

Abstract
Human hepatocellular carcinoma (HCC) is the most frequent cancer worldwide with a poor prognosis. Tumor-specific pyruvate kinase M2 (PKM2) is essential for cancer metabolism and tumorigenesis. Shikonin, a specific inhibitor of PKM2, but not PKM1, exhibits significant anticancer effect in HCC, and was deemed as a promising drug for cancer therapy. However, shikonin-mediated bypass signaling in HCC remained unclear. Here, we performed forward/reverse stable isotope labeling with amino acids in cell culture (SILAC)-based proteomics to identify the early molecular events controlled by shikonin. We demonstrated for the first time that shikonin could induce the nuclear translocation of PKM2 for recruiting Nrf2, and transcriptionally activated Nrf2 downstream target gene BAG3, therefore increasing protective effect to sustain cell survival. Knockdown of BAG3 by si-RNA significantly potentiated the anticancer effect of shikonin. These findings provided the first evidence of a new noncanonical function of inhibited PKM2 could act as a transcriptional coactivator of Nrf2 in cancer survival, highlight that shikonin in combined with BAG3 inhibitor could be a promising therapeutic strategy for HCC therapy.
Keywords: Shikonin, PKM2, Nrf2, BAG3, human hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) accounts for the most frequently occurring type of cancer in the world [1], its incidences and mortalities are steadily elevating and ranked sixth for cancer, as a result of chronic hepatitis virus infections and steatohepatitis caused by adiposity [2]. Chemotherapy remains the key approach for the treatment of HCC, some multikinase inhibitors, such as sorafenib, have been approved as the first-line targeted therapy for advanced HCC [3], however, the current chemotherapeutic agents remain unable to achieve a satisfactory effect on late phase HCC patients in clinic. Thus, it is an urgent need to investigate effective anticancer agents for HCC clinical therapy.
Pyruvate kinase was recognized as the final rate-limiting enzyme of cellular glycolysis, and its expression is dramatically increased in multiple tumor cells [4]. Different from PKM1, PKM2 is a key modulator in regulating the cancer metabolism by increasing glucose uptake and lactate production. More importantly, PKM2 was reported to be a transcriptional coactivator to promote the gene transcription in cancer cells, and therefore be considered to be a potential target to suppress the growth of tumor. Since PKMs are critical kinase that indispensable for normal physiological metabolism, many strategies to decrease PKM2 expression like using si-RNA or CRISP/Cas9 [5], are toxicity for normal tissue, which are not suitable for clinical cancer therapy. Therefore, identification of a new drug that inhibits PKM2 activity would be clinically meaningful.
Shikonin, a naphthoquinone that extracted from comfrey, was recently reported to be a specific inhibitor to PKM2, rather than PKM1 [6], and exhibited significant anticancer effect on cancer [7]. A study showed that shikonin could induce endometrioid endometrial cell apoptosis by controlling mTOR signaling [8]. In non-small cell lung cancer, shikonin stimulation could induce necroptosis and autophagy [9]. Current studies demonstrated that shikonin was employed as chemosensitizer to potentiate the anticancer effect of traditional chemotherapeutic agents of HCC, such as arsenic trioxide [10], doxorubicin and cisplatin [11]. These results suggested that shikonin that inhibiting PKM2 activity can achieve significant anticancer effect in multiple cancers, therefore, identification of the underling mechanism of action is critical needed for preclinical investigation of shikonin.
It has caught interest to investigate what proteins or bypass signaling pathways can be controlled by shikonin, which warrants proteome level study [12]. SILAC-based mass spectrometry is an effective technology to comprehensively quantify protein change in cell [13,14]. Advanced forward/reverse labelling strategy could significantly improve the accuracy of quantitation. In this study, our forward/reverse SILAC-proteomics and a series of experimental confirmation demonstrate that protective Nrf2-BAG3 signaling was upregulation in response to shikonin stimulation in HCC cells, suggesting BAG3 inhibition in combined with shikonin could be a novel therapeutic strategy against HCC.
Material and methods
Cell lines and reagents
Human hepatocellular carcinoma cell lines SMMC-7721 and HepG2 were obtained from Cell Bank, Chinese Academy of Sciences (Shanghai, China). Cells were routinely cultured in Dulbecco’s Modified Eagle Medium (DMEM) purchased from Gibco (NY, USA) containing 10% fetal bovine serum (FBS, Gibco, NY, USA) and 1% Penicillin/Streptomycin mixture (Solarbio, Beijing, China) at 37°C in 5% CO2 atmosphere. Shikonin with purity above 98% was purchased from Topscience (Shanghai, China) and was dissolved in DMEM. Antibodies against PARP (full and cleaved form), caspase3 (full and cleaved form), Bax, BCL-2, BAG3, Nrf2 and GAPDH were purchased from Proteintech Group (Wuhan, China), P62/SQSTM1 was purchased from Santa Cruz biotechnology (MA, USA). PKM2 was purchased from Cell Signaling Technology (Cell Signaling Technology, MA, USA).
Cell viability assay and cell morphology detection
Cell viability was evaluated by performing the cell counting kit 8 (CCK-8, Beyotime, Shanghai, China) assay. SMMC-7721 and HepG2 cells seeded in 96-well plates at a density of 4×103 cells/well respectively, were incubated with increasing dosages (0-10 μM) of shikonin for 12, 24, 36 and 48 h. After incubation, CCK-8 was added to the wells for another 1 hour. Finally, the absorbance for each sample was measured in a microplate reader (Tecan, Switzerland) at 450 nm. In addition, inverted microscope (DMi8, Leica, Germany) can be used to observe changes in cell morphology at different concentrations.
Colony formation
An amount of 5×103 SMMC-7721 and HepG2 cells seeded in 6-well plates respectively were incubated with shikonin at a concentration of 0, 1, 3 and 6 μM for about 7 days. Subsequently, all cell colonies in plates were fixed with ethanol for 5 min and stained with 1% crystal violet for another 5 min. The areas of cell colonies were counted by photoshop, and the statistical data were obtained from three independent experiments [15].
Detection of apoptosis by flow cytometry
For the detection of apoptotic cells, 2×105 SMMC-7721 and HepG2 cells seeded in 6-well plates respectively were incubated with increasing concentration (0, 2, 4, 8 μM) of shikonin for 24 hours. All cells were harvested and washed twice with PBS, 5-10×104 cells were dissolved in 500 μL binding buffer, where 5 µL Annexin V FITC and Propidium Iodide (PI) (Beyotime biotechnology, Shanghai, China) added respectively, and incubated together for 15 min in the dark. The apoptotic cells were analysis via C6 flow cytometer (BD Biosciences, San Diego, CA, USA), and the data obtained was further analyzed quantitatively by using FlowJo software (Version 7.6.5). In the statistical analysis, cell populations in the Q2 and Q3 quadrant (Annexin V+) was considered as apoptosis cells.
