

Abstract
Although quantum mechanical/molecular mechanics (QM/MM) methods are now routinely applied to the studies of chemical reactions in condensed phases and enzymatic reactions, they may experience technical difficulties when the reactive region is varying over time. For instance, when the solvent molecules are directly participating in the reaction, the exchange of water molecules between the QM and MM regions may occur on a time scale comparable to the reaction time. To cope with this situation, several adaptive QM/MM schemes have been proposed. However, these methods either add significantly to the computational cost or introduce artificial restraints to the system. In this work, we developed a novel adaptive QM/MM scheme and applied it to the study of a nucleophilic addition reaction. In this scheme, the configuration sampling was performed with a small QM region (without solvent molecules), and the thermodynamic properties under another potential energy function with a larger QM region (with a certain number of solvent molecules and/or different levels of QM theory) are computed via extrapolation using the reference-potential method. Our simulation results show that this adaptive QM/MM scheme is numerically stable, at least for the case studied in this work. Furthermore, this method also offers an inexpensive way to examine the convergence of the QM/MM calculation with respect to the size of the QM region.
Graphical Abstract
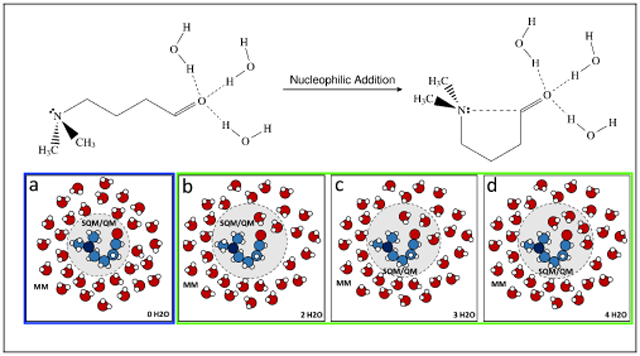
1. Introduction
Hybrid quantum mechanical/molecular mechanical (QM/MM) methods are nowadays well accepted for the simulations of chemical reactions in condensed phases and enzymatic reactions. 1-10 However, applications of these methods are always hindered by their steep computational expense and complexity in domain partitioning. First, in order to determine the reaction mechanism, a long molecular dynamics simulation at a certain ab initio level is needed, from which statistical properties can be extracted reliably. With a subfemtosecond time step for propagation, 106–109 steps of energy and force evaluations are required to reach a nanosecond to microsecond time scale. Second, the size of the QM region matters. Defining the QM region is often based on chemical intuition and is inevitably a compromise between accuracy and efficiency. A small QM region may lead to systematically biased results.11-15 Last but not least, the partitioning of the whole system into the QM and the MM regions is nontrivial, especially when solvent molecules are explicitly participating in the reactions. By including some of the solvent molecules near the solute molecule into the QM region, one can capture the quantum mechanical interaction between the solute and the solvent molecules. However, it introduces another technical difficulty in maintaining dynamic continuity when the exchange of solvent molecules between the QM and the MM regions takes place, especially when an abrupt on-the-fly repartitioning scheme of the QM and MM regions is adopted. To surmount this difficulty, various schemes of adaptive QM/MM methods have been proposed,16-19 which can be categorized broadly into restrained QM/MM schemes20-22 and adaptive QM/MM schemes.23-30 In the former class of schemes, solvent exchange between the QM and the MM regions is prevented by applying a restraining potential. However, the evolution of the system under study is no longer under a realistic Hamiltonian due to the introduction of an artificial restraint, and an unbiasing process is necessary. In the adaptive QM/MM scheme, an effective QM/MM potential is adopted by a weighted average of the potentials from multiple means of partitioning of the system with different combinations of the solute and solvent molecules. This may markably increase the computational expense.
