

Abstract
Purpose:
The ruthenium complex cis-[Ru(H-dcbpy−)2(Cl)(NO)] (DCBPY) is a nitric oxide (NO) donor and studies suggested that the ruthenium compounds can inactivate O2−. The aim of this study is to test if DCBPY can revert and/or prevent the endothelial dysfunction.
Methods:
Normotensive (2K) and hypertensive (2K-1C) wistar rats were used. To vascular reactivity study, thoracic aortas were isolated, rings with intact endothelium were incubated with: DCBPY: 0.1; 1 and 10μM, DCBPY plus hydroxocobalin (NO scavenger) or tempol during 30 minutes, and concentration effect curves to acetylcholine were performed. The potency values (pD2) and maximum effect (ME) were analyzed. The O2− was generated using hypoxantine xantine oxidase and the reduction of cytochrome c, NO consumption by O2− and the effect in avoid NO consumption was measured.
Results:
In 2K-1C DCBPY at 0.1; 1 or 10μM improved the relaxation endothelium dependent induced by acetylcholine in aortic rings compared to control 2K-1C, and also improved ME. In rings from 2K incubation with DCBPY (0.1; 1.0 and 10 μM) did not change pD2 or ME. Incubation with 0.1 μM of DCBPY plus hydroxocobalamin did not modify the potency and ME in 2K-1C compared to DCBPY (0.1 μM). DCBPY and SOD inhibits the reduction of cytochrome c and inhibited the NO consumption by O2−, showing that O2− has been removed from the solution.
Conclusion:
Our results suggest that DCBPY at a lower concentration (0.1 μM) is not an NO generator, but can inactivate superoxide and improves the endothelial function.
INTRODUTION
Endothelial dysfunction involves decreased production or bioavailability of endogenous nitric oxide (NO) and it is associated with hypertension [1, 2] as well as other cardiovascular diseases [3–6].
Nitric oxide is involved in multiple physiological and pathophysiological processes which has promoted a large amount of research involved to development of drugs that are able to modulate NO concentration in biological environment, for therapeutic purpose [7], including NO donors.
NO donors are pharmacological active substances that in-vivo or in-vitro release NO. Organic nitrates (eg, glyceryltrinitrate GTN) and sodium nitroprusside (SNP) have been used as therapeutic agents, which presents an important vasodilation effect [8]. However, despite its vasodilation effect, GNT causes headache and tolerance, while SNP releases highly cytotoxic cyanide [9–10].
Exogenous NO donors agents based on ruthenium-derived metal nitrosyl complexes have been developed as strategy to reduce side effects and cytotoxicity. They have not displayed any toxic effects and are able to induce vascular relaxation and decrease blood pressure in normotensive and hypertensive rats [11–15]. Rodrigues et al [16] showed that the compound cis-[Ru(H-dcbpy_)2(Cl)(NO)] (DCBPY) is an NO generator that promotes relaxation of rat aortic rings by increasing the cytosolic NO concentration and reduce the cytosolic Ca2+ concentration [Ca2+]c in rat aortic rings.
Hypertension is associated with impaired endothelial function that includes reduced production and/or NO release. Previous studies have reported that in aortas from renal hypertensive rats, the NO-dependent relaxation endothelium dependent [17–18] or endothelium-independent is impaired, due to several factors such as caveolae number alteration [19,14], increased production of superoxide anion among others [20–21]. An antioxidant action as well as superoxide anion scavenger action has been attributed to ruthenium compounds [22]. Thus, a compound that could remove superoxide anion and release NO would be desired to improve endothelial function in hypertension.
In this context, this study tested the hypothesis that the ruthenium DCBPY is able to revert the endothelial dysfunction by inactivate superoxide anion in aortas from hypertensive rats.
MATERIAL AND METHODS
Materials
Superoxide dismutase (SOD), Diethyltriamine NONOate (DETA/NO) were acquired from Sigma. The c DCBPY was synthesized in our laboratory, as described by Cicillini et al. (2009). Experiments carried out for cis-[Ru(H-dcbpy)2(Cl)(NO)] evidenced that the compound was converted to cis-[Ru(H-dcbpy−)2(Cl)(NO2−)] (Ru-NO2−) after its solubilization at pH 7.4 [16]. The conversion was complete after 150 min. Thus, the Ru-NO2− (DCBPY) complex was employed in the present work.
Experimental animals
Experimental protocols followed standards and policies of the Animal Care and Use Committee of the Federal University of São Carlos, and was approved by this committee (Protocol number: 012/13). Renovascular hypertension was induced in rats following the two kidney one clip (2K-1C) Goldblatt model [23]. Briefly, male Wistar rats (180–200 g) were anesthetized with tribromoethanol (2.5 mg/kg, i.p.) and after a midline laparotomy a silver clip with an internal diameter of 0.20 mm was placed around the left renal artery. Normotensive two-kidney rats (2K) were only submitted to laparotomy. Animals were maintained on standard rat chow with a 12 h light/dark cycle and given free access to both food (standard rat chow) and water. The systolic blood pressure (SBP) was measured weekly in non-anesthetized animals by an indirect tail-cuff method (MLT125R pulse transducer/pressure cuff coupled to the PowerLab 4/S analog-to-digital converter; AD Instruments Pty Ltd., Castle Hill, Australia) and rats were considered hypertensive when the SBP was higher than 160 mmH, six weeks after surgery.
Vascular reactivity study
Rats were killed by decapitation six weeks after surgery, and the thoracic aortas were isolated and placed in a Krebs solution. Aortas were carefully dissected and mounted as ring preparations (≅4 mm in length) and placed in bath chambers (5 mL) containing Krebs solution at 37° C, continuously bubbled with 95% O2 and 5% CO2, pH 7.4, in an isometric Mulvany-Halpern myograph (model 610 DMT-USA, Marietta, GA) and recorded by a PowerLab8/SP data acquisition system (AD Instruments Pty Ltd., Colorado Springs, CO). The aortic rings were submitted to a tension of 1.5 g, which was readjusted every 15 min during a 60 min equilibration period before addition of the given drug. Endothelial integrity was assessed by the degree of relaxation induced by 1μmol/L acetylcholine in the presence of contractile tone induced by phenylephrine (0.1μmol/l). The ring was discarded if relaxation with acetylcholine was lower than 80% in 2K and 60% in 2K-1C rat aortas. After the endothelial integrity test, aortic rings were pre-contracted with phenylephrine (0.1μM). When the plateau was reached, concentration–effect curves to acetylcholine (0.1nM to 0.1mM) were constructed in the 2K and 2K-1C aortic rings. The potency (pD2) and the maximum relaxant effect (ME) were evaluated. Aortic rings from 2K and 2K-1C received different treatment for 30 min, including: DCBPY (at concentrations: 0.1; 1.0 and 10μM); DETA-NO (0.1 μM); tempol (1mM). In addition, aortic rings were incubated for 10 min with hydroxicobalamin (100 μM) followed by addition of DCBPY (0.1 μM). After incubation period, aortic rings were washed 3 times to remove drugs and concentration–effect curves to acetylcholine were constructed in aortic rings, pre-contracted with phenylephrine (0.1μM). Control responses were obtained in experiments where DCBPY, DETA-NO and tempol were replaced by PBS.
