

Abstract
In psychotherapy, paradoxical interventions are characterized by a deliberate reinforcement of the pathological behavior to improve the clinical condition. Such a counter‐intuitive approach can be considered when more conventional interventions fail. The development of targeted cancer therapies has enabled the selective inhibition of activated oncogenic signaling pathways. However, in advanced cancers, such therapies, on average, deliver modest benefits due to the development of resistance. Here, we review the perspective of a ‘paradoxical intervention’ in cancer therapy: rather than attempting to inhibit oncogenic signaling, the proposed therapy would further activate mitogenic signaling to disrupt the labile homeostasis of cancer cells and overload stress response pathways. Such overactivation can potentially be combined with stress‐targeted drugs to kill overstressed cancer cells. Although counter‐intuitive, such an approach exploits intrinsic and ubiquitous differences between normal and cancer cells. We discuss the background underlying this unconventional approach and how such intervention might address some current challenges in cancer therapy.
Keywords: cancer therapy, collateral sensitivity, mitogenic signaling, stress response
Cancer cells are defective in homeostatic feedbacks. Therefore, pharmacological mitogenic stimulation disrupts the homeostasis of cancer cells and further increases their overall stress levels. Such toxic overactivation should increase the sensitivity of these cells to stress‐targeted drugs, providing opportunities for efficient drug combinations. Normal cells should more efficiently buffer such overactivation due to a more effective negative feedback network.

Abbreviations
- ALL
acute lymphoblastic leukemia
- DDR
DNA damage response
- ESFT
ewing sarcoma family tumor
- ER
endoplasmic reticulum
- MAPK
mitogen‐activated protein kinase
- PKC
protein kinase C
- ROS
reactive oxygen species
1. Introduction
Genetic and epigenetic alterations leading to oncogenic signaling underlie the hallmarks of cancer and are also responsible for drug resistance [1, 2]. Most of the current targeted therapies are focused on blocking such aberrant mitogenic signaling pathways in cancer cells. However, the natural selection occurring throughout tumorigenesis shapes cancer cells toward the most efficient, not necessarily the highest, level of mitogenic signaling activity. This notion is becoming increasingly apparent, with accumulating evidence showing that overactivation of these same pathways might also disrupt cancer cell homeostasis. For instance, EGFR and RAS mutations are frequent but not concomitant in lung cancer. It has been recently shown that synthetic lethality rather than redundancy underlies this mutual exclusivity [3, 4]. Moreover, in a Kras (G12V)/Braf (D631A)‐driven lung cancer model, the ablation of the wild‐type Braf allele further increased mitogen‐activated protein kinase (MAPK) signaling, leading to oncogenic toxicity and preventing the development of adenocarcinoma [5]. Another case in point is that an unbiased loss‐of‐function genetic screen looking for chromatin modulators that induce senescence in melanoma cells revealed SMARCB1 as the top hit [6]. The knockout of this tumor suppressor gene in BRAF mutant melanoma cells triggered senescence and apoptosis through overactivation of the EGFR‐MAPK pathway [6]. Pharmacological examples of this oncogenic overload were also found in melanomas. A subgroup of BRAFV600E ‐driven melanomas acquires resistance to BRAF inhibitors by overexpressing this protein. The inhibitor withdrawal results in tumor regression due to an overactivated MAPK [7]. In line with this, we previously showed that EGFR expression in melanoma models triggers oncogene‐induced senescence in the absence of drugs but becomes beneficial in the presence of BRAF or MEK inhibitors [8]. We later showed that BRAF inhibitor‐resistant melanoma cells have increased reactive oxygen species (ROS) levels due to overactivation in MAPK signaling. Subsequent addition of the histone deacetylase inhibitor vorinostat further increases the ROS levels, selectively killing these drug‐resistant tumor cells [9].
Collectively, these data highlight that too much oncogenic activity can be as lethal to cancer cells as the inhibition of oncogenic pathways. Based on this notion, we propose that a deliberate overactivation of mitogenic signaling pathways can be used to disrupt the fragile homeostasis of cancer cells. Such ‘paradoxical’ therapeutic intervention exploits the most basic characteristic of cancer cells, pathological oncogenic signaling, to push these cells over the viability edge.
Another ubiquitous feature of cancer cells is the increased mobilization of stress response pathways, which can be seen as an inherent cost for the oncogenic activity. Multiple cellular stress response pathways may have to be mobilized by the cancer cells to support the tumorigenic state. Together, they can be recognized collectively as the stress hallmarks of cancer [10]. The causes and consequences of these adaptations are interconnected but vary among different cancer cells. Nonetheless, the aneuploidy so characteristic to cancer cells is causally linked to proteotoxic, replication, mitotic, metabolic, and oxidative stresses, and the engagement of the respective stress response pathways [11]. Importantly, these adaptations are not merely associated with the malignant transformation but instead are intrinsically linked to it. Such increased mobilization and dependence on stress response pathways results from the upregulation of survival and mitogenic signaling in cancer cells. A dynamic and fine‐tuned balance between the hyperactivated signaling pathways and the stress response pathways induced by them supports cancer cell viability and tumor progression [10]. Indeed, the rationale for using drugs targeting stress pathways (referred to below as ‘stress‐targeted drugs’) has recently gained traction, showing encouraging results in disrupting the homeostasis of cancer cells [12, 13, 14, 15, 16].
The scenario outlined above suggests that further activation of mitogenic pathways should increase the dependency of cancer cells on stress response pathways. Therefore, we advocate that pharmacological overactivation of the same pathways driving tumorigenesis can disrupt the homeostasis of cancer cells and that such drugs may show synergy in combination with stress‐targeted drugs. A schematic model illustrating this rationale is described in Fig. 1. We address below how this paradoxical approach may have implications for different aspects of cancer treatment.
Fig. 1.
