

Summary
Current studies estimate that 1–3% of females with unexplained intellectual disability (ID) present de novo splice site, nonsense, frameshift, or missense mutations in the DDX3X protein (DEAD-Box Helicase 3 X-Linked). However, the cellular and molecular mechanisms by which DDX3X mutations impair brain development are not fully comprehended. Here, we show that the ID-linked missense mutation L556S renders DDX3X prone to aggregation. By using a combination of biophysical assays and imaging approaches, we demonstrate that this mutant assembles solid-like condensates and amyloid-like fibrils. Although we observed greatly reduced expression of the mutant allele in a patient who exhibits skewed X inactivation, this appears to be enough to sequestrate healthy proteins into solid-like ectopic granules, compromising cell function. Therefore, our data suggest ID-linked DDX3X L556S mutation as a disorder arising from protein misfolding and aggregation.
Subject areas: Molecular biology, Neuroscience, Biophysics
Graphical abstract
Highlights
-
•
DDX3X mutations skew X-inactivation and are found in 1-3% of unexplained ID in females
-
•
DDX3X mutant proteins assemble solid-like condensates and amyloid-like fibrils
-
•
Aberrant granules formed by DDX3X mutants sequestrate healthy DDX3X protein
-
•
ID-linked DDX3X L556S mutation decreases cell viability and induces apoptosis
Molecular biology; Neuroscience; Biophysics
Introduction
Intellectual disability (ID) comprises a spectrum of clinically heterogeneous disorders defined by limitations in mental functioning and adaptive behavior that is observed in at least 1% of the population (Ellison et al., 2013). Although many factors can cause ID, genetic disturbances are responsible for a significant part in its etiology, with males being more often affected than females mostly owing to the presence of X-linked genes (Goldenberg and Saugier-Veber, 2010; Migeon, 2020; Vissers et al., 2016). Back in 2015, de novo variants were identified in DDX3X in 39 females in a large cohort of individuals with unexplained ID, but no de novo variants were observed in males (Snijders Blok et al., 2015). However, three unrelated males with hemizygous missense DDX3X variants and ID were recently identified, with two established as de novo (Nicola et al., 2019). This suggests de novo DDX3X variants as a cause of syndromic ID in both males and females.
Fewer genes and mutation frequency have been implicated in X-linked ID (XLID) affecting females. When it happens, the disorders often arise from a dominant effect. The X-inactivation pattern poses additional challenges for interpreting the effects of mutant alleles in females with XLID (Ziats et al., 2020). One hundred seven X-chromosome-related genes are recognized to be associated with monogenic XLID (Migeon, 2020; Piton et al., 2013). Several of these genes have been associated with an increased frequency of skewing in the somatic cells of carrier females, with preferential selection against the chromosome carrying the mutant allele (Cantone and Fisher, 2017; Lyon, 2002; Migeon, 2020). However, many X genes escape from inactivation and can cause ID irrespective of being located on the active or inactive X chromosome (Posynick and Brown, 2019; Wainer Katsir and Linial, 2019). Although pathogenic variants located in genes that escape X inactivation can be expressed to different extents, even limited expression can contribute to the severity of brain development disturbances and ID phenotype (Lennox et al., 2020; Migeon, 2020). Therefore, studies examining the effects of mutations on protein and cellular function are critical for elucidating the mechanisms underlying brain development disturbances in XLID (Gecz et al., 2009).
Genes with pathogenic variants associated with ID encode proteins involved in different neuron-specific or ubiquitous cellular processes as illustrated by the DEAD-Box Helicase 3 X-Linked protein (DDX3X). It is estimated that 1–3% of females with unexplained ID present de novo splice site, nonsense, frameshift, or missense mutations in DDX3X (Scala et al., 2019; Wang et al., 2018). This gene maps to Xp11.4 and encodes a highly conserved, ubiquitously expressed ATP-dependent DEAD-box RNA helicase (Ross et al., 2005). The DDX3X protein functions in various cellular processes related to RNA metabolism, including transcription regulation, pre-mRNA splicing, export and translation, RNA decay, and ribosome biogenesis (Kukhanova et al., 2020). These activities have been associated with a broad spectrum of biological processes, such as cell cycle/cell growth, apoptosis, tumorigenesis, innate immunity, and virus replication (Bol et al., 2015; Kukhanova et al., 2020; Pene et al., 2015). Although ubiquitously expressed in human tissues, DDX3X is particularly abundant in the developing brain, and DDX3X-associated brain malformations overwhelmingly affect the cerebral cortex (Lennox et al., 2020).
Notably, there is also evidence for the role of DDX3X in cellular stress response and stress granules (SGs) assembly independent of its RNA helicase activity (Pene et al., 2015; Valentin-Vega et al., 2016). SGs are membrane-less cytosolic organelles formed by liquid-liquid phase separation and structurally composed of mRNAs and proteins (Anderson and Kedersha, 2006). Importantly, DDX3X is not merely present but is also essential for SG assembly through regulating the molecular dynamics within SGs (Lin et al., 2015). Additionally, some DDX3X missense variants predispose to the formation of aberrant RNA-protein granules (Lennox et al., 2020), suggesting that a toxic gain of function may also contribute to the disease mechanism of ID caused by DDX3X missense mutations. Evidence indicates that SGs are temporary structures, but persistent stresses or protein misfolding may lead to chronic SGs that act as nidi for the aggregation of cellular proteins (Wolozin, 2012; Wolozin and Ivanov, 2019). Although DDX3X is known to escape X inactivation, it remains poorly explored how DDX3X mutations contribute to the pathogenesis of ID (Scala et al., 2019; Snijders Blok et al., 2015).
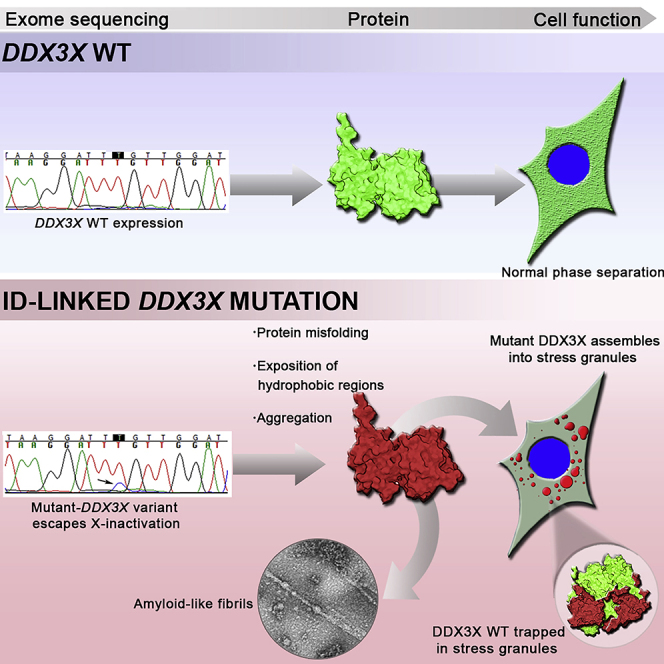
Here, we used recently described DDX3X ID-linked missense mutation, namely p.Leu556Ser (L556S) (Carneiro et al., 2018), to gain insights into the molecular mechanisms underlying neurodevelopmental abnormalities associated with pathogenic DDX3X variants. We observed that L556S mutation causes dramatic reduction of DDX3X ATPase activity, makes this protein prone to misfolding, and induces its aggregation. A combination of fluorescence spectroscopy and transmission electron microscopy reveals that the self-assembly of mutant proteins evolves from aggregates to amyloid structures in which oligomeric species play an on-pathway role. Cells expressing DDX3X-L556S mutant accumulate protein aggregates mainly owing to the resulted structural stability and solvent-exposed hydrophobic surfaces, as evidenced by biophysical and in silico approaches. Moreover, these granules are dynamically impaired, with no DDX3X-protein mobility and insensitivity to 1,6-hexanediol, implying that this pathogenic DDX3X mutant predisposes to an aberrant formation of solid-like aggregates. We propose that the aberrant phase separation of DDX3X mutants, which escape X-inactivation, can sequester wild-type (WT) DDX3X in ectopic SGs. Also, DDX3X-L556S brain organoids derived from patient-induced pluripotent stem cells (iPSCs) present several intracellular ectopic granules and reduced size and surface area compared with the control, suggesting an essential role of DDX3X in brain development.
Results
ATPase activity and cellular distribution in missense L556S pathogenic variant of DDX3X
By undertaking a systematic analysis of whole-exome sequencing (WES) of eight patients with idiopathic syndromic ID and their parents (trios), we have previously found a heterozygous de novo pathogenic variant (L556S) in the X-linked gene DDX3X in one of the patients (Figure 1A) (Carneiro et al., 2018).
Figure 1.

ID-related DDX3X L556S mutation drives the assembly of stable ectopic stress granules under basal conditions
(A) Sanger sequencing validation of the C/T substitution found in the patient from the analyzed cohort.
(B) ATPase activity of 0.5 μM purified D1D2 domain of WT, L556S, and R376C DDX3X variants (N = 5 different experiments with different batches of proteins performed in triplicate).
(C) Images of SH-SY5Y cells transfected for 48 hr with WT, or mutant DDX3X L556S-GFP mutant DDX3X R376C-GFP (green) and immunostained for TIA-1 (red). Inserts show colocalization (yellow) of DDX3X-GFP-tagged granules (green) with the stress granule marker TIA-1 (red). Intensity map on the right represents relative gray scale intensity.
(D) Quantification of percentage of transfected SH-SY5Y cells containing WT or mutant GFP-tagged-DDX3X granules (N = at least 55 cells per group; one-way ANOVA; ns.: not statistically significant; ∗p < 0.05).
(E) FRAP analysis of DDX3X L556S-GFP granule. Granule (arrow) is shown prior to photobleaching, and at 0 s, 4 s, and 40 s after. Graph shows recovery curve as an average ± SEM at each respective time point. Error bars represent mean values ± SEM (N = at least 20 granules from multiple cells per experiment from 3 individual experiments). Scale bar = 10 μm.
(F) MTS assay showing the reduced cell viability of SH-SY5Y cells expressing full human DDX3X-L556S mutant protein. Error bars represent mean values ± SEM (N = 3 individual experiments in triplicate, Student t-test; ∗p < 0.05 when compared to DDX3X WT; ∗∗p < 0.05 when compared to DDX3X WT).
(G) Apoptosis in DDX3X-GFP-positive transfected cells evaluated by TUNEL staining. Error bars represent mean values ± SEM (N = 55 from 3 individual experiments were evaluated, one-way ANOVA; ∗p < 0.05 when compared with DDX3X WT).
The phenotype associated with mutations in this gene is clinically variable and exhibits several other symptoms in addition to cognitive impairment, such as microcephaly and hearing loss, which were also found in this patient. The DDX3X protein contains a structured core composed of two RecA-like domains (D1 and D2) flanked by N- and C-terminal tails predicted to be unstructured (Figures S1A and S1B). The minimal functional core contains, besides the D1D2 domains, short N- and C-terminal extensions, spanning residues between S131 and G607 (Floor et al., 2016). L556S DDX3X mutation affects highly conserved residues in the structured helicase core mapping to an α-helix of the RecA-like D2 domain (Figure S1B). To explore the impact of the L556S mutations in the DDX3X ATPase activity in vitro, we used WT and mutant constructs of the minimal functional core, referred to as D1D2 for the ease of description. As can be seen on Figure 1B, the ATPase activity of the D1D2-L556S mutant was considerably decreased compared with the corresponding WT construct (Figure 1B).
L556S but not R376C DDX3X mutant displays aberrant phase separation in cells under no stress conditions
DDX3X protein contributes to the biophysical process of liquid-liquid phase separation (LLPS) and functions as an SG-nucleating factor in response to cellular insults such as oxidative and heat stress (Shih et al., 2012). Emerging data indicate that some pathogenic mutants of DDX3X form aberrant cytoplasmic granules in cells, what is as suggested as a potential mechanism of DDX3X mutant pathogenicity (Lennox et al., 2020; Valentin-Vega et al., 2016). To examine the possible damaging effects of L556S mutation on DDX3X cellular behavior, we monitored GFP-tagged L556S and WT full-length DDX3X in SH-SY5Y neuroblastoma cells. For the sake of comparison, we also worked with the p.Arg376Cys DDX3X mutant (R376C), which has emerged as recurrent ID-causing variant (Scala et al., 2019; Snijders Blok et al., 2015; Valentin-Vega et al., 2016). Differently from the L556, the R376 residue maps to a solvent-exposed loop of the RecA-like D1 domain (Figures S1A and S1B) and does not exhibit impaired ATPase activity (Figure S2A).
