

Abstract
Angiogenesis, the formation of new blood vessels, has become a well-established hallmark of cancer. Its functional importance for the manifestation and progression of tumors has been further validated by the beneficial therapeutic effects of angiogenesis inhibitors, most notably ones targeting the vascular endothelial growth factor (VEGF) signaling pathways. However, with the transient and short-lived nature of the patient response, it has become evident that tumors have the ability to adapt to the pressures of vascular growth restriction. Several escape mechanisms have been described that adapt tumors to therapy-induced low-oxygen tension by either reinstating tumor growth by vascular rebound or by altering tumor behavior without the necessity to reinitiate revascularization. We review here two bypass mechanisms that either instigate angiogenic and immune-suppressive polarization of intratumoral innate immune cells to facilitate VEGF-independent angiogenesis or enable metabolic adaptation and reprogramming of endothelial cells and tumor cells to adapt to low-oxygen tension.
Neovascularization, the formation of new blood vessels, has been defined by Hanahan and Weinberg (2011) as one of the six pivotal hallmarks of cancer because it is a necessary process to enable sufficient oxygen supply for an expanding tumor and to dispose of waste products. Vascular sprouting is the most prominent mechanism by which new vessels arise from preexisting capillaries or postcapillary venules. Thereby, endothelial cells become activated by and move toward a gradient of proangiogenic factors, forming expanding sprouts with proliferating endothelial stalk cells and migrating tip cells at the leading edge until tip cells anastomose with cells from neighboring sprouts to connect the newly formed vessel structures. Consequently, vascular growth is terminated and vessels mature and stabilize with the formation of a basement membrane and recruitment and embracement of pericytes (Carmeliet 2005; Adams and Alitalo 2007). Numerous activating and inhibiting factors including the vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and angiopoietin tyrosine kinase receptor families and various members of the CXC and CC chemokines orchestrate this multistep process (Bergers and Benjamin 2003; Santoni et al. 2014). However, although blood vessel formation is tightly regulated during physiological conditions, tumors have lost the appropriate balances between positive and negative angiogenic controls. Once tumor angiogenesis is induced, it remains activated leading to a continually and abnormally expanding tumor vasculature (Baluk et al. 2005; Carmeliet 2005). As a consequence, tumor vessels become leaky, aggravating tumor cell intravasation and dissemination and causing a sluggish and irregular blood flow that, together with an expanding tumor mass, increases inter-stitial pressure and hypoxia, further exacerbating an an giogenic response (Jain 2002; Carmeliet and Jain 2011; Potente et al. 2011).
There is increasing evidence that angiogenesis and inflammation, although being distinct and separable processes, are closely related events that are in part regulated by common chemokines. Indeed, VEGF, one of the most prominent angiogenic factors, which is over-expressed in numerous tumor types, not only promotes angiogenesis but also conveys suppressive effects on different immune cell constituents (Motz and Coukos 2011). VEGF blocks dendritic cell maturation, enhances the expression of the negative checkpoint regulator programmed cell death ligand 1 (PDL1) on dendritic cells, and activates antigen-specific regulatory T cells (Tregs) in a neuropilin-1 (NRP1)-dependent manner (Gabrilovich et al. 1996; Motz and Coukos 2011). Further, VEGF generates an immune-suppressive vasculature by enhancing PDL1 expression and reducing intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1) on endothelial cells to diminish leukocyte–endothelial interactions and consequently block infiltration of cytotoxic T cells into the tumor (Griffioen et al. 1996; Bouzin et al. 2007; Motz and Coukos 2013).
Recognition of tumor angiogenesis as a hallmark of cancer, together with the functional importance of VEGF in tumor angiogenesis and immune suppression, has led to the development of various VEGF and VEGF receptor inhibitors (Jayson et al. 2016) Consequently, bevacizumab (Avastin, Genentech/Roche), a VEGF ligand-trapping monoclonal antibody, was the first U.S. Federal Drug Administration (FDA)-approved antiangiogenic drug followed by the angiokinase inhibitors sorafenib (Nexavar, Bayer), sunitinib (Sutent, Pfizer), and axitinib (Inlyta, Pfizer) and others, that target the VEGF receptor (VEGFR) tyrosine kinases and other receptor tyrosine kinases (RTKs) (Ferrara 2004; Jain 2014; Vasudev and Reynolds 2014; McIntyre and Harris 2015). Although these antiangiogenic drugs have produced encouraging and favorable effects in a subset of cancer patients, they have only increased progression-free survival but have had little effect on overall survival. The basis for these treatment limitations lies in part in the development of multiple forms of adaptive resistance to angiogenic inhibitors in which tumors either reinstate growth by neovascularization (Casanovas et al. 2005; Shojaei and Ferrara 2008; Rivera and Bergers 2015), bypass the need for revascularization by co-opting blood vessels, or become more invasive or undergo metabolic reprogramming to withstand the deleterious effects of therapy (Rubenstein et al. 2000; Ebos et al. 2007; Bergers and Hanahan 2008; Paez-Ribes et al. 2009; Lu et al. 2012; Sennino et al. 2012). Importantly, several of these adaptation mechanisms are triggered by hypoxia that is generated by therapy-induced vascular pruning of tumors (Bergers and Hanahan 2008; McIntyre and Harris 2015). The molecular responses to hypoxia are predominantly mediated by a family of hypoxia-inducible factors (HIFs), which are composed of an α and a β subunit, and become stabilized and active under low-oxygen tension (Liao and Johnson 2007; Semenza 2011; Palazon et al. 2014). HIF proteins coordinate an extensive transcriptional program that ensures metabolic and vascular adaptation to low-oxygen tension by producing a plethora of factors implicated in different aspects of cellular homeostasis, including survival, metabolism, invasion, and angiogenesis. In this review, we will discuss two hypoxia-dependent adaptation mechanisms that enable tumors to escape the effects of antiangiogenic therapy: (1) the recruitment and polarization of innate immune cells to recreate a proangiogenic and immune-suppressive environment and, (2) metabolic reprogramming of tumor and host cells to endure tumor propagation in the absence of revascularization.