Fortunately, if we are interested in equilibrium thermodynamic properties only, for instance the free energy profile, instead of real dynamics, these properties can be calculated indirectly via the reference-potential approach,31-34 of which the idea has been applied to many studies.35-59 Specifically, the QM/MM partitioning with a fixed number of solvent molecules in the QM region is the target Hamiltonian state denoted by H1. The specific water molecules inside the QM region may vary from one snapshot to another, and the exchange of solvent molecules between the QM and MM regions is just a permutation of the state before the exchange takes place. Meanwhile, another partitioning scheme excluding all the solvent molecules from the QM region corresponds to Hamiltonian H0, which serves as the reference potential. The ensemble average of an operator X under H1 can be computed from the ensemble of H0 via reweighting:60
where 〈·〉 denotes the ensemble average or expectation, and the subscript “0” or “1” indicates the Hamiltonian under which the ensemble is calculated. Here, can be considered as the weight under H1 for the configurations sampled with H0. For a generalized ensemble, the equation can be slightly more complicated, but the idea is the same. Recently, Jia et al proposed a reference-potential method for the free energy calculations at an expensive level of theory using a unique Boltzmann ensemble.43 Li et al extended this method to mixed ensembles from, but not limited to, umbrella sampling (US)60 simulations.49 In these methods, a long simulation using a less expensive Hamiltonian is performed to explore the phase space, and from this simulation a free energy profile corresponding to this Hamiltonian can be estimated using well-established postprocessing methods such as the multistate Bennett acceptance ratio (MBAR)61,62 and the weighted histogram analysis method (WHAM).63-65 Next, a correction in the free energy from this inexpensive Hamiltonian to the Hamiltonian of interest is calculated by using thermodynamic perturbation (TP).66 In this way, expensive direct simulations at the high level Hamiltonian can be avoided. When the correction for a mixed ensemble is calculated, weight factors from the MBAR analysis should be used.49 Therefore, the TP should be carried out with nonuniform weights for the samples. Thermodynamic expectations of any structural properties can be computed in a similar way.49,55
In this work, we put forth a new method for the free energy calculations with an adaptive QM domain for the study of the intramolecular nucleophilic addition reaction of Me2N–(CH2)3–CH=O (NCO) molecule (Fig. 1), utilizing the idea of energy reweighting in the reference-potential methods. It has been shown in a previous study that explicit solvation matters for the thermodynamic property calculations along the reaction.67 The umbrella sampling simulations are carried out only at the hybrid semiempirical/MM level, specifically PM668/MM level, without a single water molecule in the QM region. After the simulation, the trajectories are postprocessed for semiempirical QM/MM or ab initio QM/MM energy calculations with a certain number of solvent molecules included in the QM region, from which the free energy profiles at these levels are obtained. This paper is organized in the following way. In section 2, the theory behind this method and the simulation setup are explained. The results are presented subsequently with discussion in section 3. Finally, a conclusion for this study is presented in section 4.
Figure 1:
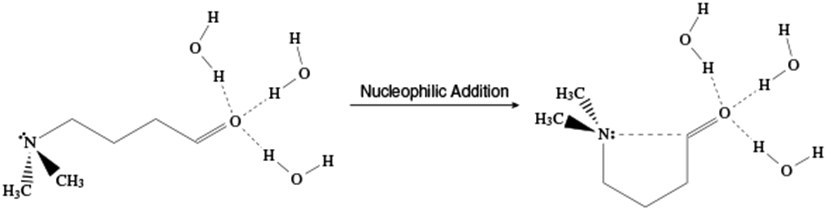
Nucleophilic addition reaction of Me2N–(CH2)3–CH=O (NCO) molecule studied in this work.
2. Method
2.1. Multistate Thermodynamic Perturbation (MsTP) Method
The MsTP method, previously known as MBAR+wTP, was proposed by Li et al.49 recently. Derivation of the MsTP method has been fully presented in ref. 49. In this method, enhanced sampling methods such as umbrella sampling simulations are conducted under a reference (and usually inexpensive) Hamiltonian, for instance, semiempirical (SE) QM/MM. Thermodynamics properties under this reference Hamiltonian can be obtained using MBAR analysis, and then they are corrected to the target Hamiltonian using the weighted thermodynamic perturbation. These steps can be integrated into the MBAR formulation as explained in the following.
With trajectories from K simulations using different potential energy functions Uk as is typically done in umbrella sampling, thermodynamic properties, which depend only on coordinates, under another potential energy function Ut can be computed via
| (1) |
in which Nk is the number of configurations extracted from the kth simulation and
| (2) |
is the unnormalized weight of configuration rn under Uk. is the total number of snapshots. Here, fk is known as the free energy corresponding to Uk and can be obtained by iteratively solving the MBAR equations
| (3) |
In US, the potential energy functions used for configuration sampling are
| (4) |
where U0(r) and Wk(r) are the unbiased potential energy function and the biasing potential for the kth simulation, respectively. Equation 2 can be rewritten as
| (5) |
with ΔUt(r) = Ut(r) – U0(r). Further defining the free energy ft corresponding to the potential energy function Ut(r)
| (6) |
we obtain the normalized weight for configuration rn under the potential energy function Ut(r)
| (7) |
and eq. 1 can be simplified as
| (8) |
It can be easily identified that for a single unbiased simulation with K = 1 and W = 0, eq. 7 can be rewritten as the normal TP equation. Therefore, the idea behind eq. 7 can be seen as multistate thermodynamics perturbation (MsTP). MsTP has been applied to the calculations of free energy profiles for chemical reactions in condensed phase rendered in both one dimensional49 and two dimensional55 reaction coordinates. The computational expense decreases by 2 orders of magnitude comparing with direct QM/MM calculations while maintaining a high accuracy.
Specifically for this adaptive QM/MM calculation, potential energy function U0(r) corresponds to the partitioning with a solvent-free QM region described by a semiempirical Hamiltonian PM6. Wk(ξ(r)) is the restraining potential on a predefined collective variable (CV) ξ(r) that may enhance the phase space sampling in a certain region. is the potential energy function for the partitioning with M solvent molecules in the QM region described by either a semiempirical QM or an ab initio QM level of theory. The prime sign here is to emphasize that the QM level of theory can be either the same as or different from the reference Hamiltonian. If A is an indication function δ of some chosen CV ξ(r)
| (9) |
we have the PMF for as
| (10) |
defined up to an additive constant, and
| (11) |
Similarly, the potential of mean force (PMF) for U0(r) is
| (12) |
in which
| (13) |
Random noise in the potential of mean force from finite sampling was eliminated by a Gaussian smoothing on the density-of-states of .69 Gaussian processes regression (GPR) method70 is used to eliminate the statistical noise in the free energy profile from the MsTP calculation.
