

Abstract
Methuosis is a novel type of non-apoptotic cell death characterized by accumulation of cytoplasmic vacuoles. Identification of molecules that induce methuosis may provide alternative therapeutics for cancers that are refractory to apoptosis. Epimedokoreanin C (EKC) is a prenylated flavonoid isolated from a Chinese herb Epimedium koreanum. In this article, we described that EKC reduced cell viability accompanied by extreme vacuolation in human lung cancer cells. The EKC-induced cell death was clarified as non-apoptosis based on the absence of apoptotic changes. The vacuoles stimulated by EKC were supposed to be derived from macropinocytosis based on the engulfment of extracellular fluid tracer, Lucifer Yellow. The vacuoles acquired some characteristics of late endosomes supported that EKC-induced cell death could be described as methuosis. Rac1 and Arf6 were found to be regulated inversely after EKC treatment. Blocking Rac1 activation with the specific Rac1 inhibitor EHT 1864 prevented the accumulation of vacuoles induced by EKC markedly, suggested that the regulation of Rac1 and Arf6 was at least partial mechanism involved in EKC induced methuosis. EKC synergized the effects of doxorubicin and etoposide, demonstrating the effectiveness of using EKC to synergize conventional chemotherapy. Collectively, EKC was demonstrated as a methuosis-like cell death inducer in lung cancer NCI-H292 and A549 cells. It has the potential to be used as an attractive prototype for developing drugs that could kill apoptosis-resistant cancer cells.
Keywords: Methuosis, macropinocytosis, epimedokoreanin C, Rac1, Arf6
Introduction
Despite the fact that remarkable progress has been achieved in the understanding of cancer therapies over the past several decades, cancer remains the second leading cause of death in the world [1]. Surgery, radiotherapy and chemotherapy are the most widely used treatments for cancers [2]. Chemotherapy is the only option for treatment of metastatic cancers [3]. Although many new chemotherapeutic drugs especially molecular-targeted approaches have been developed during the past decades, drug resistance remains a major obstacle in cancer treatments [4,5]. Apoptosis is the most recognized form of programmed cell death. As most conventional chemotherapeutic agents work by triggering apoptotic cell death [6], the discovery of alternative agents targeting nonapoptotic cell death pathway might be used as a new strategy to kill cancer cells that are resistant to apoptosis.
On the basis of distinguished morphological and molecular characteristics, several non-apoptotic forms of cell death have been identified. Cytoplasmic vacuoles are morphological features of several non-apoptotic cell deaths including autophagy, necroptosis and paraptosis [7-9]. Methuosis is a newly defined form of non-apoptotic cell death characterized by the accumulation of massive vacuoles in cytoplasm. These vacuoles are originated from macropinosomes and endosomes showing impaired recycling and trafficking [10,11]. As a novel nonapoptotic mode of cell death, methuosis has attracted particular attention as a target for cancer treatments. Several methuosis inducers have been reported, for example, indole-based chalcones MIPP and MOMIPP triggered cell death by methuosis in glioblastoma [11,12]; an ursolic acid derivative led cell death via methuosis [13]; a 4,6-disubstituted Aza-indole compound 13 induced methuosis and displayed cytotoxicities against a panel of cancer cells [14]; a quinoline derivative vacquinol also caused methuosis in glioblastoma [15] and CX-4945 induced methuosis in cholangiocarcinoma cells [16]. These reported results confirmed that the identification of methuosis inducers could provide both probes for studying the specific molecular mechanism of the non-conventional cell death pathway, and promising therapeutic agents for the treatment of cancers.
Isoprenylated flavonoids represented a subgroup of natural flavonoids possessing at least one prenyl in their structure. Increasing evidences suggested that prenylation was a determinant factor for the enhancement of cytotoxicity of flavonoids. This made prenylated flavonoids as attractive compounds for obtaining anti-cancer leading structures [17-19]. In the course of screening for bioactive anticancer molecules, Epimedokoreanin C (EKC), a prenylated flavonoid obtained from Epimedium koreanum, caught our attention because of the induction of extreme cytoplasmic vacuolization in several cancer cell lines. In the present study, we reported that EKC induced cell death with the specific characteristics of methuosis in human lung cancer NCI-H292 and A549 cells. We also discussed the regulation of Rac1, Arf6 and PIKfyve expression. This compound may be considered as a prototype for a class of therapeutic agents that could be used to treat tumors resistant to conventional apoptosis-inducing agents.
Materials and methods
Cell culture
Human lung cancer NCI-H292 and A549 cells, bronchial epithelial 16HBE cells were purchased from Shanghai Cell Bank (Shanghai, China), and were cultured in Roswell Park Memorial Institute (RPMI) 1640 (Gibco, Grand Island, NY, USA) and Dulbecco’s Modified Eagle Medium (DMEM) respectively. All culture media were supplemented with 10% (v/v) fetal bovine serum (FBS, HyClone, Logan, UT, USA), 100 U/ml penicillin and 100 U/ml streptomycin. All cells were maintained at 37°C in a humidified incubator containing 5% CO2.
Chemicals and reagents
Epimedokoreanin C and its analogues, Epicornuin A, B, F and Epimedokoreanin B were purified by our laboratory from the leaves of Epimedium koreanum.
3-(4, 5-dimethyl-2-thiazol)-2, 5-diphenyl-2-H-tetrazolium bromide (MTT), dimethyl sulfoxide (DMSO) and acridine orange (AO) were from Solarbio life sciences (Beijing, China). Annexin V-FITC/Propidium iodide (PI) apoptosis detection kit was obtained from BD Biosciences (San Jose, CA, USA). 4, 6-dianmidino-2-phenylindole (DAPI) was purchased from Genview Scientific Inc. (FL, USA). Bafilomycin A1 and Z-VAD (OMe)-FMK were purchased from MedChemExpress (MCE) (NJ, USA). MitoTracker Red was obtained from Life Technologies (MA, USA). LysoTracker Red and ERTracker Red were from Beyotime Biotechnology (Shanghai, China). Lucifer Yellow CH dilithium salt was purchased from Sigma-Aldrich. Trizol Reagent was from Invitrogen. PrimeScript RT reagent kit and SYBR Premix Ex Taq were purchased from Takara Bio Inc. (Japan). Rac1 and Arf6 pull-down activation assay kits were obtained from Cytoskeleton Inc. (Denver, CO, USA). EHT 1864 was purchased from Target Molecule Corp (Boston, MA, USA). mRFP-GFP-LC3 adenovirus was obtained from Genechem (Shanghai, China).
Antibodies
Details of the primary antibody and dilutions used for these studies are listed in Table 1. Alexa Fluor 594 and 488 anti-rabbit IgG antibodies were from Proteintech Group (Wuhan, China). GAPDH and β-actin were purchased from Santa Cruz Biotechnology.
Table 1.
List of primary antibodies
| Target | Dilution | Company and Catalog No | Predicted MW (kDa) |
|---|---|---|---|
| β-actin | 1:2000 | Santa Cruz (sc-47778) | 43 |
| GAPDH | 1:2000 | Santa Cruz (sc-25778) | 37 |
| Cleaved-caspase9 | 1:500 | Cell Signaling (#20750) | 35 |
| Caspase9 | 1:1000 | Santa Cruz (sc-8355) | 46 |
| Cleaved-caspase3 | 1:1000 | Cell Signaling (#9664) | 17, 19 |
| Caspase3 | 1:1000 | Santa Cruz (sc-7148) | 32 |
| PARP | 1:1000 | Cell Signaling (#9542) | 89, 116 |
| Rab7 | 1:1000 | Santa Cruz (sc-376362) | 22 |
| LAMP1 | 1:1000 | Santa Cruz (sc-20011) | 120 |
| Arf6 | 1:500 | Santa Cruz (sc-7971) | 26 |
| Rac1 | 1:1000 | Santa Cruz (sc-514583) | 22 |
| LC3 | 1:2000 | Proteintech (14600-1-AP) | 16-18 |
| SQSTM1 | 1:10000 | Proteintech (18420-1-AP) | 62 |
| PIKfyve | 1:1000 | Santa Cruz (sc-100408) | 262 |
| Akt | 1:1000 | Santa Cruz (sc-81434) | 62 |
| p-Akt | 1:500 | Santa Cruz (sc-271964) | 62/56/60 |
| ERK | 1:1000 | Santa Cruz (sc-514302) | 44/42 |
| p-ERK | 1:1000 | Santa Cruz (sc-7383) | 44/42 |
MTT assay
Cells were seeded in 96-well plates at a density of 1×104 cells per well and incubated overnight. The cells were then treated with DMSO or EKC for different times, after treatment, 20 μl of 2.0 mg/ml MTT solution was added in each well and incubated at 37°C for an additional 4 hr. The plates were centrifuged and the medium was removed, then 100 μl of DMSO was added to each well to dissolve the formazan crystals. The optical density value was measured at 570 nm with a microplate reader (Biorad, Model 680, USA).