Detection of NO° by electrode
An electrode selective for NO coupled with a permeable gas membrane was employed (Apollo 4000-Free Radical Analyzer, World Precision Instrument, Sarasota, FL, USA). The electrode was suspended in phosphate buffer solution at pH 7.4 (2 ml) and kept at 37° C, under constant stirring. The electrode was calibrated by using an NO donor PROLI NONOate. The NO donor diethyltryamine NONOate (DETA/NO) was selected for the assays because it can spontaneously release NO in a pH-dependent manner. The NONOate was prepared in basic solution 10 mM NaOH, in order to maintain its stability. DETA/NO (100 μM) was added to the PBS solution and the experiments were initiated when the current was stable, as verified on the recording (data not included).
To the superoxide (O2−)-scavenging effect of DCBPY, O2− was generated at 37° C by using 500 μM hypoxantine (HX) and 5mU xantine oxidase (XO) (Figure 9). To this end, HX was firstly added to the solution, followed by DETA/NO (100 μM). When the current reached a stable value, XO was added, in order to promote consumption of NO, and then DCBPY (12.5, 25, or 50 μM) or SOD (10, 20, or 30 μM) was added, to consume the superoxide.
Ultraviolet-visible (UV-Vis) spectra
The UV-Vis spectra were recorded on a Hitachi U-30501 spectrophotometer. To verify the superoxide (O2−) scavenger effect of DCBPY, O2− was generated at 37° C, pH 7.4 by using 500 μM hypoxanthine (HX) and 5mU xanthine oxidase (XO). The formation of O2− was spectrophotometrically detected by measuring the reduction of cytochrome c at 550 nm. The inhibition of the reduction of cytochrome c was determined in the presence of DCBPY (1.25, 2.5, 12.5, or 25 μM) or SOD (20 μM). The difference in absorbance (AU) was quantified, and the latter value was employed for statistical analysis.
Cell Culture
HUVEC were culture in DMEM (Dulbecco Modified Earle’s balanced salt solution) medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 units/ml) and streptomycin (100 μg/ml). Cells were grown in humidified incubator containing 5% CO2 at 37° C. For NO production experiments, cells were washed and treated with 0.1 μM of DCBPY or DETA-NO for 30 min. NO production was determined using DAF-2 DA cell membrane–permeable NO sensitive fluorescent dye, which converts to DAF-2 and reacts with NO to form a triazole form fluorescente DAF-2T [24]. The increase in fluorescence intensity was monitored with a fluorescence microplate reader (SpectraMax GeminiXS, Molecular Devices) at 485 nm excitation and 515 nm emission for 4 hours at 37° C. Alternatively, NO production was examined in DAF-stained HUVEC in fluorescence microscope (Axiovert, Zeiss, Oberkochen, Germany) at 40X objective lens magnification. The fluorescence intensity (FI) value were obtained from 3 different experiments (n=3) to all protocols performed.
STATISTICAL ANALYSIS
Data are expressed as mean ± S.E.M. In each set of experiments, n indicates the number of rats studied. Statistical analysis of the results was performed by using GraphPad Prism version 3.0. Statistical significance was tested by one-way ANOVA followed by Newman–Keuls post hoc analysis. Values of p < 0.05 were considered significant
Drugs and chemicals
Acetylcholine, phenylephrine, DETA-NO, TEMPOL, hydroxocobalamin, PROLI NONOate, were purchased from Sigma–Aldrich (St.Louis, MO, USA).
RESULTS
Systolic blood pressure
Systolic blood pressure from normotensive (2K) and hypertensive (2K-1C) rats were measured sex week after surgery, by the tail-cuff method. The systolic blood pressure was higher in 2K-1C rats (190± 4.5 mm Hg, n=20) as compared with 2K rats (112±6.1 mm Hg, n=24, p<0.001).
Vascular reactivity studies
As shown in the Figure 1, the endothelium dependent relaxation induced by acetylcholine was impaired in aortas from hypertensive rats. The potency of acetylcholine in inducing relaxation was higher in 2K (pD2: 7.57±0.12; p<0.05 n=8) compared to 2K-1C (pD2: 6.57±0.08). In addition, the maximum relaxant effect was greater in 2K (93.6 ± 3.1%; p<0.05 n=8) than in 2K-1C (78.7 ± 2.3%) rat aortas.
Figure 1.
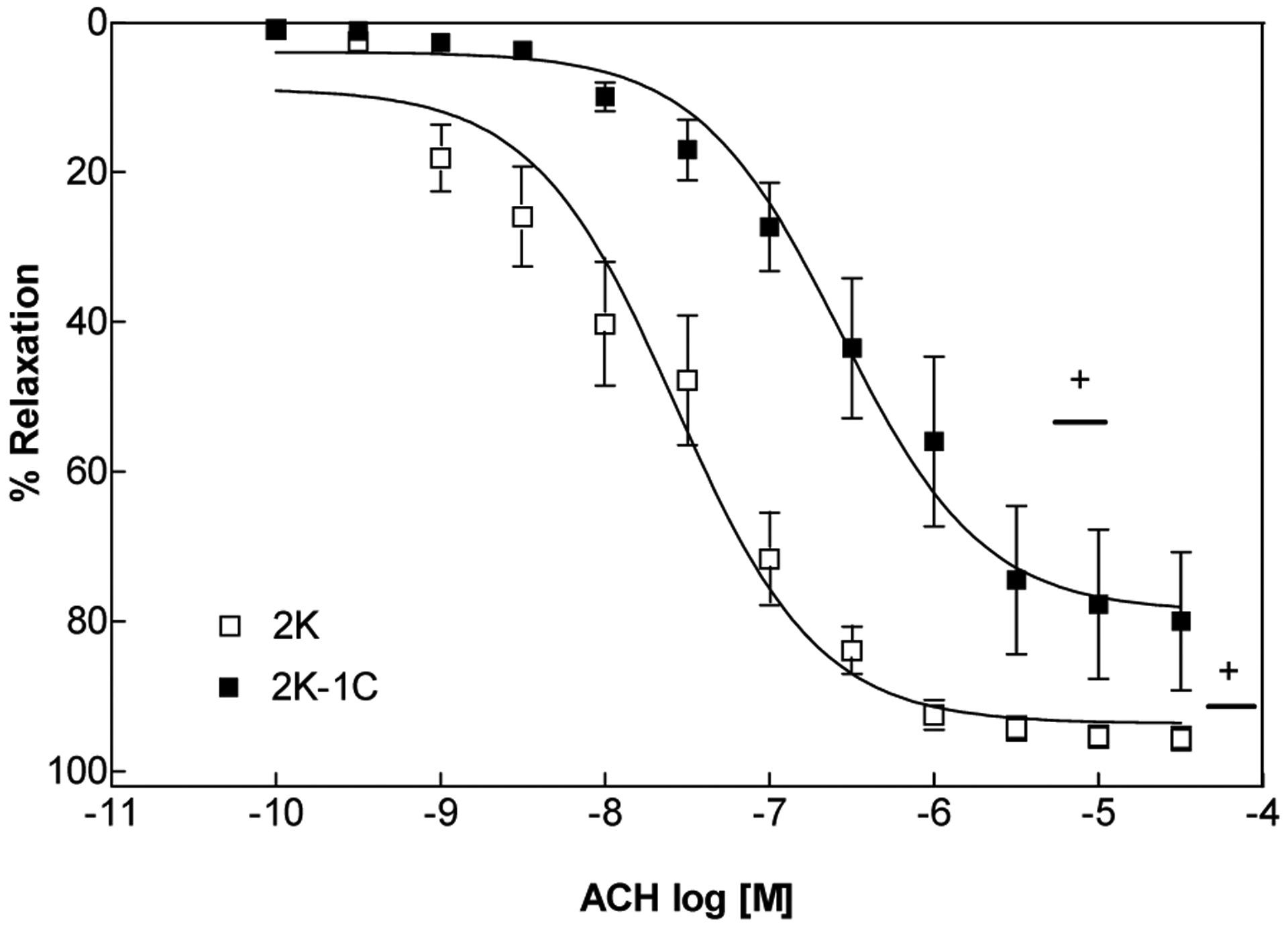
Concentration–response curves (n=8) for acetylcholine in intact endothelium- aortic rings contracted with phenylephrine. Values are mean ± S.E.M. of experiments performed on preparations obtained from different animals. + indicates significant difference (p< 0.05) in pD2 and ME values for 2K vs. 2K-1C
The treatment of aortic rings from hypertensive rats with DCBPY improved the relaxation endothelium dependent induced by acetylcholine, with no effect in aortic rings from normotensive rats. All concentration of DCBPY tested improved the relaxation just in 2K-1C aortic rings (pD2 of DCBPY 0.1μM: 7.11±0.04; 1.0 μM: 7.13±0.05; 10 μM 6.91±0.02; p<0.05 n=8) compared to aortic rings without DCBPY treatment (pD2 of PBS: 6.57±0.08), and no effect was verified in aortic rings from 2K (pD2 of DCBPY 0.1μM: 8.03±0.26; 1.0 μM: 7.64±0.14; 10 μM 7.71±0.10) (Figure 2 and Table 1). In addition, the maximum relaxant effect was improved by treatment at all concentration tested of DCBPY just in 2K-1C aorta (ME: 78.7 ±2.4%, 2K-1C 0.1 μM 93.8±1.1%, 2K-1C 1.0 μM 91.0±1.5%, 2K-1C 10.0 μM 92.4±0.7%; p<0.05 n=8), with no difference to 2K (ME: 93.6 ±3.1%, 2K-1C 0.1 μM 92.2±3.5%, 2K-1C 1.0 μM 92.1±3.2%, 2K-1C 10.0 μM 94.8±2.6%) (Figure 2 and Figure 3, Table 1). No differences were verified between the concentrations of DCBPY treatment to aortic rings from 2K and 2K-1C.
