

Abstract
The counterregulatory response to hypoglycemia is an essential survival function. It is controlled by an integrated network of glucose-responsive neurons, which trigger endogenous glucose production to restore normoglycemia. The complexity of this glucoregulatory network is, however, only partly characterized. In a genetic screen of a panel of recombinant inbred mice we previously identified Fgf15, expressed in neurons of the dorsomedial hypothalamus (DMH), as a negative regulator of glucagon secretion. Here, we report on the generation of Fgf15CretdTomato mice and their use to further characterize these neurons. We show that they were glutamatergic and comprised glucose-inhibited and glucose-excited neurons. When activated by chemogenetics, Fgf15 neurons prevented the increase in vagal nerve firing and the secretion of glucagon normally triggered by insulin-induced hypoglycemia. On the other hand, they increased the activity of the sympathetic nerve in the basal state and prevented its silencing by glucose overload. Higher sympathetic tone increased hepatic Creb1 phosphorylation, Pck1 mRNA expression, and hepatic glucose production leading to glucose intolerance. Thus, Fgf15 neurons of the DMH participate in the counterregulatory response to hypoglycemia by a direct adrenergic stimulation of hepatic glucose production while suppressing vagally induced glucagon secretion. This study provides new insights into the complex neuronal network that prevents the development of hypoglycemia.
Introduction
The central nervous system controls multiple aspects of glucose homeostasis including pancreatic islet hormone secretion and hepatic glucose production as well as glucose utilization by muscle and fat. The connection between the brain and these peripheral tissues is ensured, in large part, by the autonomic nervous system. This is activated in response to changes in the concentration of circulating hormones such as insulin, leptin, or ghrelin and of nutrients such as glucose and lipids. Glucose-responsive neurons, which increase their firing activity in response to hyperglycemia (glucose-excited [GE] neurons) or to hypoglycemia (glucose-inhibited [GI] neurons) (1–3), are thought to couple fluctuations in blood glucose concentrations to the regulation of sympathetic or parasympathetic nerve activity.
A major glucoregulatory role of the central nervous system is to maintain glycemic levels at a minimum value of ∼5 mmol/L to preserve sufficient glucose provision to the brain. Hypoglycemia does not usually occur in healthy subjects because of the rapid secretion of the counterregulatory hormones glucagon, epinephrine, norepinephrine, cortisol, and growth hormones when blood glucose concentrations fall below the euglycemic level (4,5). However, in insulin-treated patients with type 1 or type 2 diabetes, this counterregulatory response becomes blunted, leading to hypoglycemic episodes of progressively increased severity; this condition represents a major limitation in the insulin treatment of diabetes (6–8). The cellular and molecular basis for this deregulation are not understood because of the insufficient knowledge of the complex cellular network involved in hypoglycemia detection.
Glucose-responsive neurons involved in the counterregulatory response to hypoglycemia have been located in several brain areas, including the hypothalamus and the brainstem (2). In the brainstem, neurons activated by hypoglycemia or 2-deoxy-d-glucose (2DG)-induced neuroglucopenia and that stimulate glucagon secretion are present in the dorsal vagal complex, in particular in the nucleus of the tractus solitarius (9,10) and in the basolateral medulla (11). In the hypothalamus, the ventromedial nucleus (VMN) contains glucose-sensing neurons that also participate in the control of glucagon secretion (12–15). These are part of a circuit that includes upstream neurons of the parabrachial nucleus and downstream neurons of the bed nucleus of the stria terminalis (16) and of the periaqueductal gray (17). Hypoglycemia is also detected by neurons that are located at peripheral locations such as the hepatoportal vein area and that are connected to the brainstem and hypothalamus glucoregulatory centers (18–20). Thus, the currently emerging picture is that hypoglycemia sensing occurs in a distributed system of glucose-sensing neurons present not only in several brain nuclei but also at peripheral locations. These cells form a connected network that generates an integrated hormonal response to prevent hypoglycemia development (21).
To uncover new aspects of this hypoglycemia monitoring system, we previously performed a genetic screen in a panel of advanced recombinant inbred BXD mice (22) in search of hypothalamic genes regulating 2DG-induced glucagon secretion (23). We identified Fgf15, whose expression in a subpopulation of dorsomedial hypothalamus (DMH) neurons negatively correlated with glucagon secretion. We showed that intracerebroventricular injection of FGF19, the human ortholog of Fgf15, dampened 2DG-induced glucagon secretion by inhibiting vagal activity and that silencing Fgf15 expression specifically in the DMH increased 2DG-induced glucagon secretion.
Here, we further explored the role of these Fgf15 neurons in the regulation of glucose homeostasis and the counterregulatory response to insulin-induced hypoglycemia. We first generated a mouse line that expresses the Cre recombinase and tdTomato from the Fgf15 locus (Fgf15CretdT/+ mice), allowing for genetic visualization and chemogenetic activation of Fgf15 neurons as well as for the identification of the afferent input they receive and the site where they send projections. We found that Fgf15 neurons of the DMH have a dual role in blunting vagal activity and glucagon secretion while increasing sympathetic tone to increase hepatic glucose production.
Research Design and Methods
Mice
The Fgf15CretdT/+ mouse line was generated by Ozgene Pty Ltd. (Bentley, Australia) by homologous recombination in C57BL/6 embryonic stem cells, which were injected into goGermline blastocysts (24). Male chimeric mice were crossed with C57BL/6J females to establish Fgf15CretdT/+ mice. Mice were housed on a 12-h light/dark cycle and were fed a standard rodent chow diet (Diet 3436; Provimi Kliba AG, Kaiseraugst, Switzerland). Experiments were performed with 10- to 14-week-old male mice. All animal experimentations were approved by the Veterinary Office of Canton de Vaud.