Wound-healing assay
Cells were seeded in 6-well plates at a density of 5×105 cells per well and incubated overnight. The wounds were made by straight scratching the cell surface with a 20 ul pipette tip. Cells were washed with PBS gently, and then incubated in medium with 2% FBS with DMSO or EKC. Pictures of the entire scratched areas in each group were acquired at 12 h and 24 h using a phase-contrast microscope (Olympus, IX71, Japan). The wound area was quantified using Image J software. The wound healing rate was calculated as follows: (area of original wound-area of wound at different time points)/area of original wound ×100%.
Apoptosis assay
Apoptosis was detected by flow cytometry using an Annexin V-FITC/PI kit according to the manufacturer’s instruction. In brief, cells were seeded in 6-well plates at a density of 4×105 cells per well, after treatment with EKC for 48 h, cells were harvested, washed, centrifuged and then resuspended in binding buffer containing annexin V-FITC and PI. After staining, cells were analyzed using a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA). Cells treated with 15 μM cisplatin were served as positive control.
DAPI (4, 6-diamidino-2-phenylindole) staining
DNA condensation was detected with DAPI staining. Briefly, cells cultured in 24-wells plate were treated with EKC 12.5 μM or 25 μM for 24 h, after fixation, cells were incubated with 2.5 μg/ml DAPI solution for 10 min in the dark. After washing with PBS, images of cell nuclei were taken by a flurescence microscope system (Olympus, IX71, Japan).
Autophagic flux measurement
Autophagic flux was detected by using the mRFP-GFP-LC3 adenovirus. Cells cultured on coverslips were infected with mRFP-GFP-LC3 adenovirus according to the manufacture’s protocol. After infection, cells were treated with or without EKC for 2 h or 24 h. Autophagic flux was detected using a flurescence microscope system (Olympus, IX71, Japan).
mRNA extraction and real-time quantitative reverse transcription-polymerase chain reaction
Trizol reagent was used to extract the total mRNA of cells. Equal amount of RNA was reverse transcribed to cDNA using the PrimeScript RT reagent Kit (Takara) according to the manufacturer’s instruction. Quantitative real-time PCR (qRT-PCR) was performed using a SYBR Premix Ex Taq Kit (Takara) and the condition was an initial cycling for 2 min at 95°C, followed by 45 cycles of 15 s at 95°C, 15 s at 55°C and 20 s at 72°C. The data presented are relative mRNA levels normalized to GAPDH, the value from the untreated group was set as 1. The primer sequences are list in Table 2.
Table 2.
Primer sequences used in the RT-PCR
| primer sequences | |
|---|---|
| Rac1 | Forward (ATGTCCGTGCAAAGTGGTATC) |
| Reverse (CTCGGATCGCTTCGTCAAACA) | |
| Arf6 | Forward (GGGAAGGTGCTATCCAAAATCTT) |
| Reverse (CACATCCCATACGTTGAACTTGA) | |
| GAPDH | Forward (CTGACTTCAACAGCGACACC) |
| Reverse (TGCTGTAGCCAAATTCGTTGT) |
Live cell imaging with fluorescent tracers
Cells were seeded in coverglass bottom dishes (confocal dishes) and allowed to attach overnight. Cells were treated with EKC for 12 h and then incubated with MitoTracker Red, LysoTracker Red and ER-Tracker according to manufacturer’s protocol respectively. Images were collected by confocal microscope (LSM 700, Zeiss, Germany). For Lucifer Yellow (LY) staining, cells were incubated with EKC and Lucifer Yellow (0.5 mg/ml) simultaneously for 24 h, then washed with PBS and photographed with a florescence microscope. For acridine orange staining, cells cultured in 24-wells plate were treated with EKC for 8 hr, then were incubated with 10 μM acridine orange for additional 30 min in serum-free medium. Pictures were obtained by a flurescence microscope system (Olympus, IX71, Japan).
Immunofluorescence
After cells were attached on glass cover slips, EKC was added into the medium and treated cells for 12 h or 24 h. At the end of incubation, cells were fixed in methanol, then introduced Rab7, LAMP1 and Rab5 primary antibody (1:100) and incubated at 4°C overnight. Afterwards, incubated cells with secondary antibody Alexa Fluor 488 or 594-conjugated at room temperature for 1 h. Fluorescent images were taken with a fluorescence microscope.
Immunoblot analysis
After treatments, cell lysate was prepared by using a sample buffer (50 mM Tris-HCl [PH 6.8], 2% SDS, 10% glycerol, 100 mM DTT, 0.1% bromophenol blue). Equal amounts of proteins were loaded and electrophoresed on SDS PAGE gel followed by a transfer onto nitrocellulose membrane (Millipore). After blocking, the membrane was incubated with a diluted primary antibody solution at 4°C overnight, and then the membrane was exposed to a horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h. Signals were visualized using the enhanced chemiluminescence (ECL) detection reagents (Millipore, Billerica, MA, USA). GAPDH and β-actin were used as internal controls.
Cytotoxicity assay using xCELLigence system
Cytotoxicity assay was conducted using the xCELLigence system (ACEA Biosciences, San Diego, CA, USA), which allows for dynamic monitoring of cell growth in response to treatment in real-time. In this experiment, cells were seeded onto E-plates with the number of 8000 per well, after an overnight incubation, cells were exposed to EKC, doxorubicin (0.3 and 0.5 μM), etoposide (30 and 50 μM) or in combination. The impedances were measured per 30 min for 70 h and expressed as CI value.
Pull-down assays to measure Rac1 and Arf6 activation
Rac1 and Arf6 activation assays were performed using the Cytoskeleton Rac1 and Arf6 activation biochem kits. Cells were seeded in 100 mm dishes and incubated overnight. Then, cells were treated with EKC or DMSO for 12 h or 24 h. Cells were lysed and used for pull-down assays according to the manufacturer’s instructions. The assays either use PAK-PBD conjugated sepharose beads to selectively bind active Rac1 in whole cell lysates, or GGA3-PBD beads to pull down Arf6. The active Rac1 or Arf6 collected on the beads were subjected to immunoblot analysis. On a separate immunoblot, the total Rac1 or Arf6 was determined.
Transmission electron microscopy
The ultra-structure of cytoplasmic vacuolization was observed using a JEOL JEM-1200EX П Transmission Electron Microscope. In brief, cells were collected and fixed with 2.5% glutaraldehyde at 4°C overnight, washed with PBS and post-fixed with 1% OsO4 for 1.5 h. The samples were then washed with PBS and dehydrated with graded alcohol. After dehydration, the samples were embedded in epoxy resin. Ultrathin sections were cut, stained with uranyl acetate and lead citrate and then examined under the transmission electron microscope.
Statistical analysis
Results are presented as the mean ± standard deviation (SD). To determine the significant difference between two groups, one way analysis of variance (ANOVA) test and post hoc multiple comparison Bonferroni test were used. P < 0.05 was considered to be significant.