INNATE IMMUNE CELLS DRIVE RESPONSE AND RELAPSE TO ANTIANGIOGENIC THERAPY
Intratumoral hypoxia drives the mobilization and retention of various bone marrow–derived cells (BMDCs) where they become an additional source of chemokines and cytokines to support tumor progression by promoting angiogenesis and suppressing antitumor immunity (Coussens et al. 2000; Giraudo et al. 2004; Lin et al. 2006; Shojaei and Ferrara 2007b; Du et al. 2008). Under normal conditions, innate immune cells first display immune-stimulatory characteristics to eliminate potential infections when they enter sites of tissue injury but then become immune-suppressive and angiogenic to hinder excessive inflammation and allow tissue repair with the formation of blood vessels and reepithelialization (Kasuya and Tokura 2014). Tumors, however, co-opt and modify the tissue repair program to escape immune-surveillance by converting innate immune cells from an immune-stimulating to an immune-suppressive and angiogenic phenotype (Motz and Coukos 2011).
Although tumors can inherently produce myeloid cell–recruiting factors (e.g., G-CSF, CSF-1, GM-CSF), hypoxia activates many of these molecules and pathways that not only attract innate immune cells but also endorse their angiogenic and immune-suppressive characteristics (Schioppa et al. 2003; Staller et al. 2003; Murdoch et al. 2004; Grunewald et al. 2006; Du et al. 2008; Casazza et al. 2013). VEGF is one of the most prominent hypoxia-regulated angiogenic factors that in addition to affecting endothelial cells can also serve as a mobilizer and chemoattractant for myeloid cells via VEGF receptor 2 (VEGFR2) on monocytes (Dineen et al. 2008). Further, CXCL12 (SDF1α), implicated in the retention of myeloid cells, is induced by HIF-1α (Grunewald et al. 2006; Du et al. 2008), as is its chemokine receptor CXCR4 (Schioppa et al. 2003; Staller et al. 2003). Semaphorin3A (Sema3A) is another hypoxia-induced factor in tumors that is implicated in macrophage recruitment and subsequent angiogenesis. Sema3A mediates Nrp-1-dependent signaling of a PlexinA1/PlexinA4/VEGFR1 holoreceptor complex that leads to VEGFR1 activation in tumor-associated microphages (TAMs) and their subsequent migration into hypoxic regions where they secrete various immune-suppressive and proangiogenic factors. Upon TAM arrival in the hypoxic environment, Nrp1 expression is repressed, which terminates the migratory response of TAMs to Sema3A. Interestingly, hypoxia-dependent Nrp1 repression is facilitated by HIF2α -mediated activation of the nuclear factor κB (NF-κB) pathway. Loss of Nrp-1 on macrophages prevents TAM infiltration in hypoxic regions and thereby maintains an immune-stimulatory phenotype, resulting in delayed tumor growth, which is in turn characterized by impaired vascularization and improved antitumor immunity (Casazza et al. 2013).
Given that hypoxia is a major driver of myeloid-cell recruitment (Du et al. 2008; Mazzieri et al. 2011), it is conceivable that antiangiogenic therapy-induced hypoxia also induces factors that mobilize cells from the bone marrow and attract them to the tumor site. Indeed, several preclinical studies have now shown that tumor-associated innate immune cells sustain angiogenesis during anti-angiogenic therapy, by stimulating VEGF-independent pathways. For example, macrophages induce the expression of various angiogenic molecules, including FGF-1,2, MMP9, and Ang2, in response to angiogenic blockade (Casanovas et al. 2005; Fischer et al. 2007; Rigamonti et al. 2014), and Gr1+ immune cells were found to convey resistance to anti-VEGF treatment via secretion of the angiogenic PKR-1/2 ligand Bv8 (Shojaei and Ferrara 2007a,b). Notably, the different intratumoral myeloid cell populations found in tumors (i.e., macrophages, neutrophils, and myeloid-derived suppressor cells [MDSCs]) share redundant functions in angiogenesis and immune modulation and therefore can compensate for each other, leading to an oscillating pattern between immune-cell populations when specific myeloid subpopulations are targeted (Rivera et al. 2015). What these studies also revealed is that VEGF/VEGFR blockade could endorse an angiostatic and immune-stimulating phenotype in all of the intratumoral myeloid cells accompanied with the up-regulation of CXCL14 and other angiostatic chemokines, and that this ability was indeed a requirement for the efficacy of antiangiogenic therapy (Fig. 1A). Because of therapy-induced hypoxia, however, tumors were able to produce factors (e.g., SDF1a and IL-6) that activated phosphoinositide 3-kinase γ (PI3Kγ) signaling in myeloid cells and converted them back into an immune-suppressive and angiogenic state that also disabled their repolarization, which resulted in a proangiogenic tumor relapse (Fig. 1A; Rivera et al. 2015). Congruent with these findings, recent studies have shown that PI3Kγ signals through Akt and mammalian target of rapamycin (mTOR) inhibited NF-κB while stimulating C/EBPβ activation, thereby inducing a transcriptional program that promotes immune suppression (Kaneda et al. 2016). The functional significance of innate immune cells in regulating angiogenesis is further underscored by the observation that the percentage of PI3K-activated myeloid cells in tumors dictated the degree of therapeutic response to angiogenic blockade (Fig. 1B). Congruently, pharmacological inhibition of myeloid PI3Kγ/δ improved and sustained the tumor response to antiangiogenic therapy by converting innate immune cells to an angiostatic and immune-stimulatory state associated with enhanced cytotoxic T-cell infiltration and activity (Fig. 1B; Rivera et al. 2015). Emerging from these studies is the proposition that angiogenesis and inflammation are functionally interregulated and that immune cells play a pivotal role in regulating both processes. It also underscores the tumor’s vigilance to conserve an immunosuppressive and angiogenic milieu to overcome growth restrictions and escape immune surveillance.
Figure 1.