Biochemical Measurements
Blood was collected from submandibullary or tail veins. Glycemia was measured with a glucometer (Ascensia Breeze 2, Bayer Healthcare, Leverkusen, Germany). ELISAs were used to quantify glucagon (cat. no. 10-1271-01; Mercodia, Uppsala, Sweden), insulin (10-1247-10; Mercodia), and corticosterone (ADI-900-097; Enzo Life Sciences, Farmingdale, NY). Free fatty acids were measured with FUJIFILM Wako NEFA assay (Tokyo, Japan); hydroxybutyric acid was determined with the D-3-Hydroxybutyrate Ranbut reagent (Randox, Crumlin, U.K.), and plasma lactate levels were determined with lactate determination reagents (Roche, Basel, Switzerland). These measurements were performed with a Roche Diagnostics cobas c 111 automatic analyzer.
Viruses and Stereotactic Injections
Surgeries were performed under ketamine/xylazine anesthesia. Recombinant AAV2-Ef1a-DIO-EYFP was purchased from the University of North Carolina vector core. Recombinant AAV8-hSyn-DIO-mCherry, AAV8-hSyn-DIO-hM3Dq-mCherry, AAV1-syn-FLEX-splitTVA-EGFP-tTA, and AAV1-TREtight-mTagBFP2-B19G were purchased from Addgene (cat. nos. 44361, 50459, 100798, and 100799, respectively; Cambridge, MA). EnvA-G-deleted-mCherry pseudotyped rabies virus was purchased from the Salk Institute for Biological Studies. Ad-CMV-synaptophysin-mCherry was a gift from Dr. M. Myers (University of Michigan, Ann Arbor, MI). Recombinant adeno associated viruses (AAVs) (200 nL; 1012–1013 viral genomes/mL) were injected stereotactically with use of appropriate coordinates (23,25). Animals were allowed to recover for 2 weeks with daily handling and body weight monitoring before physiological measurements.
Activation of Fgf15 Neurons and Physiological Measurements
Chemogenetic activation of hM3Dq-expessing neurons was achieved by i.p. injection of clozapine (Clz) (cat. no. C6305; Sigma-Aldrich, St. Louis, MO) at a dose of 0.1 mg/kg.
Intraperitoneal glucose (2 g/kg) and pyruvate (2 g/kg) tolerance tests mice were performed in overnight-fasted mice. Insulin tolerance tests (0.8 units/kg; Actrapid) were performed in mice deprived of food for 6 h. β-2 adrenergic receptor blockade was achieved by i.p. injection of butaxamine hydrochloride (10 mg/kg, cat. no. B1385; Sigma-Aldrich). Glycemia and plasma insulin were measured from tail vein blood samples, and glucagon was measured from blood collected under anesthesia from the submandibular vein. Basal glucose turnover/hepatic glucose production rates were measured as previously described (26,27).
Quantitative RT-PCR
Tissue preparation, RNA extraction, and quantitative RT-PCR were performed as previously described (28). Forward (F) and reverse (R) primers were as follows: phosphoenolpyruvate carboxykinase (Pck-1), F5-ACAACTGTTGGCTGGCTCTC-3' and F5'- TGAGGCCAGTTTTGGGGATG-3'; glucokinase (Gck), F5'-ACTGCGGAGATGCTCTTTGA-3' and F5'-TCTCGGAGAAGTCCCACGAT-3'; and β-glucuronidase (Gusb), F5'-GTGATGGAGGAGCTGGTTCG-3' and R5'-AGCAGAGGAAGGCTCATTGG-3'. Gusb was use as housekeeping gene to normalize gene expression in all experiments.
Western Blots
Livers were homogenized in ice-cold lysis buffer (250 mmol/L sucrose; 20 mmol/L HEPES, pH 7.4; 10 mmol/L EDTA; and 2 mmol/L phenylmethylsulfonylfluoride). Homogenates were centrifuged first at 2,000 rpm for 5 min at 4°C; the supernatant was then submitted to a second centrifugation at 10,000 rpm for 15 min at 4°C. Proteins from the final supernatant were separated by electrophoresis on SDS-containing, 7.5% (Gys, phosphorylated [p]Gys, Pygl, and pPygl) or 12% (Creb1 and pCreb1) polyacrylamide gels, transferred to nitrocellulose membranes (cat. no. 10600001; GE Healthcare, Chicago, IL) with use of a Mini Trans-Blot apparatus from Bio-Rad Laboratories. The membranes were blocked in 5% BSA (w/v) (Gys, pGys, Creb1, and pCreb1) or 3% milk (w/v) (Pygl and pPygl) in Tris-buffered saline with Tween-20 (TBST) (in millimoles per liter: 15 Tris-HCl, 137 NaCl, 0.1% Tween-20, pH 7.6) at room temperature. Membranes were incubated with anti-Gys (cat. no. 3886, 1/1,000; Cell Signaling Technology, Danvers, MA), anti-pGys (cat. no. 3891, 1/1,000; Cell Signaling Technology), anti-Pygl (cat. no. 1851-1, 1/1,000; Proteintech, Rosemont, IL), anti-pPygl (1/500; University of Dundee), anti-Creb1 (cat. no. 9197, 1/1,000; Cell Signaling Technology), and anti-pCreb1 (cat. no. 9198, 1/1,000; Cell Signaling Technology) in 5% BSA/TBST or 3% milk/TBST overnight at 4 °C. After TBST 4 × 5 min washes, the membranes were incubated for 1 h at room temperature with the appropriate secondary anti-rabbit (cat. no. NA934, 1/8,000; Amersham, Little Chalfont, U.K.) or anti-sheep (cat. no. 81-8620, 1/6,000; Invitrogen, Carlsbad, CA) horseradish peroxidase–conjugated antibody in 5% BSA/TBST or 3% milk/TBST, followed by further TBST 4 × 5 min washes. Bands corresponding to the specific proteins were visualized with enhanced chemiluminescence reagent (Advansta, San Jose, CA). Digital images were acquired with FUSION FX7 system (Vilber Lourmat) and Bio-1D software (Vilber Lourmat) for quantification and normalization.
