

Summary
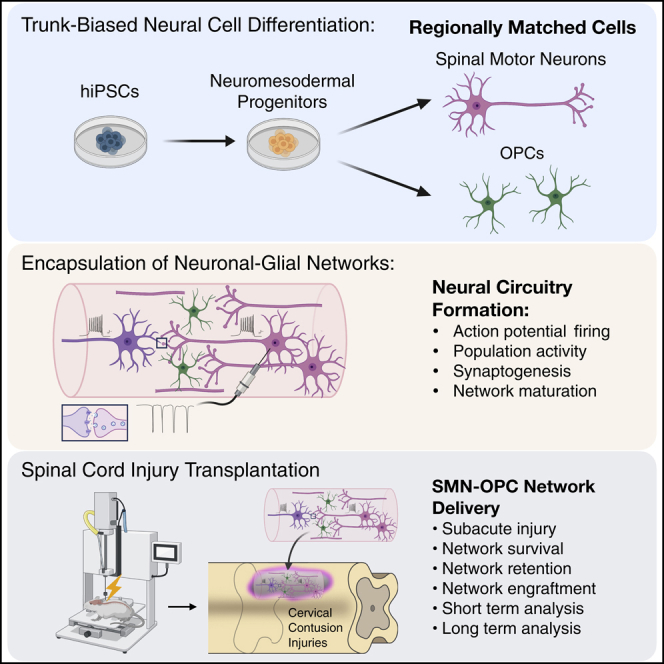
To repair neural circuitry following spinal cord injury (SCI), neural stem cell (NSC) transplantation has held a primary focus; however, stochastic outcomes generate challenges driven in part by NSC differentiation and tumor formation. The recent ability to generate regionally specific neurons and their support cells now allows consideration of directed therapeutic approaches with pre-differentiated and networked spinal neural cells. Here, we form encapsulated, transplantable neuronal networks of regionally matched cervical spinal motor neurons, interneurons, and oligodendrocyte progenitor cells derived through trunk-biased neuromesodermal progenitors. We direct neurite formation in alginate-based neural ribbons to generate electrically active, synaptically connected networks, characterized by electrophysiology and calcium imaging before transplantation into rodent models of contused SCI for evaluation at 10-day and 6-week timepoints. The in vivo analyses demonstrate viability and retention of interconnected synaptic networks that readily integrate with the host parenchyma to advance goals of transplantable neural circuitry for SCI treatment.
Subject areas: Neuroscience, Bioengineering, Tissue engineering
Graphical abstract
Highlights
-
•
Neuromesodermal progenitor derivation of human spinal neurons as therapeutic cells
-
•
Neural ribbons bridge in vitro network formation and in vivo host transplantation
-
•
In vivo visualization of encapsulated graft placement with magnetic resonance imaging
-
•
Six-week viability of human neuronal networks with OPCs in rat contusion SCI
Neuroscience; Bioengineering; Tissue engineering
Introduction
Stem cell technologies remain at the forefront for therapeutic treatment of SCI, with the potential to address some of the complex spatiotemporal parameters surrounding injuries that include neuronal and glial cell death, destruction of axons, inflammation, and variations in anatomical context. Multipotent neural stem cells provide a pathway to neurogenesis as well as paracrine signaling that support regeneration and plasticity (Yu et al., 2012). However, once transplanted, neural stem cells are largely unregulated in cell division and differentiation, which creates stochastic outcomes that are a barrier to clinical implementation (Assinck et al., 2017). Nevertheless, stem cell technologies are evolving rapidly for SCI including realization of developmental principles to better anatomically match delivered therapeutic neural cells (Olmsted and Paluh, 2021). The benefits of a neurodevelopmental approach are already being realized in groundbreaking advances to treat Parkinson’s disease (Barker et al., 2017). By focusing on a specific population of dopaminergic neurons in the substantia nigra of the midbrain that are affected (DeMaagd and Philip, 2015), benefits for robust engraftment, in vivo neuronal maturation, and circuit repair are being realized (Kikuchi et al., 2017; Xiong et al., 2021). This approach has not yet been applied to treat acute traumatic insults, such as in SCI (Silva et al., 2014) or stroke (Baker et al., 2019), in which the spectrum of severity, anatomical distribution, and breadth of neural circuits affected within a single subject and between subjects will likely require patient-customized neuronal network therapies.
Neuromesodermal progenitors are the embryological source of neurons and glia in the spinal cord (Henrique et al., 2015), and are expected to be an ideal source for deriving therapeutic cells and networks for SCI. Spinal cord developmental principles and corresponding implementation into stem cell research has enabled the generation of cell types that are regionally matched to the site of injury (Gouti et al., 2014; Sagner and Briscoe, 2019). Importantly, the caudal neuromesodermal progenitor-derived cells represent the appropriate bifurcated developmental lineage for spinal neurons (Nedelec and Martinez-Arias, 2021; Wind et al., 2021). This is a relatively recent finding and may be relevant even for neural stem cell studies in which the past use of a default CNS pathway with anterior telencephalic identity may have had an impact (Dulin et al., 2018; Kumamaru et al., 2018). With up-to-date CNS developmental information applied to stem cell protocols, the reinvestigation into use of neurons in therapeutics for SCI is warranted. Combined with biomaterials (Katoh et al., 2019), there is a need to generate and test customized 3D neuronal networks for SCI as well as for a variety of in vitro and in vivo biomedical applications.
We previously developed an improved alginate-based methodology for neural ribbon co-encapsulation of homotypic spinal cord neural stem cells and the microenvironment regulator chondroitinase ABC, designed to facilitate shipment of cells and remote SCI transplantation (Olmsted et al., 2020). Encapsulation of neural stem cells can provide neuroprotection of transplanted cells to improve survival (Tsintou et al., 2015; Ahuja and Fehlings, 2016; Liu et al., 2018; Olmsted et al., 2020). We extend this technology here as a platform for neural network formation in vitro. In this study, we generate neural ribbons of synaptically connected neuronal networks composed of functionally maturing caudal spinal motor neurons (SMNs), interneurons, and oligodendrocyte progenitor cells (OPCs) for in vitro analyses and demonstrate neuron retention in vivo following transplantation in a rat cervical hemicontusion SCI model. Our SMN-OPC neural ribbon-encapsulated networks demonstrate high synaptic density and the output of stereotypic firing in response to glutamate stimulation.
When transplanted into the subacute phase hemicontusion injury microenvironment (15 days after injury) and analyzed by biomarker immunohistochemistry of spinal cord sections 10 days and 6 weeks after transplantation, we observe robust survival and engraftment. The presence of reduced cell numbers (~5,000 cells per animal) in neural ribbon grafts (versus traditional suspension methods that deliver up to 40x more cells) allows us to readily evaluate graft survival of OPCs, neurons, and retained synapses. Human neurons and OPCs graft along the parenchyma that borders the host injury cavity, intercalated with host tissue and co-localized with synaptic biomarkers. This study is the first to investigate the formation of pre-constructed human neuronal networks in an encapsulated ribbon format and use of these in a rat SCI model. We achieve neuron-OPC graft survival and integration without the use of additional injury site modifiers, such as chondroitinase ABC.
Results
Differentiation of hiPSC-derived neuromesodermal progenitors to caudal SMNs with interneuron and glial subpopulations
SMNs were derived from the African American hiPSC line F3.5.2 (Chang et al., 2015; Tomov et al., 2016) in three stages referred to here as (1) induction, (2), patterning, and (3) neuronal maturation according to a protocol that we previously described (Figure 1A) (Olmsted et al., 2020). The hiPSCs were first used to generate trunk-biased neuromesodermal progenitors that are the developmental origin of the spinal cord in vivo (Figures 1B–1D) (Gouti et al., 2014). Neuromesodermal progenitors were differentiated to spinal cord neuroectoderm using 100 nM retinoic acid and 200 nM Hh-Ag1.5, a potent sonic hedgehog agonist. Further exposure to retinoic acid and Hh-Ag1.5 in N2B27 basal medium generates motor neuron progenitors at high efficiency that mature to SMNs (Figure 1E). Over the course of differentiation, SMNs expressed a panel of canonical biomarkers including NKX6-1 (day 17), ISL1&2 (day 25), FOXP1 (day 28), HB9 (Mnx1, day 35), and choline acetyltransferase (ChAT, day 35) by immunofluorescence (Figure 1F). The differentiated cells also expressed generic biomarkers of neuronal compartments such as somatodendritic (MAP2), axonal (SMI312), and the pre-synaptic terminal (Synapsin 1, or SYN1). A list of all biomarkers used and their significance is provided (Table S1).
Figure 1.

Differentiation of trunk-biased neuromesodermal progenitors to SMNs with interneuron and glia subpopulations
(A) Motor neuron differentiation protocol.
(B) Phase contrast image of F3.5.2 hiPSC colony in pluripotency medium.
(C) Induction of hiPSCs to SOX2/Bra neuromesodermal progenitors shown at day 2 with merged image and individual channels.
(D) Induction of hiPSCs to neuromesodermal progenitors shown at day 2 (phase contrast) and day 4 (CDX2/SOX2).
(E) Phase contrast images of motor neuron progenitors (day 22) and SMNs (day 30).
(F) Panel of canonical SMN biomarkers during neuronal differentiation.
(G) Subpopulations of spinal interneurons in day 58 SMN differentiating cultures shown with CHX10 and PAX2 counterstained with TUJ1.
(H) Histogram quantification of spinal neuron subtypes including ventral-dorsal interneurons in differentiating SMN cultures by immunofluorescence. Each biomarker is denoted according to the developing spinal cord neuronal domains along the ventral-dorsal axis (Sagner and Briscoe, 2019).
(I) CD44/GFAP astrocytes in day 58 SMN differentiating cultures.
(J) Encapsulation of differentiating SMNs in hydrogel to form neural ribbons. SMN biomarkers ChAT/TUJ1 and V2a interneuron biomarkers CHX10/TUJ1 shown with high magnification image (right). Data are reported as mean ± SEM. Scale bars are 50 μm.
Ventral and dorsal spinal interneurons and astrocytes that promote synaptogenesis are indispensable to normal neuronal circuit function in vivo. Since ventral spinal interneurons are patterned similarly to SMNs during development, we investigated the co-differentiation of both ventral and dorsal spinal interneurons in SMN cultures (Figures 1G and 1H), as well as astrocytes (Figure 1I). We provide representative images for CHX10 and PAX2 (Figure 1G) and quantified the proportion of nine generic subtypes of spinal neurons in day 58 cultures by immunofluorescence that are matched to the following biomarkers (Figure 1H): Nkx-2.2 (V3, ~2%), FOXP1+ISL1 (MN, ~52%), CHX10 (V2a, ~12%), GATA-3 (V2b, < 1%), PAX2 (V0/V1 or dI4/dI6, ~8%), LBX1 (dI4-dI6, 0%), TLX3 (dI3, ~1%), LHX9 (dI1, 0%), and BRN3A (sensory, ~25%). Low levels of LBX1, TLX3, and LHX9 in the presence of peripherin expression indicate that BRN3A+ cells were neural crest-derived sensory neurons that can be patterned through neuromesodermal progenitors (Frith et al., 2018). GFAP/CD44 astrocytes were not observed in SMN cultures until after day 40, shown here at day 58 (Figure 1I). In adherent cultures, INs developed in close proximity with SMNs, and this configuration was retained within neural ribbon-encapsulated constructs (Figure 1J). We describe neural ribbon encapsulation of differentiating neuronal cultures in the subsequent section (Figure 2). Neural ribbons generated by hydrogel encapsulation prior to day 40 do not contain astrocytes because these cells are not yet differentiated. We refer to the SMN cultures as simply “SMNs” throughout the manuscript.
Figure 2.

Neural ribbon encapsulation of a spinal neuron network
(A) Cartoon diagram of SMN encapsulation in hydrogel neural ribbons and extracellular matrix embedding platform for imaging studies (glass bottom dish). CSPG components in collagen embedding matrix can constrain axodendritic growth unidirectionally.
(B) Phase contrast images of 1.5% RGD-functionalized alginate-based ribbons formed by extrusion into 100 mM CaCl2crosslinking solution (left). Long ribbons adopt concentric configurations (middle-top) and have circular cross-sections (middle-bottom). Extrusion-based technique enables high-density encapsulation of neural cells in RGD-Col hydrogel ribbons.
(C) Directed anisotropic SMN axonal configurations templated in neural ribbon platform. Ribbon stained with SMI312 (green), SYN1 (red), DAPI (blue). DIC image is shown.
(D) SMN phenotyping within neural ribbons reveals retained expression of FOXP1 (left) and ChAT (right). Ribbons counterstained with TUJ1 and DAPI.
(E) Glutamate receptor scaffolding protein PSD-95 expression in adherent SMN cultures (left) and co-localization with SYN1 in neural ribbons (right) at day 10 post-encapsulation. Additional NCAM-1 stain highlights cell boundaries and adhesion. Scale bars are 50 μm.