Cell cycle analysis
Both HCC cells were respectively seeded into the plates with a density of 2×105 and cultured overnight. Then, the cells were incubated with different concentration (0, 2, 4, 8 μM) of shikonin. Cells were harvested via centrifugation (1500 g, 10 min) and wash with PBS twice, which was fixed in cold 70% ethanol at 4°C for 24 h. Subsequently, the cells were collected in cold PBS. Before detection, all cells were incubated with prepared propidium iodide staining mixture solution containing 500 μL staining buffer, 25 μL propidium iodide staining solution and 50 μL Rnase A (Beyotime biotechnology, Shanghai, China) in the dark at 37°C for 30 min. In the end, the samples were analyzed using C6 flow cytometer (BD Biosciences, San Diego, CA, USA), and the cycle phase were furthered analyzed via Modifit software.
Western blot analysis
After designed treatment completed, the proteins of both HCC cells were extracted via RIPA lysis buffer (CST), and the concentration of each sample was determined by Bradford assay reagent (GeneCopeia, MD, USA). Equal amounts of protein lysates were electrophoresed by 10% or 12% SDS-PAGE and then transferred to polyvinylidine fluoride (PVDF) membranes (Bio-Rad, CA, USA). The PVDF membranes were blocked with 5% fresh nonfat milk for 2 h and incubated with indicated primary antibodies at 4°C overnight, followed by incubation with corresponding secondary antibodies for 1.5 h at room temperature. Ultimately, blot bands were visualized via BeyoECL Plus reagent (Beyotime biotechnology, Shanghai, China).
SILAC labeling and protein extraction
HepG2 cells were selected for proteomics analysis. First of all, HepG2 cells (2×105) were seeded in petri dish and cultured with 12C6 (light)/ 13C6 (heavy) isotope labeled lysine (Lys) SILAC DMEM medium respectively. After about six subcultures, HepG2 cells stably labeled with corresponding amino acids were obtained. When the cell density reached the appropriate density, shikonin was added in the experimental group, and the group without shikonin was set as a control. Here, we set positive and negative labeling repeat which including two groups. In detail, in one group, cells cultured with light medium were the control group and cultured with heavy medium were the experimental group. In another group, the settings of the control and experimental groups are reversed. 24 h later, the cells were collected and then lysed at low temperature for 30 min, centrifuged at 20000 g, 4°C for 20 min, the protein lysate was obtained eventually via supernatant collection. After cell concentrations were measured using an ultraviolet spectrophotometer with the Bradford method, the light/weight (12C6/13C6) isotope-labeled proteins of the experimental and control groups were mixed in a 1:1 ratio into the same tube. Then the final concentration of 15% TFA (v/v) was added to precipitate the crude protein, wash twice with acetone (-20°C), centrifuge (20000 g, 4°C) for 10 min. Protein precipitation obtained by centrifugation was dissolved in 100 mM NH4HCO3 (PH 8.0).
Peptide acquisition and HPLC separation
Trypsin is added to the collected cell protein solution in an appropriate ratio, and digested for 16 h at 37°C. Then 10 mM dithiothreitol (DTT) was added and reacted at 37°C for 1 h. Add indole-3-acetic acid (IAA) to a final concentration of 20 mM, and react in the dark for 45 min, in which process the protein was alkylated. Finally, trypsin was added again so that the ratio of trypsin to protein was 1:100 (w/w), and digestion was continued at 37°C for 4 h to complete the digestion cycle.
The trypsin digested samples were subjected to high pH reverse HPLC by Agilent 300 Extend C18 column (5 μm particles, 250×4.6 mm) to fractionate the protein samples. Initially, protein peptides were separated in a gradient of 2% to 60% acetonitrile in 10 mM ammonium bicarbonate buffer (pH = 10) within 80 min, followed by vacuum centrifugal drying.
LC-MS/MS analysis
The EASY-nLC 1000 UPLC system (Thermo Fisher Scientific, MA, USA) was used for peptide analysis using Acclaim PepMap RSLC column (Thermo Fisher Scientific, MA, USA). The composition of mobile phase A was 0.1% FA and 5% CAN while mobile phase B was 0.1% FA and 98% CAN. The obtained peptide was dissolved in 0.1% TFA, and the sample flow rate was 400 nl/min. The linear separation gradient ranged from 4% to 80% with the elution time is 40 min. All peptides were analyzed by Q ExactiveTM hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, MA, USA). The mass spectrometer (MS) was operated in a top 20 data-dependent mode to switch automatically between MS and MS/MS acquisition with a dynamic exclusion time of 20 s. The MS scanning ranges from 350 to 1800 (m/z) and the scanning resolution is 70000.
Database search and bioinformatics analysis
The raw MS data was imported in MaxQuant software program (V.1.4.1.2), which used Andromeda search engine to search the Swissprot_human database (v.3.85). The relevant parameters of MaxQuant software are set as: fixed modification: carbamyl methylation of cysteine; optional modification: acetylation of N-term and oxidation on met; fragment ion mass error is 0.02 Da; protein, peptide and modification site FDR threshold are 1%. The following bioinformatics analysis mainly including Go analysis, KEGG analysis, String analysis and so on.
Immunofluorescence
Cells were treated with shikonin (2 μM) for 24 h, fixed in 4% paraformaldehyde and permeabilized with 0.1% TrintonX-100 for 10 min before being blocked with 10% BSA for 2 h. The cells were then stained with PKM2 and Nrf2 antibodies (proteintech) according to manufacturer’s instructions. The cells were washed 3 times with 1% TBST, and counterstained with DAPI (Beyotime) to determine the cellular nucleus, followed by laser scanning confocal microscopy observation (Leica Microsystems, Wetzlar, Germany).
RNA interference
The small-interfering RNAs (siRNA) were synthesized by GenePharma (Shanghai, China) and the sequence is listed as follows: BAG3 sense: (5’-AAGGUUCAGACCAUCUUGGAA-3’ and 3’-UUCCAAGAUGGUCUGAACCUU-5’). Cells were transfected via the LipofectamineTM RNAiMAX Transfection reagent (Invitrogen, CA, USA) according to the instruction for 48 h, and then collect transfected cells for further analysis.
Tumor xenograft experiments
Female BALB/c nude mice aged 6-8 weeks were maintained under standard conditions. HepG2 cells (1×106) in 100 μL of PBS and Matrigel were subcutaneously implanted into the flanks of mice to establish tumor xenografts. When the tumor xenografts reached ~4 mm in diameter, the mice were randomly divided into treatment and control groups (4 mices/group). The treatment group received intraperitoneal injection of shikonin at a dose of 1 mg/kg every three day for 12 days, whereas the control group received the PBS only. Tumor size was measured every three days, and the tumor volume was calculated using the following equation: V = (length × width2)/2.
Statistical analysis
All statistics are expressed as means ± SD from three independent experiments, for in vitro studies, the significant differences in means are tested by two-tailed Student’s t-test. In vivo experiment is tested by one-way ANOVA analysis. The differences with *P < 0.05, **P < 0.01, or ***P < 0.001 were considered statistically significant. GraphPad Prism 8 (GraphPad, San Diego, CA) was used to perform statistical analyses and graphic processing. Survival analysis was performed using the Kaplan-Meier method with the log-rank test by IBM SPSS Statistics v.19 (SPSS Inc., Chicago, IL).