Immunofluorescence Microscopy
For c-fos immunodetection, Fgf15CretdT/+ mice were fixed by cardiac perfusion of 4% cold paraformaldehyde in sodium phosphate buffer (0.1 mol/L, pH 7.4). Brains were dissected and kept for 2h in paraformaldehyde at 4°C, incubated overnight in sucrose 30% at 4°C, and frozen at −80°C. Serial hypothalamic 20-μm cryosections were prepared and incubated first in 0.1 mol/L phosphate buffer saline pH 7.4; 3% normal goat serum; and 0.3% Triton X-100 for 1 hour then with a rabbit c-fos monoclonal antibody (cat. no. 2250, 1/7,000; Cell Signaling Technology) for 24 h at 4°C and for 1.5 h at room temperature with an Alexa Fluor 488–conjugated goat anti-rabbit IgG antibody (cat. no. 11008, 1/400; Life Technologies, Carlsbad, CA).
For mCherry immunodetection, 30-μm cryosections were prepared as described above and incubated overnight at room temperature with an anti-mCherry rabbit polyclonal antibody (cat. no. 632496, 1/2,500; Clontech Laboratories, Mountain View, CA) and for 1 h with an Alexa Fluor 594–conjugated goat anti-rabbit IgG antibody (cat. no. A-11012, 1/200; Life Technologies). For eGFP immunodetection a chicken polyclonal antibody directed against EGFP (cat. no. A10262 1/500; Invitrogen) and an Alexa Fluor 647–conjugated rabbit anti-chicken IgY antibody (cat. no. 303-605-003, 1/500; Jackson ImmunoResearch, Ely, U.K.) were used. Nuclei were counterstained with DAPI (cat. no. D9542, Sigma-Aldrich), and the slides were mounted in Fluoromount (cat. no. 15664, Southern Biotechnology, Birmingham, AL).
Images were acquired with a Zeiss Axio Imager D1 microscope interfaced with Axiovision software or a Zeiss Axio Imager M2 microscope, equipped with ApoTome.2 and a Camera Axiocam 702 mono (Zeiss, Oberkochen, Germany). Image analysis was performed with ImageJ.
In Situ Hybridization
For in situ hybridization detection of eYFP and Vglut2 or Gad67, Fgf15CretdT/+ mouse brains were dissected with use of a mouse brain matrix with 1-mm section dividers (CellPoint Scientific, Gaithersburg, MD), fixed for 28 h in 10% formalin, and embedded in paraffin. Five-μm cryosections were used for in situ hybridization with Advanced Cell Diagnostics probes (cat. nos. 312131-C2, 319171-C1, 400951-C1, and 320701). Sections were counterstained with Mayer’s Hematoxylin Solution and mounted with Aquatex mounting medium (cat. no. 363123S; VWR, Radnor, PA). Computerized cartography of Fgf15-expressing neurons in the DMH was performed with use of the camera lucida method (29).
Autonomic Nervous System Activity Recording
The firing rates of the thoracic branch of both vagal and sympathetic nerves along the carotid artery were recorded as previously described (30,31). Unipolar nerve activity was recorded continuously under isoflurane anesthesia with the LabChart 8 software (ADInstruments, Oxford, U.K.). Data were digitized with PowerLab 16/35 (ADInstruments). Signals were amplified 105 times and filtered with 200/1,000 Hz band pass filter. Firing rate analysis was performed with LabChart 8.
Patch Clamp
Electrophysiological characterization of glucose responsiveness of Fgf15 neurons was performed on acute 250-μm brain sections. Electrophysiological recordings of eYFP-labeled neurons were performed as previously described (10). Whole-cell recordings were performed in current-clamp mode with a MultiClamp 700B amplifier (Molecular Devices, Berkshire, U.K.). Neurons with an access resistance >25 mol/LΩ were excluded. A hyperpolarization step (−20 pA, 500 ms) was applied every 30 s to measure input resistance. Signals were filtered at 2 kHz, digitized at 10 kHz, and collected online with a pClamp 10 data acquisition system (Molecular Devices).
Anterograde and Retrograde Mapping of DMH Fgf15 Neurons
For mapping of the Fgf15 neuron anterograde projections, Fgf15CretdT/+ mice were bilaterally injected in the DMH with Ad-CMV-synaptophysin-mCherry (32) and synaptic terminals were detected on cryosections prepared as described above. Identification of neurons innervating Fgf15 neurons was performed as previously described (29,33). Fgf15CretdT/+ mice were injected bilaterally in the DMH with AAV1-syn-FLEX-splitTVA-EGFP-tTA and AAV1-TREtight-mTagBFP2-B19G and 1 week later with EnvA-G-deleted-mCherry pseudotyped rabies virus. Fgf15 neuron presynaptic inputs were identified by the presence of mCherry.
Statistical Analysis
Data are expressed as mean ± SEM. Statistical analysis was performed with GraphPad Prism 7.0c with one-way ANOVA followed by an appropriate post hoc test, two-way ANOVA followed by an appropriate post hoc test, or an unpaired two-tailed Student t test. P values <0.05 were considered significant.