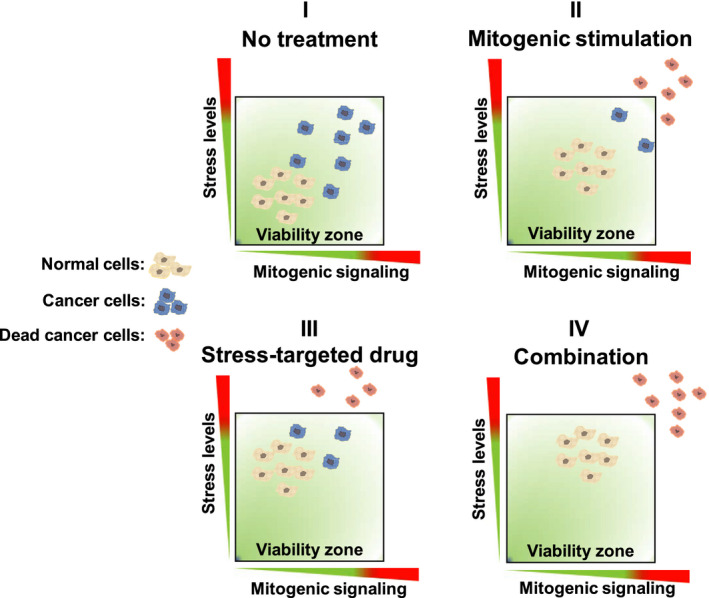
Schematic model of a paradoxical intervention for cancer treatment. (I) In untreated conditions, cancer cells show higher basal levels of mitogenic signaling and stress, inherent to the malignant phenotype, with a higher degree of heterogeneity in these levels. (II) Mitogenic stimulation takes advantage of the defective homeostatic feedbacks of cancer cells to promote overactivation of mitogenic signaling, driving part of the cancer cell population out of the viability zone and also increasing the overall stress levels. Normal cells, on the other hand, can buffer such activation due to an intact and effective negative feedback network. (III) Stress‐targeted drugs increase the overall stress levels and promote the killing of cancer cells based on their higher basal levels of stress; however, the heterogeneity of the cancer cell population allows some cancer cells to remain viable in the presence of the increased stress. (IV) The combination of mitogenic stimulation and targeting of stress response pathways should drive cancer cells to an overactivated, overstressed phenotype, out of the viability zone, promoting selective killing. Normal cells retain sufficient homeostatic feedback mechanisms to survive these perturbations.
2. Selectivity: targeting the fundamental difference between normal cells and cancer
Highly selective targeting of cancer cells is mostly an elusive enterprise because they survive and proliferate using the very same signaling and metabolic pathways that normal cells employ to maintain homeostasis. Moreover, cancer cells also show genetic instability, self‐sufficiency in growth signals, and a lack of commitment to systemic homeostasis. Together, these properties provide them with the plasticity required to rewire signaling and adapt to drug treatment. These ‘hallmarks of cancer’ [1] are promoted by the activity of the driver oncogenes and the partial loss of counteracting tumor‐suppressive mechanisms that are the basis of cancer. Therefore, pathological mitogenic signaling and disruption of homeostatic feedback mechanisms represent a cornerstone difference between cancer and normal cells, forming the basis for selective targeting. Recent data, discussed below, show that deliberate reinforcement of mitogenic signaling disrupts the labile homeostasis and increases the mobilization of stress response pathways in cancer cells.
As one example, we recently showed that FGF2 enhances replication and proteotoxic stresses in human Ewing sarcoma family tumor models and KRas‐driven murine adrenal cancer cells. Indeed, the combination of exogenous FGF2 and the proteasome inhibitor bortezomib or inhibition of the DNA damage response (DDR) with the ATR inhibitor VE‐821 synergically triggered cell death, which was dependent on Ras‐Mek‐Erk signaling [17]. It is noteworthy that FGF2 inhibits endoplasmic reticulum (ER) stress in vitro and in vivo, protecting mouse cardiomyocytes from ischemia/reperfusion injury, also through Mek‐Erk activation [18]. These findings highlight opposite outcomes resulting from exogenous mitogenic signaling activation in normal versus cancer cells. In line with this, the EGF‐EGFR signaling axis is a model of prosurvival and proliferative signaling for normal cells, playing a key role in development, homeostasis, and wound healing [19]. In contrast, exogenous administration of EGF induced oxidative stress and PTEN upregulation in non–small‐lung cancer models, resulting in terminal autophagy and apoptosis in vitro and in vivo [20].
Hyperactivation of mitogenic pathways in tumor cells for therapeutic purposes is mostly uncharted territory. Therefore, the number of molecules other than growth factors that have been used in this context is quite limited. In this respect, protein kinase C (PKC) agonists have a substantial record of cytostatic and cytotoxic effects in multiple cancer models, often linked mechanistically to overactivation of the MAPK pathways [21, 22, 23] However, sustained activation of PKC isozymes leads to the downregulation of their protein levels [24], imposing a potential handicap for the therapeutic use of these molecules. Recently, Harold Varmus' laboratory showed that the small‐molecule DUSP1/6‐inhibitor BCI leads to toxic high levels of MAPK activity and triggers cell death in a panel of Egfr‐ and Kras‐driven lung adenocarcinoma cells [25]. It was later shown that the same inhibitor sensitizes ovarian cancer cells to carboplatin and paclitaxel [26]. These findings suggest that, although aberrant MAPK‐ERK1/2 activity is frequent in cancer cells, pharmacological DUSP inhibition and the resultant hyperactivation of this pathway can, in fact, be tumor suppressive. Indeed, Markus Müschen's laboratory has previously shown that DUSP6 activity is essential to tame ERK1/2 activity in pre‐B acute lymphoblastic leukemia (ALL) cells [27]. DUSP6 inhibition increases ROS levels and DDR activation, which triggers cell death in patient‐derived ALL cells [27]. More recently, the authors proposed the use of such therapeutic activation of ERK1/2 in STAT5‐driven B‐ALL based on the increased lifespan observed in patient‐derived xenografted mice treated with the DUSP1/6 inhibitor BCI‐215 to activate ERK1/2 [28]. It appears likely that combination with drugs targeting oxidative stress or DDR would improve the overall efficacy of the pharmacological ERK1/2 activation in these models.