After transfection, the cells expressed similar amounts of the constructs, as estimated by Western blotting with anti-GFP antibody (Figures S2B and S2C). Under normal culture conditions, ∼45% of the cells expressing DDX3X-L556S showed dense granules distributed within the cytoplasm (Figures 1C and 1D). In contrast, those expressing DDX3X-R376C or DDX3X-WT exhibited diffuse signal, indicating that the two mutant isoforms bear different granule formation propensity (Figures 1C and 1D). Several granules in cells expressing DDX3X-L556S colocalize with the SG marker TIA1. Although WT DDX3X and R376C are spread throughout the cell cytoplasm, as much as TIA-1 (Figure 1C), the L556S mutant exhibits an intense concentration in SGs, suggesting that these organelles might evolve to persistent aberrant TIA-1-enriched granules that accumulate through imbalanced assembly and clearance (Figures 1C and 1D). To test this hypothesis, we explored the dynamics of DDX3X-L556S-containing granules, using fluorescence recovery after photobleaching (FRAP) analysis to determine whether this protein shuttles in and out of SGs. Confocal microscopy was used to select SGs for FRAP analysis (Figure 1E, arrow points to a representative targeted SG). Graphical representations of fluorescence intensity were obtained by scanning an argon beam in a linear pattern across the selected field. Photobleaching treatment eliminates most (∼75%) of the DDX3X-L556S-enriched-granule-associated fluorescence, as revealed by a scan taken every 0.65s after irradiation (Figure 1E). However, the bleached DDX3X-L556S variant did not recover any fluorescence (Figure 1E, bottom graph), indicating an immobile fraction of the L556S mutant protein, suggestive of protein aggregation.
A well-documented hallmark event in several neurodegenerative diseases (NDs) is the misfolding, aggregation and accumulation of proteins, causing cellular dysfunction, loss of synaptic connections, cell death, and brain damage (Soto and Pritzkow, 2018). It was recently reported that DDX3X ID-linked missense mutations disrupt RNA helicase activity and induce SGs in neural progenitors and neurons (Lennox et al., 2020). To verify if the DDX3X-L556S is toxic for cells, we performed cytotoxicity experiments using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) cell viability assay and evaluated the number of Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive cells. Although modest, but statistically significant, expression of DDX3X-L556S reduced cell viability and increased apoptosis compared with cells expressing DDX3X-WT. Interestingly, DDX3X-R376C-expressing cells, which do not assemble granules, did not present reduced viability, suggesting that the presence of cytoplasmic condensates is a key event in this process (Figures 1F and 1G).
L556S and DDX3X-R376C mutants impair stress granule dynamics
We previously observed how DDX3X mutants bear different SG assembly and aggregation propensity in unstressed cells. Next, we sought to examine the effects of L556S and R376C mutations on SG dynamics. For this purpose, we transiently treated cells transfected with GFP-tagged mutants (L556S and R376C) or WT DDX3X with 1mM sodium arsenite, a well-known stimulus used to induce SG formation (Goncalves et al., 2019; Wheeler et al., 2016). After the treatment with sodium arsenite, cells expressing either the mutants or the WT DDX3X constructs exhibited prominent DDX3X-enriched cytoplasmic granules (Figure 2A; Videos S1, S2, and S3). The same could be observed when cells were heat-stressed (43°C) (Figures S3). Again, these granules were identified as SGs via colabeling with TIA1 (Figure 2A). By performing FRAP analysis in cells expressing the WT constructs exposed to sodium arsenite, we observed that bleached granules presented a fluorescence signal recovery of approximately 70% of the basal fluorescence in 50 s (Figures 2B and 2C). The recovery of the fluorescence signal must be due to the recruitment of unbleached GFP-tagged DDX3X from outside of the photobleached field, suggesting high protein mobility within the granule. In contrast, the fluorescence of the bleached granules containing DDX3X-L556S or DDX3X-R376C did not recover at all (Figures 2B and 2C).
Figure 2.

WT and mutant DDX3X-GFP NaAsO2-induced stress granules exhibit different dynamics
(A) Representative images of SH-SY5Y cells transfected for 48 hr with GFP-tagged WT, L556S, or R376C mutant DDX3X variants, incubated for 60 min in culture medium containing 1mM NaAsO2 and immunostained with TIA-1 (red). Intensity map on the right represents relative gray scale intensity. N = 55 cells from at least 3 different experiments were imaged.
(B) FRAP analysis of DDX3X WT, L556S, and R376C-GFP granules after 60 min incubation with 1mM NaAsO2. Granules (arrows) are shown prior to photobleaching, and at 0 s, 4 s, and 40 s after.
(C) Error bars represent mean values ± SEM (N = at least 20 granules from multiple cells per experiment from 3 individual experiments).
(D) Effects of 1,6-hexanediol on NaAsO2-induced stress granules. Representative image of a transfected SH-SY5Y cells were stressed with NaAsO2 for 60 min, imaged at time point 0 s and after 30 s of the addition of 3.5% 1,6-hexanediol. N = 9 cells for each condition were continuously imaged.
(E) SH-SY5Y cells were cotransfected with plasmids for full-length mCherry-tagged WT-DDX3X and for GFP-tagged L556S or R376C DDX3X. Colocalization of expressed proteins is shown on the right merge images. N = 55 cells from at least 3 different experiments were imaged.
(F and G) Effects of 1,6-hexanediol on NaAsO2-induced stress granules. SH-SY5Y cotransfected with-full length mCherry-tagged WT-DDX3X and for (F) GFP-tagged L556S or (G) R376C DDX3X were stressed with NaAsO2 for 60 min, imaged at time point 0 s and after 30 s of the addition of 3.5% 1,6-hexanediol. Observe that neither WT nor mutant-DDX3X granules disassemble after hexanediol treatment. N = 9 cells for each condition were continuously imaged. Scale bar = 10 μm.
. One z-stack of images was performed each 20 sec during 60 min.
One z-stack of images was performed each 20 sec during 60 min.
One z-stack of images was performed each 20 sec during 60 min
The effects of the DDX3X mutants on SG dynamics prompted us to quantify the displacement of the DDX3X-enriched SGs in cells exposed to sodium arsenite. Sixty minutes after the exposure to sodium arsenite, the mobility of granules containing the mutant variants was markedly reduced when compared with WT (Figure S2D). These findings further corroborate the idea of protein aggregation inside the SGs containing the DDX3X mutants. To investigate this hypothesis, we tested the sensitivity of the SGs containing either the DDX3X-WT or the mutants to 1,6-hexanediol, a compound known to disassemble granules that exhibit liquid-like properties by perturbing weak hydrophobic interactions (Patel et al., 2007; Ribbeck and Gorlich, 2002). The addition of 3.5% 1,6-hexanediol led to an immediate disassemble (30 s) of SGs formed by DDX3X-WT in stressed cells, indicating that they behave as liquid-like condensates (Figure 2D) (Kroschwald et al., 2015). However, the SGs containing DDX3X-L556S or DDX3X-R376C did not disassemble after exposure to 1,6-hexanediol (Figure 2D), not even after 60 min of exposure to the drug (Figure S2E), indicating that they form solid-like condensates. In addition, to determine DDX3X-WT/L556S/R376C protein stability, we used cycloheximide (CHX), a potent inhibitor of protein biosynthesis, and performed CHX-chase experiments (Figures S2C and S2D). By Western blotting, we found that DDX3X-WT and R376C are degraded over time, while DDX3X-L556S was much more stable after 9 hr of CHX treatment. Altogether, our results suggest that the mutations of L556S and R376C cause changes in the SG properties, by converting them from liquid-like structures, as in SGs formed by WT DDX3X, into solid-like aggregates. Interestingly, while the mutant R376C does not present impaired ATPase activity and requires external stress conditions to trigger its aggregation process in SGs, the mutant L556S nucleates the solid-like condensates even in the absence of external stressful insults and is more stable.
WT DDX3X colocalizes with its mutant variant in the aberrant stress granules
Aberrant phase transition of SGs triggered by misfolded proteins may induce widespread recruitment of native proteins, mainly those enriched in low-complexity sequences. This process depletes cells from essential signaling proteins, which may critically contribute to the deleterious effects of mutant variants (Cao et al., 2020; Mateju et al., 2017; Snijders Blok et al., 2015; Wolozin, 2012). To explore the hypothesis that solid-like structures formed by the mutant L556S could sequester WT DDX3X, we cotransfected cells with mCherry-tagged WT DDX3X along with each one of the GFP-tagged mutant DDX3X constructs (Figure 2E). Observation of unstressed cells coexpressing mCherry-tagged DDX3X-WT and GFP-tagged DDX3X-L556S revealed that both proteins appeared colocalized in several cytoplasmic aggregates (Figure 2E, upper panel). On the other hand, when we analyzed cells coexpressing mCherry-tagged DDX3X-WT and GFP-tagged DDX3X-R376C mutant, we observed both proteins diffusely and independently distributed through the cytoplasm (Figure 2E, bottom panel). Remarkably, after treatment with sodium arsenite, cells coexpressing mCherry-tagged DDX3X-WT and GFP-tagged DDX3X-R376C or DDX3X-L556S exhibited aggregates containing both WT and mutant protein; additionally, these aggregates could not be solubilized by treatment with 1,6-hexanediol (Figures 2F and 2G). Overall, these results led to the idea that the aggregates formed in the SGs by the mutant forms L556S and also R376C of DDX3X can both trap and deplete native DDX3X stocks from the cytoplasm leading to compromised cell function.
ID-linked mutations cause DDX3X protein misfolding
Guided by previous results showing aberrant aggregation of mutant DDX3X in cells, we turned to in vitro systems to investigate how L556S and R376C mutations, which exhibit different behaviors in cells, affect the folding properties and stability of DDX3X using different biophysical and spectroscopic methods. Because of the known difficulties in working with stable full-length purified DDX3X in vitro, we used the D1D2-WT and the mutant constructs (L556S and R376C) to investigate the effects of such mutations on protein structure. Size exclusion chromatography multiangle light scattering (SEC-MALS) analysis showed that D1D2-L556S exists as a monomer with a molecular weight (MW) of 49 ± 0.01 kDa and a dimer (MW: 97.2 ± 4.4 kDa) (Figures S4A–S4D). On the other hand, the D1D2-WT and D1D2-R376C mutant constructs exist only as monomers (MW: 50.8 ± 0.01 kDa and 49.7 ± 0.1 kDa, respectively) (Figures S4A-A and S4E). An extension of the SEC's monitoring period of up to three days at 4°C showed no interconversion between monomeric and dimeric forms of D1D2-L556S (Figure S4F). We then used dynamic light scattering (DLS) to assess the hydrodynamic radius (Rh) of the D1D2 constructs in solution by an independent technique. The DLS Rh of the D1D2-WT was 4.4 ± 0.01 nm (Table S1). The Rh of the monomeric forms of the mutants D1D2-L556S (4.7 ± 0.1 nm) and D1D2-R376C (4.6 ± 0.1 nm) were similar to the values seen for the D1D2-WT construct, while the dimeric form of the D1D2-L556S mutant had an Rh of 5.5 ± 0.1 nm (Table S1). These values follow the calculated ones from the available X-ray structures of the monomeric and dimeric structure of D1D2 (Song and Ji, 2019) and support the notion that the L556S and R376C mutations do not affect the structural packing of DDX3X-D1D2. The monomeric and the dimeric forms of D1D2-L556S showed deficient ATPase activity (Figure S4G), leading us to conclude that the impairment of the enzyme activity of D1D2-L556S is independent of the oligomerization state.
Next, we used circular dichroism (CD) and differential scanning fluorimetry (DSF) to explore whether L556S and R376C mutations affect the folding properties and stability of D1D2 constructs. CD spectroscopy of the WT and mutant forms of the DDX3X-D1D2 constructs showed spectra typical of a well-folded α-helical/β-sheet secondary structure. We found subtle changes in the far UV spectra of D1D2-L556S (mainly the dimeric form) and D1D2-R376C compared with the D1D2-WT protein, suggesting that the L556S and R376C mutations have no severe impact on the D1D2 native folding (Figure 3A). We then used DSF to determine the effects of L556S and R376C mutations on the thermal stability of the D1D2 constructs. The melting curves obtained for the WT and mutant constructs were monophasic (Figure 3B). The D1D2-WT exhibited a Tm of 51.6 ± 0.1°C. The Tm of D1D2 mutant constructs shifted to the left, indicating a decrease in thermal stability. The observed Tm values of the monomeric and the dimeric D1D2-L556S were 40.6 ± 0.1°C and 44.5 ± 0.1°C, respectively, while the D1D2-R376S exhibited a Tm of 40.1 ± 0.2°C. Our analysis also identified substantially high fluorescence of monomeric and dimeric forms of D1D2-L556S at 37°C, suggesting that the L556S mutation results in a significant exposure of hydrophobic regions to solvent.
Figure 3.

Biophysical characterization of DDX3X protein and aggregation propensity induced by pathogenic mutations
(A) Circular dichroism (CD).