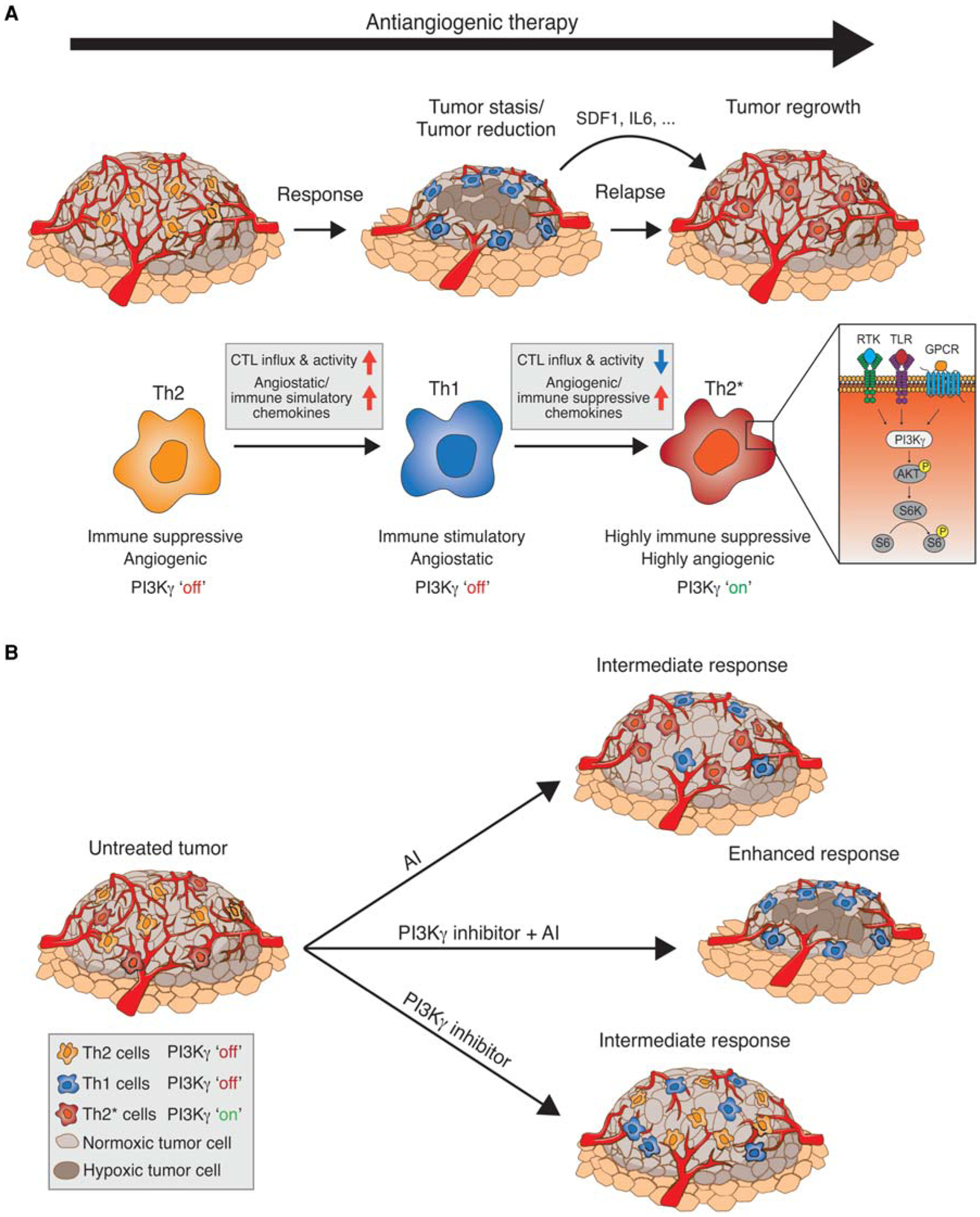
(A) Tumors responsive to antiangiogenic inhibition (AI) are infiltrated by Th2-type myeloid cells, which promote an immunosuppressive and proangiogenic phenotype. Treatment with antiangiogenic therapy leads to reduced vessel density and increased hypoxia, and cytokine release transiently shifts the population to a Th1 immune-stimulatory/angiostatic phenotype, leading to cytotoxic T-lymphocyte (CTL) influx, increased apoptosis, and tumor stasis/reduction. Activation of phosphoinositide 3-kinase γ (PI3Kγ) in myeloid cells repolarizes the immune population back to a Th2* phenotype that is now highly immune suppressive and angiogenic and unresponsive to continued antiangiogenic inhibition, leading to vascular rebound and regrowth. RTK, receptor tyrosine kinase; TLR, Toll-like receptor; GPCR, Gαi protein-coupled receptor. (B) AI-resistant tumors consist of PI3Kγ-independent, immune suppressive, and angiogenic Th2 cells (orange), and PI3Kγ-dependent, highly immune-suppressive, and angiogenic Th2* cells (red). Single AI treatment results in a shift of Th2 cells to the angiostatic and immune stimulatory Th1 cells (blue), creating an intermediate response. In addition, single PI3Kγ inhibitor treatment shifted Th2* to Th1 cells, leaving the Th2 cell population unaffected and also producing an intermediate response. In contrast, combination AI/PI3Kγ inhibition stimulated a shift of Th2 cells to Th1 cells and eradicated the highly immune-suppressive and angiogenic Th2* cells. This treatment produces an angiostatic and immune-stimulatory effect, resulting in an enhanced response.
METABOLIC ENDOTHELIAL CELL ADAPTATION DURING ANGIOGENESIS AND ANGIOGENESIS BLOCKADE
Cancer cells are highly metabolically active, thus they require an adequate vasculature to ensure a constant supply of oxygen and nutrients. In addition, endothelial cells (ECs) undergo metabolic changes when they become activated to form new vessels. Thus targeting metabolic pathways in endothelial cells may be an additional and alternative strategy, because metabolism in these cells not only fuels vascular expansion but also regulates the very formation of blood vessels (Rivera and Bergers 2014). Indeed, such a strategy has proven to inhibit angiogenesis in in vitro sprouting models and induced vessel normalization in tumors (De Bock et al. 2013; Schoors et al. 2015; Cantelmo et al. 2016). Cells use different metabolic programs to convert exogenous molecules to amino acids, nucleotides, and lipids depending on their metabolic needs (Lunt and Vander Heiden 2011; Mihaylova and Shaw 2011). Accordingly, functional differences in an giogenic and quiescent ECs mandate a different metabolic program. Angiogenic ECs require efficient biomass and adenosine triphosphate (ATP)-producing systems to proliferate and migrate (De Bock et al. 2013; Schoors et al. 2015), whereas quiescent ECs, which are exposed to the oxidative environment of the bloodstream, maintain their cellular homeostasis by optimizing the protective mechanisms involved in redox homeostasis and repair mechanisms (Wang et al. 2010; De Bock et al. 2013). Thus, it is conceivable that the angiogenic switch goes hand-in-hand with a metabolic switch, and that this metabolic switch can be an attractive therapeutic target.
In oxygen-rich conditions, the majority of nonproliferating cells rely on oxidative metabolism (Vander Heiden et al. 2009). However, despite the higher ATP yield of oxidative phosphorylation and their exposure to very high oxygen levels, ECs divert the majority of pyruvate to lactate (≈99%) in a process called “aerobic glycolysis” or the “Warburg effect” (Wang et al. 2011; De Bock et al. 2013). ECs rely on glycolysis for >85% of total ATP synthesis, and ECs lining blood vessels show glycolytic flux rates that are much higher than most healthy cells and comparable to those of various cancer cell types (Culic et al. 1997; Vander Heiden et al. 2009; De Bock et al. 2013).
