

Abstract
AST-3424/OBI-3424 (denoted by 3424) is a novel prodrug bis-alkylating agent activated by AKR1C3. AKR1C3 is overexpressed in many types of cancer, particularly in liver, non-small cell lung, gastric, renal and CRPC cancer. Currently 3424 is being studied in phase 1/2 clinical trials for the treatment of solid and hematologic cancers, and it represents potentially a novel, selective anti-cancer agent for multiple indications. In this study, AKR1C3-dependent activation of 3424 was investigated in vitro using recombinant human AKR1C3. AKR1C3-dependent cytotoxicity of 3424 was determined in a wide range of human cancer cell lines with different AKR1C3 expression levels. In addition, anti-tumor activity of 3424 was also investigated in a broad panel of CDX and PDX models. AKR1C3-dependent activation of prodrug 3424 was evident by monitoring the decrease of 3424 and generation of the active form, 2660. Kinetic analysis indicated that AKR1C3 exhibited higher catalytic efficiency towards 3424 compared to the physiological substrates. There was a strong correlation between 3424 cytotoxic potency and AKR1C3 expression. The racemic mixture induced DNA cross-linking in a concentration dependent manner. Tumor growth inhibition of 3424 was shown to be better than or comparable to the standard of care chemotherapy at clinically achievable doses as a single agent in various CDX models with high expression of AKR1C3, including liver HepG2, lung H460, castration-resistant prostate VCaP, gastric SNU-16, and kidney A498 cancer cell lines. The excellent anti-tumor efficacy of 3424 was further demonstrated in PDX models which have high level of AKR1C3 expression, but not in a model with low level of AKR1C3 expression. In the combination therapy, we showed that 3424 could enhance the efficacy of the standard care of chemotherapy in the CDX models. The results described here highlight that 3424 exhibits AKR1C3-dependent cytotoxicity in vitro and anti-tumor activity in vivo in a wide range of human cancer types, which support further development of 3424 as an anti-cancer agent for treating different types of cancers and the use of AKR1C3 as a biomarker to profile cancer patients and further guide patient selection for therapy with 3424.
Keywords: AKR1C3, prodrug, alkylating agent, cancers and anti-cancer agent
Introduction
Aldo-keto reductase 1C3 (AKR1C3) is also known as type 5, 17β-hydroxysteroid dehydrogenase (17β-HSD) and prostaglandin F synthase. AKR1C3 is one member of the 15 gene families of aldo-keto reductases (AKRs). AKR1C3 was originally cloned from human prostate [1] and placenta [2] cDNA libraries. AKR1C3 is a monomeric, cytosolic, NAD(P) (H)-dependent oxidoreductase with 323 amino acids and a molecular weight of 37 kDa [1]. AKR1C3 shares high sequence homology with the related human AKR1C family, including AKR1C1, AKR1C2, and AKR1C4. AKR1C3 catalyzes androgen, estrogen, progesterone, and prostaglandin (PG) metabolism and is subsequently involved in the regulation of nuclear receptor activities [3,4]. AKR1C3 is expressed in normal tissues including steroid hormone-dependent and steroid hormone-independent cells with an average low expression level except in liver, kidney, and small intestine [5]. Many studies have demonstrated that AKR1C3 is abnormally overexpressed in many malignant solid and hematologic tumors. The data show that more than 50% of hepatoma, bladder, renal, and gastric cancers were detected with high expression of AKR1C3 with immunohistochemistry scores (IHC score) ≥4 on a scale of 0 to 6 [6]. AKR1C3 is highly expressed in non-small cell lung cancer (NSCLC) but not in small cell-lung cancer [7].
AKR1C3 upregulation in cancer is reported to be associated with metastasis of castrate-resistant prostate cancer (CRPC, [8]) and colorectal cancer (CRC, [9]), and is also linked to poor prognosis and a low survival rate [10,11]. In addition, many types of treatment resistance are attributed to the overexpression of AKR1C3. It has been reported that chemotherapy resistance to doxorubicin [12,13], enzalutamide [14], abiraterone [15] and methotrexate [16] is directly related to high AKR1C3 expression in cells. Radiotherapy resistance in esophageal cancer [17], prostate cancer [18] and NSCL cancer cells [19] is associated with AKR1C3 overexpression. The main mechanism of action of AKR1C3 against ionizing radiation is to reduce ROS (reactive oxygen species) in cells, to increase PGF2α which subsequently leads to MAP kinase activation and PPARγ inhibition resulting in a significant reduction in DNA damage [18]. Immunotherapy resistance is also attributed to AKR1C3 high expression. One study has shown that high expression of AKR1C3 is associated with the failure of PD-1-targeted therapies in PD-L1 positive patients with advanced renal cell carcinoma (RCC) based on whole genome microarray and multiplex quantitative (q)RT-PCR gene expression analysis [20]. Due to tumor-specific overexpression of AKR1C3, the design of AKR1C3-activated prodrugs becomes an attractive approach to specifically target cancer. One such example is the AKR1C3-activated prodrug, PR104, which exhibited good anti-tumor activity in vitro and in vivo [6,21] although it was originally designed as a hypoxia-activated prodrug [22-24].
Anti-cancer prodrug 3424 is a chemically synthesized potent nitrogen mustard, which is selectively cleaved to the cytotoxic aziridine (2660) by AKR1C3 in the presence of NADPH. The active molecule, 2660, released by 3424 is similar to the standard chemotherapeutic drugs thiotepa and mitomycin C, which leads to alkylation and cross-linking of DNA at the N7 (or O6) position of guanine. Prodrug 3424 is currently under development by Ascentawits Pharmaceuticals, LTD in Asian countries and by OBI Pharma, Inc. in countries outside Asia (drug code OBI-3424) for the treatment of malignant tumors. Prodrug 3424 is currently being investigated in multiple Phase I clinical trials in the US (NCT04315324 & NCT03592264) and in China (CXHL1900137 & CXHL2000263) to treat more than 14 types of human cancer, including solid tumors and hematologic malignancies. Due to the high expression of AKR1C3 in tumors, prodrug 3424 is designed to be specifically activated in tumors but spared in normal cells which express low levels of AKR1C3 to achieve tumor-specific targeting. Furthermore, tumor-selective activation of 3424 is distinguishable from non-selective traditional alkylating agents, such as cyclophosphamide and ifosfamide, indicating that 3424 has the potential to become a broad-spectrum, highly selective anti-tumor drug. Prodrug 3424 was reported to exhibit potent efficacy against preclinical models of T-ALL in vitro and in vivo [25,26].
Given AKR1C3 is overexpressed in many types of cancer, we hypothesized that an AKR1C3-activated prodrug can be used to specifically target many types of tumor cells. To test this hypothesis, we have developed the AKR1C3 prodrug 3424, and determined its AKR1C3-dependent activation and anti-tumor activities in vitro and in vivo. In this report, the AKR1C3-dependent activation, in vitro 3424 cytotoxicity in a wide range of human cancer cell lines, and concentration-dependent DNA cross-linking of 3424 were investigated. Moreover, we also studied the in vivo anti-tumor activity of 3424 in a broad panel of CDX and PDX models.
Materials & methods
Cell lines
All human cancer cell lines were obtained from either the American Type Culture Collection (ATCC, Manassas, VA), or Japanese Collection of Research Biosources (JCRB, Osaka Japan) or Cobioer Biosciences (Nanjing, China).