Result
PKM2 overexpression predicts poor prognosis in HCC patients
To investigate the oncogenic role of PKM2 on HCC, we analyzed the PKM2 level via using TCGA (The Cancer Genome Atlas) database, and observed that PKM2 expression was significantly upregulated in tumor tissues, as compared to para-tumor tissue (Figure 1A), in these HCC patients, PKM2 expression was elevated in stage-dependent manner (Figure 1B). To evaluate the significant contribution of PKM2 in the prognosis of HCC patients, we analyzed the relevance of PKM2 expression with overall survival and disease specific survival of patients. As shown in Figure 1C, patients with PKM2 positive exhibited worse overall survival (OS, Logrank P = 3.9e-05) and lower disease specific survival (DSS, Logrank P = 0.0026) than the PKM2 low expression group. These data highlighted an oncogenic role of PKM2 in HCC, suggesting PKM2 can develop as a promising target for clinical cancer therapy. Shikonin was previous reported as the most potent and specific inhibitors of PKM2 [6], by using molecular docking, we found that shikonin could bind to PKM2 on the surface of forming dimer or tetramer (Figure 1D), the 2D digram of shikonin and PKM2 interaction was showed in Figure 1E, shikonin can form hydrogen bonds with PKM2 at Phe26, Tyr390, Leu33, Leu353, Ala388 and Lys311.
Figure 1.
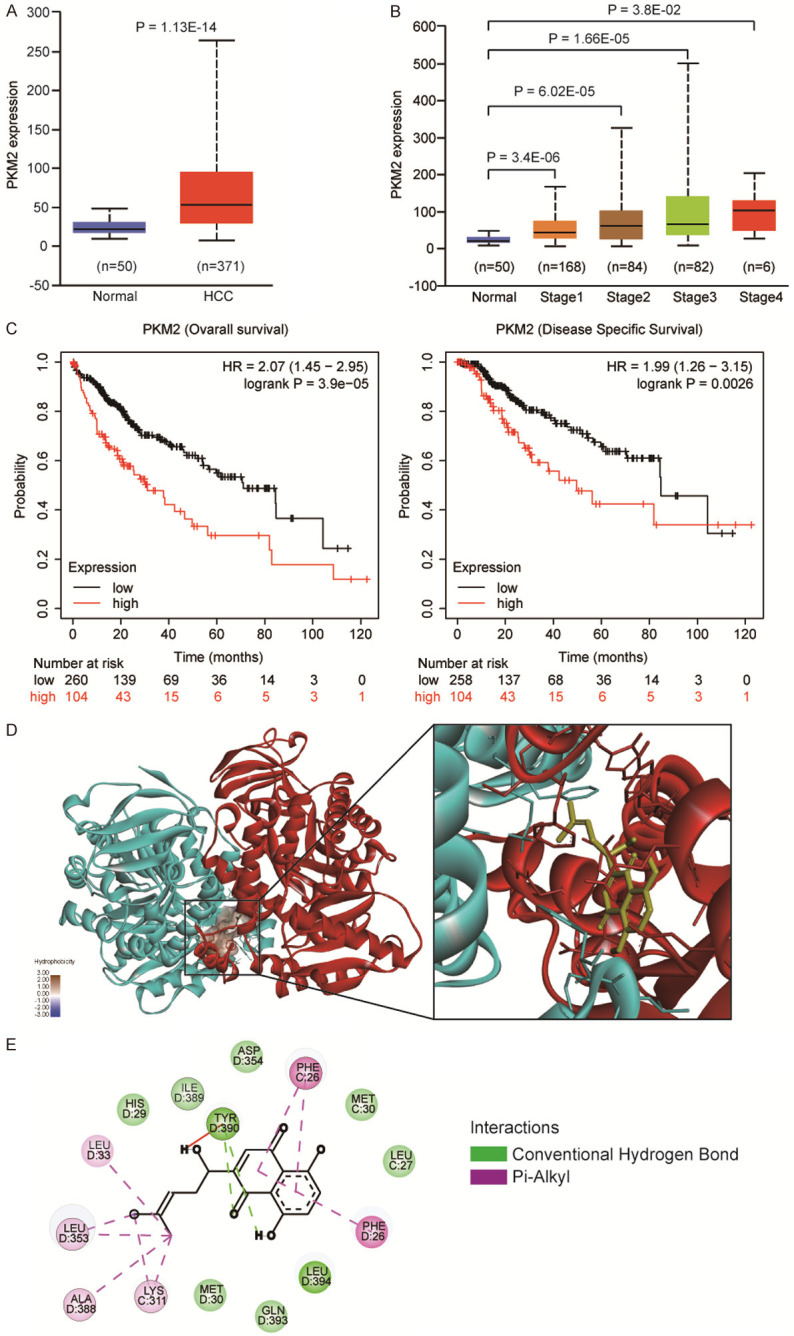
The oncogenic role of PKM2 in HCC. A. Expression level of PKM2 in TCGA HCC tumors (n = 371) compared to normal liver tissues (n = 50), were analyzed by using GEPIA (Gene Expression Profiling Interactive Analysis) webserver, showing that PKM2 was upregulated in the TCGA HCC. B. Among these patients, PKM2 expression level was positively correlated with TNM stages (normal liver samples and tumor samples classified to stage 1, stage 2, stage 3 and stage 4). C. Kaplan-Meier survival analysis showing that PKM2 overexpression was positively associated with worse Overall survival and Disease specific survival. D. The interaction of PKM2 and shikonin conducted by molecular docking. Dimerized PKM2 was coloured by cyan and red. Shikonin, yellow. The hydrophobicity of shikonin in PKM2 was showed. E. The detail interaction between shikonin-PKM2 was showed on 2D diagram. Green, conventional hydrogen bond; Pink, Pi-Alkyl interaction.
Shikonin inhibits the cell proliferation of HCC cell lines
To investigate the anti-cancer effect of shikonin in HCC, two representative HCC cell lines were then treated with increasing dosages of shikonin (0-10 µM) for 12, 24, 36 and 48 h, and the cell viability was detected by CCK8 assay. As shown in Figure 2A, shikonin suppressed the cell viability of SMMC-7721 and HepG2 cells in dose- and time-dependent manners, with IC50 values of 12.34 ± 2.03 µM and 6.82 ± 0.4 µM for 24 h treatment, and 4.28 ± 0.05 µM and 4.25 ± 0.11 µM for 48 h treatment, respectively (Tables 1, 2). Moreover, our colony formation assays were performed to detect the suppressive effect of shikonin on the cell growth of both HCC cells. As shown in Figure 2B, colony formation ability of the two HCC cells was decreased by shikonin in a dose-dependent manner. Moreover, we found that shikonin could induce cell cycle arrest in both SMMC-7721 and HepG2 cells (Figure 2C). These data suggested that shikonin inhibits the proliferation of HCC cells via triggering cell cycle arrest.