Results
The obtaining of EKC and its analogues
Epimedium koreanum is a good resource of prenylated flavonoids. Our phytochemical study on the leaves of this plant led to the isolation of EKC and other 26 prenylated flavonoids. The isolation process and structures of the isolated prenylated compounds were shown in Figure S1 and Table S1. The structure of EKC was determined by analysis of the spectroscopic data (Figure 1G). 1H NMR and 13C NMR data of EKC were listed as below. 1H-NMR (600 MHz, DMSO-d6) δppm: 12.88 (1H, br s, 5-OH), 7.50 (1H, d, J = 1.2 Hz, H-2’), 7.39 (1H, d, J = 1.2 Hz, H-6’), 6.61 (1H, s, H-3), 6.26 (1H, s, H-6), 5.24 (1H, d, J = 4.2 Hz, H-16), 5.21 (1H, t, J = 6.6 Hz, H-12), 4.22 (1H, d, J = 4.2 Hz, H-17), 3.44 (2H, d, J = 6.6 Hz, H-11), 1.77 (3H, s, CH3-14), 1.63 (6H, s, CH3-15), 1.20 (3H, s, CH3-20), 1.13 (3H, s, CH3-19); 13C-NMR (150 MHz, DMSO-d6) δppm: 182.1 (C-4), 163.9 (C-2), 162.8 (C-7), 159.4 (C-5), 154.7 (C-9), 151.7 (C-4’), 142.2 (C-3’), 132.9 (C-5’), 131.3 (C-13), 123.9 (C-1’), 122.8 (C-12), 115.2 (C-6’), 114.6 (C-2’), 106.5 (C-8), 103.6 (C-10), 103.1 (C-3), 98.8 (C-6), 98.0 (C-17), 72.0 (C-16), 70.0 (C-18), 26.2 (CH3-14), 25.8 (CH3-19), 25.4 (CH3-20), 21.7 (C-11), 18.2 (CH3-15).
Figure 1.

EKC inhibited cell proliferation and induced cytoplasmic vacuolization. (A) NCI-H292, A549 and 16HBE cells were treated with EKC as indicated, cell viability was detected by MTT assay. Data are expressed as mean ± SD, n = 3. ***P < 0.001 compared with control group. (B) NCI-H292 cells were treated with EKC at 12.5 and 25 μM, phase-contrast images were taken at indicated time points. (C) A549 cells were treated with EKC at 25 μM, phase-contrast images were taken at indicated time points. (D) 16HBE and Beas2B cells were treated with EKC at 25 μM and images were taken at the indicated time. (E) Cells were treated with EKC at 15 μM for 12 h, then placed in EKC-free medium for culturing additional 12 h. Images were captured by optical microscopy. (F) Cells were treated with EKC at 25 μM and vacuoles were observed in Calu-1, HepG2 and PANC-1 cells. (G) NCI-H292 cells were treated with four closely related compounds with > 75% structure similarity to EKC. Images were captured by optical microscopy. Bar = 50 μM (B-G).
EKC induced cell death and cytoplasmic vacuolization
During our screening of anticancer candidates from prenylated flavonoids, the effects on cell viability of these 27 compounds were determined by MTT assay in non-small cell lung cancer NCI-H292 cells first (Table S1). EKC inhibited NCI-H292 cell growth in a dose- and time-dependent manner with the IC50 value of 17.04 μM for 48 h. In another non-small cell lung cancer cell line A549, EKC also decreased cell viability in a dose-dependent manner with the IC50 value of 27.59 μM for 48 h. Meanwhile, in an immortalized normal human bronchial epithelial cell line 16HBE, the cell viability after 48 h of EKC treatment was almost unaffected (Figure 1A). Thus, EKC was more toxic to the tested lung cancer cells compared to normal bronchial epithelial cells.
In terms of cytotoxity, EKC was not really a promising candidate, but it captured our attention because of its cytoplasmic vacuoles inducing effect. We found that EKC caused a quick and striking accumulation of numerous phase-lucent cytoplasmic vacuoles within 12 h in both A549 and NCI-H292 cells, these morphological effects of EKC persisted for 24 h and beyond (Figure 1B and 1C). In contrast, 16HBE cells and the other normal human bronchial epithelial cell line, Beas2B were relatively resistant to EKC-induced vacuolization (Figure 1D). The vacuolization effects in NCI-H292 cells induced by EKC at a dose of 15 μM were demonstrated to be reversible, if cells were treated for 12 h, then placed in EKC-free medium for culturing additional 12 h, the vacuolization was almost completely resolved (Figure 1E).
The effects of EKC on other three cancer cell lines, including Calu-1, HepG2 and PANC-1 were examined. Similar to the results with NCI-H292 and A549 cells, EKC induced dramatic cytoplasmic vacuolization in all of the three cell lines (Figure 1F).
Four closely related compounds with > 75% structure similarity to EKC were isolated from E. koreanum together, they showed almost no vacuole-inducing activity or induced a small amount of vacuoles (Figure 1G). This suggested that the effect of EKC was probably due to its interaction with specific intracellular targets rather than non-specific effects.
EKC induced cell death was distinct from apoptosis
Apoptosis is the most common form of programmed cell death. To investigate if the cell death induced by EKC was related with apoptosis, annexin V-FITC/PI double staining and DAPI staining were performed in NCI-H292 cells. As shown in Figure 2A and 2B, neither significant increase of the number of apoptotic cells nor obvious morphological changes in the nuclei were observed with EKC treatment, indicating that apoptosis may not be critically involved in EKC-induced cell death. For further determination, the activation of apoptosis markers including caspase-9, caspase-3 and PARP was also examined in NCI-H292 cells. As depicted in Figure 2C, no obvious cleavage of caspase-9, caspase-3 and PARP was observed in 24 h after EKC exposure. Consistent with this assumption, pretreatment with pan-caspase inhibitor Z-VAD (OMe)-FMK (Z-VAD) for 1 h prior to EKC treatment did not block both the loss of cell viability and vacuolization induced by EKC (Figure 2D and 2E). These findings suggested that EKC-induced cell death was non-apoptotic and caspase-independent.
Figure 2.

EKC caused NCI-H292 cell death was distinct from apoptosis. A. NCI-H292 cells were treated with EKC at indicated concentrations for 48 h, stained with Annexin V/PI and followed by flow cytometry assay. Cisplatin (CDDP) 15 μM treatment was used as positive control. Data are expressed as mean ± SD, n = 3. B. NCI-H292 cells were treated with EKC at 12.5 and 25 μM for 24 h, the changes in nuclear morphology were evaluated by fluorescence microscopy after DAPI staining. Bar = 50 μM. C. NCI-H292 cells were incubated with EKC as indicated, cell lysates were subjected to immunoblotting for PARP, caspase9 and caspase3, β-actin was used as loading control. The band density was normalized to untreated group. D. Cell viability of NCI-H292 cells with or without Z-VAD (OMe)-FMK. Cells were pretreated with 20 μM Z-VAD (OMe)-FMK for 1 h before EKC treatment, cell viability was determined by MTT assay. Results were from thee separate experiments. Values are expressed as mean ± SD, n = 3. ***P < 0.001. E. NCI-H292 cells were pretreated for 1 h in the presence or absence of Z-VAD (OMe)-FMK (20 μM) prior to addition of EKC or DMSO. Phase-contrast images were captured after 12 h. Bar = 20 μM.
EKC induced methuosis in lung cancer cells
Cytoplasmic vacuolation has been observed in several types of non-apoptotic cell deaths. To determine the origin of EKC-induced vacuoles, different subcellular compartments staining dyes including acridine orange (AO), MitoTracker red, LysoTracker red and ERTracker red were used in NCI-H292 cells. Live cell imaging showed that the vacuoles could sequester AO to change to orange fluorescence (Figure 3A), which indicated the acidic nature of the vacuoles. None of MitoTracker red, LysoTracker red and ERTracker red was observed to be incorporated into the vacuoles (Figure 3B-D), indicating that the vacuoles were not derived from mitochondria, lysosome or endoplasmic reticulum.