Reagents and chemicals
Anti-human AKR1C3 monoclonal antibody, bleomycin, NADPH, lyophilized bovine serum albumin (BSA), and positive control substrates for AKR1C1/AKR1C3 (progesterone, androstenedione, and dihydrotestosterone) were purchased from Sigma (St. Louis, MO). Recombinant human AKR1C3 was purchased from Abcam (Cambridge, MA) and AKR1C1 and AKR1C4. were purchased from Sigma. Comet assay kit was purchased from Trevigen (Gaithersburg, MD). CellTiter Glo (CTG) assay kit was from Promega (Madison, WI). Racemic 2870 was synthesized by Threshold Pharmaceuticals (South San Francisco, CA). Prodrugs 3423 and 3424 were synthesized by Ascentawits Pharmaceuticals, LTD (Shenzhen, China). 3021 was synthesized based on the reported method [27]. Standard of care therapies were purchased as following: abiraterone (Bos Science, USA), prednisolone (Saen Chemical Technology, China), 5-FU (Shanhai Xudong Haipu Pharmaceutical Co., China), gemcitabine (Vianex S.A., Greece), and sunitinib (Cayman, USA).
Enzymatic activity assays
The assay mixture consisted of 10-50 μM 3424 or positive control (androstenedione or dihydrotestosterone), 100 mM phosphate buffer, pH 7.0, 300 μM NADPH, 4% ethanol and 8 µM recombinant human AKR1C3, AKR1C1 or AKR1C4 to give a total volume of 200 μL. The reaction was incubated at 25°C and terminated at various time points by adding acetonitrile and methanol (at a ratio of 9 to 1) and subjected to LC-MS/MS analysis. For enzyme kinetic analysis, the activity was determined with a SpectraMax M2e spectrophotometer (Molecular Devices, LLC, San Jose, CA) by measuring the decrease in absorbance of NADPH at 340 nm (ε = 6270 M-1cm-1). After the initiation of the reaction by addition of substrates, the reactions were monitored at 30 s intervals at 25°C. The non-substrate reaction rate was also monitored as background and its slope was used to determine the initial velocity of the reaction. The kinetic data reported were the average of triplicate measurements.
LC-MS/MS analyses
For AKR1C3-mediated 3424 metabolism, LC/MS/MS was performed using a Sciex API-4000 Qtrap (ABSciex, LLC, Framingham, MA) mass spectrometer coupled to an Agilent 1200 HPLC system (Agilent Technologies, Santa Clara, CA). For 3424 analysis, reverse phase liquid chromatographic separation was performed with a Waters Xbridge C18 column (2.1 × 100 mm, 3.5 µm, Waters Corp., Milford, MA) in a total run time of 12 min using a flow rate of 0.3 mL/min. The mobile phase A consisted of 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in ACN. The gradient was performed with an isocratic run at 15% B for 1.5 min and gradient to 50% B at 3 min, then to 95% B at 6 min and holding for 1 min, finally back to 15% B in 0.1 min and equilibrated at 15% B for 4.9 min. The column oven temperature was 40°C and the sample injection volume was 2 µL. The mass spectra were obtained in positive MRM mode. In positive ion mode, the ion spray voltage was set at 4500 V, declustering potential at 80 V, collision energy at 20 V, source temperature at 350°C, curtain gas at 10 psi and the source gas 1 and 2 both at 60 psi. The MRM pairs for 3424 and 3424-IS were m/z 461->313 and m/z 465->313, respectively. For 2660 analysis, normal phase liquid chromatographic separation was performed with Waters Atlantis HILIC Silica column (2.1 mm × 100 mm, 3 µm, Waters Corp., Milford, MA) in a total run time of 9 min using a flow rate of 0.3 mL/min. The mobile phase A consisted of 1 mM ammonium formate in water and mobile phase B using ACN. The gradient was performed from isocratic run at 89% B for 1 min and gradient to 60% B at 1.5 min, then to 40% B at 2.5 min and holding for 2 min, finally back to 89% B in 0.1 min and equilibrated at 15% B for 4.4 min. The column oven temperature was 40°C and the sample injection volume was 2 µL. The mass spectra were obtained in negative MRM mode. In negative ion mode, the ion spray voltage was set at -4500 V, declustering potential at -60 V, collision energy at -30 V, source temperature at 350°C, curtain gas at 10 psi and the source gas 1 and 2 both at 60 psi. The MRM pairs for 2660 and 2660-IS were m/z 147->63 and m/z 151->63, respectively. The peak area ratio for each MRM transition (peak area of analyte/peak area of analyte-IS) of calibration standards and samples were used for quantitative analysis using Analyst 1.6 software (ABSciex, Framingham, MA).
In vitro proliferation assay
Exponentially growing cells were seeded 24 hours before the addition of test compounds. After addition of test compounds, the plates were incubated for the indicated hours at 37°C in a standard tissue culture incubator. At the end of the experiment, the viable cells were detected using either a CellTiter Glo (CTG) assay kit or AlamarBlue [28,29]. Drug concentration resulting in growth inhibition of 50% (IC50) relative to untreated control was calculated using either XLfit (IDBS, Boston, MA) or Prism 6 (GraphPad, San Diego, CA). For 3021 experiments, cells were pretreated with 3 µM of 3021 for 2 h prior to compound treatment under air. IC50 was calculated as described above.
Western blot
Human cell extracts were prepared and protein concentrations were determined. Proteins were detected using antibodies recognizing human AKR1C3 and tubulin, or β-actin. The band densities of AKR1C3 and tubulin or β-actin were scanned and quantified using the Odyssey laser imaging system and software (LI-COR Biosciences, Lincoln, NE), or UVP ChemStudio imaging system and VisionWorks software (Analytik Jena AG), and the ratio of AKR1C3 to tubulin or actin was calculated.
Comet assay
After seeding cells for 24 hours, the test articles were added at the indicated concentrations and incubated for 2 hours. Cells were washed twice to remove compound completely. 20 µM of bleomycin was added and incubated for 1 hour under air to induce DNA strand breaks following 2 h cell resting. After washing twice with PBS, comet assay was conducted using a single-cell electrophoresis system from Trevigen (Gaithersburg, MD). The data were analyzed using Comet Assay IV software from Perceptive Instruments [29].
In vivo anti-tumor activity
All procedures related to animal handling, care, and the treatment in this study were performed according to guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of CrownBio (Beijing, China), WuXi AppTec (Shanghai, China), or Eurofins Pharmacology Discovery Services Taiwan (Taipei, Taiwan) following the guidance of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). At the time of routine monitoring, the animals were checked for any effects of tumor growth on normal behavior such as mobility, food and water consumption, body weight gain/loss, eye/hair matting and any other abnormal effect. Death and observed clinical signs were recorded on the basis of the numbers of animals within each subset.
Anti-tumor activity of 3424 in GFP expressing HepG2 and H460 CDX models was investigated at AntiCancer, Inc (Beijing, China). Female athymic nude mice (6 weeks; BALB/c-nu, Beijing HFK Bioscience Co., Ltd., Beijing, China) were used in the studies. Each mouse was implanted with a HepG2-GFP tumor chunk (~1 mm3 in diameter) in the right lobe of the liver for tumor development or inoculated subcutaneously with H460-GFP tumor chunk (~1 mm3 in diameter). About 3-10 days later, whole mouse body scan was performed using FluorVivo Model-100 fluorescence imager (INDEC Biosystems, Inc., Los Altos, CA). Mice with similar fluorescent areas were selected and randomly grouped. Prodrug 3424 was dosed intravenously (IV) at 1.25, 2.5 mg/kg or 5 mg/kg Q7D × 2 for the HepG2 orthotopic model or at 0.625, 1.25 or 2.5 mg/kg for the H460 xenograft model with a regimen of Q7D × 2, 1 week off, then Q7D × 2 again. Sorafenib was used as the positive control in the HepG2 model and was dosed orally at 30 mg/kg with a regimen of QD × 5 × 7 cycles. Taxol was used as the positive control in the H460 model and was administered at 15 mg/kg IV with a regimen of BIW × 4. During the experiments, mice were observed daily and body weight was measured twice a week. Tumor burden was monitored twice a week either by caliper measurement (H460) or FluorVivo fluorescence imager (H460-GFP and HepG2-GFP).