Figure 2.
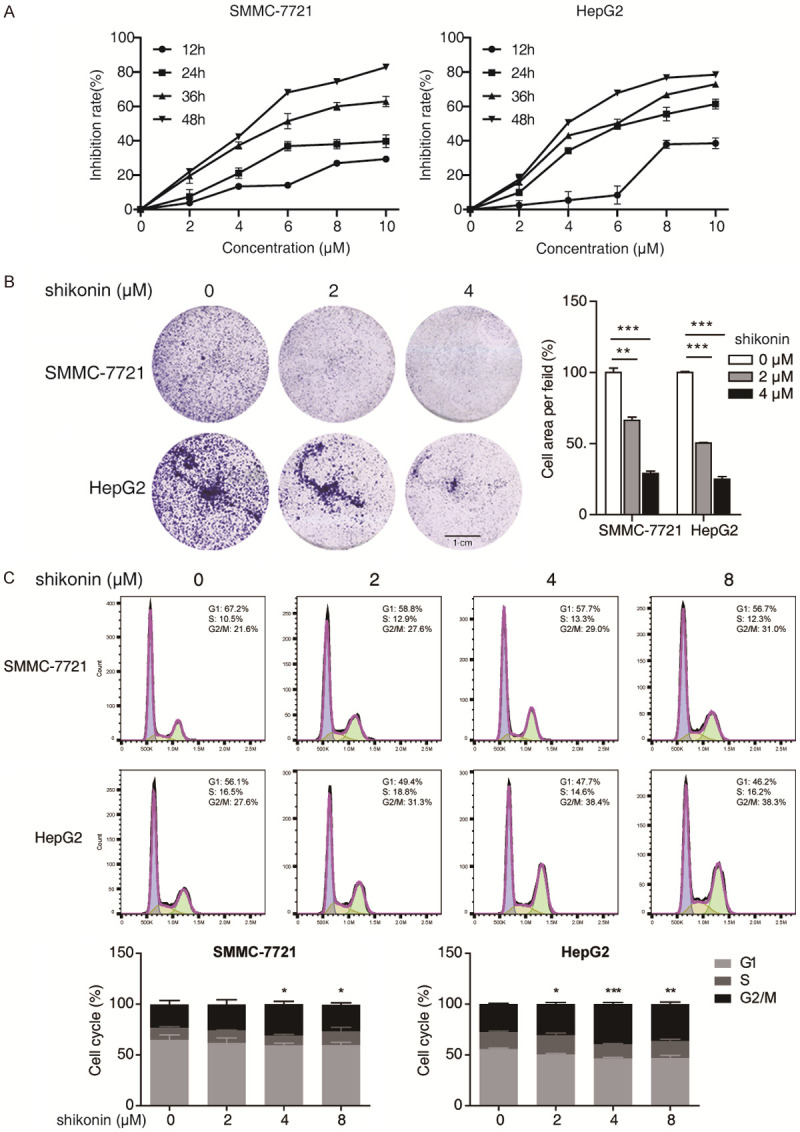
Shikonin inhibits the cell growth of HCC. A. Both SMMC-7721 and HepG2 cells were treated with increasing concentrations (0-10 μM) of shikonin for different time points (12, 24, 36, 48 h), and the cell viability was determined by CCK-8 assay. B. Both HCC cells treated with 2 and 4 μM of shikonin were compared for their abilities to form colonies; Scale bar, 1 cm. C. SMMC-7721 and HepG2 cells were treated with increasing concentrations (0-8 μM) of shikonin for 24 h, the percentages of cells in G1, S and G2/M phases were statistically presented. Mean ± SD; n = 3; *P < 0.05, **P < 0.01, ***P < 0.001 compared with DMSO-treated cells.
Table 1.
Inhibitory ratio of shikonin on SMMC-7721 cells
| Concentration (μM) | Inhibition rate (%) | |||
|---|---|---|---|---|
|
| ||||
| 12 h | 24 h | 36 h | 48 h | |
| 0 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| 2 | 3.83 ± 0.06 | 7.53 ± 3.98* | 19.53 ± 4.34** | 22.07 ± 1.33** |
| 4 | 13.47 ± 0.25** | 21.10 ± 3.01** | 37.25 ± 2.13** | 42.40 ± 1.4** |
| 6 | 14.13 ± 0.21** | 36.90 ± 2.69** | 51.30 ± 4.44** | 68.10 ± 1.35** |
| 8 | 26.95 ± 0.15** | 38.03 ± 2.65** | 60.02 ± 2.30** | 74.30 ± 1.89** |
| 10 | 29.40 ± 0.10** | 39.72 ± 3.73** | 62.83 ± 3.06** | 82.90 ± 0.89** |
indicated significant difference (P < 0.05);
indicated extremely significant difference (P < 0.01).
All concentration groups were compared with 0 μM group.
Table 2.
Inhibition ratio of shikonin on HepG2 cells
| Concentration (μM) | Inhibition rate (%) | |||
|---|---|---|---|---|
|
| ||||
| 12 h | 24 h | 36 h | 48 h | |
| 0 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| 2 | 2.42 ± 2.66 | 9.88 ± 1.55** | 15.87 ± 4.05** | 17.53 ± 3.06** |
| 4 | 5.41 ± 5.00 | 34.20 ± 1.81** | 43.00 ± 0.89** | 50.63 ± 1.01** |
| 6 | 8.40 ± 5.25 | 48.40 ± 1.44** | 50.12 ± 2.42** | 67.78 ± 0.73** |
| 8 | 37.95 ± 2.31** | 55.57 ± 3.97** | 66.76 ± 1.06** | 76.70 ± 0.69** |
| 10 | 38.55 ± 3.11** | 61.33 ± 2.86** | 72.98 ± 0.98** | 78.43 ± 0.61** |
indicated extremely significant difference (P < 0.01).
All concentration groups were compared with 0 μM group.
Shikonin induces apoptotic cell death in HCC cells via mitochondrial pathway
Since shikonin significantly decreased the cell viability of SMMC-7721 and HepG2, we further asked whether shikonin induced apoptosis in HCC cells. Initially, by investigating the morphology of both SMMC-7721 and HepG2 cells that treated with shikonin, a significant cell shrinkage and decreased cellular attachment were found (Figure 3A). Next, both SMMC-7721 and HepG2 cells were incubated with increasing dosages of shikonin (0, 2, 4, 8 µM) for 24 h, the apoptosis of HCC cells was detected by Annexin V-FITC/PI double staining assay (Figure 3B), the result showed that apoptotic rate of SMMC-7721 and HepG2 cells increased by shikonin in dose-dependent manners. Immunoblotting assay was employed and revealed that shikonin dramatically enhenced the level of cleaved-PARP and Bax, while decreased the expression of Bcl2, in a dose-dependent manner (Figure 3C), suggesting that shikonin could induce apoptotic cell death in HCC. To further determine the anticancer effect of shikonin in vivo, we established tumor xenograft model using HepG2 cells and found that intraperitoneal injection of shikonin at a dose of 1 mg/kg significantly decreased cancer growth as compared to PBS treated group (Figure 3D). These data reveal that shikonin exhibits significantly anticancer effect on HCC in vivo and in vitro.