SMNs encapsulated within hydrogels generate neural networks in ribbons
We applied an alginate hydrogel-based platform to reproducibly generate neural ribbons of encapsulated differentiating SMN cultures (Olmsted et al., 2020, previously designed for use with neural stem cells). Here, we further optimized neural ribbon methodologies in order to direct and image neuronal and glial cytoarchitecture configurations (Figure 2). Alginate biopolymer crosslinking as hydrogels included addition of Type 1 collagen as exogenous extracellular matrix to establish interpenetrating networks (IPNs) (Figure 2A). Sodium alginate (1.5% in NaCl) was further functionalized with bioactive peptide RGD that was used to promote cell adhesion and cell-cell interactions within neural ribbons (RGD-Alginate + Collagen, or RGD-Col). For immobilization, long-term culture and imaging, the extruded ribbons (Figure 2B) were rapidly formed by crosslinking in 100 mM CaCl2, rinsed, and embedded in fast-gelling collagen solution in the viewing area of a glass bottom dish. Cell-free and cell-laden ribbons each retained the circular cross-section and diameter of the needle tip template (60, 100, or 150 μm inner diameter) in CaCl2, but swelled by ~33% in culture medium. Neurite extension within 60 μm neural ribbons was constrained along the ribbon longitudinal axis by the addition of 4 μg/mL Aggrecan (chondroitin sulfate proteoglycan, CSPG) within the embedding medium. High CSPG concentrations can be inhibitory to axon penetration. Day 20 motor neuron progenitors were encapsulated as small triturated neurospheres (Figure 2C) or larger intact aggregates (Figure 2D) at 1 × 108 cells/ml concentration equivalent and cultured for up to 12 days. Cultures were transitioned to neuronal maturation medium at day 25 according to our protocol (Figure 1A). We achieved neurite anisotropic longitudinal alignment within neural ribbons and observed expression of lateral motor column SMN phenotypic biomarkers FOXP1 and ChAT, that co-localized with TUJ1 (Figure 2D). We detected pre- and post-synaptic proteins using SYN1 and PSD-95 biomarkers, respectively, as well-distributed puncta by day 10 after encapsulation (Figure 2E). Neural cell borders were visualized by co-staining with NCAM-1.
Encapsulating SMNs and OPCs into neural ribbons as purified co-cultures
OPCs facilitate neuronal damage recovery after SCI (Assinck et al., 2017), but are not co-differentiated with high efficiency by SMN protocols. We therefore generated hiPSC-derived OPCs and sorted them for co-culture with SMNs prior to neural ribbon co-encapsulation (Figure 3). OPC differentiation proceeds through spinal cord neuroectoderm (PAX6) and the motor progenitor spinal domain (Nestin/OLIG2), but is then specified by exposure to growth factors IGF1, FGF2, and PDGF-AA as well as thyroid hormone triiodothyronine (T3) at day 20 (Figure 3A) (Khazaei et al., 2017). By omitting DAPT during the first five days of differentiation, OPCs are generated as indicated by Nkx-2.2 expression that defines the developing ventral OPCs, and ultimately the OPC-specific antigen O4 (Figure 3B). Efficiency of OPC differentiation was assessed by quantifying expression of the relevant biomarker versus DAPI at each important time point as follows: PAX6 (day 6, 1,023/1,1115 cells, 92%), OLIG2 (day 20, 879/977 cells, 90%), NKX-2.5 (day 25, 1,142/1,313 cells, 87%), O4 (day 36, 593/824 cells, 72%). To ensure the greatest reproducibility of in vivo data after CNS neural ribbon delivery, two additional purification stems were used; that is, magnetic cell sorting (MACS) separation of OPCs with the O4 antigen (Figures 3C and 3D), and SMN neurosphere suspension culture. For encapsulation, OPCs were generated from hiPSCs expressing GFP under the CAG promoter (Taylor et al., 2006). Enriched GFP-OPCs were retained and mixed 1:5 with SMNs and co-cultured for 1 week in modified N2B27 bundling medium (Figure 3E). At this point, SMN-OPC co-cultures were encapsulated within 60 μm RGD-Col neural ribbons and embedded for longer term maintenance and imaging using GFP and SMI312 immunofluorescence, shown at day 7 after encapsulation (Figure 3F).
Figure 3.

Encapsulating purified SMN-OPC co-cultures
(A) Overview of differentiation protocol used to generate OPCs through neuromesodermal progenitor (NMP) and then multipotent spinal cord neural stem cell (scNSC) stages. Small molecules and growth factors used in caudo-ventral patterning and maturation are listed.
(B) Immunofluorescence images of OPC differentiation. Left-to-right: Pax6 (day 6); Nestin/OLIG2 (day 20); Nkx-2.2 (day 25); O4 (day 36). Cells counterstained with DAPI (D, nuclei).
(C) GFP-OPC MACS separation scheme (magnetic cell sorting). GFP-OPCs were isolated from differentiating mixed cultures using magnetic beads functionalized with O4 antibody. Enriched GFP-OPCs were plated for further differentiation or seeded onto SMN cultures.
(D) Adherent GFP-OPCs after MACS enrichment.
(E) GFP-OPC co-culture with SMNs prior to encapsulation.
(F) SMN-OPC connected networks were encapsulated within neural ribbons as a dual-component system. GFP and SMI312 immunostaining of neural ribbons is shown at day 7 post-encapsulation. High magnification image provided on right. Scale bars are 50 μm, and 10 μm in high magnification panel in (D).
Retained action potential firing of spinal neurons recovered from neural ribbons
Neural ribbon alginate hydrogels provide a platform for neural network formation and delivery in vivo and in cultured SMNs. To evaluate recovery and further maturation of SMNs following encapsulation, we released cells from encapsulation after 48 hr and then maintained neurons in the presence or absence of neurotrophic factors (NTFs) using slow release PLGA microbeads (Figure 4A). For encapsulation, 23 day motor neuron progenitors were passaged as aggregates and encapsulated in 60 μm neural ribbons (ProNova alginate + collagen, or NovaCol) lacking RGD peptide, or were used to generate motor neuron progenitor neurosphere cultures without encapsulation for comparison. Progenitor neural ribbons and neurospheres were cultured in suspension for 48 hr. On day 25, motor neuron progenitor aggregates were recovered from neural ribbons by dissolving alginate in 1.6% sodium citrate and seeded onto Matrigel-coated cover glass in parallel with non-encapsulated neurospheres. Cultures were further subject to SMN terminal differentiation and maintained up to day 52, analyzed at two general timepoints in differentiation that are days 32–35 and days 45-52. We provide examples of phase images of neuronal cultures (Figure 4B) and corresponding SMN biomarkers (Figure 4C) at the time that the whole-cell, patch clamp recordings were performed. Representative traces for the two timepoints are provided (Figure 4D). The recordings, in current-clamp mode, allowed us to analyze the passive and active membrane properties of SMNs at two different timepoints (total range: days 32-35 and 45-52) in naive conditions, in the presence of NTFs and in the presence of neural ribbon encapsulation (Figure 4E). The passive membrane properties (i.e. resting membrane potential, membrane resistance, membrane capacitance; Figure 4E, top) are not mediated by voltage- or ligand-gated ion channels. They describe how current flows through the membrane and how the membrane voltage responds to sub-threshold current step injections. The resting membrane potential ranged between −36 mV at d33-35 and −46 mV at d32-34 with neural ribbons (∗p = 0.023 by ANOVA). The membrane resistance (which describes how a current input can change the cell's voltage) differed between d32–34 and d51-52 only in the presence of NTFs (∗p = 0.048). The cell capacitance (which describes the ability of a cell to store electrical charges) was similar across all groups. These values are consistent with those obtained by Takazawa et al. (2012) working on SMNs derived from human embryonic stem cells (Takazawa et al., 2012). Since the membrane resistance varies with the surface density of potassium leak channels, and the cell capacitance varies with the cell surface area, these findings indicate that the different culturing conditions do not exert a major and consistent interference with the expression of potassium leak channels and with cell size regulation in these cells.
Figure 4.

Neural ribbons preserve action potential firing properties
(A) Illustrated timeframe. Differentiating SMN cultures were encapsulated into neural ribbons (NR) for 48 hr, then recovered and further matured with neurotrophic factors (NTFs) that were BDNF and GDNF before patch clamp analysis. Current clamp recordings were performed at two time points (days 32–35 and days 45-52) across N = 3-4 differentiating cultures per condition (‘n’ denotes total number of cells recorded, whereas ‘N’ represents the number of culture batches used for these experiments).
(B) NR-encapsulated SMNs (left); adherent SMNs after collection from neural ribbons and subsequent culture (right images).
(C) SMN hallmark biomarkers at the day of recording (day 35) by immunofluorescence. Scale bars for panels (C, D) are 50 μm.
(D) Differentiating SMN cultures were encapsulated into neural ribbons for 48 hr, then recovered and further matured with BDNF and GDNF before obtaining whole-cell patch clamp recordings. The traces show the voltage responses to somatic current step injections (500 ms long) at d33-35 (left) and d45-52 (right). DC current injections were used to maintain the SMNs at −65 mV.
(E) Characterization of passive and active membrane properties of different types of SMN cultures. The passive membrane properties included a measure of the resting membrane potential (Vrest), membrane resistance (Rm), and membrane capacitance (Cm). Vrest ranged between −36 mV at d33-35 and −46 mV at d32-34 with neural ribbons (∗p = 0.023 by ANOVA). Rm differed between d32–34 and d51-52 only in the presence of NTFs (∗p = 0.048). The active membrane properties included the rheobase, action potential threshold (APthr) and action potential peak (APpeak). Rheobase decreased at d32-34 after neural ribbon encapsulation, and was associated with a hyperpolarization of the AP threshold (∗p = 0.047), which also occurred between d32–35 and d45-52 SMN groups (∗p = 0.032). AP peak was reduced between these SMN groups (∗p = 0.039). A similar effect was also detected between d32–34 and d51-52 cultures treated with growth factors (∗p = 0.047). As a consequence, the AP amplitude was slightly reduced at d51-52 in the presence of growth factors (∗p = 0.048).
(F) f/I plots illustrating the firing output of SMNs in the experimental conditions described above, for 500 ms-long current step injections of increasing amplitude. The overall excitability of these SMNs did not show statistically significant differences across different types of culturing conditions. The recordings were obtained from N = 3-4 differentiating cultures per condition (‘n’ denotes total number of cells recorded, whereas ‘N’ represents the number of culture batches used for these experiments). Data represent mean ± SEM.
To study the active membrane properties of SMNs, we used DC current injections to maintain each cell at −65 mV, and applied short (5 ms) current steps to measure the rheobase, action potential (AP) threshold and peak (Figure 4E, bottom). On average, neurons fired APs in response to suprathreshold current injections, but not in all cases. The rheobase (i.e. the minimum current step amplitude required to evoke an AP) decreased at d32-34 after neural ribbon encapsulation. This effect was associated with a hyperpolarization of the AP threshold (∗p = 0.047), which also occurred between d32–35 and d45-52 SMN groups (∗p = 0.032). Together, the decreased rheobase and lower AP threshold suggest that neural ribbons increase the excitability of SMNs only in young cultures, not in older ones. This maturation trend has also been observed in developing SMNs (Gao and Ziskand-Conhaim, 1998). The AP peak was reduced between these SMN groups (∗p = 0.039), pointing to a potentially smaller driving force for Na+ (i.e. more hyperpolarized ENa) or different activation voltage for potassium channels after neural ribbon encapsulation. A similar effect was also detected between d32–34 and d51-52 cultures treated with growth factors (∗p = 0.047). As a consequence, the AP amplitude was slightly reduced at d51-52 in the presence of growth factors (∗p = 0.048).
SMNs develop the ability to fire repetitively in response to sustained suprathreshold current steps as they mature. For this reason, we investigated whether a similar change occurred as SMNs matured, depending on the conditions in which they were maintained (Figure 4F). On average, SMNs fired multiple APs in response to suprathreshold, 500 ms long depolarizing current steps. The f/I plots showed similar trends across all groups, indicating that SMNs share the same ability to respond to sustained current injections, with pronounced accommodation among consecutive APs.
Spinal neurons within 3D neural ribbons generate glutamate-responsive firing
We employed a microelectrode array (MEA) system in conjunction with calcium imaging to interrogate neuronal activity in neural ribbons (Figure 5). SMNs were encapsulated in RGD-Col neural ribbons at day 28 in differentiation and maintained in terminal differentiation medium. At day 35, we performed non-adherent, acute recordings in three separate neural ribbons positioned over hexagonal arrays of MEA electrodes and immobilized under a coverslip. Recordings were acquired before and after addition of 50 μM glutamate. Spiking activity was observed along the neural ribbon length, and was increased by glutamate stimulation in four of six electrodes (Figures 5A and 5B). Spike raster plots in the two most active electrodes (C1, D4) are provided (Figure 5C), indicating the time point of glutamate addition. To assess the degree of connectivity within the neural ribbons, we quantified both intra-electrode bursting activity in addition to network burst (NB) events and the percentage of spikes localized within a network burst (Figure 5D). The percent of spikes contained within NBs ranged from ~50 to 70%, where average NB duration was 1,011 ms. As additional validation of SMN spontaneous firing in neural ribbons, we performed live-cell calcium imaging with Fluo-4 AM on encapsulated neurons (60 μm RGD-Col), in which linear arrangements of cells were visible (Figure 5E). We also followed calcium signaling in aggregates that retain some of the neural ribbon shape during and after rapid alginate dissolution in 1.6% sodium citrate (Figure 5F) and observed retained rhythmic oscillations of somatic calcium transients (Figure 5G) with observable network bursting activity (Video S1).