The obvious initial concern for using mitogenic signaling activation in combination therapies is that normal cells could become malignant themselves. However, normal tissues display layers of control to restrain unscheduled proliferation, such as a myriad of negative feedbacks in signaling pathways, terminal differentiation, and subordination to cell‐extrinsic tumor‐suppressive signals. A number of these controls are lost in cancer cells to allow the malignant phenotype. Supporting this notion, transgenic mouse models overexpressing growth factors [29, 30, 31, 32], or displaying tonic ERK1/2 activation due to DUSP6 knockout [33] do not show increased tumorigenesis. Therefore, reinforcement of mitogenic signaling exploits a fundamental defect of cancer cells (i.e., increased oncogenic signaling) and may be combined with stress‐targeted drugs. The specific toxicities resulting from the overactivation of mitogenic pathways certainly vary among different cancer cells and contexts. Nevertheless, further increase of genotoxic, oxidative, proteotoxic, mitotic, and metabolic stresses likely result from such intervention in cancer cells, making them more sensitive to drugs targeting these stress response pathways. Figure 2 outlines the rationale for the selective targeting of cancer cells through this proposed approach.
Fig. 2.
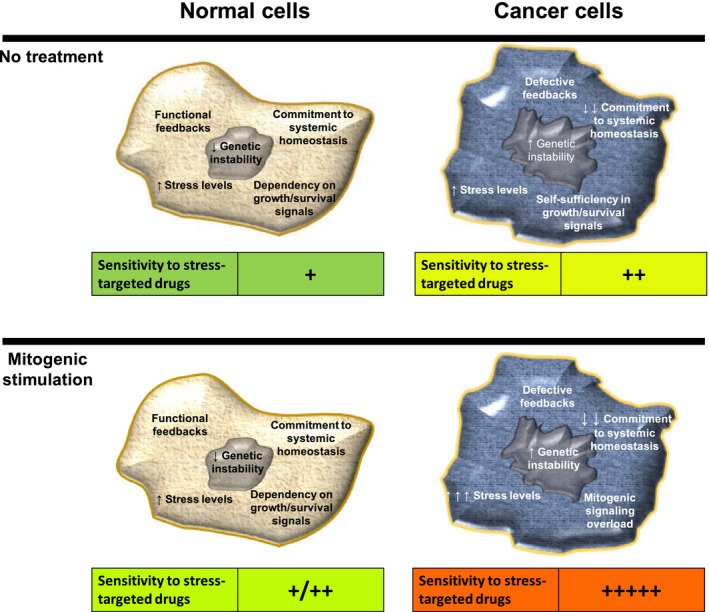
Conceptual rationale for the selective sensitization of cancer cells to stress‐targeted drugs through mitogenic signaling activation. (Top) Lack of commitment to systemic homeostasis, increased genetic instability, self‐sufficiency in growth and survival signals, and defective feedback signaling are recognized hallmarks of cancer cells and a cornerstone difference between cancer and normal cells. To support this tumorigenic state, cancer cells also show higher basal levels of stress and are therefore usually more sensitive to stress‐targeted drugs than are normal cells. (Bottom) Deliberate mitogenic stimulation would take advantage of the already increased mitogenic signaling of cancer cells and their defective feedbacks to promote mitogenic overload, while increasing the levels of stress and sensitivity to stress‐targeted drugs. On the other hand, normal cells display layers of feedback mechanisms to more efficiently tame such mitogenic activation, and lower basal levels of stress, being therefore potentially less sensitive to the combination of mitogenic activation and stress‐targeted drugs.
3. Drug resistance: exploiting ‘collateral sensitivities’ and promoting tumor‐suppressive acquired resistance
While chemo‐ and radiotherapy are based on killing rapidly dividing cells, the newer targeted therapies aim to selectively inhibit the very pathways that drive tumorigenesis [34]. Unfortunately, more often than not, targeted therapies fail to deliver long‐lasting therapeutic responses, as multiple mechanisms have been identified that can lead to resistance to these drugs [35, 36]. This is perhaps not surprising given the abundance of feedback loops and cross‐talks that operate between cellular signaling pathways, providing ample opportunity for cells to rewire their signaling under selective pressure. Massive efforts have been made to uncover powerful drug combinations, or ‘synthetic lethalities’, to tackle signaling rewiring and constrain drug resistance [37, 38]. Despite some progress in this endeavor, good long‐term prognoses are still not frequent for advanced cancer patients. The rationale behind targeted therapies is that cancer cells are dependent on specific driver signaling pathways whose inhibition would result in cell cycle arrest or death [39]. However, genetic and epigenetic plasticity at the cell level and the frequent heterogeneity at the tumor level make the bypass of these inhibitions often only a matter of time. The survival and proliferation of normal cells also rely on many of the same pathways targeted by antimitogenic drugs, but their commitment to systemic homeostasis gives them less ‘freedom’ to bypass antimitogenic therapies [40].
Exploiting the acquired vulnerabilities/fitness costs inherent to the drug‐resistant phenotype holds promise for the treatment of drug‐resistant cancers. This concept of ‘collateral sensitivity’ was recognized decades ago [41], and the recent literature has demonstrated its utility to outsmart drug resistance [42]. The attractiveness of the collateral sensitivity approach is the use of the very plasticity of cancer cells to steer them toward a predictable and targetable, highly vulnerable, phenotype. In this sense, the use of mitogenic activation to make cancer cells more targetable by stress‐related drugs may be one embodiment of the collateral sensitivity approach. The exploitation of such a frequent feature of cancer cells (i.e., overactivation of mitogenic pathways) to induce a collateral sensitivity may provide a more homogeneous sensitization of the cancer cells and thus lead to more robust responses when the collateral sensitivity is targeted. The coincident targeting of an equally ubiquitous feature (i.e., increased dependency on stress response pathways) can represent a powerful combination strategy. The specific activation signal and stress‐targeted drug in such a combination will most likely vary for different cancer phenotypes. Nonetheless, in contrast to antimitogenic therapies, this approach benefits from the fact that different mutations driving human cancers converge on a much smaller set of signaling pathways. This notion implies that different upstream or downstream activators can promote the same mitogenic signaling pathway, resulting in a similar collateral sensitivity.
One of the most attractive features of the paradoxical cancer therapy proposed above comes to light when considering how cells might develop resistance to this type of therapy. Resistance to antimitogenic targeted therapies often involves reactivation of the inhibited pathway or activation of parallel oncogenic signaling [43]. Hence, it is reasonable to assume that resistance to overactivation of mitogenic signaling pathways will develop in the opposite direction. That would include the upregulation of tumor‐suppressive mechanisms and downregulation or even loss of oncogenic signaling. Striking data in line with these assumptions come from the virtual absence of EGFR amplification in glioblastoma cell models in vitro, whereas it is found in about half of the diagnosed glioblastoma tumors [44]. Schulte and coworkers showed that the presence of EGF in the culture media purges EGFR gene amplification from patient‐derived glioblastoma cells, largely decreasing their tumorigenicity in xenografts [45]. That might be a stark example of a tumor‐suppressive effect (i.e., loss of EGFR amplification) triggered by mitogenic signaling stimulation (i.e., EGF ligand), but it is not without precedent, given that similar findings were previously described for lung [46] and breast [47] cancer models.
