

Abstract
Renal tubular epithelial cell injury is the main cause of septic acute kidney injury (AKI), which is characterized by the excessive inflammatory response and apoptosis. Numerous studies have demonstrated that miRNAs are associated with inflammatory response and apoptosis in numerous diseases. The present study mainly focuses on investigating the association between microRNA (miRNA/miR) expression and inflammatory response and apoptosis in the pathogenesis of AKI. In vitro and in vivo models of AKI were simulated using Escherichia coli lipopolysaccharide (LPS)-administrated kidney epithelial cells and mice, respectively. The miRNA expression profile was examined using miRNA microarray in kidney tissues. Next, the effects of miR-93 upregulation on the apoptosis, cytokine expression and oxidative stress in the LPS-stimulated TCMK-1 were tested. The target genes of this miRNA were investigated, and the regulatory association between miR-93 and the AKT/mTOR pathway was investigated. The results demonstrated that miR-93 was the most downregulated miRNA in mice kidney. Furthermore, in LPS-induced renal tubular epithelial cells (TECs) injury model, that upregulation of miR-93 was found to attenuate the apoptosis and inflammatory response, as well as reactive oxygen species generation. Mechanistically, phosphatase and tensin homolog deleted on chromosome 10 (PTEN) was identified as a target of miR-93. Further experiments revealed that LPS-induced the decrease of phosphorylated (p)-AKT and p-mTOR protein expression in vitro are reversed by the overexpression of miR-93. The results of the present study suggested that the protective effect of miR-93 on AKI may be associated with the activation of PTEN/AKT/mTOR pathway. miR-93 may serve as a potential therapeutic target in sepsis-induced AKI.
Keywords: acute kidney injury, microRNA-93, inflammation, apoptosis, reactive oxygen species, PTEN/AKT/mTOR pathway
Introduction
Acute kidney injury (AKI) is one common complication of sepsis with a high mortality rate in critically ill patients (1). Although great effort has been made in the mechanism underlying the development of AKI, treatment of septic AKI remains unsatisfactory (2,3). Therefore, it is necessary to investigate effective therapeutic options for sepsis-induced AKI.
Studies have reported that the loss of functional tubular epithelial cells (TECs) via apoptosis and inflammatory response are involved in the pathological process of AKI (4–6). Notably, anti-inflammatory and anti-apoptotic therapy was confirmed to be beneficial for the treatment of sepsis-induced AKI (7,8). Lipopolysaccharide (LPS) is a classical ligand for TLR4 and mediates TLR4-dependent signal transduction to activate NF-κB, leading to an increase in inflammatory cytokine expression, including interleukin-1β (IL-1β), IL-6 and tumor necrosis factor-α (TNF-α) (9). Therefore, an LPS-induced experimental model was comprehensively used to investigate and investigate the anti-inflammatory treatment of sepsis-related AKI.
MicroRNAs (miRNAs/miRs), endogenous small (~22 nucleotides) non-coding RNA, act as unique regulators of gene expression through either inducing transcript degradation or inhibiting translation (10,11). Numerous studies have highlighted the key roles of miRNAs in the pathological process of AKI (12,13). For example, Liao et al (14) demonstrated that miR-140-5p improved cisplatin-induced AKI through suppressing oxidative stress by activating the Nrf2/ARE pathway in a mouse model. Lan et al (15) reported that miR-494 may induce over-activation of inflammatory response and apoptosis in a mouse AKI model. In addition, several miRNAs, including miR-494 (16), miR-107 (17) in the serum have been proposed to service as a biomarker of AKI. Recent studies have demonstrated the protective effects of miRNAs by inhibiting apoptosis and inflammatory response in various types of renal cells (18,19). For example, Guo et al (20) reported that inhibition of miR-709 protected against cisplatin-induced the proximal tubular cell injury. Yan et al (21) reported that miR-214 ameliorated ischemic AKI through regulation of mitochondrial fragmentation and apoptosis in ATP-depleted proximal tubular cells. Therefore, it was hypothesized that miRNAs may regulate the renal TECs apoptosis and inflammatory response.
In the present study, a mouse AKI model was established and microarray analysis was conducted to determine miRNA expression profiles in kidney tissues. Subsequently, the function and possible mechanisms of candidate miRNA in regulating apoptosis, oxidative stress and inflammatory response were investigated using an AKI cell model. These results should improve awareness of miR-93 regulation following AKI and inform future direction of treatments for AKI.
Materials and methods
Animal model
A total of 20 male C57BL6/J mice, aged 10–12 weeks and weighing 20±2 g, were obtained from the Shanghai SLAC Laboratory Animal Co., Ltd. All mice were housed under standard conditions (12-h light-dark cycle, 21±2°C, ~55% humidity) with free access to food and water. In the AKI group (n=10), LPS in 200 µl saline was administrated via intraperitoneal (i.p.) injection (10 mg/kg, 0.2 ml/mouse) for 24 h to induce AKI (22), while an equal volume of saline was given to control mice (n=10). Ethical approval was obtained from the Animal Experimentation Ethics Committee of the School of Medicine, Shanghai Jiao Tong University (approval no. 201912033; Shanghai, China). For miRNA microarray analysis, the sample size was three; while for reverse transcription-quantitative polymerase chain reaction (RT-qPCR), the sample size was five.
Renal function measurement
A total of 24 h after the LPS injection, mice were humanely anesthetized by i.p. injection of pentobarbital sodium (50 mg/kg; Sigma-Aldrich; Merck KGaA). Subsequently, 0.5 ml blood samples were collected from the eyeballs of mice and placed in 1.5-ml microcentrifuge tubes for 10 min at room temperature. Serum was obtained by centrifuging at 1,500 × g for 10 min at 4°C and stored at −80°C. Serum creatinine and blood urea nitrogen (BUN) were detected by using a creatinine assay kit (cat. no. DICT-500; BioAssay Systems) and a biochemical analyzer (Roche Diagnostics GmbH), respectively. The levels of kidney injury molecule-1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) were also measured using a Mouse TIM-1/KIM-1/HAVCR Quantikine ELISA Kit (cat. no. MKM100) and a Mouse Lipocalin-2/NGAL Quantikine ELISA Kit (cat. no. MLCN20; both from R&D Systems, Inc.), respectively.
Renal histopathology
A total of 24 h after the LPS injection, mice were humanely euthanized by i.p. injection of pentobarbital sodium (50 mg/kg; Sigma-Aldrich; Merck KGaA), followed by cervical dislocation. Subsequently, the right kidney tissue was taken and fixed in 4% paraformaldehyde in PBS (pH 7.4) for 20 min at 4°C, embedded in paraffin and sectioned at 4 µm for hematoxylin and eosin (H&E) staining for 10 min at room temperature. The morphological changes were observed under a light microscope (BX-FM; Olympus Corporation; 400× magnification). Tissue damage was confirmed in a blinded manner and scored as previously described (23).