Figure 2.
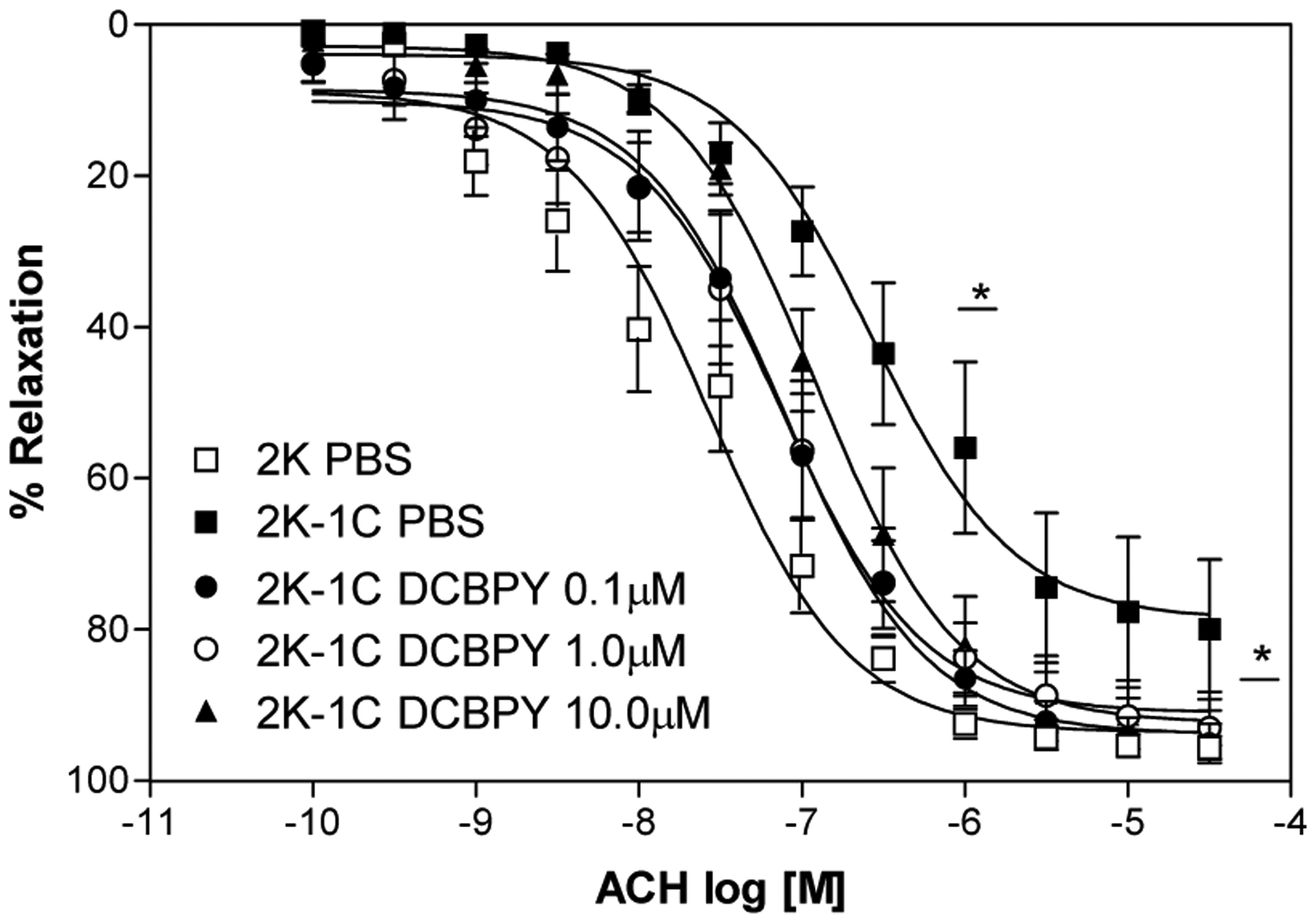
Concentration–response curves for acetylcholine in aortic rings with intact endothelium and incubated with different concentrations of DCBPY and contracted with phenylephrine. Values are mean ± S.E.M. of experiments performed on preparations obtained from different animals. * indicates significant difference (p< 0.05) in pD2 and ME values for 2K-1C PBS (n=8) vs. 2K-1C DCBPY 0.1/1.0/10.0 μM (n=8).
Table 1.
Potency (pD2) and maximum relaxant effect (ME) to acetylcholine in endothelium intact aortic rings from 2K and 2K-1C rats incubated with DCBPY (0.1;1.0;10.0 μM). 2K-1 C incubated with DCBPY 0.1 μM plus hydroxocobalamin, 2K-1C tempol or 2K-1C DETA-NO. Values are mean ± S.E.M. (n=8) of experiments performed on preparations obtained from different animals.
| pD2 | ME (%) | |
|---|---|---|
| 2K PBS | 7.57±0.12 | 93.6±3.1 |
| 2K-1C-PBS | 6.57±0.08+ | 78.7±2.3+ |
| 2K DCBPY 0.1μM | 8.03± 0.26 | 92.2±3.5 |
| 2K DCBPY 1.0 μM | 7.64 ±0.14 | 92.1±3.2 |
| 2K DCBPY 10.0 μM | 7.71±0.10 | 94.8±2.6 |
| 2K-1C DCBPY 0.1μM | 7.11±0.04* | 93.8±1.1* |
| 2K-1C DCBPY 1.0 μM | 7.13±0.05* | 91.0±1.5* |
| 2K-1C DCBPY 10 μM | 6.91±0.02* | 92.4±0.7* |
| 2K-1C TEMPOL 1 mM | 7.32±0.11*** | 86.7±2.8*** |
| 2K-1C DCBPY 0.1μM + HYDRO | 7.55±0.16 | 87.1±3.6 |
| 2K-1C DETA NO 0.1 μM | 6.99± 0.06** | 80.5±1.8 |
(pD2 and ME) p<0.05, 2K PBS vs 2K-1C PBS
(pD2 and ME) p<0.05 2K PBS vs 2K DCBPY 0.1/1.0/10.0 μM - 2K-1C PBS vs 2K-1C DCBPY 0.1/1.0/10.0 μM.
