

Abstract
Cancer cells require extensive metabolic reprograming in order to provide the bioenergetics and macromolecular precursors needed to sustain a malignant phenotype. Mutant KRAS is a driver oncogene that is well-known for its ability to regulate the ERK and PI3K signaling pathways. However, it is now appreciated that KRAS can promote the tumor growth via upregulation of anabolic metabolism. We recently reported that oncogenic KRAS promotes a gene expression program of de novo lipogenesis in non-small cell lung cancer (NSCLC). To define the mechanism(s) responsible, we focused on the lipogenic transcription factor SREBP1. We observed that KRAS increases SREBP1 expression and genetic knockdown of SREBP1 significantly inhibited the cell proliferation of mutant KRAS-expressing cells. Unexpectedly, lipogenesis was not significantly altered in cells subject to SREBP1 knockdown. Carbon tracing metabolic studies showed a significant decrease in oxidative phosphorylation and RNA-seq data revealed a significant decrease in mitochondrial encoded subunits of the electron transport chain (ETC). Taken together, these data support a novel role, distinct from lipogenesis, of SREBP1 on mitochondrial function in mutant KRAS NSCLC.
Keywords: cancer metabolism, de novo lipogenesis, electron transport chain, oxidative phosphorylation
1 |. INTRODUCTION
KRAS is the most frequently mutated oncogene in lung adenocarcinoma, present in up to 30% of cases.1–3 Lung cancer patients with tumors harboring KRAS mutations are associated with a poor prognosis and resistance to therapy.4 While there are covalent KRASG12C-specific inhibitors and KRAS-SOS interaction directed therapeutics currently in clinical trials, there are currently no successful anti-KRAS therapies.5,6 Accumulating studies have highlighted a potential for mutant KRAS to rewire cellular metabolism to promote the tumor development. Substantial evidence shows that metabolic reprograming is essential for tumor initiation and progression.7,8 The “Warburg effect” describes a propensity for cancer cells to increase glucose uptake and convert the majority of it to lactate even in the presence of oxygen.9 Originally, this increase in aerobic glycolysis displayed by cancer cells was attributed to damaged mitochondria. However, it is now appreciated that mitochondria remain functional in many tumors. In fact, mitochondrial metabolism is essential for providing the energy and precursors of protein, DNA, and lipids needed for the increased growth in cancer cells.10,11 Despite growing evidence for altered metabolism in KRAS mutant NSCLC, how KRAS drives these changes is not clearly understood.
Sterol element binding regulatory proteins (SREBPs) are key transcription factors involved in regulating lipid homeostasis in all vertebrates.12 There are three SREBP1 isoforms in mammals: SREBP1a/SREBF1-1, SREBP1b/SREBF1–2, and SREBP1c/SREBF1–3, along with the closely related SREBP2/SREBF2. SREBP1a and 1c are encoded by a single gene with alternative transcription start sites, whereas a separate gene encodes SREBP2. SREBP1c enhances the expression of genes involved in fatty acid uptake and synthesis while SREBP1a enhances the gene expression of all SREBP-responsive genes.12 SREBP2 preferentially facilitates the expression of genes required for cholesterol synthesis although it can also enhance the expression of genes involved in fatty acid synthesis through upregulation of the other SREBPs.13 Accumulating evidence suggests that SREBP1 is a critical link between oncogene signaling and metabolism in cancer.14–18 SREBP1’s ability to regulate fatty acid and cholesterol metabolism provides tumor cells with the energy, biomass, and reducing equivalents required for tumor growth and survival. Accordingly, several studies have reported on the capacity of SREBP1 to support tumor growth via increased fatty acid synthesis. For example, Guo et al reported that SREBP1 signaling is required for survival of mutant EGFR-expressing glioblastoma.19 Several other studies have highlighted the importance of SREBP1 in cancers such as pancreatic, prostate, and colorectal cancers.17,18,20 However, the role of SREBP1 in NSCLC is not clear.
We recently showed that mutant KRAS promotes a transcriptional program of de novo lipogenesis in NSCLC.21 Furthermore, mutant KRAS-expressing cells and tumors were sensitized to the growth inhibitory effects of FASN inhibitors. This prompted us to determine exactly how KRAS was driving this lipogenic transcription program. Here we show a novel function for SREBP1 in mutant KRAS-expressing NSCLC distinct from lipogenesis. We demonstrate that mutant KRAS promotes SREBP1 expression via MEK1/2 signaling, and loss of SREBP1 decreases growth. Despite this reduction in growth, de novo lipogenesis was not significantly altered. Importantly, reduction of SREBP1 led to decreased expression of the mitochondrial encoded subunits of the ETC. This decrease in mitochondrial gene expression led to impaired mitochondrial metabolism as made evident by decreased oxidative phosphorylation. These results delineate a link between mutant KRAS and SREBP1 as well as highlight a novel role for SREBP1 on mitochondrial function distinct from lipogenesis in NSCLC.
2 |. MATERIALS AND METHODS
2.1 |. [13C] isotopomer analysis
Cells were seeded in 6-cm culture dishes (800 000 cells per dish) overnight. The following day, cells were washed twice with warm 1X PBS, and medium was changed to RPMI with 10 mM [U-13C6] glucose (Cambridge Isotopes) as the only glucose source and 10% dialyzed FBS and 2 mM glutamine for 6–16 hours.
2.1.1 |. Lipid extraction and GCMS analysis
Cells were harvested in 0.9% NaCl and centrifuged at 10 000 RPM at 4°C. The pellet was resuspended in 2:1 chloroform:methanol. Before drying down under nitrogen, 50 nmoles of heptadecanoic acid was added to all samples as an internal control. Fatty acids were then saponified as previously described.21 Following saponification, metabolites were dried down under nitrogen again and methylated with boron trifluoride (Sigma, 15716). Mass spectral data were obtained on an Agilent 7890B Gas Chromatograph coupled with an Agilent 5977A MDS. The settings were as follows: GC inlet 230°C, transfer line 280°C, MS source 230°C MS Quad 150°C. An HP-5MS column (30 M length, 250 μm diameter, 0.25 μm film thickness) was used for fatty acid analysis and palmitate and its isotopomers were monitored at 270–286 m/z.
2.1.2 |. TCA cycle metabolite extraction
Intermediate metabolites were harvested in 80% methanol in water with 10 nmoles adonitol per sample as internal control. Metabolites were dried down under nitrogen and derivatized as previously described.22 In brief, cells were frozen and thawed three times, and centrifuged, and the supernatant was collected. The supernatant was then dried down and methoxi-mated using MOX (Thermo Scientific, TS-45950) and derivatized with BSTFA (TCI, B3402). All metabolite data were analyzed using Mass Hunter and abundance corrected using ISOCOR.
2.2 |. Analysis of SREBP1 expression
Publicly available data from the Cancer Genome Atlas (TCGA) were analyzed for SREBP1 expression in 720 lung tumors. Overall survival and the hazard ratio were graphed and calculated using an online tool called KmPlotter.23 All cases analyzed were adenocarcinomas.
2.3 |. Cell culture and reagents
All cells were purchased from American Type Culture Collection (Manassas, Virginia, United States) and cultured under recommended conditions. Specifically, H23, A549, H1437, and H1703 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Corning) and 293T cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Corning). All media were supplemented with 10% fetal bovine serum (FBS) (Gemini) unless indicated. Delipidated media (DL) was RPMI media supplemented 10% delipidated FBS (Gemini). Cells were transfected using Attractene reagent as per the manufacturer’s suggestions (QIAGEN). The cell lines were routinely tested for mycoplasma contamination.24 All cells were incubated at 37°C with 5% CO2. All antibodies used and corresponding dilutions are listed in Table S1.
2.4 |. Genetic manipulation of KRAS in vitro
H1437 and H1703 cells were transduced with retrovirus expressing KrasG12V (pBabe KRASG12V) or empty vector (pMSCV-Puro) to serve as controls. Retrovirus was generated using CaP transfection into phoenix cells. Cells were transfected with expression vectors for vsv and gag-pol with the retroviral vector of interest. A549 and H23 NSCLC cells were transduced with lentivirus expressing nontarget short hairpin RNA (NTshRNA) pLKO (NTshRNA) or shRNA against Kras (shKRAS;TRCN0000033262, MilliporeSigma, Burlington, MA, USA). Lentivrus was prepared as described above. Following infection, cells were selected in puromycin (1 μg/mL) for 1 week to establish stable pools.
2.5 |. Genetic manipulation of SREBP1 in vitro
For stable SREBP1 knockdown, H23, A549, H1437, and H1703 cells were infected with nontarget shRNA lentivirus (NTshRNA) or lentivirus with one of two different commercially available shRNAs against SREBP1 (shSREBP1 #1:TRCN0000020605, shSREBP1 #2: TRCN0000020607) (MilliporeSigma, Burlington, MA, USA). Following infection with virus, H23, A549, and H1703 cells were selected in 1 μg/mL of puromycin. H1437 were selected in 2 μg/mL of puromycin. All cells were grown in indicated doses of puromycin for 1 week to establish stable pools.
2.6 |. In vitro translation
In vitro translation of full-length SREBP1a and full-length SREBP1c was carried out using the TNT rabbit reticulocyte lysate translation kit (Promega, Wisconsin, USA) as per the manufacturer’s instructions. In brief, plasmids with the open reading frames of SREBP1c and SREBP1a were mixed separately with the components of the TNT rabbit reticulate lysate kit and incubated at 30°C for 90 minutes. Following incubation, the product was diluted 1:5 in water and subjected to SDS-PAGE alongside lysates of H23, A549, H1437, and H1703 cells.