Figure 5.

Glutamate-responsive functional activity in neural ribbons
(A) Left: schematic of neural ribbon recordings on hexagonal MEA array. Orange circles represent active electrodes. Right: histogram is mean firing rate (MFR, Hz) recorded in cell culture medium or medium supplemented with 50 μM glutamate.
(B) Phase contrast images (day 35) corresponding to (A).
(C) Spike rasters of electrodes with highest firing rate before and after addition of 50 μM glutamate.
(D) Quantification (N = 3 neural ribbons) of burst rate (Hz; t = 3.143, df = 4, ∗p = 0.0348), number of NBs per recording (NB; t = 7.000, df = 4, ∗∗p = 0.0022), connectivity (% spikes in NB; t = 8.212, df = 4, ∗∗p = 0.0012), and mean NB duration under spontaneous conditions versus 50 μM glutamate stimulation (t = 6.012, df = 4, ∗∗p = 0.0039). pvalues were determined with unpaired two-tailed ttest.
(E) Fluo-4 AM signal in 60 μm SMN neural ribbon (ImageJ Royal LUT).
(F) Suspended neural aggregates recovered from 1.6% sodium citrate dissolution of alginate (top) and plating on fresh substrate (bottom, day 2).
(G) Temporal oscillation of calcium transients in soma and neurites within plated aggregate from (F) plotted as ΔF/F. Scale bars 100 μm in (B), 50 μm in (E, F).
Neuron-loaded nanoparticles in neural ribbons allows tracking of in vivo linear placement
To optimize delivery and placement strategies with neural ribbons for the current study and to benefit future studies that may deliver multiple ribbons, we tested use of superparamagnetic iron oxide (SPIO) nanoparticles for tracking by magnetic resonance imaging (Figures 6A and 6B). To track neural ribbon constructs and cells, we labeled day 29 neuronal cultures with SPIOs (Figure 6C). Cultures were incubated overnight and neuron viability versus SPIO concentration was quantified to be at least ~80% (Figure 6D). Day 30 cultures labeled with ~100 μg Fe/1 x 106 cells (40 μL Miltenyi FeraTrack SPIO/2 mL culture medium) were encapsulated with Fluorescein-Dextran (500 kDa) for optical visualization (Figure 6E), and shipped overnight to Houston, TX. SPIO-Fluorescein neural ribbon segments (3-4 mm length) containing day 32 neurons (~5,000 cells) were injected first into collagen phantoms and linearly placed as visualized by stereo microscopy (Figure 6F). To extend this technique to the rat CNS, we delivered individual SPIO-labeled day 32 neural ribbon segments to the healthy spinal cord (cervical level C4). The spinal column was imaged by ultra-high field micro MRI within 24 hr after transplantation (Figures 6G–6I). One sagittal plane (Figure 6G) and an axial series (Figure 6H) are provided as raw and processed images. Processed images display the T2 map computed from the gradient-echo signal (~T2∗ values) using a bone-range enhancer color map. In processed images, the implanted ribbon is visible in bright color. The so-called ribbon tip, neck and tail are visible in the axial series wherein the entry point of the neural ribbon tip occurs in the dorsal spinal cord and extends ~1.58 mm into ventral white matter. This positioning was further validated by measurements taken from the coronal plane (Figure 6I) and provides a rapid means for optimizing injections for future strategies such as with multiple neural ribbons.
Figure 6.

Delivery and linear placement of neural ribbons by injection in vitro and in vivo
(A and B) Experimental overview and timeline for pilot in vivo transplantation studies. (A) SMN differentiation, neural ribbon encapsulation, and shipping for rat in vivo delivery. (B) SPIO-labeling and tracking. SPIO-labeled cultures were encapsulated at day 30 and shipped overnight. At day 32, neural ribbons were injected into collagen phantoms and the healthy rat spinal cord for MRI.
(C) Unlabeled (left) and SPIO nanoparticle-labeled (middle) SMN cultures at day 30 validated with Prussian blue stain (right).
(D) Neuron viability versus SPIO concentration after 24 hr incubation.
(E) Day 30 unlabeled (left; 10× magnification) and SPIO-labeled (middle; 20× magnification) SMN cultures encapsulated in neural ribbons with Fluorescein-Dextran (right).
(F) ~4 mm SPIO-Fluorescein neural ribbon segment injected into collagen phantom. Inverted LUT is shown.
(G) Sagittal plane raw (left, ribbon dark) and ~T2∗ processed (right, ribbon light) images depict neural ribbon transplant (tip, neck, tail regions; solid red arrows) and apparent direction of injection (dashed red arrow).
(H) Axial planes of raw (left, ribbon dark) and processed (right, ribbon light) images from FOV in (G), highlighting ribbon tip, neck and tail. The neural ribbon tip is located at the dorsal position, while neck and tail regions extend across white matter for ~1.58 mm.
(I) Coronal plane views of neural ribbon neck and tail. Scale bars are 50 μm, and 3 mm in (F). Data represent mean ± SEM. Scale bars in C and E are 50 microns, and 3 mm in (F).
Transplanted SMN-OPC networks retain synaptic clusters and exhibit OPC distribution along the parenchymal border of the host injury cavity
We delivered hiPSC-derived cervical SMNs and OPCs into the contused C4 SCO cavity as encapsulated neural ribbon networks (~5,000 cell dose per animal) (Figures 7 and 8). The encapsulation was performed as in Figure 3, but the ratio of OPCs to SMNs was increased from 1:5 in that experiment to 1:2 due to potential increased susceptibility of OPCs to cell death in the injury site. Neural co-cultures were encapsulated in 150 μm neural ribbons (Figure 7A) and shipped overnight on ice from Albany, NY to Houston, TX. The time of neural ribbon formation to injection was approximately 48 hr. Viability pre- and post-shipment was quantified by Live/Dead of intact neural ribbons and Trypan Blue exclusion of collected cells (Figure S1). The well-characterized, reproducible rat model of cervical SCI has been previously described (Mondello et al., 2015). A right-sided C4 hemi-contusion injury is made using an electromagnetic spinal cord injury device (ESCID) in female Long-Evans rats. At 15 days after injury, rats received cervical intraspinal injections of single neural ribbon segments 3-4 mm in length (1 × 108 cells/ml ribbon concentration, ~5,000 cell dose per animal) (N = 6 animals) (Figure 7B). Grafts were successfully placed in all 6 animals. Animals were sacrificed at 10 days after transplantation (N = 3), or at 6 weeks after transplantation (N = 3) (Figure 7C). Encapsulated SMN-OPC networks transplanted in neural ribbons showed evidence of robust survival and engraftment along the injury cavity border at both time points (Figures 7D, 7E, 8A, and 8B). The GFP-OPCs became more highly ramified and were efficiently distributed within the transplant and in host tissue with SMNs that retained robust synapses (SYN1). Graft neuronal fibers (TUJ1) were intercalated with host fibers. In sections where a portion of the ribbon appeared to remain in the cavity, GFP-OPCs and neurons remained viable at 6 weeks with dense SYN1 expression by immunostain (Figure 8C). Cavalieri analysis was used to analyze the volume of the residual lesion cavity and neural ribbon grafts at 6 weeks after transplantation (Figures 8D and 8E). Every 5 sections (40 μm thick), the area of the cord, lesion cavity, and graft (STEM121) were determined and volumes were calculated (N = 3 spinal cords analyzed). The volumes (mm3) for individual (N = 3) grafts and the corresponding residual lesions are provided in addition to the means.
Figure 7.

Histological evidence for SMN-OPC encapsulated network engraftment along the injury cavity border
(A) Neural ribbon shipped for in vivo delivery.
(B) Schematic of encapsulated SMN-OPC neural network ribbon graft (left) and of neural ribbon transplantation into the hemicontusion injury site (right).
(C) Schematic overview of SMN-OPC cervical hemicontusion grafting experiment (N = 6 animals). Animals were sacrificed 10 days (N = 3) or 6 weeks (N = 3) after transplantation.
(D) Tile scan image of SMN-OPC neural ribbon graft at 6 weeks after transplantation. Merged image is shown with STEM121 (human cytoplasm), GFP-OPCs, and GFAP (reactive gliosis).
(E) Histological evidence for neural ribbon survival and engraftment along the injury cavity border at 6 w. Merged images shown with STEM121, GFP-OPC and Synapsin 1 (SYN1). Individual scale bars are provided. Panel E top left is 500 microns, bottom left is 100 microns (lbot image) and both right side images are 500 microns (bottom right).
Figure 8.

Further characterization of SMN-OPC encapsulated network grafts
(A) Neural ribbon grafts 10 days after transplantation immunostained with STEM121 and SYN1 shown with GFP-OPCs and DAPI demonstrates early grafting along the border of the injury cavity.
(B) High magnification images at 6 weeks after transplantation robust engraftment and viability at contusion injury cavity border. Top: Merged image shown with STEM121, GFP-OPC, and TUJ1. Bottom: Merged image shown with STEM121, GFP-OPC, and GFAP. GFP-OPCs increased in number from day 10 and show evidence of ramification.
(C) Neural ribbon cross section (150 μm diameter) inside of the injury cavity 6 weeks after transplantation with high-density SYN1 puncta. Individual scale bars are provided in all image panels.
(D and E) Cavalieri volumetric analysis of the residual lesion and neural ribbon grafts. Color matching corresponds to the same animal. Shown are residual lesion versus graft volume for individual spinal cords at 6 weeks (mm3) (D), and mean ± SEM (E). N = 3 animals were sacrificed at 6 weeks after transplantation.
Discussion
Cell therapy approaches that focus on neural circuits and incorporate advanced stem cell derivation technologies have been largely unexplored. We have generated encapsulated human synaptically-connected spinal neuron networks that combine appropriately patterned SMNs, spinal interneurons and OPCs within a neural ribbon delivery platform. Transplanted neural circuits in vivo as ribbons retain synapses, and effective integration with host tissue is facilitated. This study expands our previous work with encapsulation technology for neural stem cells (Olmsted et al., 2020) to support maturing neurons and neuronal-glial co-populations as electrically-active, glutamate-responsive constructs. We apply trunk-biased neuromesodermal progenitors (Gouti et al., 2014; Lippmann et al., 2015), which are the natural origin of spinal neurons in the embryo (Henrique et al., 2015) to generate caudal hiPSC-derived SMNs and interneurons (Davis-Dusenbery et al., 2014; Sances et al., 2016; Trawczynski et al., 2019) as well as the OPCs. This is the first study to demonstrate that human neurons and their neuronal networks can be encapsulated, matured and transplanted into a SCI non-modified microenvironment and survive for a substantial timeframe of 6 weeks after transplantation.
The CNS neural circuitry for spinal cord regeneration in adulthood is regionally complex and results in challenges that span repair after injury to restoration of neural connectivity and function (Sofroniew, 2018). Neural stem cells are a traditional and prominent therapeutic choice of cell type. Studies with fetal spinal cord grafts that contained a mixture of neural stem cells, developing neurons and glia, and other non-neural cell types provided the first evidence for safety and feasibility of CNS injury repair with transplanted material (Reier et al., 1986; Bregman et al., 1993). Stem cell technologies now underlie the bulk of cell-based transplantation studies for CNS injury repair, typically using multipotent neural stem cells to differentiate into needed circuitry components such as neurons, astrocytes, and oligodendroglia (Assinck et al., 2017). However, reproducibility with neural stem cells has been a challenge. The complex cytokine-releasing injury microenvironment impacts the balance between mitotically active, dividing cells versus signaled differentiation, maturation and integration into host tissue. The use of neurotrophic factors and neuroprotective measures can increase neural stem cell viability and, therefore, time available for differentiation (Katoh et al., 2019); however, stochastic variability in outcomes using neural stem cells remains a barrier in addition to tumorigenicity concerns (Deng et al., 2018).