(pD2 and ME) p<0.05 2K-1C PBS vs 2K-1C DETA-NO
(pD2 and ME) p<0.05 2K-1C PBS vs 2K-1C TEMPOL
Figure 3.
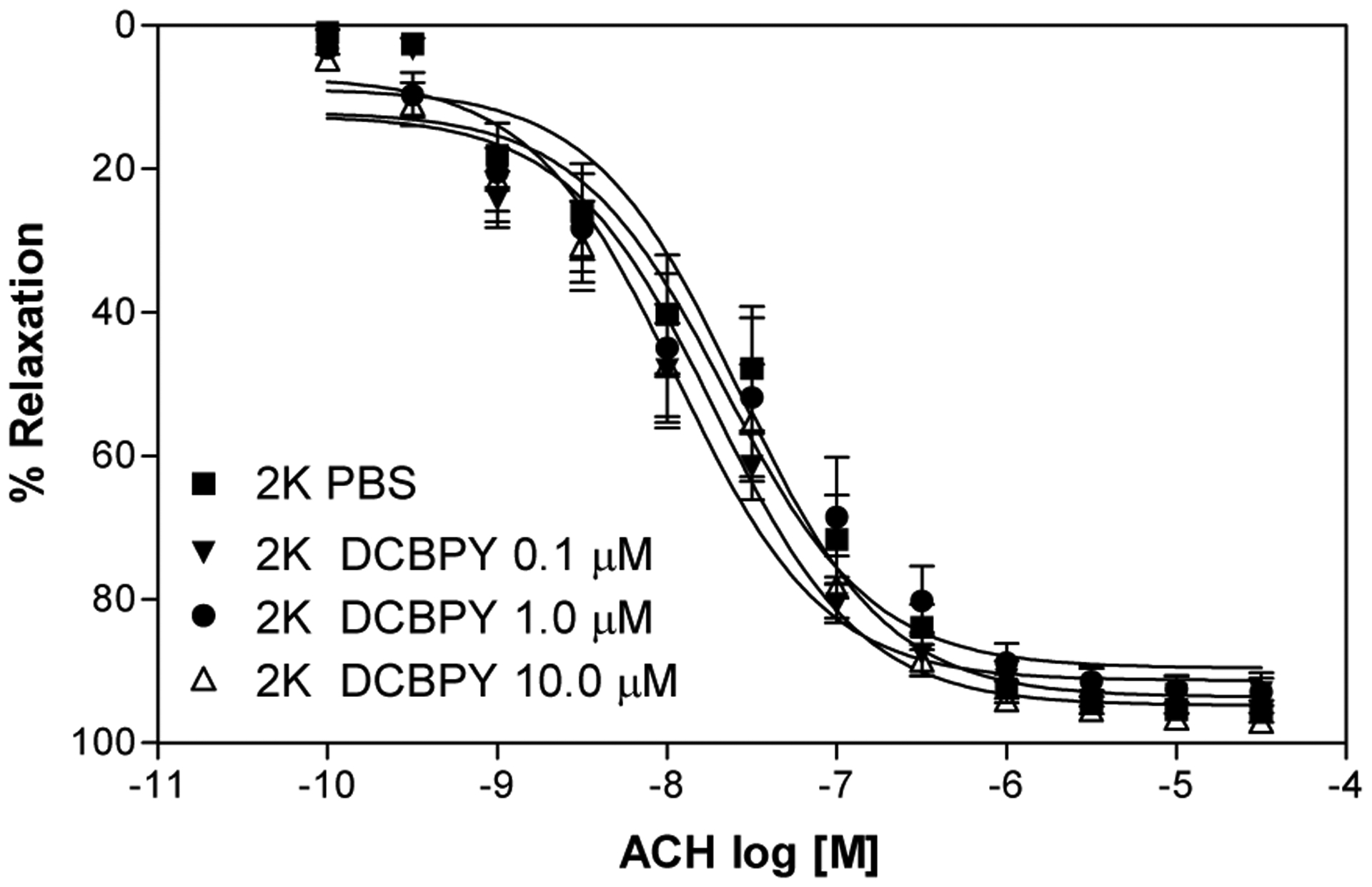
Concentration–response curves for acetylcholine in 2K aortic rings intact endothelium and incubated with different concentrations of DCBPY and contracted with phenylephrine. Values are mean ± S.E.M. of experiments performed on preparations obtained from different animals (n=8).
In order to verify if the improvement induced by 0.1 μM of DCBPY is due to NO release, we pre-incubated aortic rings from 2K-1C with hydroxocobalamin, a NO scavenger. As can be seen at Figure 4 and Table 1, the incubation with hydroxocobalamin do not modified the potency (pD2 of 2K-1C DCBPY 0.1+ Hydroxo: 7.55±0.16) and maximum effect (ME of DCBPY 0.1μM+ Hydroxo: 87.1 ±3.6%) compared to aortic rings incubated with 0.1 μM of DCBPY (pD2: 2K-1C DCBPY 0.1μM: 7.11±0.03; ME, 2K-1C DCBPY 0.1 μM 93.8±1.1%). In other words, hydroxocobalamin did not cancel the DCBPY effect, suggesting that the improvement induced by DCBPY is not dependent on NO release.
Figure 4.