The overall scenario discussed above raises the prospect that ‘tumor‐suppressive drug resistance’ might turn cancer into a chronic disease, a long‐awaited therapeutic goal. This assumption may seem unrealistic at first glance; nonetheless, deep‐sequencing investigations have recently revealed that clones carrying known driver oncogenic mutations are present in clinically normal skin, esophagus, and endometrium tissues, among others [48, 49, 50]. Moreover, it has long been shown that highly malignant teratocarcinoma cells injected into blastocysts resulted in mosaic mice, with a substantial contribution of the tumor‐derived cells to the normal functions of many tissues [51]. Similar reprogramming was also described for melanoma cells in different embryo models [52]. It remains to be addressed whether the relative higher abundance of mitogenic signals in embryos compared with aged or adult tissues is required for this tumor‐suppressive reprogramming.
4. Dormancy and relapse: restraining nonproliferative drug tolerance and resistance
The therapeutic window for cancer treatment often relies on a common trait of malignant cells: unscheduled and fast proliferation resulting from the effect of driver oncogenes on mitogenic pathways. Unfortunately, tumor heterogeneity and plasticity often make it possible for a fraction of the tumor cell population to acquire a nonproliferative drug‐tolerant phenotype under treatment. This shift is now recognized as a common mechanism underlying acquired resistance and tumor relapse [53, 54, 55]. The transition to these nonproliferative phenotypes involves rewiring signaling pathways to evade the oncogenic signaling and its proliferative consequences. Moreover, adjustments in the fine‐tuned cross‐talk between mitogenic signaling and stress response pathways support the viability of nonproliferative drug‐resistant cancer cell phenotypes (i.e., quiescent, dormant, persister, and cancer stem cells) [56, 57, 58]. Therefore, the combination of mitogenic signaling activation with stress‐targeted drugs may hinder therapy escape by restraining the exit of cancer cells from the pathologically proliferative phenotype.
In agreement with this rationale, mitogenic stimulation of quiescent drug‐resistant leukemic cells has been shown to drive them back to the proliferative phenotype and sensitize to chemo‐ and targeted therapies [59]. Even in the context of cancer stem cells, a similar approach stimulated leukemia stem cells into the cell cycle, sensitizing them to the standard‐of‐care chemotherapy in a xenotransplantation mouse model [60]. Cancer stem cells represent a formidable hurdle for cancer therapy. They share many features with normal stem cells, such as self‐renewing capability, pluripotency, and cell surface markers [61]. More importantly, they also have in common slow proliferation rates and overall metabolism, high expression of drug efflux pumps, and a consequent multidrug‐resistant phenotype [61, 62]. Not surprisingly, cancer stem cells are very elusive targets for current cancer therapies, being a reservoir for tumor relapse and metastasis after therapy withdrawal or acquired resistance [63, 64].
There is a lack of consensus about the origin, prevalence, and markers of cancer stem cells and other nonproliferative drug‐resistant phenotypes. Even so, in practical terms, they may represent a metastable state in which selective targeting of cancer cells becomes unfeasible. As such, the use of mitogenic stimulation as part of the therapy holds the promise to push cancer stem cells toward a proliferative phenotype. By doing this, we make them more different from normal cells, more prone to drug toxicity, and prevent them from hiding from therapeutic attack.
5. Side effects: targeting cancer cells while supporting tissue homeostasis and a tumor‐suppressive microenvironment
It is now clear that healthy normal cells and tissue homeostasis play a pivotal role in preventing tumor onset and progression, even when clones carrying known driver oncogenic mutations are frequent [65, 66]. This notion underlies the known correlation between tumorigenesis and aging, inflammation, chemical injuries, or other conditions leading to decreased tissue homeostasis. The fact that growth factors and mitogenic signaling drive recovery from injury is documented extensively [67, 68]; additionally, the contribution of these signaling pathways for tissue homeostasis becomes evident by the many on‐target toxicities associated with tyrosine kinase inhibitors [69]. This overall scenario suggests that therapeutic mitogenic signaling activation may not only sensitize cancer cells to stress‐targeted drugs but also favor a tumor‐suppressive microenvironment by improving tissue homeostasis.
In agreement with the assumptions above, FGF signaling promoted self‐renewing proliferation and inhibited senescence in normal stem cell pools from various tissues [70]. From the regenerative perspective, regular FGF2 injections promoted neurogenesis and increased the lifespan in neurodegenerative Huntington's disease mouse models [71]. Similarly, daily EGF administration following total body irradiation dramatically improved the recovery of the hematopoietic stem cell pools and overall survival in mice [72]. In this context of therapy models, systemic administration of human recombinant EGF in mice reduced the severity of the oral mucositis associated with chemotherapy and radiotherapy for head and neck cancers [73]. Debilitating oral mucositis is indeed a frequent side effect in patients receiving myelotoxic therapy [74]. KGF (FGF7) has been used in the clinic, decreasing the severity and shortening the recovery of the lesions [75]. Systemic homeostatic effects were described for FGF21, which protects different organs against tissue damage resulting from metabolic disorders and metaflammation, promoting tissue homeostasis [76]. It is noteworthy that the homeostatic effects of FGF21 in nonmalignant tissues also involve the activation of the very same FGFR‐MAPK axis frequently overridden in cancer cells [77].