Activation of ECs with growth factors induces proliferative and migratory capacities that require high ATP synthesis rates. EC activation increases the glycolytic flux by 35%–40% (De Bock et al. 2013) and pharmacological inhibition of glycolysis using 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO), which targets 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3), lowers glycolytic flux to prevent sprouting, and reduces migration and proliferation of ECs. Consequently, PFKFB3 inhibition targets both stalk and tip cells to impair vessel sprouting in vitro and produces vascular defects in the postnatal mouse retina (De Bock et al. 2013; Xu et al. 2014). In addition, the glycolytic intermediate, glucose-6-phosphate, can be diverted to the pentose phosphate pathway (PPP) to produce ribose-5-phosphate for nucleotide synthesis and the reducing factor nicotinamide adenine dinucleotide phosphate (NADPH). Thus, the PPP has the potential to stimulate biomass production for proliferation or to maintain redox homeostasis (Anastasiou et al. 2011; Lunt and Vander Heiden 2011). Accumulating evidence suggests that the PPP plays a regulatory role in ECs, because inhibition of glucose-6-phoshate dehydrogenase (G6PD), the rate-limiting enzyme of the oxidative branch of the PPP and a NADPH-producing enzyme, increased ROS levels in ECs (Leopold et al. 2001), whereas its overexpression had the opposite effects (Leopold et al. 2003), implying that the PPP in ECs can have a potential protective function and serve as a therapeutic target.
As alternative to glycolysis, certain cell types with high energy demand (e.g., cardiomyocytes, skeletal muscle cells, and certain cancer cells) rely on fatty acid-β-oxidation (FAO) as an energy source or to maintain redox homeostasis (Missiaen et al. 2017). Because ECs show such a high glycolytic flux, it is not surprising that ECs have a low oxidative metabolism (De Bock et al. 2013). Surprisingly, pharmacological inhibition of mitochondrial FAO with etomoxir impaired in vitro sprouting in a retinal model, suggesting an important role for FAO in ECs during angiogenesis. This effect was not due to reduced ATP synthesis or affected redox homeostasis; rather FAO inhibition selectively impaired EC proliferation but not migration (Schoors et al. 2015). 13C-palmitate tracing experiments revealed that ECs use fatty acids (FAs) to significantly fuel the tricarboxylic acid (TCA) cycle with carbons. Carbons derived from palmitate were also detected in the intracellular deoxyribonucleotide (dNTP) pool, and this labeling was impaired by FAO inhibition, indicating that FAO plays an essential role in the de novo synthesis of dNTPs in ECs, a requirement for proliferation (Schoors et al. 2015).
In tumors, excessive VEGF and hypoxia induces endothelial dysfunction, resulting in irregularly shaped, leaky, and incompletely perfused tumor blood vessels. VEGF signaling increases glycolysis in ECs by inducing GLUT1 and PFKFB3 expression and inhibition of VEGF signaling resulted in reduced glycolysis (Yeh et al. 2008; De Bock et al. 2013). Interestingly, freshly isolated tumor endothelial cells (TECs) show a higher glycolytic flux than normal endothelial cells (Cantelmo et al. 2016). In contrast to conventional antiangiogenic therapies that aim to inhibit tumor vessel growth, an emerging strategy is to normalize tumor vessels to restore blood vessel perfusion, thereby reducing metastasis and increasing chemotherapeutic delivery. Notably, partial inhibition of the glycolytic flux using low doses of 3PO induced vessel normalization, also a consequence of anti-VEGF treatment (Batchelor et al. 2013). Therefore, it is conceivable that the normalization effect of anti-VEGF treatment could in part be attributed to reduced glycolytic flux in TECs. Unfortunately, studies on the metabolic profile of tumor endothelial cells are limited, and the effect of antiangiogenic therapy on EC metabolism has yet to be investigated.
METABOLIC TUMOR CELL ADAPTATIONS TO ANTIANGIOGENIC THERAPY
As summarized in the preceding text, multiple modes of adaptive or evasive resistance to therapies targeting tumor angiogenesis have been reported, but only heighted invasiveness has been clearly implicated as an adaptive resistance mechanism in glioblastoma (GBM) (Lu and Bergers 2013, and references therein). Intriguingly, Bjerkvig and colleagues explored the metabolic response to antiangiogenic inhibition (AI) therapy using bevacizumab in xenograph models of GBM and linked therapy with increased anaerobic glycolysis and oxidative stress. In addition, they found evidence for increased glutamine consumption that could potentially be used to fuel the TCA cycle (glutaminolysis) and reduce production of reactive oxygen species (Fack et al. 2015). The authors validated their observations by studying a panel of paired human GBM biopsy samples taken before and after bev acizumab treatment that showed increased expression of a key glycolytic enzyme that converts pyruvate to lactate (LDHA), most strikingly up-regulated in the invasive margins of the treated tumors (Fack et al. 2015). It is interesting to note that others have postulated that tumor acidity/high lactate levels may promote local invasion (Estrella et al. 2013). Moreover, they found that the response of GBM to bevacizumab therapy is not mediated by clonal selection mechanisms but is rather an adaptive response. Indraccolo and colleagues studied the response to long-term antiangiogenic therapy and found that it can induce a stable glycolytic phenotype in tumor xenographs derived from cells they had classified as highly or poorly glycolytic, and that the highly glycolytic cells became more rapidly resistant to antiangiogenic therapy. They describe this phenomenon as metabolic evolution; however, it is as yet unclear whether it is the consequence of epigenetic modulation of driver genes, selection of a highly glycolytic population, or an adaptive response to therapy (Curtarello et al. 2015).
The existence of metabolic symbiosis in cancer was reported by Sonveaux, Feron, Dewhirst, and colleagues to arise spontaneously in certain transplant tumor models, and, in a series of publications, they have described a symbiotic state that conveys decreased reliance on circulating glucose and limits damage from secreted lactate (Semenza 2008; Sonveaux et al. 2008; Kennedy et al. 2013). In this model, cancer cells are compartmentalized in conditions of vascular insufficiency into comparatively hypoxic and normoxic compartments based on their proximity to functional blood vessels. In spite of their close proximity to the vasculature, normoxic cells import and catabolize lactate, thereby sparing glucose for hypoxic cells to import and catabolize glycolytically, resulting in the production and secretion of high levels of lactate. This symbiotic relationship was originally described in certain highly metabolically active tissues, including muscle and brain (Brooks 2002). In tumors, the relationship has been extended to interactions between tumor cells and cancer-associated fibroblasts (CAFs), ECs, and macrophages (Nakajima and Van Houten 2013, and references therein).