Data and Resource Availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Results
Fgf15 Neurons of the DMH and Fgf15 Reporter Mice
We first performed a detailed analysis of the distribution of Fgf15 neurons in the DMH by in situ hybridization analysis. We found that Fgf15 was expressed along the rostra-caudal axis in the dorsal, compact, and ventral parts of the DMH; some expression was also found in perifornical area (PeF) (Fig. 1A and B), as previously reported (23). Then, we created a reporter mouse line in which the Cre recombinase and the tdTomato sequences, separated by T2A sequences, were introduced just before the stop codon present in exon 3 of the Fgf15 gene (Fgf15CretdT/+ mice) (Fig. 1C). TdTomato was found to be expressed in all enterocytes lining the ileum villi, the main site of Fgf15 expression (34) (Fig. 1D–F). In brain, no fluorescent signal could be detected, suggesting that the level of tdTomato expression directed by the brain Fgf15 promoter was too low. We, thus, tested whether stereotactic injection in the hypothalamus of Fgf15CretdT/+ of a Cre-activated adenovirus (recombinant AAV2-Ef1a-DIO-eYFP) would lead to expression of eYFP in Fgf15 neurons. We found that eYFP was expressed when the virus was injected in the DMH and PeF of Fgf15CretdT/+ mice (Fig. 1H and J). No eYFP fluorescence could be detected when the virus was injected in control mice (Fig. 1G and I) or when it was injected in the VMN, cortex, hippocampus, or paraventricular nucleus of the thalamus of Fgf15CretdT/+ mice—regions that do not express Fgf15 (Fig. 2A–D). Interestingly, we found that eYFP was expressed in the medial habenula (MHb), where Fgf15 was also found to be expressed by in situ hybridization (Fig. 2E). These data, thus, indicate that Cre expression faithfully reflects the sites of Fgf15 expression.
Figure 1.

Reporter mice to characterize Fgf15 neurons. A: Camera lucida cartography of Fgf15 expression in the DMH at the indicated bregma (mm). DMC, DMH compact part; DMD, DMH dorsal part; DMV, DMH ventral part; fx, fornix; LHA, lateral hypothalamic area; 3V, third ventricle. B: In situ hybridization fluorescence detection of Fgf15 mRNA in the dorsal, compact, and ventral divisions of the DMH at bregma −1.8 mm. Scale bar = 50 μm. Inset: Scale bar = 25 μm. C: Structure of the modified Fgf15 allele for the monocistronic expression of Fgf15, Cre, and tdTomato. D–F: Immunofluorescence detection of tdTomato (A), DAPI (B), and merge signals (C) in the ileal villi of Fgf15CreTdT/+ mice. Scale bar = 100 μm. G–J: Immunofluorescence detection of eYFP in the DMH (B and C) and PeF (D and E) of Fgf15+/+ and Fgf15CretdT/+ mice injected with recombinant AAV2-EF1a-DIO-EYFP. Scale bar = 50 μm.
Figure 2.

Fgf15 neurons of the DMH are glutamatergic. A–C: Immunofluorescence detection of eYFP in the VMH (A), lateral parietal association cortex (LPtA) (B), and field CA1 of the hippocampus (CA1) (C) of Fgf15CretdT/+ mice injected with recombinant AAV2-EF1a-DIO-EYFP. fx, fornix; 3V, third ventricle. Scale bar = 100 μm. D: eYFP-positive neurons were found in the MHb after injection in the paraventricular thalamus (PVT) of Fgf15CretdT/+ mice injected with recombinant AAV2-EF1a-DIO-EYFP. Scale bar = 100 μm. E: In situ hybridization detection of Fgf15 mRNA (red dots) in the MHb. Scale bar = 50 μm. F and G: Colocalization by in situ hybridization of eYFP (red) and vGlut2 (blue) in the DMH of Fgf15CretdT/+ mice previously injected with recombinant AAV2-EF1a-DIO-EYFP. Scale bar = 50 μm (F) and 10 μm (G). H and I: Colocalization by in situ hybridization of eYFP (red) and Gad67 (blue) in the DMH of Fgf15CretdT/+ mice previously injected with recombinant AAV2-EF1a-DIO-EYFP. Scale bar = 50 μm (H) and 10 μm (I). J: Quantification of eYFP and vGlut2 or Gad67 double-positive Fgf15 neurons.
To identify the neurotransmitter expressed in DMH Fgf15 neurons, we injected recombinant AAV2-EF1a-DIO-eYFP bilaterally in the DMH and performed in situ hybridization detection of eYFP and Vglut2 or Gad67. We found that 97% of eYFP-positive neurons also expressed Vglut2 (Fig. 2F, G, and J) and only 8% expressed Gad67 (Fig. 2H–J).
Fgf15 Neurons of the DMH Negatively Regulate Insulin-Induced Glucagon Secretion
We previously found that Fgf15 expression in the DMH negatively regulated 2DG-induced glucagon secretion. Here, we investigated the role of the Fgf15 neurons in the control of glucagon secretion triggered by insulin-induced hypoglycemia with use of a chemogenetic approach. Recombinant AAV8-hSyn-DIO-mCherry or AAV8-hSyn-DIO-hM3Dq-mCherry vectors were bilaterally injected in the DMH of Fgf15CretdT/+ mice (Fig. 3A). Neuronal activation induced by intraperitoneal Clz injection was first verified by assessment of c-fos expression in mCherry-positive cells. Figure 3B shows that Clz, but not saline injection, led to expression of c-fos in most hM3Dq-mcherry–positive neurons; Clz failed to activate neurons that only expressed mCherry (Fig. 3B).
Figure 3.