MicroRNA expression profiling
Total RNA was extracted from tissues using miRNeasy mini kit (Qiagen AB). MicroRNA differential expression analysis was performed using the miRCURY LNA™ Array v. 18.0 (Exiqon; Qiagen, Inc.) as previously described (24). Briefly, total RNA was labeled using miRCURY™ Hy3™/Hy5™ power labeling kit (Exiqon; Qiagen, Inc.) and hybridized on miRCURY™ LNA Array (v 1.8.0). After washing and staining, the microarray slides were scanned in an Agilent G2565BA Microarray Scanner System (Agilent Technologies, Inc.). Scanned images were then imported into GenePix Pro 6.0 software (Molecular Devices, LLC) for grid alignment and data extraction. Differently expressed genes were then identified through fold-change (fold-change ≥2) and P-value (P<0.05). Subsequently, the miRNAs were measured by Volcano Plot filtering using GraphPad Prism 7.0 software package (GraphPad Software, Inc.). Finally, a hierarchical cluster heatmap representing expression intensity and direction was created using a method of hierarchical clustering via GeneSpring GX, version 7.3 (Agilent Technologies, Inc.).
RT-qPCR analysis
Total RNA was extracted from tissues and cells using TRIzol Reagent (Invitrogen; Thermo Fisher Scientific, Inc.). cDNA was synthesized using the PrimeScript RT reagent kit (Promega Corporation) for 1 h at 42°C. For detection of miR-93, qPCR was conducted using MicroRNAs Quantitation PCR kit (Sangon Biotech Co., Ltd.). For detection of mRNA, a SYBR Premix Ex Taq II (Takara Bio, Inc.) was used for PCR. Sequences for the primers used were as follows: MiR-93 forward, 5′-AGGCCCAAAGTGCTGTTCGT-3′ and reverse, 5′-GTGCAGGGTCCGAGGT-3′; U6 forward, 5′-GCTTCGGCAGCACATATACTAAAAT-3′ and reverse, 5′-CGCTTCACGAATTTGCGTGTCAT-3′; phosphatase and tensin homolog deleted on chromosome 10 (PTEN) forward, 5′-CCAGGACCAGAGGAAACCT-3′ and reverse, 5′-GCTAGCCTCTGGATTTGA-3′; IL-1β forward, 5′-TCTCGCAGCAGCACATCA-3′ and reverse, 5′-CACACACCAGCAGGTTAT-3′; IL-6 forward, 5′-TGGGAAATCGTGGAAATGAG-3′ and reverse, 5′-CTCTGAAGGACTCTGGCTTTG-3′; TNF-α forward, 5′-CCCGGGCTCAGCCTCTTCTCATTC-3′ and reverse, 5′-GGATCCGGTGGTTTGCTACGACGT-3′; and GAPDH forward, 5′-CGAGCCACATCGCTCAGACA-3′ and reverse, 5′-GTGGTGAAGACGCCAGTGGA-3′. U6 was used as an internal control for detecting miR-93, and GAPDH was used as an internal control for detecting PTEN. The thermocycling conditions were as follows: 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 10 min. Fold-changes in expression of each gene were calculated using the 2−∆∆Cq method (25).
Cell culture and treatment
Mouse kidney epithelial TCMK-1 cells were obtained from the American Type Culture Collection and were maintained in Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.), supplemented with 10% fetal bovine serum (FBS; Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C and 5% CO2. LPS, at concentrations of 0, 0.01, 0.1, 1 and 10 µg/ml, was used to treat TCMK-1 cells for 24 h to generate a sepsis AKI cell model (26).
Cell transfection
The miR-93 mimics (5′-CAAAGUGCUGUUCGUGCAGGUAG-3′), the mimics negative control (NC; 5′-UUCUCCGAACGUGUCACGUTT-3′), miR-93 inhibitor (5′-CUACCUGCACGAACAGCACUUUG-3′) and inhibitor NC (5′-UUGUACUACACAAAAGUACUG-3′) were purchased from Guangzhou RiboBio Co., Ltd. At 80–90% confluence, miR-93 mimics/inhibitor were transfected into TCMK-1 cells (5×105) at a final oligonucleotide concentration of 50 nmol/l. Transfection was performed using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocols. After transfection for 24 h, TCMK-1 cells were collected for subsequent experiments.
Cell proliferation
To detect cell proliferation, TCMK-1 cells were seeded onto 96-well plates (1,500 cells/well) for 24 h, and then transfected with miR-93 mimics, followed by treatment with LPS (0, 0.01, 0.1, 1 and 10 µg/ml) for 24 h at 37°C. Then, 10 µl Cell Counting Kit-8 (CCK-8) solutions (Beyotime Institute of Biotechnology) were added to cells and cells were incubated for 1.5 h at 37°C and 5% CO2. Next, the OD absorbance was detected at 450 nm by a micro-plate reader by (Infinite M200; Tecan Group, Ltd.).
Cell apoptosis
An Annexin V-FITC/PI apoptosis detection kit (Abcam) was applied to detect cells apoptosis according to the manufacturer's protocols. Briefly, 48 h after transfection, cells were centrifuged and washed with PBS, stained with Annexin V and PI for 15 min at room temperature in the dark. The results of apoptosis were measured using a FACScan flow cytometer (Beckman Coulter, Inc.) and then data were analyzed using FlowJo version 8.7.1 software (FlowJo LLC). These results showed healthy viable cells in the lower left quadrant (Q4) on the scatter plot as (FITC−/PI−). The lower right quadrant (Q3) represented the early stage apoptotic cells as (FITC+/PI−). The upper right quadrant (Q2) represented necrotic cells and late stage apoptotic cells as (FITC+/PI+). The following formula was performed to determine the apoptotic rate: Apoptotic rate=percentage of early stage apoptotic cells (Q3) + percentage of late stage apoptotic cells (Q2).
Determination of caspase 3 activity
Following cells being collected and lysed, the Caspase-3 activity was measured using the Caspase-3 assay kit (cat. no. ab252897; Abcam) according to the manufacturer's protocols. The results were determined at 450 nm using a microplate reader (Bio-Rad Laboratories, Inc.).
Detection of reactive oxygen species (ROS)
The intracellular ROS levels were tested using a total ROS detection assay kit (cat. no. K936; BioVision, Inc.), while the detection of malonaldehyde (MDA), superoxide dismutase (SOD) and glutathione peroxidase (GPx) was performed using the detection kits for SOD (cat. no. S0101), MDA (cat. no. S0131S) and GPx (cat. no. S0058; Beyotime Institute of Biotechnology), according to the manufacturer's protocols.
Measurement of IL-6, IL-1β and TNF-α
For cultured cells, the supernatant was carefully collected by centrifugation 12,000 × g for 10 min at 4°C. The concentrations of IL-6, IL-1β and TNF-α were analyzed using IL-6 (cat. no. p1330), IL-1β (cat. no. p1305) and TNF-α (cat. no. pt518) ELISA kits from Beyotime Institute of Biotechnology.
Bioinformatics analysis
miRNA target prediction tools, including PicTar version 2007 (https://pictar.mdc-berlin.de/) and TargetScan version 7.0 (http://targetscan.org/) were used to search for the putative targets of miR-93.
Luciferase assay
The luciferase reporter plasmids [wild-type (wt)-PTEN-UTR-pGL3 or mutant (mut)-PTEN-UTR-pGL3] were synthesized by Shanghai GenePharma Co., Ltd. 293T cells (8×104) (American Type Culture Collection) were co-transfected with the luciferase reporter along with miR-93 mimics/inhibitor using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.). At 48 h post-transfection, the activity of luciferase was measured using a Dual-Luciferase Reporter Assay System (Promega Corporation). Renilla luciferase expression of pRL-TK plasmids (Promega Corporation) was used for normalization.