(B) Differential scanning fluorimetry (DSF) curves for D1D2 domain of WT-DDX3X and mutants during thermal denaturation. Tm values: WT - 51.6 ± 0.1°C, L556S monomer – 40.6 ± 0.1°C, L556S dimer – 44.5 ± 0.1°C, R376C – 40.1 ± 0.2°C.
(C) ANS fluorescence at 25°C.
(D) Rayleigh light scattering (RLS) at 350 nm of WT and mutants during incubation at 37°C.
(E) ThT fluorescence of protein samples before and after incubation at 37°C. ∗p < 0.05 when compared with WT 0 hr ∗∗p < 0.005 when compared with WT 24 hr.
(F) ANS fluorescence of proteins during incubation at 37°C.
(G) CD of L556S mutant at 10°C after incubation at 37°C during 24 hr.
(H) CD of R376C mutant before and after incubation at 37°C.
(I) Negative staining image of L556S-DDX3X after 3-hr-incubation at 37°C.
(J) Negative staining image of R376C-DDX3X after 3-hr-incubation at 37°C. Scale bar = 100 nm
To confirm this observation, we compared the surface hydrophobicity of D1D2-WT and mutant DDX3X constructs, as determined by bis-ANS (8-anilino-1-naphthalenesulfonic acid) fluorescence. Bis-ANS, a hydrophobic probe, is nonfluorescent in aqueous solution and emits a fluorescent signal only when bound to the hydrophobic residues on a molecule (Lindgren et al., 2005). Consistent with the DSF assay results, the ANS fluorescence signals of both monomeric and dimeric forms of the D1D2-L556S were higher than the signal of D1D2-WT (Figure 3C). Furthermore, D1D2-R376C showed no ANS fluorescence signal, implying that this mutation does not expose hydrophobic residues. Together, these analyses led us to conclude that both L556S and R376C reduce thermodynamic stability, predisposing the D1D2 mutant constructs to misfold. However, only L556S mutation induces conformational changes within the core helicase domain that eventually led to an exposure of hydrophobic residues to the solvent in the native state.
DDX3X pathogenic mutants have a high propensity to form amyloid fibrils
Next, we used Rayleigh light scattering (RLS) measurements of the WT and mutants D1D2 constructs at 350 nm to further substantiate the hypothesis that the protein misfolding caused by L556S and R376C mutations predispose to DDX3X aggregation. The experimental approach consisted of measuring the temporal evolution of RLS peak intensity of samples containing 15 μM of the constructs at 37°C along 24 hr. The RLS intensity of samples containing D1D2-WT was constant along the experiment's period, indicating that this construct does not aggregate as a function of time (Figure 3D). However, the RLS signal of samples containing the L556S or the R376C mutant constructs increased progressively from the first hours and continued to increase up to 24 hr. These results indicate that the DDX3X mutants have an increased propensity to aggregate at physiological temperature.
To explore the nature of the aggregates formed by D1D2 mutants, we measured the progress of the aggregation reaction using ThT (Thioflavin T) fluorescence at 480 nm (excitation 440 nm), a dye that selectively binds to β-sheets which are abundant in amyloid aggregates (Lindgren et al., 2005). We monitored samples of D1D2-WT, D1D2-L556S, and D1D2-R376C mutant constructs incubated with ThT for 24 hr. Samples containing the D1D2-WT construct showed only slight increases in ThT fluorescence intensity after 24 hr of monitoring. However, for the samples of D1D2-L556S (either monomeric or dimeric forms) and D1D2-R376C mutants, the fluorescence intensities were significantly increased along the monitoring period (Figure 3E). These results suggest that both mutant constructs (D1D2-L556S and D1D2-R376C) exhibit a high propensity for self-aggregation at physiological temperature, producing amyloid-like assemblies.
Notably, we observed an increase in the ANS fluorescence intensity for both mutant constructs during protein aggregation (Figure 3F), pointing to an enhanced exposure of the mutant hydrophobic regions compared with D1D2-WT. After aggregation (24 hr at 37°C), the CD spectra showed no significant change in the secondary structure of the mutant constructs, suggesting that protein aggregation occurs without a concomitant increase in β-sheet content (Figures 3G and 3H). Nevertheless, the X-ray fibril diffraction pattern obtained from amyloid-like fibrils of mutant proteins showed the characteristic reflections at 4.7 Å and 10 Å, consistent with a cross-β fibrillary architecture (Figure S5). Accordingly, the morphology of the aggregates registered by transmission electron microscopy (TEM) showed that the mutant constructs formed fibrillary structures resembling typical amyloid-like fibrils.
Mechanism of DDX3X fibril formation induced by pathogenic mutations
Next, we used TEM to determine the morphology of the aggregates of D1D2 mutants. Samples containing 15 μM of D1D2-WT, D1D2-L556S, or D1D2-R376C were incubated at 37°C for 3 hr before application to the TEM grids for negative staining imaging. This approach was based on our observation of the significant aggregation propensity of D1D2 mutants in the RLS and ThT fluorescence assays. The micrographs show fibrillary structures only in the samples of D1D2 mutants with an average width of 25 nm (Figures 3I and 3J). To further characterize the D1D2 fibril morphology, we obtained TEM micrographs from samples of D1D2 constructs incubated at 4°C. In this condition, the dominant structures in the samples of all three constructs were spherical assemblies with a diameter ranging from 10 to 30 nm (Figure 4). The 10-nm spheres are similar in size to the trimer identified in the asymmetric unit of apo DDX3X crystallographic structure (PDB code 5e7i). The existence of larger spheres of DDX3X in TEM images suggests the assembly of higher order oligomers of DDX3X. Interestingly, similar spherical assemblies were also seen in samples of the SG protein Gle1, which is a partner of DDX3X, and in yeast and Caenorhabditis elegans DDX3X orthologous Ded1 and LAF1 (Aditi et al., 2019; Folkmann et al., 2013, 2014; Kim and Myong, 2016; Putnam et al., 2019). We reason that the formation of oligomers by proteins kept at 4°C occurred during sample preparation owing to the high local glycerol concentration. In support of this, we found spherical structures of ∼30 nm diameter in samples of D1D2-WT suspended in glycerol 40% at room temperature (Figure S6A).
Figure 4.

Transmission electron microscopy of DDX3X
Negative staining images of D1D2 domain of WT-DDX3X, L556S-DDX3X, and R376C-DDX3X before (0 hr) and after incubation at 4°C (left panel) or 37°C (right panel) for 3 hr. Scale bar = 100 nm.
Further analysis of images taken after incubation of D1D2-R376C, the monomeric and the dimeric forms of D1D2-L556S (Figures 4 and S6B), at 37°C provided compelling insights into the mechanism of fibril formation. We noticed that distinct configurations of protein aggregates coexist in the samples of D1D2 mutants incubated at 37°C, including isolated spherical assemblies, lined sequences of individual spheres, and strings of connected spherical assemblies together with the amyloid-like fibril structures. The coexistence of such structures suggests that D1D2 mutants form amyloid-like fibrils in a manner akin to nucleated conformational conversion (NCC). In this model, oligomer-like aggregates serve as precursors that gradually rearrange into amyloidogenic oligomers during the lag phase of the fibrillation process (Serio et al., 2000). Notably, in this multistate mechanism, the fibrillation reaction rate depends on the number of oligomers, but it is only weakly dependent on protein concentration.
To better understand the fibril formation mechanism, we examined the effect of mutant concentration in protein aggregation. When we carried out ThT fluorescence during a time course using the D1D2-L556S construction, we observed a prominent loss of ThT signal over time (data not shown). Loss of signal may be directly associated with protein losses due to adsorption into assay plates via hydrophobic binding (Murray et al., 2013). Reasoning that this phenomenon is related to the enhanced exposure of hydrophobic surfaces induced by the L556S mutation, we evaluated the dependence of protein concentration in protein aggregation using the D1D2-R376C construct. We performed a twofold serial dilution of D1D2-R376C to generate a set of fibrillation curves. The protein concentration spanned from 7.5 μM to 60 μM. ThT fluorescence recorded during this process displayed sigmoid-shaped curves, typical of the nucleation-elongation mechanism of fibrillation, in which fibrils are assembled through the sequential addition of monomers to a nucleus (Figure S6C) (Almeida and Brito, 2020; Librizzi and Rischel, 2005). As a first guide to determine the dominant fibril assembly mechanism, we investigated the half-time (t1/2) dependence on initial protein concentration (C0). Classical nucleation-elongation models state that t1/2 and C0 are related by the scaling law: half-time ∝ (initial monomer concentration)γ. Plotting the half-time versus the initial monomer concentration on a double logarithmic plot gives the value of γ (scaling coefficient), an indicator of the dominant microscopic mechanism responsible for generating new aggregates (Cohen et al., 2013; Linse, 2017). We obtained an experimental value of γ = −0.11 (Figure S6D), implying that mutant protein aggregation has minimal dependence on initial monomer concentration (Meisl et al., 2017). Thus, our data portray a typical secondary side fibrillation process that is affected by secondary (side) nucleation (Cohen et al., 2013; Linse, 2017; Meisl et al., 2017; Pagano et al., 2014).
Together with the enhanced exposure of hydrophobic regions during protein aggregation observed using the ANS probe (Figure 3F), we suggest that the side-by-side interactions observed for DDX3X-D1D2 oligomers by TEM (Figures 4 and S6B) may be essentially related with the process of fibril assembly (Figures 3I and 3J).
A mutation-containing domain alone is sufficient for DDX3X L556S mutant protein self-aggregation
Intrinsically disordered regions (IDRs) of RNA-binding proteins (RBPs) are drivers of granule assembly. Not surprisingly, many disease-associated genetic mutations in RBPs that promote protein aggregation occur in IDRs and prion-like domains (Da Cruz and Cleveland, 2011; Kim et al., 2013; Klar et al., 2013; Kwiatkowski et al., 2009; Molliex et al., 2015; Sreedharan et al., 2008; Vieira et al., 2014). However, the granule-assembly inducer L556S mutation is located in the helicase domain of DDX3X, not in the IDRs. The demonstration here that the D1D2 mutants, which lack the IDRs form oligomers and fibrils, suggests a distinct aggregation mechanism that does not involve the IDRs. To further explore this issue, we made constructs of WT and L556S mutant containing the D2 domain of DDX3X alone (residues 407–578), without any additional ordered or disordered regions. The D2 crystal structure (PDB code 2jgn) has a similar fold to higher ordered constructions (PDB code 5e7i). ThT assay with the D2 domain at 37°C also indicated that the L556S mutation induces amyloid-like structures (Figure S7A). Using a highly sensitive 2D-NMR technique, we compared the 1H-15N HSQC spectrum of WT and L556S D2 domain (Figures S7B and S7C). We found that while the WT construct presents a spectrum akin to a well-folded globular protein, L556S mutant spectrum corresponds to a highly aggregated protein. These results indicate that L556S mutation in the D2 domain is sufficient and capable of inducing protein aggregation, even in the absence of the intrinsically disordered regions required for LLPS.
In silico structural analysis of DDX3X mutants
To gain further insights into the impact of L556S mutation on DDX3X structure and aggregation propensity, we performed in silico structural analysis and molecular dynamics (MD) simulations using an available DDX3X crystallographic structure (PDB ID: 5E7J). L556 locates at the α-helix 18 (H18) of DDX3X domain 2 (Figure S1). This residue makes hydrophobic contacts with V419 and F545 from domain 2 β-sheet and with L518 and I522 from H17. Differently, R376 residue locates at a loop region of DDX3X domain 1 and makes hydrogen bonds with neighbors T378 and P371 residues and polar interaction with the main chain of F396, which locates at the α-helix 12 (H12) (Figure S1B).
MD simulations indicated that changing arginine to cysteine at DDX3X 376 position significantly increases the mobility of H12 (mainly residues 389 to 396) in comparison with WT, as observed by the Cα RMSF (Figure 5A, upper graph). During WT DDX3X simulations, R376 maintains putative hydrogen bonds with the main chain of P371 and F396, with an average occupancy of 64.5% and 12.3%, respectively. Also, R376 interacts with the sidechain of T378 and E366, with an average occupancy of 44.5% and 17.2%, respectively. P371 and T378 are neighbor residues of the same R376 loop, whereas F396 and E366 locate at H12 and α-helix 11 (H11), respectively (Figure 5B, upper structure). Additionally, salt bridges were observed between R376 and E366. All these contacts stabilize the R376 loop against the two helices H11 and H12 and the domain 1 β-sheet. Thus, changing the large polar residue arginine to cysteine, which is less polar and has a shorter sidechain, could abolish all these contacts (Figure 5C, lower structure). The transient destabilization of this region in MD simulations could be the origin of the R376C reduced stability observed in DSF experiments compared with the WT protein. On the other hand, the mutation of L556 to serine did not disturb H18, nor the β-sheet that contacts the α-helix (Figure 5A, lower graph, residues from 552 to 561). However, it changes its initial conformation during the simulation, making a hydrogen bond with I552 main chain from the same α-helix and avoiding the β-sheet hydrophobic environment. During the simulation, hydrophobic residues from the same α-helix, such as I552, L559, and L560, maintain contacts with the domain 2 β-sheet hydrophobic region, explaining the stability of H18. However, L556 locates at a structured α-helix, and L556S mutation could impair the helix formation during the folding process, which was not evaluated in our conventional MD simulations.