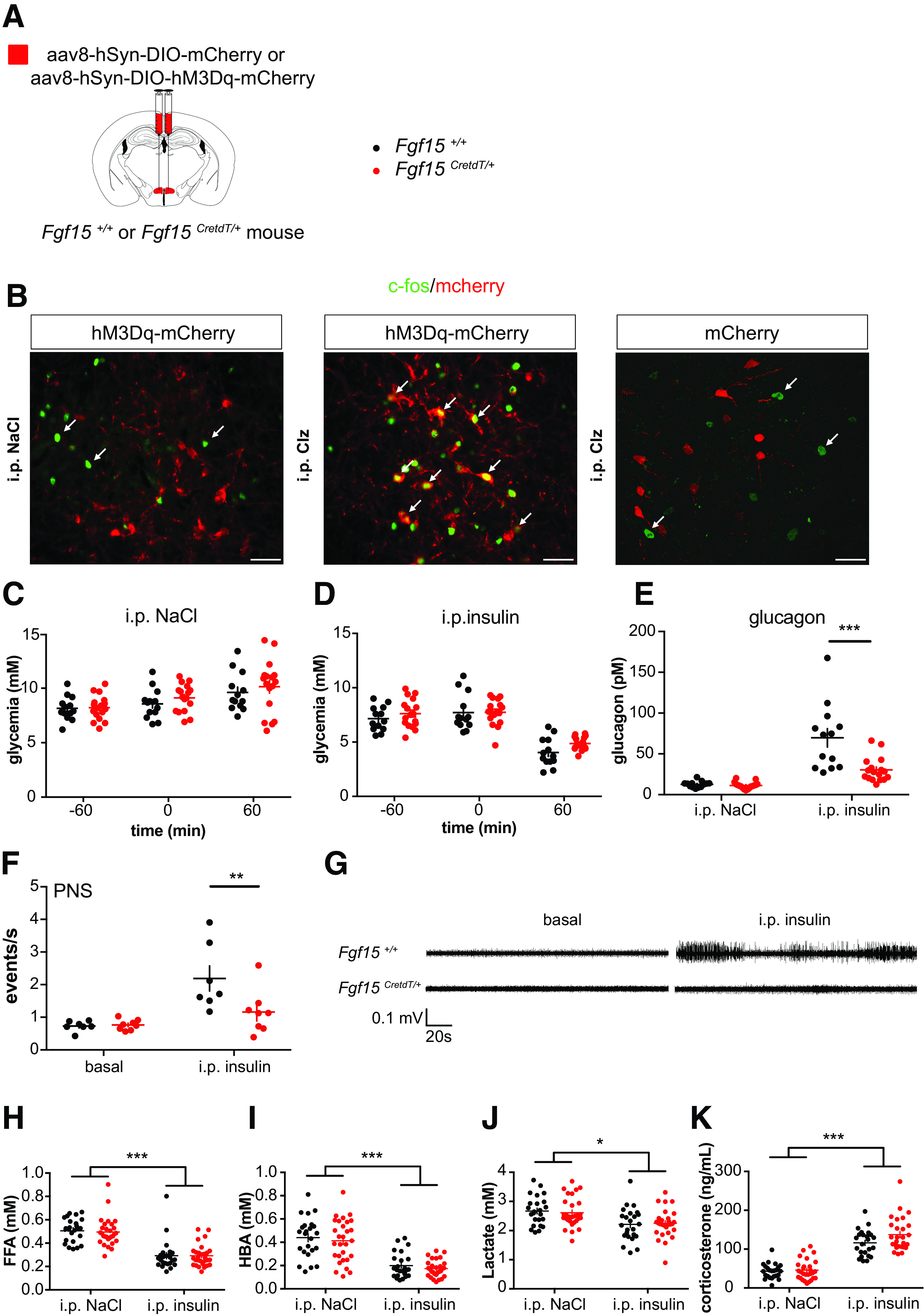
Fgf15 neurons activation negatively regulates insulin-induced glucagon secretion. A: Recombinant AAV8-hSyn-DIO-hM3Dq-mcherry injection in the DMH of Fgf15 +/+ or Fgf15CretdT/+ mice for Clz activation of Fgf15 neurons. B: Clz, but not saline intraperitoneal injection induced c-fos expression specifically in Fgf15 neurons expressing hM3Dq-mCherry. Scale bar = 50 μm. C: Glycemia at the time of Clz injection (−60 min), at the time of saline injection (0 min), and 1 h later (60 min). Fgf15 +/+ mice (black) (n = 13) and Fgf15CretdT/+ mice (red) (n = 18) previously injected as shown in A. D: Glycemia in the same conditions as for C, with insulin injection at time 0 min. E: Plasma glucagon. F and G: Vagus nerve activity was recorded in the basal state and following intraperitoneal insulin injection in Fgf15+/+ (n = 7) and Fgf15CreTdT/+ (n = 8) mice injected with Clz 30 min before the recordings were started. J: Quantification of the recorded vagal activities. K: Illustrations of activity recordings. H: Free fatty acids (FFA). I: Hydroxybutyrate (HBA). J: Lactate. K: Corticosterone levels measured 60 min after saline or insulin injections. Fgf15+/+ mice (black) (n = 24) and Fgf15CretdT/+ mice (red) (n = 29) previously injected as shown in A. Data are means ± SEM. *P < 0.05; **P < 0,005; ***P < 0,001. Two-way ANOVA followed by Bonferroni multiple comparisons test (E–J).
For determination of the effect of Fgf15 neuron activation on glucagon secretion, 6-h-fasted Fgf15CretdT/+ and Fgf15+/+ mice, which had received bilateral intra-DMH injections of recombinant AAV8-hSyn-DIO-hM3Dq-mcherry, were injected intraperitoneally with Clz and 60 min later with a saline solution. After an additional hour, the mice were bled for plasma glucagon measurements. Two weeks later the same experiment was repeated, except that instead of saline mice received insulin for induction of hypoglycemia. Glycemic levels were measured at the time of Clz, saline, or insulin injections and at the end of the experiments (Fig. 3C and D). Plasma glucagon levels were the same 60 min after saline injection in both groups of mice, but upon hypoglycemia induction, they were much less increased when Fgf15 neurons were activated (Fig. 3E). We next determined that chemogenetic activation of Fgf 15 neurons had no influence on the basal firing activity of the vagal nerve but prevented its activation by insulin-induced hypoglycemia (Fig. 3F and G). Plasma concentrations of free fatty acids, hydroxybutyric acid, lactate, and corticosterone were similarly modified by insulin-induced hypoglycemia whether Fgf15 neurons were activated or not (Fig. 3H–K). Thus, activation of Fgf15 neurons selectively dampens hypoglycemia-induced vagal activation and glucagon secretion (Fig. 8I).