Figure 3.

Shikonin induces the apoptosis of HCC. A. SMMC-7721 and HepG2 cells were treated with shikonin for 24 h. Cell morphology was photographed under a microscopy (40× magnification), Scale bar: 100 µm. B. SMMC-7721 and HepG2 cells were treated with shikonin for 24 h. The apoptosis rate of SMMC-7721 and HepG2 was determined by Annexin V-FITC/PI double staining assay. C. The expression of apoptosis-related proteins including pro-PARP, cleaved-PARP, Bax and Bcl2 were detected by immunoblotting. D. Nude mice bearing HepG2-derived xenografts were intraperitoneally injected either shikonin (1 mg/kg) or PBS every three days (n = 4 per group). Left, images of tumors; right, tumor curves showed that shikonin exerted a significant inhibitory effect on the growth of tumor xenografts. Mean ± SD; n = 3; *P < 0.05, **P < 0.01, ***P < 0.001 compared with control group.
Forward/reverse SILAC-based proteomic analysis of HCC cells treated with shikonin
To further investigate some novel molecular events regulated by shikonin, we performed an SILAC-based quantitative proteomics to globally profiling protein alteration in shikonin-treated HepG2 cells. The schematic diagram was shown in Figure 4A. HepG2 cells labeled with Heavy or Light chain were treated with DMSO or shikonin (2 μM) for 24 h, respectively. From two independent label-swap experiments (forward and reverse), a total of 702 proteins were identified in two experiments, among which 247 proteins were increased and 206 proteins were decreased, as analyzed by volcano plot (fold change > ± 1.2, P < 0.05, Figure 4C, Table S1). The 453 differential expressed proteins (DEPs) were submitted to IPA analysis to study the biological progresses that participated in the anticancer effects of shikonin in HCC. As shown in Figure 4D, shikonin-regulated proteins were remarkably enriched in several biological progresses including Nrf2-mediated oxidative stress response and protein ubiquitination pathway. Network analysis showed that BAG3 was the hub protein in the shikonin-driven network that significantly upregulated (Figure 4E). The SILAC-based quantitative proteomic result indicated that Nrf2-mediated BAG3 expression is the critical event in HCC in response to shikonin stimuli.
Figure 4.
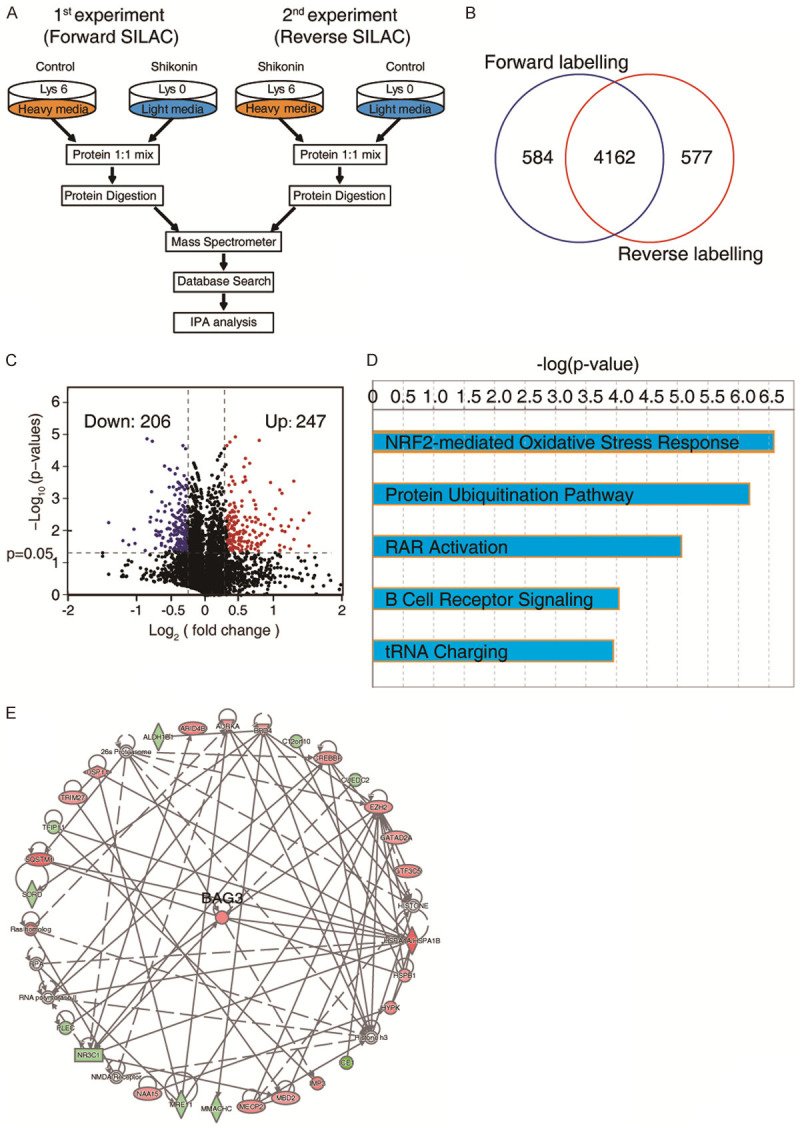
SILC-based proteomics identifies shikonin-regulated proteins. A. Experimental flow chart of the identification of shikonin regulated proteins. B. Venn diagram of the number of proteins that identified in forward and reverse experiment. C. Volcano plots of DEPs in shikonin treatment in HCC cells. D. DEPs were subjected to the IPA analysis, and the top five canonical pathways were generated according to the P values. Note that the shikonin regulated proteins were mainly enriched in Nrf2 mediated oxidative stress response. E. Network analysis from IPA showed that BAG3 is the hub protein that upregulated in shikonin regulated proteins.
Nrf2-BAG3 signaling is adaptively response for shikonin stimuli in HCC
According to the SILAC-quantitative proteomic data, shikonin dramatically induced Nrf2-signaling activation in HCC cells, which promoted the cellular adaptive response. We next performed western blotting to confirm the expression of Nrf2 in HCC cells treated with shikonin. Our data demonstrated that Nrf2 were significantly increased in the nuclear section of HCC cells treated with shikonin (Figure 5A). Previous study reported a noncanonical function of PKM2 as a transcriptional coactivator of Nrf2, which recruits Nrf2 for detoxification in cell [16]. Here, we also observed that the nuclear translocation of PKM2 in HCC cells were enhanced by shikonin stimuli. In addition, we found that shikonin treatment could enhance the interaction between Nrf2 and PKM2 (Figure 5B). To further confirm this result, we performed confocal assay and found that colocalization and nuclear translocation of Nrf2 and PKM2 were found in HCC cells treated with shikonin (Figure 5C), suggesting shikonin not only suppresses the activity of PKM2, but also induces nuclear translocation of PKM2 for recruiting Nrf2. As a downstream target of Nrf2, BAG3 was up-regulated in mRNA and protein level after shikonin treatment in dose-dependent manner (Figure 5D and 5E), suggesting that Nrf2-BAG3 signaling is response for shikonin stimulation in HCC.