To this end, neurons for the specified injury type and location are now being generated and considered to benefit therapeutic investigations. A recent study (Dulin et al., 2018) demonstrated successful graft connectivity with descending supraspinal host axons into progenitor grafts of anatomically matched neural cells suited for that injury site. In addition, interneuron transplantation to intervene in animal models of autism (Southwell et al., 2020) and neuropathic pain (Manion et al., 2020) have shown therapeutic benefits, although these are not injury site studies. Here, we generate ex vivo predefined neuronal networks using advanced stem cell differentiation protocols. These regionally matched caudal SMNs and OPCs network effectively in alginate neural ribbons and when transplanted exhibit robust long-term viability and engraftment. Pre-defined neural ribbon circuitry is an alternate strategy for transplanting neural cells in SCI. The primary method currently used for therapeutic grafting in the rodent spinal cord is delivery of high-dose dissociated neural stem cells in suspension (~200,000-2 million cells/animal), with the expectation that a substantial but unpredictable percentage of these cells will not survive or integrate. The stochastic nature of neural stem cell differentiation in the injury site as well leads to unpredictable differentiated phenotypes. In Olmsted et al. (2020), our neural ribbon alginate system was tested for the survival of small grafts containing ~5,000 neural stem cells. The encapsulated neural stem cells survived in the host, although the same small number of unprotected cells injected as a cell suspension into the host did not survive. In this current study, the engrafted spinal neurons and OPCs delivered as neural networks into the injury cavity remain viable. Importantly, the neural ribbons retain aspects of the pre-formed neural networks and the ribbon favors alignment along the injury edge, facilitating integration with host tissue. Neural network ribbons are stable shipping platforms as well that benefit collaborative efforts. We have scaled ribbon size from 60 to 150 microns that is within the constraints of nutrient and gas exchange for encapsulated cells (Raof et al., 2011; Takei et al., 2016). In the current study, the injury cavity size exceeds that of the small grafts used and so may require future experiments to fill the injury cavity. Importantly, we observed that neural ribbon grafts containing ~5,000 cells as immature neural networks delivered to the subacute phase contused injury cavity are retained and align along the edge of the injury site (Figures 7 and 8). We further demonstrate that neural ribbon linear placement and retention can be tracked using nanoparticles and MRI in the healthy spinal cord, which can be used in the continued investigations to explore technical aspects of transplantation of multiple ribbons to match cavity size dimensions.
Future transplantation therapies are expected to benefit by increased customization of delivered neural circuits that can be further developed. In this study, we focus primarily on developing the neural network ribbons and have characterized neural ribbon networks by immunofluorescence, live-cell calcium imaging, patch clamp electrophysiology, and MEAs in vitro to interrogate action potential firing, calcium signaling, population activity, and synaptogenesis. Studies included neuromesodermal progenitor-derived adherent spinal neuron cultures and in 3D neural ribbon constructs. Spontaneous firing and neurite calcium transients were observed as part of the evidence for synaptic network formation, as well as the ability of newly developed neurons to form active synapses. We observed that the three-dimensionality of ribbons improves network formation, as has been observed as well for 3D neuronal aggregates (Izsak et al., 2019) and neuromuscular trunk organoids (Faustino Martins et al., 2020) that have enhanced connectivity. In our 3D neural network ribbons, network bursts (NBs) were observed under glutamate stimulation and ~50-70% of spikes were contained within the NB, a parameter used to assess network maturation (Figure 5). We observed synaptogenesis between spinal neurons within neural ribbons, identified by immunofluorescence of pre-synaptic and post-synaptic scaffolding proteins Synapsin-1 and PSD-95, respectively (Figure 2E).
Limited studies have been done with neurons to determine the extent to which developing versus electrophysiologically mature neurons provide the greatest advantage for survival and host integration after transplantation. By whole-cell current clamp electrophysiology, we observed statistically significant differences in passive membrane properties such as resting membrane potential and input resistance, but not cell capacitance across conditions (Figure 4). High input resistance and partially depolarized resting membrane potentials are characteristic of immature SMNs (Gao and Ziskand-Conhaim, 1998; Takazawa et al., 2012) and were observed in our cultures in all conditions. Cell excitability properties changed between non-treated young and old cultures. In young cultures, they changed in the presence of neural ribbons. However, these effects did not result in significant differences in the firing output of SMNs in response to prolonged activation. The use of slow-release BDNF and GDNF neurotrophic factors did not have a discernable effect, which may be due to the action of these factors to drive neurogenesis and maintain viability, but not necessarily to promote electrophysiological maturation. As well, there may be a requirement for cycling or pulse of growth factors versus sustained concentration by the slow release beads that we apply. In general, the requisite factors that are needed for neuronal electrophysiological and morphological maturation are yet unknown. However, appropriate density of the requisite ion channel repertoire (Telezhkin et al., 2018) and other external factors (Zabolocki et al., 2020) contribute. By existing differentiation protocols, stem cell-derived SMNs resemble embryonic rather than postnatal or adult counterparts (Gao and Ziskand-Conhaim, 1998; Takazawa et al., 2012; Ho et al., 2016). In other neuronal cell therapeutic treatments, such as for Parkinson's disease (Kikuchi et al., 2017; Xiong et al., 2021) and diabetes mellitus (Tremmel et al., 2019), the transplantation of immature progenitors or terminal cell types that are then driven to maturity by the native in vivo microenvironment is observed. By morphological, functional, and biomarker analyses, we selected day 32 SMNs for downstream experiments and transplantation.
In summary, this study is unique as a first-of-its-kind initiative to test the feasibility of generating transplantable neuronal networks of electrically active, synaptically connected grafts pre-constructed in vitro in an encapsulated form. Further, we go on to demonstrate host-cavity edge interactions, even with low cell numbers. The transplanted neural network ribbons can survive and engraft within the subacute phase SCI cavity in vivo for an extended 6-week period. We expect neural ribbon constructs to also be highly useful when further developed in vitro for optimization of neural circuitry to benefit basic neuroscience and drug discovery. This study constitutes an important first step toward a neural-circuitry focused effort to provide greater control over directed delivery and repair of functional networks that support survival and engraftment in the injured CNS.
Limitations of this study
With the evolution of neurodevelopmental stem cell technologies, the re-evaluation of post-mitotic neurons in vivo is occurring and bringing with it accompanying technological challenges for in vitro and in vivo studies. This current study focused on in vitro generation and comprehensive characterization of neural circuitry and demonstrated qualitatively in vivo delivery and survival. However, future in vivo experimentation is expected to bring the technology to fruition.
The neural ribbon system has the advantage to allow faster, reproducible optimization of cell types and ratios to transplant for SCI, such as SMNs versus inhibitory and/or excitatory interneurons. In addition to SMN neuronal subtypes, the post-injury microenvironment is also known to impact oligodendroglial viability. As such, we included OPCs along with the post-mitotic SMNs to supply the potential for reconnection of NMJs and for supporting of motor neurons before they exit the spinal cord to enter the PNS. Such a cell directed approach to neural circuitry allows incredible flexibility for future studies. For example, additional spinal neuron types could be tested, such as for excitatory interneurons that are thought to play a role in functional recovery, or at different stages of maturation. It remains unknown which specific population(s) of spinal neuron subtypes and glia, and which ratios, will be important for maximizing different types of functional recovery. In the human SCI site, the unpredictable injury size and distribution across individual patients will result in damage to distinct populations of cells. Therefore, although not tested here, we anticipate that customization of regional neural populations may bring greater consistency in functional outcomes. This system allows for that customization. Future work is needed to fully demonstrate that the spinal cord regional match will lead to broader circuit matching and repair.
The survival and integration of human SMN networks with OPCs constitutes an important step toward customized neural circuitry with therapeutic potential. In one previous study (Olmsted et al., 2020), we demonstrated the ability of the neural ribbons to enable survival of low-dose (~5,000 cells) spinal cord neural stem cells after delivery. In the current study, the neural ribbon again supports the viability of an electrically-active, spinal-directed neuronal network with similar low number of transplanted cells. While we anticipate that our positive results are the combination of the appropriate differentiated cell types for the spinal cord along with encapsulation-based neuroprotection, it is unclear as to what extent the cell choices, network, and encapsulation interact to assist overall survival in the host injury environment.
We anticipate that this neural circuitry technology will drive advances for in vivo analysis of therapeutic cells and platforms. For example, driving studies beyond fixed cell queries with transmission electron microscopy, transsynaptic labeling, and patch clamp, to also include methods applied in vivo to real time monitor the complexity of networked synaptic communication between graft-delivered neural cells and host. In addition, multi-omics technologies at the single-cell and bulk population level may be informative. The use of scRNAseq could be used to increase reproducibility of differentiation protocols, and therefore of grafted contents, and may help to define ‘maturation’ sufficiency. The encapsulation of additional neural cell types can also be explored in future studies, including astrocytes that assist synapse formation and neuronal homeostasis.
In this qualitative in vivo pilot study, we interrogated two post-transplantation time points (10 days early, 6 weeks late) to determine positioning and survivability of neural circuitry transplants. With the findings of this study now in hand, it will be important in the future to perform a long-term, scaled-up study in the context of functional behavioral analyses using SCI models, which was beyond the scope of our current analysis. In particular, increased scalability to match injury size may be necessary and larger cohort numbers with requisite controls that will enable the statistical power for high quality comparative analysis over multiple time points that was limited here. In this study, we specifically generated cell populations relevant to the SMN regional site of injury, a C4 hemicontusion model. While the most commonly used animal models of SCI are at the thoracic level and often use crush or partial/complete transection injuries, the cervical contusion injury is the most prevalent type of traumatic spinal injury causing severe deficits of upper limb functions. In human SCI patients, the restoration of upper limb function has been reported as the highest priority for improving quality of life. The C4 contusion injury model specifically allows the interrogation of skilled front limb motor tasks that enables high-resolution information of specific motor tracts and volitional versus non-volitional control (Sekhon and Fehlings, 2001; Norenberg et al., 2004; Anderson, 2004). We anticipate that customization of SMN and other cell types tuned anatomically and to injury severity and functional outcome needs will be beneficial, although this remains to be explored.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-SOX2 | R&D Systems | Cat.No.: MAB2018; RRID: AB_358009 |