Three companion reports describe an unanticipated new mode of adaptive resistance to antiangiogenic therapy, metabolic symbiosis, which is induced in response to potent antiangiogenic therapies that cause vascular collapse and consequent hypoxia (Fig. 2; Allen et al. 2016; Jimenez-Valerio et al. 2016; Pisarsky et al. 2016). The reports all show induction and expression patterns of lac-tate and glucose transporters consistent with the notion of metabolic symbiosis induced in response to different antiangiogenic therapies in preclinical and clinical samples (Fig. 2). The Christofori group showed that gene expression analysis of isolated tumor cells treated with long-term antiangiogenic therapy (nintedanib) revealed a hyperglycolytic signature during the relapse phase, which was characterized by a lack of revascularization and characteristic expression patterns of lactate and glucose transporters. Resistance to nintendanib could be overcome in the PyMT orthotopic preclinical model using the glycol ysis inhibitor 3PO, which targets PFKFB3 (Pisarsky et al. 2016). Moreover, symbiosis and tumor progression was blocked in a breast cancer model by genetically suppressing lactate export in antiangiogenic-treated tumors, strengthening the rationale for the development of inhibitors targeting lactate export in the clinic (Pisarsky et al. 2016). In the study from the Hanahan laboratory using a spontaneous mouse model of pancreatic neuroendocrine tumor (PanNET), mTOR signaling was also found to be up-regulated in normoxic tumor regions, and nuclear magnetic resonance (NMR) analysis confirmed lactate catabolism in cultured normoxic tumor cells and tumors in a branched pathway involving glutamine metabolism (glutaminolysis). This pathway is enhanced in antiangiogenic (sunitinib)-treated tumors (Allen et al. 2016), reminiscent of the effects of bevacizumab on GBM (Fack et al. 2015). Moreover, normoxic PanNET cells cultured with lactate and glutamine up-regulate mTOR signaling, which can be reversed with mTOR inhibitors, impairing lactate uptake, or by inhibiting enzymes involved in glutaminolysis. In addition, combination antiangiogenic (sunitinib or axitinib)/mTOR inhibition delays tumor progression and significantly extends survival, in part by blocking lactate catabolism in conjunction with gluta-mine, and by changing the expression of glucose transporters (Fig. 2; Allen et al. 2016). Furthermore, the Cassanovas group produced evidence corroborating the existence of metabolic symbiosis as a resistance mechanism by studying renal cell carcinoma (RCC) orthoxenografts and paired human RCC biopsies samples taken before and after sunitinib treatment (Jimenez-Valerio et al. 2016). The authors found an up-regulation and redistribution of key glycolytic and lactate transporters in the antiangiogenic-treated tumors consistent with the notion of metabolic symbiosis and also found that mTOR inhibition in concert with antiangiogenic therapy with sunitinib could disrupt the symbiotic patterning and delay tumor progression; critically, the combination was associated disruption of symbiosis in a patient responding to therapy (Jimenez-Valerio et al. 2016). Thus, in the preclinical PanNET model, sunitinib is apparently inducing two distinctive modes of adaptive resistance—metabolic symbiosis, and the perivascular invasive migration of cancer cells at tumor margins (Paez-Ribes et al. 2009). It is presently unclear whether these invading cancer cells also induce symbiosis or whether secreted lactate plays a direct role in the invasion into the surrounding tissue (Estrella et al. 2013).
Figure 2.
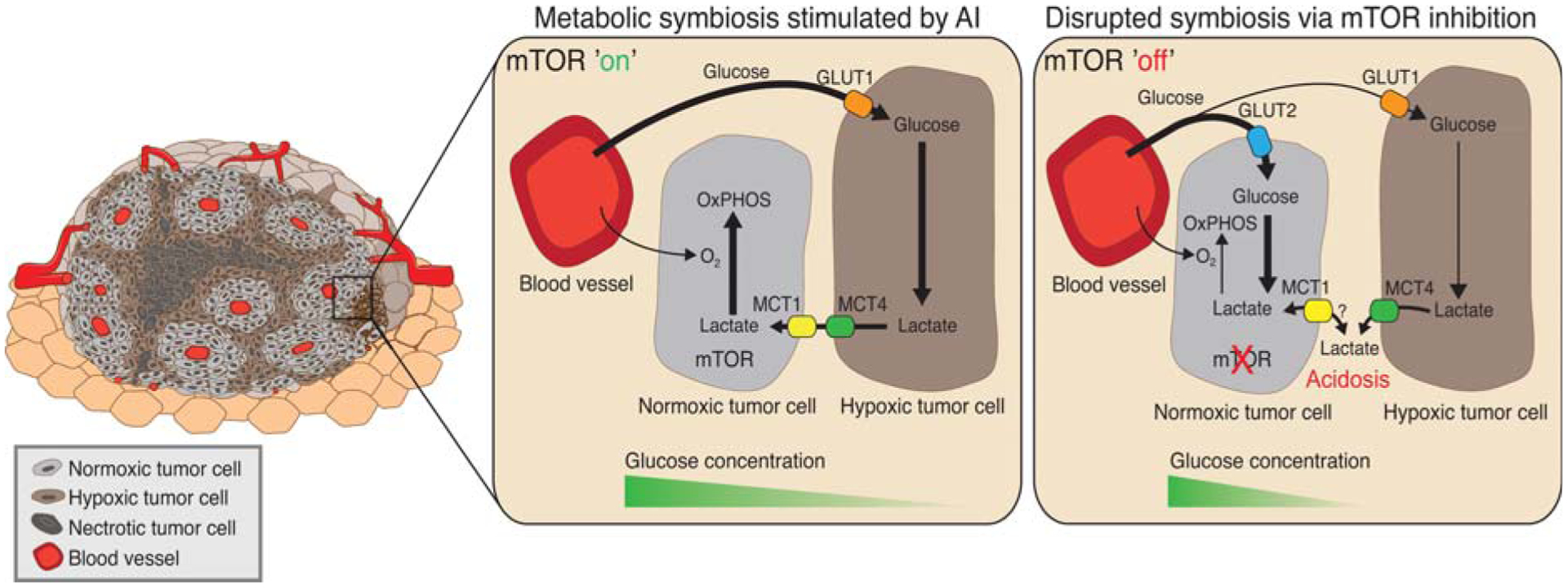
Schematic representation of angiogenesis inhibitor (AI)-induced metabolic symbiosis and the effect of mammalian target of rapamycin (mTOR) inhibition. (Left) Treatment with an AI causes vascular collapse to produce normoxic, hypoxic, and necrotic regions, based on their proximity to functional blood vessels. (Middle, mTOR”on.”) Tumor cells in hypoxic regions up-regulate expression of the glucose transporter GLUT1, increase glycolysis, and export lactate via MCT4. In parallel, mTOR-expressing normoxic tumor cells spare glucose and import lactate via MCT1 to fuel O2-dependent oxidative phosphorylation (OxPHOs). (Right, mTOR”off”) Disruption of symbiosis via mTOR inhibition disrupts lactate catabolism in normoxic cells, which can produce increased intracellular lactate concentrations, reduced extracellular lactate clearance, and increased acidosis. Furthermore, mTOR inhibition up-regulates GLUT2 expression in normoxic cells, which can increase glucose consumption by the normoxic cells to reduce its availability for the hypoxic compartment.