Figure 5.
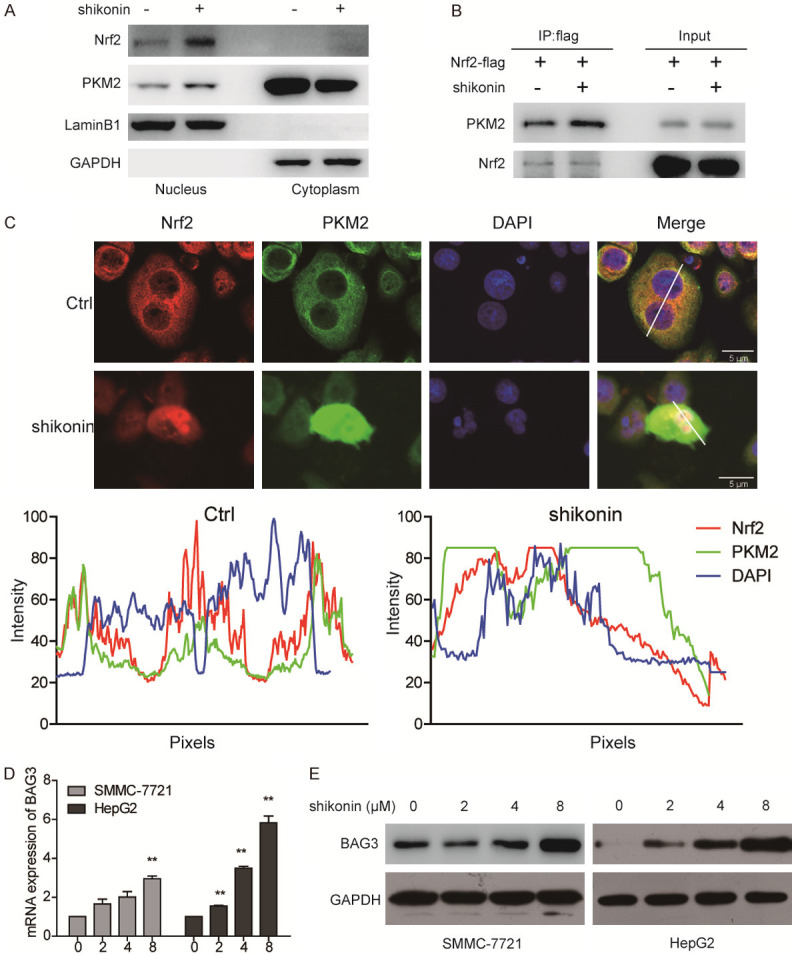
Shikonin induces BAG3 expression by enhancing the interaction of Nrf2 and PKM2. (A) HepG2 cells were treated with shikonin for 24 h, the nuclear and cytoplasmic fractions were isolated for western blot analysis. Lamin B and GAPDH was used as the marker for nuclear protein and cytoplasmic fraction, respectively. (B) HepG2 expressing Nrf2-flag was treated with shikonin (2 μM) for 24 h, the interaction of PKM2 and Nrf2 was determined by co-immunoprecipitation using anti-flag antibody. (C) HCC cells treated with shikonin (2 μM) for 24 h, the cellular localization of PKM2 and Nrf2 was determined by confocal assay. (D, E) Both SMMC-7721 and HepG2 cells were treated with indicated dosages of shikonin for 24 h, the mRNA of BAG3 was determined by qRT-PCR (D), and the protein expression of BAG3 was determined by immunoblotting (E). Bars, mean ± SD; n = 3; **P < 0.01, compared with DMSO-treated cells.
Knockdown of BAG3 enhances the anticancer effect of shikonin
Since the results above indicated that shikonin induced the upregulation of BAG3, we further asked whether blockade of BAG3 can augment the anticancer effects of shikonin. As shown in Figure 6A, both HCC cells with BAG3 knockdown showed lower cell viability after increasing shikonin treatment. We next treated HCC cells with shikonin in the presence or absence of the si-BAG3, and then detected by colony formation assay and cell cycle assay. As shown in Figure 6B, 6C, knockdown of BAG3 decreased cell proliferation and induced cell cycle arrest at G1 phase, and this anticancer effect was significantly augmented by the treatment of shikonin in both HCC cells. Moreover, we also observed that knockdown of BAG3 expression enhanced the cancer cell apoptosis induced by shikonin (Figure 6D), as indicated by Annexin V-FITC/PI double staining assay and expression for cleaved-PARP, Bax and Bcl2 (Figure 6E), suggesting a synergistic effect of shikonin stimulation and BAG3 suppression.
Figure 6.

Knockdown of BAG3 enhanced the anticancer effect of shikonin in HCC cells. (A) SMMC-7721 and HepG2 cells were transfected with siRNA against BAG3 or scramble siRNA (si-Con) for 24 h, and cells were treated with increasing dosage of shikonin (0-10 µM), and the cell viability was detected by CCK-8 assays. (B, C) SMMC-7721 and HepG2 cells were transfected with siRNA against BAG3 or scramble siRNA (si-Con) for 24 h, the BAG3 knockdown groups were treated with shikonin for 24 h, and the cell abilities to form colonies was determined by colony formation (B), and cell cycle was detected by flow cytometry (C). The apoptotic cells were determined by Annexin V-FITC/PI double staining assay (D). Mean ± SD; n = 3; *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar, 1 cm. (E) The expression of apoptosis-related proteins including pro-PARP, cleaved-PARP, Bax and Bcl2 were detected by immunoblotting.
The oncogenic role of BAG3 in HCC
It seems that BAG3 play a protective role in HCC in response to shikonin mediated cancer suppression. We further investigate the clinical significance of BAG3 on HCC by using TCGA database, and observed that BAG3 was increased in HCC tissues, as compared to para-tumor tissue (Figure 7A), in these HCC patients, BAG3 expression was elevated in stage-dependent manner (Figure 7B). To evaluate the significant contribution of BAG3 expression in the prognosis of patients with HCC, we analyzed the relevance of BAG3 expression and overall survival and disease specific survival of patients, respectively. As shown in Figure 7C, patients with high PKM2 expression exhibited worse overall survival (OS, Logrank P = 0.017) and lower disease specific survival (DSS, Logrank P = 0.042) than the PKM2 low expression group. These data highlight an oncogenic role of BAG3 in HCC, suggesting inhibition of BAG3 in combinated with PKM2 inhibitor is a promising strategy for clinical cancer therapy of HCC.
Figure 7.