2.7 |. Mitochondrial DNA copy number
Mitochondrial DNA copy number was measured as previously described.25 In brief, H23 cells expressing NTshRNA or shSREBP1 # 1 were seeded in triplicate in a 6-well culture dish (Corning). Genomic DNA was isolated using a commercially available kit (Invitrogen, K182001) as per the manufacturer’s instructions. Isolated DNA was subjected to PCR using primers for nuclear DNA (B2M) or mitochondrial DNA (ND1). The relative mitochondrial DNA content was then determined as follows:
ΔCT = (nuclear DNA CT − mito DNA CT)
Relative mitochondrial DNA content = 2 × 2ΔCT
2.8 |. Mitochondrial oxygen consumption
Oxygen consumption rates were measured using a Seahorse Bioenergetic Flux analyzer (XFe96). Basal respiration and ATP-coupled respiration, represented as OCR, were measured using a Mitochondrial Stress Test assay as per the manufacturer’s instructions (Agilent, 103015-100). 20 000 cells per well were plated in a 96-well plate in RPMI 1640 medium (Corning) and 10% FBS (Gemini). Cells were incubated overnight at 37°C and 5% CO2. The following day, Seahorse XF base medium (Agilent, 103193-100) was prepared by adding 2 mM Glutamine, and 10 mM glucose. No serum was present in XF base medium. The cells were washed twice with base medium and then, incubated at 37°C in a non-CO2 incubator for 1 hour before the start of the assay. The concentrations of oligomycin, FCCP, Rotanone, and Antimycin A used for H23 and H1437 were 1 μM, 0.25 μM, 0.5 μM, and 0.5 μM, respectively. OCR values were normalized to DNA concentration per well as measured by CyQUANT (Thermo Fisher Scientific, C7026).
2.9 |. Proliferation studies
Growth Curves: Cells were seeded into 6-well dishes (Corning) with an initial seeding density of 30 000–50 000 cells per well and counted on days indicated using a Countess Automated Cell Counter (Thermo Fisher Scientific, Waltham, MA, USA) cell automated countess (Invitrogen). For treatment with inhibitors, ROS, and nutrients, cells were treated the morning after plating and final counts were performed 3 days later.
2.10 |. Real-time RT-PCR
Total RNA was extracted from tumors and cells with the RNeasy Kit (Qiagen, Hilden, Germany). The reverse-transcription reaction was performed with a high-capacity cDNA Synthesis Kit (Applied Biosystems). Real-time quantitative PCR analyses of human genes were performed, as previously described.21 All primers used are listed in Table S2. One of three housekeeping genes, 18s, HPRT, or β-ACTIN was used for normalization.
2.11 |. RNAseq analysis
RNAseq was performed by Novagene (Novagene Sacramento, CA). Illumina HiSeq RNA sequencing of triplicate FASTQ file reads passing Illumina purity filter were aligned using TopHat2 and Cufflinks, with statistical analysis performed by CuffDiff, generating files of normalized counts for detected genes and transcripts (UCSC hg38). The Galaxy server at UCLA (galaxy.org) was used for FASTQ alignment and analysis.26 Aligned RNAs passing QC thresholds were used to calculate the transcript abundance ratios followed by log2 linear scaling. Functional association with RNA abundance changes was assessed by gene set enrichment (GSEA). As expected, multiple signatures associated with mitochondrial function were significantly enriched with normalized enrichment score (NES) P value < .05 and the false discovery rate (FDR) q value < .05.
2.12 |. Graphical packages
Analysis was performed in R version 3.6.2.27 Heatmaps were generated using the pheatmap package.28 Genes encoding subunits were retrieved from the KEGG pathway for oxidative phosphorylation (hsa:00190). Log2 ratios between shRNA/control were plotted for indicated genes and separated based on complexes.
3 |. RESULTS
3.1 |. Oncogenic KRAS increases SREBP1 expression
Mutant KRAS promotes a gene expression program which drives de novo lipogenesis in NSCLC.21,29 In order to identify the transcriptional mechanism(s) responsible, we examined cDNA microarray data from studies of lungs of wild-type and KrasLSLG12D mice and observed a significant increase in expression of SREBP1 in lung tumors compared to normal lung.21 To confirm the effect of mutant KRAS on SREBP1, we transfected HEK 293T cells with full-length (FL) SREBP1 and increasing doses of mutant KRASG12V. KRASG12V induced a dose-dependent increase in SREBP1 protein expression (Figure 1A). Classically, SREBP1 is retained in the endoplasmic reticulum. Activation of lipid sensing programs promote translocation to the Golgi and cleavage of SREBP1 (cleaved SREBP1) which can then travel to the nucleus to activate transcription of its lipogenic targets. Interestingly, cleaved SREBP1 protein levels did not further increase relative to full-length SREBP1 levels (Figure 1B) suggesting mutant KRAS is not affecting SREBP1 cleavage. To assess whether this was a mutant KRAS-dependent effect, we transfected 293T cells with SREBP1 and equal amounts of either KRASWT or KRASG12V. While KRASWT induced SREBP1 expression, the effect with KRASG12V was much greater (Figure 1C).
FIGURE 1.
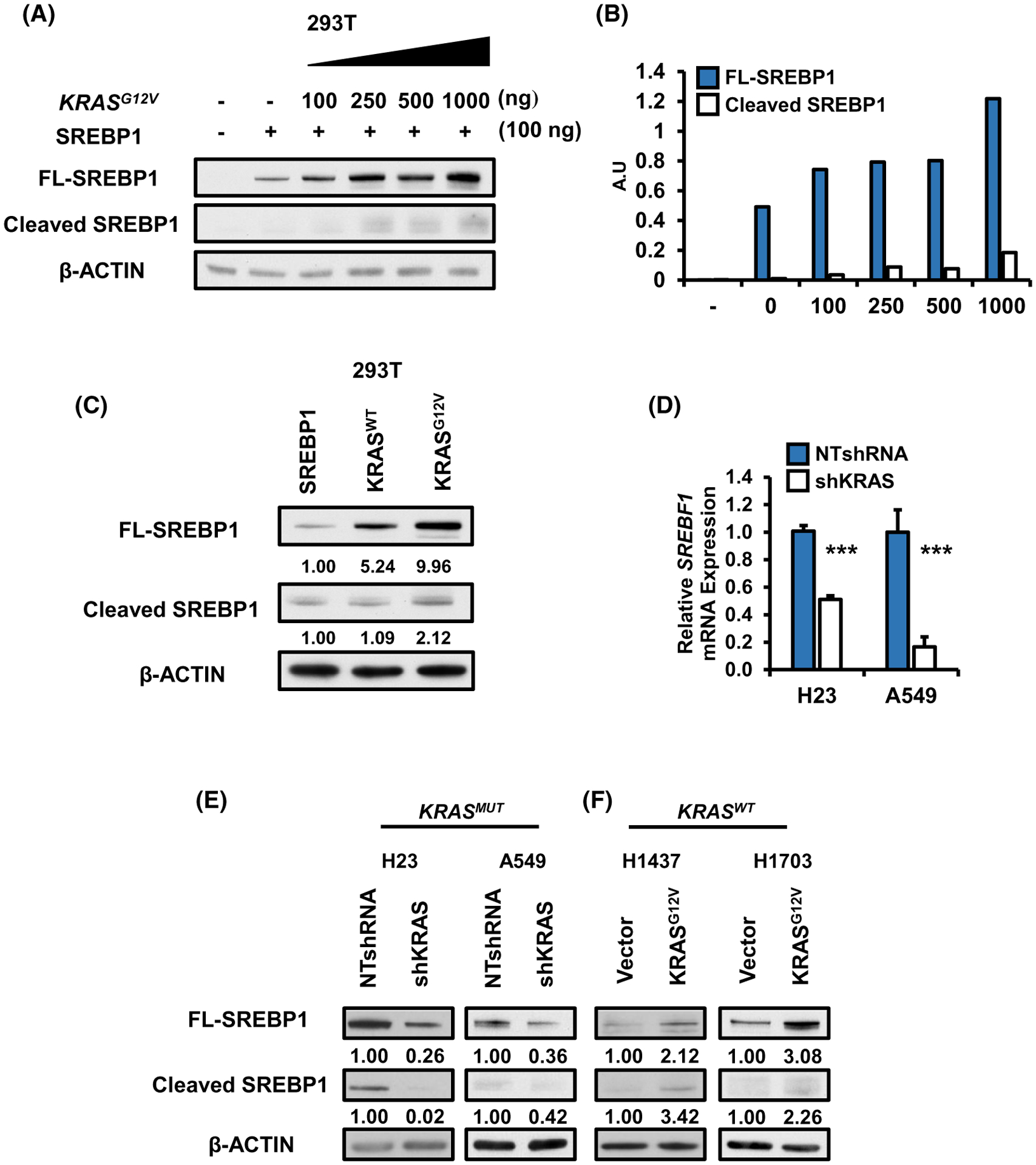
Oncogenic KRAS increases SREBP1: (A) Protein expression for 293T cells transfected with full length (FL) SREBP1 in the presence or absence of increasing amounts of KRASG12V. Protein was harvested 48 hours after transfection and analyzed via western blotting for SREBP1 and loading control, β-ACTIN. B, Densitometry analysis of FL-SREBP1 vs cleaved SREBP1 protein increase from 1a. Blots were analyzed using ImageJ. Values for SREBP1 were normalized to β-ACTIN values. C, Protein expression for SREBP1 in 293T cells transfected with FL-SREBP1 in the presence of either KRASWT or KRASG12V. SREBP1 expression for (D) mRNA and (E) protein in H23 and A549 stably expressing either NTshRNA or shKRAS. N = 3 per group. Bars represent mean ± SD.***P < .001. F, Protein expression for FL-SREBP1 and cleaved SREBP1 in H1437 and H1703 expressing either control vector, or KRASG12V. Proteins were analyzed via western blotting
Next, we investigated whether oncogenic KRAS was necessary to induce SREBP1 expression in NSCLC cells. Knockdown of KRAS in KRAS mutant H23 and A549 cells1,30 led to a reduction in SREBP1 gene expression (gene name: SREBF1) compared to non-target (NT) controls (Figure 1D). SREBP1 protein levels were similarly reduced (Figure 1E). In order to determine if KRASMUT was sufficient to induce SREBP1 expression, we ectopically over-expressed exogenous KRASG12V in wild-type KRAS NSCLC H1437 and H1703 cells. Overexpression of mutant KRAS in both wild type background increased full length and cleaved SREBP1 protein (Figure 1F). In H1437 cells KRASG12V was expressed ~fourfold over KRASWT (Figure S1A). shRNA knockdown of wild-type KRAS (~50%) had no impact on full length SREBP1 protein (Figure S1B) in the H1437 wild-type KRAS background. We asked whether correlation between KRAS mutation and SREBF1 RNA abundance could be observed in human patient samples. In lung adenocarcinomas (TCGA Provisional), mutant KRAS was significantly correlated with SREBF1 mRNA (P = .03; 566 samples) relative to wild-type KRAS.