Figure 5.

In silico structural analysis of DDX3X mutations
(A) 200-ns molecular dynamic simulations of R376C (upper graph) or L556S (lower graph) mutants and wild-type DDX3X. Simulations were performed in triplicates. The graph shows the Cα RMSF for each residue from domain 1 and 2. Lines represent mean, and vertical traces represent standard deviation.
(B) Representative frames from MD simulations of R376 or R376C showing intradomain contacts observed during simulations.
(C) Ramachandran plot of the first leucine from the wild-type sequence LLDLL and the serine from the mutated sequence SLDLL. Yellow contours represent the favored regions in Ramachandran plot (detailed information in STAR methods section).
(D) Representative frames from MD simulations showing intradomain contacts of L556S.
To investigate this hypothesis, we searched in the Protein DataBank (PDB) for available protein structures that contain the WT H18 sequence LLDLL (the first L corresponds to L556) or the mutated sequence SLDLL (S corresponds to L556S). Next, we determined the Phi and Psi dihedral angles of leucine or serine in those structures. As expected by DDX3X structure, most Phi/Psi torsional angles of the first leucine in LLDLL motif (93%) are in the favored right-handed α-helix region in Ramachandran plot (Figure 5C). However, this preference is substantially reduced for the serine in SLDLL motif. Only 36% of Phi/Psi angles are at the favored α-helix region, and most of them are sparsely distributed over a favored β-sheet region or other regions (64%) (Figures 5C and 5D). This finding may indicate that during DDX3X folding, L556S mutation could hinder the complete H18 folding, which could impair a proper binding between H18 and domain 2 β-sheet. Consequently, this hydrophobic β-sheet become solvent-exposed, which accords with the increase in solvent-exposed regions presented in ANS and DSF assays.
DDX3X-L556S escapes X-inactivation in patient-derived iPSCs cells
It has been reported that although DDX3X can escape X inactivation (Cotton et al., 2015; Lahn and Page, 1997), most of the transcripts are not fully expressed from the inactivated X chromosome, meaning that the escape is often partial and incomplete (Morleo and Franco, 2008; Shvetsova et al., 2019). We hypothesized that small amounts of the DDX3X mutated gene that escape complete X inactivation somehow impair neurodevelopment. The erythroblasts were reprogrammed to iPSCs and compared with three WT-iPSC cell lines previously established (Araujo et al., 2018). All iPSC lines used in this study expressed markers of pluripotent cells, including LIN28 and SSEA4, as demonstrated by immunofluorescence staining (Figure 6A). We used microarray comparative genomic hybridization (Array-CGH) to screen for genome-wide chromosomal imbalances in the iPSCs. Neither the control nor the mutant iPSCs showed chromosomal abnormalities (Figure 6B). Real-time quantitative reverse transcription PCR (qRT-PCR) analysis demonstrated the expression of selected endogenous pluripotent transcription factors including OCT3/4 and NANOG, in all iPSCs used in this study (Figures 6C and 6D).
Figure 6.

DDX3X-L556S patient-derived iPSCs escape X inactivation
(A) Immunofluorescence for LIN28 (red), SSEA4 (green), and DAPI (blue) in WT-iPSC and DDX3X-L556S iPSCs. Scale bar, 20 μm.
(B) Microarray comparative genomic hybridization (Array-CGH) analysis for genome-wide chromosomal imbalances shows all iPSC clones derived from the patient with L556S mutation used in this study present no chromosome copy number alteration.
(C and D) (C) RT-qPCR analysis of iPSCs for NANOG and (D) OCT3/4 expression. Error bars ± SEM. n.s: not statistically significant, Student's t t test.
(E) Immunofluorescence of WT-iPSC and DDX3X-L556S iPSCs for DDX3X (green), OCT4 (red). Scale bar, 20 μm.
(F) RT-qPCR analysis of iPSCs culture of WT and DDX3X-L556S for DDX3X expression. Error bars ± SEM, Student's t test.
(G) Sequencing chromatograms demonstrating that the mutated L556S DDX3X allele is transcribed and escapes X inactivation in iPSCs and blood cells DDX3X-L556S patient (black arrows), compared with WT blood cell sample.
To test this hypothesis, we measured the X inactivation in iPSCs from DDX3X control and L556S patients. Although no differences in DDX3X protein expression (Figure 6E) or levels of transcripts (Figure 6F) were found between WT and L556S patient-derived iPSCs, we observed an X inactivation escape of the mutant DDX3X-L556S, albeit greatly reduced compared with the WT allele, as well as in blood cells from the DDX3X-L556S carrier patient (Figure 6G). These results indicate that the skewed X inactivation of the DDX3X-L556S mutated cells favor the expression of the normal allele, as expected.
Discussion
Failure of a protein to fold correctly, or to maintain its folded state, has emerged as a hallmark and critical event triggering pathological abnormalities in many forms of neurodegenerative diseases, particularly amyotrophic lateral sclerosis, frontotemporal dementia, Parkinson's disease, Huntington's disease, and Alzheimer's disease (Hartl, 2017; Klaips et al., 2018; Ross and Poirier, 2004; Sweeney et al., 2017). Aberrant folding generally arises as a result of destabilizing mutations that generate condensates of misfolded proteins in droplets, where they acquire solid-like properties (Boeynaems et al., 2018; Verdile et al., 2019). These solid-like states of misfolded proteins may cause the loss of critical cellular functions. Apart from loss-of-function effects, these cytotoxic protein aggregates can produce a toxic gain of function (Hartl, 2017; Hipp et al., 2014; Stefani and Dobson, 2003; Winklhofer et al., 2008).
A key finding of our study, however, is that the DDX3X mutations evaluated here did not lead to dramatic changes in protein folding, as shown by our biophysical assays (Figure 4). Nevertheless, these discrete local changes caused by L556S and R376C mutations give rise to the exposure of ANS-binding hydrophobic surfaces mediating the formation of solid-like structures and interactions with other proteins, resulting in their functional impairment and sequestration (Bolognesi et al., 2010; Campioni et al., 2010; Chiti and Dobson, 2006; Olzscha et al., 2011). Accordingly, the syndromic neurodevelopmental disorder related to DDX3X missense mutations may indeed be considered a protein aggregation disease.
Through a comprehensive analysis of WT and mutant-DDX3X biophysical features, we provide evidence that the ID-linked missense mutations L556S and R376C render DDX3X prone to aggregation. Intriguingly, both mutations distinctively affect the ATPase function. While L556S reduces, R376C mutation increases DDX3X ATPase activity. By using a combination of biophysical and both in vitro and in vivo imaging approaches, we demonstrated that these mutants assemble solid-like condensates and amyloid-like fibrils through a mechanism of NCC. The formation of intracellular aggregates especially localized in SGs correlates with a reduction in the viability of SH-SY5Y cells.
The most crucial characteristic of SGs is their dynamic nature. SGs rapidly assemble in response to stresses and quickly disassemble when the stress is eliminated (Wolozin, 2012; Wolozin and Ivanov, 2019). These assemblies result from LLPS and develop properties, such as fusion, coalescence, drip, and reversibility, as observed for droplets containing DDX3X. This microscopically visible dynamic behavior is also observed at the molecular level, where the SGs-constitutive proteins are in dynamic equilibrium with the cytosol. Studies using FRAP analysis indicate that proteins shuttle in and out of SGs with residence times ranging from seconds (for example, TIA1) to minutes (for example, fragile X mental retardation protein (FMRP) (Baguet et al., 2007; Wolozin and Ivanov, 2019). However, LLPS can also undergo further transitions, formed by amyloid-like aggregates, which often are irreversible (Kato et al., 2012; Murray et al., 2017). The missense L556S mutation in DDX3X protein evaluated here strikingly portray how the SGs transition from a dynamic membrane-less organelle to a pathological cytoplasmic aggregate. The same could be observed for the DDX3X-R376C variant. It is not entirely understood how a physiological phase separation involving high solubility can be converted into a toxic solid-like phase separation. SG proteins, such as hnRNPA1 and FUS, are capable of undergoing LLPS in vitro, and protein liquid condensates can undergo liquid-to-solid transitions and nucleate aggregation (Kato et al., 2012; Kwon et al., 2013; Lin et al., 2015; Molliex et al., 2015; Murakami et al., 2015; Patel et al., 2015). The driving force for these transitions is possibly sequence related, and there is emerging evidence that individual protein sequences have evolved to use liquid-to-solid transitions for function. LLPS of proteins containing IDRs has been proposed as a mechanism underlying the formation of SGs. Fine regulation through acetylation/deacetylation, methylation, and phosphorylation are key mechanisms to guarantee that SGs formation only take places when necessary (Hofweber et al., 2018; Monahan et al., 2017; Nott et al., 2015; Saito et al., 2019). For example, it was recently shown that lysine acetylation in IDRs regulates LLPS and impacts SG maturation via DDX3X (Saito et al., 2019).
However, some mutations associated with neurodegenerative diseases, such as ALS and FTD, are capable of accelerating these liquid-to-solid transitions and increase the protein aggregation propensity (Kwon et al., 2013; Molliex et al., 2015; Murakami et al., 2015; Patel et al., 2015). These observations link phase separation with fiber formation and help to explain the emergence of aberrant phase transitions. Although well accepted for neurodegenerative diseases, this behavior had not been associated with mutations in neurodevelopmental disorders so far. Here, we provide evidence showing that ID-linked mutations in DDX3X that turns the protein into an aggregation-prone state induce the formation of aberrant phase separation, consistent with a liquid-like to a solid-like phase transition, also observed for FUS, TDP43, and Tau (Ambadipudi et al., 2017; Patel et al., 2015; Schmidt et al., 2019). However, the demonstration here that the D1D2 mutants, which lack the IDRs form oligomers and fibrils, suggests a distinct aggregation mechanism that does not involve the IDRs.
ID-linked missense mutations in DDX3X have been associated with both the formation of aberrant cytoplasmic granules and a more severe clinical phenotype than loss-of-function mutations (Lennox et al., 2020). A detailed analysis of a cohort of patients carrying different DDX3X mutations showed that de novo mutations in unrelated individuals lead to markedly similar phenotypes (Lennox et al., 2020). Those patients carrying missense mutations had more severe phenotypes. In contrast, patients with LoF mutations exhibit milder clinical phenotypes supporting the hypothesis that specific missense mutations function in a dominant-negative fashion (Lennox et al., 2020). Additionally, it was previously shown that DDX3X is required for neural progenitors to produce cortical neurons during development and even a 25% reduction in DDX3X levels strongly deregulated neurogenesis (Lennox et al., 2020). Therefore, it is reasonable to think that much of the cell toxicity portrayed by the L556S mutation may be due to the sequestration of healthy proteins into the newly formed aberrant aggregates, as we observed (Figure 2). However, it remains to be addressed whether DDX3X-L556S protein variant would reduce viability of neurons in the developing brain.
In summary, our data strongly support the hypothesis that the L556S mutation in DDX3X protein works as a trigger point to induce protein aggregation. These data offer insights into the detailed cellular and molecular mechanisms of how such missense mutation might generate solid-like structures and impair cell viability, increasing the understanding regarding the molecular mechanisms underlying intellectual disability.