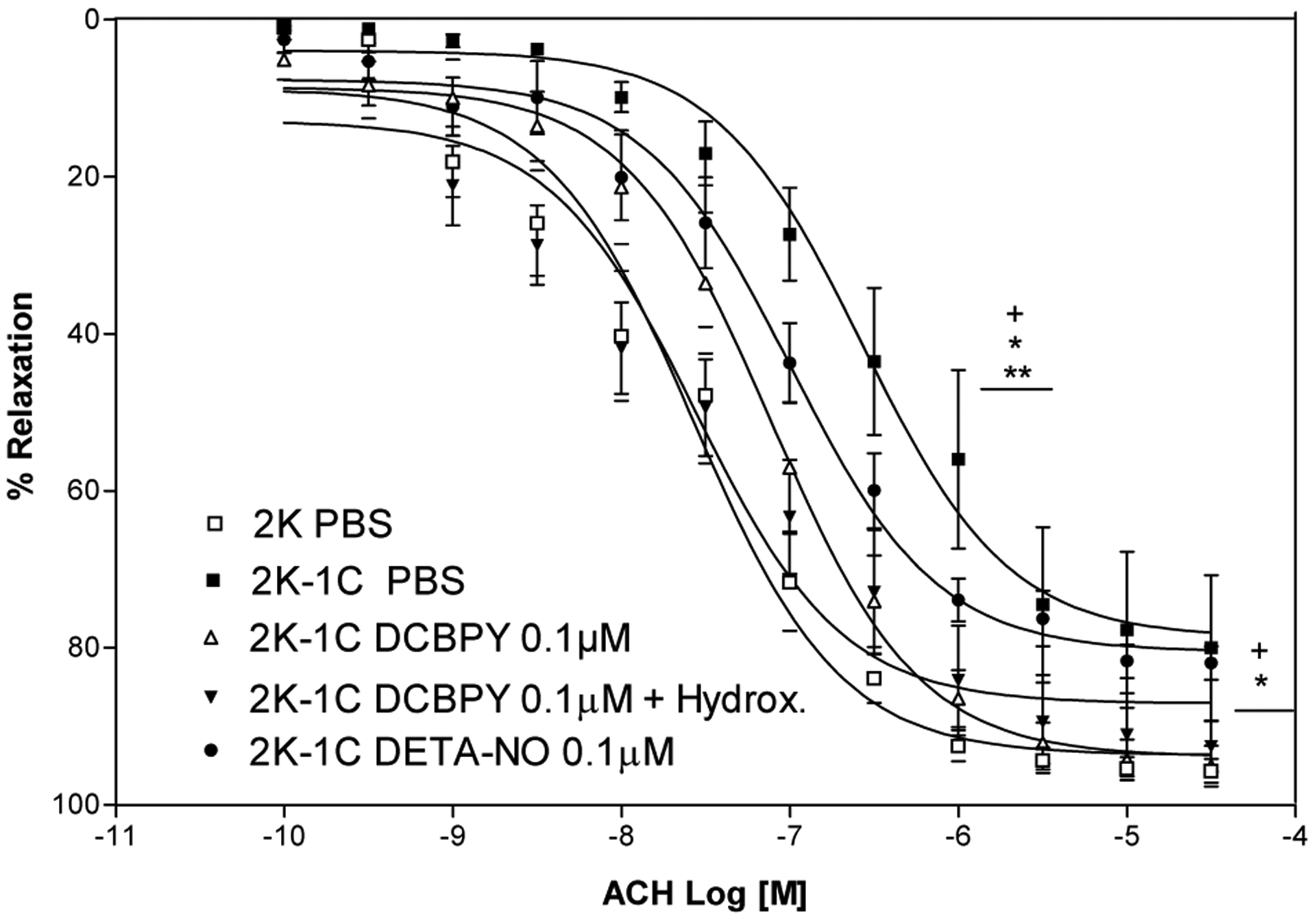
Concentration–response curves for acetylcholine after incubation with DCBPY 0,1 μM in the absence or presence of Hydroxocobalamin (100 μM) or DETA-NO, in aortic rings intact endothelium- contracted with phenylephrine. Values are mean ± S.E.M. of experiments performed on preparations obtained from different animals. + indicates significant difference (p< 0.05) in pD2 and ME values for 2K vs. 2K-1C; * indicates significant difference (p< 0.05) in pD2 and ME values for 2K-1C PBS (n=8) vs. 2K-1C DCBPY 0.1 μM (n=8); ** indicates significant difference (p< 0.05) in pD2 and ME values for 2K-1C PBS (n=8) vs DCBPY + DETA-NO (n=8).
Incubation of 2K-1C aortic rings with 0.1 μM DETA-NO have been done as a control of NO donor. The improvement of endothelium dependent relaxation was verified in 2K-1C aortas incubated with 0.1 μM of DETA-NO (pD2: 6.99±0.06; p<0.05 n=8) compared to control 2K-1C (pD2: 6.57±0.08; P<0.05 n=8), with no difference compared to incubation with 0.1 μM of DCBPY in 2K-1C aortas (pD2:7.11±0.03). In addition, the incubation with tempol that is a superoxide scavenger also improved the endothelium dependent relaxation in 2K-1C aortas (pD2: 7.32±0.11 ME: 86.7±2.8%) compared to control 2K-1C (pD2: 6.57±0.08 ME: 78.7±2.3%; p<0.05 n=8), with no difference compared to incubation with 0.1 μM of DCBPY in 2K-1C aortas (Figure 5).
Figure 5.

Concentration–response curves for acetylcholine after incubation with DCBPY 0.1 μM, and TEMPOL 10 μM in aortic rings intact endothelium- contracted with phenylephrine. Values are mean ± S.E.M. of experiments performed on preparations obtained from different animals. *** indicates significant difference (p< 0.001) in pD2 and ME values for 2K-1C PBS (n=8) vs 2K-1C TEMPOL (n=8).
Nitric oxide, superoxide detection and interaction between nitric oxide and superoxide.
To evaluate if DCBPY releases NO in endothelial cells over 30 min (which corresponds to the effects observed in aortic rings treatment), HUVEC were treated with 0.1μM of DCBPY and DETA-NO. The latter used as control of spontaneous NO donor. Figure 6A shows that DCBPY did not change in NO concentration during the time monitored (PBS: 4.32 ± 0.18 FI, n=3; compared to incubation with 0.1 μM of DCBPY: 4.43 ± 0.31 FI, n=4). However, DETA-NO showed an increase in NO in relation to its respective control (6.20 ± 0.21 FI, n=3, P<0.05). Nitric oxide production was further confirmed by fluorescence microscopy images also indicated the accumulation of DAF-2T product as green spots in HUVEC (Figure 6B).
Figure 6.

Measurement of intracellular NO production in HUVEC. Cells were treated with 0.1 μM of DCBPY and DETA-NO for 30 min. The NO production measured by fluorescence intensity of DAF-2T from stained cells. Bars (top panel) represent data of NO production are presented as means ± SEM. *p< 0.05 versus control, in 3 independent experiments of each protocol. The bottom panel represents images of NO detection by fluorescent images of DAF stained cells.
The potential effect of DCBPY with respect to scavenging superoxide was examined by means of the cytochrome c assay. The production of O2− was verified by reduction of cytochrome c (93.32 × 10−3 ± 6.10 × 10−3 AU, n=4), (Figure 7). SOD inhibited the reduction of cytochrome c (1.81 × 10−3 ± 0.94 × 10−3 AU, n=4, p<0.001), showing that O2− has been removed from the solution. The DCBPY compound suppressed the reduction of cytochrome c at all concentrations (1.25 μM: 36.73 × 10−3 ± 3.60 × 10−3 AU, n=4, 2.5 μM: 21.70 × 10−3 ± 4.60 × 10−3 AU, n=3, 12.5 μM: 6.93 × 10−3 ± 3.58 × 10−3 AU, n=3, 25 μM: 3.18 × 10−3 ± 3.44 × 10−3 AU, n=3, p<0.001) (Figure 7), and this effect is DCBPY concentration-dependent (1.25 μM < 2.5 μM, p<0.05; 1.25 μM < 12.5 μM P<0.001, and 2.5 μM < 25 μM, p<0.05) (Figure 7).
Figure 7.