2.2. Model Setup
Me2N–(CH2)3–CH=O (NCO) was solvated in a TIP3P water71 sphere with a radius of 20 Å centering at the NCO molecule, which contains 1020 water molecules. The whole system was optimized by 2000 steps of steepest descent algorithm and 3000 steps of conjugate gradient method. The optimized structure was heated up to 300 K in 1 ns and then further relaxed for 10 ns. Periodic boundary condition was not applied, and the water sphere was restrained by a soft half-harmonic potential with a force constant of 10 kcal · mol−1 · Å−2 to prevent evaporation. The integration time step was set to 2 fs, and the SHAKE algorithm was enabled to constrain all the bonds involving hydrogen atoms. The nonbonded interaction was fully counted without any truncation. The van der Waals (vdW) parameters for the NCO molecule were taken from the general AMBER force field (GAFF)72 for the ring-opening structure, and the AM1bcc charges from the reactant configuration were assigned to the NCO molecule. The temperature was regulated to 300 K using the Langevin dynamics with a collision frequency of 1 ps−1.73
2.3. Umbrella Sampling
The phase space exploration was assisted by umbrella sampling.60 The distance between the nitrogen and carbonyl carbon atoms of the NCO molecule was chosen as the CV ξ(r), which ranges from 1.50 to 5.00 Å with an increment of 0.05 Å. Overlap matrix proposed by Klimovich et al.74 was used to monitor the degree of overlap between adjacent simulation windows. Extra windows were added when the overlap between neighboring windows are insufficient, resulting in 84 windows in total. The setup of the restraint potential in each window simulation can be found in the Supporting Information (SI). The central region contains only the NCO molecule, and PM6 was used for its interaction potential. For each US window, the whole system was optimized by 1000 steepest descent steps and 1000 conjugate gradient steps. The relaxed system was heated to 300 K in 100 ps, followed by a 1-ns production simulation. The temperature was maintained at 300 K by using the Langevin dynamics with a collision frequency of 1 ps−1. The integration time step was set to 1 fs with SHAKE turned on for both the MM and QM regions. The configurations were saved every 1 ps for subsequent free energy analysis. The free energy profile at this level was computed using the MBAR analysis method. After that, single point energies under PM6/MM and ωB97X-D75/6-31+G(d,p)/MM levels were obtained for the MsTP calculations. For the single point energy calculations, the QM region was augmented with M = 0, 2, 3, or 4 water molecules that are closest to the oxygen atom in the carbonyl group. These water molecules were chosen on the fly. The propagation of the molecular dynamics simulations and single point energy calculations were carried out using the AMBER 18 package suite.76 Interfacing with Gaussian 16 package77 was utilized when wB97X-D energy calculations were requested.
3. Results and Discussion
3.1. Potential of Mean Force at the PM6/MM Levels
The free energy profiles at the PM6/MM level are shown in Fig. 2. Since in the US simulations the QM region had no water molecules included, the uncertainty of the free energy profile is very small. The free energy profile shows a shallow well at dCN = 4.54 Å for the reactant but a deep well at dCN = 1.66 Å for the product. The barrier for the forward reaction is 3.4 kcal/mol, and the reaction free energy is 4.2 kcal/mol. By including two water molecules in the QM region, the free energy profile shows only a small difference from that with a solvent-free QM region. The locations of the reactant and the product are nearly unchanged, with the new locations being dCN = 4.52 Å and dCN = 1.66 Å for the reactant and the product, respectively. The barrier for the forward reaction becomes 3.5 kcal/mol, and the reaction free energy is 4.3 kcal/mol. The PM6 free energy profile with two water molecules in the QM region has already reached convergence by comparing it with those enveloping more water molecules into the QM region. The results indicate that under the PM6/MM level of theory, the solvent molecules mainly play a role as an electrostatic per-turber to the NCO molecule that only weakly tunes the reaction. As shown in Fig. S1, the magnitude of charge transferred from the solute molecule to the solvent is smaller than 0.1e, even when the C-N bond has formed. In order to compare the adaptive simulation proposed in the current work with the restrained simulation method, we performed another round of umbrella sampling simulation, with additional restraints applied to the distances between the oxygen atoms in the NCO molecule and three water molecules nearby. The harmonic restraining potential centers at d0 = 2.8 Å and the strength kd is 40 kcal · mol−1 · Å−2. The results are plotted in Fig. S2, which shows a nice agreement between the adaptive simulation method and the restrained simulation method.
Figure 2:

Free energy profiles at the PM6/MM levels with different numbers of water molecules in the QM region. The shaded areas are the 95% confidence intervals.