Limitations of the study
In this work, we analyzed the molecular and cellular effects of pathogenic missense variants of DDX3X protein using in vitro approaches. By using a combination of biophysical assays, and in vivo imaging approaches, we demonstrate that this mutant assembles solid-like condensates and amyloid-like fibrils that sequestrate healthy protein, compromising cell viability. However, it remains to be addressed whether these mutants would reduce viability of neurons in the developing brain.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat polyclonal anti-TIA-1 | Santa Cruz Biotechnology | Cat# sc-1751, RRID:AB_2201433 |
| Mouse monoclonal andti-MAP2 | Thermo Fisher Scientific | Cat# MA5-12826, RRID:AB_10976831 |
| Mouse monoclonal anti-DDX3 | Santa Cruz Biotechnology | Cat# sc-81247, RRID:AB_2092867 |
| Mouse monoclonal anti-GFP | Santa Cruz Biotechnology | Cat# sc-9996, RRID:AB_627695 |
| Mouse monoclonal anti-LIN28A | Cell Signaling Technology | Cat# 5930, RRID:AB_1903976 |
| Mouse monoclonal anti-SSE4-A | Cell Signaling Technology | Cat# 5930, RRID:AB_1903976 |
| Mouse monoclonal anti-β-actin | Santa Cruz Biotechnology | Cat# sc-47778 HRP, RRID:AB_2714189 |
| Rabbit polyclonal anti-SOX2 | Abcam | Cat# ab97959, RRID:AB_2341193 |
| Bacterial and virus strains | ||
| Rosetta™ 2(DE3) Singles™ Competent Cells | Novagen | Novagene US171400-3 |
| Biological samples | ||
| Human fetal brain cDNA library | Clontech | Cat# HL3003a |
| Chemicals, peptides, and recombinant proteins | ||
| Thioflavin T | Sigma-Aldrich | T3516; CAS:2390-54-7 |
| Bis-ANS | Sigma-Aldrich | D4162; CAS: 65664-81-5 |
| 15NH4Cl | Cambridge Isotope Laboratories | Cat# 39466-62-1 |
| SyproOrange | Thermo Fisher | S6650 |
| NaAsO2 | Sigma-Aldrich | S7400; CAS : 7784-46-5 |
| 1,6 Hexanediol | Sigma-Aldrich | 240117; CAS: 629-11-8 |
| FuGene HD | Promega | E2311 |
| D1D2 DDX3X WT | This paper | N/A |
| D1D2 DDX3X L556S | This paper | N/A |
| D1D2 DDX3X R376C | This paper | N/A |
| D2 DDX3X WT | This paper | N/A |
| D2 DDX3X L556S | This paper | N/A |
| Critical commercial assays | ||
| CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) | Promega | G3582 |
| RNeasy Mini Kit | Qiagen | 74106 |
| cDNA using Superscript II | Thermo Fisher | 11904018 |
| Power Track SYBR Green Master Mix | Thermo Fisher | A46012 |
| AmpliSeq Exome library | Thermo Fisher | A38264 |
| QuikChange site-directed mutagenesis kit | Agilent | Cat# 200523 |
| In Situ Cell Death Detection Kit, POD | Roche | 11684817910 |
| Experimental models: Cell lines | ||
| SH-SY5Y | ATCC | Cat# CRL-2266, RRID:CVCL_0019 |
| Inactivated mouse embryonic fibroblasts | Thermo Fisher | Cat# A34180 RRID:CVCL_RB05 |
| Control Human Erythroblasts | (Araujo et al., 2018) | N/A |
| DDX3X-L556S mutant Human Erythroblasts | This paper | N/A |
| Oligonucleotides | ||
| dsRNA 5'-ACGUCGAUCCGAAACUAUACUUAAUUUUAA-3' and 5'-GUUUCGGAUCGACGU-3' |
Integrated DNA Technology | This paper |
| Primer hsDDX3X.D1D2 forward: 5'-ATTCGCATATGTCAGATG AAGATGATTGGTCAAAA-3'; reverse: 5'-AAGTGCTCGAGTT AACCGCTACTTTGTCGGTAGTC -3'; |
This paper | N/A |
| Primer: hDDX3X.D2.forward: 5'-cagtcGGATCCAGAGTTGGCT CTACCTCTGAAAAC- 3'; reverse: 5'-tcagcGAATTCTTAGTGTT CATAAGCCATGTTTTC-3' |
This paper | N/A |
| Primer: hsDDX3X.M1 forward: 5'GATCGCTCGAGCGATGAGTC ATGTGGCAGTGGAAA 3'; hDDX3X. reverse: 5-GTCAGGAATT CTCAGTTACCCCACCAGTCAACCCC-3'. |
This paper | N/A |
| Primer: hsDDX3X.L556S forward: 5'-AGAGGAACATAAATATTAC TAAGGATTCGTTGGATCTTCTTGTTGAAG-3';hDDX3X.L556S reverse: 5'-CTTCAACAAGAAGATCCAACGAATCCTTAGTAAT ATTTATGTTCCTCT-3'; |
This paper | N/A |
| Primer: hsDDX3X.R376C. forward: 5’- GCCTCCAAAGGGTGT CTGCCACACTATGATGTTTA-3’; hsDDX3X.R376C. reverse: 5’- TAAACATCATAGTGTGGCAGACACCCTTTGGAGGC-3’. |
This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pEGFP-C1 | Addgene | 6084-1 |
| Plasmid: pmCherry-C1 | Addgene | 632524 |
| Plasmid: pEGFP-DDX3X | This paper | N/A |
| Plasmid: pET28a-D1D2-DDX3X | This paper | N/A |
| Plasmid: pETSUMO-D2-DDX3X | This paper | N/A |
| Software and algorithms | ||
| Image J | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Graphpad Prism 8 | GraphPad Software, Inc | https://www.graphpad.com/scientific-software/prism/ |
| NMRPipe/NMRDraw | John Marino Group | https://www.nist.gov/ |
| NMRView | (Johnson, 2004) | http://www.onemoonscientific.com/nmrview |
| VarSeq® software | Golden Helix | https://www.goldenhelix.com/products/VarSeq/index.html |
| Protein Thermal Shift Software v1.1 | Thermo Fisher | 4466038 |
| Bio3D | Grant Lab (Grant et al., 2006) | http://thegrantlab.org/bio3d_v2/webapps |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Matheus de Castro Fonseca (matheus.fonseca@lnbio.cnpem.br).
Materials availability
All developed expression plasmids produced in this study can be made available upon request to the lead contact.
Data and code availability
All data are available within the article or its supplementary materials. Any additional information is available from the lead contact on request.
Experimental models and subject details
Cell lines
SH-SY5Y cell was obtained from the America Type Culture Collection (ATCC). SH-SY5Y cell line was grown in DMEM/F12 - GlutaMAX medium supplemented with 10% (v/v) FBS, 100 IU/ml of penicillin and 100 μg/ml of streptomycin sulfate, and the cells were grown under an atmosphere of 5% CO2 and 95% air. Cells were passaged at ∼80% confluence and seeded as explained for the individual assays.
iMEFs and erythroblasts: Inactivated mouse embryonic fibroblasts (iMEFs, Thermo Fisher) were maintained in DMEM HG (Life Technologies) containing 10% FBS (Sigma-Aldrich). Erythroblasts were expanded from peripheral blood mononuclear cells (PBMC) and then CD71+ cells were magnetically sorted (130-046-201, 1:25, Miltenyi) following the manufacturer's instructions. Erythroblasts were maintained in StemSpan (StemCell Technologies) containing 50 ng/ml SCF, 2 U/ml EPO, 1 μM dexamethasone, 40 ng/ml IGF1, 10 ng/ml IL3 (all from Peprotech) and 10 μg/ml of gentamicin (Gibco). iPSC clones were normally expanded on iMEF feeders using hES medium comprising DMEM-F12, 10% KSR (Invitrogen), 100 mM Non-Essential Amino acids (Invitrogen), 100 mM L-Glutamine (Invitrogen), 100 μM beta-mercaptoethanol (Gibco) and basic 4 ng/ml FGF (Peprotech) and passaged by using dispase (Life Technologies). For feeder- free culturing, cells were grown on hESC-qualified matrigel (Corning) coated dishes, maintained in mTeSR1 (StemCell technologies) and passaged by using dispase.
All work with iPSC lines was carried out in accordance with institutional, national, and international guidelines, and were approved by the Institutional Review Board from the Research and Ethics Committee of the Institute of Biosciences of the University of São Paulo. Blood samples from control and affected individuals were collected with informed written consent with approval from the University of São Paulo. This manuscript does not contain data from animal studies or from clinical studies using human subjects.
Method details
Whole exome sequencing
DNA samples were prepared using the AmpliSeq Exome library, according to the manufacturer's specifications. The AmpliSeq libraries were single-end sequenced on an Ion ProtonTM System at Beijing Genomics Institute (BGI, Beijing, China). The raw reads were aligned to the reference genome (GRCh37/hg19), using BWA (Li and Durbin, 2009), and pre-processed, according to GATK toolkit (DePristo et al., 2011). Filtering and prioritization of variants were conducted using VarSeq® software (Golden Helix, Bozeman, MT, USA). The variants were filtered per population frequency (<0.01), quality (Phred quality ≥ 20, genotype quality ≥ 20), read depth (≥ 10).
X-inactivation analysis
X-inactivation, tested by determining the methylation status of the androgen receptor gene (AR), was evaluated as described previously.
Sanger sequencing
Sanger sequencing was used to confirm the presence of the variants considered potentially pathogenic and segregation studies (primer sequences available upon request).
Cloning and mutagenesis
The human wild-type DDX3X gene comprising residues S131-G607 (D1D2) was amplified by PCR from blood cDNA and cloned into pET28a vector (Novagen), using NdeI and XhoI restriction sites. The D2 domain of DDX3X (residues R407-H578) was cloned into a modified version of pETSUMO vector (Invitrogen) using BamHI and EcoRI. The L556S point mutation was created using basic PCR-based site-directed mutagenesis and confirmed by DNA sequencing. The primers used were: hsDDX3X.D1D2.forward: 5′-TTCGCATATGTCAGATGAAGATGATTGGTCAAAA-3′; hDDX3X.D1D2.reverse: 5′- AAGTGCTCGAGTTAACCGCTACTTTGTCGGTAGTc -3′; hDDX3X.D2.forward: 5′-CAGTCGGATCCAGAGTTGGCTCTACCTCTGAAAAC- 3′ ; hDDX3X.D2.reverse: 5′-TCAGCGAATTCTTAGTGTTCATAAGCCATGTTTTC-3′.
For cellular experiments, the gene region encoding for DDX3X (residues 1-662; NP_001347.3) was amplified by PCR from a human fetal brain cDNA library with the cDNA library construction kit (Clontech, Mountain View, CA) and cloned into pEGFP- C1 or pcDNA-mCherry vector between the XhoI and EcoRI restriction sites. The primers used for amplification of the full construct were: hsDDX3X.forward: 5'GATCGCTCGAGCGATGAGTCATGTGGCAGTGGAAA 3'; hDDX3X.reverse:
5′GTCAGGAATTCTCAGTTACCCCACCAGTCAACCCC 3′. DDX3X (L556S and R376C) mutants were generated using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Primers used for mutagenesis were: hsDDX3X.L556S forward: 5'- AGAGGAACATAAATATTACTAAGGATTCGTTGGATCTTCTTGTTGAAG-3';
hDDX3X.L556S reverse:
5'-CTTCAACAAGAAGATCCAACGAATCCTTAGTAATATTTATGTTCCTCT-3';
hsDDX3X.R376C. forward: 5’- GCCTCCAAAGGGTGTCTGCCACACTATGATGTTTA-3’; hsDDX3X.R376C. reverse: 5’- TAAACATCATAGTGTGGCAGACACCCTTTGGAGGC-3’.
Protein expression and purification
DDX3X proteins (D1D2 – WT, L556S and R376C) were expressed in E. coli Rosetta II (Novagen). Pre-cultures were used to express the proteins in LB medium at 37°C for 5 hr at 200 rpm. The culture temperature was turned down to 18°C for 1 hr before induction with 0.5 mM IPTG (isopropyl β -D-1-thiogalactopyranoside) and cells were grown at 18°C for 16 h, then harvested by centrifugation (5,000 × g for 10 min at 4°C). Cells were disrupted by sonication in lysis buffer (50 mM Tris pH 7.5, 500 mM NaCl, 10% glycerol, 10 mM imidazole, 2 mM β-mercaptoethanol, 0.5 mM benzamidine and 0.5 mM phenylmethylsulfonyl fluoride) supplemented with 0.2 mg/mL lysozyme and 25 units/mL Benzonase nuclease (Sigma) and were centrifuged to remove cellular debris (40 000 x g for 60 min at 4 °C). The supernatants were passed by gravity through columns containing TALON metal affinity resin (Clontech) pre-equilibrate with binding buffer (50 mM Tris pH 7.5, 500 mM NaCl, 10% glycerol, 2 mM β-mercaptoethanol), and after washing with binding buffer, the protein was eluted with the same buffer containing 250 mM imidazole. The His6-tag was cleaved using thrombin protease (overnight at 4°C) and the proteins were further purified on a Superdex 200 column (GE Healthcare) with SEC buffer containing (20 mM HEPES pH 7.5, 500 mM NaCl, 10% glycerol and 1 mM DTT).
D2 domain of DDX3X was expressed in the same way that D1D2 and purified as previously described.
SEC-MALS
Size exclusion chromatography coupled to multiple angle light scattering (SEC- MALS) was performed to determine the oligomeric state of DDX3X studied variants. 1mg/mL of purified DDX3X protein was used for an SEC run on a Superdex 200 (10/300) column connected to a Viscotek (GPC-Max) triple detector system. The SEC column was equilibrated in SEC-buffer. 1 mg/ml BSA was used as the protein standard. The chromatograms were analyzed using Omnisec (Malvern), and the data was plotted using the Origin 9.1 software.