Figure 8.

Retrograde mapping of DMH Fgf15 neuron innervation. A: AAV1-syn-FLEX-splitTVA-EGFP-tTA and AAV1-TREtight-mTagBFP2-B19G were injected bilaterally in the DMH of Fgf15CretdT/+ and an EnvA-G-deleted-mCherry pseudotyped rabies virus 1 week later. B: Immunofluorescence detection of EYFP/mCherry double-positive neurons in the DMH. Scale bar = 50 μm. Immunofluorescence detection of mCherry-positive presynaptic neurons in the septohypothalamic nucleus (SHy), MPO, MPA, ventrolateral preoptic nucleus (VLPO), and lateral preoptic area (LPO) (C) (scale bar = 50 μm); the paraventricular nucleus of the hypothalamus (PVH) (D) (scale bar = 50 μm); and the VMH, ARH, and lateral hypothalamic area (LHA) (E and F) (scale bar = 50 μm). G: Quantitative assessment of the neurons innervating DHM Fgf15 neurons based on their brain localization. AHN, anterior nucleus of the hypothalamus; AVPe, anteroventral periventricular nucleus; MM, medial mammillary nucleus, medial part; PMv, premammillary nucleus, ventral part; pfx, perifornical area; Pv, periventricular nucleus of the hypothalamus; RCH, retrochiasmatic area; SCh, suprachiasmatic nucleus; TU, tuberal nucleus. H: Graphical summary of the afferent inputs into Fgf15 neurons and their efferent projections. I: Graphical summary of the impact of Fgf15 neurons activation on autonomic nervous activity, glucagon secretion, and hepatic glucose production. DVC, dorsal vagal complex; IML, intermediolateral cell column; LC, locus coeruleus; PNS, parasympathetic nerve, SNS, sympathetic nerve; 3V, third ventricle.
Fgf15 Neurons Control Liver Neoglucogenesis Through a Creb1/Pepck Axis
We found that chemogenetic activation of Fgf15 neurons induced glucose intolerance (Fig. 4A). This deregulation was not caused by insufficient secretion of insulin (Fig. 4B) or insulin resistance (Fig. 4C). However, glucose turnover measurements in mice deprived of food for 3 h showed that activation of Fgf15 neurons significantly increased endogenous glucose production (Fig. 4D). Consistent with this observation, activation of Fgf15 neurons in overnight-fasted mice increased glycemia (5.27 ± 0.14 mmol/L glucose vs. 6.13 ± 0.24 mmol/L glucose for Fgf15+/+ and Fgf15CretdT/+ mice, respectively) (Fig. 4E). To obtain further evidence that activation of Fgf15 neurons stimulated hepatic glucose production, we analyzed the expression of Pck1 (encoding phosphoenolpyruvate carboxykinase) and Gck (glucokinase) at 60, 90, and 120 min after Clz injection in mice deprived of food for 3 h. At 60 min a significant induction of Pck1 was observed, which was reduced at later time points (Fig. 4F). Gck expression was nonsignificantly decreased at the three time points tested (Fig. 4G); however, the ratio of Pck1/Gck was significantly increased 60 and 90 min after Fgf15 neurons activation (Fig. 4H), indicating a shift of hepatic glucose metabolism toward neoglucogenesis. As Pck1 transcription is regulated by cAMP-responsive element–binding protein (Creb)1 (35), which is activated by phosphorylation on Ser133 by cAMP-dependent protein kinase A, we assessed Creb1 phosphorylation by Western blot analysis of liver protein extracts prepared 60 min after Clz injection. Activation of Fgf15 neurons indeed promoted Creb1 phosphorylation (Fig. 4I and J). We then quantified total and phosphorylated glycogen synthase (Gys) and glycogen phosphorylase (Pygl) by Western blot analysis. No differences were observed in the ratios of pGys to Gys and pPygl to Pygl 60 min after activation of Fgf15 neurons (Fig. 4K and L), indicating that glycogen degradation was not regulated. Finally, to test whether these modifications were inducing gluconeogenesis, we performed a pyruvate tolerance test. This showed higher glycemic excursion following pyruvate injection when Fgf15 neurons had been activated 60 min earlier (Fig. 4M and N).
Figure 4.
