence microscopy of plasma uptake across a coronal brain tile. Scale bar, 1,000 μm. e, Punctated plasma+ vasculature. Scale bar, 20 μm. f, Abluminal plasma beyond CD31+ brain endothelial cells (BECs) and aquaporin 4 (AQP4)+ astrocyte endfeet. Histogram fluorescence intensity projections are along the yellow line and are normalized per channel (plasma is in black). Scale bars, 5 μm (left), 1 μm (right). g, Plasma in capillary basement membranes (BMs) and pericytes (arrowheads). Scale bar, 10 μm. h, Plasma in hippocampal parenchymal cells. Scale bar, 50 μm. i, Plasma in NeuN+ neurons. Scale bar, 20 μm. j, Plasma in hippocampal dentate gyrus neurons, beyond and interacting with (arrowheads) CD31+ vasculature. Scale bars, 50 μm (left), 20 μm (right). k, Orthogonal projection of plasma in neuronal cytoplasm. Scale bar, 10 μm. l, Plasma in Iba1+ microglia. Scale bar, 10 μm. m, Neurons with both transferrin and plasma (left), and only plasma (right). Scale bars, 20 μm. Fluorescence images after transfer of 150 μl of plasma and waiting for 4 h (d–f), 6 h (f, g) or 20 h (h–m).
Thus, we chemoselectively labelled the mouse plasma proteome with various small tags to enable a panel of studies following transfer into recipient young or aged mice (Fig. 1a). After optimizing conditions for N-hydroxysuccinimide (NHS) ester chemistry, we confirmed labelling of hundreds of plasma proteins across abundance, size and function using antibody array and mass spectrometry proteomics, with coverage limited by detection method (Extended Data Fig. 1d–g). Twenty hours after transfer into healthy mice, radiotracer 64Cu-labelled plasma depleted of albumin and IgG accumulated at significantly higher levels than IgG control in both the leakier, fenestrated circumventricular organs and within BBB-protected regions, even after extensive transcardial perfusion (Fig. 1b, c, Extended Data Figs. 2, 3a, b).
To study plasma uptake at a cellular resolution, we labelled whole plasma with the small but bright far-red Atto 647N fluorophore. Fluorescence imaging bore a strong resemblance to autoradiography (Fig. 1d). Plasma administration did not induce hyperproteinaemia or perturb the cerebrovasculature, measured functionally by various fixable and non-fixable tracers and transcriptionally (Extended Data Fig. 3c–h). Plasma uptake was detectable across injection volumes down to 10 μl (about 0.5% of the circulatory volume), was seen with various ex vivo and in vivo labelling chemistries, was largely not colocalized with albumin or IgG (Extended Data Fig. 3i–k), and was not appreciably confounded by residual free label or degradation products (Extended Data Figs. 4, 5q).
At greater magnification, we noticed two cardinal features: a punctate vasculature and plasma-containing (plasma+) parenchymal cells (Fig. 1e–m, Extended Data Fig. 5). A punctate vasculature suggests a high level of endocytosis by the brain endothelium to endogenous plasma proteins that was not reported with exogenous tracers8. Plasma transcytosed across the endothelium, suffusing the basement membrane and thin-strand pericytes (Fig. 1f, g, Extended Data Figs. 5j, k, 6h–k). Plasma accumulated in the choroid plexus, subarachnoid space and perivascular spaces, signifying contributions by the blood–CSF barriers (BCSFBs) (Extended Data Figs. 5l, m, 6a–g, Supplementary Video 1). While permeability for select proteins such as transferrin and insulin have been investigated in crude brain homogenate or isolated vessels9, we could visualize plasma+ neurons and microglia in situ across brain regions (Fig. 1i–l). In the hippocampal dentate gyrus, plasma traced the vasculature and adjoining neuronal processes (Fig. 1i, j). By staining for transferrin, the basis of central nervous system (CNS) drug delivery programmes10,11, we observed that most plasma+ neurons were transferrin-negative (transferrin−), implicating other possible uncharacterized brain-permeable circulatory factors (Fig. 1m, Extended Data Fig. 3l). Together, these data show that endogenous circulatory proteins physiologically permeate the healthy brain.
Regulation of plasma uptake by the BBB
We sought to reveal the complex system of genetic regulators that facilitate enhanced plasma blood-to-brain transport. We developed a ‘functional transcriptomics’ platform that records plasma uptake in endothelial cells via flow cytometry and index sorts them for deep, single-cell RNA sequencing (scRNA-seq) (about 1.5 million reads per cell). Linking transcriptomic data and plasma uptake for every cell enables unbiased and high-throughput correlations between each gene’s expression and the degree of plasma uptake. Brain endothelial cells (n = 745) were processed from healthy mice 4 h after administration of fluorescently labelled plasma to ensure measurement during protein transcytosis11,12 (Fig. 2a, Extended Data Fig. 7a–d, Supplementary Table 2).
Fig. 2|. Regulation of plasma uptake by the brain vasculature.
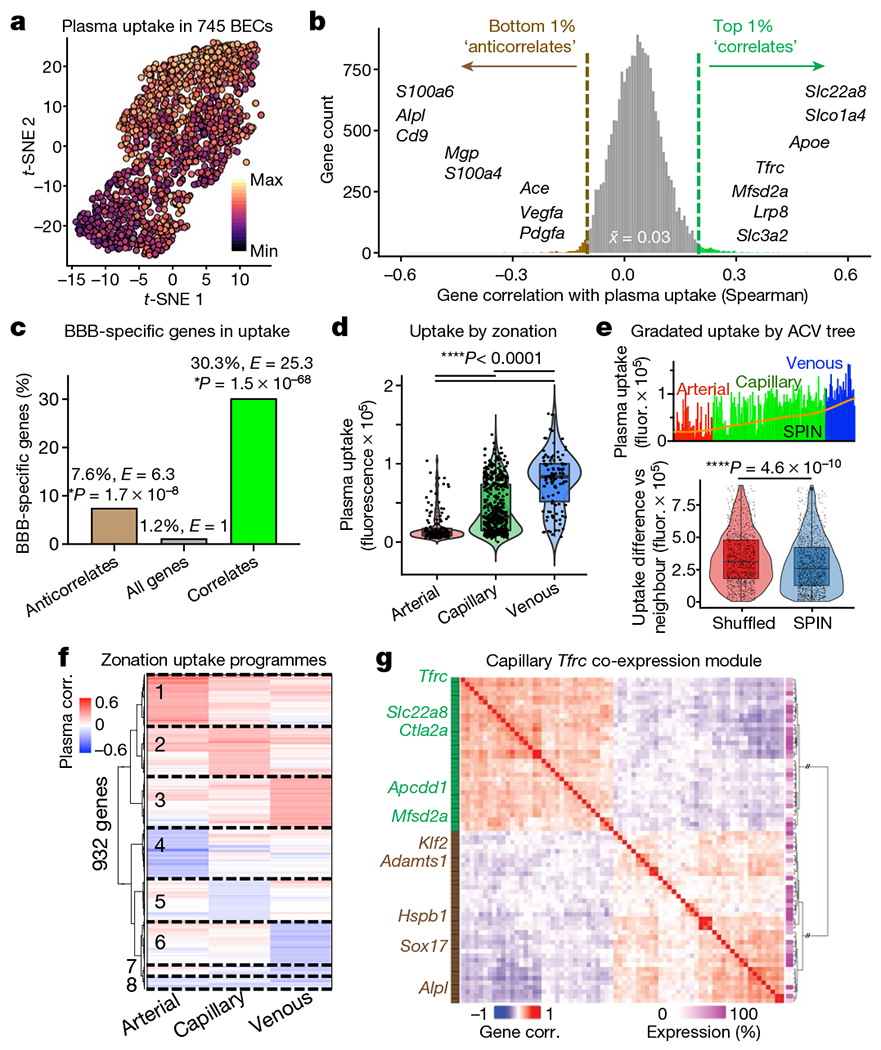
a, t-Distributed stochastic neighbour embedding (t-SNE) projection of plasma uptake across cortical and hippocampal BECs. b, Correlating 19,899 BEC-expressed genes with plasma uptake (Spearman’s). The top and bottom 1% are indicated as ‘correlates’ and ‘anticorrelates’, respectively, with examples. The distribution median is 0.033. c, Per cent and enrichment (enrichment E, hypergeometric distribution) of BBB-specific genes15 among correlates and anticorrelates. d, Plasma uptake by endothelial zonation. e, Plasma uptake across arteriovenous (ACV) SPIN axis (top), with moving average (LOWESS, orange). The pairwise difference in uptake between neighbouring cells organized by SPIN or randomly shuffled is also shown (bottom). In d and e, the box plots show the median and the 25–75th percentiles, and the whiskers indicate the minimum and maximum values. f, Zonation-specific transcriptional programmes of plasma uptake (Spearman’s). g, Co-expression of correlate (green) and anticorrelate (brown) genes in capillaries expressing Tfrc (encoding transferrin receptor) (Spearman’s). The matrix dendrogram is ordered by Euclidian distance, presenting genes expressed in over 15% of capillaries. Results from 745 BECs were isolated from n = 3 young (3-month-old) mice.
Although the vast majority of the 19,899 genes sequenced showed no effect, specific genetic correlates of enhanced or inhibited uptake emerged (Fig. 2b, Supplementary Table 3). Certain genes, such as transferrin receptor (Tfrc) and CD98hc (Slc3a2)11, were validating controls and, as expected, genes that correlated with enhanced plasma uptake (‘correlates’) were enriched in the vesicle-mediated transport pathway (Extended Data Fig. 7f, g). Several correlates support cerebrovascular integrity, such as Apoe13 and Mfsd2a14, suggesting that plasma uptake is not simply from breakdown. Finally, we found a strong enrichment of BBB-specific genes15 among correlates and anticorrelates, suggesting that regulators of plasma uptake and transcytosis may have distinctively evolved in the brain endothelium (Fig. 2c).
Plasma uptake varied significantly by vessel segment, with venous cells endocytosing the most, arterial cells the least, and capillaries in between but with the most heterogeneity (Fig. 2d). We ordered endothelial cells into a 1D sorting points into neighbourhoods (SPIN) axis16. Capillaries more transcriptionally similar to arterial cells endocytosed plasma similar to an arterial cell, and vice versa for venous-like capillaries (Fig. 2e). This suggests a spatial gradient of plasma uptake along the cerebral arteriovenous tree. Each vessel segment exhibited a unique transcriptional programme for plasma uptake (Fig. 2f). Within capillaries, Tfrc expression paralleled other correlates, such as Mfsd2a, forming a co-expression module that suggests common upstream orchestration (Fig. 2g). Expression of this Tfrc module was mutually exclusive with expression of strong anticorrelates such as Alpl. Together, these data suggest that plasma uptake is mediated by specific brain endothelial transcriptional programmes and may be modifiable by pharmacologically targeting identified correlates or anticorrelates.
Shift in BBB protein transcytosis with age
With age, the BBB has been reported to be leakier to various tracers17. We detected brain uptake of the 64Cu-labelled radiotracer at levels consistent with previous work9, and concordantly, observed an increase in IgG accumulation with age by autoradiography and quantitative gamma counting (Fig. 3a, b, Extended Data Fig. 2f). In agreement, imaging revealed endogenous IgG uptake in the aged but not the young brain endothelium (Fig. 3c, d). The opposite was true for plasma depleted of albumin and IgG: plasma uptake diminished by nearly half in the aged brain (Fig. 3e, f, Extended Data Fig. 2f). This held true in both whole brains and devascularized cortical and hippocampal parenchymal homogenate (Fig. 3f). Plasma uptake consistently exceeded that of IgG across multiple time points (Fig. 3g), suggesting a distinct mechanism mediating enhanced plasma transport to the brain. Time-point studies also enabled approximations of daily IgG and plasma brain uptake (Methods).
Fig. 3|. Age-related shift in blood-to-brain protein transcytosis.
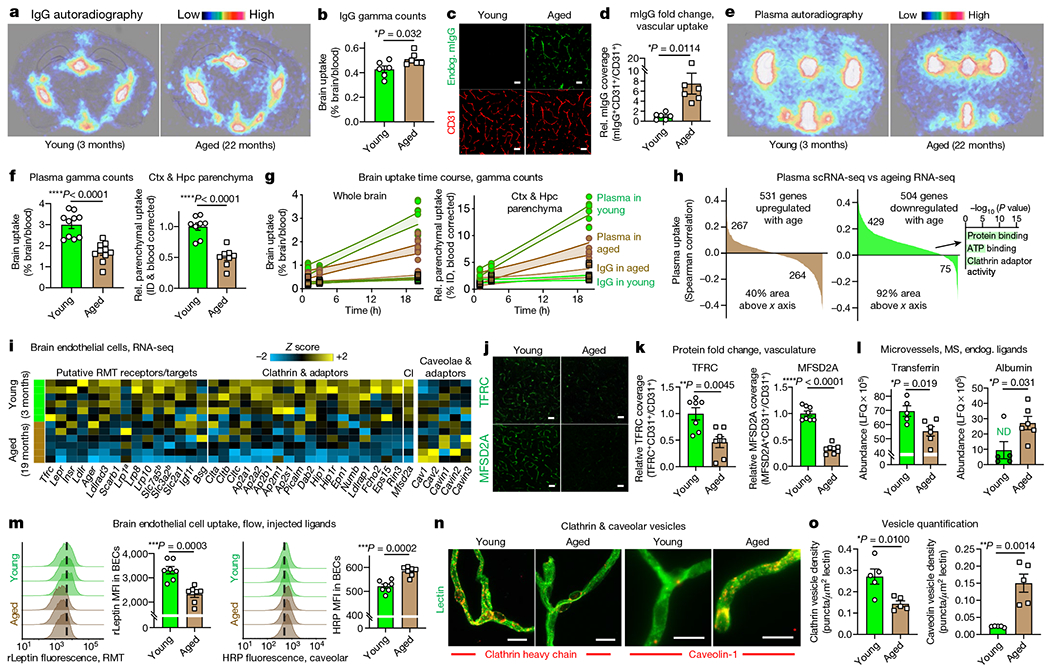
a, Brain autoradiography of 64Cu-labelled IgG (7.7 MBq matched dose, about 20 μg) injected intravenously and assessed 20 h later. Images are overlaid with Nissl stain. b, Gamma counting IgG in whole brains (left) following the paradigm shown in a. Same young IgG data as in Fig. 1c (n = 6; two-sided t-test; mean ± s.e.m.). c, Endogenous mouse IgG (mIgG) in CD31+ cortical vasculature. Scale bar, 40 μm. d, Quantification of c (n = 6; two-sided t-test; mean ± s.e.m.). e, Brain autoradiography of 64Cu-labelled Alb/IgG-depleted plasma (7.7 MBq matched dose, about 20 μg) as in a. f, Gamma counting Alb/IgG-depleted plasma in whole brains (left) and devascularized hippocampal (Hpc) and cortical (Ctx) parenchyma (right) following the paradigm shown in e. The same young plasma data as in Fig. 1c (n = 8 whole brain and n = 10 parenchyma; two-sided t-test; mean ± s.e.m.). g, Gamma counting across time (1,3 and 20 h) following the paradigms shown in a, b, e and f. Parenchymal uptake is relative to the average ‘IgG in young’ 1-h time point. The fill area represents the 95% confidence interval. h, BEC genes that change with age versus their correlation with plasma uptake (GeneAnalytics GO molecular function analysis, n = 3 young mice for plasma scRNA-seq and n = 6 for ageing RNA-seq, from i). i, BECgene expression of putative RMT receptors and clathrin, and caveolae and caveolae inhibitor (CI)18. aAbluminal expression32. bGenes form a heterodimer. Relevant genes with an average FPKM of >1 in either age shown (exception for Lrp1 and Ager). j, TFRC and MFSD2A cortical vascular expression. Scale bars, 40 μm. k, Quantification of j (n = 7 TFRC and n = 8 MFSD2A; two-sided t-test; mean ± s.e.m.). l, Label-free quantification (LFQ) of endogenous transferrin and albumin in perfused microvessels by mass spectrometry (MS). Albumin imputed for four young samples (green, ND) (n = 5 young and 6 aged; two-sided t-test; mean ± s.e.m.). m, Flow histograms and mean fluorescence intensity (MFI) recording the BEC uptake of recombinant mouse leptin (rLeptin) and HRP 4 h after administration (n = 7; two-sided t-test; mean ± s.e.m.). The dashed line approximates the median young MFI. n, Clathrin and caveolar vesicles in acutely isolated microvessels. Scale bars, 10 μm. o, Quantification of n (n = 5; two-sided t-test; mean ± s.e.m.).