| Goat anti-SOX2 | R&D Systems | Cat.No.: AF2018; RRID: AB_355110 |
| Goat anti-Brachyury | R&D Systems | Cat.No.: AF2085; RRID: AB_2200235 |
| Mouse anti-CDX2 | R&D Systems | Cat.No.: MAB3665 |
| Mouse anti-Nestin | R&D Systems | Cat.No.: MAB2736; RRID: AB_2282664 |
| Mouse anti-PAX6 | DSHB | Cat.No.: PAX6; RRID: AB_528427 |
| Mouse anti-NKX6.1 | DSHB | Cat.No.: F55A12; RRID: AB_532379 |
| Goat anti-OLIG2 | R&D Systems | Cat.No.: AF2418; RRID: AB_2157554 |
| Rabbit anti-β-tubulin III/TUJ1 (TUBB3) | BioLegend | Cat.No.: 802001; RRID: AB_2564645 |
| Mouse anti-β-tubulin III/TUJ1 (TUBB3) | Abcam | Cat.No.: ab7751; RRID: AB_306045 |
| Goat anti-NCAM1 | R&D Systems | Cat.No.: AF2408; RRID: AB_442152 |
| Rabbit anti-MAP2 | Abcam | Cat.No.: ab32454; RRID: AB_776174 |
| Mouse SMI312 cocktail | BioLegend | Cat.No.: 837904; RRID: AB_2566782 |
| Mouse anti-ISL1&2 | DSHB | Cat.No.: 39.4D5; RRID: AB_2314683 |
| Rabbit anti-ISL1 | Sigma-Aldrich | Cat.No.: HPA057416; RRID: AB_2683431 |
| Mouse anti-HB9 (MNR2/MNX1) | DSHB | Cat.No.: 81.5C10; RRID: AB_2145209 |
| Mouse anti-ChAT | R&D Systems | Cat.No.: MAB3447 |
| Mouse anti-FOXP1 | Santa Cruz Biotechnologies | Cat.No.: sc-398811 |
| Mouse anti-Peripherin | Santa Cruz Biotechnologies | Cat.No.: sc-377093 |
| Rabbit anti-Synapsin 1 (SYN) | Millipore Sigma | Cat.No.: AB1543; RRID: AB_2200400 |
| Rabbit anti-Synapsin 1 (SYN) | Abcam | Cat.No.: ab64581; RRID: AB_1281135 |
| Mouse anti-PSD95 (DLG4) | Antibodies Inc. | Cat.No.: #75-028; RRID: AB_2292909 |
| Mouse anti-GAP43 | Encor | Cat.No.: MCA-5E8; RRID: AB_2572287 |
| Mouse anti-NKX2.2 | DSHB | Cat.No.: 74.5A5; RRID: AB_531794 |
| Mouse anti-O4 | R&D Systems | Cat.No.: MAB1326; RRID: AB_357617 |
| Mouse anti-O4 | EMD Millipore | Cat.No.: MAB345; RRID: AB_94872 |
| PE-O4 | R&D Systems | Cat.No.: FAB1326P; RRID: AB_664169 |
| Goat anti-GFAP | Abcam | Cat.No.: PAB7137; AB_1575164 |
| Chicken anti-GFAP | Abcam | Cat.No.: ab4674; AB_304558 |
| Mouse anti-CD44 | R&D Systems | Cat.No.: MAB7045; RRID: AB_10973152 |
| Mouse anti-CHX10 | Santa Cruz Biotechnologies | Cat.No.: sc-374151 |
| Mouse anti-GATA3 | R&D Systems | Cat.No.: MAB6330; RRID: AB_10640512 |
| Rabbit anti-PAX2 | Invitrogen | Cat.No.: 716000; RRID: AB_2533990 |
| Rabbit anti-LBX1 | Invitrogen | Cat.No.: PA564884; RRID: AB_2662958 |
| Rabbit anti-TLX3 | Abcam | Cat.No.: ab184011 |
| Rabbit anti-LHX9 | Abcam | Cat.No.: ab224357 |
| Rabbit anti-BRN3A | EMD Millipore | Cat.No.: AB5945; RRID: 92154 |
| Mouse anti-STEM121 | Takara | Cat.No.: Y40410; RRID: AB_2801314 |
| Rabbit anti-GFP | Invitrogen | Cat.No.: A-6455; RRID: AB_221570 |
| Bacterial and virus strains | ||
| LV-CAG-eGFP lentivirus | Kerafast | Cat.No.: FCT149 |
| Chemicals, peptides and recombinant proteins | ||
| mTeSR Plus | STEMCELL Technologies | Cat.No.: 05825 |
| mFreSR | STEMCELL Technologies | Cat.No.: 05854 |
| DMEM/F-12 | Thermo Fisher Scientific | Cat.No.: 11320033 |
| Neurobasal Plus Medium | Thermo Fisher Scientific | Cat.No.: A3582901 |
| N-2 supplement (100X) | Thermo Fisher Scientific | Cat.No.: 17502048 |
| B-27 supplement (50X) | Thermo Fisher Scientific | Cat.No.: 17504044 |
| GlutaMAX | Thermo Fisher Scientific | Cat.No.: 35050061 |
| MEM Non-Essential Amino Acids | Thermo Fisher Scientific | Cat.No.: 11140050 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat.No.: 15140122 |
| hESC-qualified Matrigel | Corning | Cat.No.: 08-774-552 |
| Y-27632 | Tocris Bioscience | Cat.No.: 1254 |
| CHIR99021 | Tocris Bioscience | Cat.No.: 4423 |
| LDN193189 | Tocris Bioscience | Cat.No.: 6053 |
| SB431542 | Tocris Bioscience | Cat.No.: 1614 |
| DAPT | Tocris Bioscience | Cat.No.: 2634 |
| Retinoic acid | Sigma-Aldrich | Cat.No.: R2625 |
| Hh-Ag1.5 | Cellagen Technology | Cat.No.: C4412-2s |
| bFGF/FGF2 | R&D Systems | Cat.No.: 233-FB |
| FGF8 | R&D Systems | Cat.No.: 423-F8 |
| BDNF | R&D Systems | Cat.No.: 248-BDB-005 |
| GDNF | R&D Systems | Cat.No.: 212-GD-010 |
| StemBeads BDNF | StemCultures | Cat.No.: SBBD1 |
| StemBeads GDNF | StemCultures | Cat.No.: SBGD1 |
| NGF | R&D Systems | Cat.No.: 256-GF-100 |
| IGF-1 | R&D Systems | Cat.No.: 291-G1 |
| PDGF-AA | R&D Systems | Cat.No.: 221-AA-010 |
| Triiodothyronine (T3) | Millipore Sigma | Cat.No.: 64245 |
| Insulin | Sigma Aldrich | Cat.No.: 91077C |
| Heparin | Sigma Aldrich | Cat.No.: 60800-63-7 |
| Ascorbic acid | Sigma-Aldrich | Cat.No.: 1043003 |
| Dibutyryl-cyclic AMP | Sigma-Aldrich | Cat.No.: D0260 |
| Puromycin | Sigma-Aldrich | Cat.No.: P9620 |
| Accutase | STEMCELL Technologies | Cat.No.: 07920 |
| Gentle Cell Dissociation Reagent | STEMCELL Technologies | Cat.No.: 07174 |
| Fluo-4 AM | Thermo Fisher Scientific | Cat.No.: F14201 |
| HBSS | Thermo Fisher Scientific | Cat.No.: 14025076 |
| HBSS CM-free | Thermo Fisher Scientific | Cat.No.: 14175079 |
| Polybrene | Millipore Sigma | Cat.No.: TR-1003-G |
| Bovine Serum AlbuminFraction V | Fisher Scientific | Cat.No.: BP1600-100 |
| Triton X-100 | Electron Microscopy Sciences | Cat.No.: 221440 |
| NucBlue fixed cell stain | Thermo Fisher Scientific | Cat.No.: R37606 |
| SLM100 ProNova Alginate | Novamatrix | Cat.No.: 4202301 |
| Novatach LVM GRGDSP-coupled alginate | Novamatrix | Cat.No.: 4270321 |
| PureCol EZ gel Type 1 Collagen solution | Advanced Biomatrix | Cat.No.: #5074 |
| Tacrolimus pellet | Innovative Research of America | http://www.innovrsrch.com/product/productsmain.asp |
| Critical commercial assays | ||
| Midi MACS | Miltenyi Biotec | Cat.No.: 130-090-312 |
| FeraTrack superparamagnetic iron oxide particles | Miltenyi Biotec | Cat.No.: 130-104-185 |
| Anti-PE MicroBeads | Miltenyi Biotec | Cat.No.: 130-048-801 |
| Experimental models: Cell lines | ||
| F3.5.2 | Tomov et al. (2016) | https://doi.org/10.1038/srep37637 PMID: 27917881 |
| Experimental models: Organisms/strains | ||
| Female Long Evans rats (4 months) | The Jackson Laboratory | https://www.jax.org/ |
| Software and algorithms | ||
| Keynote | Apple | https://www.apple.com/keynote/ |
| GraphPad Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Fiji | Schindelin et al. (2012) | https://imagej.net/Fiji |
| Illustrator CC2020 | Adobe | https://www.adobe.com/products/illustrator.html |
| cellSens | Olympus | https://www.olympus-lifescience.com/en/software/cellsens/ |
| PhysImage | Hayes et al. (2018) | http://physimage.sourceforge.net/ |
| Multi Channel Experimenter | Multi Channel Systems | https://www.multichannelsystems.com/downloads |
| Multi Channel Analyzer | Multi Channel Systems | https://www.multichannelsystems.com/downloads |
| NeuroExplorer | Plexon | https://plexon.com/products/neuroexplorer/ |
| Igor Pro 6.36 | Wavemetrics | https://www.wavemetrics.com/ |
| Horos Medical Image Viewer | Horos Project, 2020 | https://horosproject.org/ |
| Stereo Investigator | MBF Bioscience | https://www.mbfbioscience.com/stereo-investigator |
| Other | ||
| 6 well plate | CELLTREAT | Cat.No.: 229105 |
| 12 well plate | CELLTREAT | Cat.No.: 229111 |
| Luer-Lok dispensing tips | Nordson EFD | Cat.No.: 7018433; https://www.nordson.com/en/divisions/efd/products/dispense-tips/general-purpose-tips |
| Luer-Lok Syringe (3 ml) | BD | Cat.No.: 309657 |
| Lab-Tek II chambered coverglass | Thermo Fisher Scientific | Cat.No.: 155382 |
| 9.4 T ultra-high field vertical MRI scanner | Bruker | https://www.bruker.com/ |
| MX-M microplate mixer | Scilogex | Cat.No.: 822000049999 |
| Cryostar NX50 Cryomicrotome | Thermo Fisher Scientific | https://assets.thermofisher.com/TFS-Assets/APD/brochures/D21987∼.pdf |
| ESCID injury system | The Ohio State University | https://link.springer.com/protocol/10.1007/978-1-60327-185-1_37 |
| MEA 2100 | Multi Channel Systems | https://www.multichannelsystems.com/products |
| MCS-IFB Interface | Multi Channel Systems | https://www.multichannelsystems.com/products |
| 60 Hexa MEA40/10 devices | Multi Channel Systems | https://www.multichannelsystems.com/products |
| Multiclamp 700B amplifier | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system/amplifiers/axon-instruments-patch-clamp-amplifiers |
| 10 kHz low pass filter | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system/amplifiers/axon-instruments-patch-clamp-amplifiers |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Janet L. Paluh (jpaluh@sunypoly.edu).
Materials availability
This study did not generate unique reagents.
Data and code availability
The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Information files or from the corresponding author upon reasonable request. This study did not generate a unique code.
Experimental model and subject details
Cell Lines
We previously reprogrammed and characterized the F3.5.2 hiPSC line from Coriell deidentified human fibroblast samples from consenting donors (Chang et al., 2015). Line F3.5.2 was reprogrammed with Yamanaka factors by the laboratories of Dr. Paluh and Dr. Jose Cibelli from the Coriell donor fibroblasts. F3.5.2 was previously comprehensively characterized for pluripotency (immunofluorescence, RT-PCR), G-band karyotype, teratoma formation, multi-lineage differentiation, bulk RNA-Seq, ChIP-Seq, was free of mycoplasma contamination and pathogens (Chang et al., 2015; Tomov et al., 2016). G-band karyotype was performed by Cell Line Genetics (Madison, WI).
Method details
Human iPSC maintenance and neuronal differentiation
The African American hiPSC line F3.5.2 was developed in our laboratory (Chang et al., 2015) and is extensively characterized (Tomov et al., 2016; Olmsted et al., 2020). F3.5.2 was cultured in mTeSR Plus (STEMCELL Technologies) supplemented with 1x penicillin-streptomycin (P-S; Gibco) on hESC-qualified Corning Matrigel (1:100) at 37οC, 5% CO2. hiPSC colonies were passaged 1:6 every 4 to 7 days with Gentle Cell Dissociation Reagent (STEMCELL Technologies). Genomic integrity was validated by G-band karyotyping (Cell Line Genetics, Madison, WI). On day 0 of differentiation, hiPSC colonies at 60-70% confluency were rinsed in DMEM/F-12 medium and neuromesodermal progenitor medium (NMPM) was added. N2B27 basal medium: 1:1 DMEM/F-12:Neurobasal Plus medium, 2% (vol/vol) B-27 Plus supplement, 1% (vol/vol) N-2 supplement, 1x GlutaMAX, 1x MEM Non-Essential Amino Acids, 1x P-S (Gibco); NMPM: N2B27 supplemented with 40 ng/ml recombinant human (rh) FGF2 (R&D Systems), 40 ng/ml rhFGF8 (R&D Systems), 2 μM CHIR99021 (Tocris Bioscience), 10 μM DAPT (Millipore Sigma), 10 μM SB431542 (Tocris Bioscience), 100 nM LDN193189 (Tocris Bioscience), 0.36 U/ml heparin (Millipore Sigma). NMPM was changed daily. At day 5, neuromesodermal progenitors were passaged with Gentle Cell Dissociation Reagent as 50-100 μm aggregates in caudo-ventral patterning medium (CVPM). CVPM: N2B27 supplemented with 100 nM Retinoic Acid (RA; Millipore Sigma), 200 nM Hh-Ag1.5. 10 μM Y-27632 (Tocris Bioscience) was added during passages and replaced with fresh medium. Spinal cord neural stem cell cultures were passaged 1:3 in 12-well plates and maintained at 90-100% confluency for 2-3 days before subsequent high-density passages (1:2). Neural cells were cultured on Matrigel for the entirety of differentiation. Spinal cord neural stem cells were patterned in CVPM to day 25 and transitioned to terminal differentiation medium (TDM). TDM: N2B27 supplemented with 10 ng/ml rhBDNF, 10 ng/ml rhGDNF, 1 μM dibutyryl cyclic-AMP (dbcAMP; Millipore Sigma). For culture purification during motor neuron progenitor and SMN differentiation, 24-48 h neurosphere (NS) cultures were formed in 6-well plates (CELLTREAT) using a Scilogex MX-M microplate mixer at ~120 rpm and freshly seeded. The differentiation protocol was repeated >60 times over the course of this study.
Lentivirus transduction
GFP labeling of cells using lentivirus transduction was performed as described (Taylor et al., 2006) at the hiPSC pluripotent stage. In brief, hiPSCs were seeded onto freshly coated 6-well plates 3 days prior to transduction with premade LV-CAG-eGFP lentivirus (Kerafast FCT149, 1 × 108 CFU/ml). Polybrene (2 μg/ml; Millipore Sigma) was added to 2 ml mTeSR Plus. hiPSCs were transduced at 5x magnification of infection by centrifugation with the mTeSR-PB-LV medium for 1 h at 32°C, 1,200 ×g and subsequent incubation at 37°C, 5% CO2 for 18 h. Cultures were rinsed 2x with DMEM/F-12 and cultured in mTeSR Plus for 48 h prior to visual inspection by fluorescence microscopy. Transduced hiPSCs were selected and expanded in the presence of 2 μM puromycin and cryo-stored in mFreSR (STEMCELL Technologies) or further differentiated to OPCs as described below.