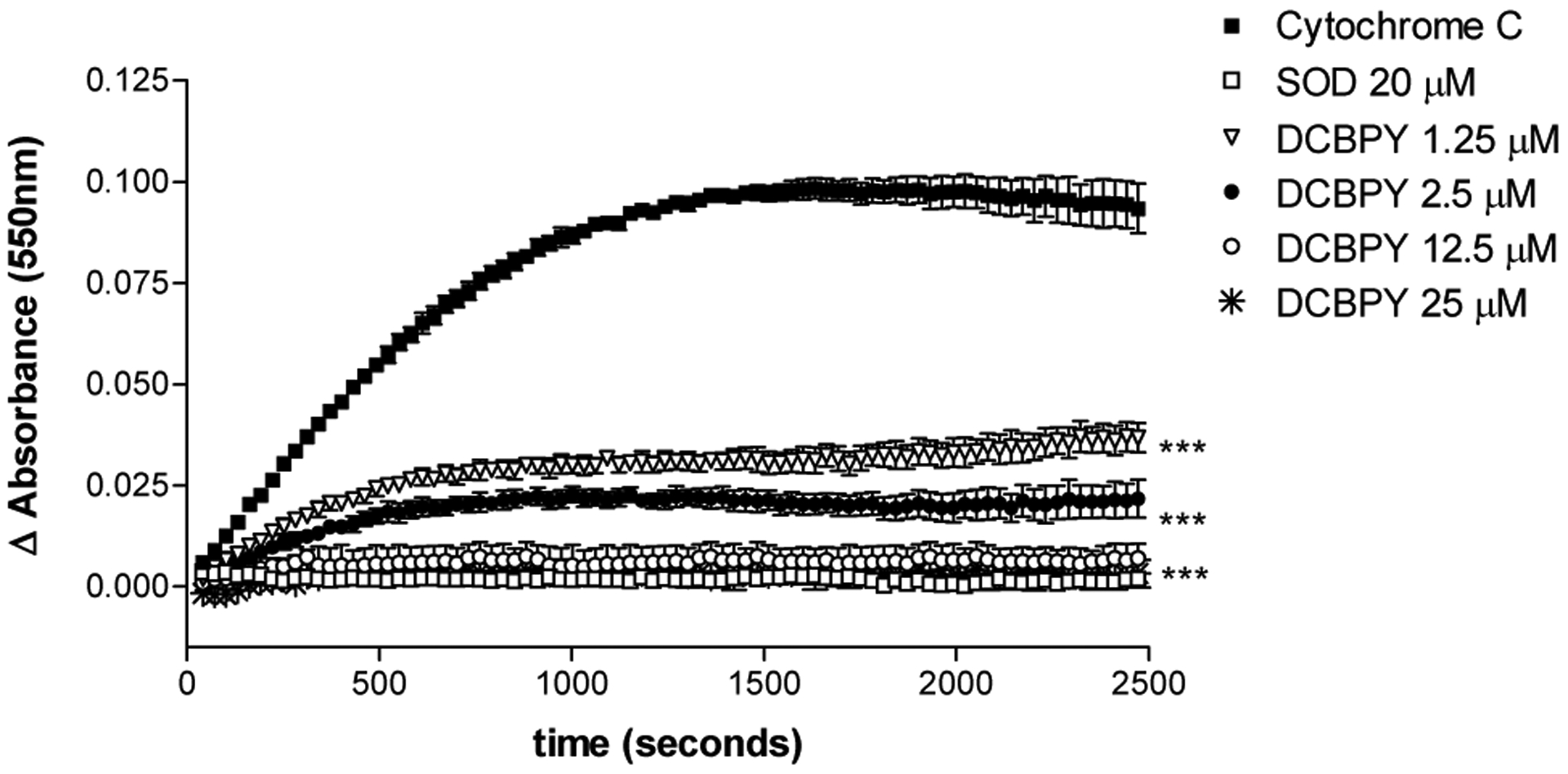
Measurement of the reduction of cytochrome c by superoxide (O2−), at 550nm. Difference (Δ) in the absorbance (AU) was obtained, and the superoxide-scavenging effect of the compound DCBPY and superoxide dismutase (SOD) was verified. Each point represents mean ± SEM of at least 3 absorbance values. *indicates significant (p<0.05) difference for Ru-NO2− 1.25 μM vs 2.5 μM, 2.5 μM vs 25 μM. ***indicates significant (p<0.001) difference for DCBPY and SOD vs control (cytochrome c), and DCBPY 1.25 μM vs 12.5 μM .
The effect of the interaction between superoxide and NO was investigated. A steady state concentration of NO can be detected over time in a solution containing DETA-NO. The addition of XO (2.70 ± 0.02 μM, n=10) evokes consumption of the NO released from DETA-NO, as predicted by the consumption of NO by O2− (Figure 8). The DCBPY compound avoids the consumption of NO in a DCBPY-concentration-dependent manner (12.5 μM: 0.92 ± 0.02 μM, n=3; 25 μM: 1.49 ± 0.05 μM, n=4; 50 μM: 3.50 ± 0.12 μM, n=4, p<0.001) (Figure 8C). SOD also prevents the consumption of NO in an SOD-concentration-dependent manner (10 μM: 0.57 ± 0.02 μM, n=3; 20 μM: 1.06 ± 0.03 μM, n=3; 30 μM: 1.36 ± 0.1 μM, n=4, p<0.001) (Figure 8C). To make sure that in the presence of XO/HX there is no NO release from Ru-NO2−, the same experiment was conducted in the absence of DETA-NO, and no release of NO was verified, as can be verified in the Figure 8B.
Figure 8.
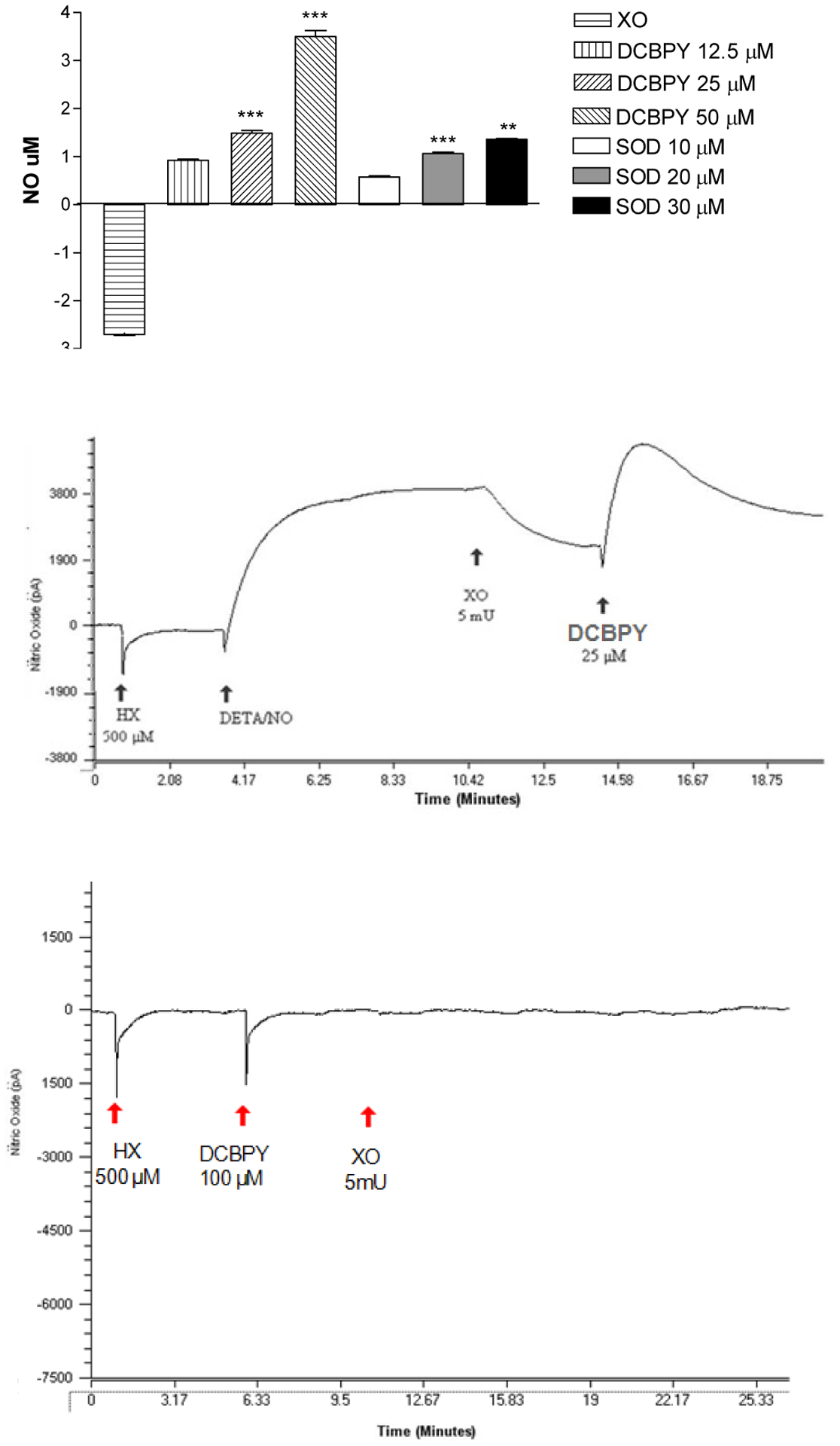
Top panel: Quantification of NO that was not consumed by superoxide (O2−) generated by xanthine oxidase (XO). Superoxide-scavenging effect of DCBPY and superoxide dismutase (SOD). Bars represent mean ± SEM of NO concentration. ***indicates significant (p<0.001) difference for DCBPY between the concentrations 12.5 vs 25 μM, 25 vs 50 μM, and for SOD between the concentrations 10 vs 20 μM, 20 vs 30 μM. Middle panel: Representative recording of experiments using the selective electrode for NO. Experimental sequence to measure the NO consumption by superoxide (O2−) generated by xanthine oxidase (XO). Bottom panel: Positive control to measure if DCBPY releases NO in the presence on hypoxanthine (HX) and xanthine oxidase (XO).