3.2. Potential of Mean Force at the DFT/MM Levels
Extrapolation to the DFT/MM level using the US trajectories from the PM6/MM simulations is also possible, and the free energy profiles are shown in Fig. 3. Similar to the results at the PM6/MM level, the locations of the reactant, the products and the transition states are nearly independent of the number of water molecules in the QM region. Without water molecules in the QM region, the free energy barrier for the forward reaction and the reaction free energy are 2.0 kcal/mol and −1.6 kcal/mol, respectively. Both of them are much smaller than those under the PM6/MM level of theory in their absolute values. An early transition state can be observed, which has a longer C–N bond length than that under the PM6/MM level of theory. This is not unexpected, since the basis set in the DFT calculation was much more diffusive than that in the PM6 calculations. When two water molecules are added to the QM region, the free energy profile shows large deviations from the one at the same level of theory but with no solvent molecules in the QM region, especially at the product side. When solvent molecules in the MM region are represented as background charges that polarize the electronic structure of the QM region, charge transfer is not allowed between the QM solute molecule and the MM solvent molecules. When two nearest solvent molecules are bracketed into the QM region, the water molecules can accommodate the extra electrons around the oxygen atom in the carbonyl group, especially after the formation of the C–N bond. Therefore, the product is stabilized by 1.6 kcal/mol, and the reaction free energy becomes −3.3 kcal/mol. The free energy barrier for the forward reaction increases to 2.4 kcal/mol. Adding more water molecules into the QM region does not significantly change the profile. With three water molecules in the QM region, the reactant and the product are located at dCN = 4.52 Å and 1.64 Å, and the free energy barrier and the reaction free energy are 2.3 kcal/mol and −3.3 kcal/mol. With four water molecules in the QM region, the reactant and the product are located at dCN = 4.50 Å and 1.66 Å, and the free energy barrier and the reaction free energy are 2.1 kcal/mol and −3.1 kcal/mol. Considering the uncertainties in the free energy profiles, these numbers are statistically identical. Therefore, when two water molecules are included in the QM region, the free energy profile has converged.
Figure 3:

Free energy profiles at the DFT/MM levels with different numbers of water molecules in the QM region. The shaded areas are the 95% confidence intervals.
The variations of the CM5 charges78 of the polar atoms in the NCO molecule during the reaction are shown in Fig. 4, with the atomic charges of the bonded hydrogen atoms merged into those of the heavy atoms. This shows that when the nitrogen atom approaches the carbon atom in the carbonyl group, the lone pair electrons of the nitrogen atom become shared electrons between the nitrogen atom and the carbon atom, and push the shared electrons in the carbonyl group to the oxygen side. Some portion of the electrons drifts away from oxygen atom in the carbonyl group to the water molecules hydrogen-bonded to the carbonyl group. As a result, the CM5 charge of the nitrogen atom rises (becomes less negative) by about 0.187e, that of the oxygen atom in the carbonyl group declines by about 0.162e, and the four water molecules near the carbonyl oxygen atom accept about 0.228 electron altogether. Since the carbonyl carbon atom accepts electrons from the nitrogen atom but donates electrons to the oxygen atom, its CM5 charge decreases by only 0.074e. This agrees with the previous observation of the N+∣C–O− pattern.79 Since the bond order between the carbon atom and the oxygen atom in the carbonyl group decreases, the bond length increases during the reaction, as can be seen in Fig. 5. In the reactant region, the CO bond distance remains around 1.22 Å. When the distance between the nitrogen atom and the carbon atom is smaller than 3.0 Å (on the product side), the CO bond length gets elongated quickly.
Figure 4:

Alternations of atomic CM5 charges along the reaction at the DFT/MM level with four water molecules in the QM region. The shaded areas are the 95% confidence intervals.
Figure 5:

Variations of the C=O bond length along the reaction at the DFT/MM level with four water molecules in the QM region. The shaded area is the 95% confidence interval.
If we define a buffer region between the QM and the MM regions, this method can be straightforwardly combined with the permuted adaptive partitioning (PAP) scheme24 by setting the PAP Hamiltonian at a certain QM level of theory as the target Hamiltonian. In spite of its rigor, the PAP partition scheme is computationally very demanding and scales poorly with the size of the buffer region. Therefore, only one permutation is considered in this work, in which the nearest M solvent molecules are bracketed into the QM region. The consequence is the loss of the detailed balance and the continuity of the trajectory because of the introduction of human intervention in picking the water molecules to be included in the QM region. Other partition schemes can be adopted. However, as shown in Fig. S3, the permutation we have used (bracketing the nearest M solvent into the QM region) in this work has the largest impact on the free energy change from the reactant to the product, since it can best relax the extra electron density around the oxygen atom in the NCO molecule once the C–N bond has formed. The other scheme (with the second and third nearest water molecules in the QM region) underestimates the reaction free energy, due to that the magnitude of the electron transfer from the NCO molecule to the solvent is underestimated, especially at the product side.
4. Conclusion
In this work, we proposed a novel method for adaptive QM/MM simulations of chemical reaction in a homogeneous environment, which is based on the reference-potential method and can be easily implemented. With this method, extrapolations to a different level of theory and/or to a different size of the QM region are made possible. The uncertainty increases with the “aggressiveness” of the extrapolation. Increasing the number of water molecules being bracketed into the QM region increases the standard deviation of the potential of mean force. Fortunately, this numerical difficulty can be easily solved by extending the length of the simulation at the low level of theory. This method also offers a convenient way to check the convergence of the QM/MM calculations with respect to the size of the QM region even in a heterogeneous but invariant embedding environment. Semiempirical methods such as PM6 should be used with care, due to the difficulty in handling the charge transfer effect with minimum basis sets.