The expression of BAG3 on HCC. A. Expression level of BAG3 in TCGA HCC tumors (n = 371) compared to normal liver tissues (n = 50), were analyzed by using GEPIA webserver, showing that BAG3 was upregulated in the TCGA HCC. B. Among these patients, BAG3 expression level was positively correlated with TNM stages (normal liver samples and tumor samples classified to stage 1, stage 2, stage 3 and stage 4). C. Kaplan-Meier survival analysis showing that BAG3 overexpression was positively associated with worse Overall survival and Disease specific survival. D. Schematic diagram of the apoptotic pathway induced by shikonin.
Discussion
This study firstly discovered a novel mechanism of PKM2 suppression induced by specific inhibitor shikonin, by which shikonin could induced PKM2 nucleus translocation for recruiting Nrf2, the activated Nrf2 transcriptionally promoted the expression of downstream target gene BAG3, which plays a protective role in HCC. We thus proposed inhibition of BAG3 could increase the anticancer effect of shikonin on HCC (Figure 7D).
HCC is an emerging problem worldwide due to the rising incidences and lack of effective preventive drugs. Therefore, the investigation for new anticancer agents that are attracted a great of interest. Natural products extracted from medicinal plants are critical sources for chemotherapeutic agent discovery [17,18]. For example, camptothecin, a quinoline type of alkaloid isolated from Camptotheca acuminata (Nyssaceae), was found to exhibit strong anticancer effect by targeting nuclear DNA topoisomerase I, resulting DNA damage in cancer cell [19]. Shikonin is the major bioactive component isolated from the dried plant roots of Lithospermum erythrorhizon, which was previously been found to exert significant anticancer effects on various carcinomas. For example, shikonin could induce necrosis or apoptotic cell death in gastrointestinal cancer by generating reactive oxygen species [20]. It could suppress the metastatic activity of breast cancer cells through reducing the promoter activity of activator protein-1 (AP-1) that suppressed MMP-9 transcription [21]. In this study, we found that shikonin not only induced cell cycle arrest at G2/M phase, but also trigger cell apoptosis in HCC, suggesting shikonin can be a potential anticancer compound for HCC therapy.
It is well recognized that PKM2 has an critical function in boosting cancer cell metabolism and proliferation by providing major energy source for cancer [22]. Inhibition of PKM2 expression by siRNA could dramatically reduce the tumor growth, therefore, PKM2 could be a promising target for cancer therapy. Shikonin was reported to be a specific inhibitor that binds to PKM2, but not PKM1 [6]. We here further demonstrated that shikonin could bind to the surface of PKM2 dimer or tetramer by forming hydrogen bonds with PKM2 at Phe26, Tyr390, Leu33, Leu353, Ala388 and Lys311. To further investigate the cellular molecular events caused by shikonin in HCC, SILAC-based quantitative proteomics were employed to characterize the shikonin-regulated protein change in HCC, and then evaluated the high relevant signaling pathways guided by the proteomic. Our data demonstrated that shikonin treatment could activate Nrf2-BAG3 signaling to protect cancer cell from cytotoxicity, as evidenced by suppression of BAG3 using siRNA could dramatically enhance the anticancer effect of shikonin. Nrf2 is a transcription factor that can enhance adaptive protection gene expression, to offset cellular oxidative and proteotoxic stress [23]. Recently study reported a new noncanonical function of PKM2 as a transcriptional coactivator of Nrf2, to generate sufficient GSH for ROS detoxification [16]. Interestingly, our study novelly found that PKM2 suppressed by shikonin could translocate to nucleus, and recruit Nrf2 for initiating downstream target gene BAG3 expression, an anti-apoptotic protein that sustaining cellular homoeostasis [24]. Therefore, we proposed abolishment of BAG3 function in HCC could enhance the anticancer effect of shikonin.
Multifunctional protein BAG3 is constitutively expressed in various tumor types, which plays an essential role in sustaining cell survival in cancer cells [25]. In glioblastoma, BAG3 was reported to bind with Bax in cytoplasm, and inhibited its translocation to mitochondria, therefore preventing cells from apoptosis [26]. The anti-apoptosis activity of BAG3 was evidenced by its involvement of ERK1/2-dependent pathway and IKKα transactivation in castration-resistant prostate cancer cells [27]. Our results showed that shikonin could induced BAG3 expression via Nrf2 signaling, which exerts protect role in offsetting the antitumor effect of shikonin.
In conclusion, this study revealed that shikonin possesses strong anticancer activity via inducing apoptotic cell death in SMMC-7721 and HepG2 cells. On the other hand, shikonin induces the PKM2-mediated Nrf2 transactivation to activate Nrf2-BAG3 signaling for sustain the survival of HCC cells. This finding reveals noncanonical function of PKM2-Nrf2-BAG3 signaling in shikonin treatment, highlighting BAG3 inhibition in combined with shikonin may be a potential antitumor strategy against HCC.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81760500 and 31360459) and the Natural Science Foundation of Guangxi Province (No. 2017GXNSFAA198362 and NO. 2018GXNSFDA050009). China Postdoctoral Science Foundation (2018M643372, 2020T130252), Postdoctoral Fund of the First Affiliated Hospital, Jinan University (801325), Guangdong Natural Science Research Grant (2019A1515010196, 2019A1515110597) and The Fundamental Research Funds for the Central Universities (11619303).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Liu T, Li S, Wu L, Yu Q, Li J, Feng J, Zhang J, Chen J, Zhou Y, Ji J, Chen K, Mao Y, Wang F, Dai W, Fan X, Wu J, Guo C. Experimental study of hepatocellular carcinoma treatment by shikonin through regulating PKM2. J Hepatocell Carcinoma. 2020;7:19–31. doi: 10.2147/JHC.S237614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paur J, Valler M, Sienel R, Taxauer K, Holzmann K, Marian B, Unterberger A, Mohr T, Berger W, Gvozdenovich A, Schimming J, Grusch M, Grasl-Kraupp B. Interaction of FGF9 with FGFR3-IIIb/IIIc, a putative driver of growth and aggressive behaviour of hepatocellular carcinoma. Liver Int. 2020;40:2279–2290. doi: 10.1111/liv.14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng J, Dai W, Mao Y, Wu L, Li J, Chen K, Yu Q, Kong R, Li S, Zhang J, Ji J, Wu J, Mo W, Xu X, Guo C. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1alpha/PPAR-gamma/PKM2-mediated glycolysis. J Exp Clin Cancer Res. 2020;39:24. doi: 10.1186/s13046-020-1528-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zahra K, Dey T, Ashish , Mishra SP, Pandey U. Pyruvate kinase M2 and cancer: the role of PKM2 in promoting tumorigenesis. Front Oncol. 2020;10:159. doi: 10.3389/fonc.2020.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen X, Song X, Yue W, Chen D, Yu J, Yao Z, Zhang L. Fibulin-5 inhibits Wnt/beta-catenin signaling in lung cancer. Oncotarget. 2015;6:15022–15034. doi: 10.18632/oncotarget.3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011;30:4297–4306. doi: 10.1038/onc.2011.137. [DOI] [PubMed] [Google Scholar]