The anti-tumor activity of 3424 was evaluated in castration-resistant prostate cancer (CRPC) VCaP, gastric cancer SNU-16, and renal cell carcinoma (RCC) A498 xenograft models. Studies were conducted at WuXi AppTec (VCaP and SNU-16 models) and Eurofins Pharmacology Discovery Services Taiwan (A498 model). Male BALB/c nude, female BALB/c nude and female SCID mice were used in the CDX models of CRPC, gastric cancer, and RCC, respectively. Human cancer cells at 5 × 106 or 1 × 107 with 1:1 matrigel were injected subcutaneously into the right flank of mice. Vehicle and test articles were administered when tumor volumes reached 150-200 mm3 (denoted as Day 1, or Day 0 in SNU-16 model). Vehicle or 3424 was administered intravenously once weekly for a total of 4 or 5 doses dependent on the model. Standard of care therapies including abiraterone/prednisolone (for CRPC), 5-fluorouracil (for gastric cancer), and sunitinib and gemcitabine (both for RCC) were administered as recommended in the literature [15,30-32]. On days of co-administration, IV injection of 3424 was done first, followed by the combined agent within 1 hour.
Anti-tumor activity of 3424 in PDX models was assessed at CrownBio Bioscience (Beijing, China) Inc. using female BALB/c nude mice (6-7 weeks old, Beijing Anikeeper Biotech Co., Ltd, Beijing, China). Tumor fragments (PA1280, GA6201, LU2057 and LU2505) from stock mice inoculated with selected primary human cancer tissues (pancreatic, gastric cancer, and lung) were harvested and used for inoculation into BALB/c nude mice. Each mouse was inoculated subcutaneously for tumor development. Mice were allocated randomly into experimental groups when the average tumor size reached ~100 mm3) by using StudyDirectorTM Ver 3.1.399.19 (Studylog Systems, Inc., S. San Francisco, CA, USA). Prodrug 3424 was administered IV at the indicated doses with a regimen of Q7D × 3. Each group consisted of 5-6 mice. The grouping day was denoted as Day 0. Prodrug 3424 was administrated to the tumor-bearing mice from Day 0 through Days as indicated for each study.
For all the animal studies, drug efficacy was assessed by tumor growth inhibition. Tumor volume (mm3) was measured twice weekly following the prolate ellipsoid formula: Length (mm) × [Width (mm)]2 × 0.5. Percent Tumor Growth Inhibition (%TGI) was determined twice weekly during the dosing period by the formula: %TGI = (1 - [(T - T0)/(C - C0)]) × 100 where T = mean tumor volume of treated group, T0 = mean tumor volume of treated group at study start, C = mean tumor volume of control group and C0 = mean tumor volume of control group at study start. Two-way ANOVA and Bonferroni test were used to assess the statistical significance of groups compared to respective vehicle control using SPSS Statistics 23 (IBM, Armonk, NY) or R (version 3.3.1). P-values of <0.05 were regarded as statistically significant.
Results
Structure and mechanism of 3424 activation
The structures of 3424, 3423 and 2870 are presented in Figure 1A. Prodrug 3424 is the S-enantiomer, 3423 is the R-enantiomer, and 2870 is a racemic mixture of 3424 and 3423 at 1:1 ratio. In the presence of NADPH, reduction of 3424 is mediated by AKR1C3 to release the cytotoxic moiety, 2660 (Figure 1B), which is an aziridine bis-alkylating agent, leading to cross-linking of DNA at the N7 (or O6) position of guanine, and subsequent cell death.
Figure 1.
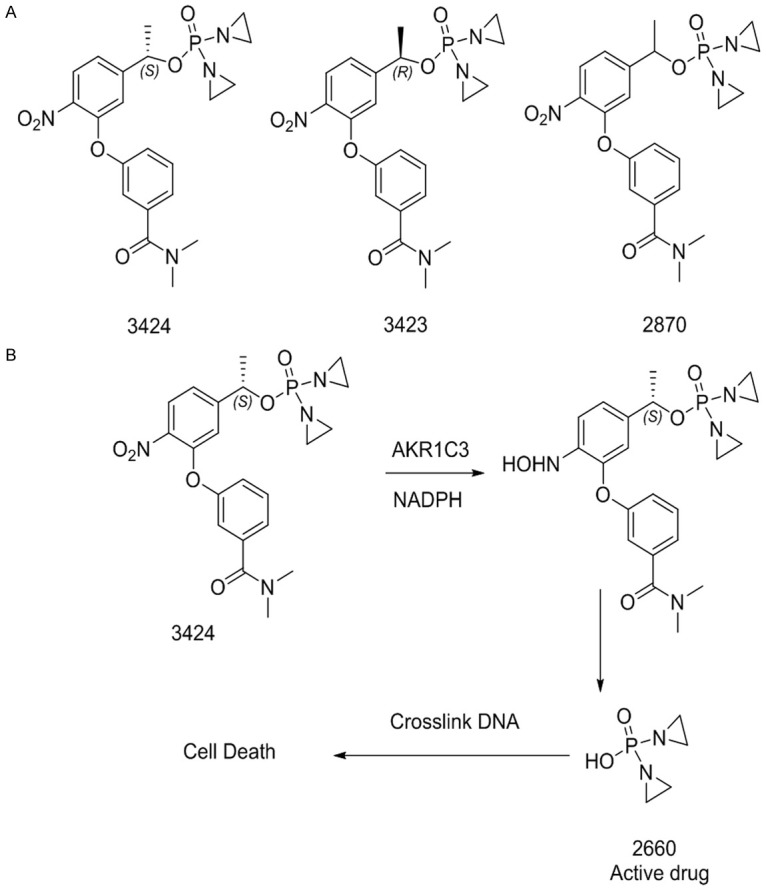
Structure and 3424 activation. A. Chemical structure of 3424 (S-enantiomer), 3423 (R-enantiomer) and 2870 (racemic mixture of 3424 and 3423 at 1:1 ratio). B. Schema of 3424 reductive activation pathway.
AKR1C3-dependent activation of 3424
The activation of 3424 by AKR1C3 was monitored by the reduction of 3424 and the generation of the active form, 2660, using LC/MS-MS. As shown in Figure 2, recombinant human AKR1C3 was able to activate 3424 into 2660 in 60 min (Figure 2A and 2B). In contrast, 3424 was not metabolized by AKR1C1 or AKR1C4, two members of the AKR1C family (Table 1). AKR1C3 exhibited similar catalytic efficiency towards 3424 (S-enantiomer) and its R-enantiomer 3423 (Table 2). Compared to the physiological substrates 4-androstenedione and 5-α dihydrotestosterone, 3424 was activated by AKR1C3 at a higher rate.
Figure 2.
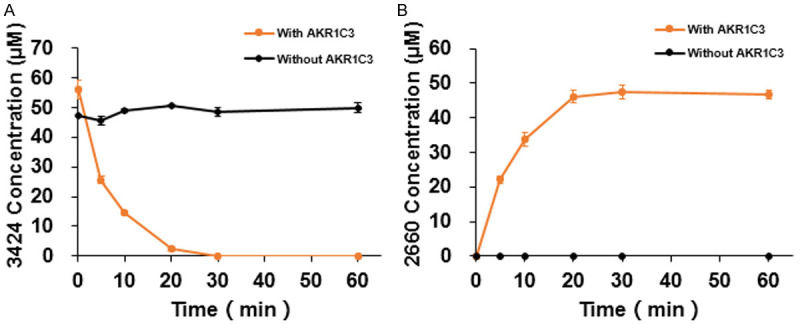
AKR1C3-dependent 3424 activation. A. 3424 reduction. B. 2660 production.
Table 1.