Supplementary Material
Acknowledgement
Y. Mei wants to express his gratitude to Dr. Wei Yang at Florida State University and Dr. Hai Lin at University of Colorado Denver for some insightful discussions. Y. Mei is supported by the National Natural Science Foundation of China (Grant Nos. 21773066 and 22073030). Y. Mo is supported by the National Natural Science Foundation of China (Grant No. 21973030). W.H. is supported by the Fundamental Research Funds for the Central Universities. Y.S. is supported by the National Institutes of Health (Grant No. R01GM135392). CPU time was provided by the Supercomputer Center of East China Normal University (ECNU Public Platform for Innovation No. 001).
References
- (1).Warshel A; Levitt M Theoretical Studies of Enzymic Reactions: Dielectric, Electrostatic and Steric Stabilization of the Carbonium Ion in the Reaction of Lysozyme. J. Mol. Biol 1976, 103, 227–249. [DOI] [PubMed] [Google Scholar]
- (2).Field MJ; Bash PA; Karplus M A Combined Quantum Mechanical and Molecular Mechanical Potential for Molecular Dynamics Simulations. J. Comput. Chem 1990, 11, 700–733. [Google Scholar]
- (3).Monard G; Merz KM Combined Quantum Mechanical/Molecular Mechanical Methodologies Applied to Biomolecular Systems. Acc. Chem. Res 1999, 32, 904–911. [Google Scholar]
- (4).Friesner RA; Guallar V Ab Initio Quantum Chemical and Mixed Quantum Mechanics/Molecular Mechanics (QM/MM) Methods for Studying Enzymatic Catalysis. Annu. Rev. Phys. Chem 2005, 56, 389–427. [DOI] [PubMed] [Google Scholar]
- (5).Gao J; Ma S; Major DT; Nam K; Pu J; Truhlar DG Mechanisms and Free Energies of Enzymatic Reactions. Chem. Rev 2006, 106, 3188–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lin H; Truhlar DG QM/MM: What Have We Learned, Where Are We, and Where Do We Go from Here? Theor. Chem. Acc 2006, 117, 185. [Google Scholar]
- (7).Hu H; Yang W Free Energies of Chemical Reactions in Solution and in Enzymes with ab initio Quantum Mechanics/Molecular Mechanics Methods. Annu. Rev. Phys. Chem 2008, 59, 573–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Senn HM; Thiel W QM/MM Methods for Biomolecular Systems. Angew. Chem., Int. Ed 2009, 48, 1198–1229. [DOI] [PubMed] [Google Scholar]
- (9).Brunk E; Rothlisberger U Mixed Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulations of Biological Systems in Ground and Electronically Excited States. Chem. Rev 2015, 115, 6217–6263. [DOI] [PubMed] [Google Scholar]
- (10).Chung LW; Sameera WMC; Ramozzi R; Page AJ; Hatanaka M; Petrova GP; Harris TV; Li X; Ke Z; Liu F; Li H-B; Ding L; Morokuma K The ONIOM Method and Its Applications. Chem. Rev 2015, 115, 5678–5796. [DOI] [PubMed] [Google Scholar]
- (11).Solt I; Kulhánek P; Simon I; Winfield S; Payne MC; Csányi G; Fuxreiter M Evaluating Boundary Dependent Errors in QM/MM Simulations. J. Phys. Chem. B 2009, 113, 5728–5735. [DOI] [PubMed] [Google Scholar]
- (12).Sumowski CV; Ochsenfeld C A Convergence Study of QM/MM Isomerization Energies with the Selected Size of the QM Region for Peptidic Systems. J. Phys. Chem. A 2009, 113, 11734–11741. [DOI] [PubMed] [Google Scholar]
- (13).Liao R-Z; Thiel W Convergence in the QM-only and QM/MM Modeling of Enzymatic Reactions: A Case Study for Acetylene Hydratase. J. Comput. Chem 2013, 34, 2389–2397. [DOI] [PubMed] [Google Scholar]
- (14).Kulik HJ; Zhang J; Klinman JP; Martínez TJ How Large Should the QM Region Be in QM/MM Calculations? The Case of Catechol O-Methyltransferase. J. Phys. Chem. B 2016, 120, 11381–11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mehmood R; Kulik HJ Both Configuration and QM Region Size Matter: Zinc Stability in QM/MM Models of DNA Methyltransferase. J. Chem. Theory Comput 2020, 16, 3121–3134. [DOI] [PubMed] [Google Scholar]
- (16).Bulo RE; Michel C; Fleurat-Lessard P; Sautet P Multiscale Modeling of Chemistry in Water: Are We There Yet? J. Chem. Theory Comput 2013, 9, 5567–5577. [DOI] [PubMed] [Google Scholar]
- (17).Pezeshki S; Lin H In Quantum Modeling of Complex Molecular Systems; Rivail J-L, Ruiz-Lopez M, Assfeld X, Eds.; Springer International Publishing: Cham, 2015; pp 93–113. [Google Scholar]
- (18).Zheng M; Waller MP Adaptive Quantum Mechanics/Molecular Mechanics Methods. WIREs Comput. Mol. Sci 2016, 6, 369–385. [Google Scholar]