Collectively, the findings above highlight that normal cells rely on the abundance of mitogenic signaling to maintain tissue homeostasis and recover from injury, and imply that pharmacological mitogenic signaling activation might be clinically safe. This dependency on mitogenic signaling in normal cells contrasts with the self‐sufficiency in survival and proliferation signals characteristic to cancer cells. Different conditions leading to decreased mitogenic signaling levels in normal cells should, therefore, impair tissue homeostasis and cell‐extrinsic tumor‐suppressive mechanisms. In line with this, the progressive loss of tissue homeostasis with aging is associated with decreased levels of mitogenic signals [78, 79, 80, 81]. These findings indicate that mitogenic signaling is inversely correlated with the major risk factor for cancer incidence [82]. These observations may puzzle our notion of the role of growth factors and mitogenic signaling for cancer onset and progression. However, this overall decline in mitogenic signals, detrimental to normal cells, likely drives the competition in favor of precancer clones carrying oncogenic mutations. Hence, if cancer is ‘a wound that does not heal’ [83], combining ‘healing signaling’ and stress‐targeted therapies might revert this pernicious dynamic, improving several aspects of cancer treatment.
6. Future perspectives
While counter‐intuitive at first glance, there appears to be a therapeutic window to selectively kill cancer cells based on the further activation of an already activated oncogenic signaling pathway. This approach opens a number of ‘out of the box’ treatment options in cases in which conventional therapies have failed. Here, we have focused on mitogenic signaling pathways, because these pathways are often the target of oncogenic mutations. However, the concept of ‘making a bad situation worse’ can also be applied to other treatment modalities. For instance, along the same lines discussed above for mitogenic signaling, it has been proposed that the level of genetic instability, a common feature of malignant transformation, is optimal, not maximal, in cancer cells [84, 85]. Deliberate overactivation of DDR signaling has been proposed to decrease the ability of cancer cells to deal with DNA damage [86]. Moreover, because cancer is a disease that results from mutations, it would seem illogical to conceive of a cancer therapy that is based on increasing the mutation burden of a cancer cell. Nevertheless, there is emerging evidence that responses to checkpoint immunotherapies can be enhanced by increasing the mutation load of the cancer. This approach is based on the notion that there is a striking correlation between the mutation burden of a given cancer and the response to these checkpoint therapies [87]. Thus, microsatellite instable colon cancers, which have a higher mutation burden due to a defect in DNA mismatch repair, show superior responses to immunotherapies as compared to microsatellite stable colon tumors [88, 89]. One paradoxical intervention may therefore be to increase the mutation burden in a microsatellite stable colon cancer by disabling the mismatch repair machinery to increase the neo‐antigen load of the cancer. Indeed, Germano and coworkers showed that genetic inactivation of the MutL homolog MLH1 leads to increased mutation burden, immune recognition, and response to checkpoint therapies [90]. Along these same lines, Bartok et al. [91] recently demonstrated that amino acid starvation can result in ‘ribosomal frameshifting’, leading to generation of neoantigens that can be seen by immune cells.
Finally, it has been suggested that deliberate interference with mRNA splicing can serve as a source of cancer neoantigens [92]. This is potentially interesting, given that drugs exist that interfere with mRNA splicing [93]. Thus, in the context of the immune response, the paradoxical intervention could be the reinforcement of the ‘non‐self’ character of tumor cells while favoring an active cytotoxic phenotype in T cells. This scenario would increase the overall immunogenicity of cancer cells, potentially acting to enhance the effects of current immunotherapies.
We have argued above that such paradoxical interventions may have only modest effects on normal cells. However, it should be borne in mind that most of the experiments to test such interventions on normal cells have been carried out in mice. Two crucial differences between mice and humans are relevant in this context: mice weigh 2500‐fold less than an average human (and have correspondingly fewer cells) and their lifespan is 40x shorter than that of humans. Thus, it may be dangerous to translate the effects of mitogenic insults on normal cells observed in mice to the human situation. There may be late effects in humans of this type of paradoxical therapy that were never seen in simple animal models. Notwithstanding this limitation, we argue here that there are compelling data from a number of independent models to seriously consider this as a novel approach to cancer therapy.
Conflict of interest
The authors declare no conflict of interest.
References
- 1. Hanahan D & Weinberg RA (2000) The hallmarks of cancer review Douglas. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D & Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 3. Ambrogio C, Barbacid M & Santamaría D (2017) In vivo oncogenic conflict triggered by co‐existing KRAS and EGFR activating mutations in lung adenocarcinoma. Oncogene 36, 2309–2318. [DOI] [PubMed] [Google Scholar]
- 4. Unni AM, Lockwood WW, Zejnullahu K, Lee‐Lin SQ & Varmus H (2015) Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. ELife 4, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nieto P, Ambrogio C, Esteban‐Burgos L, Gómez‐López G, Blasco MT, Yao Z, Marais R, Rosen N, Chiarle R, Pisano DG et al. (2017) A Braf kinase‐inactive mutant induces lung adenocarcinoma. Nature 548, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang L, Leite de Oliveira R, Wang C, Fernandes Neto JM, Mainardi S, Evers B, Lieftink C, Morris B, Jochems F, Willemsen L et al. (2017) High‐throughput functional genetic and compound screens identify targets for senescence induction in cancer. Cell Rep 21, 773–783. [DOI] [PubMed] [Google Scholar]