In order to identify which SREBP1 isoform (SREBP1a or SREBP1c) is predominately expressed in our NSCLC cell lines, we in vitro translated SREBP1a and SREBP1c from plasmid DNA and subjected the products to SDS-PAGE in parallel with protein lysates for H23, A549, H137, H1703 (Figure S1C). SREBP1a is 24 amino acids bigger than SREBP1c and they can be distinguished by size on a western blot. SREBP1c (hereafter referred to as simply SREBP1) appeared to be the predominant isoform in all four NSCLC cell lines, followed by SREBP1b. Thus, we attribute any effects from SREBP1 loss to the reduction of SREBP1c/b isoforms. SREBP1c/SREBF1–3 also was shown to be the predominant transcript by RNAseq (Figure S1D).
3.2 |. Loss of SREBP1 decreases cell proliferation in mutant KRAS NSCLC
Next, we sought to determine the role of SREBP1 on cell expansion in NSCLC. We generated KRAS mutant and KRAS wild-type NSCLC cell lines, which stably expressing lentiviral based NTshRNA or one of two different shRNAs targeting SREBP1, shSREBP1 # 1, and shSREBP1 # 2. shRNAs targeting SREBF1 impacted the three major isoforms SREBP1a/SREBF1-1, SREBP1b/SREBF1–2, and SREBP1c/SREBF1–3 (Figure S1E). We confirmed reduced expression of full length SREBP1 in shSREBP1-expressing mutant KRAS H23 and A549 cells (Figure 2A) and wild-type KRAS H1437 and H1703 (Figure 2B) by western blot. Strikingly, loss of SREBP1 resulted in a marked reduction in cell proliferation of KRAS mutant cells. (Figure 2B,C) but had little effect or enhanced the proliferation of KRASWT-expressing cells (Figure 2E,F). To determine whether the decrease in cell number observed in cells expressing shSREBP1 was due to increased cell death, we analyzed protein levels of cleaved PARP (marker for apoptosis) in H23, A549, and H1437 cells expressing NTshRNA one of two shSREBP1s (Figure 2G). There was no change in cleaved PARP levels in any of theSREBP1 knockdown cells relative to the NTshRNA controls which is consistent with the hypothesis that loss of SREBP1 is decreasing cell proliferation and not increasing cell death. These data suggest that SREBP1 plays a role in the proliferation of mutant KRAS-expressing NSCLC cells. Given the increased expression of SREBP1 in NSCLC, we examined the effect of SREBP1 transcript levels on overall survival in patients with lung adenocarcinoma using Kmplotter with stage, gender and smoking history in multivariate analysis.23 Overall survival was significantly lower in patients with tumors that have high expression of SREBP1 (P < .02) (Figure 2H). It is important to note however that the survival curve does not consider mutant KRAS expression. Nonetheless, these data suggest SREBP1 expression is negatively associated with survival in patients with lung adenocarcinoma.
FIGURE 2.
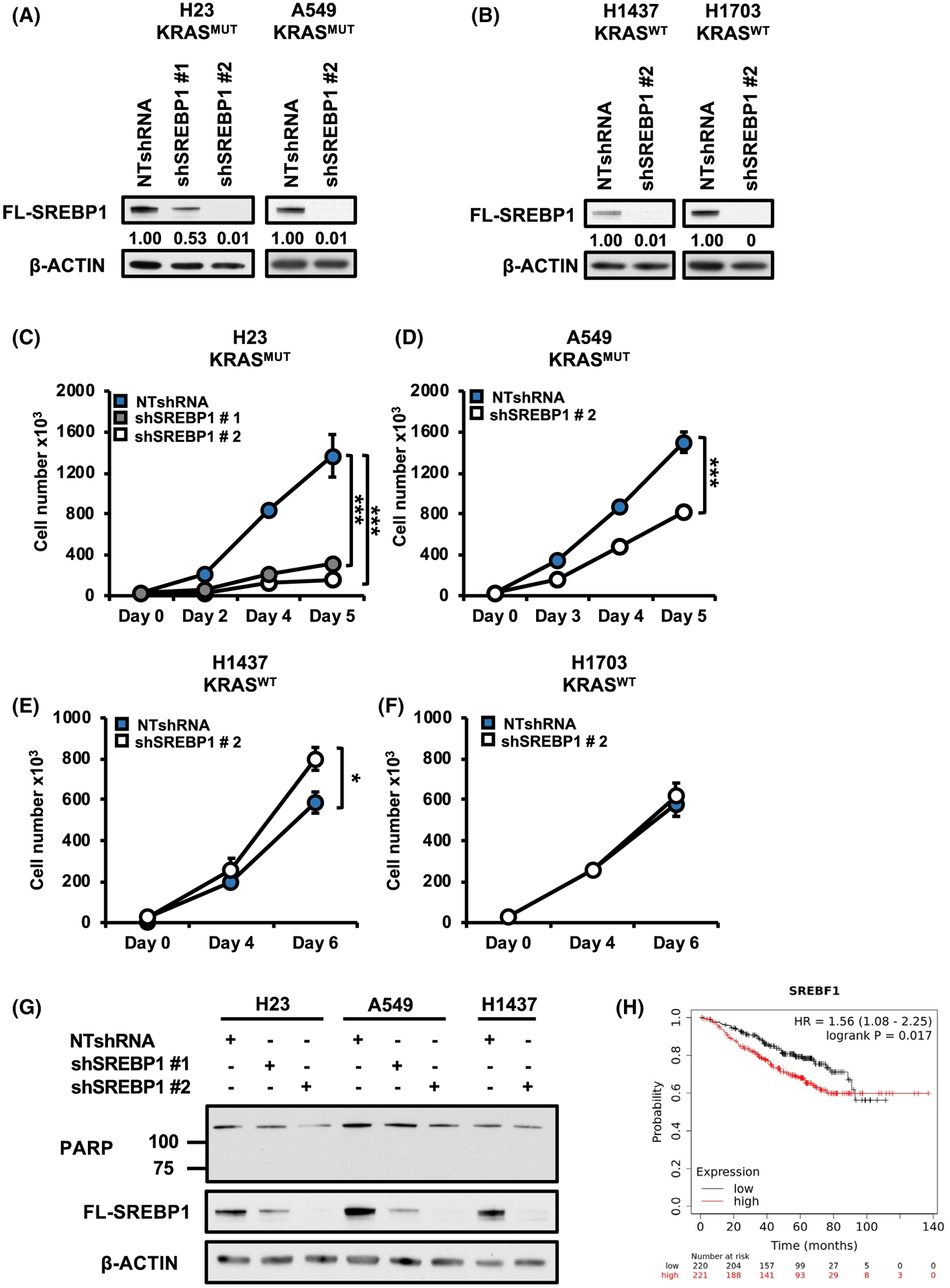
Loss of SREBP1 decreases cell proliferation in mutant KRAS-expressing NSCLC cells. A, SREBP1 protein expression for H23 (left), A549 (right) and (B) H1437 (left), H1703 (right). Cells expressed either NTshRNA or one of two shSREBP1. Proteins were analyzed via western blot analysis. Cell number for mutant KRAS-expressing cells (C) H23, (D) A549 and KRASWT expressing cells (E) H1437 (F) H1703. H23 were expressing either NTshRNA or one of two shRNAs against SREBP1; shSREBP1 #1 or shSREBP1 #2. All other cells expressed either NTshRNA or shSREBP1 #2. Cells were seeded in 6-well plates on day 0 and counted on indicated days using an automatic cell counter. N = 3 per group. Bars show ± SD. *P < .05, ***P < .005. G, Protein expression for PARP in H23, A549, and H1437 cells expressing NTshRNA or either shSREBP1 #1 or shSREBP1 #2. H, Kaplan-Meier multivariate (SREBF1, Stage, Gender, Smoking history) analysis of an integrative lung cancer microarray database showing expression levels of SREBP1 transcript (red:high, black:low) and association with overall survival. Data obtained from Kmplotter website
3.3 |. Oncogenic KRAS regulates SREBP1 protein expression via MEK1/2 signaling
Our data demonstrate that mutant KRAS increases SREBP1 expression in NSCLC and in turn, SREBP1 promotes growth in mutant KRAS-expressing NSCLC cells. KRAS asserts many of its effects through the MEK/ERK pathway.31,32 Therefore, we treated 293T cells transfected with SREBP1 and KRAS with the MEK inhibitor AZD6244.33–35 MEK inhibition greatly blunted the effect of KRAS on SREBP1 protein expression compared to vehicle control (Figure 3A; with long exposure in Figure S1F to additionally show cleaved SREBP1). In contrast, MEK inhibition did not alter SREBP1 protein levels in 293T cells not expressing KRASG12V (Figure 3B). Given that MEK inhibition reduced SREBP1 protein expression in a mutant KRAS preferential manner, we next sought to determine whether activation of the MEK/ERK pathway would increase SREBP1 protein levels. We transfected 293T cells with a constitutively active MEK1D218,D222 allele (MEKDD) or KRASG12V.36 MEKDD expression led to increased SREBP1 protein expression mimicking the effect of KRASG12V (Figure 3C). To further confirm that KRASG12V is regulating SREBP1 protein expression through the MEK/ERK pathway in NSCLC, we compared the effect of AZD6244 on SREBP1 levels to KRAS knockdown in H23 and A549 cells. MEK inhibitor treatment led to a significant decrease in protein levels of SREBP1 comparable to KRAS knockdown cells (Figure 3D). Collectively, these data suggest that mutant KRAS increases SREBP1 protein expression in part via the MEK/ERK pathway.