Noel and colleagues performed a compelling study assessing the metabolic consequences of withdrawing AI therapy in preclinical breast cancer models, which revealed enhanced lipid metabolism concomitant with reduced glycolysis (Sounni et al. 2014), implicating altered lipid metabolism as a potential mechanism of resistance during “rest periods” for patients on antiangiogenic therapy. Both knockdown and pharmacological inhibition of fatty acid synthase (FASN) activity in tumor cells and pharmacological inhibition of FASN with orlistat reversed the tumor rebound and metastasis upon sunitinib withdrawal. In another series of experiments that highlights the role of fatty acid (FA) metabolism in response to hypoxia/antiangiogenic treatment, Harris and colleagues (Bensaad et al. 2014) showed that hypoxia or bevacizumab treatment could induce formation of lipid droplets in breast and glioblastoma xenograph models under the control of HIF1α signaling. Critically, they found that oxidation of these FA stores could provide ATP to tumor cells under conditions of hypoxia or reoxygenation, and reduce levels of toxic reactive oxygen species. In this case, increased FA uptake, rather than de novo synthesis, was essential to this process and controlled by two different fatty acid binding proteins, FABP3 and FABP7. Furthermore, knockdown of these genes impairs tumor growth (Bensaad et al. 2014), providing the rationale for another potential drug target.
CONCLUSION
Although drugs targeting the VEGF pathway did not fulfill initial expectations, their development has been a seminal first step in targeting tumor vessels to impair tumor growth, providing pivotal information about how tumors react to vessel growth restriction. They further have revealed several escape mechanisms that open new avenues for the assessment of combinatorial treatment modalities aimed at targets within these evasive pathways. For example, various antiangiogenic immune therapies are currently tested in the clinic, specifically in combination with checkpoint inhibitors such as CTLA4 or PD1/PDL1 antibodies, showing encouraging results (Garber 2014; Schoenfeld and Dranoff 2011; NCT02669173, NCT03066427). Ongoing and future studies will reveal which antiangiogenic immunotherapies will more robustly promote an enduring immune-stimulatory milieu and inhibit tumor angiogenesis that could lead to prolonged survival in cancer patients. In line with the concept of targeting evasion mechanisms, targeting endothelial cell metabolism is a new and promising avenue that may circumvent growth factor–mediated resistance, and some of the agents currently used in the clinic, like a next-generation derivative of 3PO, ACT-PFK-158, which targets the glycolytic activator PFKFB3, may have enduring antiangiogenic and vasculature normalization effects. In addition, inhibitors of FAO/CPT1 are currently in the clinic for other indications, most prominently angina, including oxfenicine, perhexiline, and ranolazine (Missiaen et al. 2017, and references therein). Moreover, disruption of induced metabolic pathways might also have value in extending the duration of efficacious responses by traditional antiangiogenic therapies, or potentially antiangiogenic therapy could be used to create a “conditional lethal” metabolic state in tumors, leading to selection and targeting of the newly adapting variants (Bensaad and Harris 2013). Although metabolic symbiosis induced by sunitinib/axitinib can be disrupted by mTOR or glycolysis inhibitors, the significant reductions in tumor burden translate into a relatively modest survival benefit in preclinical studies. In spite of the positive outcome of combination therapy using sunitinib with the rapalog everolimus in the aforementioned clinical study (Jimenez-Valerio et al. 2016), other clinical studies using this combination have shown limited benefit, which may be attributable to high toxicity (Molina et al. 2012). Alternative and potentially attractive strategies may be to use antiangiogenics in combination with compounds that directly inhibit lactate transport, such as AZD3965 and 7ACC2 (Bola et al. 2014; Draoui et al. 2014; Polanski et al. 2014), or glutaminolysis, like CB-839 (Momcilovic et al. 2017). There are increasing numbers of agents in the clinic to target FA metabolism, including those that target FA synthase like orlistat and TVB-2640 (Jones and Infante 2015), and doubtless many more in the pipeline in coming years.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (NIH) (R01 CA201537 and R01 CA188404).