To investigate the mechanisms that mediate plasma transport and its impairment with age, we combined our scRNA-seq data with a published brain endothelial cell transcriptomic ageing dataset18 (Fig. 3h). Of the genes downregulated with age, 85% correlated with plasma uptake in youth (92% of area under the curve). Enrichment analysis highlighted a loss of clathrin adaptor activity and ATP utilization, hinting at defective receptor-mediated transcytosis (RMT). Indeed, we observed a global shift from ligand-specific RMT to non-specific caveolar transcytosis with age in brain endothelial cells (Fig. 3i, Extended Data Fig. 7i, j). This shift was not observed in BCSFB choroid plexus or lymphatic endothelial cells (Extended Data Fig. 6l). Most genes encoding putative RMT receptors, including Tfrc, decreased with age, as did downstream clathrin components and its adaptors. Expression of Cav1, which encodes the principal component of caveolae, trended upwards with age, whereas Mfsd2a, the product of which suppresses caveolae formation19, decreased.
We sought to functionally validate this age-related shift in endothelial transcytosis. First, we confirmed gene expression changes at the protein level and found diminished TFRC and MFSD2A abundances with age (Fig. 3j, k). Similarly, several MFSD2A-regulated docosahexaenoic acid glycerophospholipids19 trended down with age (Extended Data Fig. 8, Supplementary Table 4). Next, we assessed receptor-mediated and caveolar transport using their canonical ligands, endogenous and injected. With mass spectrometry, we found a decrease in endogenous, RMT-transported transferrin and a concomitant increase in caveolae-transported albumin within perfused, aged microvessels (Fig. 3l). This is consistent with an age-related decline in TFRC and clathrin expression, and an increase in ligand-non-specific caveolae. We observed similar patterns using the injected RMT ligand leptin and the caveolae-transported ligand horseradish peroxidase (HRP), with the young endothelium taking up more of the leptin but the aged endothelium taking up more of the HRP (Fig. 3m).
Finally, having measured an age-related change in functional transport consistent with the predicted shift from RMT (exemplified by transferrin and leptin) to caveolar transcytosis (IgG, albumin and HRP), we sought to quantify the frequency of clathrin and caveolar vesicles with age. Through high-resolution microscopy of antibody-stained microvessels, we indeed observed a greater density of clathrin vesicles in the young brain endothelium but more caveolar vesicles in the aged brain endothelium (Fig. 3n, o). Together, from geneexpression profiling to functional transport measurements to vesicle density quantification, these data suggest that the healthy BBB constitutively transports circulatory proteins via ligand-specific RMT, but that this shifts with age to ligand-non-specific caveolar transcytosis.
Restoring RMT across the aged BBB
To discover overt anatomical changes associated with the shift in transport, we assessed vascular density, astrocyte coverage and pericyte coverage with age (Fig. 4a, b, Extended Data Fig. 9a–e). We observed a specific loss of pericyte coverage (Fig. 4a, b), with the expression of several pericyte-induced genes in the endothelium, such as Tfrc, Lrp8 and Mfsd2a, decreasing both in pericyte-deficient Pdgfret/ret models20 and normal ageing (Fig. 3j, k, Extended Data Fig. 9f). This suggests a role for pericytes in maintaining mature BBB function, including ligand-specific transport. Pericyte-deficient transgenic models display ectopic calcifications, which manifest as bone-dense nodules and vascular spheroids21,22. Such calcifications are phenocopied in normal, disease-free ageing (Fig. 4c, Extended Data Fig. 9g, h). Thus, pericyte loss and concomitant ectopic brain calcifications and BBB dysfunction may be normal features of brain ageing (Extended Data Fig. 9k). Molecularly, calcifications arise from the colocalization of type I collagen and the alkaline phosphatase ALPL23, both of which increase in parallel with age (Fig. 4d, e, Extended Data Fig. 9i).
Fig. 4|. Inhibiting an age-related negative regulator of blood-to-brain transport.
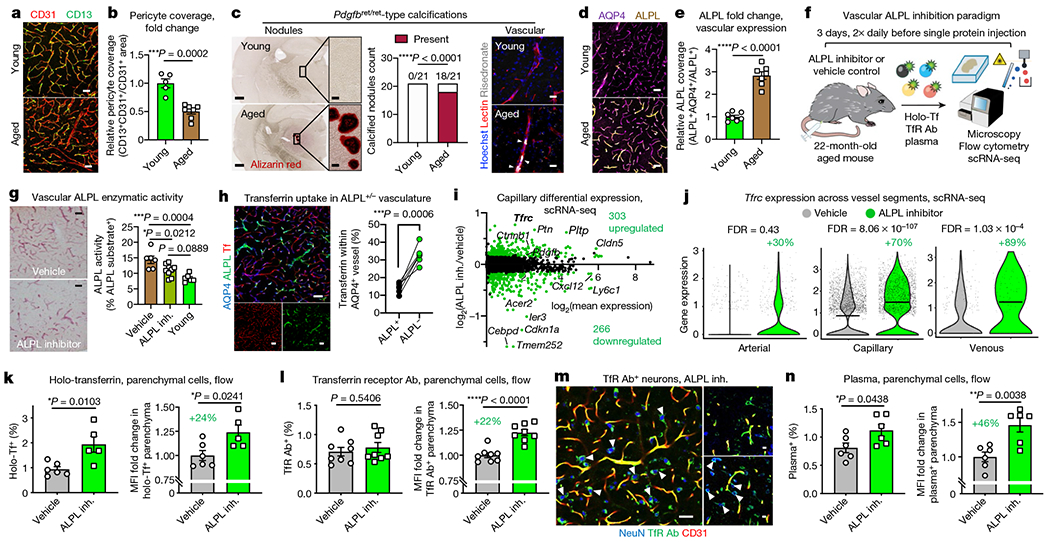
a, Cortical CD13+ vascular pericytes and CD31+ endothelial cells. Scale bars, 40 μm. b, Quantification of a (n = 5 young and n = 7 aged mice; two-sided t-test; mean ± s.e.m.). c, Calcified nodules in aged (23-month-old) mice, phenocopying pericyte-deficient Pdgfbret/ret mice21 (left). The frequency of nodules (n = 21 and 3 cohorts, chi-squared test) (centre). Vascular calcifications (arrowheads) (right). Scale bars, 500 μm (left) and 50 μm (right). d, ALPL expression in cortical AQP4+ vasculature. Scale bars, 40 μm. e, Quantification of d (n = 7; two-sided t-test; mean ± s.e.m.). f, Cerebrovascular ALPL inhibition and protein uptake measurement. Tf, transferrin. g, ALPL activity (red) in cortical vasculature of aged mice treated with an ALPL inhibitor (inh.) or vehicle and in young mice (n = 6 vehicle and young and n = 9 ALPL inhibitor; one-way ANOVA with Tukey’s multiple comparisons correction; mean ± s.e.m.). Scale bars, 50 μm. h, Holo-transferrin in ALPL+/− cortical vasculature (n = 6; paired two-sided t-test). i, MA plot of differentially expressed genes (false discovery rate (FDR) of <0.05) from aged mice after an ALPL inhibitor and vehicle (7,036 capillary cells, n = 4 mice pooled per group; model-based analysis of single-cell transcriptomics (MAST) with Bonferroni correction). j, Tfrc expression across vessel segments, following the paradigm shown in i (1,171 arterial, 7,036 capillary and 430 venous cells; MAST with Bonferroni correction). The box plots show the median and the 25–75th percentiles, and the whiskers indicate the minimum and maximum values. The violin plot midlines indicate the median. k,l, Flow cytometry quantification of brain parenchymal (CD31−CD45−) cell uptake of holo-transferrin (k) and transferrin receptor antibody (TfR Ab) in aged mice (l) after an ALPL inhibitor and vehicle, assessed by the per cent and relative MFI of tracer+ parenchymal cells (holo-Tf: n = 5 vehicle and n = 6 ALPL inhibitor; TfR Ab: n = 9; two-sided t-test; mean ± s.e.m.). m, Transferrin receptor antibody in neurons (arrowheads) following the paradigm shown in l. Scale bars, 40 μm (left) and 10 μm (right). n, As in k and l, but for plasma (n = 6; two-sided t-test; mean ± s.e.m.).
Coincidentally, ALPL arose in our search for molecular targets to enhance transcytosis across the aged cerebrovasculature (Extended Data Fig. 9j). Several drug development programmes to enhance CNS drug delivery rely on RMT, many targeting TFRC10. We filtered for genes that were: (1) BBB-specific (Extended Data Figs. 9j, 10a, b); (2) expressed on the endothelial cell surface (Fig. 4d); (3) upregulated with age (Fig. 4e, Extended Data Fig. 10d, e); (4) anticorrelates of plasma uptake (Fig. 2b, Extended Data Fig. 10c); and (5) amenable to inhibition with US Food and Drug Administration-approved molecules and improved derivatives. Only Alpl (encoding ALPL) fulfilled these criteria.
Adopting a previous paradigm20 (Fig. 4f), we first confirmed that intravenous administration of a selective, small-molecule ALPL inhibitor24 decreased vascular ALPL activity (Fig. 4f, g). Consistent with the non-overlapping expression of Alpl and Tfrc in capillaries (Fig. 2g), we noticed that holo-transferrin was preferentially taken up by ALPL-deficient capillaries (Fig. 4h). To determine whether this correlation hinted at a causal mechanism, we performed scRNA-seq of aged brain endothelial cells after pharmacological ALPL inhibition. The top upregulated gene after ALPL inhibition was Tfrc, with an approximately 70% expression increase in capillaries and approximately 89% increase in venous cells (Fig. 4i, j, Extended Data Fig. 10f–h, Supplementary Table 5).
Thus, we wondered whether ALPL inhibition could enhance transferrin and TFRC antibody transport across the aged brain endothelium. We indeed observed increased parenchymal uptake for both holo-transferrin and a high-affinity TFRC antibody (Fig. 4k–m, Extended Data Fig. 11a–e). Enhancement of TFRC antibody uptake was less pronounced than that of holo-transferrin, which is consistent with previous work showing that high-affinity antibody variants are preferentially trapped in the endothelium10. Parenchymal access of holo-transferrin and TFRC antibody after ALPL inhibition was confirmed in situ, with accumulation in cell types such as cortical NeuN+ neurons (Fig. 4m, Extended Data Fig. 11c). ALPL inhibition did not perturb vascular morphology or paracellular tight junctions (Extended Data Fig. 11f, g).
We also noticed increased plasma uptake—beyond that of transferrin—in aged brain parenchymal cells after ALPL inhibition (Fig. 4n, Extended Data Fig. 11h). This suggests mechanisms of enhanced uptake beyond upregulating Tfrc (Fig. 4i). ALPL inhibition trended towards but did not significantly enhance plasma transcytosis in young mice (Extended Data Fig. 11i). ALPL expression in youth is restricted to arterial cells, which may be insufficient in number to confer enhanced transcytosis upon ALPL inhibition (Fig. 4d, e). Together, these data suggest that pericyte loss and ectopic brain calcifications are attributes of normal ageing, and that inhibition of age-upregulated vascular ALPL can enhance transcytosis of plasma and therapeutically relevant biologic agents.
Discussion
Complementing leakiness from focal BBB breakdown17,25,26, a global age-related shift in protein transcytosis provides a molecular basis for widespread BBB dysfunction that may provoke neurodegenerative disease (Extended Data Fig. 9k). Specifically, this shift alters the composition of transcytosing plasma proteins and permits neurotoxic proteins such as albumin, fibrinogen and autoantibodies that lack canonical transporting receptors–and thus minimally enter in youth–ready access to the aged parenchyma25. Their dysregulated entry could then trigger neuroinflammation27. It will be important to further understand how specific circulatory proteins–whether via their loss or gain of brain exposure with age–contribute to disease. Diminished RMT transport with age also predicts a smaller therapeutic index in CNS drug delivery. Our findings suggest potential benefits from targeting putative RMT receptors in combination, exploiting increased caveolar transport, and/or inhibiting negative regulators of transport, such as ALPL.
Although loss of pericyte coverage in normal ageing has previously been debated28, Pdgfret/ret-phenocopying calcifications provide evidence in the affirmative. Pericyte loss has also been linked to non-specific macromolecule transport20,29, impaired blood flow30 and neuronal dysfunction25,26. Thus, pericyte loss and the concomitant shift in transcytosis with normal, disease-free ageing support a two-hit vascular hypothesis for Alzheimer’s disease26, as both vascular dysfunction and pericyte degeneration do not depend on, but could be exacerbated by and in turn exacerbate, amyloid-β accumulation. Whether ALPL inhibition has clinical utility remains to be seen. Within the CNS, human ALPL is enriched in the endothelium as a sheddable glycosylphosphatidylinositol-anchored protein (Extended Data Fig. 11j–l) and is upregulated in the brains and plasma of patients with Alzheimer’s disease, with increased plasma levels inversely correlated with cognitive performance31.
The development of sensitive proteomics techniques will accelerate efforts to catalogue and characterize BBB-permeable plasma proteins. Because labelled plasma is simply a surrogate for endogenous plasma, direct visualization of uptake in the healthy brain is just a snapshot of constitutive blood plasma protein binding, signalling and trafficking across the brain endothelium throughout life. Thus, the BBB–although less permeable than the peripheral endothelium–may be more aptly characterized as an active interface that mediates communication between the periphery and the CNS. Understanding the principles of this communication, and how they change with age, may be fundamental to understanding brain function in health and disease.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-020-2453-z.
Methods
Animals
Aged C57BL/6 mice (20–24-months old) were obtained from the National Institute on Aging rodent colony. Young male C57BL/6 mice (3-months old) were obtained from Jackson Laboratories or Charles River Laboratories. All experiments used male mice. All mice were kept on a 12-h light/dark cycle and provided ad libitum access to food and water. All animal care and procedures complied with the Animal Welfare Act and were in accordance with institutional guidelines and approved by the V.A. Palo Alto Committee on Animal Research and the institutional administrative panel of laboratory animal care at Stanford University.
Plasma collection and labelling
Blood was collected with 250 mM EDTA (Millipore Sigma) as anticoagulant by terminal intracardial bleeding. EDTA plasma was isolated by centrifugation at 1,000g for 15 min at 4 °C before pooling, dividing into aliquots, flash freezing in liquid nitrogen and storage at −80 °C. Haemolysed plasma was discarded. Before labelling, frozen plasma was thawed on ice, gently mixed and inspected for precipitates. Plasma protein molarity was approximated to be 750 μM. Labelling relied on amine-reactive NHS ester moieties, and labelling ratios and times were determined empirically for each specific label: for radiotracing, NHS-DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, Macrocyclics) was added at a 10× molar ratio; for fluorescence, NHS-Atto 647N (ATTO-TEC, Millipore Sigma) was added at 0.8–1.2× molar ratio; and for characterizing the labelled plasma proteome, NHS-biotin (Thermo Fisher) and NHS-trans-Cyclooctene (Click Chemistry Tools) added at 12.5× molar ratio. NHS-Atto 647N was incubated with plasma for 1.5 h rotating at room temperature before PBS dialysis overnight. The next day, 50 mM Tris pH 8.0 was added for 10 min at room temperature, and the labelled plasma was washed extensively three times with Amicon Ultra-15 Centrifugal Filter Unit, 3 kDa cutoff (Millipore Sigma; 10 kDa for NHS-Atto 647N), and was subsequently washed four times with Zeba Spin Desalting Columns, 7K MWCO cutoff (Thermo Fisher) in chilled PBS. Other conjugations proceeded overnight at 4 °C before washing. These steps expanded on washing that consistently yielded >99% radiochemical purity of 64Cu-labelled DOTA (below) to minimize contamination from residual-free label. Throughout the labelling and washing steps, plasma samples were monitored for signs of aggregation and flocculation. Plasma concentrations were measured using the Nanodrop spectrophotometer (Thermo Fisher), and approximately 10–15 mg (0.5 mg/g body weight, in line with previous studies using HRP8,14,19) was administered intravenously (retro-orbital) per mouse in a volume less than or equal to 150 μl.