FIGURE 3.
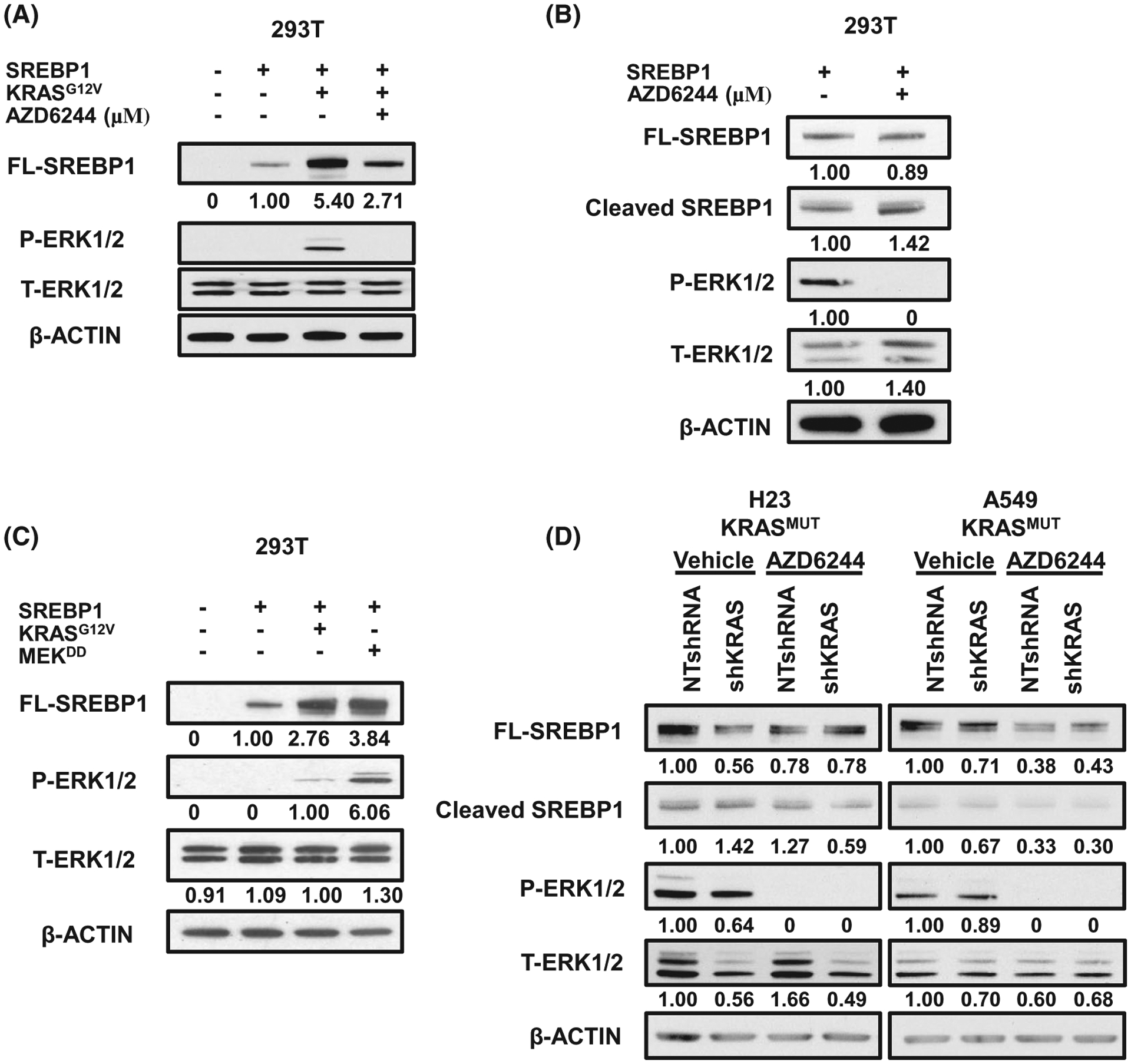
Oncogenic KRAS regulates SREBP1 protein expression via MEK1/2 signaling. A, Protein expression for 293T treated with 2.5 μM of MEK inhibitor, AZD6244, for 16 hours. 293T were transfected with either control vector or FL-SREBP1 in the presence or absence of KRASG12V, 24 hours prior to drug treatment. B, Protein expression for 293T transfected with FL-SREBP1 and treated with AZD6244. Cells were treated 24 hours post transfections and collected 16 hours after treatment. C, Protein expression for 293T cells transfected with FL-SREBP1 and either KRASG12V or MEKDD. D, Protein expression for H23 (left) and A549 (right) treated with 10 μM of AZD6244 for 16 hours. Cells were stably expressing either NTshRNA or shKRAS. E, Densitometry of A549 western blot from (D). FL-SREBP1 was normalized to ACTIN. Blots were analyzed using ImageJ and intensity values are placed under corresponding band. All bands are normalized to corresponding loading control, β ACTIN
3.4 |. Knockdown of SREBP1 has minimal impacts on lipogenesis or glycolysis in mutant KRAS NSCLC
SREBP1 is known to induce the expression of key lipogenic enzymes including ATP Citrate lyse (ACLY), Acetyl-CoA carboxylase (ACC), and Fatty Acid Synthase (FASN), which in turn, promote cell growth by providing fatty acids, which are essential for the synthesis of membranes, energy storage, and signaling in cancer cells.12,16,37 Our lab has previously shown that mutant KRAS promotes the expression of these genes in NSCLC.21 We sought to determine whether SREBP1’s canonical role in lipogenesis might explain the decrease in cell proliferation observed following KRAS knockdown in KRASMUT-expressing cells. We began by examining the expression of lipogenic genes in mutant KRAS cells with stable SREBP1 knockdown. Interestingly, knockdown of SREBP1 caused only modest or no decreases in ACLY, ACACA1 (ACC,) and FASN RNAs in H23 and A549 mutant KRAS backgrounds (Figure 4A,B). Similarly, SREBP1 knockdown caused only a modest decrease in FASN protein (Figure 4C) and in ACLY and ACC protein (Figure S2A), with slight increase in KRAS protein (Figure S2B). Little change in lipogenic RNA or protein were observed with SREBP1 knockdown in wild-type KRAS lines H1437 and H1703 (Figure S2C,D). We looked at this more broadly in 84 CCLE non-small cell adenocarcinoma lines measuring co-correlation pairs KRAS-SREBF1, FASN-SREBF1, ACC-SREBF1 (Figure S2E). KRAS-SREBF1 RNAs co-correlated (P = .03) only in mutant KRAS lines, while FASN-SREBF1 and ACC-SREBF1 RNAs co-correlated (P < .001) in the KRAS wild-type lines. Taken together, these findings suggest that SREBP1 coupling to lipogenic genes in NSC lines is only modest, and that KRAS mutations may further decrease coupling between SREBP1 and lipogenic gene expression.
FIGURE 4.