Figure 3.
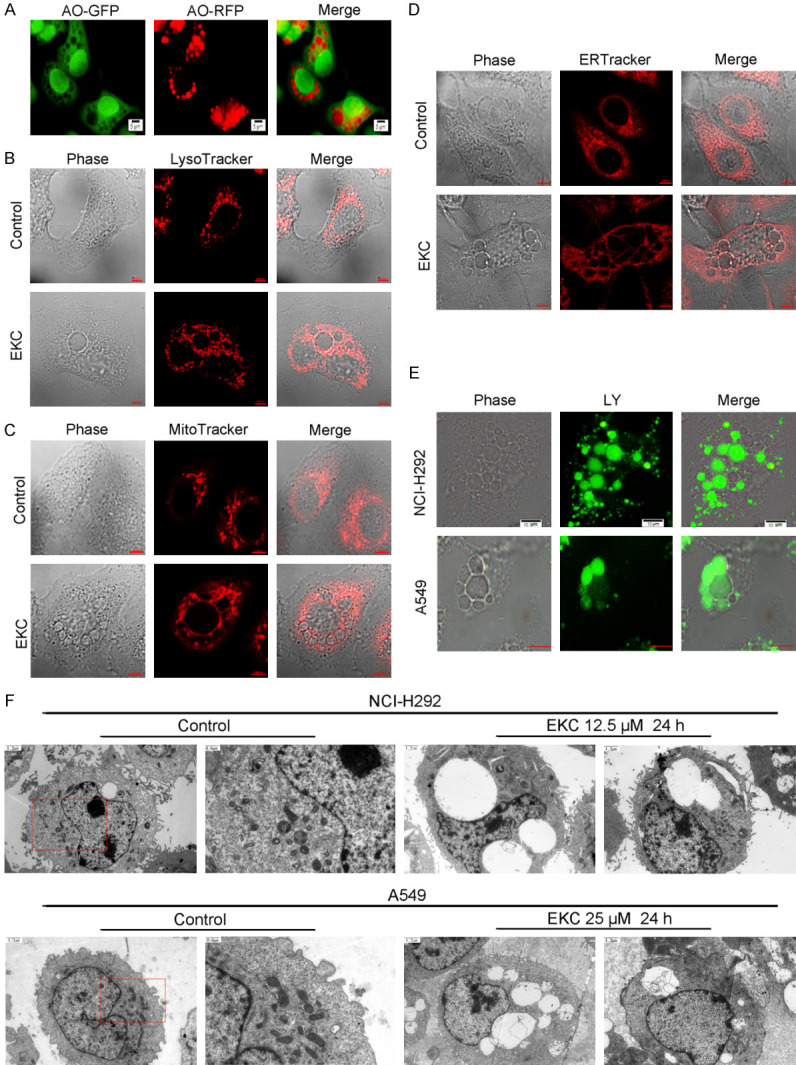
EKC induced vacuoles were correlated with macropinocytosis. (A) NCI-H292 cells were treated with 15 μM EKC for 8 h, then incubated with 10 μM acridine orange (AO) for additional 30 min. Pictures were obtained by flurescence microscope. NCI-H292 cells were treated with 15 μM EKC for 12 h, and further incubated with 50 nM LysoTracker Red (B), 75 nM MitoTracker Red (C), and 0.5 μM ERTracker Red (D) for 30 min at 37°C. Phase-contrast and fluorescent images were captured by confocal microscope. (E) NCI-H292 and A549 cells were treated with 15 μM EKC and Lucifer Yellow (0.5 mg/ml) for 24 h. Phase-contrast and fluorescent images were captured by fluorescence microscopy. Bar = 10 μM. (F) Transmission electron microscopy of NCI-H292 and A549 cells treated with EKC for 24 h.
The formation of large vacuoles is one of the most important characteristics of macropinocytosis. We next turned our attention to determine whether the vacuoles induced by EKC were correlated with macropinocytosis. One of the typical features of macropinocytosis is the incorporation of extracellular-phase fluid tracers. Lucifer Yellow (LY) is a well-established fluorescent tracer used to define macropinocytosis [20]. As shown in Figure 3E, LY incorporated into most of the phase-lucent vacuoles induced by EKC in NCI-H292 and A549 cells, suggested that EKC-induced vacuoles might be derived from macropinosomes.
These results were further confirmed by transmission electron microscopy examination. Compared with the untreated NCI-H292 and A549 cells, EKC treatment led to the formation of large and empty vacuoles bounded by a single membrane. Other cellular organelles such as the mitochondrial and endoplasmic reticulum remained unaffected. Moreover, the nuclear chromatin remained diffuse and the nuclear membrane appeared intact (Figure 3F). Collectively, these data further illustrated that EKC induced lung cancer cell death was related to methuosis.
Normally, once internalized into cells, macropinosomes pesist for only about 5 to 20 minutes, during which they either recycle back to the cell surface or merge with lysosomes [20,21]. While in methuosis, macropinosomes bypass the normal endosomal trafficking pathway, immediately recruit Rab7 and form progressively larger LAMP1-positive structures [22]. For further investigation of whether EKC induced methuosis, immunofluorescence assay against two late endosome markers, Rab7 and LAMP1, was performed. As seen in Figure 4A, Rab7 and LAMP1 were detected on the membrane of EKC-induced vacuoles in both NCI-H292 and A549 cells. Rab5, a marker of early endosome, was also detected, but it was not labeled on the membrane of the vacuoles (Figure 4A). The protein levels of Rab7 and LAMP1 were also detected, as shown in Figure 4B, EKC treatment increased the levels of Rab7 and LAMP1 in dose- and time-dependent manners.
Figure 4.
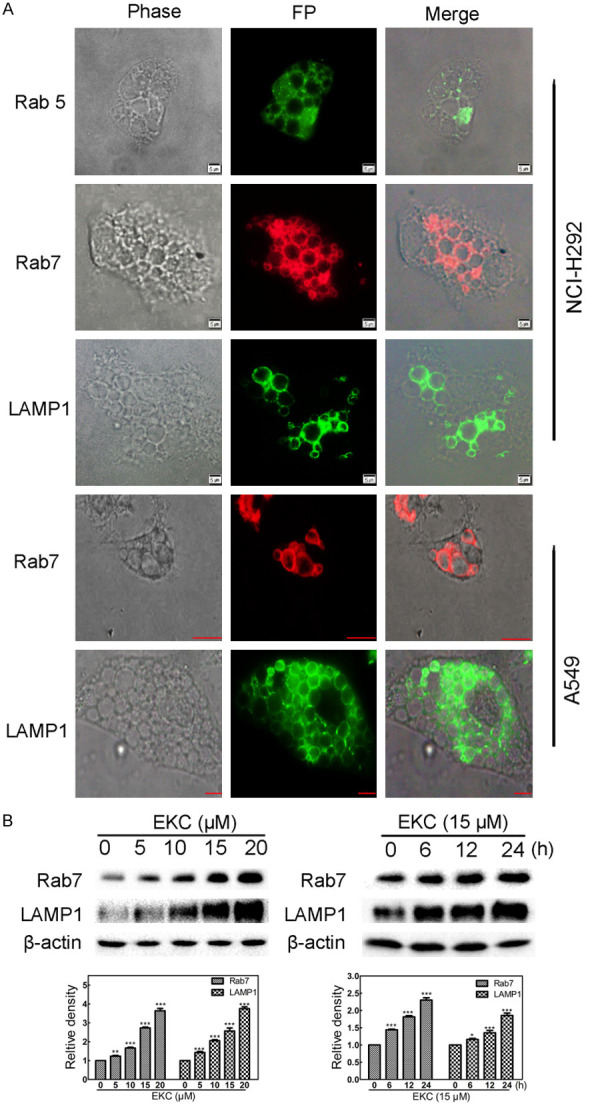
EKC induced vacuoles acquired characteristics of late endosomes. A. Cells were treated with 15 μM EKC for 12 h, then stained with anti-Rab5, anti-LAMP1 and anti-Rab7, images were taken by fluorescent microscopy. Bar = 5 μM. B. Cells were incubated with EKC as indicated, cell lysates were subjected to immunoblotting for Rab7 and LAMP1. The band density was normalized to untreated group. *P < 0.05, **P < 0.01, and ***P < 0.001.