Labelled plasma characterization by mass spectrometry and protein microarray
For characterization by mass spectrometry, labelled plasma was depleted of albumin and IgG using the ProteoPrep Immunoaffinity Albumin & IgG Depletion Kit (Millipore Sigma), enriched with tetrazine agarose overnight at 4 °C (Click Chemistry Tools) and extensively washed, as previously described33. In parallel, unlabelled plasma was processed using paramagnetic beads, termed single-pot solid-phase-enhanced sample preparation (SP3), as previously described34. Plasma was fractionated using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo Fisher) and concatenated fractions were cleaned using C18-based STAGE Tips and lyophilized. Peptides were analysed on an LTQ Orbitrap Fusion Lumos Tribrid Mass Spectrometer (Thermo Fisher Scientific). Peptides were separated by capillary reverse-phase chromatography for 180 min on a 24-cm reversed-phase column (inner diameter of 100 μm, packed in-house with ReproSil-Pur C18-AQ 3.0 m resin (Dr. Maisch)). A four-step linear gradient was achieved using a Dionex Ultimate 3000 LC-system (Thermo Fisher Scientific) as follows: 97% A + 3% B for 15 min, 75% A + 25% B for 135 min, 55% A + 45% B for 15 min, and 5% A + 95% B for 15 min, where buffer A is 0.1% formic acid in water and buffer B is 0.1% formic acid in acetonitrile. Full MS scans were acquired in the Orbitrap mass analyser at a resolution of 120,000 (FWHM) and m/z scan ranges of 340–1,540 in a data-dependent mode. The AGC targets were 4 × 105 and the maximum injection time for FTMS1 was 50 ms. The most intense ions were then selected for sequencing and fragmented in the Orbitrap mass analyser using higher-energy collisional dissociation (HCD) with a normalized collision energy of 30% and resolution of 15,000 (FWHM). Monoisotopic precursor selection was enabled, and singly charged ion species and ions with no unassigned charge states were excluded from MS2 analysis. Dynamic exclusion was enabled with a repeat count of two and ions within ±10 ppm m/z window around ions selected for MS2 were excluded from further selection for fragmentation for 30 s. AGC targets were 5 × 104 and the maximum injection time was 200 ms. The raw data files were processed and analysed using MaxQuant and Perseus (Max Planck)35,36. In brief, spectra were matched to a Mus musculus database downloaded from uniprot.org, and a contaminant and decoy database. Precursor mass tolerance was set to 4.5 ppm, fragment ion tolerance to 10 ppm, with fixed modification of Cys residues (carboxyamidomethylation +57.021 Da) and variable modifications of Met residues (Ox +15.995 Da), Lys residues (acetylation +42.011 Da), Asn and Gln residues (deamidation +0.984 Da) and of N termini (carbamylation +43.006 Da). Peptide identifications were calculated with an FDR of <0.01, and for protein quantification, the minimum ratio count was set to one, with both unique and razor peptides used for quantification.
For characterization by protein microarrays, ELISA-grade antibodies to several hundred secreted and potentially cleaved transmembrane mouse or human signalling proteins were obtained from commercial sources and printed in five replicates onto SuperEpoxy2 glass slides (Arrayit) with a robotic microarrayer (NanoPrint LM210, Arrayit), as previously described37. Arrays were then blocked with 3% (w/v) casein solution before incubation with biotinylated plasma samples overnight at 4 °C. Following washes, arrays were incubated with Alexa Fluor 647-conjugated streptavidin secondary antibodies (Thermo Fisher) and the fluorescent signal was detected using a GenePix4400A scanner and GenePix Pro7 software (Molecular Devices). Data processing and analysis followed previously described methods37.
Brain perfusion
Mice were euthanized with 2.5% (v/v) Avertin and transcardially perfused with at least 50 ml of chilled PBS. Perfusion was performed using a peristaltic pump, with the flow rate not exceeding 10 ml/min to not exceed the physiological pressure of the mouse’s circulatory system38–40. Mice with perfusate leaking out of the nostrils were not processed further for analysis. For immunohistochemistry (below), mice were subsequently perfused with 4% PFA.
Immunohistochemistry
Brain tissue processing, immunohistochemistry and immunofluorescence experiments were performed as described previously37,41. Hemibrains were isolated and post-fixed in 4% (w/v) PFA overnight at 4 °C before preservation in 30% (w/v) sucrose in PBS. Hemibrains were sectioned coronally or sagittally at a thickness of 50 μm on a freezing–sliding microtome, and sections were stored in cryoprotective medium at −20 °C. Free-floating sections were blocked with appropriate serum before incubation at 4 °C with primary antibodies at the following concentrations for confocal microscopy: goat monoclonal anti-CD31 (1:100, AF3628, R&D), fluorescein-labelled lectin (1:200, Vector Laboratories), rabbit monoclonal anti-AQP4 (1:500, AB2218, Millipore Sigma), rat anti-CD13 (1:100, MCA2183EL, Bio-Rad), goat anti-ALPL (1:100, AF2909, R&D), mouse anti-NeuN (1:400, MAB377, Millipore), goat anti-albumin (1:100, NB600-41532, Novus), rabbit anti-transferrin (1:100, ab82411, Abcam or 1:100, AF3987, R&D), rabbit anti-collagen I (1:100, ab21286, Abcam), goat anti-Ibal (1:500, ab5076, Abcam), rabbit anti-MFSD2A (1:300, gift from C. Gu), rat anti-TFRC (1:100, Novus, NB100-64979) and rabbit anti-phospho-STAT3 (1:100, 9145S, CST). Sections were washed, stained with Alexa Fluor-conjugated secondary antibodies (1:250), mounted and coverslipped with ProLong Gold (Life Technologies) before imaging on a confocal laser scanning microscope (Zeiss LSM880). Age-related autofluorescence was quenched with 1 mM CuSO4 in 50 mM ammonium acetate buffer (pH 5), as previously described42. National Institutes of Health ImageJ software was used to quantify the percentage of vasculature (CD31 or AQP4) covered by mIgG, MFSD2A, TFRC, CD13, AQP4 or ALPL, as described previously18. All analyses were performed by a blinded observer. Alizarin red staining was performed as described previously21, with minor adaptations: sections were incubated for 1 h in 40 mM Alizarin red in PBS (pH 7.4) at room temperature, and extensively washed overnight with PBS before mounting. Images of brain sections were acquired by conventional light microscopy to detect calcified nodules. Sections with biotinylated plasma were blocked overnight in 6% BSA at room temperature, detected with streptavidin-Alexa Fluor 647 (1:1,500, Thermo Fisher) for 2 h and washed overnight before mounting. Sections containing L-azidohomoalanine-labelled plasma were blocked overnight in 6% BSA at room temperature, incubated in 45 mM iodoacetamide (Millipore Sigma) in 100% methanol for 1 h, washed, detected with 1.2 μM sDIBO (Thermo Fisher Scientific) in 100% methanol, and washed overnight before mounting. Vascular ALPL activity was measured using the Red Alkaline Phosphatase Substrate Kit (SK-5100, Vector Laboratories) with 20-min incubation. Risedronate-647 (RIS-647, BioVinc, BV500101) was either injected intravenously (retro-orbital) at 8 nmol per mouse and brains were collected 18 h later, or brain sections were stained with 0.1 nM RIS-647 for 2 h at room temperature alongside fluorescein-labelled lectin (1:200, Vector Laboratories).
High-resolution microscopy
Brain microvessels were isolated from young (3-month-old) and aged (22-month-old) mice as previously described11,19,43–47. To generate sufficient numbers of microvessels, 20 mice of each age were used, with microvessels from four mice pooled in each group (n = 5 groups per age). Microvessels were permeabilized, blocked, stained and mounted as described previously48. In brief, acutely isolated microvessels were fixed with 4% PFA in PBS at room temperature for 20 min, washed, blocked and permeabilized, and stained overnight at 4 °C with fluorescein-labelled lectin (1:200, Vector Laboratories), mouse anti-clathrin heavy chain Alexa Fluor 647 (X22, 1:100, MA1-065, Thermo Fisher Scientific), and rabbit anti-caveolin-1 (1:200, 3267T, CST) pre-complexed with donkey anti-rabbit Alexa Fluor 555 at a 1:1 ratio. Stained microvessels were subsequently washed with PBS with 1 mM EDTA and mounted with Prolong Gold. Imaging was performed on either a confocal laser scanning microscope (×63 magnification, Zeiss LSM880) or a 3D-structured structured illumination microscope (×100, OMX BLAZE). Microvessels were located at low resolution using only lectin fluorescence; and specific, high-resolution fields of single-plane microvessels were chosen at random for blinded quantification of clathrin and caveolar vesicles normalized to an area of lectin.
Gamma counting, autoradiography and PET scanning
Conjugation of rat IgG2a (400501, BioLegend) and albumin/IgG-depleted plasma with DOTA was performed using metal-free buffers, as previously described37,49,50. Conjugations proceeded overnight at 4 °C with a 10× molar ratio of DOTA-NHS ester in PBS buffer (pH 7.4) and quenched with 50 mM Tris-HCl (Millipore Sigma). Excess DOTA-NHS was removed by Zeba SPIN desalting columns (0.5 ml, 7K molecular weight cutoff, Thermo Fisher), and the resulting solution was buffer-exchanged into ammonium acetate buffer (0.1 M, pH 5.5) for 64Cu labelling. DOTA-conjugate solutions were further washed and concentrated by ultrafiltration (Amicon 3 kDa MWCO, Millipore Sigma) and kept on ice. Radiolabelling with 64Cu (half-life = 12.7 h) was performed as previously described37. In brief, IgG2a–DOTA or plasma–DOTA (in 25 μg aliquots in 50 μl) was mixed with pH-balanced [64Cu]Cl2 solution (pH 4.5–5.5, University of Wisconsin, Madison) at 37 °C with gentle shaking at 400 rpm. After incubation (30 min for IgG2a–DOTA and 60 min for plasma–DOTA), 0.1 M EDTA (0.5 M, pH 8.0) was added to a final concentration of 0.01 M and incubated at ambient temperature for 15 min to scavenge unchelated [64Cu]Cl2 before purification by G25 Sephadex size-exclusion columns (GE Life Sciences). Radiochemical purity was determined using instant thin-layer TEC-control chromatography strips (iTLC, Biodex Medical Systems) developed in saline, and size-exclusion liquid chromatography with a SEC 3000 column (Phenomenex).
Mice were randomized to receive intravenous injections (retro-orbital unless tail vein indicated) of 7.7 ± 1.5 MBq of 64Cu-labelled IgG2a–DOTA or 64Cu-labelled plasma–DOTA (radiochemical purity of >99%). After 20 h, mice were placed in a dual microPET/CT scanner (Inveon, Siemens) to capture static images (10 min) for subsequent analysis with Vivo-Quant software (version 4.0, inviCRO), as previously described51,52. After anaesthetization, blood samples (100–200 μl) were collected by cardiac puncture immediately before transcardial perfusion. Tissue-related radioactivity (dose and decay-corrected to time of injection) in blood and brain was assessed following the literature37 procedure with an automated gamma counter (Hidex Oy; calibrated using aliquots of the initial administered dose as standards). Activity in cardiac blood samples at time of death was used to correct for differential in vivo stability and clearance of plasma versus IgG. Brain tissue was embedded in optimal-cutting temperature compound (Tissue-Tek), and coronal sections (40 μm) were obtained for ex vivo autoradiography. Autoradiography was conducted using previously described methods37,53: 40-μm sections were mounted, air-dried for 10 min and then exposed to a digital storage phosphor screen (Amersham Biosciences) for 72 h at −20 °C. The image plate was analysed using a Typhoon 9410 Variable Mode Imager (Perkin Elmer). Slides were then Nissl stained and scanned using a Nanozoomer 2.0-RS (Hamamatsu) to enable anatomical colocalization. Images were visualized, processed and quantified blinded with ImageJ. For quantification, at least ten consistently sized hippocampal and cortical areas were drawn for each mouse. The mean pixel intensity was dose and decay corrected for each mouse. Analyses were performed blinded to tracer and age group.
Blood half-lives were calculated assuming first-order kinetics. Brain uptake rates were calculated assuming two compartments (brain and blood) and first-order elimination kinetics from the blood. Moreover, the injected substance was assumed to distribute across circulation nearly instantaneously after injection. Hence, the rate of brain uptake can be described as:
| (1) |
in which kin is the rate of substance entry from blood to brain; and kout is the rate of substance exit from brain back out to blood. Because concentrations in the brain are much lower than in the blood, we assume that kout[Substance]Brain is negligible, especially at the early time points after injection of the radiolabelled substance. To approximate kin, we calculate the amount of brain uptake at 1 h, yielding:
| (2) |
kin was approximated as: 2.2 × 10−3 h−1 for IgG in young, 2.5 × 10−3 h−1 for IgG in aged, 8.6 × 10−3 h−1 for plasma in young and 6.1 × 10−3 h−1 for plasma in aged mice. kinrates for the other time points can be similarly approximated.
Estimations of IgG or plasma binding and uptake by the brain were made by first calculating the blood–brain partition for each substance in young and aged mice over 24 h. Brain uptake was then calculated as the product of the blood–brain partition value and the concentration of the substance in blood at physiological baseline and the total circulatory volume (equations below). IgG was estimated at a concentration of about 12 mg/ml and albumin/IgG-depleted plasma at about 15 mg/ml (50 mg/ml total × 30% non-albumin/IgG). Blood circulatory volumes have been estimated as about 2 ml in young and about 2.5 ml in aged mice, with the plasma volume approximately half of the blood volume (about 1 ml in young and about 1.25 ml in aged mice). To calculate the blood–brain partition over 24 h, the slope arising from time-point studies was integrated, accounting for the intercept (instantaneous/pre-1-h time-point brain uptake):
| (3) |
| (4) |
Blood–brain partitions were estimated as: 3.46% plasma in young, 0.46% IgG in young, 1.92% plasma in aged and 0.57% IgG in aged mice. This resulted in approximate daily brain uptake values of: about 0.52 mg of plasma in young, about 0.06 mg of IgG in young, about 0.36 mg plasma in aged and about 0.09 IgG in aged mice.
Primary CNS cell isolation for flow cytometry analysis
CNS cell isolation adopted previously described methods11,18,54. In brief, cortices and hippocampi were microdissected, minced and digested using the Neural Dissociation Kit (Miltenyi). Suspensions were filtered through a 100-μm strainer and myelin removed by centrifugation in 0.9 M sucrose. The remaining myelin-depleted cell suspension was blocked for 10 min with Fc preblock (CD16/CD32, 553141, BD) on ice and stained for 30 min with antibodies to distinguish brain endothelial cells (CD31+CD45−), astrocytes (ACSA-2+) and neurons (NeuN+ or CD90.2/Thy1.2+). For analysis of plasma uptake, at least 1,000 cells of each population of interest were analysed per sample. Antibody dilutions: rat anti-CD31-PE/CF594 (1:100, clone MEC 13.3, cat. no. 563616, BD), rat anti-CD45-PE/Cy7 (1:200, clone 30-F11, cat. no. 103114, BioLegend), rat anti-ACSA-2-PE (1:200, clone IH3-18A3, cat. no. 130-102-365, Miltenyi), mouse anti-NeuN-PE (1:100, clone A60, cat. no. FCMAB317PE, Millipore Sigma) and rat anti-CD90.2-FITC (1:100, clone 30-H12, cat. no. 105305, BioLegend,). MFI refers to the geometric mean of tracer+/tracerhi cells. Supplementary Fig. 1 provides the gating strategy. For the experiment in Fig. 3m, aged mice received intravenous injection of fluorescently labelled 7.5 mg/kg recombinant leptin (98-OB-05M, R&D) or 150 mg/kg HRP (31491, Thermo) before being killed 4 h later. Subsequent analyses and quantification were performed blinded to group.
Flow index sorting and scRNA-seq
Cell lysis, first-strand synthesis and cDNA synthesis were performed using the Smart-seq-2 protocol asdescribed previously18,54,55 in 384-well format, with some modifications. In brief, brain endothelial cells (CD31+CD45−) were sorted using SH800S (Sony) sorters on the highest purity setting (‘Single cell’). Plasma-647 fluorescence was recorded for each cell and corresponding sorted well. cDNA synthesis was performed using the Smart-seq2 protocol54–56. After cDNA amplification (23 cycles), concentrations were determined via the PicoGreen quantitation assay. Cells passing quality control were selected through published scripts57,58 and cDNA concentrations normalized to about 0.2 ng/μl using the TPPLabtech Mosquito HTS and Mantis (Formulatrix) robotic platforms. Libraries were prepared and pooled using the Nextera XT kits (Illumina), following the manufacturer’s instructions. Libraries were then sequenced on the Nextseq or Novaseq (Illumina) using 2 × 75-bp paired-end reads and 2 × 8-bp index reads with a 200-cycle kit (20012861, Illumina). Samples were sequenced at an average of 1.5 million reads per cell. Raw sequencing files were demultiplexed with bcl2fastq, reads were aligned using STAR and gene counts were made using HTSEQ version 0.6.1p1. Downstream analysis was performed as previously described16,59, and plasma fluorescence of each cell correlated (Spearman’s) with gene expression and zonation57.