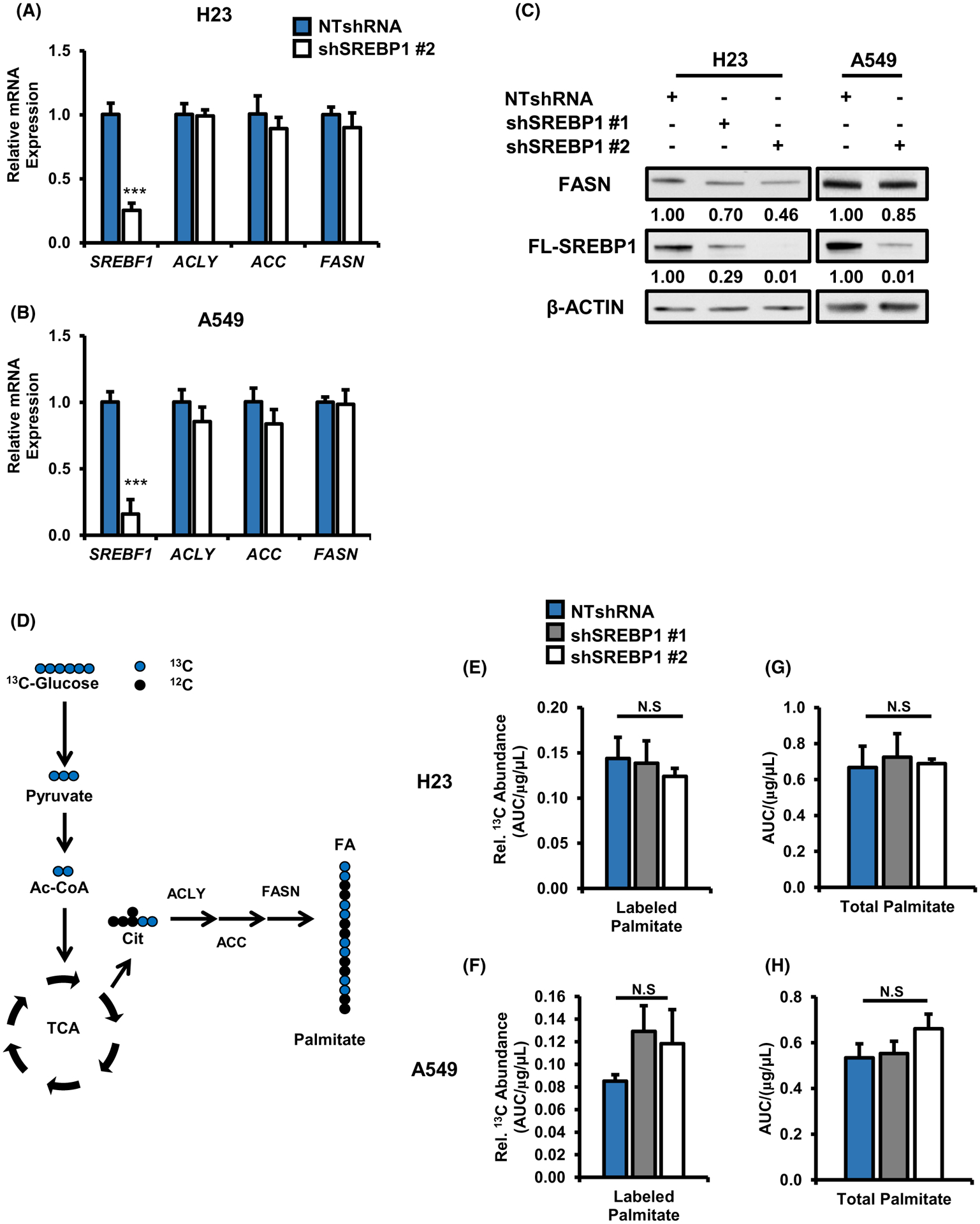
SREBP1 knockdown does not decrease de novo lipogenesis in mutant KRAS expressing NSCLC cells. Gene expression for SREBP1 and its lipogenic targets ACLY, ACC, and FASN in (A) H23, (B) A549. Cells expressed either NTshRNA or shSREBP1 #2. N = 3 per group. Bars indicate mean ± SD. ***P < .005. C, Protein expression for FASN in H23 and A549 expressing NTshRNA or one of two different shSREBP1s. D, Schematic for 13C glucose tracer analysis on de novo lipogenesis. Total 13C glucose labeled palmitate in (E) H23, (F) A549. Total palmitate levels in (G) H23 (H) A549 cells. Palmitate was measured via GS/MS and total counts were normalized to protein concentration of cells on day of collection (μg/μL). Cells expressed NTshRNA or one of two different shSREBP1 for all metabolite tracing experiments. N = 5 per group. Bars indicate mean ± SD. *P < .05, ***P < .005
We next investigated the functional effect of SREBP1 knockdown on de novo lipogenesis by performing 13C stable isotope analysis to measure the incorporation of 13C glucose into palmitate, which requires ACLY, ACC and FASN (Figure 4D). 13C enrichment into palmitate was not reduced following SREBP1 knockdown, demonstrating reduced SREBP1 levels neither altered lipogenic gene expression nor lipogenesis (Figure 4E,F). We also measured total levels of palmitate and saw no significant difference in palmitate levels in SREBP1 knockdown cells compared to nontarget controls (Figure 4G,H). Although SREBP2 preferentially activates expression of genes involved in cholesterol biosynthesis, it has been shown to activate the expression of genes involved in fatty acid synthesis.38,39 Importantly, we did not observe a compensatory increase in SREBF2 levels (Figure S3A) to rescue de novo lipogenesis in shSREBP1 expressing cells. Finally, KRAS wild-type cells subject to stable SREBP1 knockdown did not exhibit altered de novo lipogenesis (Figure S3B). KRAS mRNA and protein levels change slightly when SREBP1 expression is reduced however it does not correlate with the much greater decrease in SREBP1 mRNA and protein in the SREBP1 knockdown models. Importantly, we mined in vivo data from lung conditional knockout models Scap−/− (reducing SREBP1 activity) and Insig−/− (increasing SREBP1 activity),40 and observed little or no change in KRAS RNA (Figure S3C). The data suggest the effect of SREBP1 knockdown are not due to varying KRAS protein levels. Together, these data argue that SREBP1 maintains cell proliferation in mutant KRAS-expressing cells independent of its canonical role in lipogenesis.
Enhanced glycolysis is a common alteration in cancer cells which allows for increased cell proliferation.41 To explore whether loss of SREBP1 leads to significant alterations in glycolysis, we mined the H23 RNAseq data for differential expression in key glycolytic enzymes such as hexokinase (HK), phosphofructokinase (PFK), and pyruvate kinase (PK) between NTshRNA and shSREBP1 expressing cells (Figure S3D). Only slight decreases in HK1 and HK2 were observed, with no changes in RNA levels of 19 other glycolytic enzymes. HK2 catalyzes the committing step of glycolysis and has been shown to be upregulated in multiple cancers, including lung at both RNA and protein levels42,43 Loss of SREBP1 did not decrease HK2 protein in H23 cells and had an inconsistent effect on HK2 in A549 measured by western blot (Figure S3E). To determine the functional consequences of SREBP1 loss on glycolysis in A549 cells, we performed 13C glucose tracer analysis and measured total 13C labeled lactate (Figure S3F,G). We observed a ~40% (H23) and ~10% (A549) decrease in total labeled lactate levels in shSREBP1 expressing cells. Altogether, the data suggest loss of SREBP1 in the presence of mutant KRAS, results in a minor decrease in lipogenesis and in glycolysis.
3.5 |. Loss of SREBP1 decreases mitochondrial-encoded electron transport chain (ETC) genes in mutant KRAS cells
The lack of changes to lipogenic gene expression in SREBP1 knockdown cells prompted us to performed RNA-seq analysis in NTshRNA and SREBP1 knockdown H23 cells. Strikingly, we observed significant decreases in mitochondrial-encoded, but not nuclear-encoded, ETC genes (Figure 5A) and confirmed these findings in H23 and in A549 by qRT-PCR (Figure 5B–E). The statistical significance of the thirteen mitochondrial encoded ETC decreases are shown in (Figure S4A) with 10 of 13 showing fold change P < .005 for both SREBP1 shRNA hairpins. The findings were not simply related to encoded protein localization (Figure S4B) for example with no RNA abundance differences between cytosolic, mitochondrial or nuclear encoded protein locations. Consistent with RNA levels, protein levels for mitochondrial-encoded cytochrome c oxidase I (MT-CO1) were also reduced in H23 and A549 cells expressing shSREBP1, whereas protein levels for nuclear-encoded ATP5A did not change (Figure 5F). In contrast, SREBP1 knockdown in KRASWT cells resulted in only minor declines to mitochondrial-encoded ETC gene expression in H1437 (Figure 5G) and significant increases in H1703 (Figure 5H). Geneset enrichment analysis (GSEA) also showed significant association (FDR q < .001) with TCA cycle and oxidative phosphorylation signatures (data not shown). These data suggest that SREBP1’s effect on mitochondrial gene expression might be facilitated by mutant KRAS.
FIGURE 5.