- 7. Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M & Stuart DD (2013) Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun C, Wang L, Huang S, Heynen GJJE, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM et al. (2014) Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508, 118–122. [DOI] [PubMed] [Google Scholar]
- 9. Wang L, Leite de Oliveira R, Huijberts S, Bosdriesz E, Pencheva N, Brunen D, Bosma A, Song J‐Y, Zevenhoven J, Los‐de Vries GT et al. (2018) An acquired vulnerability of drug‐resistant melanoma with therapeutic potential. Cell 173, 1413–1425.e14. [DOI] [PubMed] [Google Scholar]
- 10. Luo J, Solimini NL & Elledge SJ (2009) Principles of cancer therapy: oncogene and non‐oncogene addiction the current state of cancer research. Cell 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu J, Tsai H‐J, Gordon MR & Li R (2018) Cellular stress associated with aneuploidy. Dev Cell 44, 420–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meister S, Schubert U, Neubert K, Herrmann K, Burger R, Gramatzki M, Hahn S, Schreiber S, Wilhelm S, Herrmann M et al. (2007) Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Can Res 67, 1783–1792. [DOI] [PubMed] [Google Scholar]
- 13. Yap TA, Plummer R, Azad NS & Helleday T (2019) The DNA damaging revolution: PARP inhibitors and beyond. Am Soc Clin Oncol Educ Book 39, 185–195. [DOI] [PubMed] [Google Scholar]
- 14. Trachootham D, Alexandre J & Huang P (2009) Targeting cancer cells by ROS‐mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8, 579–591. [DOI] [PubMed] [Google Scholar]
- 15. Janssen A & Medema RH (2011) Mitosis as an anti‐cancer target. Oncogene 30, 2799–2809. [DOI] [PubMed] [Google Scholar]
- 16. Luengo A, Gui DY & Vander Heiden MG (2017) Targeting metabolism for cancer therapy. Cell Chem Biol 24, 1161–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dias MH, Fonseca CS, Zeidler JD, Albuquerque LL, da Silva MS, Cararo‐Lopes E, Reis MS, Noël V, Santos EO, Prior IA et al. (2019) Fibroblast growth factor 2 lethally sensitizes cancer cells to stress‐targeted therapeutic inhibitors. Mol Oncol 13, 290–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Z, Wang Y, Ye J, Lu X, Cheng Y, Xiang L, Chen L, Feng W, Shi H, Yu X et al. (2015) bFGF attenuates endoplasmic reticulum stress and mitochondrial injury on myocardial ischaemia/reperfusion via activation of PI3K/Akt/ERK1/2 pathway. J Cell Mol Med 19, 595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen J, Zeng F, Forrester SJ, Eguchi S, Zhang M‐Z & Harris RC (2016) Expression and function of the epidermal growth factor receptor in physiology and disease. Physiol Rev 96, 1025–1069. [DOI] [PubMed] [Google Scholar]
- 20. Ryu JW, Choe SS, Ryu SH, Park EY, Lee BW, Kim TK, Ha CH & Lee S‐W (2017) Paradoxical induction of growth arrest and apoptosis by EGF via the up‐regulation of PTEN by activating Redox factor‐1/Egr‐1 in human lung cancer cells. Oncotarget 8, 4181–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cozzi SJ, Parsons PG, Ogbourne SM, Pedley J & Boyle GM (2006) Induction of senescence in diterpene ester‐treated melanoma cells via protein kinase C‐dependent hyperactivation of the mitogen‐activated protein kinase pathway. Can Res 66, 10083–10091. [DOI] [PubMed] [Google Scholar]
- 22. Oliva JL, Caino MC, Senderowicz AM & Kazanietz MG (2008) S‐phase‐specific activation of PKC induces senescence in non‐small cell lung. Cancer Cells 283, 5466–5476. [DOI] [PubMed] [Google Scholar]
- 23. Serova M, Ghoul A, Benhadji KA, Faivre S, le Tourneau C, Cvitkovic E, Lokiec F, Lord J, Ogbourne SM, Calvo F et al. (2008) Effects of protein kinase C modulation by PEP005, a novel ingenol angelate, on mitogen‐activated protein kinase and phosphatidylinositol 3‐kinase signaling in cancer cells. Mol Cancer Ther 7, 915–922. [DOI] [PubMed] [Google Scholar]
- 24. Newton AC (2018) Protein kinase C: perfectly balanced. Crit Rev Biochem Mol Biol 53, 208–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Unni AM, Harbourne B, Oh MH, Wild S, Ferrarone JR, Lockwood WW, & Varmus H (2018) Hyperactivation of ERK by multiple mechanisms is toxic to RTK‐RAS mutation‐driven lung adenocarcinoma cells. ELife 7, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. James NE, Beffa L, Oliver MT, Borgstadt AD, Emerson JB, Chichester CO, , Yano N, Freiman RN, DiSilvestro PA & Ribeiro JR (2019) Inhibition of DUSP6 sensitizes ovarian cancer cells to chemotherapeutic agents via regulation of ERK signaling response genes. Oncotarget 10, 3315–3327. [PMC free article] [PubMed] [Google Scholar]
- 27. Shojaee S, Caeser R, Buchner M, Park E, Swaminathan S, Hurtz C, Geng H, Chan LN, Klemm L, Hofmann W‐K et al. (2015) Erk negative feedback control enables pre‐B cell transformation and represents a therapeutic target in acute lymphoblastic leukemia. Cancer Cell 28, 114–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chan LN, Murakami MA, Robinson ME, Caeser R, Sadras T, Lee J, Cosgun KN, Kume K, Khairnar V, Xiao G et al. (2020) Signalling input from divergent pathways subverts B cell transformation. Nature 583, 845–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chan SY & Wong RW (2000) Expression of epidermal growth factor in transgenic mice causes growth retardation. J Biol Chem 275, 38693–38698. [DOI] [PubMed] [Google Scholar]
- 30. Coffin JD, Florkiewicz RZ, Neumann J, Mort‐Hopkins T, Dorn GW 2nd, Lightfoot P, German R, Howles PN, Kier A & O'Toole BA (1995) Abnormal bone growth and selective translational regulation in basic fibroblast growth factor (FGF‐2) transgenic mice. Mol Biol Cell 6, 1861–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Santoni‐Rugiu E, Preisegger KH, Kiss A, Audolfsson T, Shiota G, Schmidt EV & Thorgeirsson SS (1996) Inhibition of neoplastic development in the liver by hepatocyte growth factor in a transgenic mouse model. Proc Natl Acad Sci USA 93, 9577–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y, Xie Y, Berglund ED, Coate KC, He TT, Katafuchi T, Xiao G, Potthoff MJ, Wei W, Wan Y et al. (2012) The starvation hormone, fibroblast growth factor‐21, extends lifespan in mice. ELife 1, e00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maillet M, Purcell NH, Sargent MA, York AJ, Bueno OF & Molkentin JD (2008) DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J Biol Chem 283, 31246–31255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baudino TA (2015) Targeted cancer therapy: the next generation of cancer treatment. Curr Drug Discov Technol 12, 3–20. [DOI] [PubMed] [Google Scholar]