REFERENCES
- Adams RH, Alitalo K. 2007. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8: 464–478. [DOI] [PubMed] [Google Scholar]
- Allen E, Mieville P, Warren CM, Saghafinia S, Li L, Peng MW, Hanahan D. 2016. Metabolic symbiosis enables adaptive resistance to anti-angiogenic therapy that is dependent on mTOR signaling. Cell Rep 15: 1144–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, et al. 2011. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334: 1278–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluk P, Hashizume H, McDonald DM. 2005. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev 15: 102–111. [DOI] [PubMed] [Google Scholar]
- Batchelor TT, Gerstner ER, Emblem KE, Duda DG, Kalpathy-Cramer J, Snuderl M, Ancukiewicz M, Polaskova P, Pinho MC, Jennings D, et al. 2013. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc Natl Acad Sci 110: 19059–19064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Harris AL. 2013. Cancer metabolism as a therapeutic target: Metabolic synthetic lethality. Oncology (Williston Park) 27: 467, 473–474. [PubMed] [Google Scholar]
- Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, et al. 2014. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep 9: 349–365. [DOI] [PubMed] [Google Scholar]
- Bergers G, Benjamin LE. 2003. Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3: 401–410. [DOI] [PubMed] [Google Scholar]
- Bergers G, Hanahan D. 2008. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8: 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bola BM, Chadwick AL, Michopoulos F, Blount KG, Telfer BA, Williams KJ, Smith PD, Critchlow SE, Stratford IJ. 2014. Inhibition of monocarboxylate transporter-1 (MCT1) by AZD3965 enhances radiosensitivity by reducing lactate transport. Mol Cancer Ther 13: 2805–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzin C, Brouet A, De Vriese J, Dewever J, Feron O. 2007. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: Identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J Immunol 178: 1505–1511. [DOI] [PubMed] [Google Scholar]
- Brooks GA. 2002. Lactate shuttles in nature. Biochem Soc Trans 30: 258–264. [DOI] [PubMed] [Google Scholar]
- Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, Chaturvedi P, Hol J, Thienpont B, Teuwen LA, et al. 2016. Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell 30: 968–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P 2005. VEGF as a key mediator of angiogenesis in cancer. Oncology 69 (Suppl 3): 4–10. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. 2011. Molecular mechanisms and clinical applications of angiogenesis. Nature 473: 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanovas O, Hicklin DJ, Bergers G, Hanahan D. 2005. Drug resistance by evasion of anti-angiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8: 299–309. [DOI] [PubMed] [Google Scholar]
- Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, Deschoemaeker S, Van Ginderachter JA, Tamag-none L, Mazzone M. 2013. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 24: 695–709. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Tinkle CL, Hanahan D, Werb Z. 2000. MMP-9 supplied by bone marrow–derived cells contributes to skin carcinogenesis. Cell 103: 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culic O, Gruwel ML, Schrader J. 1997. Energy turnover of vascular endothelial cells. Am J Physiol 273: C205–C213. [DOI] [PubMed] [Google Scholar]
- Curtarello M, Zulato E, Nardo G, Valtorta S, Guzzo G, Rossi E, Esposito G, Msaki A, Pasto A, Rasola A, et al. 2015. VEGF-targeted therapy stably modulates the glycolytic phenotype of tumor cells. Cancer Res 75: 120–133. [DOI] [PubMed] [Google Scholar]
- De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, et al. 2013. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154: 651–663. [DOI] [PubMed] [Google Scholar]
- Dineen SP, Lynn KD, Holloway SE, Miller AF, Sullivan JP, Shames DS, Beck AW, Barnett CC, Fleming JB, Brekken RA. 2008. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res 68: 4340–4346. [DOI] [PubMed] [Google Scholar]
- Draoui N, Schicke O, Seront E, Bouzin C, Sonveaux P, Riant O, Feron O. 2014. Antitumor activity of 7-aminocarboxycou-marin derivatives, a new class of potent inhibitors of lactate influx but not efflux. Mol Cancer Ther 13: 1410–1418. [DOI] [PubMed] [Google Scholar]
- Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, et al. 2008. HIF1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 13: 206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. 2007. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci 104: 17069–17074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, et al. 2013. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res 73: 1524–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fack F, Espedal H, Keunen O, Golebiewska A, Obad N, Harter PN, Mittelbronn M, Bahr O, Weyerbrock A, Stuhr L, et al. 2015. Bevacizumab treatment induces metabolic adaptation toward anaerobic metabolism in glioblastomas. Acta Neuropathol 129: 115–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N 2004. Vascular endothelial growth factor as a target for anticancer therapy. Oncologist 9 (Suppl 1): 2–10. [DOI] [PubMed] [Google Scholar]
- Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M, De Mol M, et al. 2007. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 131: 463–475. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP. 1996. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med 2: 1096–1103. [DOI] [PubMed] [Google Scholar]
- Garber K 2014. Promising early results for immunotherapyantiangiogenesis combination. J Natl Cancer Inst 106: dju392. [DOI] [PubMed] [Google Scholar]
- Giraudo E, Inoue M, Hanahan D. 2004. An amino-bisphospho-nate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest 114: 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G. 1996. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: The role of angiogenic factors. Cancer Res 56: 1111–1117. [PubMed] [Google Scholar]
- Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Jung S, Chimenti S, Landsman L, Abramovitch R, Keshet E. 2006. VEGF-induced adult neovascularization: Recruitment, retention, and role of accessory cells. Cell 124: 175–189. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: The next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Jain RK. 2002. Angiogenesis and lymphangiogenesis in tumors: Insights from intravital microscopy. Cold Spring Harb Symp Quant Biol 67: 239–248. [DOI] [PubMed] [Google Scholar]
- Jain RK. 2014. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 26: 605–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayson GC, Kerbel R, Ellis LM, Harris AL. 2016. Anti-angiogenic therapy in oncology: Current status and future directions. Lancet 388: 518–529. [DOI] [PubMed] [Google Scholar]
- Jimenez-Valerio G, Martinez-Lozano M, Bassani N, Vidal A, Ochoa-de-Olza M, Suarez C, Garcia-Del-Muro X, Carles J, Vinals F, Graupera M, et al. 2016. Resistance to anti-angiogenic therapies by metabolic symbiosis in renal cell carcinoma PDX models and patients. Cell Rep 15: 1134–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SF, Infante JR. 2015. Molecular pathways: Fatty acid synthase. Clin Cancer Res 21: 5434–5438. [DOI] [PubMed] [Google Scholar]
- Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, et al. 2016. PI3Kγ is a molecular switch that controls immune suppression. Nature 539: 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuya A, Tokura Y. 2014. Attempts to accelerate wound healing. J Dermatol Sci 76: 169–172. [DOI] [PubMed] [Google Scholar]
- Kennedy KM, Scarbrough PM, Ribeiro A, Richardson R, Yuan H, Sonveaux P, Landon CD, Chi JT, Pizzo S, Schroeder T, et al. 2013. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS One 8: e75154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopold JA, Cap A, Scribner AW, Stanton RC, Loscalzo J. 2001. Glucose-6-phosphate dehydrogenase deficiency promotes endothelial oxidant stress and decreases endothelial nitric oxide bioavailability. FASEB J 15: 1771–1773. [DOI] [PubMed] [Google Scholar]