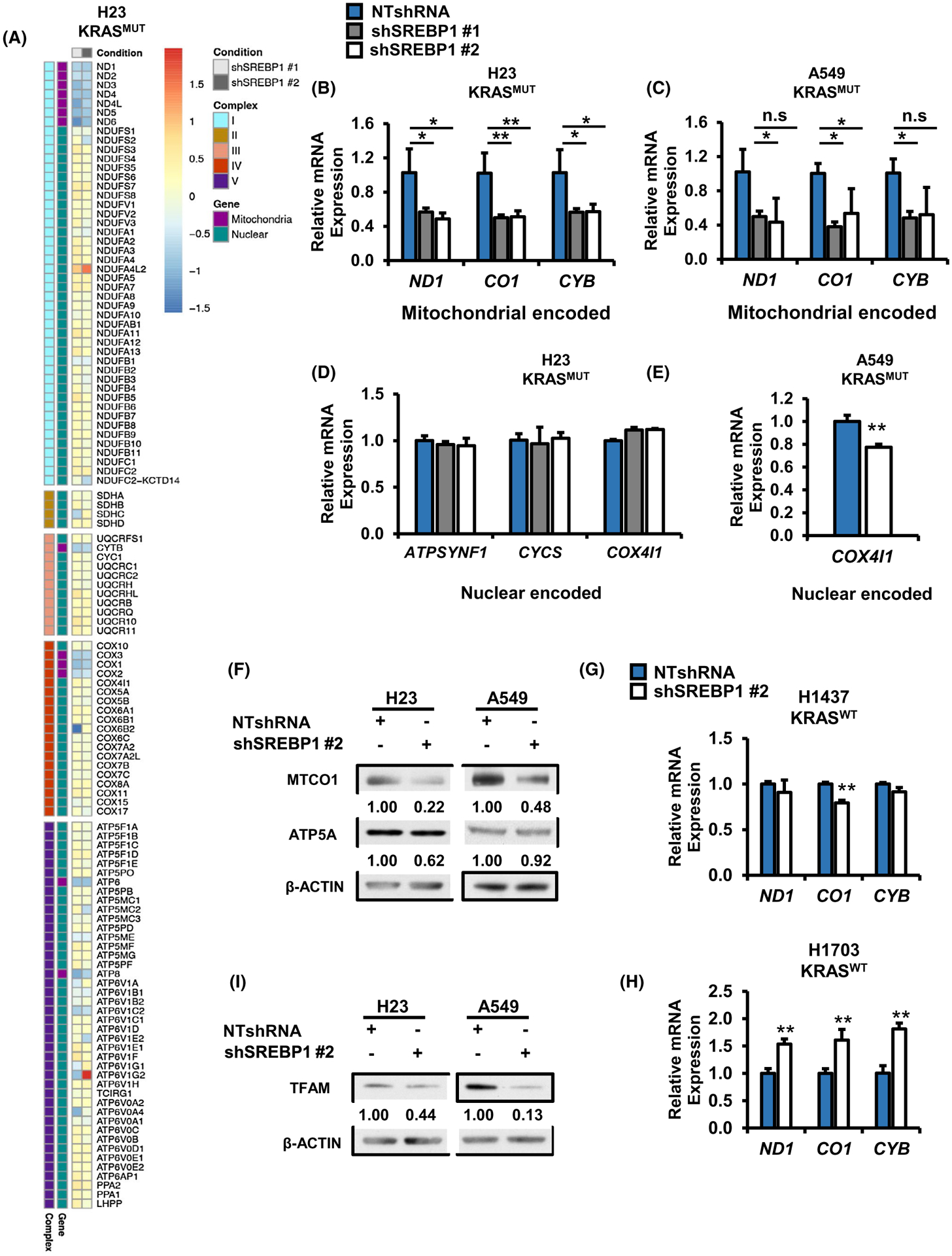
Loss of SREBP1 decreases expression of mitochondrial encoded ETC genes in mutant KRAS NSCLC cells. A, RNA seq analysis for mitochondrial encoded and nuclear encoded mitochondrial proteins in H23 cells expressing either NTshRNA, shSREBP1 #1 or shSREBP1 #2. 122 genes matching the KEGG pathway for oxidative phosphorylation (hsa00190) were extracted from RNA seq data for H23 expressing NTshRNA or one of two shSREBP1s. Log2FC for shSREBP1 #1 and shSREBP1 #2 were plotted as a heat map and separated by ETC complex, with nuclear or mitochondrial encoded genes indicated by color. N = 3 per group. Heat map was generated using the R package pheatmap. Gene expression for mitochondrial encoded ETC genes in (B) H23, and (C) A549 cells expressing either NTshRNA shSREBP1 #1 or shSREBP1 #2. N = 3 per group. Bars indicate ± SD. *P < .05. Gene expression for nuclear encoded ETC genes in (D) H23 and (E) A549 cells expressing either NTshRNA or one of two different shSREBP1. N = 3 per group. Bars indicate ± SD. *P < .05. F, Protein expression for H23 (left) and A549 (right) cells expressing either NTshRNA or shSREBP1 #2. Proteins were analyzed via western blotting. and densitometry was performed using ImageJ. Gene expression for mitochondrial encoded genes in KRASWT (G) H1437 and (H) H1703 cells expressing either NTshRNA or shSREBP1 #2. N = 3 per group. Bars indicate ± SD. **P < .01. Protein expression for TFAM in H23 (left) and A549 (right) cells expressing NTshRNA or shSREBP1 #2. Proteins were analyzed via western blotting and densitometry was performed using ImageJ
3.6 |. Loss of SREBP1 does not alter mitochondrial mass and slightly decreases copy number
Our data suggested that SREBP1 plays a role in mitochondrial biology specifically in KRAS mutant NSCLC cells. Indeed, mitochondrial transcription factor A (TFAM), which is required for mitochondrial DNA replication and transcription44,45 was reduced following SREBP1 knockdown in H23 and A549 by western blot (Figure 5I) and by RNAseq (log2FC = −0.52; P = .01). To determine if the loss of ETC gene and protein expression was due in part to a decrease in number of mitochondria, we stained cells with Mitotracker Green, a cell permeable dye which localizes and binds to mitochondria. There was no difference in GFP intensity, quantified by flow cytometry, between NTshRNA control and shSREBP1 cells (Figure S4C). To further confirm that reduced SREBP1 expression is not affecting mitochondrial number, we measured mitochondrial copy number relative to nuclear DNA using RT-PCR, as previously described.25 There was a ~17% decrease (P < .05) in mitochondrial DNA copy number in SREBP1 knockdown cells compared to NTshRNA controls (Figure S4D). Suggesting the decrease in mitochondrial gene transcription could be in part due to lower mitochondrial DNA content. Taken together, these data suggest that SREBP1 knockdown results in decreased expression of mitochondrial-encoded ETC genes and mitochondrial DNA copy number. Furthermore, there were no alterations to mitochondrial mass suggesting SREBP1 knockdown is affecting transcription not mitochondrial biogenesis. Lastly, nuclear encoded genes that make up subunits of the ETC do not significantly change, suggesting this is not due to SREBP1’s transcriptional activity in the nucleus.
3.7 |. Loss of SREBP1 decreases oxidative phosphorylation in mutant KRAS-expressing NSCLC cells
Given the effect of SREBP1 knockdown on mitochondrial ETC gene expression, we wanted to determine the effects on mitochondrial function. Knockdown of SREBP1 resulted in a significant decrease in basal oxygen consumption rate (OCR) (~80%) and maximal respiration (~70%) (Figure 6A–C) compared to NTshRNA cells expressing mutant KRAS. In contrast, we did not see any difference in basal OCR in KRASWT cells (H1437) with stable knockdown of SREBP1 (Figure S5A,B). A major fuel for oxidative phosphorylation via the TCA cycle is glucose. Therefore, we performed 13C glucose tracer analysis to determine whether SREBP1 knockdown alters glucose utilization by the TCA cycle. We measured enrichment of m + 2 metabolites into the TCA, since they would be derived from labeled glucose (Figure 6D). We observed a significant decrease in m + 2 citrate (~56%), fumarate (~43%), and malate (~40%) when SREBP1 was knocked down in H23 cells (Figure 6E). In contrast, we did not see a significant change in these metabolites in KRASWT cells (H1437) with SREBP1 knockdown (Figure 6F). These data suggest that knockdown of SREBP1 impairs oxidative phosphorylation from glucose in mutant KRAS-expressing cells.
FIGURE 6.
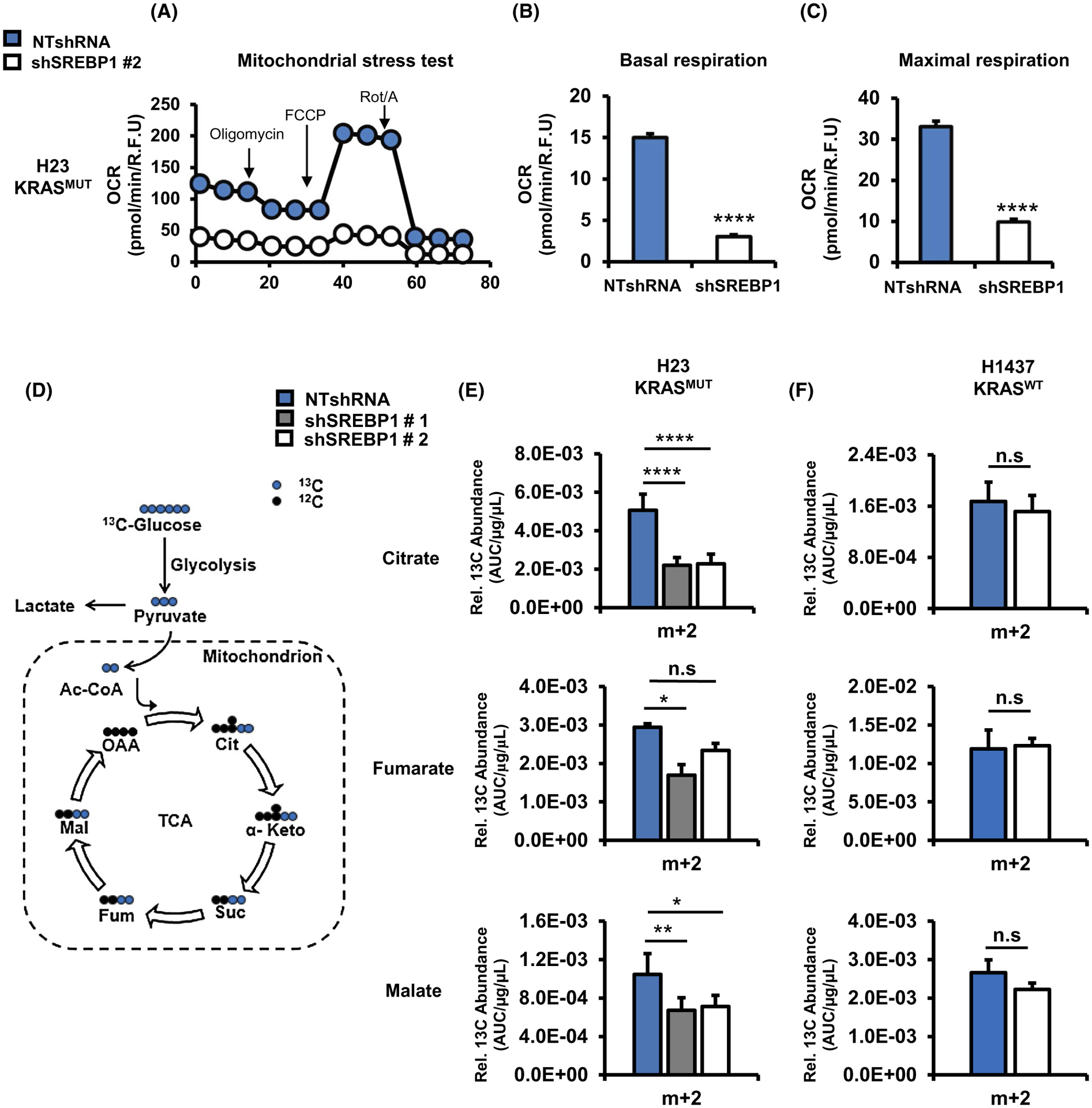
Loss of SREBP1 decreases oxidative phosphorylation in mutant KRAS-expressing NSCLC cells. A, Representative mitochondria stress test performed on H23 expressing NTshRNA or shSREBP1 #2. The stress test provides (B) basal respiration and (C) maximal respiration. OCR was measured using a Seahorse Bioenergetic Flux Analyzer. N ≥ 10 per group. Bars indicate mean ± SE. ****P < .0001. D, Schematic of glucose utilization by the TCA cycle into m + 2 intermediates. Relative amount of 13C labeled m + 2 citrate, fumarate, and malate in (E) H23 and (F) H1437 cells. Cells expressed NTshRNA or one of two different shSREBP1. Cells were labeled with 13C [U6] glucose, and harvested after 6 hours, and analyzed by GC/MS for TCA cycle metabolites. N = 5 per group. Bars indicate ± SD. *P < .01, **P < .001
4 |. DISCUSSION
Activating mutations of KRAS drive the metabolic alterations that promote tumor growth in NSCLC.21,29,31,46–49 However, the detailed molecular mechanisms by which KRAS regulates metabolism in NSCLC are not well understood. Here, we report a novel role for SREBP1 distinct from lipogenesis in KRAS-expressing NSCLC. Oncogenic KRAS increases SREBP1 expression and loss of SREBP1 leads to decreased cell proliferation independent of its role in lipogenesis. Importantly, high SREBP1 expression correlates with poor survival in patients with lung adenocarcinoma. Most interestingly, we report for the first time, to our knowledge, that loss of SREBP1 in mutant KRAS-expressing NSCLC leads to reduction of mitochondrial-encoded ETC subunits, resulting in deficient mitochondrial metabolism. Studies investigating the role of mutant KRAS on mitochondrial content and function of cells have produced contradictory results with some reports showing mutant KRAS pushing a more glycolytic phenotype (aerobic glycolysis) while other studies have shown mutant KRAS expressing tumors to have increased glycolytic and oxidative phosphorylation activity. For example, using a spontaneous KrasG12D/+;p53−/− lung tumor model, Kerr et al reported that tumors homozygous for KrasG12D had increased shuttling of glucose carbons into the TCA cycle relative to tumors heterozygous for KrasG12D.48 In contrast, Humpton et al, recently reported that mutant KRAS promotes a program of mitophagy to inhibit shunting of glucose carbons into the TCA cycle in PDAC cells.50 Altogether, the data suggest mutant KRAS regulation of mitochondrial metabolism and content is context dependent, with at least tissue type playing a significant role. How exactly mutant KRAS regulates mitochondrial content and respiratory capacity in NSCLC and the role of SREBP1 in this process is the focus of our future studies.