- 35. Gillies RJ, Verduzco D & Gatenby RA (2012) Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer 12, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Groenendijk FH & Bernards R (2014) Drug resistance to targeted therapies: déjà vu all over again. Mol Oncol 8, 1067–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beijersbergen RL, Wessels LFA & Bernards R (2017) Synthetic lethality in cancer therapeutics. Annu Rev Cancer Biol 1, 141–161. [Google Scholar]
- 38. Huang A, Garraway LA, Ashworth A & Weber B (2020) Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discovery 19, 23–38. [DOI] [PubMed] [Google Scholar]
- 39. Torti D & Trusolino L (2011) Oncogene addiction as a foundational rationale for targeted anti‐cancer therapy: promises and perils. EMBO Mol Med 3, 623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gatenby RA & Brown JS (2020) Integrating evolutionary dynamics into cancer therapy. Nat Rev Clin Oncol 17, 675–686. [DOI] [PubMed] [Google Scholar]
- 41. Hutchison DJ (1963) Cross resistance and collateral sensitivity studies in cancer chemotherapy. Adv Cancer Res 7, 235–250. [DOI] [PubMed] [Google Scholar]
- 42. Wang L & Bernards R (2018) Taking advantage of drug resistance, a new approach in the war on cancer. Front Med 12, 490–495. [DOI] [PubMed] [Google Scholar]
- 43. Nussinov R, Tsai C‐J & Jang H (2017) A new view of pathway‐driven drug resistance in tumor proliferation. Trends Pharmacol Sci 38, 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liffers K, Lamszus K & Schulte A (2015) EGFR amplification and glioblastoma stem‐like cells. Stem Cells Int 2015, 427518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schulte A, Günther HS, Martens T, Zapf S, Riethdorf S, Wülfing C, Stoupiec M, Westphal M & Lamszus K (2012) Glioblastoma stem‐like cell lines with either maintenance or loss of high‐level EGFR amplification, generated via modulation of ligand concentration. Clin Cancer Res 18, 1901–1913. [DOI] [PubMed] [Google Scholar]
- 46. Kawamoto T, Mendelsohn J, Le A, Sato GH, Lazar CS & Gill GN (1984) Relation of epidermal growth factor receptor concentration to growth of human epidermoid carcinoma A431 cells. J Biol Chem 259, 7761–7766. [PubMed] [Google Scholar]
- 47. Filmus J, Pollak MN, Cailleau R & Buick RN (1985) MDA‐468, a human breast cancer cell line with a high number of epidermal growth factor (EGF) receptors, has an amplified EGF receptor gene and is growth inhibited by EGF. Biochem Biophys Res Commun 128, 898–905. [DOI] [PubMed] [Google Scholar]
- 48. Colom B, Alcolea MP, Piedrafita G, Hall MWJ, Wabik A, Dentro SC, Fowler JC, Herms A, King C, Ong SH et al. (2020) Spatial competition shapes the dynamic mutational landscape of normal esophageal epithelium. Nat Genet 52, 604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moore L, Leongamornlert D, Coorens THH, Sanders MA, Ellis P, Dentro SC, Dawson KJ, Butler T, Rahbari R, Mitchell TJ et al. (2020) The mutational landscape of normal human endometrial epithelium. Nature 580, 640–646. [DOI] [PubMed] [Google Scholar]
- 50. Martincorena I, Roshan A, Gerstung M, Ellis P, van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM et al. (2015) High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mintz B & Illmensee K (1975) Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci USA 72, 3585–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hendrix MJC, Seftor EA, Seftor REB, Kasemeier‐Kulesa J, Kulesa PM & Postovit L‐M (2007) Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer 7, 246–255. [DOI] [PubMed] [Google Scholar]
- 53. Jahanban‐Esfahlan R, Seidi K, Manjili MH, Jahanban‐Esfahlan A, Javaheri T & Zare P (2019) Tumor cell dormancy: threat or opportunity in the fight against cancer. Cancers 11, 1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vallette FM, Olivier C, Lézot F, Oliver L, Cochonneau D, Lalier L, Cartron P‐F & Heymann D (2019) Dormant, quiescent, tolerant and persister cells: four synonyms for the same target in cancer. Biochem Pharmacol 162, 169–176. [DOI] [PubMed] [Google Scholar]
- 55. Sharma SV, , Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA et al. (2010) A chromatin‐mediated reversible drug‐tolerant state in cancer cell subpopulations. Cell 141, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Manic G, Sistigu A, Corradi F, Musella M, de Maria R & Vitale I (2018) Replication stress response in cancer stem cells as a target for chemotherapy. Semin Cancer Biol 53, 31–41. [DOI] [PubMed] [Google Scholar]
- 57. Senft D & Ronai ZEA (2016) Adaptive stress responses during tumor metastasis and dormancy. Trends Cancer 2, 429–442. [DOI] [PubMed] [Google Scholar]
- 58. Vera‐Ramirez L & Hunter KW (2017) Tumor cell dormancy as an adaptive cell stress response mechanism. F1000Research 6, 2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Essers MAG & Trumpp A (2010) Targeting leukemic stem cells by breaking their dormancy. Mol Oncol 4, 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa‐Murasawa M, Sone A, Najima Y, Takagi S, Aoki Y, Wake A et al. (2010) Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol 28, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen K, Huang Y & Chen J (2013) Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin 34, 732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Moore N & Lyle S (2011) Quiescent, slow‐cycling stem cell populations in cancer: a review of the evidence and discussion of significance. J Oncol 2011, 396076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Medema JP (2013) Cancer stem cells: the challenges ahead. Nat Cell Biol 15, 338–344. [DOI] [PubMed] [Google Scholar]
- 64. Peitzsch C, Tyutyunnykova A, Pantel K & Dubrovska A (2017) Cancer stem cells: the root of tumor recurrence and metastases. Semin Cancer Biol 44, 10–24. [DOI] [PubMed] [Google Scholar]
- 65. Bissell MJ & Hines WC (2011) Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 17, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liggett LA & DeGregori J (2017) Changing mutational and adaptive landscapes and the genesis of cancer. Biochim Biophys Acta 1867, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Barrientos S, Stojadinovic O, Golinko MS, Brem H & Tomic‐Canic M (2018) Growth factors and cytokines in wound healing. Wound Repair Regen 16, 585–601. [DOI] [PubMed] [Google Scholar]