- 7.Chandimali N, Sun HN, Kong LZ, Zhen X, Liu R, Kwon T, Lee DS. Shikonin-induced apoptosis of colon cancer cells is reduced by peroxiredoxin V expression. Anticancer Res. 2019;39:6115–6123. doi: 10.21873/anticanres.13819. [DOI] [PubMed] [Google Scholar]
- 8.Huang C, Hu G. Shikonin suppresses proliferation and induces apoptosis in endometrioid endometrial cancer cells via modulating miR-106b/PTEN/AKT/mTOR signaling pathway. Biosci Rep. 2018;38:BSR20171546. doi: 10.1042/BSR20171546. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Kim HJ, Hwang KE, Park DS, Oh SH, Jun HY, Yoon KH, Jeong ET, Kim HR, Kim YS. Shikonin-induced necroptosis is enhanced by the inhibition of autophagy in non-small cell lung cancer cells. J Transl Med. 2017;15:123. doi: 10.1186/s12967-017-1223-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song J, Zhao Z, Fan X, Chen M, Cheng X, Zhang D, Wu F, Ying X, Ji J. Shikonin potentiates the effect of arsenic trioxide against human hepatocellular carcinoma in vitro and in vivo. Oncotarget. 2016;7:70504–70515. doi: 10.18632/oncotarget.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin SP, Fako V, Dang H, Dominguez DA, Khatib S, Ma L, Wang H, Zheng W, Wang XW. PKM2 inhibition may reverse therapeutic resistance to transarterial chemoembolization in hepatocellular carcinoma. J Exp Clin Cancer Res. 2020;39:99. doi: 10.1186/s13046-020-01605-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Zhang J, Li B, He QY. Advances of proteomics in novel PTM discovery: applications in cancer therapy. Small Methods. 2019:3. [Google Scholar]
- 13.Wang Y, Yu RY, Zhang J, Zhang WX, Huang ZH, Hu HF, Li YL, Li B, He QY. Inhibition of Nrf2 enhances the anticancer effect of 6-O-angeloylenolin in lung adenocarcinoma. Biochem Pharmacol. 2017;129:43–53. doi: 10.1016/j.bcp.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Zhang J, Li B, He QY. Proteomic analysis of mitochondria: biological and clinical progresses in cancer. Expert Rev Proteomics. 2017;14:891–903. doi: 10.1080/14789450.2017.1374180. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Sun Y, Zhong LY, Yu NN, Ouyang L, Fang RD, Wang Y, He QY. Structure-based discovery of neoandrographolide as a novel inhibitor of Rab5 to suppress cancer growth. Comput Struct Biotechnol J. 2020;18:3936–3946. doi: 10.1016/j.csbj.2020.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei Y, Lu M, Mei M, Wang H, Han Z, Chen M, Yao H, Song N, Ding X, Ding J, Xiao M, Hu G. Pyridoxine induces glutathione synthesis via PKM2-mediated Nrf2 transactivation and confers neuroprotection. Nat Commun. 2020;11:941. doi: 10.1038/s41467-020-14788-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao C, Zheng K, Li Y, Xu H, Kang Q, Fan L, Hu X, Jin Z, Zeng Y, Kong X, Zhang J, Wu X, Wu H, Liu L, Xiao X, Wang Y, He Z. Gypenoside L inhibits autophagic flux and induces cell death in human esophageal cancer cells through endoplasm reticulum stress-mediated Ca2+ release. Oncotarget. 2016;7:47387–47402. doi: 10.18632/oncotarget.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Zhou Y, Li N, Liu WT, Liang JZ, Sun Y, Zhang WX, Fang RD, Huang SL, Sun ZH, Wang Y, He QY. Curcumol overcomes TRAIL resistance of non-small cell lung cancer by targeting NRH: quinone oxidoreductase 2 (NQO2) Adv Sci (Weinh) 2020;7:2002306. doi: 10.1002/advs.202002306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beretta GL, Gatti L, Perego P, Zaffaroni N. Camptothecin resistance in cancer: insights into the molecular mechanisms of a DNA-damaging drug. Curr Med Chem. 2013;20:1541–1565. doi: 10.2174/0929867311320120006. [DOI] [PubMed] [Google Scholar]
- 20.Lee MJ, Kao SH, Hunag JE, Sheu GT, Yeh CW, Hseu YC, Wang CJ, Hsu LS. Shikonin time-dependently induced necrosis or apoptosis in gastric cancer cells via generation of reactive oxygen species. Chem Biol Interact. 2014;211:44–53. doi: 10.1016/j.cbi.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 21.Jang SY, Lee JK, Jang EH, Jeong SY, Kim JH. Shikonin blocks migration and invasion of human breast cancer cells through inhibition of matrix metalloproteinase-9 activation. Oncol Rep. 2014;31:2827–2833. doi: 10.3892/or.2014.3159. [DOI] [PubMed] [Google Scholar]
- 22.Liu M, Zhang Z, Wang H, Chen X, Jin C. Activation of AMPK by metformin promotes renal cancer cell proliferation under glucose deprivation through its interaction with PKM2. Int J Biol Sci. 2019;15:617–627. doi: 10.7150/ijbs.29689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Zhang J, Huang ZH, Huang XH, Zheng WB, Yin XF, Li YL, Li B, He QY. Isodeoxyelephantopin induces protective autophagy in lung cancer cells via Nrf2-p62-keap1 feedback loop. Cell Death Dis. 2017;8:e2876. doi: 10.1038/cddis.2017.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kogel D, Linder B, Brunschweiger A, Chines S, Behl C. At the crossroads of apoptosis and autophagy: multiple roles of the co-chaperone BAG3 in stress and therapy resistance of cancer. Cells. 2020;9:574. doi: 10.3390/cells9030574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Marco M, Basile A, Iorio V, Festa M, Falco A, Ranieri B, Pascale M, Sala G, Remondelli P, Capunzo M, Firpo MA, Pezzilli R, Marzullo L, Cavallo P, De Laurenzi V, Turco MC, Rosati A. Role of BAG3 in cancer progression: a therapeutic opportunity. Semin Cell Dev Biol. 2018;78:85–92. doi: 10.1016/j.semcdb.2017.08.049. [DOI] [PubMed] [Google Scholar]
- 26.Festa M, Del Valle L, Khalili K, Franco R, Scognamiglio G, Graziano V, De Laurenzi V, Turco MC, Rosati A. BAG3 protein is overexpressed in human glioblastoma and is a potential target for therapy. Am J Pathol. 2011;178:2504–2512. doi: 10.1016/j.ajpath.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ammirante M, De Laurenzi V, Graziano V, Turco MC, Rosati A. BAG3 is required for IKKalpha nuclear translocation and emergence of castration resistant prostate cancer. Cell Death Dis. 2011;2:e139. doi: 10.1038/cddis.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.