Production of 2660 (active form) following incubation of 3424 (prodrug) with various AKR1C enzymes
| AKR1C1 | AKR1C4 | AKR1C3 | |
|---|---|---|---|
| 3424 Concentration (µM) | 2660 Concentration (μM) | 2660 Concentration (μM) | 2660 Concentration (μM) |
| 0 | BQL | BQL | BQL |
| 10 | - | - | 10.71±0.53 |
| 20 | - | - | 14.77±1.44 |
| 30 | - | - | 33.96±1.90 |
| 40 | - | - | 34.30±6.63 |
| 50 | BQL | BQL | 34.59±7.44 |
Table 2.
Kinetic analysis of AKR1C3 towards various substrates
| Enzyme | Substrate | Vmax (μm/min) | Km (μm) | Kcat (min-1) | T1/2 (min) | Kcat/Km (min-1mM-1) |
|---|---|---|---|---|---|---|
| AKR1C3 | 4-Androstenedione | 0.49±0.04 | 1.98±0.43 | 0.07±0.01 | 165.97±14.69 | 35.35 |
| 5α-DHT | 0.85±0.01 | 10.18±0.62 | 0.12±0.00 | 95.58±0.99 | 11.79 | |
| 3424 (S-form) | 4.45±0.07 | 12.55±0.28 | 0.64±0.01 | 18.17±0.29 | 50.99 | |
| 3423 (R-form) | 5.03±0.29 | 12.88±1.71 | 0.72±0.04 | 16.13±0.99 | 59.90 |
AKR1C3-dependent 3424 cytotoxicity
Cytotoxicity of 3424 was evaluated in a panel of liver cancer cell lines and non-small cell lung cancer (NSCLC) cell lines. AKR1C3 protein expression in liver cancer cell lines was determined using Western blot and tubulin was used as a loading control. A ratio of AKR1C3 to tubulin was calculated and defined as normalized AKR1C3 expression in Table 3. AKR1C3 RNA expression data was obtained from the CrownBio (Beijing, China) database. As shown in Table 3, after 96 h exposure to 3424, liver cancer cell lines with high AKR1C3 expression at both the protein and RNA levels were more sensitive to 3424 with IC50 values in the low nanomolar range. On the other hand, cells expressing low AKR1C3 were less sensitive to 3424 with IC50 values higher than 1000 nM. Similarly, NSCLC cells also exhibited an AKR1C3-dependent cytotoxic profile after 72 h exposure to 3424 (Table 4). There was a high correlation between 3424 IC50 and the level of AKR1C3 protein (R2 = 0.71, Figure 3A, left) and RNA expression (R2 = 0.87, Figure 3A, middle) in liver cancer cells and in NSCLC cells (R2 = 0.80, Figure 3A, right), respectively. These results demonstrated that 3424-mediated cytotoxicity was highly correlated with the level of AKR1C3 expression in both liver cancer and NSCLC cell lines.
Table 3.
In vitro cytotoxicity of 3424 against a panel of human liver cancer cell lines after 96 hours of drug exposure
| Liver cancer cell line | Normalized AKR1C3 expression | AKR1C3 protein expression level | AKR1C3 RNA expression level (Log2 FPKM) | 3424 IC50 (nM) |
|---|---|---|---|---|
| SNU-475 | 1.22 | High | 9.19 | 15±3.3 |
| SNU-449 | 1.19 | High | 8.3 | 45±0.2 |
| C3A | 0.95 | High | 4.58 | 5±0.3 |
| SNU-387 | 0.64 | Medium | 6.42 | 103±2.2 |
| PLC/PRF/5 | 0.63 | Medium | 4.66 | 167±12.8 |
| HLE | 0.3 | Medium | 4.36 | 113±16.0 |
| HuCCT1 | 0.21 | Low | 0.19 | 696±6.6 |
| SNU-182 | 0.17 | Low | -0.05 | >1000 |
| HLF | 0.15 | Low | -1.69 | >1000 |
| SK-HEP-1 | 0.13 | Low | -1.39 | >1000 |
| SNU-398 | 0.08 | Low | -1.33 | >1000 |
Table 4.
In vitro cytotoxicity of 3424 against a panel of human non-small cell lung cancer cell lines (NSCLC) after 72 hours of drug exposure
| NSCLC cancer cell line | AKR1C3 RNA expression level (Log2 FPKM) | 3424 IC50 (nM) |
|---|---|---|
| NCI-H1944 | 11.06 | 2.3±0.07 |
| NCI-H2228 | 9.25 | 0.21±0.01 |
| NCIH1755 | 9 | 8.2±0.31 |
| NCI-H1563 | 8.61 | 2.5±0.19 |
| NCI-H2110 | 8.23 | 1.1±0.06 |
| NCI-H1792 | 8.07 | 4.5±0.18 |
| CAL12T | 3.86 | 29.1±1.12 |
| NCIH2106 | 2.68 | >1000 |
| NCI-H23 | -1.98 | >1000 |
| NCI-H522 | -1.88 | >1000 |
Figure 3.
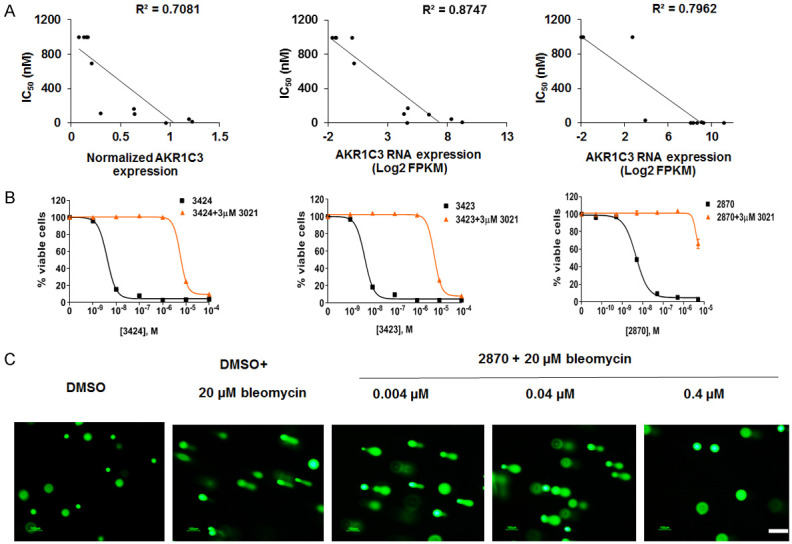
AKR1C3-dependent in vitro cytotoxicity of 3424. A. Correlation between AKR1C3 protein expression and 3424 IC50 in liver cancer cells (left); Correlation between AKR1C3 RNA expression and 3424 IC50 in liver cancer cells (middle); Correlation between AKR1C3 RNA expression and 3424 IC50 in NSCLC cancer cells (right). B. AKR1C3-specific inhibitor, 3021, efficiently inhibited cytotoxicity of 3424 (left), 3423 (middle) and racemic mixture 2870 (right). The data are the representative of three independent experiments. C. Compound 2870 induced concentration-dependent DNA cross-linking. The data are the representative images of cell nuclei stained with SYBR Green after electrophoresis of two independent experiments. Original magnification: 10 ×, scale bar: 100 μm.
AKR1C3-mediated specific activation of 3424 was confirmed using the AKR1C3 inhibitor 3021 in H460 cells. After 2 h pretreatment of H460 cells with 3 µM 3021 followed by co-treatment with 3424 for 2 h, the AKR1C3-specific inhibitor, 3021, was able to effectively inhibit 3424 cytotoxicity in H460 with an IC50 of 6.3 µM as compared to an IC50 of 4 nM in the absence of 3021 (Figure 3B, left), which was also reported by Evans et al. [25]. Here we also profiled the cytotoxicity of the R-enantiomer, 3423, and a racemic mixture of R- and S-enantiomers at 1:1 ratio, 2870, in the absence or presence of 3021 in H460 cells. As shown in Figure 3B, 3423 (middle) and 2870 (right) exerted equally potent cytotoxicity to 3424 with IC50 values of 5 nM and 4 nM, respectively. Similar to 3424, the cytotoxicity of 3423 and 2870 was highly AKR1C3-dependent and 3021 inhibited their cytotoxicity at concentrations over 1000-fold greater than in the absence of the inhibitor with IC50 values of 6.3 µM and >5 µM, respectively. Inhibitor 3021 alone did not exert any cytotoxicity up to 100 µM (data not shown).