Mutant KRAS activates over a dozen downstream targets to assert its pro-tumorigenic effects and the RAF/MEK/ERK pathway is among one of the most well-characterized15 and many approaches to targeting mutant KRAS cancers involve the utilization of MEK/ERK inhibitors.33–35,51–53 Our work revealed that mutant KRAS regulates SREBP1 expression via MEK/ERK activation. MEK inhibition using AZD6244 greatly reduced the effect of mutant KRAS on SREBP1 protein expression in 293T cells. Furthermore, activation of MEK pathway with constitutively active MEK1 mutant, MEKDD, was sufficient to increase SREBP1 protein expression. Similarly, MEK inhibition in NSCLC cells (H23 and A549) reduced SREBP1 levels similar to KRAS knockdown. Multiple ERK1/2 phosphorylation sites have been mapped on SREBP1,54 and ERKs 1/2 are the only known targets of MEKs 1/2, implicating ERK in mutant KRAS mediated regulation of SREBP1. Interestingly, mutant KRAS does not appear to regulate SREBP1 cleavage, suggesting KRAS controls SREBP1 activity independent of cleavage. However, additional inhibitor studies need to be performed to fully elucidate the mechanism(s) responsible for KRAS regulation of SREBP1. Beyond characterizing ERK1/2 phosphorylation sites on SREBP1 and their functional consequences, further work should also include analysis of the mutant KRAS pathway in regulating translation and stability of SREBP1. Preliminary data in H1703 cells using a cycloheximide translational block suggest SREBP1 may be regulated by translation and/or stability (Figure S5D), which should be pursued in future studies. GSEA also suggest modulation of TORC1 signaling with SREBP1 knockdown in the mutant KRAS H23 background, suggesting that KRAS-Erk-Rsk2-TORC1 pathway may contribute to translational control and should be examined together with phospho-proteomic analysis of posttranslational modifications of the Erk-Rsk2-TORC1 pathway on SREBP1 in the presence and absence of mutant KRAS.
Our results support the notion that SREBP1 is important for mutant KRAS expressing NSCLC cell proliferation16–18,55; however we were surprised to find that loss of SREBP1 did not significantly alter gene expression of classic lipogenic targets ACLY, ACACA1, and FASN.21 Furthermore, using 13C tracer analysis with GC/MS, we found that mutant KRAS-expressing NSCLC with reduced levels of SREBP1 could still make sufficient levels of saturated fatty acids such as palmitate. Williams et al showed that an essential requirement for SREBP1 is to maintain the ratio of monosaturated vs monounsaturated fatty acids.56 In their study, loss of SREBP1 did not lead to decrease in palmitate but instead lead a significant decrease in monounsaturated fatty acids such as oleate, which ultimately resulted in lipotoxicity and cell death. However, these studies were carried out in glioma cells, which are not mutant KRAS-dependent. Additionally, oleate levels did not decrease in our models when SREBP1 was knocked down (data not shown), suggesting an alternative mechanism for loss of cell proliferation.
Earlier studies focused on SREBP1’s role in lipid homeostasis and regulation via cleavage in low-cholesterol environments.12,14,19,57 Recently, however, multiple studies have unraveled novel roles for SREBP1 in unexpected pathways linked to diabetes, cancer, the immune system, and autophagy.15,58–62 Using RNA-seq analysis, we discovered loss of SREBP1 resulted in decreased mitochondrial gene expression in NSCLC cells. Loss of SREBP1 also reduced protein levels of mitochondrial transcription factor A (TFAM), which is one of three key transcription factors required for mitochondrial DNA replication and transcription.44 Decreased mitochondrial gene expression resulted in impaired mitochondrial function characterized by reduced TCA cycle activity and oxygen consumption. It is not clear whether decreased proliferation in SREBP1 knockdown cells is due to SREBP1’s effects on the mitochondria. Furthermore, our work did not establish whether SREBP1’s effect on the mitochondria is strictly mutant KRAS-dependent. While mutant KRAS expression was sufficient to enhance SREBP1’s effect on the mitochondria as shown by genetic knockdown and overexpression experiments, further studies are required to determine to what extent KRAS is important in SREBP1-mediated mitochondrial metabolism and transcription. Additionally, it remains to be seen whether other prominent oncogenes in lung cancer, such as mutant EGFR which also activates ERK1/2, might similarly alter SREBP1 function.
Citrate which provides carbon substrates in the form of Acetyl-CoA to the de novo lipogenesis pathway is decreased in shSREBP1 cells, yet it does not appear to be rate-limiting for palmitate production. We hypothesize that KRASMUT-expressing cells maintain their free fatty acid levels by decreasing lipid degradation programs such as beta-oxidation. Peroxisome proliferator-activated receptor gamma co-activator 1-alpha (PGC-1α) is a transcriptional co-activator that regulates a plethora of cellular metabolic pathways including oxidative phosphorylation and beta-oxidation.63,64 Given the striking deficiency in cellular respiration observed in SREBP1 knockdown cells, we measured PGC1a (Gene name: PPARGC1A) mRNA expression in H23 and A549 expressing NTshRNA or one of two shSREBP1s (Figure S5C). PGC1a levels decreased substantially in cells with loss of SREBP1. This is consistent with decreased cellular respiration and suggest a potential decrease in beta oxidation in SREBP1 knockdown cells. However, the role of PGC1a and beta oxidation in SREBP1 regulation of mitochondrial metabolism will have to be further investigated in future studies.
Our results also suggest an alternative pathway for KRAS-mediated lipogenesis in NSCLC since loss of SREBP1 showed no significant decrease in de novo lipogenesis. Further studies are required to illuminate how KRAS is regulating fatty acid synthesis which could potentially be via other lipogenic transcription factors implicated in cancer such as carbohydrate responsive element-binding protein (ChREBP).65 Finally, the finding that SREBP1 plays a role in mitochondrial homeostasis presents a novel opportunity for targeted therapy in KRAS mutant lung cancers.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Martha B. Furie PhD, and Mandar Muzumdar, MD, for critical editing of the manuscript. This work was supported in part by funds from the Ruth L. Kirschstein National Research Service Award (NRSA) F31 Grant 1F31CA210626-01. AJB received support from the National Cancer Institute (NCI) Predoctoral to Postdoctoral Fellow Transition Award, K00CA212445.
Funding information
HHS | NIH | National Cancer Institute (NCI), Grant/Award Number: F31 Grant 1F31CA210626-01 and K00CA212445
Abbreviations:
- ACACA1/ACC
acetyl coenzyme A carboxylase
- acetyl-CoA
acetyl-Co enzyme A
- ACLY
ATP-citrate lyase
- ATP
adenosine triphosphate
- CO1
cytochrome c oxidase subunit I
- CYB
cytochrome b
- ETC
electron transport chain
- FASN
fatty acid synthase
- GC/MS
gas chromatography-mass spectrometry
- GSEA
gene set enrichment analysis
- KRAS
Kirsten rat sarcoma viral oncogene homolog
- LUAD
lung adenocarcinoma
- ND1
NADH:Ubiquinone Oxidoreductase Core Subunit 1
- NSCLC
non-small cell lung cancer
- NTshRNA
non-target shRNA
- RNASeq
RNA sequencing
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
- OxPhos
oxidative phosphorylation
- TCA
tricarboxylic acid cycle
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the Supporting Information section.