EKC induced PIKfyve phosphoinositide kinase inhibition and Akt suppression
PIKfyve, a class Ш phosphoinositide (PI) kinase, plays crucial role in the regulation of trafficking events associated with the endocytic pathway [23,24]. It has been reported that PIKfyve was one candidate protein target of a methuosis inducer, MOMIPP [25]. To explore whether EKC inhibited PIKfyve expression, the protein level of PIKfyve was detected by immunoblot assay. As depicted in Figure 5A, EKC treatment decreased the protein levels of PIKfyve dose- and time-dependently. Akt suppression was required for the cytotoxicity of PIKfyve inhibitors, the phosphorylation of Akt was shown to be down-regulated concurrently (Figure 5A).
Figure 5.

EKC induced PIKfyve phosphoinositide kinase inhibition and Akt suppression. Cells were incubated with EKC as indicated, cell lysates were subjected to immunoblotting for PIKfyve and Akt (A); LC3 and SQSTM1 (B). The band density was normalized to untreated group. (C) Cells were transfected with mRFP-GFP-LC3 adenovirus for 24 h prior to addition of 15 μM EKC or DMSO. Images were captured by fluorescence microscope. Bar = 10 μM. (D) NCI-H292 cells were pretreated for 1 h in the presence or absence of Baf A1 (20 nM) prior to addition of 15 μM EKC or DMSO. Images were captured by optical microscopy after 24 h treatment. Bar = 20 μM. (E) NCI-H292 cells were pretreated for 1 h with 20 nM Baf A1 prior to addition of EKC. Cell viability was detected by MTT assay after 48 h. Results were from three separate experiments. Values are expressed as mean ± SD, n = 3. *P < 0.05, **P < 0.01, and ***P < 0.001.
PIKfyve also plays a crucial role in ensuring phagosomal maturation [26]. Under the same conditions in which EKC induced the accumulation of cytoplasmic vacuoles, immunoblotting analysis revealed a dose- as well as time-dependent increase of two biomarkers of autophagosome, LC3-П and SQSTM1, indicating the accumulation of autophagosomes (Figure 5B). To determine whether the accumulation of autophagosomes resulted from induction of autophagy or from disruption of autophagic flux, cells were transfected with mRFP-GFP-LC3 adenovirus, as shown in Figure 5C, distinct yellow puncta were observed in EKC treated cells. Therefore, EKC disrupted autophagic flux by preventing autolysosome formation.
Bafilomycin A1 (Baf A1), an H+-vacuolar ATPase inhibitor, has been shown to prevent formation of cytoplasmic vacuoles that are induced by inhibition of PIKfyve PI kinase [27]. To determine whether or not Baf A1 also prevented induction of the cytoplasmic vacuoles that were induced by EKC, NCI-H292 cells were treated by Baf A1, EKC, or both Baf A1 and EKC, the results showed that Baf A1 prevented EKC-induced cytoplasmic vacuolation almost completely (Figure 5D), suggesting that EKC inhibited PIKfyve activity.
EKC affected the activities of Rac1 and Arf6 GTPases
It has been reported that methuosis triggered by over-expression of activated H-Ras required activation of the Rac1 GTPase with a concomitant deactivation of another GTPase, Arf6 [28]. We first detected the total protein level and mRNA level of Rac1 and Arf6 in NCI-H292 cells. The immunoblot results revealed that EKC treatment had no significant effect on the expression of Rac1, while decreased the total level of Arf6 (Figure 6A). The qPCR results were consistent, the mRNA level of Rac1 was not changed, while the mRNA level of Arf6 decreased (Figure 6B). Futhermore, the active GTP-bound forms of Rac1 and Arf6 were measured in pull-down assays using fusion proteins that bind specifically to the activated forms of Rac1 and Arf6. The results showed that the amounts of active Rac1 (Rac1-GTP) increased although the total level of Rac1 did not change in EKC treated NCI-H292 and A549 cells. In contrast, EKC had the opposite effect on Arf6, both total level and active Arf6 decreased, however Arf6-GTP decreased more significant than total Arf6 after EKC treatment, that is the ratio of active/total Arf6 decreased (Figure 6C and 6F). These results indicated that EKC treatment induced activation of the Rac1 GTPase, with a concomitant reduction in the activation state of Arf6 GTPase. For further exploration, NCI-H292 and A549 cells were pretreated with a highly specific Rac inhibitor EHT 1864 for 24 h, then incubated with EKC in the presence of EHT 1864 for additional 24 h, as illustrated in Figure 6D, EKC induced cell viability decrease was antagonized by EHT 1864; in Figure 6E and 6G, most of the vacuoles were precluded by EHT 1864. These results suggested that the mechanisms of vacuolization induced by EKC is probably similar with that induced by Ras, that is, might be depended on reverse regulation of Rac1 and Arf6.
Figure 6.
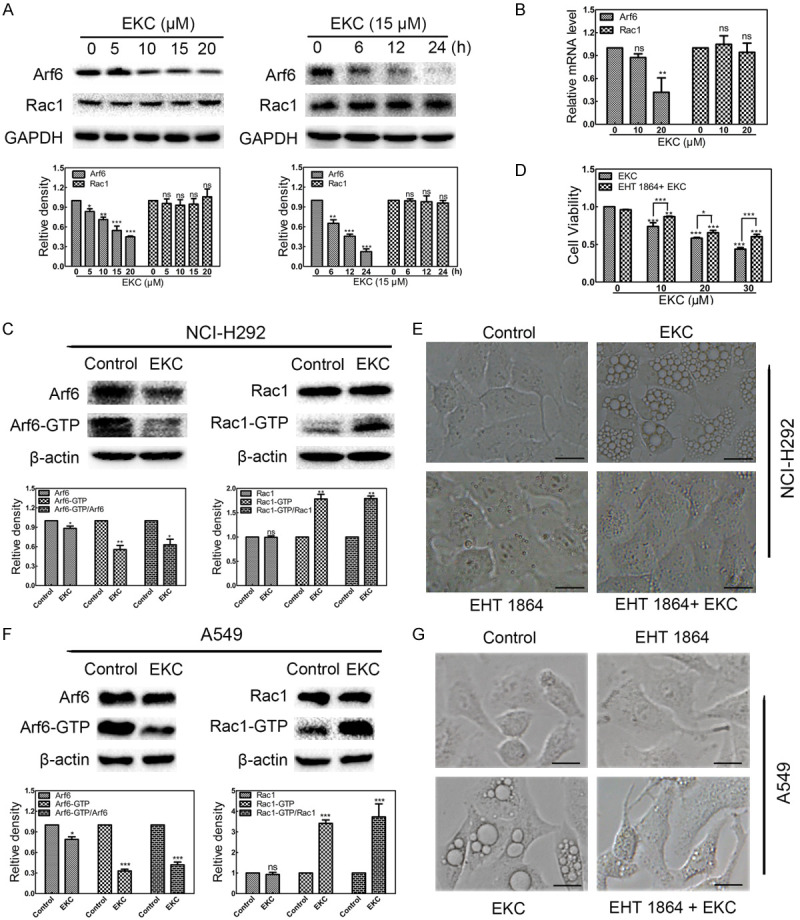
EKC affected the activities of Rac1 and Arf6 GTPases. A. Cells were incubated with EKC as indicated, cell lysates were subjected to immunoblotting for Arf6 and Rac1. GAPDH was used as loading control. Bar graphs represented the relative express level of proteins. B. EKC downregulated the mRNA level of Arf6, but did not affect the mRNA level of Rac1. mRNA was extracted from NCI-H292 cells treated with EKC as indicated, and subjected to qRT-PCR analysis. C. NCI-H292 cells were incubated with DMSO or 12.5 μM EKC for 12 h. Pull-down assays were performed for activated Rac1 (Rac1-GTP) and Arf6 (Arf6-GTP). D. NCI-H292 cells were pretreated for 24 h with 10 μM EHT 1864 prior to addition of EKC or DMSO. Cell viability was tested by MTT assay after 48 h treatment. Values are expressed as mean ± SD, n = 3. *P < 0.05, **P < 0.01, and ***P < 0.001. E. NCI-H292 cells were pretreated for 24 h in the presence or absence of 10 μM EHT 1864 prior to addition of 15 μM EKC or DMSO for additional 16 h treatment. Images were captured by optical microscopy. Bar = 20 μM. F. A549 cells were incubated with DMSO or 25 μM EKC for 24 h. Pull-down assays were performed for activated Rac1 (Rac1-GTP) and Arf6 (Arf6-GTP). G. A549 cells were pretreated for 24 h in the presence or absence of 10 μM EHT 1864 prior to addition of 25 μM EKC or DMSO for additional 16 h treatment. Images were captured by optical microscopy. Bar = 20 μM.