To evaluate whether 3424 cross-linked DNA, a direct biochemical assay for DNA cross-linking, the single-cell electrophoresis-based comet assay was employed using H460 cells. In this assay, the racemic mixture, 2870, was used. As shown in Figure 3C, there was no detectable double-strand breaks with tail moment 0.3 in vehicle DMSO-treated H460 cells. Bleomycin was used to induce DNA strand breaks which was evident by increased tail moment to 35. DNA cross-linking induced by 2870 was tested using three concentrations under identical conditions that were used for bleomycin. Compound 2870-induced concentration-dependent DNA-crosslinking was evident by a decreased tail moment from 36 to 1.6.
Anti-tumor efficacy of 3424 in various CDX models
In vivo anti-tumor activity of 3424 was evaluated using GFP-expressing cancer cell lines in an orthotopic liver cancer model (HepG2) and a subcutaneous lung cancer model (H460). Both cell lines expressed high levels of AKR1C3 (Table 5). AKR1C3 RNA expression data was obtained from the CrownBio (Beijing, China) database. Using the HepG2 orthotopic xenograft model, we investigated the dose-dependent anti-tumor activity of 3424 by whole body fluorescence imaging. When 3424 was administered via IV injection at doses of 1.25, 2.5 or 5 mg/kg once a week for 2 weeks, the tumor growth inhibition (TGI) at Day 34 was 52.4%, 91.5% and 101.2%, respectively (Table 5). The anti-tumor efficacy of 3424 was observed in a dose-dependent manner (Figure 4A and Table 5). Compared to the vehicle group, tumor inhibition induced by 3424 at 2.5 mg/kg and 5 mg/kg was statistically significant with a complete regression rate of 80% and 100%, respectively. At the end of the experiment, the data from tumor fluorescent images (Figure 4B) and tumor weight (data not shown) were consistent with the measurement of fluorescence area (Figure 4A). Sorafenib, a first-line treatment of hepatocellular carcinoma (HCC), was administered orally at 30 mg/kg and reduced tumor growth by 52.1% with no statistical significance (Figure 4A). With this dosing regimen, sorafenib resulted in body weight loss (-11%, data not shown). In contrast, no body weight loss was observed in 3424-treated groups at all tested doses in the current study (Table 5).
Table 5.
Summary of 3424 in vivo efficacy in HepG2 and H460 CDX models
| CDX model | Cancer type | Implantation | Route | Regimen | 3424 Dose (mg/kg) | TGI% | Body weight change % | AKR1C3 LOG2 (FPKM) |
|---|---|---|---|---|---|---|---|---|
| HepG2 | Liver | Orthotopic | i.v. | Q7D × 2 | 1.25 | 52.4 | 22.8 | 6.9 |
| 2.5 | 91.5 | 19.5 | ||||||
| 5 | 101.2 | 17.1 | ||||||
| H460 | Lung | Subcutaneous | i.v. | Q7D × 2; 1 week off; Q7D × 2 | 0.625 | 60.2 | -1 | 8.6 |
| 1.25 | 67.2 | -1 | ||||||
| 2.5 | 88.0 | -14 |
Figure 4.
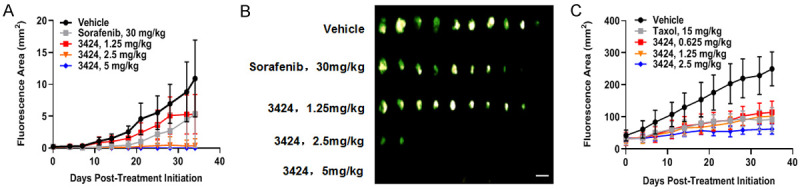
In vivo anti-tumor activity of 3424 in HepG2 and H460 CDX models. A. Fluorescence area of 3424 and sorafenib in GFP-HepG2 orthotopic CDX model. B. GFP-HepG2 tumor fluorescent images at the end of the experiment by using FluorVivo Model-100 fluorescence imager. Scale bar: 10 mm. C. Fluorescence area of 3424 and Taxol in GFP-H460 subcutaneous CDX model. Animals were monitored daily and tumor growth was quantified twice a week. Data are expressed as Mean ± SEM of 10 or 8 animals per group in HepG2 and H460, respectively.
In the H460 subcutaneous model, prodrug 3424 was given weekly for 2 cycles; with one week off, and another 2 cycles of weekly dosing at 0.625, 1.25, and 2.5 mg/kg. As shown in Figure 4C and Table 5, prodrug 3424 exhibited dose-dependent anti-tumor activity with TGI of 60.2%, 67.2% and 88%, respectively. The anti-tumor efficacy of 3424 was comparable to paclitaxel at 15 mg/kg showing a TGI of 64%. At the end of the study (Day 35), the treatment of 3424 resulted in minimal body weight loss at low- and mid-doses, whereas a statistically significant 14% loss at the high dose was observed (Table 5). Paclitaxel treatment caused a statistically significant 11% body weight loss (Data not shown).
In vivo anti-tumor efficacy of 3424, as monotherapy or in combination with standard of care therapy, was evaluated in castration-resistant prostate cancer (CRPC) VCaP, gastric cancer SNU-16, and renal cell carcinoma A498 xenograft models. These three human cancer cell lines expressed high levels of AKR1C3 at both levels of protein and RNA. AKR1C3 protein expression in VCap, SNU-16 and A498 was determined by Western Blot with a ratio of AKR1C3 to β actin at 1.6, 1.3, and 8.9, respectively. AKR1C3 RNA expression (LOG2 (FPKM) in VCap, SNU-16 and A498 was 5.2, 8.0 and 10.0, respectively (CrownBio database). In the CRPC model, castrated male BALB/c nude mice were treated with 3424 (weekly IV injection for 5 doses), abiraterone plus prednisolone (daily oral gavage), or the combination (Figure 5A). At 5 mg/kg, 3424 showed significant TGI of 148%, which was further enhanced to 158% when combined with abiraterone/prednisolone. At terminal sacrifice on Day 32, animals receiving the combination showed a corresponding reduction in serum prostate specific antigen (PSA) (Figure 5B). In the gastric cancer SNU-16 model, female BALB/c mice were treated with 3424 (weekly IV injection for 4 doses), 5-flurouracie (5-FU) (IP injection, twice a week), or 3424 combined with 5-FU (Figure 5C). Animals treated with 3424 at 2.5 or 5 mg/kg showed remarkable TGI of 87.8% or 96.2%, respectively, whereas 5-FU had no anti-tumor activity at the level of 30 mg/kg. Synergistic effect was noted when 3424 was combined with 5-FU. In the renal cell carcinoma A498 model, female SCID mice were administered 3424 (weekly IV injection for 4 doses), sunitinib (25 mg/kg, daily oral gavage), or the combination (Figure 5D). At 2.5 mg/kg, 3424 showed a significant TGI of 73%, compared to a modest 52% TGI by sunitinib. When animals were treated with a combination of 3424 and sunitinib, the TGI was further increased to 88%. The efficacy of 3424 combined with gemcitabine was also tested in A498 xenograft model, where gemcitabine at 80 mg/kg (weekly IP injection for 4 doses) offered 19% TGI only but in combination with 2.5 mg/kg 3424, the TGI was increased to 87% (Figure 5E). In all three models, 3424 was well tolerated in mice and there was no significant body weight loss during the treatment (data not shown).
Figure 5.