Circular dichroism
Experiments were performed with the proteins at 15 μM in SEC, using a Jasco J- 810 spectropolarimeter equipped with a Peltier system PFD 425S for temperature control. CD spectra were acquired at 10°C or 37°C using a 0.1-mm path length cell at 1-nm intervals, response time of 4 s and scanning speed of 100 nm/min, over the wavelength range from 195 to 260 nm. Each data point was generated by averaging ten accumulations.
Differential scanning fluorimetry (DSF)
DSF analysis was performed to investigate the changes in thermal stability due to disease-related mutation, by monitoring the fluorescence intensity of the environmentally sensitive dye SyproOrange (Thermo Fisher Scientific). Protein samples at 5 μM in SEC buffer were mixed with 20X SyproOrange and heated from 25 to 95°C using an Applied Biosystems 7500 Cycler RT PCR. Curves were analyzed using the Protein Thermal Shift Software v1.1 (Applied Biosystems).
DDX3X ATPase assay
The ATPase activity of DDX3X was determined using the EnzChek Phosphate Assay Kit (Thermo Life Sciences), according to the manufacturer's instructions. Reactions were performed at 25°C using 2 μM purified DDX3X and 800 nM dsRNA, in a final volume of 200 μl. dsRNA with a 3' overhang was purchase (Integrated DNA Technology) containing the following sequences: 5'- ACGUCGAUCCGAAACUAUACUUAAUUUUAA-3′ and 5'-GUUUCGGAUCGACGU-3′(Celik et al., 2015). The reactions were started by the addition of 0.5 mM ATP, and the absorbance values were measured at 360 nm using an EnSpire® Multimode Plate Reader (PerkinElmer). Standard curve reactions were also prepared following the manufacturer's instructions.
Thioflavin T and 1,8-ANS measurements
Protein samples at 15 μM in SEC buffer were incubated at 37°C in the presence of 20 μM ThT or ANS (freshly prepared and quantified by absorbance). Fluorescence measurements were recorded on a plate reader spectrofluorometer (Enspire, Perkin Elmer) using a 96-well plate (Greiner) every 60 s for 4 hrs. Excitation wavelength: 435 nm for ThT and 370 nm for ANS; emission wavelength: 455-600 nm for ThT and 400- 600 nm for ANS.
Fibril formation experiment
An aliquot of DDX3X (D1D2 – WT, L556S and R376C) with a starting concentration of 60 μM was used to prepare a set of four two-fold serial dilutions. For each sample, ThT was added to a final concentration of 20 μM. Fibril formation was followed by fluorescence measurements on a plate reader spectrofluorometer (EnSpire Multimode Plate Reade, PerkinElmer) using a 96-well plate (Greiner). Excitation and emission wavelengths were 435 nm and 490 nm, respectively. Plate holder was pre-heated and kept at 37 °C. Measurements were performed every 60 s for 24hr. The dependence of half-time (t1/2, time at which half the soluble protein has been converted into aggregates) on the initial protein concentration (C0) follows a scaling law in which t1/2 is proportional to [C0] γ. We plotted the half-time against the initial monomer concentration in a double logarithmic plot. The slope of this plot corresponds to γ, as log(t1/2) = γ log(C0)+ constant (Pagano et al., 2014; Serio et al., 2000).
X-ray fiber diffraction
The fibrils solutions (L556S monomer, L556S dimer and R376C) were dialyzed overnight against 1L of water to remove salts and buffer and then lyophilized for 48 h. For each sample, a certain amount of lyophilized material (enough to cover the tip of the loop) was mounted on a MiTeGen MicroLoop previous wetted in ethylene glycol. The diffraction images were collected at 100 K using a Rigaku MicroMax-007HR generator (copper rotating anode, wavelength 1.5418 Å) equipped with VariMax-HR optics and R- AXIS IV imaging plate detector. Each sample was rotated 1° during 20 min of exposure, while keeping the sample-to-detector distance of 250mm.
Dynamic light scattering (DLS)
Pure DDX3X at 15 μM in SEC buffer or SEC buffer supplemented with 40% glycerol were centrifuged (13000 rpm, 10 minutes at 4°C) and submitted to Dynamic Light Scattering (DLS) measurements. Data were recorded at 25°C on a Malvern Zetasizer Nano ZS90 (Malvern Instruments, Worcestershire, UK), equipped with 632,8 nm He-Ne laser, in a quartz cell with a scattering angle of 90°. For each sample, one measurement corresponds to 15-100 acquisitions (according to sample concentration) of 10 seconds, with an automatic attenuator (Attenuation 11). The intensity size distribution, the Z-average radius (Z-ave) and the polydispersity index (PdI) were obtained from the autocorrelation function using the “Protein Analysis mode” for the protein sample. The software used to collect and analyze the data was the Malvern ZetaSizer Software version 7.11.
Rayleigh light scattering (RLS)
Rayleigh Scattering measurements were performed on K2 Multifrequency Phase Fluorometer (ISS), at 350 nm, using a cell with a 5 mm light path. Protein samples were measured at 15 μM in SEC buffer, at 37°C for 24 hr.
Transmission electron microscopy
For TEM analysis, 3 μL of samples at 15 μM were applied to glow-discharged (15 mA, negative charge for 25 s), 400-mesh copper grids covered by a thin layer of continuous carbon film (Ted Pella). After 1 min, the excess sample was removed using a filter paper. Uranyl acetate solution (2%) was applied twice to the grids for 30 sec each and the excess solution was removed. Specimens were analyzed with a defocus range of −1 to −2 μm, using a Jeol JEM-1400Plus instrument operating at 120 kV. Images were acquired using an OneView 4K camera (Gatan). Sample preparation and data acquisition were performed at the Electron Microscopy Laboratory (LME) at Brazilian Nanotechnology National Laboratory (LNNano).
NMR spectroscopy
15N-labeled D2 domain of DDX3X WT and L556S was produced in M9 minimal medium supplemented with 1g/L 15NH4Cl (Cambridge Isotope Laboratories). The expression and purification protocols were performed as described for the unlabeled protein. The gel-filtration final buffer contained 10 mM Hepes, 500 mM ammonium sulfate and 1 mM DTT. Labeled protein was concentrated and 10% D2O was added. NMR spectra were recorded at 12°C on an Agilent Inova 500-MHz spectrometer. The spectra were processed with NMRPipe/NMRDraw and analyzed with NMRView (Johnson, 2004).
Molecular dynamics simulations and in silico structural analysis
Molecular dynamics simulations were performed using Amber 18 suit of programs with the ff03ws force field. The crystallographic DDX3X structure (PDB code 5e7j) was used for wild-type and mutant initial coordinates. Each protein was neutralized and solvated in a truncated octahedral box (spacing distance of up to 15 A from the solute) filled with TIP4P/2005 water. The systems were minimized by 2500 steps of steepest descent minimization followed by 2500 steps of conjugate gradient. Next, we equilibrated the systems using the NVT ensemble (200 ps) followed by NPT ensemble for 200 ps, both with Cα restraints, and an additional NPT equilibration step (500 ps) without any restraints. The production runs were performed using NPT ensemble for 200 ns and 0.002 fs time step in each triplicate, totaling 600 ns for each protein. Hydrogen-containing bonds were constrained using SHAKE. Long-range electrostatic interactions were calculated using particle Mesh Ewald and short-range nonbonded interactions were calculated with a 9 A cutoff. In the simulation analysis step, Cα RMSF calculation was performed using the R package Bio3D whereas the hydrogen bond formation was evaluated in VMD software using the distance and angle cutoff of 3.5 A and 30°, respectively.
For structural analysis of L556S mutation, we searched in the Protein Data Bank for atomic structures that contain LLDLL (DDX3X WT) or SLDLL (L556S mutation) sequences using the motif sequence advanced search. We filtered results by X-ray crystallography or Cryo-EM methods and resolution better than 4A. A total of 261 and 148 structures containing the sequences LLDLL or SLDLL, respectively, was found. The structures were processed using in-house made R scripts and the Bio3D package (Grant et al., 2006) to determine dihedral rotational angles Psi and Phi. For general case Ramachandran plot, the Phi/Psi density profile was based on the more recent dataset of 8000 top structures and the favored Ramachandran boundary was set at 0.02 (Laskowski et al., 1996).
Cell transfection
SH-SY5Y cell line was transfected with full-length human mCherry/GFP-tagged DDX3X WT or mutant variants using FuGene HD (Promega) according to manufacturer's instructions. Cells were used 48 hr after transfection.
Immunofluorescence
SH-SY5Y and iPSCs cultures grown on coverslips were fixed with 4% PFA followed by permeabilization with 0.1% Triton X-100 and for 15 mins at RT and then, blocked with 5% donkey serum for 1 hr at RT. Primary antibodies (TIA-1: SC-1751; SSEA4 - Ab16287 – 1:250; LIN28 - 8641S – 1:250) diluted in antibody diluent (1% Donkey serum + 0.1% Triton X-100) were incubated overnight at 4°C. Secondary antibodies (Goat-anti-mouse Alexa 488 - A11001 - 1:500; Goat-anti-rabbit Alexa 594 - A11005 - 1:500) were incubated for 1h at RT. Cells were washed three times with PBS + 0.1% Tween-20. Coverslips were mounted on slides using VectaShield with DAPI mounting medium (Vector laboratories).
Western blotting
Cell proteins were extracted with RIPA buffer supplemented with inhibitors of proteases and phosphatases followed by centrifugation at 10000 rpm for 10 min at 4⁰ C. For the western blotting, proteins were separated by SDS-PAGE in 12% Bis-Tris gels and transferred onto 0.45 μm nitrocellulose membranes (BioRad). The blots were incubated overnight at 4°C with anti-GFP (Santa Cruz sc-9996, 1:1000), and β-actin (Santa Cruz) antibodies followed by incubation with horseradish peroxidase (HRP)-conjugated secondary IgGs (GE Healthcare). Chemiluminescence signals were quantified using Image J software. β -actin was used as a loading control (Guerra et al., 2019).
Stressing cells
For heat stress, cells were incubated at 43°C/5% CO2 for 20 min. For NaAsO2-induced stress, cells were treated with 1 mM NaAsO2 at 37°C/5% CO2 (unless otherwise stated).
FRAP analysis of SH-SY5Y transfected cells
SH-SY5Y cells were cultured on glass-bottom 35 mm dishes. FRAP experiments were performed at indicated times, as previously described. At least 20 granules from multiple cells per experiment from 3 individual experiments were used.
1,6-hexanediol treatment
For treatment with 1,6-Hexanediol, complete media (DMEM/F12 supplemented with 10% FBS, 1% penicillin/streptomycin) was prepared containing 3.5% 1,6- Hexanediol. Media was exchanged and replaced with 3.5% 1,6-Hexanediol and cells were continuously imaged for at least 60 min.
Cell viability
Cellular viability was measured using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) (Promega) according to the manufacturer's protocol. SH- SY5Y cells were transfected and cultivated for 48 hr. Following this period, media was exchanged, and cells were incubated in the presence of MTS Tetrazolium Compound for 2 hr at 37°C. Absorbance measurements (490nm) were performed using a plate reader (PerkinElmer; Waltham, MA).
TUNEL staining
We conducted TUNEL by using an In Situ Cell Death Detection Kit, POD (Roche, Germany), according to the manufacturer’s instructions. After 48hs of transfection, the cells were submitted to TUNEL staining. The fluorescent nuclear signal was converted to DAB-staining using the Converter POD also provided by the kit. At least 50 GFP-positive cells were analyzed for each one of the DDX3X constructs.
Protein stability measurement
Assessment of DDX3X protein stability was assessed as described previously After 48 hr of transfection with DDX3X-WT-GFP or mutant variants, cells were stimulated with 50 μg/ml of cycloheximide (CHX) (Sigma-Aldrich) and harvested at the indicated time points. Densitometric analysis of DDX3X-GFP levels over β-actin protein levels was performed with ImageJ software.
Generation of iPSCs
Erythroblasts from three normal control (Wild Type - WT) and one patient (DDX3X-L556S) were infected with Sendai virus (obtained from life technologies- A16517), accordingly to manufacturer's instructions. Four days post-infection erythroblasts were plated on the iMEF feeders and cultured using hESC medium. After 3–4 weeks, iPSC clones were manually picked and were further propagated clonally on feeders. WT iPSC-1 (F10) and 2 (F3) derived from a normal, healthy male donor. The WT iPSC-3 (8799) was derived from a normal, healthy female donor. For the DDX3X- L556S iPSC (M1005T - C5, C6, and C7) three clones were derived from a female patient.
Array CGH
Array CGH (Pinkel et al., 1998; Solinas-Toldo et al., 1997) was performed as described previously. In brief, DNA from healthy or diseased patient derived-IPSC clones were sonicated and labeled by random priming (Bioprime Array CGH, Invitrogen, Carlsbad, CA) with Cy3 and Cy5 (Amersham Biosciences, Piscataway, NJ), respectively, and hybridized onto a tiling path BAC array (Fiegler et al., 2003). Analysis was performed according to the protocol provided by the manufacturer and analyzed using the company’s software CGH Analytics (Agilent, Santa Clara, CA).