OPC differentiation, sorting, and SMN co-culture
Similarly to SMNs, OPCs were patterned through neuromesodermal progenitors and spinal cord neural stem cell stages, but excluding DAPT (Goldman and Kuypers, 2015). For neural ribbon encapsulation, LV-CAG-eGFP transduced hiPSCs were used as starting material. At day 19, 24 h short-term NS suspension culture was used as a purification strategy. NS were seeded at day 20 in OPC medium (OPCM) (Khazaei et al., 2017). OPCM: N2B27 (no Vitamin A) supplemented with 10 ng/ml rhIGF-1 (R&D Systems), 20 ng/ml rhFGF2 (R&D Systems), 20 ng/ml rhPDGF-AA (R&D Systems), 60 ng/ml tri-iodothyronine (T3; Millipore Sigma), 0.36 U/ml heparin (Millipore Sigma). At day 36, cultures were transitioned to OPC maturation medium (OPMM). OPMM: 10 ng/ml rhIGF-1, 25 μg/ml insulin, 1 μM dbcAMP, 20 μg/ml ascorbic acid, 60 ng/ml T3. For co-culture with SMNs, GFP-OPCs at day 35 were sorted manually using the MidiMACS kit (Miltenyi Biotec 130-090-312) with LS columns and Anti-PE MicroBeads (Miltenyi Biotec 130-048-801) according to manufacturer’s instructions. Cells were labeled with IgM PE-O4 antibody (R&D Systems FAB1326P) at 10 μl/106 cells. GFP-OPCs were retained and mixed 1:5 with SMNs. Co-cultures were seeded and maintained for 1 week in modified N2B27 medium, deemed bundling medium, prior to neural ribbon co-encapsulation. Bundling Medium: N2B27 supplemented with 10 ng/ml rhBDNF, 10 ng/ml rhGDNF, 10 ng/ml rhNGF, 20 ng/ml rhPDGF-AA, 10 ng/ml rhIGF-1, 60 ng/ml T3, 100 μM dbcAMP, 25 μg/ml insulin, 20 μg/ml ascorbic acid. For in vivo grafting experiments with SMNs and OPCs, the ratio was increased to 1:2 OPC to SMN.
Phase contrast imaging and immunofluorescence
Phase contrast images were acquired using a Nikon Eclipse TS100 microscope (10x/0.25 Ph1 ADL and 20x/0.40 Ph1 ADL objectives; Qimaging Retiga 2000R camera) or Zeiss Invertoskop 40C (5x/0.12 CP-Apochromat, 10x/0.25 Ph1 A-Plan and 20x/0.30 Ph1 LD A-Plan objectives; Olympus DP22 color camera). For immunofluorescence (IF) imaging, cells were seeded into Matrigel-coated glass-bottom chambers (Lab-Tek II 4-chambered cover glass; Nunc, #155382) and fixed with 10% buffered formalin for 30 min at specified time points. Samples were permeabilized for 5 min in 0.1% Triton-X-100 and blocked for 30 min in 1% BSA fraction V (1x PBS). Primary antibodies were applied in 1 ml fresh blocking buffer and incubated at 4οC overnight. Samples were rinsed thoroughly in 1x PBS before applying immunoglobulin- and species-matched AlexaFluor secondary antibodies (Invitrogen) for 1 h in the dark with DAPI (4οC). Cells were imaged directly in chambered cover glass wells. Fluorescence microscopy was performed using a Zeiss Axio Observer.Z1 inverted fluorescence microscope (20x/0.8 air and 63x/1.4 oil Plan-Apochromat DIC objectives). Images were acquired using a Hamamatsu ORCA ER CCD camera and Zeiss AxiovisionRel software (ver. 4.8.2). For adherent cultures, 6- to 30-slice Z-stacks were gathered at 1 μm separation distance and compressed using the Extended Focus feature. Uniform exposure times were maintained across samples for identical antibodies. If necessary, images were adjusted linearly for brightness in Keynote or ImageJ. A Leica confocal TCS SP5 II system was used for lower magnification images (10x/0.30 HCX PL FLUOTAR air objective lens), acquired using Leica Application Suite Advanced Fluorescence software.
Calcium imaging
Neural ribbons were incubated at 37oC with 5 μM Fluo-4 AM (Invitrogen) for 30 min in Neurobasal Plus medium (no supplements). Cultures were then recovered in fresh TDM for 30 min and imaged in HBSS using the Zeiss Axio Observer.Z1 system with the 20x air objective described above. Time lapse series were captured at an exposure time of 50 ms obtained in 200 ms intervals for a total duration of 1.5 or 2.5 min.ΔF/F over time was determined using PhysImage that is an ImageJ plug-in (Hayes et al., 2018; Faustino Martins et al., 2020).
Neural ribbon encapsulation and dissolution
Lyophilized ultrapure sodium alginate (DuPont Novamatrix) was resuspended in sterile 0.9% NaCl (w/w) to 1.5% (w/w) and stored at 4°C. Two alginate products were used that are ProNova SLM100 (MWT: 150-250 kDa; G/M ratio <1; viscosity 170 mPa∗s; endotoxin <25 EU/g) or Novatach LVM GRGDSP-coupled alginate (G/M ratio <1, endotoxin <10; peptide 0.015 μmol/mg). Peptide-conjugated alginate was used for in vitro assays while unconjugated alginate was used for in vivo grafting studies. 5 mg/ml PureCol EZ gel Type 1 Collagen solution (Advanced Biomatrix #5074) was included 1:3 with 1.5% alginate to provide initial ECM in neural ribbons prior to extrusion. ProNova-Collagen and RGD-Alginate-Collagen hydrogels are referred to here as NovaCol and RGD-Col, respectively. Differentiating cultures were gently lifted as semi-continuous aggregates using Accutase diluted 1:1 in HBSS and distributed to 1.5 ml Eppendorf tubes in DMEM/F-12. A subset of aggregates was dissociated to single cells and counted. Cells were centrifuged at 350 ×g for 5 min and the supernatant was aspirated. Cell pellets were combined first with non-gelled PureCol solution and resuspended gently at room temperature. 1.5% alginate solution was then added slowly while mixing, avoiding bubbles, to a final cell suspension concentration equivalent of 1× 108 cells/ml. 40 μl of this suspension was loaded directly into a luer lock steel tip (34G, inner diameter 60 μm, 32G, inner diameter 100 μm, or 30G, inner diameter 150 μm, Nordson EFD) and attached to a sterile 3 ml BD syringe. Contents were dispensed directly into 100 mM CaCl2crosslinking solution normal to the liquid surface for immediate templated formation of 60 μm neural ribbons. CaCl2 was aspirated completely after 1 min and the neural ribbons were resuspended in cell culture medium. Suspended neural ribbons were subsequently incubated (37°C, 5% CO2) to drive interpenetrating network formation by collagen gelation. Ribbon quality was monitored by visual inspection. Cell suspensions were excluded when forming empty hydrogel ribbons. Neural ribbons were either used immediately for downstream experiments or retained in culture.
Neural ribbon immobilization and fixed imaging
To template and image neural ribbon cytoarchitecture, RGD-Col ribbons were formed as described above directly into the 12 mm viewing area of Nunc 35 mm glass bottom dishes (ThermoFisher #150680). CaCl2 was aspirated after crosslinking and the ribbons were manually aligned. To immobilize ribbons in position for imaging assays, 100 μl of PureCol EZ gel solution (5 mg/ml in DMEM) was gently applied over the ribbons, filling the viewing area, and incubated for 45 min at 37οC. After gelation, 1.5 ml of culture medium was added. To constrain neurite outgrowth to within the neural ribbon body, 4 μg/ml Aggrecan containing CSPGs was included in the collagen embedding matrix prior to gelation. This cytoarchitecture was further promoted by culturing in bundling medium, described above. Cultures were maintained for 7-12 days in this platform before collagen gel began to degrade. For IF experiments, collagen solution was diluted 1:1 with DMEM/F-12 to enhance antibody permeability and wash steps were extended to 15 min each at room temperature. The IF protocol was modified for neural ribbons and was performed in 25 mM PIPES/10 mM HEPES/2.5 mM CaCl2 buffer to prevent dissolution by high phosphate concentration and Ca2+ leaching.
Patch clamp electrophysiology
hiPSCs were differentiated as described above to day 23, when the adherent cultures were passaged using Gentle Cell Dissociation Reagent as aggregates and encapsulated in neural ribbons (1 x 108 cells/ml equivalent) or as neurospheres in suspension culture. Neural ribbon and NS suspension cultures were cultured for 48 h. On day 25, NS were plated onto 18 mm Matrigel-coated cover glass (#1.5 thickness, Warner Instruments). Neural ribbons were dissolved in 1.6% sodium citrate and seeded at an equivalent cell density. We further compared neurons cultured in patterning versus maturation medium using 5 μl/ml BDNF and 5 μl/ml GDNF slow release PLGA microbeads (StemCultures; 5 ng/ml steady-state concentrations) at two time points, for a total of six conditions (Table S2). The experiments were performed on multiple differentiations per condition (N = 3-4). We added 10 ng/ml soluble rhBDNF and rhGDNF (R&D Systems) to the culturing medium on days of TDM media changes. For patch clamp studies, SMNs were transferred to a recording chamber perfused at 2.5 ml/min with a solution saturated with 95% O2/5% CO2 and containing (in mM): 119 NaCl, 2.5 KCl, 1.2 CaCl2, 1 MgCl2, 26.2 NaHCO3, 1 NaH2PO4, 22 glucose; 300 mOsm; pH 7.4. SMNs were visually identified under infrared differential interference contrast (IR-DIC) using an upright microscope (Olympus BX51), based on their trapezoidal shape, and the presence of more than two dendritic processes and high-contrast deposition in the cell cytoplasm (Sareen et al., 2013). Whole-cell patch clamp recordings were made with a pipette solution containing (in mM): 120 KCH3SO3, 10 EGTA, 20 HEPES, 2 MgATP, 0.2 NaGTP, 290 mOsm, pH 7.2. The resistance of the recording electrode was ~5 MΩ and was monitored throughout the experiments by applying a -50 pA, 10 ms-long current step. Data were discarded if the input resistance of the recorded SMN changed more than 20% during the course of the experiment. All recordings were obtained under current-clamp configuration using a Multiclamp 700B amplifier and a 10 KHz low-pass filter (Molecular Devices). The resting membrane potential was measured immediately after breaking into the whole-cell configuration. After this, a DC current injection was used to maintain cells at -65 mV. All traces were digitized at 10 KHz. Each cell was subject to a series of 5 ms (short) and 500 ms (long) current step injections. The amplitude of the short current step ranged between 50 and 350 pA. The amplitude of the long current step ranged between -100 and 350 pA. The amplitude of the short and long current injections were changed in steps of 10 pA. Each protocol was applied for 10 consecutive times. The data analysis was performed off-line with custom-made software (A.S.) written in IgorPro 6.36 (Wavemetrics). All experiments were performed at room temperature.
Microelectrode array electrophysiology recordings from neural ribbons
For microelectrode array (MEA) recordings, we used an MEA2100 head stage system connected to an MCS-IFB-in-vitro interface board (Multi Channel Systems). Temperature was maintained at 37oC during recordings using a temperature controller. One electrode per MEA chip served as a reference electrode. To investigate spiking, bursts, and networks bursts in 3D neural ribbons, we used 60HexaMEA40/10 single-well MEA chips. Each hexagonal array contained 60 electrodes, each of 10 μm diameter with 40 μm inter-electrode spacing that is suitable to neural ribbon geometry. Neural ribbon recordings were taken as acute measurements using day 35 SMNs encapsulated at day 28. To ensure electrode contact, neural ribbons were immobilized under a 12-mm coverslip compressed by a 5 g stainless steel weight. N = 3 neural ribbons were recorded under spontaneous conditions and after addition of 50 μM glutamate for a minimum of 6 min. Recordings were acquired Multi Channel Experimenter software (v2.14.0) sampled at 25 kHz for at least 5 min. Spike threshold for detection was automatically set to 5x above standard deviation over background noise (1 ms pre trigger, 2 ms post trigger). Spike and burst analyses were performed using Multi Channel Analyzer (v2.14.0) in conjunction with NeuroExplorer (v5.022). For statistical analysis on the day of recording, glutamate addition and washout was performed three times for the same neural ribbon wherein recordings were separated by 30 min each to account for inactivation of glutamate ionotropic channels. Burst detection parameters were identical to adherent recordings described above. For network burst analysis, detection was set to minimum active channels of 4 and minimum simultaneous channels of 4, due to incomplete electrode coverage by the neural ribbons. Generation of raw data traces and statistical analyses were performed using GraphPad Prism and NeuroExplorer in conjunction with Microsoft Excel.