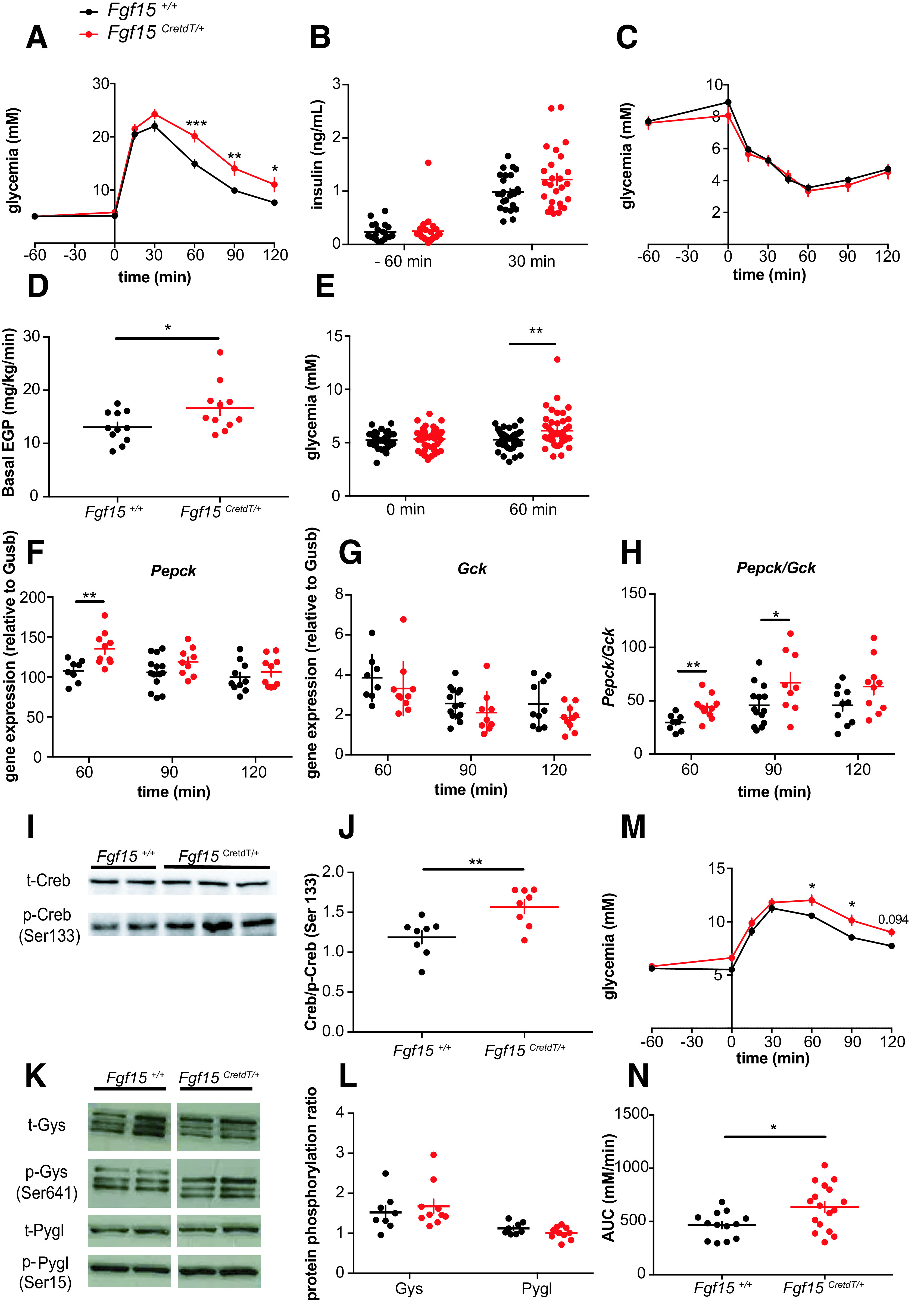
Activated Fgf15 neurons increase liver gluconeogenesis through a Creb1/Pepck axis. A: Intraperitoneal glucose tolerance test performed 1 h after Clz injection (Fgf15+/+ mice, n = 26, and Fgf15CretdT/+ mice, n = 26). B: Plasma insulin at the time of Clz injection (−60 min) and 30 min after intraperitoneal glucose injection (Fgf15+/+ mice, n = 26, and Fgf15CretdT/+ mice, n = 26). C: Insulin tolerance test performed 1 h after Clz injection (Fgf15+/+ mice, n = 9, and Fgf15CretdT/+ mice, n = 12). D: Endogenous glucose production following Clz injection (Fgf15+/+ mice, n = 11, and Fgf15CretdT/+ mice, n = 11). E: Glycemia before and 1 h following Clz injection in overnight-fasted Fgf15+/+ mice (n = 39) and Fgf15CretdT/+ mice (n = 43). F–H: Liver expression levels of Pepck (E) and Gck (F) mRNAs and Pepck-to-Gck ratio (G) at the indicated times after Clz injection (Fgf15+/+ mice, n = 8–14, and Fgf15CretdT/+ mice, n = 9–10). I–J: Western blot analysis of hepatic pCreb1 and total (t-)Creb1 at 1 h after Clz injection (Fgf15+/+ mice, n = 8, and Fgf15CretdT/+ mice, n = 8). K and L: Western blot analysis of pGys, pPygl, and total Gys and Pygl 1 h after Clz injection (Fgf15+/+ mice, n = 8, and Fgf15CretdT/+ mice, n = 10). M: Pyruvate tolerance in response to an intraperitoneal bolus of pyruvate 1 h after Clz injection (Fgf15+/+ mice, n = 13, and Fgf15CretdT/+ mice, n = 17). N: Areas under the curve (AUC) of the pyruvate tolerance tests. Data are means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Two-way ANOVA followed by Bonferroni multiple comparisons test: A, E, and M. Two-way ANOVA followed by Sidak multiple comparisons test: F and H. Student t test: D, I, and N.
Fgf15 Neurons Activate the Sympathetic Nervous System
Activation of hepatic glucose production could be due to an increase in glucagon secretion and activation of hepatic glucagon receptor or to an adrenergic stimulation of the liver β-2 adrenergic receptors (Adrb2). We found that activation of Fgf15 neurons did not impact plasma glucagon levels measured 30 min after an intraperitoneal glucose injection (Fig. 5A). However, administration of the selective Adrb2 blocker butaxamine abolished the glucose intolerance induced by chemogenetic activation of Fgf15 neurons (Fig. 5B [compare with Fig. 4A]). Direct recording of sympathetic nerve firing confirmed that chemogenetic activation of Fgf15 neurons increased sympathetic activity in the basal state and prevented the suppressive effect of glucose injection (Fig. 5C).
Figure 5.