DISCUSSION
The main finding of the present manuscript was that the DCPBY aortic treatment improves the relaxation endothelium dependent in aortic rings with endothelium dysfunction, which is not induced by NO release.
Our results have shown that the endothelium-dependent relaxation induced by acetylcholine is impaired in aortic rings from hypertensive rats (2K-1C). These results are in accordance with previous investigation [25,26,27,14]. In endothelial cells, muscarinic receptor activation by acetylcholine induces NO production by increase cytosolic Ca2+ concentration with consecutive activation of NO synthase (NOS) enzyme. The NO produced in endothelial cells migrates to smooth muscle cells and activate the soluble guanylyl cyclase (sGC) enzyme, producing cyclic GMP (cGMP), which activates cGMP-dependent protein kinase (PGK), inducing a decrease in cytosolic Ca2+− concentration with vascular relaxation [28].
Produced in low concentrations, NO in the blood is rapidly converted to nitrate and nitrite after 10 seconds of its formation, [29]. This short half-life is even more shortened in various cardiovascular diseases, as in the hypertension, in which oxidative stress reduces the NO availability. Several models of hypertension stimulate the deficiency in the production / availability of NO, among other alterations that are typical of endothelial dysfunction [30].
The aortic rings treatment with 0.1μM of DCBPY improved the relaxation endothelium dependent induced by acetylcholine just in hypertensive aortic rings. Also, the aortic rings treatment from hypertensive rats with 0.1μM of DETA/NO improved the relaxation endothelium-dependent, suggesting that a NO donor can improve this response. In order to evaluate if DCBPY effect is due to NO release, we have used a NO scavenger (hydroxocobalamin) together with DCBPY. Our results have suggested that this effect is not due to NO release, considering that hydroxocobalamin did not cancel the DCBPY effect. In addition, our results in HUVECs cells confirm that at concentration 0.1μM (incubation for 30 min) the compound DCBPY does not release NO. In previous publication [14] it was verified that the relaxation induced by DCPPY in aortic rings was abolished in the presence of hydroxocobalamin, indicating that hydroxocobalamin is able to scavenge NO release from DCBPY.
Reactive oxygen species (ROS) are important molecules regulating numerous physiological processes. However, their overproduction is harmful as these species could participate in several pathological processes. ROS could contribute to the mechanism of hypertension and are responsible for the cardiovascular diseases and atherosclerosis [31] it increases formation of superoxide radicals, lipid peroxidation and protein oxidation, which can directly promote damage of cellular organelles and DNA [32–33]. The anion superoxide (O2−) is an important member of the ROS family, which has been found in elevated concentration in the vascular smooth muscle cells from renal hypertensive rats [21, 20]. The NO bioavailability could be significantly reduced in the presence of O2− because NO reacts with O2− to form peroxynitrite (ONOO−) [34]. It has been suggested that ruthenium complex can react with O2− and inactivate or scavenge this ROS [22]. Thus, our results suggest that the improvement in endothelial function verified in response to DCBPY treatment is related to O2− inactivation, considering that this improvement was verified just in hypertensive aortic rings. In positive control, with tempol (SOD mimetic) aortic treatment, the endothelial function improvement also was verified just in hypertensive aortic rings, suggesting that the same mechanism is present in response to DCBPY and tempol.
The next step was to evaluate the potential action of DCBPY with respect to scavenging superoxide, by means of the cytochrome c assay. Our results suggested that DCBPY inactivate O2− in a concentration dependent manner. In addition, the effect of DCBPY on NO consumption by O2− was evaluated. Our results indicated that DCBPY prevents the NO consumption by O2− in a DCBPY-concentration-dependent manner. Taken together, our results indicate that DCBPY inactivates O2−.
In conclusion, our results suggest that the DCBPY at a lower concentration (0.1 μM) is not a NO generator, but it can inactivate superoxide in aortic rings and improves the endothelial function.