Western blotting
Proteins were extracted from cells using RIPA lysis buffer (EMD Millipore) containing protease inhibitors and phosphatase inhibitors. In brief, the protein samples (40 µg/lane) were separated by 12% SDS-PAGE gel, and subsequently transferred to polyvinylidene difluoride membranes (EMD Millipore). Then, the membrane was blocked with 5% skimmed milk for 2 h at room temperature, followed by incubation with antibodies against PTEN (cat. no. 9559; 1:2,000), phosphorylated (p)-AKT (cat. no. 4060; 1:1,000), AKT (cat. no. 4685; 1:1,000), p-mTOR (cat. no. 5536; 1:1,000), mTOR (cat. no. 2983; 1:2,000) and β-actin (cat. no. 3700; 1:2,000) for an additional 2 h at room temperature. Next, the membranes were incubated with a goat anti-mouse HRP-conjugated secondary antibody (cat. no. 91196; 1:2,000) at room temperature for 1 h. All antibodies were obtained from Cell Signaling Technology. Detection was performed using ECL reagents (Advansta, Inc.) and blots were semi-quantified with ImageJ software (version 1.46; National Institutes of Health).
Statistical analysis
SPSS 19.0 software package (IBM Corp.) was used to analyze the data. All data are presented as the mean ± standard deviation. Comparisons between multiple groups were analyzed by one-way analysis of variance, followed by Tukey's post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
miR-93 is downregulated in kidney tissues from AKI mice
In the present study, a mouse AKI model was established, and H&E staining was used to evaluate the morphological changes of kidney tissues. As shown in Fig. 1A, substantial pathological changes were observed in the LPS group, including edema of renal TECs, tubular necrosis and inflammatory cell infiltration, accompanied by a marked increase in the renal injury scores. Subsequently, the renal function in the mouse AKI model was investigated. It was demonstrated that the levels of BUN and Cre, urine KIM-1 and urine NGAL, which are commonly used biochemical indicators for detecting renal function were also higher in the AKI group than in the control group (Fig. 1B-E). This suggested that LPS-induced AKI animal models were successfully established.
Figure 1.

miR-93 is downregulated in kidney tissues from AKI mice. (A) H&E staining was used to examine the histopathological changes of the kidney and evaluation of renal injury scores was assessed following H&E staining under an optical microscope (magnification, ×200). The levels of (B) serum BUN, (C) serum Cre, (D) urine KIM-1 and (E) urine NGAL in mice were measured using commercial kits. Data are presented as the mean ± SD of three independent experiments. **P<0.01 vs. Control group. (F) Heatmap of miRNA profiles represented the significantly regulated miRNAs. The color code in the heatmaps is linear with green as the lowest and red as the highest. (G) Volcano plot presenting the differentially expressed miRNAs. Y-axis represents log transformed P-value, and x-axis indicates the mean expression differences of miRs between AKI group and control group. |log2FoldChange| >2 was set as the cut-off criteria. (H) miR-93 expression was validated by reverse transcription-quantitative polymerase chain reaction in mice following AKI (n=10). Data are presented as the mean ± SD of three independent experiments. **P<0.01 vs. Control group. miR/miRNA, micro RNA; AKI, acute kidney injury; H&E, hematoxylin and eosin; BUN, blood urea nitrogen; Cre, creatinine; KIM-1, kidney injury molecule-1; NGAL, neutrophil gelatinase-associated lipocalin; SD, standard deviation.
To investigate the potential involvement of miRNA in AKI, microarray analysis was performed to determine miRNA expression levels in kidney tissues. It was observed that 22 miRNAs were significantly downregulated and 26 miRNAs were markedly upregulated in AKI group, compared with the control group (Fig. 1F). The volcano plot demonstrates all the differentially expressed miRNAs between the AKI group and the control group (Fig. 1G). Of these aberrant miRNAs, miR-93, miR-140 and miR-21 were decreased, while miR-182 was increased, which was consistent with the results of previous studies (14,27,28), indicating the reliability of the microarray used in the present study. Notably, miR-93 exhibited the most markedly downregulated expression in the present study. Notably, a previous study reported that the expression level of miR-93 was correlated with the severity of oxalic acid-induced AKI (29). In addition, several studies have demonstrated that miR-93 exerts anti-inflammatory and anti-apoptotic abilities in several disease models (30,31). Therefore, RT-qPCR was used to further verify the miR-93 expression level in kidney tissues of 10 AKI mice and it was observed that miR-93 expression was significantly decreased in the AKI group, compared with that in the control group (Fig. 1H). All data indicated that AKI results in miRNA aberrant expression in kidney tissues and miR-93 may serve an important role in the pathogenesis of AKI.
Overexpression of miR-93 suppresses LPS-induced renal cell apoptosis
To investigate the roles of miR-93 in AKI, mouse kidney epithelial TCMK-1 cells were applied for construction of an AKI cell model under LPS simulation (32). To begin with, expression of miR-93 in TCMK-1 cells was detected under different concentrations of LPS. It was demonstrated that miR-93 expression was dose-dependently decreased in TCMK-1 cells and was minimal at 10 µg/ml LPS treatment (Fig. 2A). Therefore, 10 µg/ml LPS was selected for the subsequent experiments, which is also consistent with a previous study (33).
Figure 2.

Overexpression of miR-93 suppresses LPS-induced renal cell apoptosis. (A) TCMK-1 cells were treated with different concentrations of LPS (0, 0.01, 0.1, 1 and 10 µg/ml) for 24 h, and the expression of miR-93 was detected by RT-qPCR. (B) The transfection efficiency of miR-93 mimics was detected by RT-qPCR. (C) miR-93 mimics were added to the cultured TCMK-1 cells for 24 h, and treated with LPS, and then cell viability was assessed by Cell Counting Kit-8 assay. (D) Activity of Caspase-3 was measured using a Caspase-3 activity assay kit. (E) Apoptosis was detected by flow cytometry. Data are presented as the mean ± standard deviation of three independent experiments. *P<0.05, **P<0.01 vs. Control group; ##P<0.01 vs. LPS group. miR, microRNA; LPS, lipopolysaccharide; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; NC, negative control.
To investigate the role of miR-93 in LPS-induced cell injury, miR-93 mimics were transfected into TCMK-1 cells. Compared with the mimics NC group, miR-93 was significantly increased in TCMK-1 cells, indicating that miR-93-overexpression was successful (Fig. 2B). CCK-8 assay demonstrated that, compared with the control group, LPS treatment resulted in a significant decrease in cell viability; however, this decrease was reversed by overexpression of miR-93 (Fig. 2C). Furthermore, it was investigated whether miR-93 modulates cell apoptosis following AKI induction. As shown in Fig. 2D, the Caspase-3 activity in the LPS group was significantly upregulated compared with that in the control group and this increase was attenuated by overexpression of miR-93. Cell apoptosis was further analyzed by flow cytometry, and the results demonstrated that LPS induced a significant increase in cell apoptosis. However, the increase in apoptosis was significantly decreased by miR-93 mimics (Fig. 2E). The aforementioned results indicated that overexpression of miR-93 alleviated cell apoptosis in the AKI cell model.
Overexpression of miR-93 suppresses LPS-induced ROS generation
It has been demonstrated that oxidative damage to tubular cells and renal tissue is associated with renal injury (34–36). Therefore, the influence of miR-93 on oxidative stress in LPS-treated TCMK-1 cells was further investigated. As shown in Fig. 3A, LPS treatment led to a marked increase in ROS generation; however, this increase was attenuated by overexpression of miR-93. Additionally, the levels of MDA, and activities of SOD and GPx were measured. It was demonstrated that LPS clearly increased the level of MDA, and decreased the activities of SOD and GPx in the LPS group, compared with the control group. However, these effects caused by LPS were reversed by miR-93 upregulation (Fig. 3B-D). All these data indicated that overexpression of miR-93 may mitigate renal damage through suppressing oxidative stress.