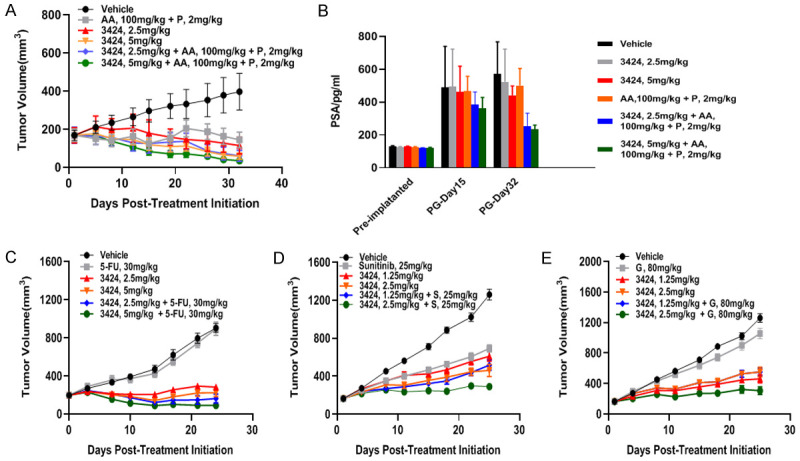
In vivo anti-tumor efficacy of 3424 as monotherapy or in combination with standard of care therapy in VCap, SNU-16 and A498 CDX models. A. Tumor growth of 3424 and abiraterone plus prednisolone as a monotherapy or in combination in VCaP. B. Measurement of serum prostate specific antigen (PSA) after treatment at the times indicated in VCap. C. Tumor growth of 3424 and 5-flurouracie as a monotherapy or in combination in SNU-16. D. Tumor growth of 3424 and sunitinib as monotherapy or in combination in A498. E. Tumor growth of 3424 and gemcitabine as a monotherapy or in combination in A498. Data are expressed as Mean ± SEM of 8 animals per group. AA, abiraterone acetate; P, prednisolone; 5-FU, 5-flurouracie; S, sunitinib; G, Gemcitabine.
In vivo anti-tumor efficacy of 3424 in PDX models
In vivo anti-tumor activity of 3424 was further evaluated in a panel of patient-derived xenograft (PDX) models, including pancreatic cancer PA1280, gastric cancer GA6201, and two lung cancers, LU2505 with high AKR1C3 expression and LU2057 with low AKR1C3 expression. AKR1C3 RNA expression data was obtained from the CrownBio (Beijing, China) database. Prodrug 3424 was administered weekly for 3 cycles via IV injection. Patient information for the four PDX models is shown in Table 6. The validation data of each PDX model are presented in Table 7. In the pancreatic PDX model (Figure 6A and Table 8), 3424 at 2.5 mg/kg exhibited statistically significant anti-tumor activity of 77.4% TGI. In the gastric PDX model, 5 mg/kg 3424 displayed a remarkable anti-tumor inhibition of 110% TGI (Figure 6B and Table 8). Tumor volume continued to decrease and remained low or unmeasurable even 1 month after discontinuation of therapy. At the end of the study, 60% of mice remained tumor free. Two lung cancer PDX models with differential levels of AKR1C3 expression were chosen to study if the in vivo anti-tumor activity of 3424 is AKR1C3-dependent. LU2505 PDX model expressed higher AKR1C3 RNA (log2 = 6.03) whereas LU2057 model expressed low AKR1C3 RNA (log2 = 2.08). As shown in Figure 6C and 6D and Table 8, at the same dose of 2.5 mg/kg, 3424 exerted excellent tumor growth inhibition in LU2505 with high AKR1C3 expression (TGI 105.2%) but no inhibition was observed in LU2057 PDX tumor (TGI-20.9%) with low AKR1C3 expression, indicating an AKR1C3-dependent in vivo anti-tumor activity of 3424. Even at a lower dose of 1.25 mg/kg, 3424 still exhibited statistically significant inhibition of LU2505 tumor growth (TGI 105.0%). In all PDX models, prodrug 3424 was well-tolerated with no body weight loss observed at all tested doses (Table 8).
Table 6.
PDX patient information
| HuPrime Model ID | Patient information | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Cancer type | Subtype | Race | Gender | Age | Pathology diagnosis | |
| PA1280 | Pancreatic cancer | ADC | Asian | F | 82 | Pancreatic cancer |
| GA6201 | Gastric cancer | ADC | Asian | M | 60 | Gastric cancer |
| LU2505 | Lung cancer | NSCLC, ADC | Asian | F | 69 | Lung cancer |
| LU2057 | Lung cancer | SCLC | Western | M | NA | Lung cancer |
Table 7.
PDX model validation
| HuPrime Model ID | affy snp6 data | RNA Seq | Exome Seq | Growth curve | Soc data |
|---|---|---|---|---|---|
| PA1280 | Yes | Yes | Yes | Yes | Yes |
| GA6201 | NA | Yes | Yes | Yes | Yes |
| LU2505 | Yes | Yes | NA | Yes | Yes |
| LU2057 | Yes | Yes | Yes | Yes | NA |
Figure 6.
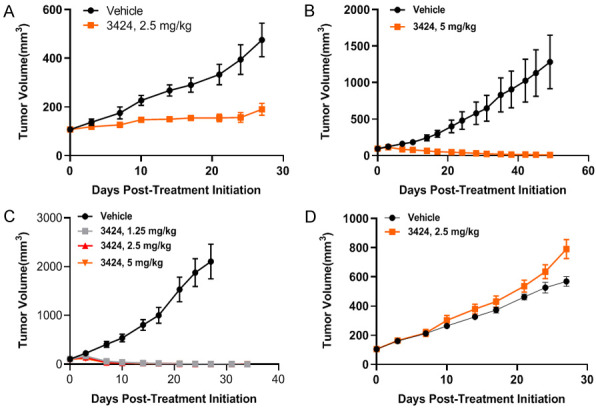
In vivo anti-tumor activity of 3424 against a panel of PDXs. A. Tumor growth of 3424 in pancreatic cancer PA1280. B. Tumor growth of 3424 in gastric cancer GA6201. C. Tumor growth of 3424 in lung cancer LU2505 with higher AKR1C3 expression. D. Tumor growth of 3424 in lung cancer LU2057 with low AKR1C3 expression. Animals were monitored daily and tumor growth was quantified twice a week. Data are expressed as Mean ± SEM of 5-6 animals per group.
Table 8.
Summary of 3424 in vivo efficacy in PDX models
| PDX model | Cancer type | Implantation | Route | Regimen | 3424 Dose (mg/kg) | TGI% | Body weight change % | AKR1C3 LOG2 (FPKM) | P Value (Compared to Vehicle) |
|---|---|---|---|---|---|---|---|---|---|
| PA1280 | Pancreas | Subcutaneous | i.v. | Q7D × 3 | 2.5 | 77.4 | 4.72 | 9.04 | 0.004 |
| GA6201 | Gastric | Subcutaneous | i.v. | Q7D × 3 | 5 | 110 | 4 | 6.78 | 0.00000265 |
| LU2057 | Lung | Subcutaneous | i.v. | Q7D × 3 | 2.5 | -20.9 | 7.69 | 2.08 | 0.023 |
| LU2505 | Lung | Subcutaneous | i.v. | Q7D × 3 | 1.25 | 105 | -0.54 | 6.03 | 0.001 |
| 2.5 | 105.2 | 3.37 | 0.001 | ||||||
| 5 | 105.2 | 1.1 | 0.001 |
Discussion
Selective overexpression of AKR1C3 in tumors provides a unique opportunity for the development of novel anti-cancer therapies. Prodrug 3424 is an AKR1C3-activated aziridine bis-alkylating agent that aims to achieve tumor-specific targeting and to improve therapeutic index.