ALPL inhibitor treatment
Mice were randomized to receive six doses of ALPL inhibitor (613810, Millipore Sigma)24 or ‘vehicle’ control over 3 days, injected intraperitoneally twice per day (8.75 mg/kg). ‘Vehicle’ treatments consisted of PBS with a matching concentration of DMSO (<4% vol/vol). After the sixth treatment, mice were injected intravenously with 150 μl of Atto 647N-labelled plasma (as above), human holo-transferrin (40 mg/kg; T4132, Millipore Sigma), transferrin receptor antibody (20 mg/kg; BE0175, BioXcell) or 3 kDa dextran–FITC (10 mg/kg; Thermo Fisher). Plasma, human holo-transferrin and transferrin receptor antibody were infused 20 h before being killed, while 3 kDa dextran–FITC was given 2 h before being killed, as previously described60. After extensive perfusion, CNS cells were isolated as described above. Cell pellets were additionally incubated in red blood cell lysis buffer (R7757, Millipore Sigma) for 2 min to ensure high CNS cell purity. Brain endothelial cells were distinguished by CD31+CD45− staining and parenchymal cells by CD31−CD45− staining. Flow cytometry analyses were performed blinded to group.
scRNA-seq after ALPL inhibitor treatment
Brain endothelial cells (as above; n = 5,000–10,000) from ALPL inhibitor-treated or vehicle-treated aged (22-month-old) mice (pool of four mice per group) were sorted into PBS with 0.1% BSA. Cells were immediately counted using a Neubauer haemocytometer and loaded on a Chromium Single-Cell Instrument (10x Genomics) to generate single-cell Gel Bead-in-Emulsion (GEMs). The 10× Genomics v3 libraries were prepared as per the manufacturer’s instructions. Libraries were sequenced on an Illumina NextSeq 550 (paired-end; read 1:28 cycles; i7 index: 8 cycles, i5 index: 0 cycles; read 2:91 cycles). Demultiplexing was performed using the Cellranger toolkit (v.3.0.0) ‘cellranger mkfastq’ command and the ‘cellranger count’ command for alignment to the mouse transcriptome, cell barcode partitioning, collapsing unique molecular identifier (UMI) to transcripts and gene-level quantification. Approximately 70% sequencing saturation (>20,000 reads per cell) was achieved, for a median of about 2,000 genes detected per cell and about 16,500 genes detected in total. Downstream analysis using the Seurat package (v3)61 was performed as previously described16, applying standard algorithms for cell filtration, feature selection and dimensionality reduction. In brief, samples with fewer than 1,000 and more than 4,000 unique feature counts, samples with more than 15% mitochondrial RNA, samples with more than 15% small subunit ribosomal genes (Rps) and counts of more than 10,000 were excluded from the analysis. Counts were log-normalized. Next, the Integration function was used to align data from ALPL inhibitor and vehicle samples with default settings. Genes were projected into principal component (PC) space using the PC analysis (RunPCA). The first 30 dimensions were used as inputs into Seurat’s FindNeighbours and RunTsne functions. In brief, a shared-nearest-neighbour graph was constructed based on the Euclidean distance metric in PC space, and cells were clustered using the Louvain method. RunTsne functions with default settings were used to calculate 2D t-SNE coordinates and search for distinct cell populations. Cells and clusters were then visualized using 3D t-SNE on the same distance metric. Differential gene expression analysis was done by applying MAST. Raw P values were adjusted via Bonferroni correction. Analyses and results were reproduced by an individual blinded to the identity of the treatment groups.
Co-injection of plasma into the CSF and blood
Plasma was labelled and subsequently washed of free dye (as above) with NHS ester AF647 or NHS ester AF488 (Click Chemistry Tools). Mice were intravenously injected (retro-orbital) with 150 μl plasma (about 7.5% of circulatory volume), and 2–3 h later, anaesthetized with 2% isofluorane for injections directly into the CSF–either intraventricular (ICV) or cisterna magnal (CM). For ICV injections, the mouse was placed in a stereotactic apparatus, the skull was exposed and a hole was drilled at the injection site (+1 mm lateral, 0 mm anterior–posterior and −2.5 mm deep relative to bregma) using aseptic technique62. For CM injections, 10 μl of plasma was injected as previously reported63,64. For both CSF injection routes, the needle was left in place for an additional 30 min to allow for diffusion, then mice were killed and perfused with ice-cold PBS and 4% PFA. Brain sections were processed for immunohistochemistry (as above) or whole brains were cleared via iDISCO65 for downstream Light Sheet Ultramicroscope imaging (Lavision Biotec) and image reconstruction (arivis).
Fluorescent gel detection of transferrin in brain homogenates
Human holo-transferrin (T4132, Millipore Sigma) was labelled with NHS ester AF 647 (Click Chemistry Tools) at a 5:1 molar ratio overnight at 4 °C. Free label was removed and protein was washed four times with Zeba Spin Desalting Columns, 40K MWCO cutoff (Thermo Fisher). Transferrin was injected intravenously (retro-orbital) at 10 mg/kg and brains were extracted 3–4 h later. Brains were sonicated to homogenates in 4% SDS, centrifuged at 14,000g for 20 min at 4 °C, and supernatant was collected to measure protein concentrations with the BCA Protein Assay Kit (Pierce). A 4× stock solution of NuPAGE LDS (Thermo) was added to brain homogenates and plasma samples before heating at 95 °C for 10 min. Proteins were briefly spun and separated by electrophoresis in 12% Bis-Tris polyacrylamide gels (Invitrogen). Gels were washed twice in distilled water for 10 min before Alexa Fluor 647 imaging in the 700-nm channel of an Odyssey CLx (LI-COR).
Brain endothelial cell lipidomics, mass spectrometry
Brain microvessels were isolated from young (3-month-old) and aged (20-month-old) perfused mice as previously described11,19,43–47, after overnight fasting (about 12 h). Sixteen mice of each age were used, with microvessels from four mice pooled in each group (n = 4 groups). Lipids were extracted in a randomized order via biphasic separation with cold methyl tert-butyl ether (MTBE), methanol and water, as previously described66. In brief, 260 μl of methanol and 40 μl of water were added to the brain microvessels and vortexed for 20 s. A lipid internal standard mixture was spiked in each sample (EquiSPLASH LIPIDOMIX (330731, Avanti Polar Lipids) and d17-oleic acid (9000432, Cayman chemicals)) to control for extraction efficiency, evaluate LC–MS performance and normalize LC–MS data. Samples were diluted with 1,000 μl of MTBE, vortexed for 10 s, sonicated for 30 s three times in a water bath and incubated under agitation for 30 min at 4 °C. After addition of 250 μl of water, the samples were vortexed for 1 min and centrifuged at 14,000g for 5 min at 20 °C. The upper phase containing the lipids was collected and dried down under nitrogen. The dry extracts were reconstituted with 150 μl of 9:1 methanol to toluene.
Lipid extracts were analysed in a randomized order using an Ultimate 3000 RSLC system coupled with a Q Exactive mass spectrometer (Thermo Fisher Scientific) as previously described67. Each sample was run twice in positive and negative ionization modes. Lipids were separated using an Accucore C18 column 2.1 × 150 mm, 2.6 μm (Thermo Fisher Scientific) and mobile-phase solvents comprised 10 mM ammonium acetate and 0.1% formic acid in 60/40 acetonitrile/water (A) and 10 mM ammonium acetate and 0.1% formic acid in 90/10 isopropanol/acetonitrile (B). The gradient profile used was 30% B for 3 min, 30–43% B for 2 min, 43–55% B for 6 s, 55–65% B for 10 min, 65–85% B for 6 min, 85–100% B for 2 min and 100% B for 5 min. Lipids were eluted from the column at 0.4 ml/min, the oven temperature was set at 45 °C and the injection volume was 5 μl. Autosampler temperature was set at 20 °C to prevent lipid aggregation. The Q Exactive was equipped with a HESI-II probe and operated in data-dependent acquisition mode for all of the samples. To maximize the number of identified lipids, the 100 most abundant peaks found in blanks were excluded from MS/MS events. External calibration was performed using an infusion of Pierce LTQ Velos ESI Positive Ion Calibration Solution or Pierce ESI Negative Ion Calibration Solution.
LC–MS peak extraction, alignment, quantification and annotation were performed using LipidSearch software version 4.2.21 (Thermo Fisher Scientific). Lipids were identified by matching the precursor ion mass to a database and the experimental MS/MS spectra to a spectral library containing theoretical fragmentation spectra. The following lipid species were used for quantification: [M+H]+ for lysophosphati-dylcholine (LPC), lysophosphatidylethanolamine (LPE), phosphatidylcholine (PC), phosphatidylethanolamine (PE) and sphingomyelin (SM), [M−H]− for phosphatidylinositol (PI), phosphatidylserine (PS) and phosphatidylglycerol (PG), and [M+NH4]+ for diacylglycerol (DG) and triacylglycerol (TG). To reduce the risk of misidentification, MS/MS spectra from lipids of interest were validated as follows: (1) both positive-mode and negative-mode MS/MS spectra match the expected fragments, (2) the main lipid adduct forms detected in positive and negative modes are in agreement with the lipid class identified, (3) the retention time is compatible with the lipid class identified, and (4) the peak shape is acceptable. The fragmentation pattern of each lipid class was experimentally validated using lipid internal standards.
Single-point internal standard calibrations were used to estimate absolute concentrations for 332 unique identified lipids using one internal standard for each lipid class. Importantly, we ensured linearity within the range of detected endogenous lipids using serial dilutions of internal standards (IS) spanning four orders of magnitude. Subsequently, sum normalization was employed on lipid molar concentrations to correct for differential quantity of starting material. Lipid molar concentrations for a given class were calculated by summing individual lipid species molar concentrations belonging to that class68. Mol% by class of lipid species was also reported. Fatty acid composition analysis was performed in each lipid class. Fatty acid composition was calculated by taking the ratio of the sum molar concentration of a given fatty acid over the sum molar concentration across fatty acids found in the lipids of the class.
Brain endothelial cell proteomics, mass spectrometry
Brain microvessels were isolated from young (3-month-old) and aged (20-month-old) perfused mice as previously described11,19,43–47 SP3-prepared and STAGE tip cleaned brain microvessel peptides were resuspended in 0.1% formic acid and analysed by online capillary nanoLC–MS/MS. Samples were separated on an in-house made 20-cm reversed phase column (inner diameter of 100 μm, packed with ReproSil-Pur C18-AQ 3.0 μm resin (Dr. Maisch GmbH)) equipped with a laser-pulled nanoelectrospray emitter tip. Peptides were eluted at a flow rate of 400 nl/min using a four-step linear gradient including 2–4% buffer B for 1 min, 4–25% buffer B for 120 min and 25–40% B for 30 min, 40–98% buffer B for 2 min (buffer A: 0.2% formic acid and 5% DMSO in water; buffer B: 0.2% formic acid and 5% DMSO in acetonitrile) in a Dionex Ultimate 3000 LC-system (Thermo Fisher Scientific). Peptides were then analysed using an LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). Data acquisition was executed in data-dependent mode with full MS scans acquired in the Orbitrap mass analyser with a resolution of 60,000 and m/z scan range of 340–1,600. The top 20 most abundant ions with an intensity threshold above 500 counts and charge states of two and above were selected for fragmentation using collision-induced dissociation (CID) with an isolation window of 2 m/z, normalized collision energy of 35%, activation Q of 0.25 and activation time of 5 ms. The CID fragments were analysed in the ion trap with rapid scan rate. Dynamic exclusion was enabled with a repeat count of 1 and an exclusion duration of 30 s. The AGC target was set to 1,000,000 and 5,000 for full FTMS scans and ITMSn scans, respectively. The maximum injection time was set to 250 ms and 100 ms for full FTMS scans and ITMSn scans, respectively. The raw files were analysed as above, but with a precursor mass tolerance set to 20 ppm, the fragment ion tolerance set to 0.6 Da, and for protein quantification, the minimum ratio count was set to two, with unique peptides used for quantification.
BBB permeability and perfusion assessment
Following intravenous injections of 150 μl of saline or plasma (prepared as above), tracers were injected 20 h later as previously described18,20,60. In brief, mice were injected with fixable 3 kDa dextran–FITC (10 μg/g; Thermo Fisher), 70 kDa dextran–TMR (100 μg/g; Thermo Fisher) or 2 mDa dextran–FITC (100 μg/g; Thermo Fisher) dissolved in saline. After 2 h (for 3 kDa dextran–TMR) or 4 h (for 70 kDa dextran–TMR), mice were anaesthetized and perfused, and cortices and hippocampi were microdissected. Frozen tissue was thawed and suspended in 300 μl of a custom lysis buffer (200 mM Tris, 4% CHAPS, 1 M NaCl and 8 M urea, pH 8.0). Tissues were then homogenized using a Branson Digital Sonifier sonicator set to 20% amplitude for 3 s, allowed to rest for 30 s on ice and repeated three times. Samples were centrifuged at 14,000g for 20 min at 4 °C, and supernatant was extracted for analysis. Fluorescence for FITC and Texas Red were measured with a Varioskan Flash microplate reader (Thermo Fisher Scientific). The fluorescence signal was standardized to the quantity of protein per sample using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Brains from separate mice were processed for immunohistochemistry as described above, and endogenous mouse IgG was imaged and quantified. As a positive control, cortical mild traumatic brain injury (also referred to as concussion) injury was induced in mice using a Benchmark Stereotaxic Impactor (MyNeurolab), as previously described69.
Statistics and reproducibility
Box plots show the median and 25–75th percentiles, and the whiskers indicate the minimum and maximum. Violin plot midlines indicate the median. Imaging is representative of perfused n = 4 young (3-month-old) and aged (20–23-month-old) mice, or as quantified for fluorescence images or noted otherwise. Enrichment analysis is on the cumulative distribution function of the hypergeometric distribution from the Graeber lab (https://systems.crump.ucla.edu/hypergeometric/). Across experiments, mice were selected by their age. Selection of specific mice within the age group was completely random. All other criteria were not considered and as such, were randomized. Data in Figs. 1, 2g, 3, 4a–h, k–n and Extended Data Figs. 1d, e, 2–5, 6a–k, 7a–h, 9a–c, g–i, 10c, e and 11i were successfully replicated in two independent experiments.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Extended Data
Extended Data Fig. 1|. Endogenous circulatory proteins detected in the brain parenchyma and characterization of the labelled plasma proteome.
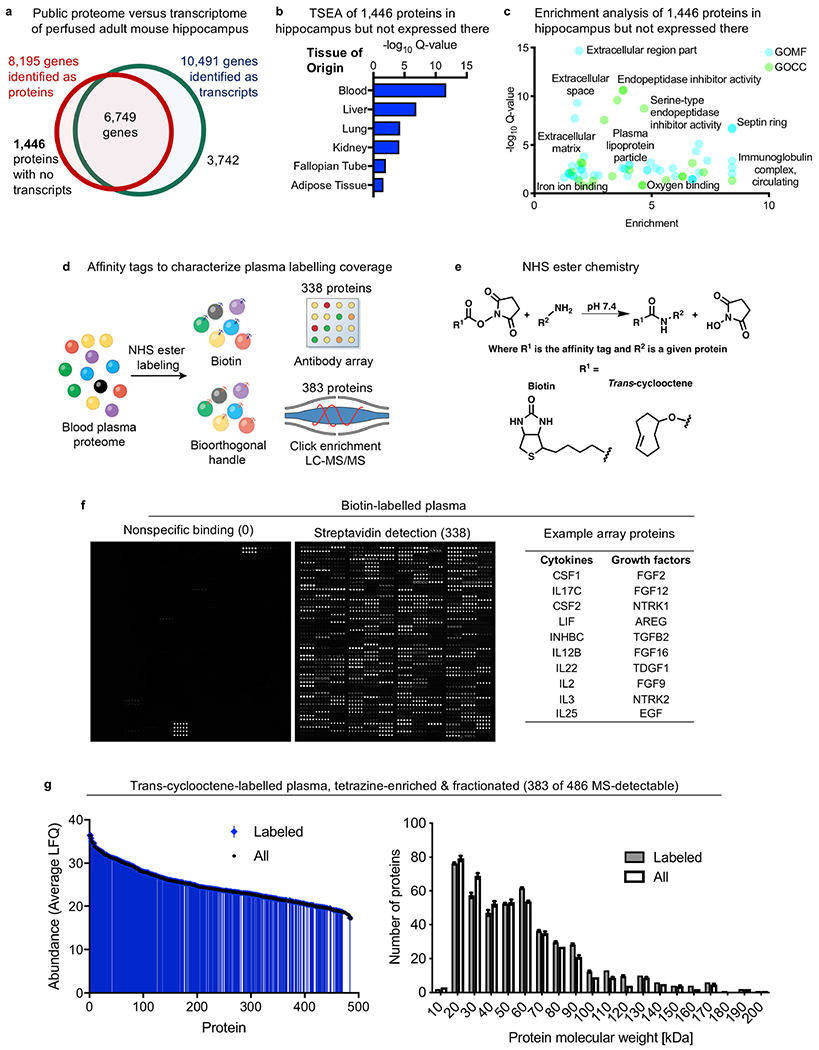
a, Comparison of published perfused brain RNA-seq70 and mass spectrometry-based proteomics71 datasets reveals that 1,446 proteins are present in the hippocampus but are not expressed in the hippocampus. These 1,446 proteins then probably migrate from the periphery into the hippocampus. b, c, The 1,446 proteins present but not expressed in the hippocampus in a are probably derived from the blood, as assessed by tissue-specific expression analysis (TSEA)72 (b) and Gene Ontology Molecular Function (GOMF) and Cell Compartment (GOCC)73,74 (c) (Fisher’s exact test with Benjamini–Hochberg correction). d, Overview of plasma labelling chemistries and detection methods to confirm labelling of hundreds of distinct plasma proteins, limited by detection method. e, Chemoselective labelling of plasma proteins via NHS ester chemistry under non-denaturing conditions (top). Conditions were optimized for each tag to achieve broad and non-perturbative protein labelling. Structures for the small affinity tags biotin and trans-cyclooctene are also shown (bottom). f, Plasma proteins labelled with biotin were incubated on an antibody array and probed with streptavidin. Hundreds of biotinylated proteins were detected across protein groups (n = 6 mice). Specific protein–antibody binding indicates that the labelling did not interfere with protein structure. The signal in the unlabelled plasma array (left) corresponds to biotinylated positive controls to ensure proper antibody printing. g, Plasma proteins labelled with the click moiety trans-cyclooctene were enriched on tetrazine beads before fractionation and mass spectrometry (MS)-based identification. Labelled proteins spanned abundance and sizes, and show no overt bias compared to the overall, unlabelled and detectable plasma proteome (FDR of <0.05, n = 3 mice per group, two-way ANOVA; mean ± s.e.m.).