Figure 3.

Overexpression of miR-93 suppresses LPS-induced ROS generation. miR-93 mimics were added to the cultured TCMK-1 cells for 24 h and subjected to LPS treatment, and then cells were harvested for subsequent experiments. (A) Effects of miR-93 on the intracellular ROS levels in LPS-treated TCMK-1 cells (magnification, ×400). Effects of miR-93 on the (B) MDA, (C) SOD and (D) GPx levels in LPS-treated TCMK-1 cells. Data are presented as the mean ± standard deviation of three independent experiments. *P<0.05, **P<0.01 vs. Control group; ##P<0.01 vs. LPS group. miR, microRNA; LPS, lipopolysaccharide; ROS, reactive oxygen species; MDA, malonaldehyde; SOD, superoxide dismutase; GPx, glutathione peroxidase; NC, negative control.
Overexpression of miR-93 suppresses LPS-induced inflammatory response
A previous study has suggested that the production of pro-inflammatory cytokines in renal TECs are the main pathological features of AKI (37). Therefore, the present study further investigated the influence of miR-93 on the LPS-induced inflammatory response. As shown in Fig. 4A-C, LPS stimulation markedly promoted the mRNA levels of IL-1β, TNF-α and IL-6, compared with the control group, but these promoting effects of LPS were abolished by miR-93-overexpression. Similar results were observed in the levels of IL-1β, TNF-α and IL-6, as determined by ELISA (Fig. 4D-F). These data suggested that miR-93 suppressed the inflammatory response in LPS-treated TCMK-1 cells.
Figure 4.

Overexpression of miR-93 suppresses LPS-induced inflammatory response. miR-93 mimics were added to the cultured TCMK-1 cells 24 h prior to LPS treatment and incubated for 24 h and then cells were harvested for subsequent experiments. Effects of miR-93 on the protein levels of (A) IL-1β, (B) TNF-α and (C) IL-6 in LPS-treated TCMK-1 cells. Effects of miR-93 on mRNA levels of (D) IL-1β, (E) TNF-α and (F) IL-6 in LPS-treated TCMK-1 cells. Data are presented as the mean ± standard deviation of three independent experiments. *P<0.05, **P<0.01 vs. Control group; ##P<0.01 vs. LPS group. miR, microRNA; LPS, lipopolysaccharide; IL, interleukin; TNF, tumor necrosis factor; NC, negative control.
PTEN is a direct target of miR-93
To elucidate the molecular mechanisms involved in the protective role of miR-93 in AKI, the target genes of miR-93 were predicted using PicTar and TargetScan, and it was revealed that miR-93 may target PTEN. PTEN sequences in the 3′-UTR region of miR-93 are shown in Fig. 5A. To investigate whether miR-93/PTEN was involved in the pathogenesis of LPS-induced TCMK-1 injury, a luciferase reporter assay was performed in TCMK-1 cells to validate PTEN as a direct target of miR-93. To begin with, it was confirmed that the expression of miR-93 was significantly decreased following miR-93 inhibitor transfection in TCMK-1 cells (Fig. 5B). The dual-luciferase reporter assay demonstrated that the luciferase activity was significantly decreased when PTEN 3′-UTR wt plasmids were co-transfected with miR-93 mimics in TCMK-1 cells, but markedly increased when co-transfected with miR-93 inhibitors (Fig. 5C). However, the luciferase activity showed no obvious change following TCMK-1 co-transfection with miR-93 mimics/inhibitor and PTEN 3′-UTR mut plasmids. To investigate whether PTEN levels were regulated by miR-93, TCMK-1 cells were transfected with miR-93 mimics/inhibitor and the levels of PTEN mRNA were measured by RT-qPCR. It was demonstrated that PTEN was significantly downregulated when miR-93 was overexpressed in TCMK-1 cells, but upregulated following miR-93-knockdown (Fig. 5D). Subsequent experiments demonstrated that PTEN levels were significantly increased in the kidney tissues of the AKI group compared with that in the control group (Fig. 5E). Furthermore, whether miR-93 regulates the expression of PTEN was determined in an AKI cell model. As shown in Fig. 5E, LPS stimulation upregulated the mRNA levels of PTEN and this increase was attenuated by overexpression of miR-93, but miR-93-knockdown enhanced the LPS-induced upregulation of PTEN protein expression (Fig. 5F). All data indicated that miR-93 may exert its protective effects by targeting PTEN in the AKI cell model.
Figure 5.

PTEN is a direct target of miR-93. (A) Predicted miR-93 targeting sequence in PTEN 3′-UTR (wt PTEN 3′-UTR). Target sequences of PTEN 3′-UTR were mutated (mut PTEN 3′-UTR). (B) Expression of miR-93 was measured by RT-qPCR in TCMK-1 cells following miR-93 inhibitor transfection. **P<0.01 vs. inhibitor NC group. (C) Luciferase assay of TCMK-1 cells co-transfected with firefly luciferase constructs containing the PTEN wt or mut 3′-UTRs and miR-93 mimics, mimic NC, miR-93 inhibitor or inhibitor NC, as indicated (n=3). **P<0.01 vs. mimics NC group; ##P<0.01 vs. inhibitor NC group. (D) Expression of PTEN mRNA following transfection with miR-93 mimics or miR-93 inhibitor was measured by RT-qPCR. Data are presented as the mean ± SD of three independent experiments. **P<0.01 vs. mimics NC group; ##P<0.01 vs. inhibitor NC group. (E) PTEN expression was measured by RT-qPCR in mouse kidneys following AKI (n=5). (F) The miR-93 mimics/inhibitor and corresponding NC were added to TCMK-1 cells, followed by 10 ng/ml LPS stimulation for 24 h, and then the mRNA levels of PTEN were detected by RT-qPCR. Data are presented as the mean ± SD of three independent experiments. *P<0.05, **P<0.01 vs. Control group; ##P<0.01 vs. LPS + mimics NC group; &&P<0.01 vs. LPS + inhibitor NC group. PTEN, phosphatase and tensin homolog deleted on chromosome 10; miR, microRNA; UTR, untranslated region; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; NC, negative control; SD, standard deviation; wt, wild-type; mut, mutant; AKI, acute kidney injury; LPS, lipopolysaccharide.
miR-93 reactivates the AKT/mTOR pathway in the AKI cell model
It is well-known that PTEN is a negative regulator of AKT/mTOR pathway, which is directly associated with apoptosis (38,39). Therefore, the present study further investigated whether miR-93 affected the activation of AKT/mTOR in the AKI cell model. The results of western blotting demonstrated that the protein levels of p-AKT and p-mTOR were decreased in LPS-treated TCMK-1 cells, compared with that in the control group, but these inhibitory effects of LPS on the protein levels of p-AKT and p-mTOR were reversed by miR-93 upregulation (Fig. 6A and B). These findings suggested that miR-93 may re-activate the AKT/mTOR pathway through suppressing PTEN.
Figure 6.