- Leopold JA, Zhang YY, Scribner AW, Stanton RC, Loscalzo J. 2003. Glucose-6-phosphate dehydrogenase overexpression decreases endothelial cell oxidant stress and increases bio-available nitric oxide. Arterioscler Thromb Vasc Biol 23: 411–417. [DOI] [PubMed] [Google Scholar]
- Liao D, Johnson RS. 2007. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev 26: 281–290. [DOI] [PubMed] [Google Scholar]
- Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. 2006. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res 66: 11238–11246. [DOI] [PubMed] [Google Scholar]
- Lu KV, Bergers G. 2013. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol 2: 49–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, Isachenko N, Fouse SD, Phillips JJ, Cheresh DA, et al. 2012. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 22: 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. 2011. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27: 441–464. [DOI] [PubMed] [Google Scholar]
- Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L, et al. 2011. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 19: 512–526. [DOI] [PubMed] [Google Scholar]
- McIntyre A, Harris AL. 2015. Metabolic and hypoxic adaptation to anti-angiogenic therapy: A target for induced essentiality. EMBO Mol Med 7: 368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. 2011. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13: 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiaen R, Morales-Rodriguez F, Eelen G, Carmeliet P. 2017. Targeting endothelial metabolism for anti-angiogenesis therapy: A pharmacological perspective. Vascul Pharmacol 90: 8–18. [DOI] [PubMed] [Google Scholar]
- Molina AM, Feldman DR, Voss MH, Ginsberg MS, Baum MS, Brocks DR, Fischer PM, Trinos MJ, Patil S, Motzer RJ. 2012. Phase 1 trial of everolimus plus sunitinib in patients with metastatic renal cell carcinoma. Cancer 118: 1868–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Magyar C, Braas D, Graeber T, Jackson NJ, Czernin J, Emberley E, et al. 2017. Targeted inhibition of EGFR and glutaminase induces metabolic crisis in EGFR mutant lung cancer. Cell Rep 18: 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motz GT, Coukos G. 2011. The parallel lives of angiogenesis and immunosuppression: Cancer and other tales. Nat Rev Immunol 11: 702–711. [DOI] [PubMed] [Google Scholar]
- Motz GT, Coukos G. 2013. Deciphering and reversing tumor immune suppression. Immunity 39: 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch C, Giannoudis A, Lewis CE. 2004. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 104: 2224–2234. [DOI] [PubMed] [Google Scholar]
- Nakajima EC, Van Houten B. 2013. Metabolic symbiosis in cancer: Refocusing the Warburg lens. Mol Carcinog 52: 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. 2009. Anti-angiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15: 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazon A, Goldrath AW, Nizet V, Johnson RS. 2014. HIF transcription factors, inflammation, and immunity. Immunity 41: 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarsky L, Bill R, Fagiani E, Dimeloe S, Goosen RW, Hagmann J, Hess C, Christofori G. 2016. Targeting metabolic symbiosis to overcome resistance to anti-angiogenic therapy. Cell Rep 15: 1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanski R, Hodgkinson CL, Fusi A, Nonaka D, Priest L, Kelly P, Trapani F, Bishop PW, White A, Critchlow SE, et al. 2014. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer Res 20: 926–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potente M, Gerhardt H, Carmeliet P. 2011. Basic and therapeutic aspects of angiogenesis. Cell 146: 873–887. [DOI] [PubMed] [Google Scholar]
- Rigamonti N, Kadioglu E, Keklikoglou I, Wyser Rmili C, Leow CC, De Palma M. 2014. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep 8: 696–706. [DOI] [PubMed] [Google Scholar]
- Rivera LB, Bergers G. 2014. Targeting vascular sprouts. Science 344: 1449–1450. [DOI] [PubMed] [Google Scholar]
- Rivera LB, Bergers G. 2015. Tumor angiogenesis, from foe to friend. Science 349: 694–695. [DOI] [PubMed] [Google Scholar]
- Rivera LB, Meyronet D, Hervieu V, Frederick MJ, Bergsland E, Bergers G. 2015. Intratumoral myeloid cells regulate responsiveness and resistance to anti-angiogenic therapy. Cell Rep 11: 577–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF, Shuman MA. 2000. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia 2: 306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoni M, Bracarda S, Nabissi M, Massari F, Conti A, Bria E, Tortora G, Santoni G, Cascinu S. 2014. CXC and CC chemokines as angiogenic modulators in nonhaematological tumors. Biomed Res Int 2014: 768758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, et al. 2003. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med 198: 1391–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfeld JD, Dranoff G. 2011. Anti-angiogenesis immuno-therapy. Hum Vaccin 7: 976–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, Zecchin A, Cantelmo AR, Christen S, Goveia J, et al. 2015. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520: 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. 2008. Tumor metabolism: Cancer cells give and take lactate. J Clin Invest 118: 3835–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. 2011. Oxygen sensing, homeostasis, and disease. N Engl J Med 365: 537–547. [DOI] [PubMed] [Google Scholar]
- Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, Tabruyn SP, You WK, Chapman HA, Christensen JG, et al. 2012. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov 2: 270–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shojaei F, Ferrara N. 2007a. Antiangiogenesis to treat cancer and intraocular neovascular disorders. Lab Invest 87: 227–230. [DOI] [PubMed] [Google Scholar]
- Shojaei F, Ferrara N. 2007b. Anti-angiogenic therapy for cancer: An update. Cancer J 13: 345–348. [DOI] [PubMed] [Google Scholar]
- Shojaei F, Ferrara N. 2008. Role of the microenvironment in tumor growth and in refractoriness/resistance to anti-angiogenic therapies. Drug Resist Updat 11: 219–230. [DOI] [PubMed] [Google Scholar]
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, et al. 2008. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 118: 3930–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounni NE, Cimino J, Blacher S, Primac I, Truong A, Mazzucchelli G, Paye A, Calligaris D, Debois D, De Tullio P, et al. 2014. Blocking lipid synthesis overcomes tumor regrowth and metastasis after anti-angiogenic therapy withdrawal. Cell Metab 20: 280–294. [DOI] [PubMed] [Google Scholar]
- Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. 2003. ChemokinereceptorCXCR4downregulatedbyvonHippel-Lindau tumour suppressor pVHL. Nature 425: 307–311. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 324: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudev NS, Reynolds AR. 2014. Anti-angiogenic therapy for cancer: Current progress, unresolved questions and future directions. Angiogenesis 17: 471–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YY, Chen SM, Li H. 2010. Hydrogen peroxide stress stimulates phosphorylation of FoxO1 in rat aortic endothelial cells. Acta Pharmacol Sin 31: 160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Liang B, Shirwany NA, Zou MH. 2011. 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS One 6: e17234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, An X, Guo X, Habtetsion TG, Wang Y, Xu X, Kandala S, Li Q, Li H, Zhang C, et al. 2014. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler Thromb Vasc Biol 34: 1231–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh WL, Lin CJ, Fu WM. 2008. Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia. Mol Pharmacol 73: 170–177. [DOI] [PubMed] [Google Scholar]