- (19).Duster AW; Wang C-H; Garza CM; Miller DE; Lin H Adaptive Quantum/Molecular Mechanics: What Have We Learned, Where Are We, and Where Do We Go from Here? WIREs Comput. Mol. Sci 2017, 7, e1310. [Google Scholar]
- (20).Rowley CN; Roux B The Solvation Structure of Na+ and K+ in Liquid Water Determined from High Level ab Initio Molecular Dynamics Simulations. J. Chem. Theory Comput 2012, 8, 3526–3535. [DOI] [PubMed] [Google Scholar]
- (21).Shiga M; Masia M Boundary Based on Exchange Symmetry Theory for Multilevel Simulations. I. Basic Theory. J. Chem. Phys 2013, 139, 044120. [DOI] [PubMed] [Google Scholar]
- (22).Takahashi H; Kambe H; Morita A A Simple and Effective Solution to the Constrained QM/MM Simulations. J. Chem. Phys 2018, 148, 134119. [DOI] [PubMed] [Google Scholar]
- (23).Kerdcharoen T; Morokuma K ONIOM-XS: An Extension of the ONIOM Method for Molecular Simulation in Condensed Phase. Chem. Phys. Lett 2002, 355, 257–262. [Google Scholar]
- (24).Heyden A; Lin H; Truhlar DG Adaptive Partitioning in Combined Quantum Mechanical and Molecular Mechanical Calculations of Potential Energy Functions for Multiscale Simulations. J. Phys. Chem. B 2007, 111, 2231–2241. [DOI] [PubMed] [Google Scholar]
- (25).Bulo RE; Ensing B; Sikkema J; Visscher L Toward a Practical Method for Adaptive QM/MM Simulations. J. Chem. Theory Comput 2009, 5, 2212–2221. [DOI] [PubMed] [Google Scholar]
- (26).Bernstein N; Várnai C; Solt I; Winfield SA; Payne MC; Simon I; Fuxreiter M; Csányi G QM/MM Simulation of Liquid Water with an Adaptive Quantum Region. Phys. Chem. Chem. Phys 2012, 14, 646–656. [DOI] [PubMed] [Google Scholar]
- (27).Takenaka N; Kitamura Y; Koyano Y; Nagaoka M The Number-adaptive Multi-scale QM/MM Molecular Dynamics Simulation: Application to Liquid Water. Chem. Phys. Lett 2012, 524, 56–61. [DOI] [PubMed] [Google Scholar]
- (28).Waller MP; Kumbhar S; Yang J A Density-Based Adaptive Quantum Mechanical/Molecular Mechanical Method. ChemPhysChem 2014, 15, 3218–3225. [DOI] [PubMed] [Google Scholar]
- (29).Watanabe HC; Kubař T; Elstner M Size-Consistent Multipartitioning QM/MM: A Stable and Efficient Adaptive QM/MM Method. J. Chem. Theory Comput 2014, 10, 4242–4252. [DOI] [PubMed] [Google Scholar]
- (30).Field MJ An Algorithm for Adaptive QC/MM Simulations. J. Chem. Theory Comput 2017, 13, 2342–2351. [DOI] [PubMed] [Google Scholar]
- (31).Gao J Absolute Free Energy of Solvation from Monte Carlo Simulations Using Combined Quantum and Molecular Mechanical Potentials. J. Phys. Chem 1992, 96, 537–540. [Google Scholar]
- (32).Gao J; Xia X A Priori Evaluation of Aqueous Polarization Effects through Monte Carlo QM-MM Simulations. Science 1992, 258, 631–635. [DOI] [PubMed] [Google Scholar]
- (33).Muller RP; Warshel A Ab Initio Calculations of Free Energy Barriers for Chemical Reactions in Solution. J. Phys. Chem 1995, 99, 17516–17524. [PubMed] [Google Scholar]
- (34).Bentzien J; Muller RP; Florián J; Warshel A Hybrid ab initio Quantum Mechanics/Molecular Mechanics Calculations of Free Energy Surfaces for Enzymatic Reactions: The Nucleophilic Attack in Subtilisin. J. Phys. Chem. B 1998, 102, 2293–2301. [Google Scholar]
- (35).Rod TH; Ryde U Quantum Mechanical Free Energy Barrier for an Enzymatic Reaction. Phys. Rev. Lett 2005, 94, 138302. [DOI] [PubMed] [Google Scholar]
- (36).Beierlein FR; Michel J; Essex JW A Simple QM/MM Approach for Capturing Polarization Effects in Protein-Ligand Binding Free Energy Calculations. J. Phys. Chem. B 2011, 115, 4911–4926. [DOI] [PubMed] [Google Scholar]
- (37).König G; Boresch S Non-Boltzmann Sampling and Bennett’s Acceptance Ratio Method: How to Profit from Bending the Rules. J. Comput. Chem 2011, 32, 1082–1090. [DOI] [PubMed] [Google Scholar]
- (38).Heimdal J; Ryde U Convergence of QM/MM Free-Energy Perturbations Based on Molecular-Mechanics or Semiempirical Simulations. Phys. Chem. Chem. Phys 2012, 14, 12592–12604. [DOI] [PubMed] [Google Scholar]
- (39).Polyak I; Benighaus T; Boulanger E; Thiel W Quantum Mechanics/Molecular Mechanics Dual Hamiltonian Free Energy Perturbation. J. Chem. Phys 2013, 139, 064105. [DOI] [PubMed] [Google Scholar]