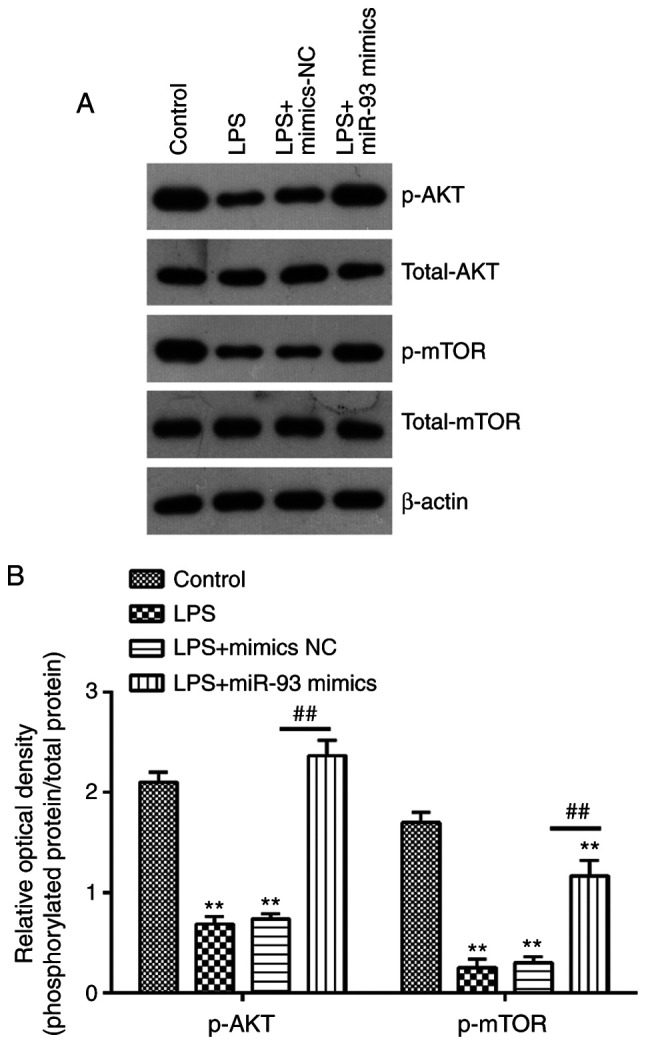
miR-93 reactivates the AKT/mTOR pathway in an acute kidney injury cell model. (A) Protein expression of AKT, p-AKT, mTOR and p-mTOR were determined by western blotting. (B) The bands were semi-quantitatively analyzed using ImageJ software, p-protein was normalized to total protein. Data are presented as the mean ± standard deviation of three independent experiments. **P<0.01 vs. Control group; ##P<0.01 vs. LPS group. miR, microRNA; p-, phosphorylated; LPS, lipopolysaccharide; NC, negative control.
Discussion
In the present study, miR-93 was revealed to be significantly downregulated in kidney tissues from an AKI mouse model. These results demonstrated that overexpression of miR-124 alleviates LPS-induced TEC cell injury by suppressing apoptosis, oxidative stress and inflammation. In addition, it was demonstrated that overexpression of miR-93 may exert protective effects against AKI by reactivating the PTEN/AKT/mTOR pathway. The results of the present study suggested that miR-93 may serve as a potential future therapeutic target for AKI treatment.
There are several AKI animal models that have been generated and widely used in research, which have provided significant information on the post-AKI pathophysiological changes and molecular mechanisms (40,41). All types of models of AKI have their own advantages in clinical difficulty, stability and feasibility, and at the same time, there are certain deficiencies in the process of modeling. The animal models of renal ischemia-reperfusion injury-induced AKI generated by temporary unilateral or bilateral clamping of renal pedicles or renal arteries have been well established and broadly used in the study of AKI pathogenesis and drug efficacy evaluation (42,43). Furthermore, the cecal ligation and puncture mouse model of sepsis is also used to investigate the pathogenesis of septic AKI (44). Of note, the LPS model differs substantially from the ischemia and maleate models in that LPS does not induce significant proximal tubule necrosis. A previous study has reported that the loss of functional TECs via inflammatory response in the pathological process of AKI (4). Notably, anti-inflammatory and anti-apoptotic therapy was confirmed to be beneficial for the treatment of sepsis-induced AKI (5,6). LPS is a classical ligand for TLR4 and mediates TLR4-dependent signal transduction to activate NF-κB, leading to an increase in inflammatory cytokine expression, including IL-1β, IL-6 and TNF-α (7). Therefore, an LPS-induced experimental model was comprehensively used to investigate and evaluate the anti-inflammatory treatment of sepsis-related AKI. Future studies should aim to further verify the results of the present study using different AKI models.
Growing evidence has revealed that miRNAs are abnormally expressed in renal tissues and are often associated with renal injury responses, including apoptosis, oxidative stress and inflammation (45–48). For example, Li et al (49) demonstrated that miR-25-overexpression may ameliorate high glucose (HG)-induced oxidative stress and apoptosis in renal TECs. Qu and Zhang (50) reported that downregulation of miR-122 alleviated renal ischemic reperfusion injury through inhibiting apoptosis and ROS generation in rat renal TECs. Song et al (27) reported that miR-21-overexpression protects against AKI by preventing epithelial cell apoptosis. Therefore, clarification of the role and regulation of miRNAs in AKI may generate a potential therapeutic strategy for AKI. In the present study, using an miRNA microarray revealed that a large set of miRNAs were abnormally expressed; in particular, miR-93 was identified as the most downregulated miRNA in kidney tissues from AKI mice, suggesting that miR-93 may be involved in the development of AKI.
Several studies have demonstrated that miR-93 exerts protective effects in various injury models. For example, Ma et al (51) reported that miR-93 decreased the cardiac microvascular endothelial cell injury via inactivation of the NF-κB signaling pathway. Yan et al (52) demonstrated that miR-93 inhibition ameliorated oxygen-glucose deprivation/reoxygenation injury in cardiomyocytes by targeting Nrf2. Xiong et al (30) reported that overexpression of miR-93 alleviated hepatic injury by suppressing apoptosis and inflammatory response. In a clinical study, miR-93 has been proposed to serve as a biomarker for early detection of AKI (29). However, the regulatory roles of miR-93 in AKI remain unknown. In the present study, an LPS-induced TCMK-1 cell model revealed that overexpression of miR-93 suppressed the apoptosis, oxidative stress and inflammatory response. Taken together, these data revealed that miR-93 upregulation has a protective effect in AKI via suppressing the apoptosis, oxidative stress and inflammatory response, indicating that miR-93 may be a potential candidate target for AKI therapy. However, the molecular mechanism requires further clarification.
It is recognized that identifying the downstream target gene(s) is a key to elucidating the pathophysiological role of an miRNA. In the present study, using computational algorithms, PTEN was identified as a target of miR-93. PTEN has been found to serve important roles in renal injury (53). For example, Li et al (49) demonstrated that overexpression of PTEN promoted the apoptosis of renal TECs in a HG-induced cell damage model. Zhang et al (54) reported that activation of PTEN/AKT signaling alleviated tubular cell apoptosis, thereby protecting animals from Cis-induced AKI. Notably, PTEN has been reported to be a direct target of miR-93 in myocardial ischemia/reperfusion (I/R) injury (55). The present study demonstrated that PTEN is also a direct target of miR-93 in renal TECs and its protein expression was negatively regulated by miR-93 in TCMK-1 cells and in kidney tissues of AKI mice. These results suggested that miR-93 may exert its protective effect in AKI via reactivating the PTEN/AKT/mTOR pathway.