We first confirmed that 3424 reduction is AKR1C3 dependent. Recombinant human AKR1C3, but not AKR1C1 or AKR1C4, reduced 3424 to its active aziridine nitrogen mustard moiety 2660. Moreover, we demonstrated that 3424-mediated cytotoxicity is highly AKR1C3-dependent. By using more than 20 cell lines from either liver cancer or NSCLC, we found that 3424 in all cell lines with high expression of AKR1C3 exhibited enhanced cytotoxicity compared to cells expressing low or no detectable AKR1C3. The IC50 values of 3424 in cell lines expressing high AKR1C3 were in the low nanomolar range, indicating a high potency that is characteristic of a potent nitrogen mustard. Of particular note was the finding of an enhanced cytotoxicity up to 5000-fold in NCI-H2228 NSCLC line expressing high AKR1C3. This result is consistent with targeting tumors with high expression of AKR1C3 while sparing low AKR1C3 expressing regions found in normal tissues.
The AKR1C3-dependent cytotoxicity was further confirmed in animal anti-tumor efficacy studies. We tested 3424 anti-tumor activity in a broad panel of different tumor type CDX and PDX xenograft models. Prodrug 3424 exhibits excellent anti-tumor activity as a single agent in all models expressing AKR1C3 at high levels of protein and RNA, including CRPC, gastric, renal, liver, pancreatic, and lung cancers. At clinically achievable doses, 3424 showed dose-dependent anti-tumor activities and significant TGI. A key finding described here is the correlation between 3424 anti-tumor activity and AKR1C3 expression present in lung PDX xenograft models (Figure 6C and 6D; Table 8). At equal dose, prodrug 3424 exhibited superior anti-tumor activity in LU2505 with high AKR1C3 expression whereas no activity was observed in LU2057 with relatively low expression of AKR1C3, confirming that 3424 in vivo activity is AKR1C3-dependent. In combination therapy, we have demonstrated that 3424 could enhance the efficacy of the standard of care in the CDX models of CRPC, gastric cancer, and renal cell carcinoma.
Prodrugs designed to target cancer cells have emerged as an attractive strategy for cancer therapy in recent years; however, many prodrugs failed in Phase 3 clinical trials due to a lack of valid biomarkers to select patients [33]. Given that the AKR1C3 expression can be assessed using RT-PCR or immunohistochemistry, 3424 can be developed in a clinically efficient manner by selecting patients who have high AKR1C3 expression and are most likely to respond to the prodrug. AKR1C3 has been demonstrated to be overexpressed upon acquisition of chemoresistance [13,14], radioresistance [19] and immunoresistance [20], indicating that 3424 may act as an effective therapy for cancers resistant to conventional chemo- and radio-treatment. In addition, cancers with homologous recombination deficiency (HRD) such as ovarian, breast, and pancreatic cancers, are known to be sensitive to DNA damaging agents [34]. As a DNA alkylator, 3424 may also be a good candidate drug to treat HRD cancers that have AKR1C3 expression.
In conclusion, the results described here highlight that 3424 exhibits AKR1C3-dependent cytotoxicity in vitro and anti-tumor activity in vivo in a wide range of human cancer types, which support further development of 3424 as a single anti-cancer agent or in combination with chemotherapeutic agents for treating different types of cancer and the use of AKR1C3 as a biomarker to profile cancer patients and further guide patient selection for therapy with 3424.
Disclosure of conflict of interest
All authors are current employees paid by either Ascentawits Pharmaceuticals, LTD or OBI Pharma, Inc.
References
- 1.Lin HK, Jez JM, Schlegel BP, Peehl DM, Pachter JA, Penning TM. Expression and characterization of recombinant type 2 3 alpha-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3 alpha/17 beta-HSD activity and cellular distribution. Mol Endocrinol. 1997;11:1971–1984. doi: 10.1210/mend.11.13.0026. [DOI] [PubMed] [Google Scholar]
- 2.Dufort I, Rheault P, Huang XF, Soucy P, Luu-The V. Characteristics of a highly labile human type 5 17beta-hydroxysteroid dehydrogenase. Endocrinology. 1999;140:568–574. doi: 10.1210/endo.140.2.6531. [DOI] [PubMed] [Google Scholar]
- 3.Penning TM, Drury JE. Human aldo-keto reductases: function, gene regulation, and single nucleotide polymorphisms. Arch Biochem Biophys. 2007;464:241–250. doi: 10.1016/j.abb.2007.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizner TL, Penning TM. Role of aldo-keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids. 2014;79:49–63. doi: 10.1016/j.steroids.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang TS, Lin HK, Rogers KA, Brame LS, Yeh MM, Yang Q, Fung KM. Expression of aldo-keto reductase family 1 member C3 (AKR1C3) in neuroendocrine tumors & adenocarcinomas of pancreas, gastrointestinal tract, and lung. Int J Clin Exp Pathol. 2013;6:2419–2429. [PMC free article] [PubMed] [Google Scholar]
- 6.Guise CP, Abbattista MR, Singleton RS, Holford SD, Connolly J, Dachs GU, Fox SB, Pollock R, Harvey J, Guilford P, Donate F, Wilson WR, Patterson AV. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res. 2010;70:1573–1584. doi: 10.1158/0008-5472.CAN-09-3237. [DOI] [PubMed] [Google Scholar]
- 7.Miller VL, Lin HK, Murugan P, Fan M, Penning TM, Brame LS, Yang Q, Fung KM. Aldo-keto reductase family 1 member C3 (AKR1C3) is expressed in adenocarcinoma and squamous cell carcinoma but not small cell carcinoma. Int J Clin Exp Pathol. 2012;5:278–289. [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao J, Zhang M, Liu J, Liu Z, Shen P, Nie L, Guo W, Cai D, Liu J, Armstrong CM, Sun G, Chen J, Zhu S, Dai J, Zhang H, Zhao P, Zhang X, Yin X, Zhu X, Ni Y, Chen N, Zeng H. AKR1C3 expression in primary lesion rebiopsy at the time of metastatic castration-resistant prostate cancer is strongly associated with poor efficacy of abiraterone as a first-line therapy. Prostate. 2019;79:1553–1562. doi: 10.1002/pros.23875. [DOI] [PubMed] [Google Scholar]
- 9.Nakarai C, Osawa K, Akiyama M, Matsubara N, Ikeuchi H, Yamano T, Hirota S, Tomita N, Usami M, Kido Y. Expression of AKR1C3 and CNN3 as markers for detection of lymph node metastases in colorectal cancer. Clin Exp Med. 2015;15:333–341. doi: 10.1007/s10238-014-0298-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu NY, Ho HC, Chow KC, Lin TY, Shih CS, Wang LS, Tsai CM. Overexpression of dihydrodiol dehydrogenase as a prognostic marker of non-small cell lung cancer. Cancer Res. 2001;61:2727–2731. [PubMed] [Google Scholar]
- 11.Powell K, Semaan L, Conley-LaComb MK, Asangani I, Wu YM, Ginsburg KB, Williams J, Squire JA, Maddipati KR, Cher ML, Chinni SR. ERG/AKR1C3/AR constitutes a feed-forward loop for AR signaling in prostate cancer cells. Clin Cancer Res. 2015;21:2569–2579. doi: 10.1158/1078-0432.CCR-14-2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heibein AD, Guo B, Sprowl JA, Maclean DA, Parissenti AM. Role of aldo-keto reductases and other doxorubicin pharmacokinetic genes in doxorubicin resistance, DNA binding, and subcellular localization. BMC Cancer. 2012;12:381. doi: 10.1186/1471-2407-12-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong T, Xu F, Xu J, Liu L, Chen Y. Aldo-keto reductase 1C3 (AKR1C3) is associated with the doxorubicin resistance in human breast cancer via PTEN loss. Biomed Pharmacother. 2015;69:317–325. doi: 10.1016/j.biopha.2014.12.022. [DOI] [PubMed] [Google Scholar]