Extended Data Fig. 2|. Enhanced uptake of plasma into the brain.
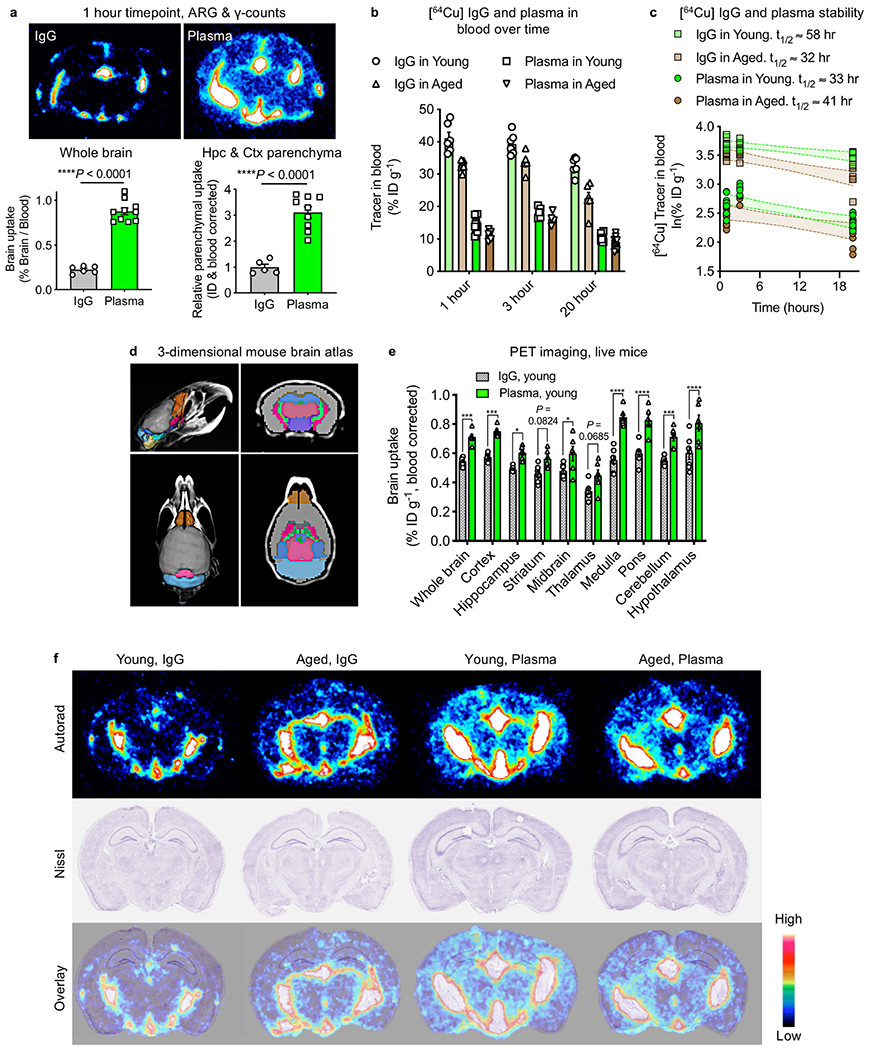
a, Autoradiography (ARG) and gamma counter quantification of 64Cu-labelled IgG and Alb/IgG-depleted plasma in whole brains (top) and the devascularized hippocampal and cortical parenchyma (bottom) from young (3-month-old) mice 1 h after intravenous ID (7.7 MBq matched dose, about 20 μg) (n = 6 IgG whole, n = 10 plasma whole brain; n = 5 IgG parenchyma, n = 9 plasma parenchyma, two-sided t-test; mean ± s.e.m.). b, 64Cu-labelled IgG and Alb/IgG-depleted plasma detected in the blood of young (3-month-old) and aged (22-month-old) mice at 1, 3 and 20 h after ID (IgG in young n = 6 for all three time points; IgG in aged n = 6 for 1 h and 20 h time points and n = 5 for 3 h time point; plasma in young and plasma in aged n = 10 for 1 h and 20 h time points and n = 5 for 3 h time point; mean ± s.e.m.). c, Time points (1, 3 and 20 h) for gamma counter quantification of 64Cu-labelled IgG and Alb/IgG-depleted plasma in circulating blood from young (3-month-old) and aged (22-month-old) mice after intravenous ID (7.7 MBq matched dose, about 20 μg). t1/2 represents the estimated blood half-life for each condition (n = 5–10). d, Brain regions demarcated by a 3D mouse brain atlas rendering51,52 for in vivo PET signal detection. e, In vivo PET signal detected across brain regions from a in young (3-month-old) mice 20 h after tail-vein intravenous ID of 7.7 MBq (about 20 μg) of 64Cu-labelled IgG or Alb/IgG-depleted plasma. The signal is corrected for activity in a corresponding cardiac blood sample at 20 h (n = 7 young IgG, n = 6 young plasma; *P < 0.05, ***P < 0.001, ****P < 0.0001, two-way ANOVA; mean ± s.e.m.). f, Ex vivo autoradiography assessment of 64Cu-labelled IgG or Alb/IgG-depleted plasma localization in coronal brain sections from tail-vein injected young (-month-old) and aged (22-month-old) mice. Nissl staining (middle) and radioactive signal/Nissl overlay (bottom) to examine anatomical co-registration (the colour bar indicates the radioactive signal from low (black) to high (white) (n = 4 per group).
Extended Data Fig. 3|. BBB permeability and homeostasis after plasma injections, and brain uptake of plasma across injection volumes and chemistries.
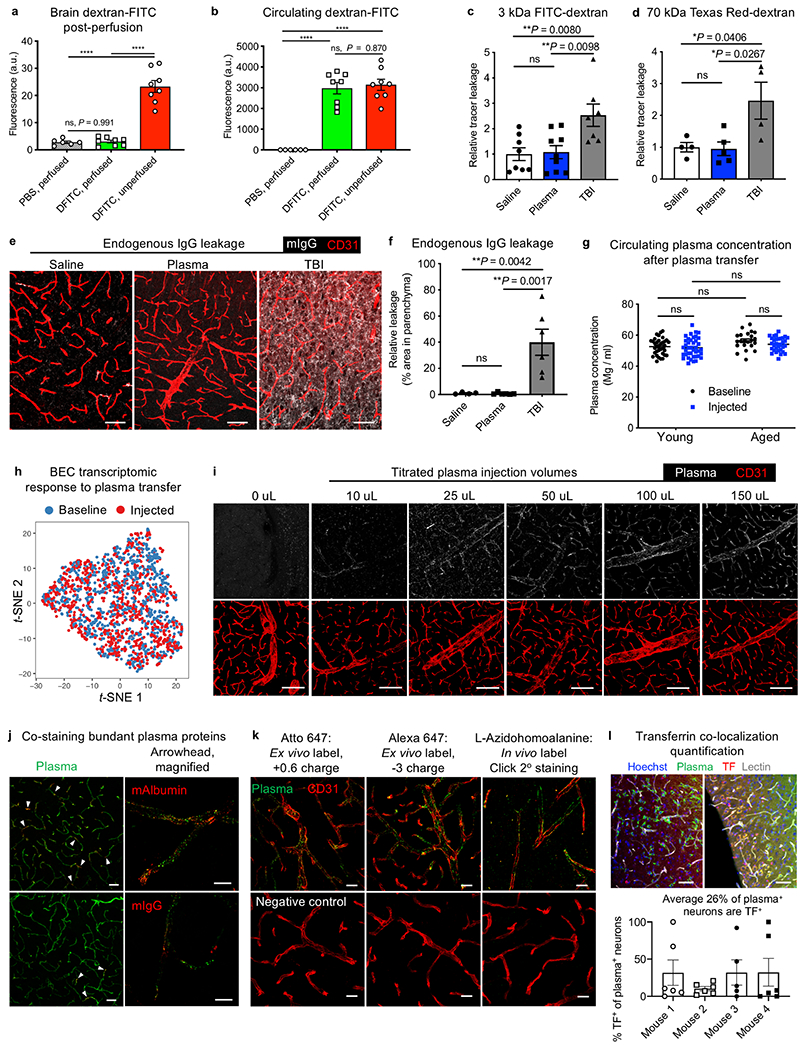
a, Assessment of perfusion completeness by measuring residual, intravenously injected 2 mDa dextran-FITC (DFITC) in brain tissue. No significant difference between dextran-FITC-injected and PBS-injected young (3-month-old) mice after perfusion (n = 6 PBS unperfused and n = 8 for the others; ****P < 0.0001, one-way ANOVA with Tukey’s multiple comparisons correction; mean ± s.e.m.). b, Circulating 2 mDa dextran-FITC in plasma at the time of death in d (n = 6 PBS unperfused and n = 8 for the others; ****P < 0.0001, one-way ANOVA with Tukey’s multiple comparisons correction; mean ± s.e.m.). c, d, BBB permeability after exposure to 150 μl saline or plasma was probed the next day by injections of 3-kDa (c) and 70-kDa (d) dextran tracers for quantification by fluorescence plate reader. Mild traumatic brain injury (TBI) served as a positive control for BBB leakage (for 3 kDa dextran: n = 8 saline, n = 9 plasma, n = 7 TBI; for 70 kDa dextran: n = 4 saline and TBI, n = 5 plasma; one-way ANOVA with Tukey’s multiple comparisons correction; mean ± s.e.m.). e, Representative images of endogenous immunoglobulin (white) extravasation into the parenchyma after exposure to 150 μl saline or plasma, or TBI75. Scale bars, 50 μm. f, Quantification of endogenous immunoglobulin (IgG) extravasation into the brain parenchyma after exposure to 150 μl saline or plasma, or TBI (n = 4 saline, n = 6 plasma and TBI; ns P = 0.9995, one-way ANOVA with Tukey’s multiple comparisons correction; mean ± s.e.m.). g, Plasma protein concentration of mice injected with 150 μl of plasma, 20 h later at time of death in young (3-month-old) and aged (20–24-month-old) mice used throughout the study compared to plasma concentrations at baseline (n = 35 young baseline, n = 35 young injected, n = 22 aged baseline, n = 32 aged injected; ns (left to right) P = 0.9923, P = 0.0657, P = 0.0657, P = 0.4302, P = 0.5430, two-way ANOVA with Sidak’s multiple comparisons test; mean ± s.e.m.). h, t-SNE plot of brain endothelial cells from young (3-month-old) mice at baseline and after exposure to 150 μl of plasma, demonstrating no significant perturbation of the cerebrovascular transcriptome upon plasma transfer (n = 3 mice). i, Representative brain images of Atto 647N-labelled plasma injected at various volumes, assayed 4 h later. 150 μl corresponds to 10–15 mg (0.5 mg/g body weight). CD31 marks brain endothelial cells. Scale bars, 100 μm. j, Representative brain images after Atto 647N-labelled plasma transfusion and stained for mouse albumin (mAlbumin; top) and mIgG (bottom). Minority of capillaries are albumin and IgG+ capillaries (arrowheads). Scale bars, 40 μm (left) and 10 μm (right). k, Atto 647N-labelled plasma (left), Alexa Fluor 647-labelled plasma (middle) and l-azidohomoalanine-labelled plasma, detected by sDIBO-647 click strain-promoted alkyne-azide cycloadditions (SPAAC) slice staining. In vivo labelled l-azidohomoalanine plasma76,77 was extracted and transfused into separate mice under normal (no l-azidohomoalanine) conditions. Scale bars, 20 μm. l, Representative images of after Atto 647N-labelled plasma transfusion and stained for mouse transferrin (top). Scale bars, 50 μm. In addition, quantification of the per cent of plasma+ neurons that also contain transferrin is shown (bottom, n = 4).
Extended Data Fig. 4|. Plasma signal in the brain does not arise from free label or degradation product.
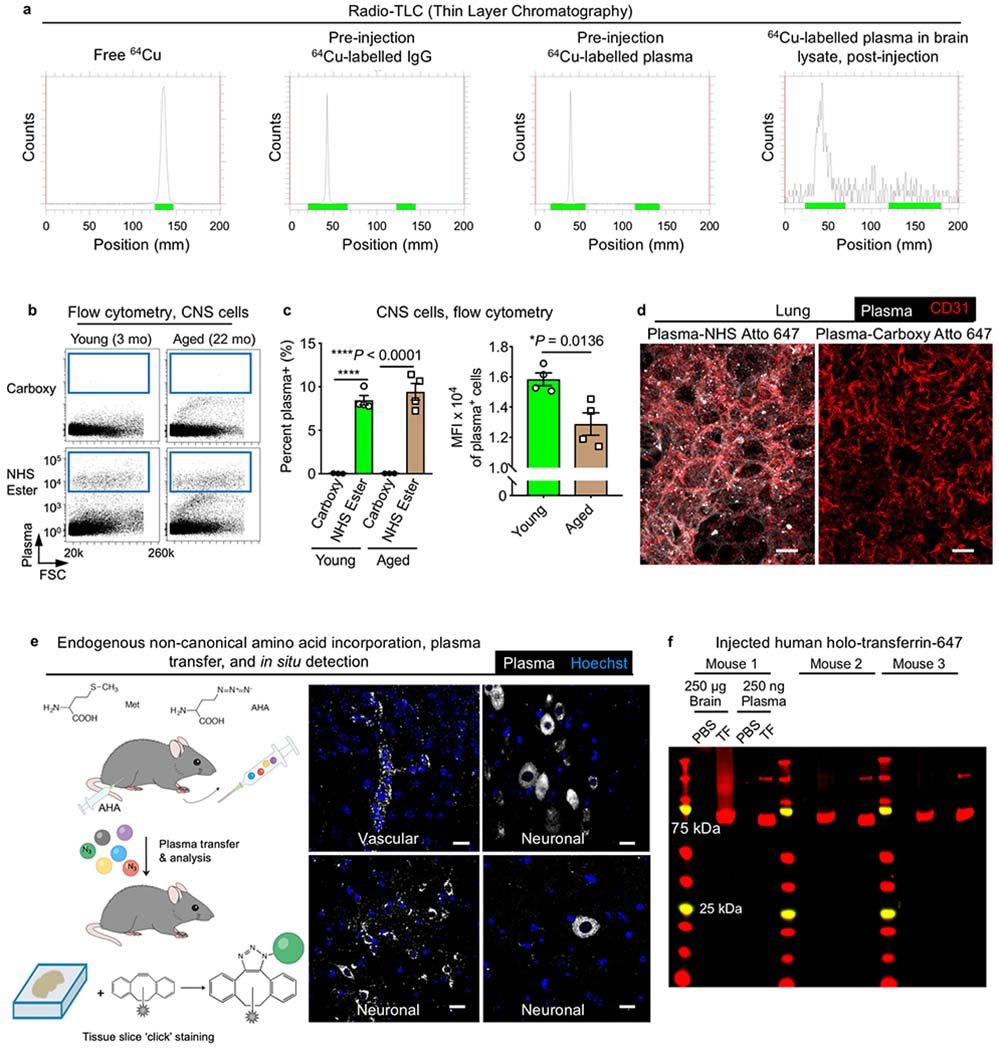
a, Representative radio-TLC (thin layer chromatography) of free 64Cu, pre-injection 64Cu-labelled IgG, pre-injection 64Cu-labelled plasma and brain lysates after intravenous injection of 64Cu-labelled plasma. Note the migration of free 64Cu relative to protein. Brain lysates are near the limit of detection, hence the background noise. b, Flow cytometry of fluorescent plasma uptake across all CNS cells. Plasma fluorescence appears only if plasma is appropriately labelled via NHS ester chemistry before intravenous injection (bottom), compared to plasma similarly incubated with fluorophore without a covalent conjugative moiety (carboxy; top). mo, month old. c, Flow cytometry quantification of plasma uptake by CNS cells from young (3-month-old) and aged (22-month-old) mice, as assessed by the per cent (%) of cells that are plasma+ and the MFI of plasma+ cells, 4 h after intravenous injection of 150 μl plasma. Note the lack of signal from carboxy-fluorophore-injected plasma, indicating no detectable confound from non-specifically bound free label (n = 4; two-sided t-test; mean ± s.e.m.). d, Representative images of plasma accumulation in lung tissue and endothelium (CD31+), showing no detectable signal from residual, non-specifically bound free dye6 in lung tissue lacking a canonical barrier. Scale bars, 20 μm. e, Schematic and representative images of azidohomoalanine (AHA)-incorporated plasma detected in brain vasculature and neurons. Unlike NHS ester labelling, there is no protein-reactive ‘free label’ with AHA-incorporated plasma, as AHA substitutes for methionine at a low rate via the methionyl-tRNA synthetase. AHA-incorporated plasma is dialysed and transfused into recipient mice and brain tissue collected for copper-free click chemistry. Scale bars, 20 μm. f, Fluorescent gels showing minimal protein degradation of an example BBB-permeable ligand, holo-transferrin, 3–4 h after intravenous injection. Human holo-transferrin runs at the expected approximately 75 kDa in size, with its dimer visible in collected plasma samples of injected mice.