Numerous signaling pathways are downstream of PTEN. One important pathway, the endogenous PI3K/AKT pathway, regulates negative feedback in response to LPS stimuli (56,57). The PI3K/AKT/mTOR pathway is ubiquitous in cells and is involved in the modulation of a series of physiological activities associated with AKI, including cell apoptosis, oxidative indices and inflammatory response (58,59). A previous study has demonstrated that AKT/mTOR signaling activation markedly attenuated inflammation, mitochondrial damage and apoptosis caused by ischemia/reperfusion (I/R)-induced AKI (60). Zhang et al (61) demonstrated that activation of the PI3K/AKT/mTOR pathway may prevent the apoptosis and inflammation in tubular epithelial cells following I/R injury. Of note, miR-93 may protect against I/R-induced cardiomyocyte apoptosis by inhibiting the PTEN/AKT/mTOR signaling pathway (55). Therefore, the present study aimed to investigate whether miR-93 affects the PTEN/AKT/mTOR signaling pathway in an AKI cell model. In the present study, it was demonstrated that overexpression of miR-93 decreased the increased protein levels of p-AKT and p-mTOR induced by LPS in TCMK-1 cells. Taken together, these results suggested that miR-93 may exert its protective effects on AKI by promoting the PTEN/AKT/mTOR signaling pathway.
However, there are certain limitations to the present study. For example, only the AKT/mTOR signaling pathway was investigated, while other pathways may also be associated with the pathogenesis of AKI. Additionally, the number of experimental animals was limited. In the future, further systematic and in-depth studies investigating the pathogenesis of AKI will be conducted.
In conclusion, the results of the present study demonstrated that upregulation of miR-93 protects against LPS-induced TEC apoptosis, oxidative stress and inflammatory response in an in vitro model of AKI. The underlying molecular mechanism is mediated via promoting the activation of the PTEN/AKT/mTOR pathway. These findings may provide a novel direction for the treatment of AKI.
Acknowledgements
Not applicable.
Funding Statement
The present study was supported by the Clinical Innovation and Multi-discipline Integrated Medical Construction Project of South Campus, Renji Hospital, School of Medicine, Shanghai Jiaotong University (grant no. 2014MDT02) and Cultivating Funds of Medicine, Shanghai Jiaotong University (grant no. 2017PYQB05).
Funding
The present study was supported by the Clinical Innovation and Multi-discipline Integrated Medical Construction Project of South Campus, Renji Hospital, School of Medicine, Shanghai Jiaotong University (grant no. 2014MDT02) and Cultivating Funds of Medicine, Shanghai Jiaotong University (grant no. 2017PYQB05).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
YZ, MZ, SL, JL and ZN performed all the experiments and collected the data. YZ, HC and WZ confirm the authenticity of all the raw data. YZ, HC and WZ conceived and designed the study. YZ HC and WZ wrote the main manuscript and analyzed the data. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
The present study was approved by the Animal Experimentation Ethics Committee of the School of Medicine, Shanghai Jiao Tong University (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moreth K, Frey H, Hubo M, Zeng-Brouwers J, Nastase MV, Hsieh LT, Haceni R, Pfeilschifter J, Iozzo RV, Schaefer L. Biglycan-triggered TLR-2- and TLR-4-signaling exacerbates the pathophysiology of ischemic acute kidney injury. Matrix Biol. 2014;35:143–151. doi: 10.1016/j.matbio.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7:189–200. doi: 10.1038/nrneph.2011.16. [DOI] [PubMed] [Google Scholar]
- 4.Scharnweber T, Alhilali L, Fakhran S. Contrast-induced acute kidney injury: Pathophysiology, manifestations, prevention, and management. Magn Reson Imaging Clin N Am. 2017;25:743–753. doi: 10.1016/j.mric.2017.06.012. [DOI] [PubMed] [Google Scholar]
- 5.Peng X, Wang Y, Li H, Fan J, Shen J, Yu X, Zhou Y, Mao H. ATG5-mediated autophagy suppresses NF-κB signaling to limit epithelial inflammatory response to kidney injury. Cell Death Dis. 2019;10:253. doi: 10.1038/s41419-019-1483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhi D, Zhang M, Lin J, Liu P, Duan M. GPR120 ameliorates apoptosis and inhibits the production of inflammatory cytokines in renal tubular epithelial cells. Inflammation. 2021;44:493–505. doi: 10.1007/s10753-020-01346-2. [DOI] [PubMed] [Google Scholar]
- 7.Souza AC, Volpini RA, Shimizu MH, Sanches TR, Camara NO, Semedo P, Rodrigues CE, Seguro AC, Andrade L. Erythropoietin prevents sepsis-related acute kidney injury in rats by inhibiting NF-κB and upregulating endothelial nitric oxide synthase. Am J Physiol Renal Physiol. 2012;302:F1045–F1054. doi: 10.1152/ajprenal.00148.2011. [DOI] [PubMed] [Google Scholar]
- 8.Xie C, Liu L, Wang Z, Xie H, Feng Y, Suo J, Wang M, Shang W, Feng G. Icariin improves sepsis-induced mortality and acute kidney injury. Pharmacology. 2018;102:196–205. doi: 10.1159/000487955. [DOI] [PubMed] [Google Scholar]
- 9.Wang C, Sun H, Song Y, Ma Z, Zhang G, Gu X, Zhao L. Pterostilbene attenuates inflammation in rat heart subjected to ischemia-reperfusion: Role of TLR4/NF-kappaB signaling pathway. Int J Clin Exp Med. 2015;8:1737–1746. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Thomson DW, Dinger ME. Endogenous microRNA sponges: Evidence and controversy. Nat Rev Genet. 2016;17:272–283. doi: 10.1038/nrg.2016.20. [DOI] [PubMed] [Google Scholar]
- 11.Dragomir MP, Knutsen E, Calin GA. SnapShot: Unconventional miRNA functions. Cell. 2018;174:1038–1038.e1. doi: 10.1016/j.cell.2018.07.040. [DOI] [PubMed] [Google Scholar]
- 12.Ren GL, Zhu J, Li J, Meng XM. Noncoding RNAs in acute kidney injury. J Cell Physiol. 2019;234:2266–2276. doi: 10.1002/jcp.27203. [DOI] [PubMed] [Google Scholar]
- 13.Bhatt K, Mi QS, Dong Z. MicroRNAs in kidneys: Biogenesis, regulation, and pathophysiological roles. Am J Physiol Renal Physiol. 2011;300:F602–F610. doi: 10.1152/ajprenal.00727.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao W, Fu Z, Zou Y, Wen D, Ma H, Zhou F, Chen Y, Zhang M, Zhang W. MicroRNA-140-5p attenuated oxidative stress in Cisplatin induced acute kidney injury by activating Nrf2/ARE pathway through a Keap1-independent mechanism. Exp Cell Res. 2017;360:292–302. doi: 10.1016/j.yexcr.2017.09.019. [DOI] [PubMed] [Google Scholar]