Real-time quantitative PCR (RT-qPCR)
Total RNA was isolated from iPSCs culture using the RNeasy Mini Kit (Qiagen), following the manufacturer's recommendations. Briefly, 1μg of total RNA was converted into cDNA using Superscript II (Life Technologies) and oligo-dT primers according to the manufacturer's specifications. RT-qPCR reactions were performed with Power SYBR Green Master Mix (Life Technologies). Fluorescence was detected using the Applied Biosystem 7500 Real-Time PCR System, under standard temperature protocol. Primer pairs were designed with Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). Quantitative analyses were performed using the relative quantification curve with positive control and GAPDH as an endogenous control. Primer pairs used were as follows: NANOG forward: 5'-CCTGAAGAAAACTATCCATCC-3’, reverse: 5'- CCTTGTCTTCCTTTT TTGCGA-3'; OCT3/4 forward: 5'- CCTGAAGCAGAAGAGGATCACC-3′, reverse: 5'- AAAGCGGCAGATGGTCGTTTGG-3′; DDX3X forward: 5'- ACGAGAGAGTTGGCAGTACAG-3′ , reverse: 5'-ATAAACCACGCAAGGACGAAC-3′; GAPDH forward: 5'- GAGCCAAAAGGGTCATCATCTC -3', reverse: 5'- AGGAGGCATTGCTGATGATCT-3'.
Live cell imaging
Live cell imaging was performed using a Leica TCS SP8 Confocal microscope outfitted with an environmental chamber with O2, CO2, temperature, and humidity control. All images were acquired using a 63X objective with a CCD camera by exciting for 400 ms using a 488 nm laser at a gain of 100 (unless otherwise stated). Cells were imaged for 50s (FRAP experiments) or at least 60 min (live imaging of granule assembly with sodium arsenite or disassembly with hexanediol). All images were analyzed using ImageJ software (Schneider et al., 2012).
Quantification and statistical analysis
All statistical analyses were performed in Prism 8 (Graphpad). All experiments were performed in triplicate and repeated as indicated in the figure legends. Data was analyzed statistically using unpaired Student’s t-test when two groups were being compared or by one-way ANOVA for multiple comparisons. The error bars in the figures refer to mean ± SEM. The specific statistical tests used are listed in the figure legends. Experiments were evaluated by statistical significance when p < 0.05.
Acknowledgments
We thank LNLS, LNNano, and LNBio/CNPEM for access to core facilities. This work was supported by the Coordination of Superior Level Staff Improvement (CAPESP), National Program to Support Health Care for Persons with Disabilities (PRONAS/PCD, K.G.F. 25000.016966/2018-27), and the Brazilian National Council for Scientific and Technological Development (CNPq, A.K., 305806/2019; K.G.F, 307498/2019-0) and grants from São Paulo Research Foundation (FAPESP, C.R., A.K., 2013/08028-1; M.C.F, 2018/20014-0, 2019/24511-0). Summary Figure was created with BioRender.com.
Author contributions
M.C.F., J.F.O., C.R, A.K., and K.G.F. conceived, initiated, and coordinated the project. M.C.F., B.H.S.A., C.C., P.F.V.P., D.P.A.N., B.P.B., P.V.R., J.V.P.G., K.T, H.V.R.F., A.F.Z.N., A.S., C.C.C.T., and F.A.H.B. designed and performed the experimental work. M.C.F and K.G.F. supplied reagents. P.S.L.O., A.C.F., S.S.C., C.R., A.K., H.W. Jr., and A.J.R.S. provided essential discussion and advice. M.C.F. and K.G.F. performed the main fundraising. M.C.F., J.F.O., P.F.V.P., and and K.G.F. wrote the manuscript. All authors discussed the experiments and results, read, and approved the manuscript.
Declaration of interests
Nothing to disclose.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Published: August 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102841.
Contributor Information
Matheus de Castro Fonseca, Email: matheus.fonseca@lnbio.cnpem.br.
Kleber Gomes Franchini, Email: kleber.franchini@lnbio.cnpem.br.
Supplemental information
References
- Aditi, Mason A.C., Sharma M., Dawson T.R., Wente S.R. MAPK- and glycogen synthase kinase 3-mediated phosphorylation regulates the DEAD-box protein modulator Gle1 for control of stress granule dynamics. J. Biol. Chem. 2019;294:559–575. doi: 10.1074/jbc.RA118.005749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida Z.L., Brito R.M.M. Structure and aggregation mechanisms in amyloids. Molecules. 2020;25:1195. doi: 10.3390/molecules25051195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambadipudi S., Biernat J., Riedel D., Mandelkow E., Zweckstetter M. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017;8:275. doi: 10.1038/s41467-017-00480-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P., Kedersha N. RNA granules. J. Cell Biol. 2006;172:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo B.H.S., Kaid C., De Souza J.S., Gomes da Silva S., Goulart E., Caires L.C.J., Musso C.M., Torres L.B., Ferrasa A., Herai R. Down syndrome iPSC-derived astrocytes impair neuronal synaptogenesis and the mTOR pathway in vitro. Mol. Neurobiol. 2018;55:5962–5975. doi: 10.1007/s12035-017-0818-6. [DOI] [PubMed] [Google Scholar]
- Baguet A., Degot S., Cougot N., Bertrand E., Chenard M.P., Wendling C., Kessler P., Le Hir H., Rio M.C., Tomasetto C. The exon-junction-complex-component metastatic lymph node 51 functions in stress-granule assembly. J. Cell Sci. 2007;120:2774–2784. doi: 10.1242/jcs.009225. [DOI] [PubMed] [Google Scholar]
- Boeynaems S., Alberti S., Fawzi N.L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L. Protein phase separation: a new phase in cell biology. Trends Cell Biol. 2018;28:420–435. doi: 10.1016/j.tcb.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bol G.M., Xie M., Raman V. DDX3, a potential target for cancer treatment. Mol. Cancer. 2015;14:188. doi: 10.1186/s12943-015-0461-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi B., Kumita J.R., Barros T.P., Esbjorner E.K., Luheshi L.M., Crowther D.C., Wilson M.R., Dobson C.M., Favrin G., Yerbury J.J. ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 2010;5:735–740. doi: 10.1021/cb1001203. [DOI] [PubMed] [Google Scholar]
- Campioni S., Mannini B., Zampagni M., Pensalfini A., Parrini C., Evangelisti E., Relini A., Stefani M., Dobson C.M., Cecchi C. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010;6:140–147. doi: 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- Cantone I., Fisher A.G. Human X chromosome inactivation and reactivation: implications for cell reprogramming and disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017;372:20160358. doi: 10.1098/rstb.2016.0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X., Jin X., Liu B. The involvement of stress granules in aging and aging-associated diseases. Aging cell. 2020;19:e13136. doi: 10.1111/acel.13136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro T.N., Krepischi A.C., Costa S.S., Tojal da Silva I., Vianna-Morgante A.M., Valieris R., Ezquina S.A., Bertola D.R., Otto P.A., Rosenberg C. Utility of trio-based exome sequencing in the elucidation of the genetic basis of isolated syndromic intellectual disability: illustrative cases. Appl. Clin. Genet. 2018;11:93–98. doi: 10.2147/TACG.S165799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik H., Sajwan K.P., Selvanathan S.P., Marsh B.J., Pai A.V., Kont Y.S., Han J., Minas T.Z., Rahim S., Erkizan H.V. Ezrin Binds to DEAD-Box RNA Helicase DDX3 and Regulates Its Function and Protein Level. Molecular and cellular biology. 2015;35:3145–3162. doi: 10.1128/MCB.00332-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Cohen S.I., Linse S., Luheshi L.M., Hellstrand E., White D.A., Rajah L., Otzen D.E., Vendruscolo M., Dobson C.M., Knowles T.P. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U S A. 2013;110:9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton A.M., Price E.M., Jones M.J., Balaton B.P., Kobor M.S., Brown C.J. Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum. Mol. Genet. 2015;24:1528–1539. doi: 10.1093/hmg/ddu564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Cruz S., Cleveland D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011;21:904–919. doi: 10.1016/j.conb.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison J.W., Rosenfeld J.A., Shaffer L.G. Genetic basis of intellectual disability. Annu. Rev. Med. 2013;64:441–450. doi: 10.1146/annurev-med-042711-140053. [DOI] [PubMed] [Google Scholar]
- Fiegler H., Carr P., Douglas E.J., Burford D.C., Hunt S., Scott C.E., Smith J., Vetrie D., Gorman P., Tomlinson I.P. DNA microarrays for comparative genomic hybridization based on DOP-PCR amplification of BAC and PAC clones. Genes, chromosomes & cancer. 2003;36:361–374. doi: 10.1002/gcc.10155. [DOI] [PubMed] [Google Scholar]
- Floor S.N., Condon K.J., Sharma D., Jankowsky E., Doudna J.A. Autoinhibitory interdomain interactions and subfamily-specific extensions redefine the catalytic core of the human DEAD-box protein DDX3. J. Biol. Chem. 2016;291:2412–2421. doi: 10.1074/jbc.M115.700625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkmann A.W., Collier S.E., Zhan X., Aditi, Ohi M.D., Wente S.R. Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell. 2013;155:582–593. doi: 10.1016/j.cell.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkmann A.W., Dawson T.R., Wente S.R. Insights into mRNA export-linked molecular mechanisms of human disease through a Gle1 structure-function analysis. Adv. Biol. Regul. 2014;54:74–91. doi: 10.1016/j.jbior.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gecz J., Shoubridge C., Corbett M. The genetic landscape of intellectual disability arising from chromosome X. Trends Genet. 2009;25:308–316. doi: 10.1016/j.tig.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Goldenberg A., Saugier-Veber P. [Genetics of mental retardation] Pathol. Biol. 2010;58:331–342. doi: 10.1016/j.patbio.2009.09.013. [DOI] [PubMed] [Google Scholar]
- Goncalves A.C., Towers E.R., Haq N., Porco J.A., Jr., Pelletier J., Dawson S.J., Gale J.E. Drug-induced stress granule formation protects sensory hair cells in mouse cochlear explants during ototoxicity. Sci. Rep. 2019;9:12501. doi: 10.1038/s41598-019-48393-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra M.T., Florentino R.M., Franca A., Lima Filho A.C., Dos Santos M.L., Fonseca R.C., Lemos F.O., Fonseca M.C., Kruglov E., Mennone A. Expression of the type 3 InsP3 receptor is a final common event in the development of hepatocellular carcinoma. Gut. 2019;68:1676–1687. doi: 10.1136/gutjnl-2018-317811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B.J., Rodrigues A.P., ElSawy K.M., McCammon J.A., Caves L.S. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- Hartl F.U. Protein misfolding diseases. Annu. Rev. Biochem. 2017;86:21–26. doi: 10.1146/annurev-biochem-061516-044518. [DOI] [PubMed] [Google Scholar]
- Hipp M.S., Park S.H., Hartl F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014;24:506–514. doi: 10.1016/j.tcb.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Hofweber M., Hutten S., Bourgeois B., Spreitzer E., Niedner-Boblenz A., Schifferer M., Ruepp M.D., Simons M., Niessing D., Madl T. Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell. 2018;173:706–719 e713. doi: 10.1016/j.cell.2018.03.004. [DOI] [PubMed] [Google Scholar]
- Johnson B.A. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- Kato M., Han T.W., Xie S., Shi K., Du X., Wu L.C., Mirzaei H., Goldsmith E.J., Longgood J., Pei J. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.J., Kim N.C., Wang Y.D., Scarborough E.A., Moore J., Diaz Z., MacLea K.S., Freibaum B., Li S., Molliex A. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Myong S. RNA remodeling activity of DEAD box proteins tuned by protein concentration, RNA length, and ATP. Mol. Cell. 2016;63:865–876. doi: 10.1016/j.molcel.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaips C.L., Jayaraj G.G., Hartl F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018;217:51–63. doi: 10.1083/jcb.201709072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar J., Sobol M., Melberg A., Mabert K., Ameur A., Johansson A.C., Feuk L., Entesarian M., Orlen H., Casar-Borota O. Welander distal myopathy caused by an ancient founder mutation in TIA1 associated with perturbed splicing. Hum. Mutat. 2013;34:572–577. doi: 10.1002/humu.22282. [DOI] [PubMed] [Google Scholar]