- (40).König G; Hudson PS; Boresch S; Woodcock HL Multiscale Free Energy Simulations: An Efficient Method for Connecting Classical MD Simulations to QM or QM/MM Free Energies Using Non-Boltzmann Bennett Reweighting Schemes. J. Chem. Theory Comput 2014, 10, 1406–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hudson PS; Woodcock HL; Boresch S Use of Nonequilibrium Work Methods to Compute Free Energy Differences Between Molecular Mechanical and Quantum Mechanical Representations of Molecular Systems. J. Phys. Chem. Lett 2015, 6, 4850–4856. [DOI] [PubMed] [Google Scholar]
- (42).Hudson PS; White JK; Kearns FL; Hodoscek M; Boresch S; Woodcock HL Efficiently Computing Pathway Free Energies: New Approaches Based on Chain-of-Replica and Non-Boltzmann Bennett Reweighting Schemes. Biochim. Biophys. Acta, Gen. Subj 2015, 1850, 944–953. [DOI] [PubMed] [Google Scholar]
- (43).Jia X; Wang M; Shao Y; Koenig G; Brooks BR; Zhang JZH; Mei Y Calculations of Solvation Free Energy through Energy Reweighting from Molecular Mechanics to Quantum Mechanics. J. Chem. Theory Comput 2016, 12, 499–511. [DOI] [PubMed] [Google Scholar]
- (44).Shen L; Wu J; Yang W Multiscale Quantum Mechanics/Molecular Mechanics Simulations with Neural Networks. J. Chem. Theory Comput 2016, 12, 4934–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Dybeck EC; König G; Brooks BR; Shirts MR Comparison of Methods To Reweight from Classical Molecular Simulations to QM/MM Potentials. J. Chem. Theory Comput 2016, 12, 1466–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kearns FL; Hudson PS; Woodcock HL; Boresch S Computing Converged Free Energy Differences between Levels of Theory via Nonequilibrium Work Methods: Challenges and Opportunities. J. Comput. Chem 2017, 38, 1376–1388. [DOI] [PubMed] [Google Scholar]
- (47).Wang M; Li P; Jia X; Liu W; Shao Y; Hu W; Zheng J; Brooks BR; Mei Y Efficient Strategy for the Calculation of Solvation Free Energies in Water and Chloroform at the Quantum Mechanical/Molecular Mechanical Level. J. Chem. Inf. Model 2017, 57, 2476–2489. [DOI] [PubMed] [Google Scholar]
- (48).Li P; Liu F; Jia X; Shao Y; Hu W; Zheng J; Mei Y Efficient Computation of Free Energy Surfaces of Diels-Alder Reactions in Explicit Solvent at Ab Initio QM/MM Level. Molecules 2018, 23, 2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Li P; Jia X; Pan X; Shao Y; Mei Y Accelerated Computation of Free Energy Profile at ab Initio Quantum Mechanical/Molecular Mechanics Accuracy via a Semi-Empirical Reference Potential. I. Weighted Thermodynamics Perturbation. J. Chem. Theory Comput 2018, 14, 5583–5596. [DOI] [PubMed] [Google Scholar]
- (50).Wang M; Mei Y; Ryde U Predicting Relative Binding Affinity Using Nonequilibrium QM/MM Simulations. J. Chem. Theory Comput 2018, 14, 6613–6622. [DOI] [PubMed] [Google Scholar]
- (51).Hudson PS; Boresch S; Rogers DM; Woodcock HL Accelerating QM/MM Free Energy Computations via Intramolecular Force Matching. J. Chem. Theory Comput 2018, 14, 6327–6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Konig G; Brooks BR; Thiel W; York DM On the Convergence of Multi-Scale Free Energy Simulations. Mol. Simulat 2018, 44, 1062–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wang M; Mei Y; Ryde U Host-guest Relative Binding Affinities at Density-Functional Theory Level from Semiempirical Molecular Dynamics Simulations. J. Chem. Theory Comput 2019, 15, 2659–2671. [DOI] [PubMed] [Google Scholar]
- (54).Pan X; Li P; Ho J; Pu J; Mei Y; Shao Y Accelerated Computation of Free Energy Profile at ab initio Quantum Mechanical/Molecular Mechanical Accuracy via a Semi-Empirical Reference Potential. II. Recalibrating Semi-Empirical Parameters with Force Matching. Phys. Chem. Chem. Phys 2019, 21, 20595–20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Li P; Liu F; Shao Y; Mei Y Computational Insights into Endo/Exo Selectivity of Diels-Alder Reaction in Explicit Solvent at ab Initio Quantum Mechanical/Molecular Mechanical Level. J. Phys. Chem. B 2019, 123, 5131–5138. [DOI] [PubMed] [Google Scholar]
- (56).Hudson PS; Woodcock HL; Boresch S Use of Interaction Energies in QM/MM Free Energy Simulations. J. Chem. Theory Comput 2019, 15, 4632–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Giese TJ; York DM Development of a Robust Indirect Approach for MM → QM Free Energy Calculations That Combines Force-Matched Reference Potential and Bennett’s Acceptance Ratio Methods. J. Chem. Theory Comput 2019, 15, 5543–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Piccini G; Parrinello M Accurate Quantum Chemical Free Energies at Affordable Cost. J. Phys. Chem. Lett 2019, 10, 3727–3731. [DOI] [PubMed] [Google Scholar]