Extended Data Fig. 5|. Plasma uptake across the healthy brain.
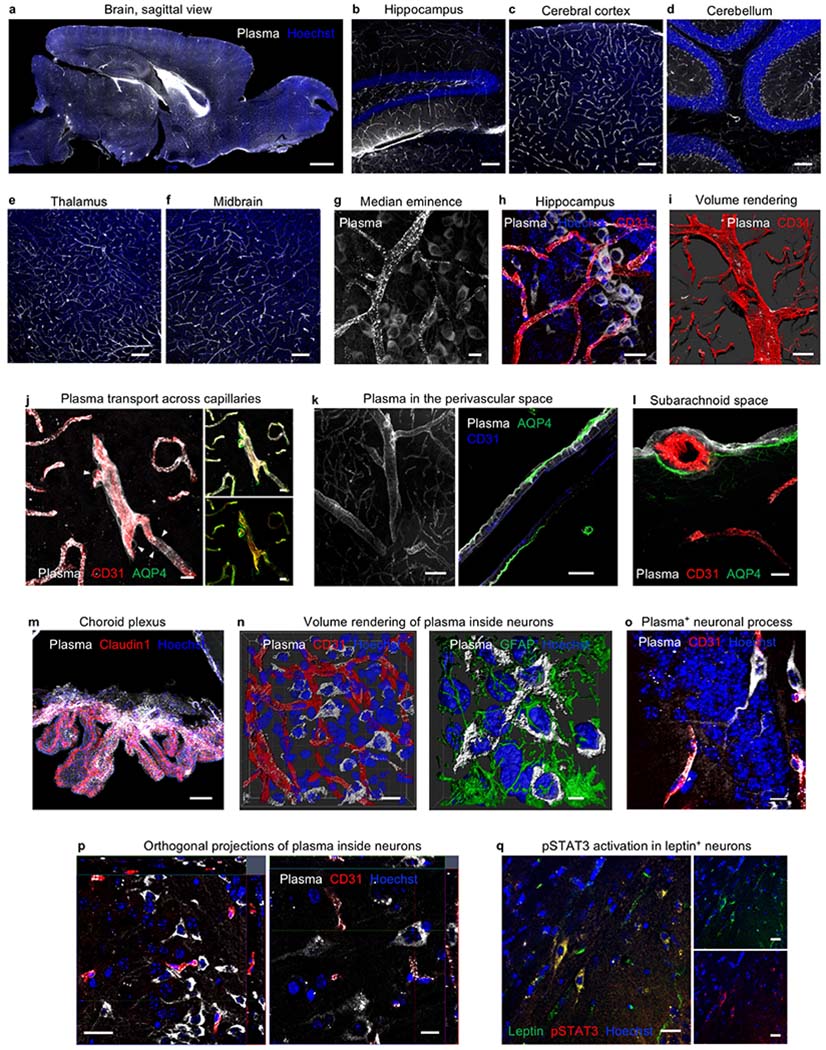
a, Representative sagittal image of plasma suffused in the healthy adult brain. Scale bar, 1,000 μm. b–f, Plasma permeates the vasculature and parenchyma across brain regions, including the hippocampus (b), the cerebral cortex (c), the cerebellum (d), the thalamus (e) and the midbrain (f). Scale bars, 100 μm. g, Representative image of plasma in the median eminence. Scale bar, 10 μm. h, Representative image of plasma (white) uptake in neurons associated with CD31+ vasculature (red) in the hippocampal dentate gyrus. Scale bar, 20 μm. i, Representative volume rendering of plasma distribution in a large artery. Plasma accumulates in focal clusters, characteristic of clathrin-mediated endocytosis78. Scale bar, 50 μm. j, Plasma accumulation in capillary basement membranes and uptake by thin-strand pericytes (arrowheads) 6 h after intravenous injection of 150 μl plasma. Scale bars, 10 μm (representative image of n = 4 young mice). k, Representative image of plasma in the perivascular space, with plasma tracing the outlines of smooth muscle cells. Scale bars, 50 μm (left), 20 μm (right). l, Representative image of plasma suffusing the subarachnoid space, localizing beyond endothelial cells (CD31) but within the glia limitans (AQP4). Scale bar, 10 μm. m, Representative image of a strong plasma signal in the choroid plexus epithelium (Claudin1+), suggesting plasma entry into the CSF and a potential role of the glymphatic system in plasma uptake and clearance. Scale bar, 50 μm. n, 3D reconstruction of plasma (white) uptake specifically in the cytoplasm of neurons, not colocalized with the CD31+ vasculature (red) or GFAP+ astrocytes (green). Scale bars, 15 μm (left), 5 μm (right). o, Plasma uptake by both neuronal soma and processes in the dentate gyrus of the hippocampus, 20 h after intravenous injection of 150 μl plasma. Scale bar, 10 μm (representative image of n = 4 young mice). p, Orthogonal view projections of plasma (white) uptake in the cytoplasm of cortical and hippocampal neurons, beyond the CD31+ vasculature (red). Scale bars, 20 μm (left), 10 μm (right). q, Detection of pSTAT3 (red) in a leptin+ (green) neuron, indicating neuronal binding and response to full-length, intravenously injected leptin. Scale bars, 20 μm.
Extended Data Fig. 6|. Contributions to plasma uptake by the BCSFB and the vascular BBB.
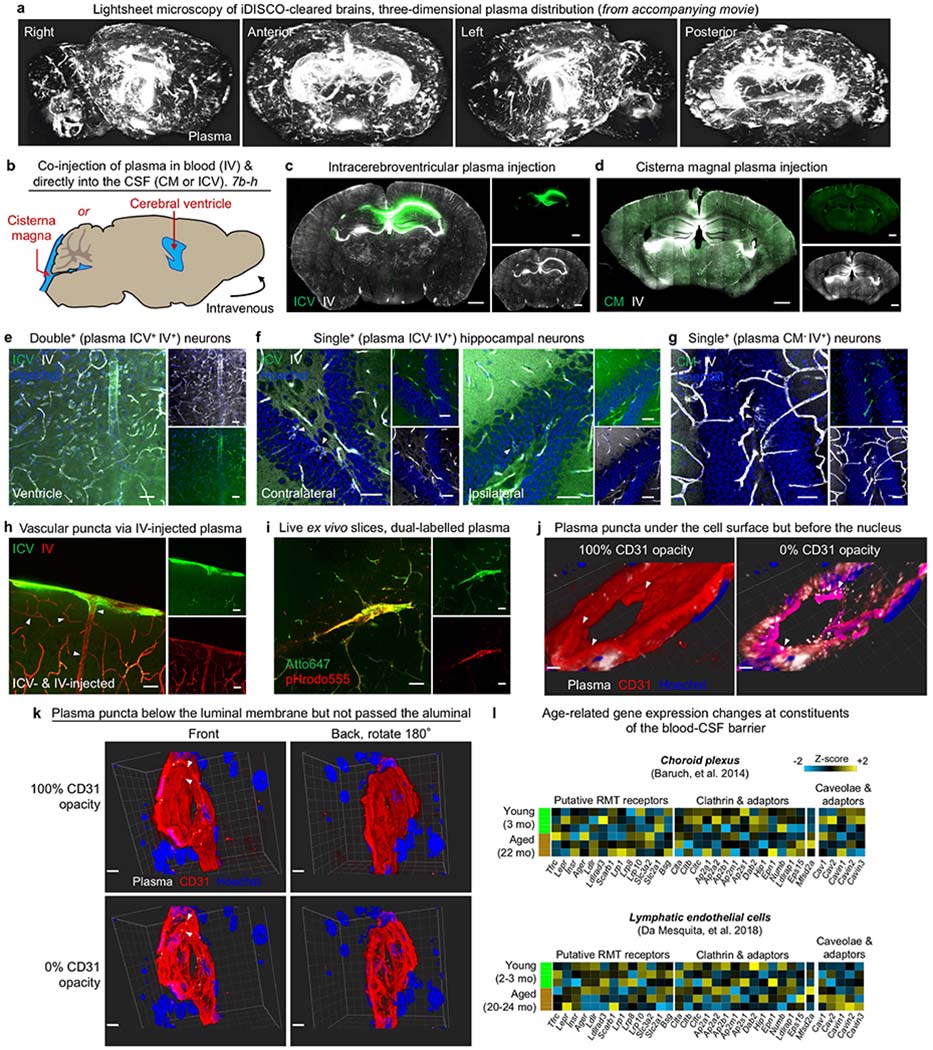
a, Lightsheet microscopy of iDISCO-cleared brains to reveal the three-dimensional distribution of plasma (white) uptake across the BCSFB and vascular BBB. b, Co-injection paradigm of plasma into the CSF via either CM or intraventricular injections (ICV), both alongside intravenous (IV) injections. Plasma injected into the CSF (10 μl) and blood (150 μl) were labelled with distinct fluorophores. Injection volumes were based on previous studies41,63,64, representing 25% (CSF) and 7.5% (blood) of total reservoir volumes (for example, 150 μl in 2 ml total circulatory volume). c, Representative coronal image of ICV-injected (green) and IV-injected (white) plasma in the healthy adult brain. ICV-injected plasma is localized near the targeted ventricle. Scale bars, 1,000 μm. d, Representative coronal image of CM-injected (green) and IV-injected (white) plasma in the healthy adult brain. Scale bars, 1,000 μm. e, Representative image of double+ (plasma ICV+ IV+) neurons, where plasma uptake could have arisen via both the BCSFB and the vascular BBB. Scale bars, 40 μm. f, Representative image of single4 (plasma ICV− IV+) neurons, where plasma uptake probably occurred across the vascular BBB. Scale bars, 40 μm. g, Representative image of single+ (plasma CM− IV+) neurons, where plasma uptake probably occurred across the vascular BBB. Scale bars, 40 μm. h, Representative image of cortical vasculature with punctated vesicles (arrowheads) arising only from IV-injected plasma (red). The diffuse signal in subarachnoid and perivascular spaces arise from IV-injected plasma (green). Scale bars, 40 μm. i, Representative image of an acutely generated ex vivo brain slice demonstrating intracellular lysosomal uptake (pHrodo555) of a subset of total IV-injected plasma (Atto647) in the brain vasculature. Scale bars, 40 μm. j, 3D reconstruction of plasma (white) within CD31+ (red) vasculature. Plasma puncta are under the endothelial CD31+ cell surface (left) but in front of intracellular nuclei (right), indicating cytoplasmic localization of plasma proteins within the brain vasculature. Scale bars, 3 μm. k, 3D reconstruction of plasma (white) within CD31+ (red) vasculature, as in j. Plasma puncta are under the luminal endothelial CD31+ cell surface (left) but not visible from the abluminal side (right), indicating cytoplasmic localization of plasma proteins within the brain vasculature. Scale bars, 5 μm. l, Gene expression of putative RMT receptors, clathrin components, and caveolar components and their inhibitor in choroid plexus and lymphatic endothelial cells of young (2–3 mo) and aged (20–24 mo) mice79,80. Relative Z-scored values are indicated in graded yellow (high) or blue (low).
Extended Data Fig. 7|. Purity and consistency of sorted BECs for scRNA-seq and correlative plasma uptake analysis.
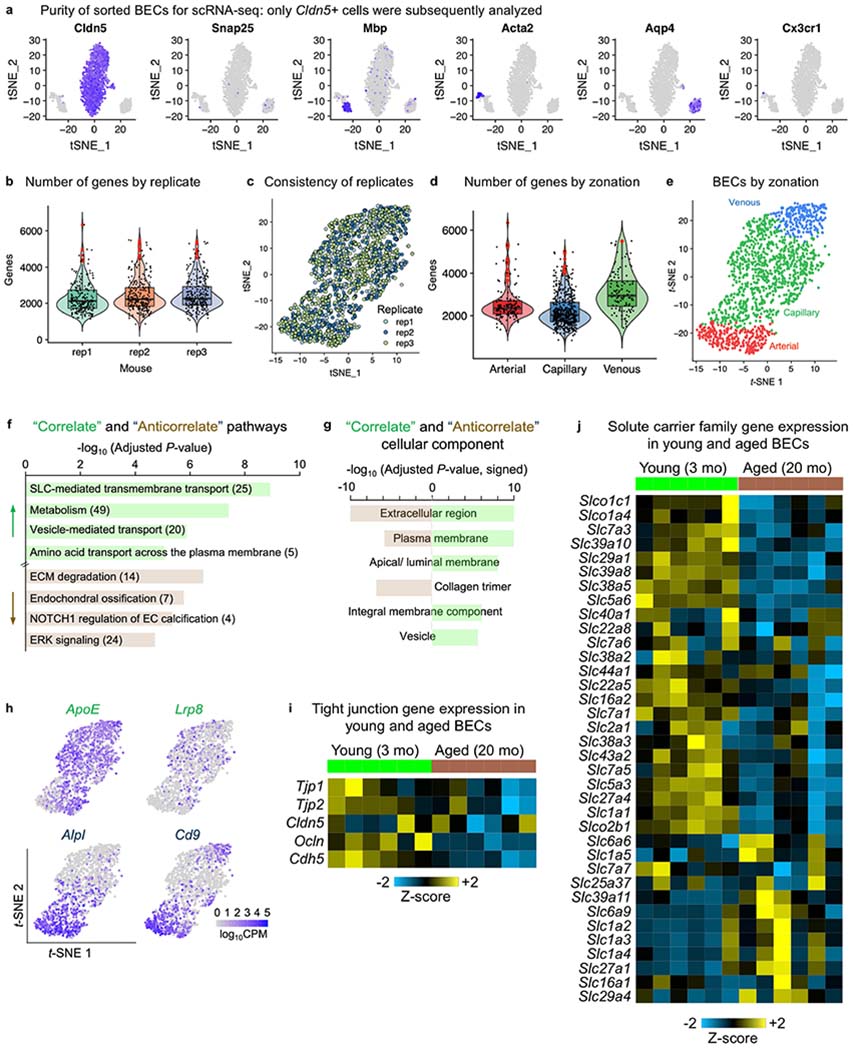
a, Sorted BECs from young (3-month-old) mice were further filtered to purity using scRNA-seq analysis using canonical cell-type markers (n = 3 young mice). b, Violin plot of the number of genes expressed per BEC within each biological replicate. Cells in red are outliers (745 total BECs from n = 3 young mice). Violin box plots show the median and the 25th to 75th percentiles, and the whiskers indicate the minimum and maximum. c, t-SNE plot with BECs from separate mice coloured distinctly, showing consistency in transcriptomes across replicates (745 total BECs from n = 3 young mice). d, Violin plot of the number of genes expressed per BEC by zonation. Cells in red are outliers (745 total BECs from n = 3 young mice). Violin box plots show the median and the 25th to 75th percentiles, and the whiskers indicate the minimum and maximum. e, t-SNE plot demonstrating BEC separation by arterial, capillary and venous zonation (n = 3 young mice). f, Pathway enrichment analysis of ‘Correlate’ and ‘Anticorrelate’ genes, with the number of genes in each pathway listed (from n = 3 young mice, GeneAnalytics analysis)81. ECM, extracellular matrix; SLC, solute carrier. g, Gene Ontology cellular component enrichment analysis of ‘Correlate’ and ‘Anticorrelate’ genes (from n = 3 young mice, GOrilla exact mHG P-value computation)82. h, Robust PCA plots of example ‘Correlate’ and ‘Anticorrelate’ gene expression across BECs. Each dot is an individual cell, and gene expression levels are indicated by the colour spectrum (log10 CPM, 745 total BECs from n = 3 young mice). i, Tight junction gene expression from RNA-seq of BECs from young (3 mo) and aged (20 mo) mice (n = 6). Relative Z-scored values are indicated in graded yellow (high) or blue (low). j, SLC transporter gene expression from RNA-seq of BECs from young (3 mo) and aged (20 mo) mice (n = 6). Relative Z-scored values are indicated in graded yellow (high) or blue (low). The SLC genes shown are derived from two comprehensive reviews83,84.