- 15.Lan YF, Chen HH, Lai PF, Cheng CF, Huang YT, Lee YC, Chen TW, Lin H. MicroRNA-494 reduces ATF3 expression and promotes AKI. J Am Soc Nephrol. 2012;23:2012–2023. doi: 10.1681/ASN.2012050438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu R, Wu Y, Yang L, Deng Y, Chen D. Value of serum level of microRNA-494 in predicting prognosis of acute renal injury after cardiac surgery in children. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2019;31:1469–1473. doi: 10.3760/cma.j.issn.2095-4352.2019.12.007. (In Chinese) [DOI] [PubMed] [Google Scholar]
- 17.Wang S, Zhang Z, Wang J, Miao H. MiR-107 induces TNF-α secretion in endothelial cells causing tubular cell injury in patients with septic acute kidney injury. Biochem Biophys Res Commun. 2017;483:45–51. doi: 10.1016/j.bbrc.2017.01.013. [DOI] [PubMed] [Google Scholar]
- 18.Lv LL, Feng Y, Wu M, Wang B, Li ZL, Zhong X, Wu WJ, Chen J, Ni HF, Tang TT, et al. Exosomal miRNA-19b-3p of tubular epithelial cells promotes M1 macrophage activation in kidney injury. Cell Death Differ. 2020;27:210–226. doi: 10.1038/s41418-019-0349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang L, Liu XQ, Ma Q, Yang Q, Gao L, Li HD, Wang JN, Wei B, Wen J, Li J, et al. hsa-miR-500a-3P alleviates kidney injury by targeting MLKL-mediated necroptosis in renal epithelial cells. FASEB J. 2019;33:3523–3535. doi: 10.1096/fj.201801711R. [DOI] [PubMed] [Google Scholar]
- 20.Guo Y, Ni J, Chen S, Bai M, Lin J, Ding G, Zhang Y, Sun P, Jia Z, Huang S, et al. MicroRNA-709 mediates acute tubular injury through effects on mitochondrial function. J Am Soc Nephrol. 2018;29:449–461. doi: 10.1681/ASN.2017040381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan Y, Ma Z, Zhu J, Zeng M, Liu H, Dong Z. MiR-214 represses mitofusin-2 to promote renal tubular apoptosis in ischemic acute kidney injury. Am J Physiol Renal Physiol. 2020;318:F878–F887. doi: 10.1152/ajprenal.00567.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, Zhang X, Xu J, Zhao Y, Bao J, Zheng Z, Han J. AZD4547 attenuates lipopolysaccharide-induced acute kidney injury by inhibiting inflammation: The Role of FGFR1 in renal tubular epithelial cells. Drug Des Devel Ther. 2020;14:833–844. doi: 10.2147/DDDT.S224343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z, He L, Tan J, Liu Y, Liu H, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy. 2018;14:880–897. doi: 10.1080/15548627.2017.1405880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mei LL, Wang WJ, Qiu YT, Xie XF, Bai J, Shi ZZ. MiR-125b-5p functions as a tumor suppressor gene partially by regulating HMGA2 in esophageal squamous cell carcinoma. PLoS One. 2017;12:e0185636. doi: 10.1371/journal.pone.0185636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Xu J, Ma X, Yu K, Wang R, Wang S, Liu R, Liu H, Gao H, Yu K, Wang C. Lactate up-regulates the expression of PD-L1 in kidney and causes immunosuppression in septic acute renal injury. J Microbiol Immunol Infect. 2019;(S1684-1182):30168–30169. doi: 10.1016/j.jmii.2019.10.006. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 27.Song N, Zhang T, Xu X, Lu Z, Yu X, Fang Y, Hu J, Jia P, Teng J, Ding X. MiR-21 protects against ischemia/reperfusion-induced acute kidney injury by preventing epithelial cell apoptosis and inhibiting dendritic cell maturation. Front Physiol. 2018;9:790. doi: 10.3389/fphys.2018.00790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Ma Y, Chen B, Shi J. MiR-182 enhances acute kidney injury by promoting apoptosis involving the targeting and regulation of TCF7L2/Wnt/β-catenins pathway. Eur J Pharmacol. 2018;831:20–27. doi: 10.1016/j.ejphar.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 29.Shihana F, Joglekar MV, Raubenheimer J, Hardikar AA, Buckley NA, Seth D. Circulating human microRNA biomarkers of oxalic acid-induced acute kidney injury. Arch Toxicol. 2020;94:1725–1737. doi: 10.1007/s00204-020-02679-5. [DOI] [PubMed] [Google Scholar]
- 30.Xiong L, Yu KH, Zhen SQ. MiR-93 blocks STAT3 to alleviate hepatic injury after ischemia-reperfusion. Eur Rev Med Pharmacol Sci. 2018;22:5295–5304. doi: 10.26355/eurrev_201808_15729. [DOI] [PubMed] [Google Scholar]
- 31.Wang P, Liang X, Lu Y, Zhao X, Liang J. MicroRNA-93 downregulation ameliorates cerebral ischemic injury through the Nrf2/HO-1 defense pathway. Neurochem Res. 2016;41:2627–2635. doi: 10.1007/s11064-016-1975-0. [DOI] [PubMed] [Google Scholar]
- 32.Shen Y, Yu J, Jing Y, Zhang J. MiR-106a aggravates sepsis-induced acute kidney injury by targeting THBS2 in mice model. Acta Cir Bras. 2019;34:e201900602. doi: 10.1590/s0102-865020190060000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Y, Wei SW, Ding A, Zhu WP, Mai MF, Cui TX, Yang H, Zhang H. The long noncoding RNA ANRIL promotes cell apoptosis in lipopolysaccharide-induced acute kidney injury mediated by the TLR4/nuclear factor-kappa B pathway. Kidney Blood Press Res. 2020;45:209–221. doi: 10.1159/000505154. [DOI] [PubMed] [Google Scholar]
- 34.Paller MS, Hoidal JR, Ferris TF. Oxygen free radicals in ischemic acute renal failure in the rat. J Clin Invest. 1984;74:1156–1164. doi: 10.1172/JCI111524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baliga R, Ueda N, Walker PD, Shah SV. Oxidant mechanisms in toxic acute renal failure. Drug Metab Rev. 1999;31:971–997. doi: 10.1081/DMR-100101947. [DOI] [PubMed] [Google Scholar]
- 36.Brezniceanu ML, Lau CJ, Godin N, Chénier I, Duclos A, Ethier J, Filep JG, Ingelfinger JR, Zhang SL, Chan JS. Reactive oxygen species promote caspase-12 expression and tubular apoptosis in diabetic nephropathy. J Am Soc Nephrol. 2010;21:943–954. doi: 10.1681/ASN.2009030242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li GS, Chen XL, Zhang Y, He Q, Wang F, Hong DQ, Zhang P, Pu L, Zhang Y, Yang XC, Wang L. Malnutrition and inflammation in acute kidney injury due to earthquake-related crush syndrome. BMC Nephrol. 2010;11:4. doi: 10.1186/1471-2369-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang P, Peairs JJ, Tano R, Jaffe GJ. Oxidant-mediated Akt activation in human RPE cells. Invest Ophthalmol Vis Sci. 2006;47:4598–4606. doi: 10.1167/iovs.06-0140. [DOI] [PubMed] [Google Scholar]
- 39.Byeon SH, Lee SC, Choi SH, Lee HK, Lee JH, Chu YK, Kwon OW. Vascular endothelial growth factor as an autocrine survival factor for retinal pigment epithelial cells under oxidative stress via the VEGF-R2/PI3K/Akt. Invest Ophthalmol Vis Sci. 2010;51:1190–1197. doi: 10.1167/iovs.09-4144. [DOI] [PubMed] [Google Scholar]