- (59).Chung S; Choi SM; Lee W; Cho KH; Rhee YM Free Energy Level Correction by Monte Carlo Resampling with Weighted Histogram Analysis Method. Chin. J. Chem. Phys 2020, 33, 183–195. [Google Scholar]
- (60).Torrie GM; Valleau JP Nonphysical Sampling Distributions in Monte Carlo Free-energy Estimation: Umbrella Sampling. J. Comput. Phys 1977, 23, 187–199. [Google Scholar]
- (61).Shirt MR; Chodera JD Statistically Optimal Analysis of Samples from Multiple Equilibrium States. J. Chem. Phys 2008, 129, 124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Shirts MR Reweighting from the Mixture Distribution as a Better Way to Describe the Multistate Bennett Acceptance Ratio. arXiv.org 2017, 1704.00891, https://arxiv.org/abs/1704.00891. [Google Scholar]
- (63).Ferrenberg AM; Swendsen RH Optimized Monte Carlo Data Analysis. Phys. Rev. Lett 1989, 63, 1195–1198. [DOI] [PubMed] [Google Scholar]
- (64).Souaille M; Roux B Extension to the Weighted Histogram Analysis Method: Combining Umbrella Sampling with Free Energy Calculations. Comput. Phys. Commun 2001, 135, 40–57. [Google Scholar]
- (65).Gallicchio E; Andrec M; Felts AK; Levy RM Temperature Weighted Histogram Analysis Method, Replica Exchange, and Transition Paths. J. Phys. Chem. B 2005, 109, 6722–6731. [DOI] [PubMed] [Google Scholar]
- (66).Zwanzig RW High-Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases. J. Chem. Phys 1954, 22, 1420. [Google Scholar]
- (67).Boereboom JM; Fleurat-Lessard P; Bulo RE Explicit Solvation Matters: Performance of QM/MM Solvation Models in Nucleophilic Addition. J. Chem. Theory Comput 2018, 14, 1841–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Stewart JJP Optimization of Parameters for Semi-Empirical Methods V: Modification of NDDO Approximations and Application to 70 Elements. J. Mol. Model 2007, 13, 1173–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Hu W; Li P; Wang J-N; Xue Y; Mo Y; Zheng J; Pan X; Shao Y; Mei Y Accelerated Computation of Free Energy Profile at Ab Initio Quantum Mechanical/Molecular Mechanics Accuracy via a Semiempirical Reference Potential. 3. Gaussian Smoothing on Density-of-States. J. Chem. Theory Comput 2020, doi: 10.1021/acs.jctc.0c00794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Rasmussen C; Williams C Gaussian Processes for Machine Learning; MIT Press, 2006; pp 7–32. [Google Scholar]
- (71).Jorgensen WL; Chandresekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (72).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and Testing of a General Amber Force Field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (73).Langevin P Sur la Théorie du Mouvement Brownien. C. R. Acad. Sci. (Paris) 1908, 146, 530–533. [Google Scholar]
- (74).Klimovich PV; Shirts MR; Mobley DL Guidelines for the Analysis of Free Energy Calculations. J. Comput. Aid. Mol. Des 2015, 29, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Chai J-D; Head-Gordon M Long-range Corrected Hybrid Density Functionals with Damped Atom-atom Dispersion Corrections. Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- (76).Case DA; Ben-Shalom IY; Brozell SR; Cerutti DS; Cheatham TE III; Cruzeiro VWD; Darden TA; Duke RE; Ghoreishi D; Gilson MK; Gohlke H; Goetz AW; Greene D; Harris R; Homeyer N; Izadi S; Kovalenko A; Kurtzman T; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein DJ; Merz KM; Miao Y; Monard G; Nguyen C; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe DR; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling CL; Smith J; Salomon-Ferrer R; Swails J; Walker RC; Wang J; Wei H; Wolf RM; Wu X; Xiao L; York DM; Kollman PA AMBER 18, University of California, San Francisco. 2018. [Google Scholar]
- (77).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Hender-son T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16 Revision B.01 2016; Gaussian Inc. Wallingford CT. [Google Scholar]
- (78).Marenich AV; Jerome SV; Cramer CJ; Truhlar DG Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases. J. Chem. Theory Comput 2012, 8, 527–541. [DOI] [PubMed] [Google Scholar]
- (79).Pilmé J; Berthoumieux H; Robert V; Fleurat-Lessard P Unusual Bond Formation in Aspartic Protease Inhibitors: A Theoretical Study. Chem. - Eur. J 2007, 13, 5388–5393. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.