EKC inhibited cell migration
It has been demonstrated that Arf6 regulates cancer cell invasion through the activation of MEK/ERK signaling pathway, and downregulation of Arf6 correlate with impaired cell migration [29,30]. We evaluated the effect of EKC on the migration of NCI-H292 cells by performing a scratch wound-healing assay. As shown in Figure 7A, EKC treatment at the dose of 6.5 and 12.5 μM could significantly reduce cell migration in NCI-H292 cells. We also examined the phosphorylation as well as total ERK levels by immunoblot analysis. EKC treatment decreased the levels of phosphorylation ERK in dose- and time-dependent manners, while no obvious change of total ERK was observed (Figure 7B).
Figure 7.
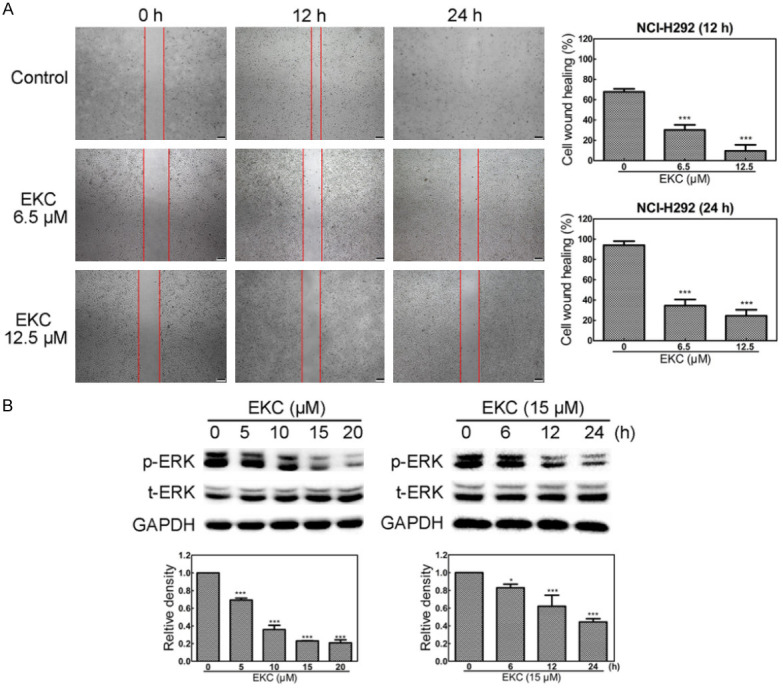
EKC inhibited NCI-H292 cell migration. A. Cells were treated with EKC as indicated, scratch wound assay was performed after treatment. Images were captured by optical microscopy. B. Cells were incubated with EKC at different concentrations or times and cell lysates were subjected to immunoblotting for p-ERK, t-ERK. The band density was normalized to untreated group. *P < 0.05, **P < 0.01, and ***P < 0.001. Bar = 200 μM.
EKC sensitized lung cancer cells to doxorubicin and etoposide
To determine the effects of EKC on the sensitivity to conventional chemotherapy agents, NCI-H292 and A549 cells were exposed to doxorubicin or etoposide in the presence of 6.5 μΜ of EKC, the viability of cells was monitored using a real time cell analyzer (RTCA). As shown in Figure 8A and 8B, both doxorubicin and etoposide displayed a reduction in cell growth, whereas treatment with 6.5 μΜ of EKC alone resulted in slight growth inhibition. However, co-treatment of EKC with doxorubicin/etoposide significantly reduced cell growth as judged by decreased cell index (CI) compared with doxorubicin and etoposide treatment alone.
Figure 8.
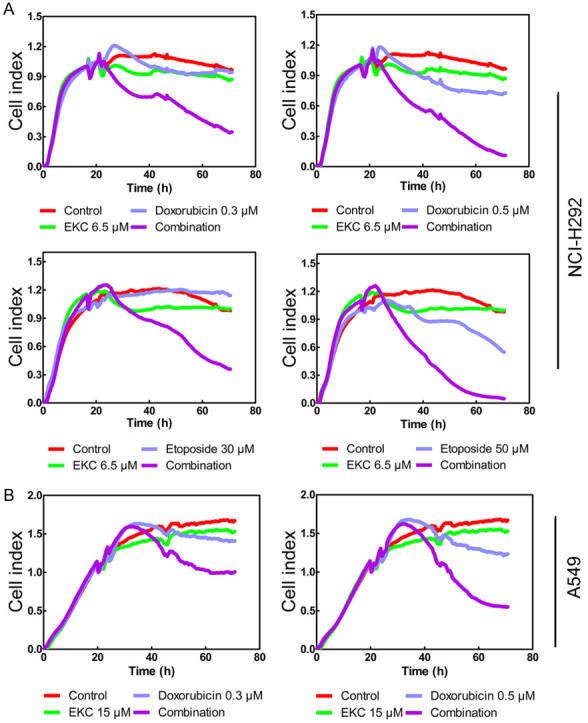
EKC sensitized lung cancer cells to doxorubicin/etoposide treatment. A. NCI-H292 cells were pretreated with 6.5 μM EKC for 4 h before the addition of DMSO, doxorubicin (0.3 and 0.5 μM) or etoposide (30 and 50 μM). B. A549 cells were pretreated with 15 μM EKC for 4 h before the addition of DMSO, doxorubicin (0.3 and 0.5 μM). Cell index was measured using an xCELLigence system for 70 h.
Discussion
Apoptosis is a mechanism of programmed cell death and is essential for cell development and homeostasis. Apoptotic cells undergo characteristic changes in cell morphology, including cell rounding, plasma membrane blebbing and nuclear fragmentation [31,32]. In cancer cells, modulation of apoptosis plays a significant role in the progression of malignancy. Alongside the discoveries of the mechanisms underlying apoptosis, targeting apoptotic pathways has been approved as a primary therapeutic strategy [33]. However, many cancer cells developed resistance to most of the apoptosis-based cancer therapies. In this sense, targeting non-apoptotic forms of cell death therapeutics may have important implications.
Several forms of non-apoptotic programmed cell death including autophagy, paraptosis, necroptosis, oncosis and ferroptosis has been reported in the past three decades [34]. Methuosis is one of the most recent additions to the non-apoptotic cell death forms. As a unique form of non-apoptotic cell death, methuosis was initially defined in glioblastoma cells after overexpression of Ras [35]. In recent years, some methuosis inducers were identified and all of these compounds displayed meaningful effects in killing cancer cells [11-16]. This indicated that small molecules with the capacity to induce methuosis may be of considerable interest as potential therapeutics for cancers that are resistant to apoptosis.