- 68. Kulebyakin KY, Nimiritsky PP & Makarevich PI (2020) Growth factors in regeneration and regenerative medicine: “the cure and the cause”. Front Endocrinol 11, 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shen X, Xiong GL, Jing Y, Xiao H, Cui Y, Zhang YF, Shan Y‐J, Xing S, Yang M, Liu X‐L et al. (2015) The protein kinase C agonist prostratin induces differentiation of human myeloid leukemia cells and enhances cellular differentiation by chemotherapeutic agents. Cancer Lett 356, 686–696. [DOI] [PubMed] [Google Scholar]
- 70. Yeoh JSG & de Haan G (2007) Fibroblast growth factors as regulators of stem cell self‐renewal and aging. Mech Ageing Dev 128, 17–24. [DOI] [PubMed] [Google Scholar]
- 71. Jin K, LaFevre‐Bernt M, Sun Y, Chen S, Gafni J, Crippen D, Logvinova A, Ross CA, Greenberg DA & Ellerby LM (2005) FGF‐2 promotes neurogenesis and neuroprotection and prolongs survival in a transgenic mouse model of Huntington’s disease. Proc Natl Acad Sci USA 102, 18189–18194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Doan PL, Himburg HA, Helms K, Russell JL, Fixsen E, Quarmyne M, Harris JR, Deoliviera D, Sullivan JM, Chao NJ et al. (2013) Epidermal growth factor regulates hematopoietic regeneration after radiation injury. Nat Med 19, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ryu S‐H, Kang KM, Moon SY, Chai GY, Hong JP, Cho K‐O, Kang M‐I, Choi EK & Lee S‐W (2010) Therapeutic effects of recombinant human epidermal growth factor (rhEGF) in a murine model of concurrent chemo‐ and radiotherapy‐induced oral mucositis. J Radiat Res 51, 595–601. [DOI] [PubMed] [Google Scholar]
- 74. Epstein JB, Thariat J, Bensadoun R‐J, Barasch A, Murphy BA, Kolnick L, , Popplewell L & Maghami E (2012) Oral complications of cancer and cancer therapy. CA Cancer J Clin 62, 400–422. [DOI] [PubMed] [Google Scholar]
- 75. Logan RM, Al‐Azri AR, Bossi P, Stringer AM, Joy JK, Soga Y, Ranna V, Vaddi A, Raber‐Durlacher JE, Lalla RV et al. (2020) Systematic review of growth factors and cytokines for the management of oral mucositis in cancer patients and clinical practice guidelines. Support Care Cancer 28, 2485–2498. [DOI] [PubMed] [Google Scholar]
- 76. Luo Y, Ye S, Chen X, Gong F, Lu W & Li X (2017) Rush to the fire: FGF21 extinguishes metabolic stress, metaflammation and tissue damage. Cytokine Growth Factor Rev 38, 59–65. [DOI] [PubMed] [Google Scholar]
- 77. Minard AY, Tan S‐X, Yang P, Fazakerley DJ, Domanova W, Parker BL, Humphrey SJ, Jothi R, Stöckli J & James DE (2016) mTORC1 is a major regulatory node in the FGF21 signaling network in adipocytes. Cell Rep 17, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chung JH, Kang S, Varani J, Lin J, Fisher GJ & Voorhees JJ (2000) Decreased extracellular‐signal‐regulated kinase and increased stress‐activated MAP kinase activities in aged human skin in vivo . J Invest Dermatol 115, 177–182. [DOI] [PubMed] [Google Scholar]
- 79. Enwere E, Shingo T, Gregg C, Fujikawa H, Ohta S & Weiss S (2004) Aging results in reduced epidermal growth factor receptor signaling, diminished olfactory neurogenesis, and deficits in fine olfactory discrimination. J Neurosci 24, 8354–8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kang W & Hébert JM (2015) FGF signaling is necessary for neurogenesis in young mice and sufficient to reverse its decline in old mice. J Neurosci 35, 10217–10223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shiraha H, Gupta K, Drabik K & Wells A (2000) Aging fibroblasts present reduced epidermal growth factor (EGF) responsiveness due to preferential loss of EGF receptors. J Biol Chem 275, 19343–19351. [DOI] [PubMed] [Google Scholar]
- 82. White MC, Holman DM, Boehm JE, Peipins LA, Grossman M & Henley SJ (2014) Age and cancer risk: a potentially modifiable relationship. Am J Prev Med 46, S7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dvorak HF (1986) Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315, 1650–1659. [DOI] [PubMed] [Google Scholar]
- 84. Cahill DP, Kinzler KW, Vogelstein B & Lengauer C (1999) Genetic instability and darwinian selection in tumours. Trends Cell Biol 9, M57–60. [PubMed] [Google Scholar]
- 85. Janssen A & Medema RH (2013) Genetic instability: tipping the balance. Oncogene 32, 4459–4470. [DOI] [PubMed] [Google Scholar]
- 86. Girard P‐M, Berthault N, Kozlac M, Ferreira S, Jdey W, Bhaskara S, Alekseev S, Thomas F & Dutreix M (2020) Evolution of tumor cells during AsiDNA treatment results in energy exhaustion, decrease in responsiveness to signal, and higher sensitivity to the drug. Evol Appl 13, 1673–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Samstein RM, Lee C‐H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ et al. (2019) Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 51, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mandal R, Samstein RM, Lee K‐W, Havel JJ, Wang H, Krishna C, Sabio EY, Makarov V, Kuo F, Blecua P et al. (2019) Genetic diversity of tumors with mismatch repair deficiency influences anti‐PD‐1 immunotherapy response. Science (New York, NY) 364, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Roudko V, Bozkus CC, Orfanelli T, McClain CB, Carr C, O’Donnell T, Chakraborty L, Samstein R, Huang K‐L, Blank SV et al. (2020) Shared immunogenic poly‐epitope frameshift mutations in microsatellite unstable tumors. Cell 183, 1634–1649.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Germano G, Lamba S, Rospo G, Barault L, Magrì A, Maione F, Maione F, Russo M, Crisafulli G, Bartolini A et al. (2017) Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 552, 116–120. [DOI] [PubMed] [Google Scholar]
- 91. Bartok O, Pataskar A, Nagel R, Laos M, Goldfarb E, Hayoun D, Levy R, Körner P‐R, Kreuger IZM, Champagne J et al. (2021) Anti‐tumour immunity induces aberrant peptide presentation in melanoma. Nature 590, 332–337. [DOI] [PubMed] [Google Scholar]
- 92. Shen L, Zhang J, Lee H, Batista MT & Johnston SA (2019) RNA transcription and splicing errors as a source of cancer frameshift neoantigens for vaccines. Sci Rep 9, 14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, Xie Y, Williams NS & Nijhawan D (2017) Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356, eaal3755. [DOI] [PubMed] [Google Scholar]