Extended Data Fig. 8|. Lipidomics companion to the decrease in MFSD2A expression in the aged brain vasculature.
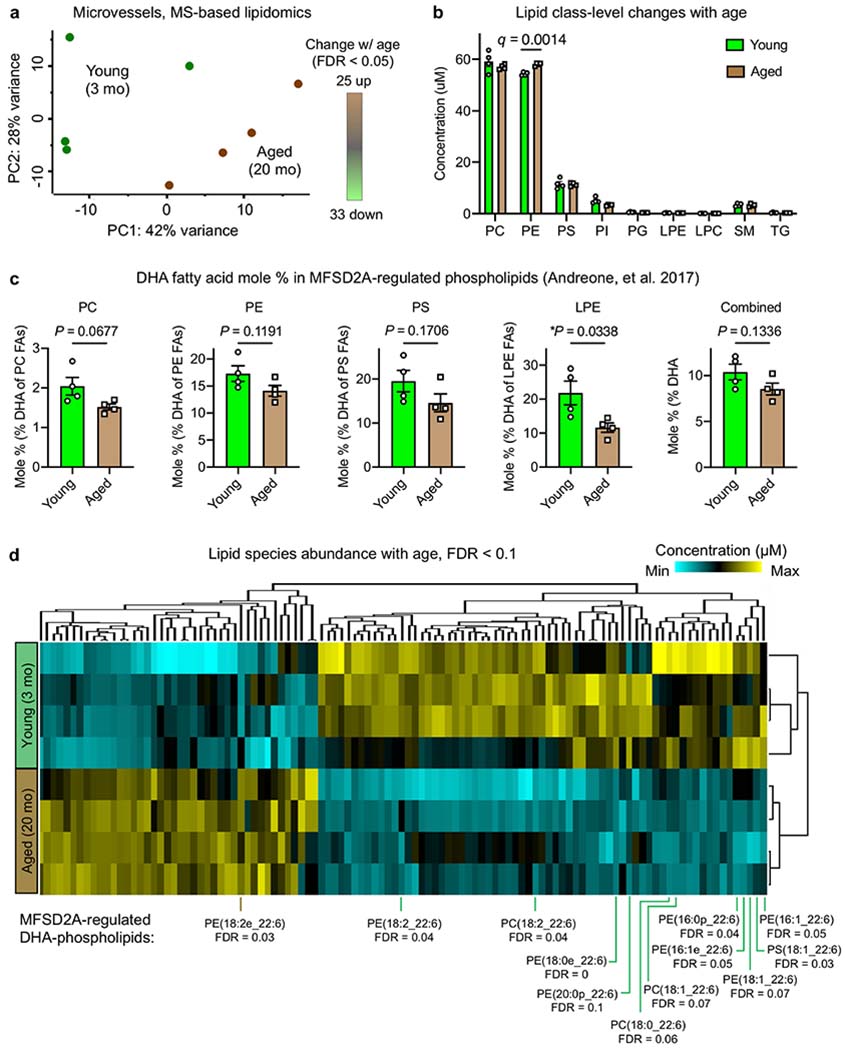
a, PC analysis plot of brain microvessel lipidomes from young (3 mo) and aged (20 mo) mice. There were 25 lipids significantly upregulated and 33 lipids downregulated with age (n = 4, each n is a pool of 4 mice (16 young and 16 aged total), FDR-corrected q < 0.05, Permutation-based FDR of two-sided t-test). b, Absolute abundance (concentrations) of measured microvessel lipid classes with age (n = 4, each n is a pool of 4 mice, Benjamini, Krieger and Yekutieli FDR-corrected two-sided t-test; mean ± s.e.m.). DG is not listed as only one DG species was detected. c, Mole % of MFSD2A-regulated phospholipids19 (PC, PE, PS and LPE) across and within phospholipid classes. Concentrations of individual lipid species were derived with spike-in standards and presented as a quantitative mole % of DHA fatty acid (FA) of all fatty acids in the given class68 or across PC, PE, PS and LPE classes (right, ‘Combined’) (n = 4, each n is a pool of 4 mice, two-sided t-test; mean ± s.e.m.). d, Hierarchical clustering of lipids (by concentration) of microvessels from young (3 mo) and aged (20 mo) mice (n = 4; each sample is a pool of 4 mice) normalized by Z-score. All lipids differentially abundant with an FDR-corrected q < 0.1 are shown (permutation-based FDR of two-sided t-test), and MFSD2A-regulated DHA-phospholipids are labelled.
Extended Data Fig. 9|. Specificity and effect of pericyte loss in the aged brain vasculature and working model of BBB transcytosis with age.
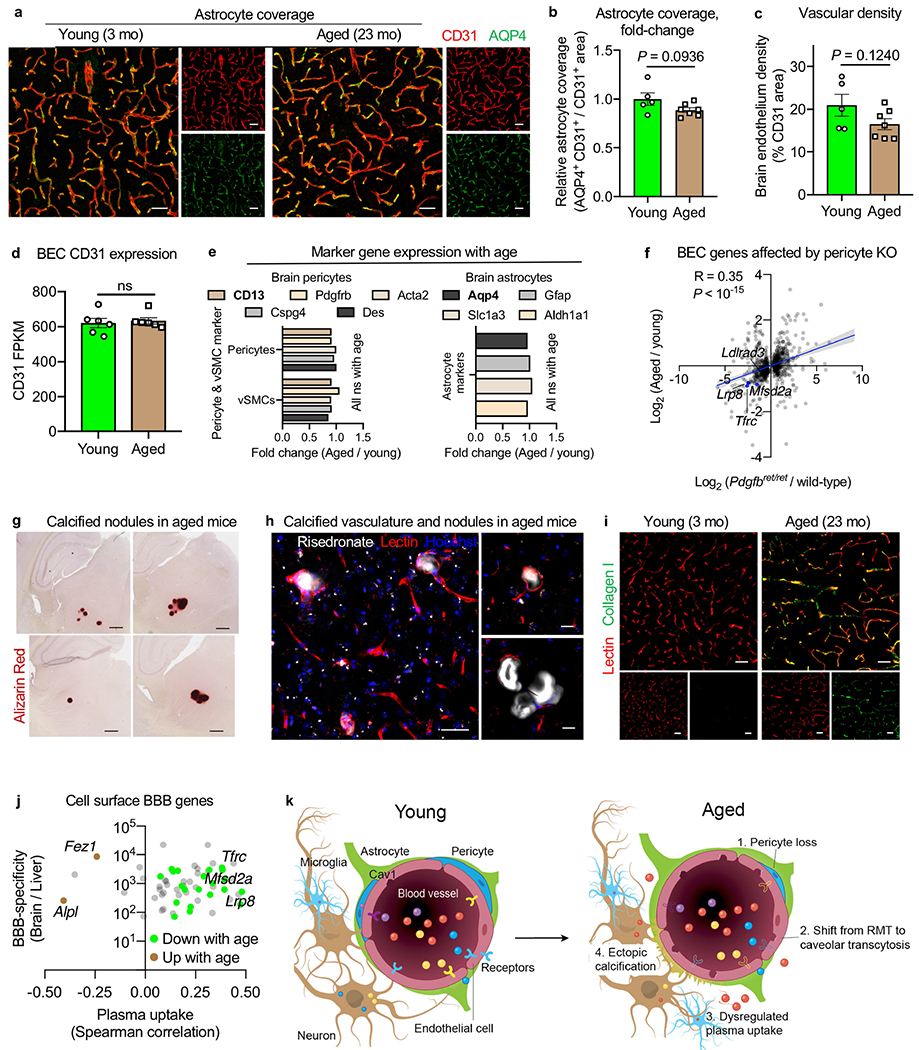
a, Representative images of AQP4+ vascular astrocytes (green) and CD31+ endothelial cells in the cerebral cortex of young (3 mo) and aged (23 mo) mice. Scale bars, 40 μm. b, Quantification of vascular astrocyte coverage in the cerebral cortex vasculature of young (3 mo) and aged (23 mo) mice (n = 5 young and n = 7 aged; two-sided t-test; mean ± s.e.m.). c, Quantification of endothelial cell density in the cerebral cortex vasculature of young (3 mo) and aged (23 mo) mice (n = 5 young and n = 7 aged; two-sided t-test; mean ± s.e.m.). d, CD31 gene expression from RNA-seq of BECs from young (3 mo) and aged (20 mo) mice (ns P = 0.699, two-sided t-test; mean ± s.e.m.). e, Gene expression of brain pericyte and astrocyte markers from young (2–3 mo) and aged (21–22 mo) mice85 (ns (exact P-values can be found in Supplementary Table 6 of ref.85), n = 8 young and aged, FDR-corrected MAST test). f, Correlation of changes in gene expression between BECs from aged (20 mo) and young (3 mo) and BECs from pericyte knockout Pdgfbret/ret mice20 and age-matched wild-type mice (R = 0.35, P < 10−15; the blue line is the simple linear regression and the fill is the 95% confidence interval, Spearman’s correlation; n = 4 Pdgfbret/ret mice and n = 3 controls, and n = 6 young and aged mice). g, Representative images of calcified nodules (Alizarin Red staining) detected in aged (23 mo) but not young (3 mo) mice, recapitulating nodules seen in pericyte-deficient Pdgfbret/ret mice (representative image of n = 21 mice per age). Scale bars, 330 μm. h, Representative images of vascular calcifications in the aged (23 mo) brain. Risedronate (white) accumulation in lectin+ (red) vasculature by immunofluorescence staining. i, Representative images of type I collagen expression (green) in lectin+ vasculature (red) in the cerebral cortex of young (3 mo) and aged (23 mo) mice. Scale bars, 40 μm (left), 20 μm (right). j, Dot plot representation of cell-surface (from Uniprot), druggable gene candidates on BECs. Genes are plotted by their degree of BBB specificity15 and correlation with plasma uptake. Genes are coloured by their upregulation (brown) or downregulation (green) with age. k, Working model of BBB transcytosis with age. In healthy adults, BECs express higher levels of receptors and components of clathrin-coated pits86,87 to transport select circulatory proteins via RMT. With age, pericytes degenerate, promoting vascular calcification and a shift in endothelial transport from ligand-specific RMT to caveolar transcytosis. Caveolar transcytosis is non-specific87, rendering the aged BBB ‘leaky’ to neurotoxic proteins excluded in health, such as fibrin(ogen), thrombin and autoantibodies25,27.
Extended Data Fig. 10|. Rationale for ALPL inhibitor treatment in aged brains and purity of sorted aged BECs for scRNA-seq after pharmacological ALPL inhibition.
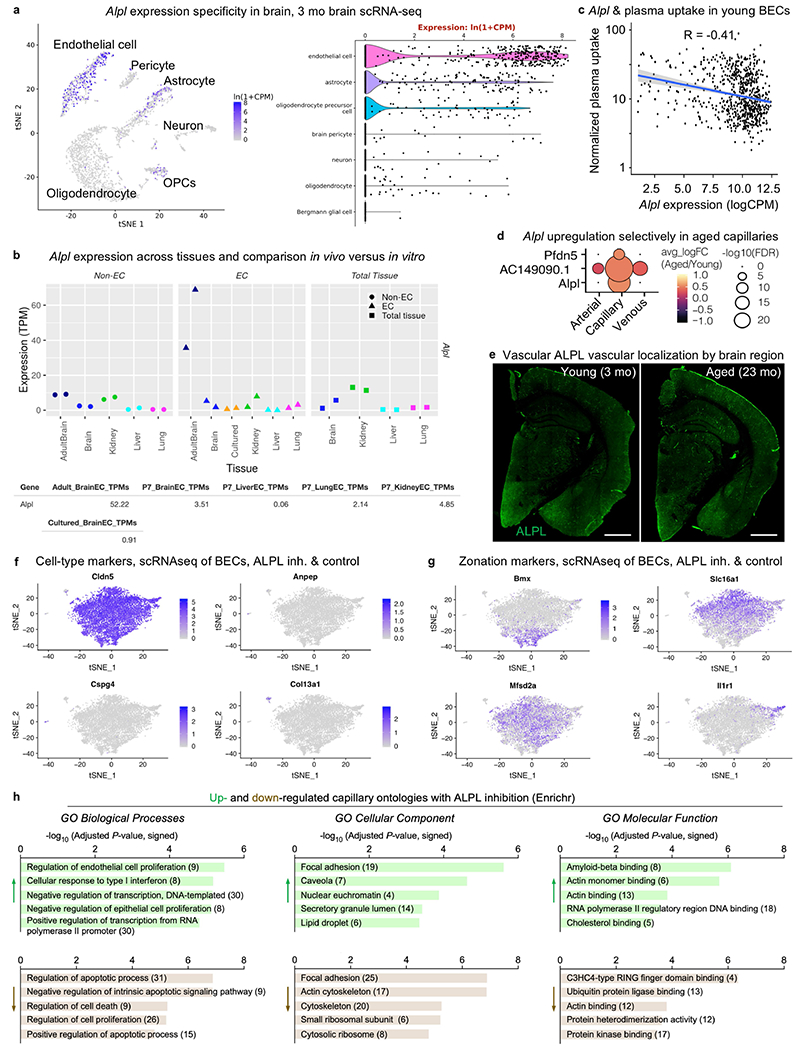
a, t-SNE and violin plot showing scRNA-seq analysis of Alpl expression in CNS cells, log-normalized counts per million reads (CPM). Alpl is expressed mainly in BECs (n = 7 young mice). Data from the Tabula Muris Consortium54. OPC, oligodendrocyte precursor cell. b, Alpl expression across endothelial cells (ECs) and non-ECs in the CNS, periphery and tissue culture (transcripts per million (TPM)). Alpl is specific to the brain endothelial and expression is lost upon culture. Data from the Vascular Endothelial Cell Trans-omics Resource Database (https://markfsabbagh.shinyapps.io/vectrdb/)88. c, Correlation between Alpl expression in young BECs and plasma uptake, from combined flow cytometry index sorting and scRNA-seq. The blue line denotes linear regression and the fill denotes the confidence interval, Spearman’s correlation. Non-Alpl-expressing BECs were excluded (n = 3 young (3 mo) mice). d, Alpl expression increases specifically in capillaries with age (n = 6 mice, FDR-corrected Mann-Whitney U-test). Analysis based on published data57. e, ALPL protein expression in young (3 mo) and aged (23 mo) brains. Scale bars, 1,000 μm. f, Sorted BECs from aged (22 mo) mice treated with an ALPL inhibitor or vehicle were further filtered to purity using scRNA-seq analysis using canonical cell-type markers (n = 4 mice per group). g, BECs were assigned to arterial, capillary and venous vessel segments based on vasculature zonation markers (n = 4 mice per group)16,57. h, Gene Ontologies (GO) enriched in differentially expressed genes in capillaries upon ALPL inhibitor versus vehicle treatment (Enrichr analysis; the number of genes in the pathway are shown in brackets)82.
Extended Data Fig. 11|. Validation of ALPL inhibitor treatment and relevance to the human vasculature and Alzheimer’s disease.