REFERENCES
- 1.Puddu P, Puddu GM, Zaca F, Muscari A. Endothelial dysfunction in hypertension, Acta Cardiol. 2000; 55(4):221–32. [DOI] [PubMed] [Google Scholar]
- 2.Lobysheva I, Rath G, Sekkali B, Bouzin C, Feron O, Gallez B. Moderate caveolin-1 downregulation prevents NADPH oxidase-dependent endothelial nitric oxide synthase uncoupling by angiotensin II in endothelial cells. Arterioscler Thromb Vasc Biol. 2011. September;31(9):2098–105. doi: 10.1161/ATVBAHA.111.230623. Epub 2011 Jun [DOI] [PubMed] [Google Scholar]
- 3.Ignarro LJ, Cirino G, Casini A, Napoli C. Nitric oxide as a signaling molecule in the vascular system: an overview, J Cardiovasc Pharmacol. 1999; 34:879–886. [DOI] [PubMed] [Google Scholar]
- 4.Taddei S, Virdis A, Ghiadoni L, Salvetti A. Endothelial dysfunction in hypertension: fact or fancy? J Cardiovasc Pharmacol. 1998; 32 (3):S41–7. [PubMed] [Google Scholar]
- 5.Munk PS, Butt N, Larsen AI. Endothelial dysfunction predicts clinical restenosis after percutaneous coronary intervention, Scand Cardiovasc J. 2011;45(3):139–45. [DOI] [PubMed] [Google Scholar]
- 6.Bonaventura D, Lima RG, Silva RS, Bendhack LM. NO donors-relaxation is impaired in aorta from hypertensive rats due to a reduced involvement of K+ channels and sarcoplasmic reticulum Ca2+−ATPase, Life Sci. 2011; (89):595–602. [DOI] [PubMed] [Google Scholar]
- 7.Lunardi CN, Silva RS, Bendhack LM. New nitric oxide donors based on ruthenium complexes, Braz. J. Med. Biol. Res 2009; 42(1):87–93. [DOI] [PubMed] [Google Scholar]
- 8.Clozel M, Kuhn H, Hefti F. Effects of angiotensin converting enzyme inhibitors and of hydralazine on endothelial function in hypertensive rats, Hypertension 1990; 16(5): 532–40. [DOI] [PubMed] [Google Scholar]
- 9.Friederich JA, Butterworth JF. Sodium nitroprusside: Twenty years and counting. Anesth. Analg, 1995; 85: 152–162. [DOI] [PubMed] [Google Scholar]
- 10.Lima RG, Silva BR, Da Silva RS, Bendhack LM. Ruthenium complexes as NO donors for vascular relaxation induction, Molecules 2014;19:9628–9654; doi: 10.3390/molecules19079628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonaventura D, Oliveira FS, Da Silva RS, Bendhack LM. Decreased vasodilation induced by a new nitric oxide donor in 2K–1C hypertensive rats is due to impaired K+ channel activation, Clin. Exp. Pharmacol. Physiol 2005; 32: 478–481. [DOI] [PubMed] [Google Scholar]
- 12.Lunardi CN, Vercesi JA, Da Silva RS, Bendhack LM. Vasorelaxation induced by the new nitric oxide donor cis-[Ru(Cl)(bpy)(2)(NO)](PF(6)) is due to activation of K(Ca) by a cGMP-dependent pathway, Vasc. Pharmacol 2007; 47:139–144. [DOI] [PubMed] [Google Scholar]
- 13.Castro PFS, Pereira AC, Rogrigues GJ, Batista AC, Da Silva RD, Bendhack LM, Rocha ML. A new nitrosyl ruthenium complex nitric oxide donor presents higher efficacy than sodium nitroprusside on relaxation of airway smooth muscle, Eur. J. Pharm. Sci 2011;43:370–377. [DOI] [PubMed] [Google Scholar]
- 14.Guedes PMM, Oliveira FS, Gutierrez FRS, Silva GK, Rodrigues GJ, Bendhack LM, Franco DW, Matta MV, Zamboni DS, Silva RS, Silva JS. Nitric oxide donor trans-[RuCl([15]aneN)NO] as a possible therapeutic approach for Chagas’ disease, Br J Pharmacol. 2010; 160 (2):270–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodrigues GJ, Pereira AC, Vercesi JA, Lima RG, Da Silva RS, Bendhack LM. Long-lasting hypotensive effect in renal hypertensive rats induced by nitric oxide released from a ruthenium complex, J Cardiovasc Pharmacol. 2012;60(2):193–198. [DOI] [PubMed] [Google Scholar]
- 16.Rodrigues GJ, Cicillini SA, Da Silva RS, Bendhack LM. Mechanisms underlying the vascular relaxation induced by a new nitric oxide generator. Nitric Oxide. 2011; 30:25(3)331–337. [DOI] [PubMed] [Google Scholar]
- 17.Callera GE, Varanda WA, Bendhack LM. Impaired relaxation to acetylcholine in 2K-1Chypertensive rat aortas involves changes in membrane hyperpolarization instead of an abnormal contribution of endothelial factors, Gen Pharmacol. 2000; 34(6): 379–389. [DOI] [PubMed] [Google Scholar]
- 18.Callera GE, Yogi A, Tostes RC, Rossoni LV, Bendhack LM. Ca2+-activated K+ channels underlying the impaired acetylcholine-induced vasodilation in 2K-1C hypertensive rats, J Pharmacol Exp Ther. 2004; 309(9): 1036–1042. [DOI] [PubMed] [Google Scholar]
- 19.Rodrigues GJ, Restini CB, Lunardi CN, Moreira JE, Lima RG, Da Silva RS, Bendhack LM. Caveolae dysfunction contributes to impaired relaxation induced by nitric oxide donor in aorta from renal hypertensive rats, J. Pharmacol. Exp. Ther 2007;323: 831–837. [DOI] [PubMed] [Google Scholar]
- 20.Heitzer T, Wenze U, Hink U. Krollner D, Skatchkov M, Stahl RA, MacHarzina R, Bräsen JH, Meinertz T, Münzel T. Increased NAD(P)H oxidase mediated superoxide production in renovascular hypertension: evidence for an involvement of protein kinase C. Kidney Int. 1999; 55:252–260. [DOI] [PubMed] [Google Scholar]
- 21.Rodrigues GJ, Lunardi CN, Lima RG, Santos CX, Laurindo FR, da Silva RS, Bendhack LM. Vitamin C improves the effect of a new nitric oxide donor on the vascular smooth muscle from renal hypertensive rats. Nitric Oxide. 2008; 18(3): 176–183. [DOI] [PubMed] [Google Scholar]
- 22.Stambury DM, Mulac WA, Sullivan JC, Taube H. Superoxide reactions with (isonicotinamide) pentaammineruthenium(II) and -(III). Inorg Chem. 1980; 19:3735. [Google Scholar]
- 23.Goldblatt H, Lynch J, Hanzal RF, Summerville WW. Studies on experimental hypertension: I: The production of persistent elevation of systolic blood pressure by means of renal ischemia, J Exp Med. 1934; 28;59(3):347–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kojima S, Yoshitomi Y, Sugi T, Matsumoto Y, Yano M, Kuramochi M. Changes in plasma levels of cyclic nucleotides during low-density lipoprotein apheresis. Ther Apher. 1998;2(4):263–267. [DOI] [PubMed] [Google Scholar]
- 25.Angus JA, Dyke AC, Jennings GL, Korner PI, Sudhir K, Ward JE, Wright CE. Release of endothelium-derived relaxing factor from resistance arteries in hypertension. Kidney Int Suppl. 1992;37:S73–78. [PubMed] [Google Scholar]
- 26.Vega GW, Roson MI, Bellver A, Celentano MM, de la Riva IJ, Nitric oxide and superoxide anions in vascular reactivity of renovascular hypertensive rats. Clin Exp Hypertens 1995;17(5):817–835 [DOI] [PubMed] [Google Scholar]
- 27.Sendao Oliveira AP, Bendhack LM. Relaxation induced by acetylcholine involves endothelium-derived hyperpolarizing factor in 2-kidney 1-clip hypertensive rat carotid arteries. Pharmacology 2004;72(4):231–239. [DOI] [PubMed] [Google Scholar]
- 28.Bryan NS, Bian K, Murad E. Discovery of the nitric oxide signaling pathway and targets for drug development. Front Biosci (Landmark Ed). 2009;1(14):1–18. [DOI] [PubMed] [Google Scholar]
- 29.Hall CN, Garthwaite J. What is the real physiological NO concentration in vivo? Nitric Oxide 2009; 21: 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silva BR, Pernomian L, Bendhack LM. Contribution of oxidative stress to endothelial dysfunction in hypertension. Front. Physiol 2012; 3: 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Victor VM, Rocha M, Sola E, Bañuls C, Garcia-Malpartida K, Hernández- Mijares A. Oxidative stress, endothelial dysfunction and atherosclerosis, Curr. Pharm. Des 2009.15(26):2988–3002. [DOI] [PubMed] [Google Scholar]
- 32.Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogenspecies: role in inflammatory disease and progression to cancer. Biochem J. 1996; 313: 7–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imlay JA, Pathways of oxidative damage. Annu Rev Microbiol. 2003; 57: 395–418. [DOI] [PubMed] [Google Scholar]
- 34.Hamilton CA, Brosnan MJ, McIntyre M, Graham D, Dominiczak AF. Superoxide excess in hypertension and aging: a common cause of endothelial dysfunction. Hypertension. 2001; 37(2):529–534. [DOI] [PubMed] [Google Scholar]