- 14.Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, Evans CP, Gao AC. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 2015;75:1413–1422. doi: 10.1158/0008-5472.CAN-14-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, Gao AC. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol Cancer Ther. 2017;16:35–44. doi: 10.1158/1535-7163.MCT-16-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao J, Xiang Y, Xiao C, Guo P, Wang D, Liu Y, Shen Y. AKR1C3 overexpression mediates methotrexate resistance in choriocarcinoma cells. Int J Med Sci. 2014;11:1089–1097. doi: 10.7150/ijms.9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiong W, Zhao J, Yu H, Li X, Sun S, Li Y, Xia Q, Zhang C, He Q, Gao X, Zhang L, Zhou D. Elevated expression of AKR1C3 increases resistance of cancer cells to ionizing radiation via modulation of oxidative stress. PLoS One. 2014;9:e111911. doi: 10.1371/journal.pone.0111911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun SQ, Gu X, Gao XS, Li Y, Yu H, Xiong W, Yu H, Wang W, Li Y, Teng Y, Zhou D. Overexpression of AKR1C3 significantly enhances human prostate cancer cells resistance to radiation. Oncotarget. 2016;7:48050–48058. doi: 10.18632/oncotarget.10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie L, Yu J, Guo W, Wei L, Liu Y, Wang X, Song X. Aldo-keto reductase 1C3 may be a new radioresistance marker in non-small-cell lung cancer. Cancer Gene Ther. 2013;20:260–266. doi: 10.1038/cgt.2013.15. [DOI] [PubMed] [Google Scholar]
- 20.Ascierto ML, McMiller TL, Berger AE, Danilova L, Anders RA, Netto GJ, Xu H, Pritchard TS, Fan J, Cheadle C, Cope L, Drake CG, Pardoll DM, Taube JM, Topalian SL. The intratumoral balance between metabolic and immunologic gene expression is associated with anti-PD-1 response in patients with renal cell carcinoma. Cancer Immunol Res. 2016;4:726–733. doi: 10.1158/2326-6066.CIR-16-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moradi Manesh D, El-Hoss J, Evans K, Richmond J, Toscan CE, Bracken LS, Hedrick A, Sutton R, Marshall GM, Wilson WR, Kurmasheva RT, Billups C, Houghton PJ, Smith MA, Carol H, Lock RB. AKR1C3 is a biomarker of sensitivity to PR-104 in preclinical models of T-cell acute lymphoblastic leukemia. Blood. 2015;126:1193–1202. doi: 10.1182/blood-2014-12-618900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guise CP, Abbattista MR, Tipparaju SR, Lambie NK, Su J, Li D, Wilson WR, Dachs GU, Patterson AV. Diflavin oxidoreductases activate the bioreductive prodrug PR-104A under hypoxia. Mol Pharmacol. 2012;81:31–40. doi: 10.1124/mol.111.073759. [DOI] [PubMed] [Google Scholar]
- 23.Guise CP, Wang AT, Theil A, Bridewell DJ, Wilson WR, Patterson AV. Identification of human reductases that activate the dinitrobenzamide mustard prodrug PR-104A: a role for NADPH: cytochrome P450 oxidoreductase under hypoxia. Biochem Pharmacol. 2007;74:810–820. doi: 10.1016/j.bcp.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 24.Patterson AV, Ferry DM, Edmunds SJ, Gu Y, Singleton RS, Patel K, Pullen SM, Hicks KO, Syddall SP, Atwell GJ, Yang S, Denny WA, Wilson WR. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin Cancer Res. 2007;13:3922–3932. doi: 10.1158/1078-0432.CCR-07-0478. [DOI] [PubMed] [Google Scholar]
- 25.Evans K, Duan J, Pritchard T, Jones CD, McDermott L, Gu Z, Toscan CE, El-Zein N, Mayoh C, Erickson SW, Guo Y, Meng F, Jung D, Rathi KS, Roberts KG, Mullighan CG, Shia CS, Pearce T, Teicher BA, Smith MA, Lock RB. OBI-3424, a novel AKR1C3-activated prodrug, exhibits potent efficacy against preclinical models of T-ALL. Clin Cancer Res. 2019;25:4493–4503. doi: 10.1158/1078-0432.CCR-19-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Liu Y, Zhou C, Wang C, Zhang N, Cao D, Li Q, Wang Z. An AKR1C3-specific prodrug with potent anti-tumor activities against T-ALL. Leuk Lymphoma. 2020:1–9. doi: 10.1080/10428194.2020.1728746. [DOI] [PubMed] [Google Scholar]
- 27.Flanagan JU, Atwell GJ, Heinrich DM, Brooke DG, Silva S, Rigoreau LJ, Trivier E, Turnbull AP, Raynham T, Jamieson SM, Denny WA. Morpholylureas are a new class of potent and selective inhibitors of the type 5 17-beta-hydroxysteroid dehydrogenase (AKR1C3) Bioorg Med Chem. 2014;22:967–977. doi: 10.1016/j.bmc.2013.12.050. [DOI] [PubMed] [Google Scholar]
- 28.Meng F, Bhupathi D, Sun JD, Liu Q, Ahluwalia D, Wang Y, Matteucci MD, Hart CP. Enhancement of hypoxia-activated prodrug TH-302 anti-tumor activity by Chk1 inhibition. BMC Cancer. 2015;15:422. doi: 10.1186/s12885-015-1387-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng F, Evans JW, Bhupathi D, Banica M, Lan L, Lorente G, Duan JX, Cai X, Mowday AM, Guise CP, Maroz A, Anderson RF, Patterson AV, Stachelek GC, Glazer PM, Matteucci MD, Hart CP. Molecular and cellular pharmacology of the hypoxia-activated prodrug TH-302. Mol Cancer Ther. 2012;11:740–751. doi: 10.1158/1535-7163.MCT-11-0634. [DOI] [PubMed] [Google Scholar]
- 30.Lee Y, Vassilakos A, Feng N, Jin H, Wang M, Xiong K, Wright J, Young A. GTI-2501, an antisense agent targeting R1, the large subunit of human ribonucleotide reductase, shows potent anti-tumor activity against a variety of tumors. Int J Oncol. 2006;28:469–478. [PubMed] [Google Scholar]
- 31.Oksala R, Moilanen A, Riikonen R, Rummakko P, Karjalainen A, Passiniemi M, Wohlfahrt G, Taavitsainen P, Malmstrom C, Ramela M, Metsankyla HM, Huhtaniemi R, Kallio PJ, Mustonen MV. Discovery and development of ODM-204: a novel nonsteroidal compound for the treatment of castration-resistant prostate cancer by blocking the androgen receptor and inhibiting CYP17A1. J Steroid Biochem Mol Biol. 2019;192:105115. doi: 10.1016/j.jsbmb.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Zhang L, O’Neill A, Bahamon B, Alsop DC, Mier JW, Goldberg SN, Signoretti S, Atkins MB, Wood CG, Bhatt RS. Cox-2 inhibition enhances the activity of sunitinib in human renal cell carcinoma xenografts. Br J Cancer. 2013;108:319–326. doi: 10.1038/bjc.2012.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spiegelberg L, Houben R, Niemans R, de Ruysscher D, Yaromina A, Theys J, Guise CP, Smaill JB, Patterson AV, Lambin P, Dubois LJ. Hypoxia-activated prodrugs and (lack of) clinical progress: the need for hypoxia-based biomarker patient selection in phase III clinical trials. Clin Transl Radiat Oncol. 2019;15:62–69. doi: 10.1016/j.ctro.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heeke AL, Pishvaian MJ, Lynce F, Xiu J, Brody JR, Chen WJ, Baker TM, Marshall JL, Isaacs C. Prevalence of homologous recombination-related gene mutations across multiple cancer types. JCO Precis Oncol. 2018;2018 doi: 10.1200/PO.17.00286. PO.17.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]