- Kroschwald S., Maharana S., Mateju D., Malinovska L., Nuske E., Poser I., Richter D., Alberti S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. eLife. 2015;4:e06807. doi: 10.7554/eLife.06807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukhanova M.K., Karpenko I.L., Ivanov A.V. DEAD-box RNA helicase DDX3: functional properties and development of DDX3 inhibitors as antiviral and anticancer drugs. Molecules. 2020;25:1015. doi: 10.3390/molecules25041015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski T.J., Jr., Bosco D.A., Leclerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Kwon I., Kato M., Xiang S., Wu L., Theodoropoulos P., Mirzaei H., Han T., Xie S., Corden J.L., McKnight S.L. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell. 2013;155:1049–1060. doi: 10.1016/j.cell.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahn B.T., Page D.C. Functional coherence of the human Y chromosome. Science. 1997;278:675–680. doi: 10.1126/science.278.5338.675. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Rullmannn J.A., MacArthur M.W., Kaptein R., Thornton J.M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. Journal of biomolecular NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Lennox A.L., Hoye M.L., Jiang R., Johnson-Kerner B.L., Suit L.A., Venkataramanan S., Sheehan C.J., Alsina F.C., Fregeau B., Aldinger K.A. Pathogenic DDX3X mutations impair RNA metabolism and neurogenesis during fetal cortical development. Neuron. 2020;106:404–420 e408. doi: 10.1016/j.neuron.2020.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librizzi F., Rischel C. The kinetic behavior of insulin fibrillation is determined by heterogeneous nucleation pathways. Protein Sci. 2005;14:3129–3134. doi: 10.1110/ps.051692305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Protter D.S., Rosen M.K., Parker R. formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell. 2015;60:208–219. doi: 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren M., Sorgjerd K., Hammarstrom P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys. J. 2005;88:4200–4212. doi: 10.1529/biophysj.104.049700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linse S. Monomer-dependent secondary nucleation in amyloid formation. Biophys. Rev. 2017;9:329–338. doi: 10.1007/s12551-017-0289-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon M.F. X-chromosome inactivation and human genetic disease. Acta Paediatr. 2002;91:107–112. doi: 10.1111/j.1651-2227.2002.tb03120.x. [DOI] [PubMed] [Google Scholar]
- Mateju D., Franzmann T.M., Patel A., Kopach A., Boczek E.E., Maharana S., Lee H.O., Carra S., Hyman A.A., Alberti S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017;36:1669–1687. doi: 10.15252/embj.201695957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisl G., Yang X., Dobson C.M., Linse S., Knowles T.P.J. Modulation of electrostatic interactions to reveal a reaction network unifying the aggregation behaviour of the Abeta42 peptide and its variants. Chem. Sci. 2017;8:4352–4362. doi: 10.1039/c7sc00215g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migeon B.R. X-linked diseases: susceptible females. Genet. Med. 2020;22:1156–1174. doi: 10.1038/s41436-020-0779-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A., Temirov J., Lee J., Coughlin M., Kanagaraj A.P., Kim H.J., Mittag T., Taylor J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163:123–133. doi: 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan Z., Ryan V.H., Janke A.M., Burke K.A., Rhoads S.N., Zerze G.H., O'Meally R., Dignon G.L., Conicella A.E., Zheng W. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017;36:2951–2967. doi: 10.15252/embj.201696394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morleo M., Franco B. Dosage compensation of the mammalian X chromosome influences the phenotypic variability of X-linked dominant male-lethal disorders. J. Med. Genet. 2008;45:401–408. doi: 10.1136/jmg.2008.058305. [DOI] [PubMed] [Google Scholar]
- Murakami T., Qamar S., Lin J.Q., Schierle G.S., Rees E., Miyashita A., Costa A.R., Dodd R.B., Chan F.T., Michel C.H. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron. 2015;88:678–690. doi: 10.1016/j.neuron.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray A.N., Palhano F.L., Bieschke J., Kelly J.W. Surface adsorption considerations when working with amyloid fibrils in multiwell plates and Eppendorf tubes. Protein Sci. 2013;22:1531–1541. doi: 10.1002/pro.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray D.T., Kato M., Lin Y., Thurber K.R., Hung I., McKnight S.L., Tycko R. Structure of FUS protein fibrils and its Relevance to self-assembly and phase separation of low-complexity domains. Cell. 2017;171:615–627 e616. doi: 10.1016/j.cell.2017.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola P., Blackburn P.R., Rasmussen K.J., Bertsch N.L., Klee E.W., Hasadsri L., Pichurin P.N., Rankin J., Raymond F.L., Study D.D.D. De novo DDX3X missense variants in males appear viable and contribute to syndromic intellectual disability. Am. J. Med. Genet. A. 2019;179:570–578. doi: 10.1002/ajmg.a.61061. [DOI] [PubMed] [Google Scholar]
- Nott T.J., Petsalaki E., Farber P., Jervis D., Fussner E., Plochowietz A., Craggs T.D., Bazett-Jones D.P., Pawson T., Forman-Kay J.D. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell. 2015;57:936–947. doi: 10.1016/j.molcel.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzscha H., Schermann S.M., Woerner A.C., Pinkert S., Hecht M.H., Tartaglia G.G., Vendruscolo M., Hayer-Hartl M., Hartl F.U., Vabulas R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144:67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- Pagano R.S., Lopez Medus M., Gomez G.E., Couto P.M., Labanda M.S., Landolfo L., D'Alessio C., Caramelo J.J. Protein fibrillation lag times during kinetic inhibition. Biophys. J. 2014;107:711–720. doi: 10.1016/j.bpj.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A., Lee H.O., Jawerth L., Maharana S., Jahnel M., Hein M.Y., Stoynov S., Mahamid J., Saha S., Franzmann T.M. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Patel S.S., Belmont B.J., Sante J.M., Rexach M.F. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell. 2007;129:83–96. doi: 10.1016/j.cell.2007.01.044. [DOI] [PubMed] [Google Scholar]
- Pene V., Li Q., Sodroski C., Hsu C.S., Liang T.J. Dynamic interaction of stress granules, DDX3X, and IKK-alpha Mediates multiple functions in Hepatitis C virus Infection. J. Virol. 2015;89:5462–5477. doi: 10.1128/JVI.03197-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piton A., Redin C., Mandel J.L. XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am. J. Hum. Genet. 2013;93:368–383. doi: 10.1016/j.ajhg.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkel D., Segraves R., Sudar D., Clark S., Poole I., Kowbel D., Collins C., Kuo W.L., Chen C., Zhai Y. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nature genetics. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- Posynick B.J., Brown C.J. Escape from X-chromosome inactivation: an evolutionary perspective. Front. Cell Dev. Biol. 2019;7:241. doi: 10.3389/fcell.2019.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam A., Cassani M., Smith J., Seydoux G. A gel phase promotes condensation of liquid P granules in Caenorhabditis elegans embryos. Nat. Struct. Mol. Biol. 2019;26:220–226. doi: 10.1038/s41594-019-0193-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribbeck K., Gorlich D. The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. EMBO J. 2002;21:2664–2671. doi: 10.1093/emboj/21.11.2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross C.A., Poirier M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004;10:S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Ross M.T., Grafham D.V., Coffey A.J., Scherer S., McLay K., Muzny D., Platzer M., Howell G.R., Burrows C., Bird C.P. The DNA sequence of the human X chromosome. Nature. 2005;434:325–337. doi: 10.1038/nature03440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M., Hess D., Eglinger J., Fritsch A.W., Kreysing M., Weinert B.T., Choudhary C., Matthias P. Acetylation of intrinsically disordered regions regulates phase separation. Nat. Chem. Biol. 2019;15:51–61. doi: 10.1038/s41589-018-0180-7. [DOI] [PubMed] [Google Scholar]
- Scala M., Torella A., Severino M., Morana G., Castello R., Accogli A., Verrico A., Vari M.S., Cappuccio G., Pinelli M. Three de novo DDX3X variants associated with distinctive brain developmental abnormalities and brain tumor in intellectually disabled females. Eur. J. Hum. Genet. 2019;27:1254–1259. doi: 10.1038/s41431-019-0392-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H.B., Barreau A., Rohatgi R. Phase separation-deficient TDP43 remains functional in splicing. Nat. Commun. 2019;10:4890. doi: 10.1038/s41467-019-12740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serio T.R., Cashikar A.G., Kowal A.S., Sawicki G.J., Moslehi J.J., Serpell L., Arnsdorf M.F., Lindquist S.L. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- Shih J.W., Wang W.T., Tsai T.Y., Kuo C.Y., Li H.K., Wu Lee Y.H. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem. J. 2012;441:119–129. doi: 10.1042/BJ20110739. [DOI] [PubMed] [Google Scholar]
- Shvetsova E., Sofronova A., Monajemi R., Gagalova K., Draisma H.H.M., White S.J., Santen G.W.E., Chuva de Sousa Lopes S.M., Heijmans B.T., van Meurs J. Skewed X-inactivation is common in the general female population. Eur. J. Hum. Genet. 2019;27:455–465. doi: 10.1038/s41431-018-0291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijders Blok L., Madsen E., Juusola J., Gilissen C., Baralle D., Reijnders M.R., Venselaar H., Helsmoortel C., Cho M.T., Hoischen A. Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on Wnt signaling. Am. J. Hum. Genet. 2015;97:343–352. doi: 10.1016/j.ajhg.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas-Toldo S., Lampel S., Stilgenbauer S., Nickolenko J., Benner A., Dohner H., Cremer T., Lichter P. Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes, chromosomes & cancer. 1997;20:399–407. [PubMed] [Google Scholar]
- Song H., Ji X. The mechanism of RNA duplex recognition and unwinding by DEAD-box helicase DDX3X. Nat. Commun. 2019;10:3085. doi: 10.1038/s41467-019-11083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C., Pritzkow S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018;21:1332–1340. doi: 10.1038/s41593-018-0235-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J., Blair I.P., Tripathi V.B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J.C., Williams K.L., Buratti E. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani M., Dobson C.M. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- Sweeney P., Park H., Baumann M., Dunlop J., Frydman J., Kopito R., McCampbell A., Leblanc G., Venkateswaran A., Nurmi A. Protein misfolding in neurodegenerative diseases: implications and strategies. Transl. Neurodegen. 2017;6:6. doi: 10.1186/s40035-017-0077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Vega Y.A., Wang Y.D., Parker M., Patmore D.M., Kanagaraj A., Moore J., Rusch M., Finkelstein D., Ellison D.W., Gilbertson R.J. Cancer-associated DDX3X mutations drive stress granule assembly and impair global translation. Sci. Rep. 2016;6:25996. doi: 10.1038/srep25996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdile V., De Paola E., Paronetto M.P. Aberrant phase transitions: side effects and Novel Therapeutic strategies in human disease. Front. Genet. 2019;10:173. doi: 10.3389/fgene.2019.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira N.M., Naslavsky M.S., Licinio L., Kok F., Schlesinger D., Vainzof M., Sanchez N., Kitajima J.P., Gal L., Cavacana N. A defect in the RNA-processing protein HNRPDL causes limb-girdle muscular dystrophy 1G (LGMD1G) Hum. Mol. Genet. 2014;23:4103–4110. doi: 10.1093/hmg/ddu127. [DOI] [PubMed] [Google Scholar]
- Vissers L.E., Gilissen C., Veltman J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016;17:9–18. doi: 10.1038/nrg3999. [DOI] [PubMed] [Google Scholar]
- Wainer Katsir K., Linial M. Human genes escaping X-inactivation revealed by single cell expression data. BMC Genomics. 2019;20:201. doi: 10.1186/s12864-019-5507-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Posey J.E., Rosenfeld J.A., Bacino C.A., Scaglia F., Immken L., Harris J.M., Hickey S.E., Mosher T.M., Slavotinek A. Phenotypic expansion in DDX3X - a common cause of intellectual disability in females. Ann. Clin. Transl. Neurol. 2018;5:1277–1285. doi: 10.1002/acn3.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler J.R., Matheny T., Jain S., Abrisch R., Parker R. Distinct stages in stress granule assembly and disassembly. eLife. 2016:e18413. doi: 10.7554/eLife.18413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklhofer K.F., Tatzelt J., Haass C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008;27:336–349. doi: 10.1038/sj.emboj.7601930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B. Regulated protein aggregation: stress granules and neurodegeneration. Mol. Neurodegen. 2012;7:56. doi: 10.1186/1750-1326-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B., Ivanov P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019;20:649–666. doi: 10.1038/s41583-019-0222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziats C.A., Schwartz C.E., Gecz J., Shaw M., Field M.J., Stevenson R.E., Neri G. X-linked intellectual disability: phenotypic expression in carrier females. Clin. Genet. 2020;97:418–425. doi: 10.1111/cge.13667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
. One z-stack of images was performed each 20 sec during 60 min.
One z-stack of images was performed each 20 sec during 60 min.
One z-stack of images was performed each 20 sec during 60 min
Data Availability Statement
All data are available within the article or its supplementary materials. Any additional information is available from the lead contact on request.