Natural compounds are good resources of methuosis inducers, as most of aforementioned methuosis inducers are natural compounds related. Therefore natural compounds have gained increasing interests for the future discovery of non-apoptotic cell death based anticancer therapeutics [36,37]. Epimedium koreanum is a traditional medicinal plant recorded in Chinese Pharmacopoeia. The aerial part of this plant has been used for alleviating a variety of disorders [38]. Previous phytochemical investigations revealed that prenylated flavonoids are major constituents of E. koreanum [39]. During our exploration of cytotoxic prenylated flavonoids, we performed a systematic isolation of Epimedium koreanum, as a result, 27 prenylated flavonoids were obtained. In the cell viability assay, EKC caught our attention because of the induction of catastrophic vacuolization in human lung cancer NCI-H292 and A549 cells. The phenotype of EKC-induced cell death was identified as methuosis based on the following features: (1) Shortly after being exposed to EKC, extreme cytoplasmic vacuolization was observed (Figure 1B and 1C); (2) The vacuoles were decorated with late endosomal markers Rab7 and LAMP1, but did not sequester with lysosomes (Figures 3B and 4A); (3) Consistent with a non-apoptotic mechanism, cell death could not be prevented by caspase inhibitor, and nuclear chromatin condensation was not observed (Figure 2B and 2D).
Small G proteins in the Rab and Arf family are present on endosomes and coordinate the trafficking of cargo proteins. It was reported that methuosis induced by overexpression of Ras required the activation of Rac1 and inactivation of Arf6, two GTPases implicated in macropinocytosis and endosome recycling [28]. Our findings indicated that Rac1 was activated while Arf6 was inactivated under EKC treatment, a Rac1 inhibitor EHT 1864 precluded the vacuole-inducing effect of EKC, suggesting that inversed regulation of Rac1 and Arf6 was involved in EKC-induced methuosis (Figure 6C-G).
PIKfyve is a critical regulator that has been implicated in various trafficking events associated with the endocytic pathway [40]. PIKfyve is required for the maturation of both endosome and phagosome [26]. Our results indicated that EKC treatment decreased the expression level of PIKfyve, also blocked the formation of autophagolysosome (Figure 5C). Considering the function of PIKfyve, we hypothesized that EKC-induced vacuoles were blocked to merge with lysosomes might be involved with down-regulation of PIKfyve.
Based on our findings, we suggested a working model of EKC depicted in Figure 9. For the normal process of macropinocytosis in mammalian cells, macropinosomes are internalized first, then the macropinosomes rapidly acquire Rab5, EEA1 and sorting nexins, which promote fusion with early endosomes. The latter retain the capacity to recycle to the cell surface, but after several additional minutes they lose Rab5 and acquire Rab7 and LAMP1, taking on the characteristics of late endosomes. Ultimately, the vesicles dissipate as they merge with lysosomes [11]. EKC induced vacuoles were suggested to bypass the normal endosomal trafficking pathway and undergo abnormal homotypic fusions to form progressively larger LAMP1-positive structures. The possible reasons were that the decline of Arf6 and PIKfyve blocked vacuoles recycling and maturation to late endosomes.
Figure 9.
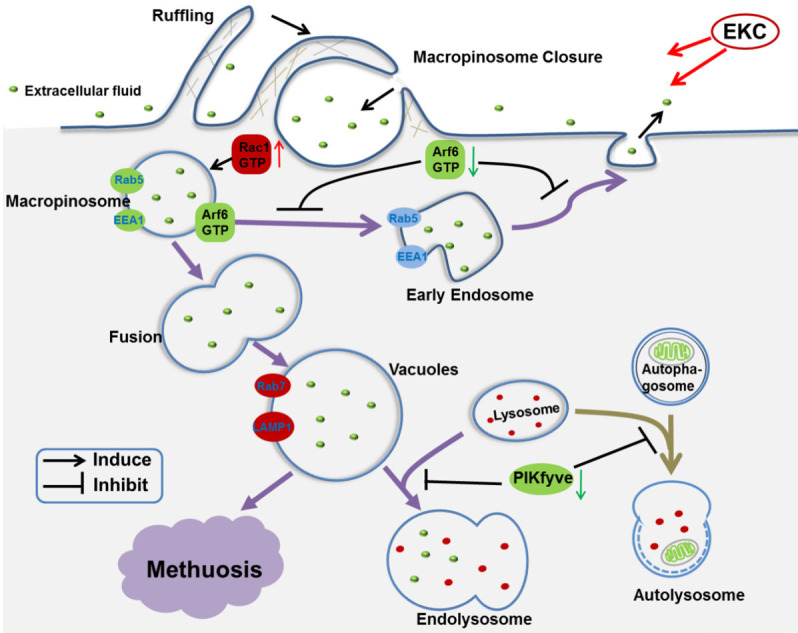
A possible pathway for EKC-induced methuosis suggested by the present studies: EKC caused abnormal macropinocytosis. In the presence of EKC, Rac1 was activated and resulted in the stimulation of macropinosome formation. At the same time, Arf6 was inactivated which impeded the process of endosome returning to the plasma membrane. The abnormal macropinocytosis underwent homotypic fusion and formed huge vacuoles. These vacuoles failed to fuse with lysosomes and were degraded, resulting in methuosis ultimately.
The key observation that EKC effectively induced methuosis, as well as synergized with doxorubicin or etoposide in human lung cancer cells, raises the possibility that further development of this compound could lead to useful therapeutic agents for treating cancers that are resistant to drugs that commonly work by inducing apoptosis.
Conclusions
In conclusion, EKC was identified as an inducer of methuosis, an unconventional non-apoptotic cell death form. Inverse regulation of Rac1 and Arf6 contributed to EKC induced cytoplasmic vacuolization, this might be related to the mechanism of action of EKC. Combined treatment of EKC with doxorubicin and etoposide enhanced the efficacy of chemotherapeutic drugs dramatically. EKC could be considered as a candidate for the development of methuosis-based therapies against cancers which are resistant to chemo-drugs that commonly work by inducing apoptosis.