Neural ribbon long-distance shipping and receiving
In order to perform transplantation experiments, neural ribbons were shipped overnight from SUNY Polytechnic (Albany, NY) to Houston Methodist (Houston, TX). They were encapsulated as previously described above, using alginate only. Day 30 spinal neuron cultures were encapsulated in neural ribbons in 12-well plates, resuspended in TDM supplemented with 10 μM ROCK inhibitor, and incubated at 37oC for 1 h. Ribbon suspensions were then transferred to 1.8 ml cryovials using 1,000-μl pipette tips and immediately shipped on ice (4oC) via FedEx (20-22 h total in transit). At the destination laboratory, neural ribbons were recovered in a 37oC humidified cell culture incubator prior to the day of transplantation in rat.
SPIO labeling and collagen phantom injections
At day 29, confluent neuronal cultures were incubated with FeraTrack MRI SPIO nanoparticles (Miltenyi Biotec) overnight in 12-well plates. Cell viability versus SPIO concentration analysis was performed after 24 h incubation. ~100 μg Fe/1 x 106 cells (40 μl Miltenyi Biotec FeraTrack SPIO/2 ml culture medium) was chosen according to cell viability and manufacturer’s instructions. SPIO uptake was confirmed using a Prussian blue iron assay kit (Millipore Sigma) according to manufacturer’s instructions. Day 30 SPIO-labeled cultures were encapsulated in neural ribbons plus Fluorescein-Dextran (500 kDa, 1:30 in alginate) and shipped on ice overnight as described above. Collagen phantom slabs were generated from 5 mg/ml PureCol EZ gel Type 1 Collagen solution (Advanced Biomatrix #5074) at RT by plating the solution in 3 cm dish. 60 μm diameter neural ribbons were manually severed to single 3-4 mm segments containing ~5,000 cells. Single ribbon segments were individually drawn into a 26G custom made curved needle Hamilton syringe with a ~45 degree bend, ~2 mm proximal to the bevel and injected by hand into collagen phantoms. Fluorescein-Dextran in neural ribbon segments was visualized using a Leica M165 FC stereo microscope.
Rat cervical spinal cord hemi-contusion injury and transplants
A total of N = 6 four-month-old Female Long-Evans rats were used in SCI experiments. We induced cervical level C4 hemi-contusion injuries as previously described (Mondello et al., 2015), wherein C4 contusion injuries were made using the electromagnetic spinal cord injury device (ESCID). In short, right-sided hemi-laminectomy was performed at C4 level in anesthetized animals that were then placed in a spinal frame to clamp the lateral processes of C3, C5. The ESCID electromagnetic probe was positioned at the surface of the dura with an initial sensing force followed by rapid displacement (0.8 mm with 14 ms dwell). Muscles were then sutured in layers and the skin was clipped. One week prior transplantation of human cells, the immunosuppressant tacrolimus was administrated at the dosage of 3.4 mg/kg/day through gradual release from subcutaneous implanted pellet (Innovative Research of America). Near the time of cell transplantation, 150 μm diameter neural ribbons containing day 30 human spinal neurons were shipped and received in Houston one day prior to injections. Constructs were suspended in fresh N2B27 and maintained in a humidified tissue culture incubator overnight. Cell number and viability was determined with an automatic counter (Invitrogen) before transplantation. For transplants, the spinal cord was re-exposed at injury level 15 days after injury in anesthetized animals. To stabilize the animal, the lateral process of C3 was secured in a custom spinal frame. Grafts were delivered in HBSS vehicle with the addition of 5 mM glucose, 20 ng/ml FGF and EGF for metabolic and neurotrophic support. Neural ribbon segments (N = 6 animals, ~5,000 cells per segment) were individually drawn into a 26G curved needle Hamilton syringe with a ~45 degree bend ~2 mm proximal to the bevel. The syringe was placed in a Kopf micromanipulator attached to the spinal frame and positioned 0.5 mm lateral to midline ipsilaterally. The syringe tip was used to puncture the dura, and advanced to a depth of 0.7 mm. The single ribbon segment was injected into the injury cavity over 1 min in 5 μl of HBSS per animal. The needle was maintained in position for 2 min dwell, followed by 100 μm retraction and a second 2 min dwell, and then was slowly retracted while monitoring for efflux of fluid or the ribbon segment onto the surface of the dura. After injection, the dura was sealed using a fibrin sealant made by mixing a 1:1 ratio of fibrinogen solution (20 mg/ml) with a bovine thrombin solution (10 U/ml) (Millipore Sigma). Muscle and skin layers were closed, and animals received appropriate post-operative care. For visualization of graft placement by MRI, detailed in the section below, one additional uninjured control animal was injected with a neural ribbon segment containing SPIO-labeled cells at the C4 level. All rat experimental protocols were approved by Houston Methodist Research Institute IACUC and carried out in accordance with relevant ethical guidelines and regulations. This includes animal comfort, veterinary care, methods and reasons for euthanasia, and materials and hiPSC-derived neural injections.
Ex vivo spinal column MRI
Spinal columns were imaged in a 9.4 T ultra-high field vertical MRI scanner (Bruker) using an adjusted multi-echo T2 rapid acquisition refocused echo sequence with acceleration factor of 4, 10 averages and single excitation cartesian k-space scan trajectory. Pixel bandwidth of 40,761 Hz/px, time-to-repetition of 1800 ms, and time-to-echo of 25, 37 and 45 ms were used. For in silica characterization of the neural ribbon transversal relaxation (T2) signal, samples were imaged in 200 μL plastic tubes containing physiologic saline solution. For in situ imaging, in-plane resolution of 25 μm and slice thickness of 1 mm was used for axial acquisitions with anterior-posterior phase encoding and FOV of 12.5 mm. 13 x 21 μm resolution and 3 mm slice thickness were used for coronal and sagittal acquisitions with rostral-caudal phase encoding directions and FOV of 50 mm. For quantitative analysis in situ, T2 relaxation maps were generated pixelwise by applying a single exponential fit to image intensity measured over four time-to-echo values. Transplanted neural ribbon constructs were visualized in the rodent spinal column using Horos Medical Image Viewer (Horos Project, 2020). Raw images acquired at time-to-echo = 25 ms and computed T2 maps were considered. An inverse log-scale colormap, typically applied to imaging of bone perfusion, was chosen for enhanced visualization of the ribbons.
Tissue processing and immunohistochemistry
Spinal cords were harvested at two time points for short term (10 days after transplantation, N = 3) and long term (6 weeks after transplantation, N = 3) analyses. Rats were sedated with isoflurane and transcardially perfused first with cold 0.1 M PBS with 10,000 U of heparin followed by 4% paraformaldehyde in 0.1 M PBS (pH 7.4). Spinal cords were harvested for storage in 4% paraformaldehyde at 4°C overnight. The spinal cords were passed through a sucrose buffer gradient (10%, 20%, 30%; 24 h per solution) at 4°C for cryoprotection. Cords were then aligned longitudinally in a Tissue Tek OCT block (Sakura, Nederland) using dry ice, and stored at -80°C. 40 μm thick spinal cord sagittal sections were prepared using a Cryostar NX50 cryomicrotome (ThermoFisher). Serial sections of the entire spinal cord were then mounted onto positively charged slides (Fisher Scientific). For immunohistochemistry, slides were first blocked with 10% goat serum in 0.2% Triton-PBS (T-PBS) for 1 h at room temperature followed by primary antibody incubation in 1% serum, 0.2% T-PBS overnight at 4°C. Primary antibodies used were mouse anti-STEM121 (1:500; Takara), chicken anti-GFAP (1:2,000; Abcam), rabbit anti-Synapsin 1 (1:200; Abcam) and rabbit anti-TUJ1 (1:1,000; BioLegend). After three additional wash steps with 1% serum T-PBS (1-2 min/wash), slides were incubated with secondary antibodies in PBS for 1 h. GFP-OPC signal was visualized without an antibody (fixed, native GFP signal). Secondary antibodies used were goat anti-mouse AF488 (1:1,000; Invitrogen), goat anti-chicken Cy5 (1:1,000; Invitrogen), goat anti-rabbit AF568 (1:1000; Invitrogen). Slides were mounted with DAPI-Permount (Invitrogen) and imaged with a Leica DMi8 confocal microscope. Graft volume and area at the lesion site were calculated with the stereological probe Cavalieri using the software StereoInvestigator (MBF Bioscience). The lesion area of the cord (4 mm), cavity and graft size were determined on every 5 sections by tracing the different areas in the cords transplanted with cell suspensions and sacrificed at 6 weeks (N = 3). Transplant area was defined in STEM121/DAPI longitudinal sections 40μm thick using a Zeiss Axioskop 2 microscope. For the Cavalieri method, the following parameters were chosen: grid size (μm): 100; Associated Area (μm2): 10,000; Section Cut Thickness (μm): 40; Section Evaluation Interval: 5; Associated Volume (μm³): 2,000,000; Number of Sections: 4.
Quantification and statistical analysis
Statistical analysis and reproducibility
Raw data were compiled in Microsoft Excel (v16.16.16) and exported to GraphPad Prism (v8.3.0) for plotting and statistical analysis. Data are reported as (mean ± s.e.m.) and analyzed using unpaired two-tailed t-test unless otherwise specified. Cells were manually counted in ImageJ to quantify immunofluorescence data. One-way ANOVA test was used to compare patch clamp electrophysiology experimental conditions. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05, n.s. not significant (α = 0.05). Power analysis was not performed for grafting studies. Detailed information for each experiment is provided in figure legends.
Figures
Figures for this manuscript were made in Keynote (v9.2.1), Canvas Draw (v4.0.1), Adobe Illustrator CC (v25.0.1), and BioRender.com. Data plots were generated using GraphPad Prism (v9) or Microsoft Excel (v16.16.19).
Acknowledgments
This work was funded by the New York State Department of Health (NYS DOH) Spinal Cord Injury Research Board (NYSCIRB), Projects to Accelerate Research Translation (PART) award C33278GG and SUNY Polytechnic SEED award 917035-21 and used a published line developed through previous New York State stem cell research (NYSTEM) funding. This work was also supported by NSF/IOS1655365 and SUNY Albany Research Foundation grants to A.S.
Author contributions
J.P. and Z.O. designed experiments and wrote the manuscript with input from all co-authors. Z.O. compiled figures, and performed hiPSC maintenance, neural differentiation and analysis, neural ribbon studies and shipping, in vitro imaging, electrophysiology sample preparation, and MEA experiments. A.S. performed patch clamp experiments and analysis. C.S. and P.H. conducted in vivo grafting studies and data acquisition. T.W. assisted with MRI. The F3.5.2 hiPSC line was previously derived in collaboration with J.C., who also provided key input for experiments and the final manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
Multiple authors on this paper received support from a program NYSTEM designed to increase minority representation in science. The research in this paper includes work with ethnically diverse human iPS lines generated from that support. In order to bring increased representation of the diverse U.S. and global populations to stem cell research, multiple authors on this paper have played an integral role in the reprogramming, characterization, and application of iPSC lines from self-reported African American, Hispanic Latino, and Asian fibroblast donors. These lines represented the first such lines of their kind, and the African American line F3.5.2 is applied here in this iScience study. Furthermore, lines from all three donors have been used in numerous other publications, including generation of the first gastruloids, termed ‘elongating multi-lineage organized (EMLO) gastruloids’, published in Nature Communications (PMID: 34021144).
Published: August 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102827.