- 40.Skrypnyk NI, Voziyan P, Yang H, de Caestecker CR, Theberge MC, Drouin M, Hudson B, Harris RC, de Caestecker MP. Pyridoxamine reduces postinjury fibrosis and improves functional recovery after acute kidney injury. Am J Physiol Renal Physiol. 2016;311:F268–F277. doi: 10.1152/ajprenal.00056.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zager RA, Johnson AC, Becker K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and ‘end-stage’ kidney disease. Am J Physiol Renal Physiol. 2011;301:F1334–F1345. doi: 10.1152/ajprenal.00431.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gall JM, Wong V, Pimental DR, Havasi A, Wang Z, Pastorino JG, Bonegio RG, Schwartz JH, Borkan SC. Hexokinase regulates Bax-mediated mitochondrial membrane injury following ischemic stress. Kidney Int. 2011;79:1207–1216. doi: 10.1038/ki.2010.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee HT, Park SW, Kim M, Ham A, Anderson LJ, Brown KM, D'Agati VD, Cox GN. Interleukin-11 protects against renal ischemia and reperfusion injury. Am J Physiol Renal Physiol. 2012;303:F1216–F1224. doi: 10.1152/ajprenal.00220.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351:159–169. doi: 10.1056/NEJMra032401. [DOI] [PubMed] [Google Scholar]
- 45.Saikumar J, Hoffmann D, Kim TM, Gonzalez VR, Zhang Q, Goering PL, Brown RP, Bijol V, Park PJ, Waikar SS, Vaidya VS. Expression, circulation, and excretion profile of microRNA-21, −155, and −18a following acute kidney injury. Toxicol Sci. 2012;129:256–267. doi: 10.1093/toxsci/kfs210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szeto CC, Ching-Ha KB, Ka-Bik L, Mac-Moune LF, Cheung-Lung CP, Gang W, Kai-Ming C, Kam-Tao LP. Micro-RNA expression in the urinary sediment of patients with chronic kidney diseases. Dis Markers. 2012;33:137–144. doi: 10.1155/2012/842764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen W, Ruan Y, Zhao S, Ning J, Rao T, Yu W, Zhou X, Liu C, Qi Y, Cheng F. MicroRNA-205 inhibits the apoptosis of renal tubular epithelial cells via the PTEN/Akt pathway in renal ischemia-reperfusion injury. Am J Transl Res. 2019;11:7364–7375. [PMC free article] [PubMed] [Google Scholar]
- 48.Wu H, Huang T, Ying L, Han C, Li D, Xu Y, Zhang M, Mou S, Dong Z. MiR-155 is involved in renal ischemia-reperfusion injury via direct targeting of FoxO3a and regulating renal tubular cell pyroptosis. Cell Physiol Biochem. 2016;40:1692–1705. doi: 10.1159/000453218. [DOI] [PubMed] [Google Scholar]
- 49.Li H, Zhu X, Zhang J, Shi J. MicroRNA-25 inhibits high glucose-induced apoptosis in renal tubular epithelial cells via PTEN/AKT pathway. Biomed Pharmacother. 2017;96:471–479. doi: 10.1016/j.biopha.2017.10.019. [DOI] [PubMed] [Google Scholar]
- 50.Qu XH, Zhang K. MiR-122 regulates cell apoptosis and ROS by targeting DJ-1 in renal ischemic reperfusion injury rat models. Eur Rev Med Pharmacol Sci. 2018;22:8830–8838. doi: 10.26355/eurrev_201812_16651. [DOI] [PubMed] [Google Scholar]
- 51.Ma SX, Bai ZF, Wang W, Wu HY. Effects of microrna-93 on mouse cardiac microvascular endothelial cells injury and inflammatory response by mediating SPP1 through the NF-ΚB pathway. J Cell Biochem. 2019;120:2847–2858. doi: 10.1002/jcb.26567. [DOI] [PubMed] [Google Scholar]
- 52.Yan LJ, Fan XW, Yang HT, Wu JT, Wang SL, Qiu CG. MiR-93 inhibition ameliorates OGD/R induced cardiomyocyte apoptosis by targeting Nrf2. Eur Rev Med Pharmacol Sci. 2017;21:5456–5461. doi: 10.26355/eurrev_201712_13935. [DOI] [PubMed] [Google Scholar]
- 53.Gao S, Zhu Y, Li H, Xia Z, Wu Q, Yao S, Wang T, Yuan S. Remote ischemic postconditioning protects against renal ischemia/reperfusion injury by activation of T-LAK-cell-originated protein kinase (TOPK)/PTEN/Akt signaling pathway mediated anti-oxidation and anti-inflammation. Int Immunopharmacol. 2016;38:395–401. doi: 10.1016/j.intimp.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 54.Zhang W, Chen C, Jing R, Liu T, Liu B. Remote ischemic preconditioning protects cisplatin-induced acute kidney injury through the PTEN/AKT signaling pathway. Oxid Med Cell Longev. 2019;2019:7629396. doi: 10.1155/2019/7629396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ke ZP, Xu P, Shi Y, Gao AM. MicroRNA-93 inhibits ischemia-reperfusion induced cardiomyocyte apoptosis by targeting PTEN. Oncotarget. 2016;7:28796–28805. doi: 10.18632/oncotarget.8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhong J, Qiu X, Yu Q, Chen H, Yan C. A novel polysaccharide from Acorus tatarinowii protects against LPS-induced neuroinflammation and neurotoxicity by inhibiting TLR4-mediated MyD88/NF-κB and PI3K/Akt signaling pathways. Int J Biol Macromol. 2020;163:464–475. doi: 10.1016/j.ijbiomac.2020.06.266. [DOI] [PubMed] [Google Scholar]
- 57.Liu F, Huang X, He JJ, Song C, Peng L, Chen T, Wu BL. Plantamajoside attenuates inflammatory response in LPS-stimulated human gingival fibroblasts by inhibiting PI3K/AKT signaling pathway. Microb Pathog. 2019;127:208–211. doi: 10.1016/j.micpath.2018.11.034. [DOI] [PubMed] [Google Scholar]
- 58.Meng L, Li L, Lu S, Li K, Su Z, Wang Y, Fan X, Li X, Zhao G. The protective effect of dexmedetomidine on LPS-induced acute lung injury through the HMGB1-mediated TLR4/NF-κB and PI3K/Akt/mTOR pathways. Mol Immunol. 2018;94:7–17. doi: 10.1016/j.molimm.2017.12.008. [DOI] [PubMed] [Google Scholar]
- 59.Liu HB, Meng QH, Huang C, Wang JB, Liu XW. Nephroprotective effects of polydatin against ischemia/reperfusion injury: A role for the PI3K/Akt signal pathway. Oxid Med Cell Longev. 2015;2015:362158. doi: 10.1155/2015/362158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yingjie K, Haihong Y, Lingwei C, Sen Z, Yuanting D, Shasha C, Liutong P, Ying W, Min Z. Apoptosis repressor with caspase recruitment domain deficiency accelerates ischemia/reperfusion (I/R)-induced acute kidney injury by suppressing inflammation and apoptosis: The role of AKT/mTOR signaling. Biomed Pharmacother. 2019;112:108681. doi: 10.1016/j.biopha.2019.108681. [DOI] [PubMed] [Google Scholar]
- 61.Zhang G, Wang Q, Wang W, Yu M, Zhang S, Xu N, Zhou S, Cao X, Fu X, Ma Z, et al. Tempol protects against acute renal injury by regulating PI3K/Akt/mTOR and GSK3β signaling cascades and afferent arteriolar activity. Kidney Blood Press Res. 2018;43:904–913. doi: 10.1159/000490338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.