Acknowledgements
This research was funded by the National Natural Science Foundation of China (No. 81973202) and the Key Research and Development Program of Shandong Province (No. 2018GSF118085).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Cortes J, Perez-Garcia JM, Llombart-Cussac A, Curigliano G, El Saghir NS, Cardoso F, Barrios CH, Wagle S, Roman J, Harbeck N, Eniu A, Kaufman PA, Tabernero J, Garcia-Estevez L, Schmid P, Arribas J. Enhancing global access to cancer medicines. CA Cancer J Clin. 2020;70:105–124. doi: 10.3322/caac.21597. [DOI] [PubMed] [Google Scholar]
- 2.Perez-Herrero E, Fernandez-Medarde A. Advanced targeted therapies in cancer: drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm. 2015;93:52–79. doi: 10.1016/j.ejpb.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 3.Szakacs G, Paterson J, Ludwig J, Booth-Genthe C, Gottesman M. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 4.Peeper DS. Cancer drug resistance: old concept, novel solutions required. Mol Oncol. 2014;8:1064–1066. doi: 10.1016/j.molonc.2014.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao XL, Tu Y, Xu YL, Guo YY, Yao F, Zhang XH. Endoplasmic reticulum stress confers 5-fluorouracil resistance in breast cancer cell via the GRP78/OCT4/lncRNA MIAT/AKT pathway. Am J Cancer Res. 2020;10:838–855. [PMC free article] [PubMed] [Google Scholar]
- 6.Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D’Orazi G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY) 2016;8:603–619. doi: 10.18632/aging.100934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diederich M, Cerella C. Non-canonical programmed cell death mechanisms triggered by natural compounds. Semin Cancer Biol. 2016;40-41:4–34. doi: 10.1016/j.semcancer.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Lee D, Kim IY, Saha S, Choi KS. Paraptosis in the anti-cancer arsenal of natural products. Pharmacol Ther. 2016;162:120–133. doi: 10.1016/j.pharmthera.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Maltese WA, Overmeyer JH. Non-apoptotic cell death associated with perturbations of macropinocytosis. Front Physiol. 2015;6:38. doi: 10.3389/fphys.2015.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Overmeyer JH, Young AM, Bhanot H, Maltese WA. A chalcone-related small molecule that induces methuosis, a novel form of non-apoptotic cell death, in glioblastoma cells. Mol Cancer. 2011;10:69. doi: 10.1186/1476-4598-10-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson MW, Overmeyer JH, Young AM, Erhardt PW, Maltese WA. Synthesis and evaluation of indole-based chalcones as inducers of methuosis, a novel type of nonapoptotic cell death. J Med Chem. 2012;55:1940–1956. doi: 10.1021/jm201006x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun L, Li B, Su X, Chen G, Li Y, Yu L, Li L, Wei W. An ursolic acid derived small molecule triggers cancer cell death through hyperstimulation of macropinocytosis. J Med Chem. 2017;60:6638–6648. doi: 10.1021/acs.jmedchem.7b00592. [DOI] [PubMed] [Google Scholar]
- 14.Huang W, Sun X, Li Y, He Z, Li L, Deng Z, Huang X, Han S, Zhang T, Zhong J, Wang Z, Xu Q, Zhang J, Deng X. Discovery and identification of small molecules as methuosis inducers with in vivo antitumor activities. J Med Chem. 2018;61:5424–5434. doi: 10.1021/acs.jmedchem.8b00753. [DOI] [PubMed] [Google Scholar]
- 15.Cerny J, Feng Y, Yu A, Miyake K, Borgonovo B, Klumperman J, Meldolesi J, McNeil PL, Kirchhausen T. The small chemical vacuolin-1 inhibits Ca(2+)-dependent lysosomal exocytosis but not cell resealing. EMBO Rep. 2004;5:883–888. doi: 10.1038/sj.embor.7400243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lertsuwan J, Lertsuwan K, Sawasdichai A, Tasnawijitwong N, Lee KY, Kitchen P, Afford S, Gaston K, Jayaraman PS, Satayavivad J. CX-4945 induces methuosis in cholangiocarcinoma cell lines by a CK2-independent mechanism. Cancers (Basel) 2018;10:283. doi: 10.3390/cancers10090283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartmanska A, Tronina T, Poplonski J, Milczarek M, Filip-Psurska B, Wietrzyk J. Highly cancer selective antiproliferative activity of natural prenylated flavonoids. Molecules. 2018;23:2922. doi: 10.3390/molecules23112922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venturelli S, Burkard M, Biendl M, Lauer UM, Frank J, Busch C. Prenylated chalcones and flavonoids for the prevention and treatment of cancer. Nutrition. 2016;32:1171–1178. doi: 10.1016/j.nut.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Liu X, Zheng H, Liu Q, Zhang H, Wang X, Shen T, Wang S, Ren D. Morusin induces apoptosis and autophagy via JNK, ERK and PI3K/Akt signaling in human lung carcinoma cells. Chem Biol Interact. 2020;331:109279. doi: 10.1016/j.cbi.2020.109279. [DOI] [PubMed] [Google Scholar]
- 20.Lim JP, Gleeson PA. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol. 2011;89:836–843. doi: 10.1038/icb.2011.20. [DOI] [PubMed] [Google Scholar]
- 21.Wong AO, Marthi M, Mendel ZI, Gregorka B, Swanson MS, Swanson JA. Renitence vacuoles facilitate protection against phagolysosomal damage in activated macrophages. Mol Biol Cell. 2018;29:657–668. doi: 10.1091/mbc.E17-07-0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maltese WA, Overmeyer JH. Methuosis: nonapoptotic cell death associated with vacuolization of macropinosome and endosome compartments. Am J Pathol. 2014;184:1630–1642. doi: 10.1016/j.ajpath.2014.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, Szigeti K, Shy ME, Li J, Zhang X, Lupski JR, Weisman LS, Meisler MH. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, Xu H. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun. 2010;1:38. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho H, Geno E, Patoor M, Reid A, McDonald R, Hild M, Jenkins JL. Indolyl-pyridinyl-propenone-induced methuosis through the inhibition of PIKFYVE. ACS Omega. 2018;3:6097–6103. doi: 10.1021/acsomega.8b00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krishna S, Palm W, Lee Y, Yang W, Bandyopadhyay U, Xu H, Florey O, Thompson CB, Overholtzer M. PIKfyve regulates vacuole maturation and nutrient recovery following engulfment. Dev Cell. 2016;38:536–547. doi: 10.1016/j.devcel.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma G, Guardia CM, Roy A, Vassilev A, Saric A, Griner LN, Marugan J, Ferrer M, Bonifacino JS, DePamphilis ML. A family of PIKFYVE inhibitors with therapeutic potential against autophagy-dependent cancer cells disrupt multiple events in lysosome homeostasis. Autophagy. 2019;15:1694–1718. doi: 10.1080/15548627.2019.1586257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhanot H, Young AM, Overmeyer JH, Maltese WA. Induction of nonapoptotic cell death by activated Ras requires inverse regulation of Rac1 and Arf6. Mol Cancer Res. 2010;8:1358–1374. doi: 10.1158/1541-7786.MCR-10-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tague SE, Muralidharan V, D’Souza-Schorey C. ADP-ribosylation factor 6 regulates tumor cell invasion through the activation of the MEK/ERK signaling pathway. Proc Natl Acad Sci U S A. 2004;101:9671–9676. doi: 10.1073/pnas.0403531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Z, Xu R, Liu J, Zhang Y, Du J, Li W, Zhang W, Li Y, Zhu Y, Gu L. GEP100 regulates epidermal growth factor-induced MDA-MB-231 breast cancer cell invasion through the activation of Arf6/ERK/uPAR signaling pathway. Exp Cell Res. 2013;319:1932–1941. doi: 10.1016/j.yexcr.2013.05.028. [DOI] [PubMed] [Google Scholar]
- 31.Blaho JA. Oncoapoptosis: a novel molecular therapeutic for cancer treatment. IUBMB Life. 2010;62:87–91. doi: 10.1002/iub.274. [DOI] [PubMed] [Google Scholar]
- 32.Fulda S. Cross talk between cell death regulation and metabolism. Methods Enzymol. 2014;542:81–90. doi: 10.1016/B978-0-12-416618-9.00004-2. [DOI] [PubMed] [Google Scholar]
- 33.Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17:395–417. doi: 10.1038/s41571-020-0341-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nunez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Overmeyer JH, Kaul A, Johnson EE, Maltese WA. Active Ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol Cancer Res. 2008;6:965–977. doi: 10.1158/1541-7786.MCR-07-2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye J, Zhang RN, Wu F, Zhai LJ, Wang KF, Xiao M, Xie T, Sui XB. Non-apoptotic cell death in malignant tumor cells and natural compounds. Cancer Lett. 2018;420:210–227. doi: 10.1016/j.canlet.2018.01.061. [DOI] [PubMed] [Google Scholar]
- 37.Guaman-Ortiz LM, Orellana MI, Ratovitski EA. Natural compounds as modulators of non-apoptotic cell death in cancer cells. Curr Genomics. 2017;18:132–155. doi: 10.2174/1389202917666160803150639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pharmacopoeia Commission of People’s Republic of China: pharmacopoeia of the People’s Republic of China, Part 1. Beijing, China: Chinese Medical Science and Technology Press; 2015. p. 486. [Google Scholar]
- 39.Zhang H, Wu X, Wang J, Wang M, Wang X, Shen T, Wang S, Ren D. Flavonoids from the leaves of Epimedium Koreanum Nakai and their potential cytotoxic activities. Nat Prod Res. 2020;34:1256–1263. doi: 10.1080/14786419.2018.1560283. [DOI] [PubMed] [Google Scholar]
- 40.de Lartigue J, Polson H, Feldman M, Shokat K, Tooze SA, Urbe S, Clague MJ. PIKfyve regulation of endosome-linked pathways. Traffic. 2009;10:883–893. doi: 10.1111/j.1600-0854.2009.00915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.