Supplemental information
References
- Ahuja C.S., Fehlings M. Concise review: bridging the gap: novel neuroregenerative and neuroprotective strategies in spinal cord injury. Stem Cells Transl. Med. 2016;5:914–924. doi: 10.5966/sctm.2015-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K.D. Targeting recovery: priorities of the spinal cord-injured population. J. Neurotrauma. 2004;21:1371–1383. doi: 10.1089/neu.2004.21.1371. [DOI] [PubMed] [Google Scholar]
- Assinck P., Duncan G.J., Hilton B.J., Plemel J.R., Tetzlaff W. Cell transplantation therapy for spinal cord injury. Nat. Neurosci. 2017;20:637–647. doi: 10.1038/nn.4541. [DOI] [PubMed] [Google Scholar]
- Baker E.W., Kinder H.A., West F.D. Neural stem cell therapy for stroke: a multimechanistic approach to restoring neurological function. Brain Behav. 2019;9:e01214. doi: 10.1002/brb3.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker R.A., Parmer M., Studer L., Takahashi J. Human trials of stem cell-derived dopamine neurons for Parkinson’s disease: dawn of a new era. Cell Stem Cell. 2017;21:569–573. doi: 10.1016/j.stem.2017.09.014. [DOI] [PubMed] [Google Scholar]
- Bregman B.S., Kunkel-Bagden E., Reier P.J., Dai H.N., McAtee M., Gao D. Recovery of function after spinal cord injury: mechanisms underlying transplant-mediated recovery of function differ after spinal cord injury in newborn and adult rats. Exp. Neurol. 1993;123:3–16. doi: 10.1006/exnr.1993.1136. [DOI] [PubMed] [Google Scholar]
- Chang E.A., Tomov M.L., Suhr S.T., Luo J., Olmsted Z.T., Paluh J.L., Cibelli J. Derivation of ethnically diverse human induced pluripotent stem cell lines. Sci. Rep. 2015;5:15234. doi: 10.1038/srep15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Dusenbery B.N., Williams L.A., Klim J.R., Eggan K. How to make spinal motor neurons. Development. 2014;141:491–501. doi: 10.1242/dev.097410. [DOI] [PubMed] [Google Scholar]
- DeMaagd G., Philip A. Parkinson’s disease and its management: part 1: disease entity, risk factors, pathophysiology, clinical presentation, and diagnosis. P T. 2015;40:504–532. [PMC free article] [PubMed] [Google Scholar]
- Deng J., Zhang Y., Xie Y., Zhang L., Tang P. Cell transplantation for spinal cord injury: tumorigenicity of induced pluripotent stem cell-derived neural stem/progenitor cells. Stem Cells Int. 2018;2018:5653787. doi: 10.1155/2018/5653787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulin J.N., Adler A.F., Kumamaru H., Poplawski G.H.D., Lee-Kubli C., Strobl H., Gibbs D., Kadoya K., Fawcett J.W., Lu P., Tuszynski M.H. Injured adult motor and sensory axons regenerate into appropriate organotypic domains of neural progenitor grafts. Nat. Commun. 2018;9:84. doi: 10.1038/s41467-017-02613-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustino Martins J.M., Fischer C., Urzi A., Vidal R., Kunz S., Ruffault P.-R., Kabuss L., Hube I., Gazzerro E., Birchmeier C. Self-organizing 3D human trunk neuromuscular organoids. Cell Stem Cell. 2020;26:172–186. doi: 10.1016/j.stem.2019.12.007. [DOI] [PubMed] [Google Scholar]
- Frith T.J.R., Granata I., Wind M., Stout E., Thompson O., Neumann K., Stavish D., Heath P.R., Ortmann D., Hackland J.O. Human axial progenitors generate trunk neural crest cells in vitro. eLife. 2018;7:e35786. doi: 10.7554/eLife.35786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B.X., Ziskand-Conhaim L. Development of ionic currents underlying changes in action potential waveforms in rat spinal motoneurons. J. Neurophysiol. 1998;80:3047–3061. doi: 10.1152/jn.1998.80.6.3047. [DOI] [PubMed] [Google Scholar]
- Goldman S.A., Kuypers N.J. How to make an oligodendrocyte. Development. 2015;142:3983–3995. doi: 10.1242/dev.126409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouti M., Tsakiridis A., Wymeersch F.J., Huang Y., Kleinjung J., Wilson V., Briscoe J. In vitro generation of neuromesodermal progenitors reveals distinct roles for Wnt signaling in the specification of spinal cord and paraxial mesoderm identity. PLoS Biol. 2014;12:e1001937. doi: 10.1371/journal.pbio.1001937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes J.A., Papagiakoumou E., Ruffault P.-L., Emiliani V., Fortin G. Computer-aided neurophysiology imaging with open-source PhysImage. J. Neurophysiol. 2018;120:23–36. doi: 10.1152/jn.00048.2017. [DOI] [PubMed] [Google Scholar]
- Henrique D., Abranches E., Verrier L., Storey K.G. Neuromesodermal progenitors and the making of the spinal cord. Development. 2015;142:2864–2875. doi: 10.1242/dev.119768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho R., Sances S., Gowing G., Amoroso M.W., O’Rourke J.G., Sahabian A., Wichterle H., Baloh R.H., Sareen D., Svendsen C.N. ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat. Neurosci. 2016;19:1256–1267. doi: 10.1038/nn.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izsak J., Seth H., Andersson M., Vizlin-Hodzic D., Theiss S., Hanse E., Agren H., Funa K., Illes S. Robust generation of person-specific, synchronously active neuronal networks using purely isogenic human iPSC-3D neural aggregate cultures. Front Neurosci. 2019;13:351. doi: 10.3389/fnins.2019.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh H., Yokota K., Fehlings M.G. Regeneration of spinal cord connectivity through stem cell transplantation and biomaterial scaffolds. Front.Cell. Neurosci. 2019;13:248. doi: 10.3389/fncel.2019.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazaei M., Ahuja C.S., Fehlings M.G. Generation of oligodendrogenic spinal neural progenitor cells from human induced pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 2017;42:2D.20.1–2D.20.14. doi: 10.1002/cpsc.31. [DOI] [PubMed] [Google Scholar]
- Kikuchi T., Morizane A., Doi D., Magotani H., Onoe H., Hayashi T., Mizuma H., Takara S., Takashi R., Inoue H. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature. 2017;548:592–596. doi: 10.1038/nature23664. [DOI] [PubMed] [Google Scholar]
- Kumamaru H., Kadoya K., Adler A.F., Takashima Y., Graham L., Coppola G., Tuszynski M.H. Generation and post-injury integration of human spinal cord neural stem cells. Nat. Methods. 2018;15:723–731. doi: 10.1038/s41592-018-0074-3. [DOI] [PubMed] [Google Scholar]
- Lippmann E.S., Williams C.E., Ruhl D.A., Esteves-Silva M.C., Chapman E.R., Coon J.J., Ashton R.S. Deterministic HOX patterning in human pluripotent stem cell-derived neuroectoderm. Stem Cell Reports. 2015;4:632–644. doi: 10.1016/j.stemcr.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Schackel T., Weidner N., Puttagunta R. Biomaterial-supported cell transplantation treatments for spinal cord injury: challenges and perspectives. FrontCell Neurosci. 2018;11:430. doi: 10.3389/fncel.2017.00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manion J., Khuong T., Harney D., Littleboy J.B., Ruan T., Loo L., Costigan M., Larance M., Caron L., Neely G.G. Human induced pluripotent stem cell-derived GABAergic interneuron transplants attenuate neuropathic pain. Pain. 2020;161:379–387. doi: 10.1097/j.pain.0000000000001733. [DOI] [PubMed] [Google Scholar]
- Mondello S.E., Sunshine M.D., Fischedick A.E., Mortiz C.T., Horner P.J. A cervical hemi-contusion spinal cord injury model for the investigation of novel therapeutics targeting proximal and distal forelimb functional recovery. J. Neurotrauma. 2015;32:1994–2007. doi: 10.1089/neu.2014.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelec S., Martinez-Arias A. In vitro models of spinal motor circuit’s development in mammals: achievements and challenges. Curr. Opin. Neurobiol. 2021;66:240–249. doi: 10.1016/j.conb.2020.12.002. [DOI] [PubMed] [Google Scholar]
- Norenberg M.D., Smith J., Marcillo A. The pathology of human spinal cord injury: defining the problems. J. Neurotrauma. 2004;21:429–440. doi: 10.1089/089771504323004575. [DOI] [PubMed] [Google Scholar]
- Olmsted Z.T., Stigliano C.S., Badri A., Zhang F., Williams A., Koffas M.A.G., Xie Y., Linhradt R.J., Cibelli J., Horner P.J., Paluh J.L. Fabrication of homotypic neural ribbons as a multiplex platform optimized for spinal cord delivery. Sci. Rep. 2020;10:12939. doi: 10.1038/s41598-020-69274-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmsted Z.T., Paluh J.L. Stem cell neurodevelopmental solutions for restorative treatments of the human trunk and spine. Front.Cell. Neurosci. 2021;15:667590. doi: 10.3389/fncel.2021.667590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raof N.A., Padgen M.R., Gracias A.R., Bergkvist M., Xie Y. One dimensional self-assembly of mouse embryonic stem cells using an array of hydrogel microstrands. Biomaterials. 2011;32:4498–4505. doi: 10.1016/j.biomaterials.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reier P.J., Bregman B.S., Wujek J.R. Intraspinal transplantation of embryonic spinal cord tissue in neonatal and adult rats. J. Comp. Neurol. 1986;247:275–296. doi: 10.1002/cne.902470302. [DOI] [PubMed] [Google Scholar]
- Sagner A., Briscoe J. Establishing neuronal diversity in the spinal cord: a time and a place. Development. 2019;146:dev182154. doi: 10.1242/dev.182154. [DOI] [PubMed] [Google Scholar]
- Sances S., Bruijn L.I., Chandran S., Eggan K., Ho R., Klim J.R., Livesey M.R., Lowry E., Macklis J.D., Rushton D. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat. Neurosci. 2016;19:542–553. doi: 10.1038/nn.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareen D., O’Rourke J.G., Meera P., Muhammad A.K.M.G., Grant S., Simpkinson M., Bell S., Carmona S., Ornelas L., Sahabian A. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saaflfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhon L.H., Fehlings M.G. Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine. 2001;26:S2–S12. doi: 10.1097/00007632-200112151-00002. [DOI] [PubMed] [Google Scholar]
- Silva N.A., Sousa N., Reis R.L., Salgado A.J. From basics to clinical: a comprehensive review on spinal cord injury. Prog. Neurobiol. 2014;114:25–27. doi: 10.1016/j.pneurobio.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Sofroniew M.V. Dissecting spinal cord regeneration. Nature. 2018;557:343–350. doi: 10.1038/s41586-018-0068-4. [DOI] [PubMed] [Google Scholar]
- Southwell D.G., Seifikar H., Malik R., Lavi K., Vogt D., Rubenstein J.L., Sohal V.S. Interneuron transplantation rescues social behavior deficits without restoring wild-type physiology in a mouse model of autism with excessive synaptic inhibition. J. Neurosci. 2020;40:2215–2227. doi: 10.1523/JNEUROSCI.1063-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takazawa T., Croft G.F., Amoroso M.W., Studer L., Wichterle H., MacDermott A.B. Maturation of spinal motor neurons derived from human embryonic stem cells. PLoS One. 2012;7:e40154. doi: 10.1371/journal.pone.0040154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei T., Kitazono J., Tanaka S., Nishimata H., Yoshida M. Necrotic regions are absent in fiber-shaped cell aggregates, approximately 100 um in diameter. Artif. Cells Nanomed. Biotechnol. 2016;44:62–65. doi: 10.3109/21691401.2014.909824. [DOI] [PubMed] [Google Scholar]
- Taylor L., Jones L., Tuszynski M.H., Blesch A. Neurotrophin-3 gradients established by lentiviral gene delivery promote short-distance axonal bridging beyond cellular grafts in the injured spinal cord. J. Neurosci. 2006;26:9713–9721. doi: 10.1523/JNEUROSCI.0734-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telezhkin V., Straccia M., Yarova P., Pardo M., Yung S., Vinh N.-N., Hancock J.M., Barriga G.G.-D., Brown D.A., Rosser A.E. Kv7 channels are upregulated during striatal neuron development and promote maturation of human iPSC-derived neurons. Eur. J. Physiol. 2018;470:1359–1376. doi: 10.1007/s00424-018-2155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomov M.L., Olmsted Z.T., Dogan H., Gongorurler E., Tsompana M., Otu H.H., Buck M., Chang E.-H., Cibelli J., Paluh J.L. Distinct and shared determinants of cardiomyocyte contractility in multi-lineage competent ethnically diverse iPSCs. Sci. Rep. 2016;6:37637. doi: 10.1038/srep37637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trawczynski M., Liu G., David B.T., Fessler R.G. Restoring motor neurons in spinal cord injury with induced pluripotent stem cells. FrontCell Neurosci. 2019;13:369. doi: 10.3389/fncel.2019.00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremmel D.M., Mitchell S.A., Sackett S.D., Odorico J.S. Mimicking nature-made beta cells: recent advances towards stem cell-derived islets. Curr. Opin. Organ Transplant. 2019;24:574–581. doi: 10.1097/MOT.0000000000000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsintou M., Dalamagkas K., Seifalian A.M. Advances in regenerative therapies for spinal cord injury: a biomaterials approach. Neural Regen. Res. 2015;10:726–742. doi: 10.4103/1673-5374.156966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wind M., Gogolou A., Manipur I., Granata I., Butler L., Andrews P.W., Barbaric I., Ning K., Guarracino M.R., Placzek M., Tsakiridis A. Defining the signaling determinants of a posterior ventral spinal cord identity in human neuromesodermal progenitor derivatives. Development. 2021;148:dev194415. doi: 10.1242/dev.194415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong M., Tao Y., Gao Q., Feng B., Yan W., Zhou Y., Kotsonis T.A., Yuan T., You Z., Wu Z. Human stem cell-derived neurons repair circuits and restore neural function. Cell Stem Cell. 2021;28:112–126.e6. doi: 10.1016/j.stem.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., Mahairaki V., Koliatsos V.E. Host induction by transplanted neural stem cells in the spinal cord: further evidence for an adult spinal cord neurogenic niche. Regen. Med. 2012;7:785–797. doi: 10.2217/rme.12.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabolocki M., McCormack K., van den Hurk M., Milky B., Shoubridge A.P., Adams R., Tran J., Mahadevan-Jansen A., Reineck P., Thomas J. BrainPhys neuronal medium optimized for imaging and optogenetics in vitro. Nat. Commun. 2020;11:5550. doi: 10.1038/s41467-020-19275-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Information files or from the corresponding author upon reasonable request. This study did not generate a unique code.