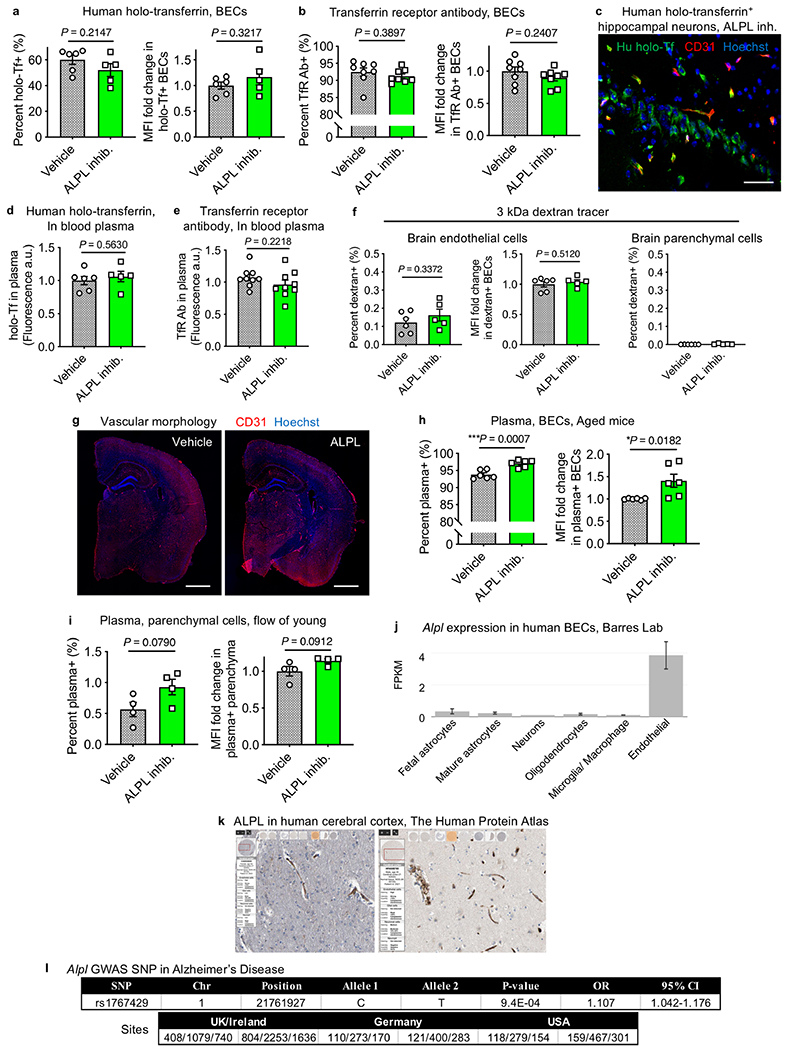
a, b, Flow cytometry quantification of BEC uptake of human holo-transferrin (hu-Tf) from aged (a) or TfR Ab from aged (b) mice treated with vehicle (grey) or ALPL inhibitor (green) via IP injections twice daily for 3 days, as assessed by the percent of BECs that are tracer+ and the relative MFI of tracer+ BECs (for hu-Tf: n = 6 mice vehicle, n = 5 ALPL inhibitor; for TfR Ab: n = 8; two-sided t-test; mean ± s.e.m.). Tracer refers to hu-Tf or TfR Ab. c, Representative images of human holo-Tf+ neurons detected in the cortex of mice treated with ALPL inhibitor. Scale bar, 40 μm. d, e, Circulating human holo-Tf (d) and TfR Ab (e) in blood plasma upon time of death to control for and ensure no differences in the injected amount (for hu-Tf+: n = 6 mice vehicle, n = 5 ALPL inhibitor; for TfR Ab: n = 9; two-sided t-test; mean ± s.e.m.). f, As in a and b, but for the 3-kDa dextran tracer that probes for disruptions in paracellular permeability (n = 6 mice vehicle, n = 5 ALPL inhibitor; two-sided t-test; mean ± s.e.m.). g, Overall cerebrovascular morphology, as assayed by CD31 staining, from aged mice (22 mo) treated with vehicle or an ALPL inhibitor. Scale bars, 1,000 μm. h, i, As in a and b, but for plasma uptake in BECs from aged mice (22 mo, n = 6) (h) and parenchymal cells from young mice (3 mo, n = 4; two-sided t-test; mean ± s.e.m.) (i). j, Alpl gene expression in various cell types of the human CNS, showing exclusive expression in endothelial cells. Data from Barres lab RNA-seq (http://www.brainrnaseq.org; mean ± s.e.m.)89. k, ALPL protein expression localized to the human brain vasculature in The Human Protein Atlas (http://www.proteinatlas.org)90,91. l, The Alpl single-nucleotide polymorphism (SNP) rs1767429 shows association with Alzheimer’s disease (over 160,000 individuals, genome-wide association study (GWAS) analysis)92.
Supplementary Material
Acknowledgements
We thank K. Brewer, O. Leventhal, N. Schaum, S. R. Shuken, C. Munson, J. V. Pluvinage, C. A. Maat, N. Khoury and other members of the Wyss-Coray, James, Snyder and Elias laboratories for feedback and support; H. Zhang and K. Dickey for laboratory management; B. Carter for flow cytometry technical expertise; J. Mulholland and Y. Lim of the Stanford Cell Sciences Imaging Facility for help with high-resolution microscopy; and U. Langen and C. Gu for the MFSD2A antibody. This work was funded by the Department of Veterans Affairs (to T.W.-C.), the National Institute on Aging (DP1-AG053015 to T.W.-C., T32-AG0047126 to A.C.Y. and 1RF1AG059694 to T.W.-C. and J.L.), the National Institute of General Medical Sciences (R01-GM059907 to C.R.B.), the NOMIS Foundation (to T.W.-C.), the Glenn Foundation for Aging Research (to T.W.-C.), the National Institutes of Health (3P50HG00773505S1 to M.P.S.), and the Big Idea Brain Rejuvenation Project from the Wu Tsai Neurosciences Institute (to T.W.-C., J.E.E. and C.R.B.). A.C.Y was supported by a Siebel Scholarship. M.Y.S. is a Wallenberg Foundation postdoctoral fellow. S.R.Q. is a Chan Zuckerberg Investigator. We acknowledge the National Institutes of Health shared equipment grants, 1S10OD025091-01 and 1S10OD01227601 from the National Center for Research Resources (NCRR). This study was supported by the AHA-Allen Initiative in Brain Health and Cognitive Impairment: 19PABHI34580007. The statements in this work are solely the responsibility of the authors and do not necessarily represent the views of the American Heart Association (AHA) or the Paul G. Allen Frontiers Group.
Footnotes
Competing interests C.R.B. is a co-founder and Scientific Advisory Board member of Palleon Pharmaceuticals, Enable Bioscience, InterVenn Bio and Redwood Bioscience (a subsidiary of Catalent), and a member of the Board of Directors of Eli Lilly and Company. T.W.-C. is a co-founder and scientific advisor of Alkahest Inc. T.W.-C. and A.C.Y. are co-inventors on a patent application related to the work published in this paper. N.O. is affiliated with Calico Life Sciences and has no financial interests to declare.
Supplementary information is available for this paper at https://doi.org/10.1038/s41586-020-2453-z.
Peer review information Nature thanks Peter Meikle, Christopher Walsh, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data Availability
Raw sequencing data are deposited under NCBI GEO: GSE134058 and GSE142500. Raw lipidomics data are available from figshare (https://doi.org/10.6084/m9.figshare.6025748). Source data are provided with this paper.
References
- 1.Obermeier B, Daneman R & Ransohoff RM Development, maintenance and disruption of the blood–brain barrier. Nat. Med 19, 1584–1596 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chow BW & Gu C The molecular constituents of the blood–brain barrier. Trends Neurosci 38, 598–608 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Profaci CP, Munji RN, Pulido RS & Daneman R The blood–brain barrier in health and disease: important unanswered questions. J. Exp. Med 217, e20190062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conboy IM et al. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433, 760–764 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Villeda SA et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saunders NR, Daneman R, Dziegielewska KM & Liddelow SA Transporters of the blood–brain and blood–CSF interfaces in development and in the adult. Mol. Aspects Med 34, 742–752 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Saunders NR et al. The rights and wrongs of blood–brain barrier permeability studies: a walk through 100 years of history. Front. Neurosci 8, 404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reese TS & Karnovsky MJ Fine structural localization of a blood–brain barrier to exogenous peroxidase. J. Cell Biol 34, 207–217 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poduslo JF, Curran GL & Berg CT Macromolecular permeability across the blood–nerve and blood–brain barriers. Proc. Natl Acad. Sci. USA 91, 5705–5709 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu YJ & Watts RJ Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 10, 459–472 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuchero YJY et al. Discovery of novel blood–brain barrier targets to enhance brain uptake of therapeutic antibodies. Neuron 89, 70–82 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Niewoehner J et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 81, 49–60 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Bell RD et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ben-Zvi A et al. Mfsd2a is critical for the formation and function of the blood–brain barrier. Nature 509, 507–511 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daneman R et al. The mouse blood–brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS ONE 5, e13741 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanlandewijck M et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Montagne A et al. Blood–brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yousef H et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat. Med 25, 988–1000 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andreone BJ et al. Blood–brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron 94, 581–594.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Armulik A et al. Pericytes regulate the blood–brain barrier. Nature 468, 557–561 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Keller A et al. Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat. Genet 45, 1077–1082 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Zarb Y et al. Ossified blood vessels in primary familial brain calcification elicit a neurotoxic astrocyte response. Brain 142, 885–902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murshed M, Harmey D, Millan JL, McKee MD & Karsenty G Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev 19, 1093–1104 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dahl R et al. Discovery and validation of a series of aryl sulfonamides as selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP). J. Med. Chem 52, 6919–6925 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montagne A, Zhao Z & Zlokovic BV Alzheimer’s disease: a matter of blood–brain barrier dysfunction? J. Exp. Med 214, 3151–3169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zlokovic BV Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci 12, 723–738 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petersen MA, Ryu JK & Akassoglou K Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat. Rev. Neurosci 19, 283–301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erdő F, Denes L & de Lange E Age-associated physiological and pathological changes at the blood–brain barrier: a review. J. Cereb. Blood Flow Metab 37, 4–24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daneman R, Zhou L, Kebede AA & Barres BA Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 468, 562–566 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall CN et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508, 55–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vardy ERLC, Kellett KAB, Cocklin SL & Hooper NM Alkaline phosphatase is increased in both brain and plasma in Alzheimer’s disease. Neurodegener. Dis 9, 31–37 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Zlokovic BV, Deane R, Sagare AP, Bell RD & Winkler EA Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer’s amyloid β-peptide elimination from the brain. J. Neurochem 115, 1077–1089 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang AC et al. Multiple click-selective tRNA synthetases expand mammalian cell-specific proteomics. J. Am. Chem. Soc 140, 7046–7051 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hughes CS et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol 10, 757 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cox J & Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Tyanova S, Temu T & Cox J The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc 11, 2301–2319 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Castellano JM et al. Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature 544, 488–492 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Treweek JB et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nat. Protoc 10, 1860–1896 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tournoux F et al. Validation of noninvasive measurements of cardiac output in mice using echocardiography. J. Am. Soc. Echocardiogr 24, 465–470 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Domínguez E et al. Non-invasive in vivo measurement of cardiac output in C57BL/6 mice using high frequency transthoracic ultrasound: evaluation of gender and body weight effects. Int. J. Cardiovasc. Imaging 30, 1237–1244 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Villeda SA et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med 20, 659–663 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schnell SA, Staines WA & Wessendorf MW Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J. Histochem. Cytochem 47, 719–730 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Triguero D, Buciak J & Pardridge WM Capillary depletion method for quantification of blood–brain barrier transport of circulating peptides and plasma proteins. J. Neurochem 54, 1882–1888 (1990). [DOI] [PubMed] [Google Scholar]
- 44.Singh I et al. Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl Acad. Sci. USA 110, 14771–14776 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paris-Robidas S, Brouard D, Emond V, Parent M & Calon F Internalization of targeted quantum dots by brain capillary endothelial cells in vivo. J. Cereb. Blood Flow Metab 36, 731–742 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Preston JE, al-Sarraf H & Segal MB Permeability of the developing blood–brain barrier to 14C-mannitol using the rat in situ brain perfusion technique. Brain Res. Dev. Brain Res 87, 69–76 (1995). [DOI] [PubMed] [Google Scholar]
- 47.Dan M, Cochran DB, Yokel RA & Dziubla TD Binding, transcytosis and biodistribution of anti-PECAM-1 iron oxide nanoparticles for brain-targeted delivery. PLoS ONE 8, e81051 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boulay AC, Saubaméa B, Declèves X & Cohen-Salmon M Purification of mouse brain vessels. J. Vis. Exp 105, e53208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferreira CL et al. Comparison of bifunctional chelates for 64Cu antibody imaging. Eur. J. Nucl. Med. Mol. Imaging 37, 2117–2126 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Ilovich O et al. Development and validation of an immuno-PET tracer as a companion diagnostic agent for antibody–drug conjugate therapy to target the CA6 epitope. Radiology 276, 191–198 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chaney A et al. 11C-DPA-713 versus 18F-GE-180: a preclinical comparison of translocator protein 18 kDa PET tracers to visualize acute and chronic neuroinflammation in a mouse model of ischemic stroke. J. Nucl. Med 60, 122–128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaney AM, Johnson EM, Cropper HC & James ML PET imaging of neuroinflammation using [11C]DPA-713 in a mouse model of ischemic stroke. J. Vis. Exp 136, 57243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.James ML et al. New positron emission tomography (PET) radioligand for imaging σ-1 receptors in living subjects. J. Med. Chem 55, 8272–8282 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tabula Muris Consortium. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Picelli S et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Wu AR et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods 11, 41–46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen MB et al. Brain endothelial cells are exquisite sensors of age-related circulatory cues. Cell Rep 30, 4418–4432.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Q et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 101, 207–223.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsafrir D et al. Sorting points into neighborhoods (SPIN): data analysis and visualization by ordering distance matrices. Bioinformatics 21, 2301–2308 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Yanagida K et al. Size-selective opening of the blood–brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc. Natl Acad. Sci. USA 114, 4531–4536 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pluvinage JV et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 568, 187–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iliff JJ et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med 4, 147ra111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xavier ALR et al. Cannula implantation into the cisterna magna of rodents. J. Vis. Exp 135, 57378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Renier N et al. iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 159, 896–910 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Cajka T, Smilowitz JT & Fiehn O Validating quantitative untargeted lipidomics across nine liquid chromatography-high-resolution mass spectrometry platforms. Anal. Chem 89, 12360–12368 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Contrepois K et al. Cross-platform comparison of untargeted and targeted lipidomics approaches on aging mouse plasma. Sci. Rep 8, 17747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zemski Berry KA, Murphy RC, Kosmider B & Mason RJ Lipidomic characterization and localization of phospholipids in the human lung. J. Lipid Res 58, 926–933 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luo J et al. Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Front. Neurol 5, 12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kadakkuzha BM et al. Transcriptome analyses of adult mouse brain reveal enrichment of lncRNAs in specific brain regions and neuronal populations. Front. Cell. Neurosci 9, 63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharma K et al. Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci 18, 1819–1831 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dougherty JD, Schmidt EF, Nakajima M & Heintz N Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res 38, 4218–4230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ashburner M et al. Gene Ontology: tool for the unification of biology. Nat. Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harris MA et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 32, D258–D261 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang XD et al. Traumatic brain injury imaging in the second near-infrared window with a molecular fluorophore. Adv. Mater 28, 6872–6879 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kiick KL, Saxon E, Tirrell DA & Bertozzi CR Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl Acad. Sci. USA 99, 19–24 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Calve S, Witten AJ, Ocken AR & Kinzer-Ursem TL Incorporation of non-canonical amino acids into the developing murine proteome. Sci. Rep 6, 32377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu AP, Aguet F, Danuser G & Schmid SL Local clustering of transferrin receptors promotes clathrin-coated pit initiation. J. Cell Biol 191, 1381–1393 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baruch K et al. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Da Mesquita S et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 560, 185–191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fuchs SBA et al. GeneAnalytics: an integrative gene set analysis tool for next generation sequencing, RNAseq and microarray data. OMICS 20, 139–151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eden E, Navon R, Steinfeld I, Lipson D & Yakhini Z GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Saunders NR, Dziegielewska KM, Møllgård K & Habgood MD Physiology and molecular biology of barrier mechanisms in the fetal and neonatal brain. J. Physiol. (Lond.) 596, 5723–5756 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhao Z, Nelson AR, Betsholtz C & Zlokovic BV Establishment and dysfunction of the blood–brain barrier. Cell 163, 1064–1078 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ximerakis M et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci 22, 1696–1708 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Simionescu M et al. The cerebral microvasculature of the rat: structure and luminal surface properties during early development. J. Submicrosc. Cytol. Pathol 20, 243–261 (1988). [PubMed] [Google Scholar]
- 87.Hervé F, Ghinea N & Scherrmann J-M CNS delivery via adsorptive transcytosis. AAPS J 10, 455–472 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sabbagh MF et al. Transcriptional and epigenomic landscapes of CNS and non-CNS vascular endothelial cells. eLife 7, e36187 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thul PJ et al. A subcellular map of the human proteome. Science 356, eaal3321 (2017). [DOI] [PubMed] [Google Scholar]
- 91.Uhlén M et al. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 92.Harold D et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet 41, 1088–1093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data are deposited under NCBI GEO: GSE134058 and GSE142500. Raw lipidomics data are available from figshare (https://doi.org/10.6084/m9.figshare.6025748). Source data are provided with this paper.