REFERENCES
- 1.Sunaga N, Shames DS, Girard L, et al. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther. 2011;10(2):336–346. 10.1158/1535-7163.MCT-10-0750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karachaliou N, Mayo C, Costa C, et al. KRAS mutations in lung cancer. Clin Lung Cancer. 2013;14(3):205–214. 10.1016/j.cllc.2012.09.007 [DOI] [PubMed] [Google Scholar]
- 3.Suda K, Tomizawa K, Mitsudomi T. Biological and clinical significance of KRAS mutations in lung cancer: an oncogenic driver that contrasts with EGFR mutation. Cancer Metastasis Rev. 2010;29(1):49–60. 10.1007/s10555-010-9209-4 [DOI] [PubMed] [Google Scholar]
- 4.Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2(1):e17. 10.1371/journal.pmed.0020017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223. 10.1038/s41586-019-1694-1 [DOI] [PubMed] [Google Scholar]
- 6.Mullard A Cracking KRAS. Nat Rev Drug Discov. 2019;18(12):887–891. 10.1038/d41573-019-00195-5 [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 8.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. 10.1126/sciadv.1600200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez-Reyes I, Diebold LP, Kong H, et al. TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol Cell. 2016;61(2):199–209. 10.1016/j.molcel.2015.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018;20(7):745–754. 10.1038/s41556-018-0124-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131. 10.1172/JCI15593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madison BB. Srebp2: a master regulator of sterol and fatty acid synthesis. J Lipid Res. 2016;57(3):333–335. 10.1194/jlr.C066712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des. 2014;20(15):2619–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Assmann N, O’Brien KL, Donnelly RP, et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat Immunol. 2017;18(11):1197–1206. 10.1038/ni.3838 [DOI] [PubMed] [Google Scholar]
- 16.Griffiths B, Lewis CA, Bensaad K, et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013;1(1):3. 10.1186/2049-3002-1-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Y, He W, Luo M, et al. SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumour Biol. 2015;36(6):4133–4141. 10.1007/s13277-015-3047-5 [DOI] [PubMed] [Google Scholar]
- 18.Wen YA, Xiong X, Zaytseva YY, et al. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018;9(3):265. 10.1038/s41419-018-0330-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo D, Reinitz F, Youssef M, et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1(5):442–456. 10.1158/2159-8290.CD-11-0102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao J, Zhu L, Zhu Q, Su J, Liu M, Huang W. SREBP-1 is an independent prognostic marker and promotes invasion and migration in breast cancer. Oncol Lett. 2016;12(4):2409–2416. 10.3892/ol.2016.4988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh A, Ruiz C, Bhalla K, et al. De novo lipogenesis represents a therapeutic target in mutant Kras non-small cell lung cancer. FASEB J. 2018;32(12):7018–7027. 10.1096/fj.201800204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montal ED, Dewi R, Bhalla K, et al. PEPCK coordinates the regulation of central carbon metabolism to promote cancer cell growth. Mol Cell. 2015;60(4):571–583. 10.1016/j.molcel.2015.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013;8(12):e82241. 10.1371/journal.pone.0082241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Young L, Sung J, Stacey G, Masters JR. Detection of mycoplasma in cell cultures. Nat Protoc. 2010;5:929–934. 10.1038/nprot.2010.43 [DOI] [PubMed] [Google Scholar]
- 25.Rooney JP, Ryde IT, Sanders LH, et al. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol Biol. 2015;1241:23–38. 10.1007/978-1-4939-1875-1_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Afgan E, Baker D, Batut B, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46(W1):W537–W544. 10.1093/nar/gky379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Team RC. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2019. [Google Scholar]
- 28.Kolde R pheatmap: Pretty Heatmaps. R package version 1.0.12 ed2019.
- 29.Gouw AM, Eberlin LS, Margulis K, et al. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc Natl Acad Sci U S A. 2017;114(17):4300–4305. 10.1073/pnas.1617709114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh A, Greninger P, Rhodes D, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15(6):489–500. 10.1016/j.ccr.2009.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39(2):91–100. 10.1016/j.tibs.2013.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36(6):320–328. 10.1016/j.tibs.2011.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janne PA, Shaw AT, Pereira JR, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14(1):38–47. 10.1016/S1470-2045(12)70489-8 [DOI] [PubMed] [Google Scholar]
- 34.Carter CA, Rajan A, Keen C, et al. Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced non-small-cell lung cancer. Ann Oncol. 2016;27(4):693–699. 10.1093/annonc/mdw008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Park D, Shin DM, Deng X. Targeting KRAS-mutant non-small cell lung cancer: challenges and opportunities. Acta Biochim Biophys Sin (Shanghai). 2016;48(1):11–16. 10.1093/abbs/gmv118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brunet A, Pages G, Pouyssegur J. Constitutively active mutants of MAP kinase kinase (MEK1) induce growth factor-relaxation and oncogenicity when expressed in fibroblasts. Oncogene. 1994;9(11):3379–3387. [PubMed] [Google Scholar]
- 37.Shimano H Sterol regulatory element-binding proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Prog Lipid Res. 2001;40(6):439–452. [DOI] [PubMed] [Google Scholar]
- 38.Pai JT, Guryev O, Brown MS, Goldstein JL. Differential stimulation of cholesterol and unsaturated fatty acid biosynthesis in cells expressing individual nuclear sterol regulatory element-binding proteins. J Biol Chem. 1998;273(40):26138–26148. 10.1074/jbc.273.40.26138 [DOI] [PubMed] [Google Scholar]
- 39.Horton JD, Shah NA, Warrington JA, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci. 2003;100(21):12027–12032. 10.1073/pnas.1534923100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bridges JP, Schehr A, Wang Y, et al. Epithelial SCAP/INSIG/SREBP signaling regulates multiple biological processes during perinatal lung maturation. PLoS One. 2014;9(5):e91376. 10.1371/journal.pone.0091376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41(3):211–218. 10.1016/j.tibs.2015.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng Y, Lu J. Targeting hexokinase 2 in castration-resistant prostate cancer. Mol Cell Oncol. 2015;2(3):e974465. 10.4161/23723556.2014.974465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patra KC, Wang Q, Bhaskar PT, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24(2):213–228. 10.1016/j.ccr.2013.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gustafsson CM, Falkenberg M, Larsson NG. Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem. 2016;85:133–160. 10.1146/annurev-biochem-060815-014402 [DOI] [PubMed] [Google Scholar]
- 45.Barshad G, Marom S, Cohen T, Mishmar D. Mitochondrial DNA transcription and its regulation: an evolutionary perspective. Trends Genet. 2018;34(9):682–692. 10.1016/j.tig.2018.05.009 [DOI] [PubMed] [Google Scholar]
- 46.White E Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 2013;27(19):2065–2071. 10.1101/gad.228122.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kerr EM, Martins CP. Metabolic rewiring in mutant Kras lung cancer. FEBS J. 2018;285(1):28–41. 10.1111/febs.14125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531(7592):110–113. 10.1038/nature16967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Min HY, Lee HY. Oncogene-driven metabolic alterations in cancer. Biomol Ther (Seoul). 2018;26(1):45–56. 10.4062/biomolther.2017.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Humpton TJ, Alagesan B, DeNicola GM, et al. Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 2019;9(9):1268–1287. 10.1158/2159-8290.CD-18-1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13(12):928–942. 10.1038/nrd4281 [DOI] [PubMed] [Google Scholar]
- 52.Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–575. 10.1038/nature11005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang J, Nannapaneni S, Wang D, et al. Phenformin enhances the therapeutic effect of selumetinib in KRAS-mutant non-small cell lung cancer irrespective of LKB1 status. Oncotarget. 2017;8(35):59008–59022. 10.18632/oncotarget.19779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knebel B, Lehr S, Hartwig S, et al. Phosphorylation of sterol regulatory element-binding protein (SREBP)-1c by p38 kinases, ERK and JNK influences lipid metabolism and the secretome of human liver cell line HepG2. Arch Physiol Biochem. 2014;120(5):216–227. 10.3109/13813455.2014.973418 [DOI] [PubMed] [Google Scholar]
- 55.Porstmann T, Santos CR, Griffiths B, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8(3):224–236. 10.1016/j.cmet.2008.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williams KJ, Argus JP, Zhu Y, et al. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res. 2013;73(9):2850–2862. 10.1158/0008-5472.CAN-13-0382-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Daemen S, Kutmon M, Evelo CT. A pathway approach to investigate the function and regulation of SREBPs. Genes Nutr. 2013;8(3):289–300. 10.1007/s12263-013-0342-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ide T, Shimano H, Yahagi N, et al. SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat Cell Biol. 2004;6(4):351–357. 10.1038/ncb1111 [DOI] [PubMed] [Google Scholar]
- 59.Im SS, Yousef L, Blaschitz C, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 2011;13(5):540–549. 10.1016/j.cmet.2011.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park HJ, Georgescu SP, Du C, et al. Parasympathetic response in chick myocytes and mouse heart is controlled by SREBP. J Clin Invest. 2008;118(1):259–271. 10.1172/JCI32011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M. Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genet. 2008;4(7):e1000133. 10.1371/journal.pgen.1000133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rome S, Lecomte V, Meugnier E, et al. Microarray analyses of SREBP-1a and SREBP-1c target genes identify new regulatory pathways in muscle. Physiol Genomics. 2008;34(3):327–337. 10.1152/physiolgenomics.90211.2008 [DOI] [PubMed] [Google Scholar]
- 63.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J Biol Chem. 2005;280(16):16456–16460. 10.1074/jbc.M501485200 [DOI] [PubMed] [Google Scholar]
- 64.Tan Z, Luo X, Xiao L, et al. The role of PGC1alpha in cancer metabolism and its therapeutic implications. Mol Cancer Ther. 2016;15(5):774–782. 10.1158/1535-7163.MCT-15-0621 [DOI] [PubMed] [Google Scholar]
- 65.Tong X, Zhao F, Mancuso A, Gruber JJ, Thompson CB. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc Natl Acad Sci U S A. 2009;106(51):21660–21665. 10.1073/pnas.0911316106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.