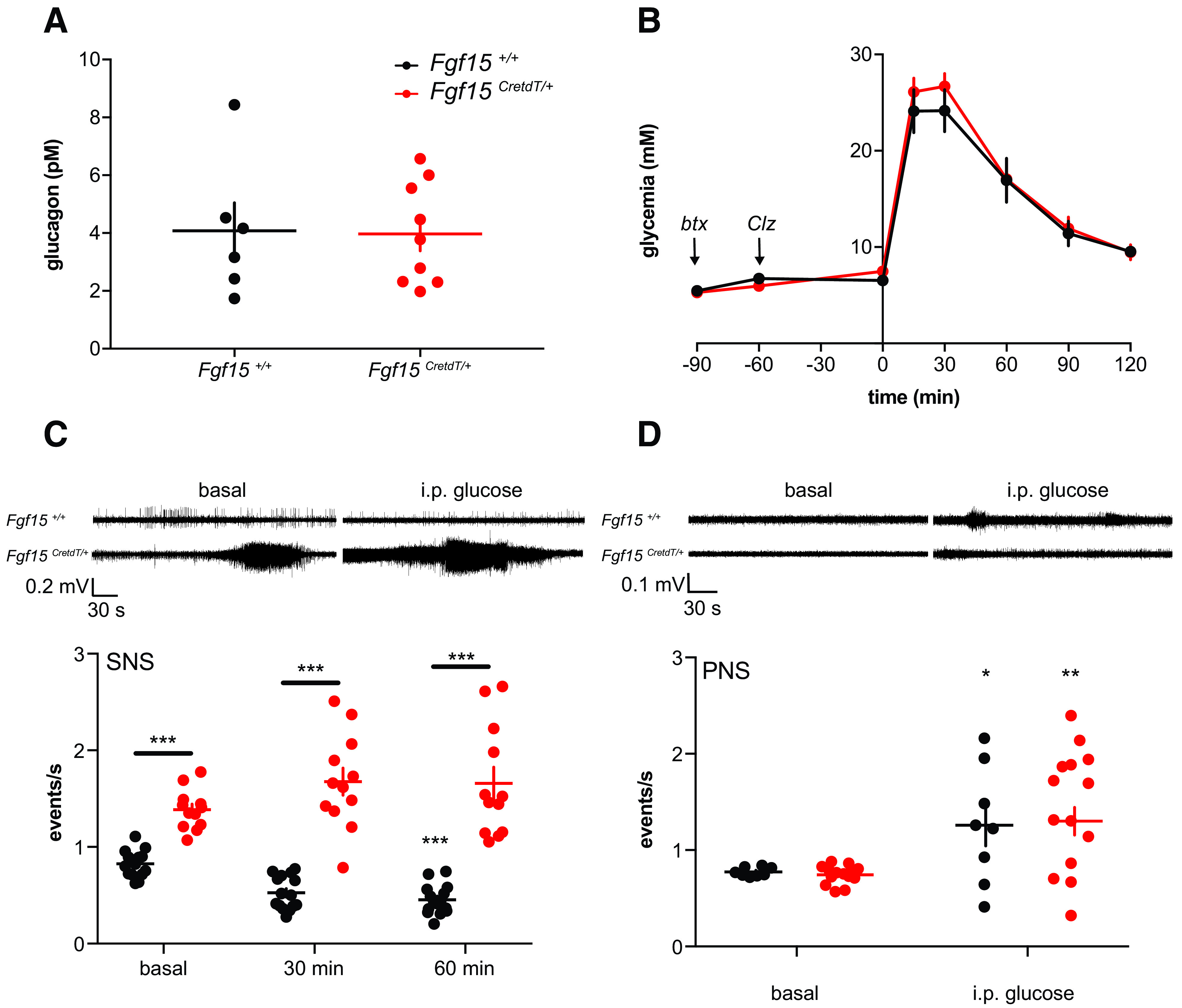
Activated Fgf15 neurons induce glucose intolerance by increasing β-2 adrenergic signaling. A: Plasma glucagon 30 min after intraperitoneal injection of glucose in mice previously injected with Clz (Fgf15+/+ mice, n = 6, Fgf15CreTdT/+ mice, n = 9). B: Intraperitoneal glucose tolerance test in mice injected with the Adrb2 blocker butaxamine (btx) (−90 min) and Clz (−60 min) and with glucose (0 min) (Fgf15+/+ mice, n = 6, and Fgf15CretdT/+ mice, n = 6). C: Sympathetic nerve (SNS) firing rates after intraperitoneal injection of Clz in the basal state and at the indicated times after intraperitoneal glucose injection (Fgf15+/+ mice, n = 15, and Fgf15CreTdT/+ mice, n = 12). D: Parasympathetic nerve (PNS) firing rate after intraperitoneal injection of Clz in the basal state and 30 min after glucose injection (Fgf15+/+ mice, n = 8, and Fgf15CreTdT/+ mice, n = 14). Data are means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Two-way ANOVA followed by Bonferroni multiple comparisons test (C and D).
Finally, we also measured whether Fgf15 neurons activation would impact on the response to glucose of the vagal nerve. As expected, glucose increased vagal nerve activity but Fgf15 neuron activation did not change basal or glucose-activated firing activity (Fig. 5D). This is to be compared with the data of Fig. 3 that showed that Fgf15 neurons activation prevented the stimulation of vagal nerve by insulin-induced hypoglycemia.
Collectively, our data show that Fgf15 neurons of the DMH control sympathetic nerve activity and Adrb2-dependent induction of a Creb1/Pepck axis to increase hepatic gluconeogenesis; the consequence of activating this DMH-liver axis is induction of glucose intolerance (Fig. 8I).
Glucose Responsiveness of DMH Fgf15 Neurons
To assess whether the Fgf15 neurons were directly responsive to changes in extracellular glucose concentrations, we injected recombinant AAV2-Ef1a-DIO-eYFP in the DMH of Fgf15CretdT/+ mice (Fig. 6A) and performed patch clamp analysis of eYFP-expressing neurons exposed to 2.5 mmol/L or 0.5 mmol/L glucose (14,15). We found that of 23 recorded neurons, 15 were glucose-nonresponding neurons (NR) (Fig. 6C–E); 6 were GE neurons, which had increased membrane potential and membrane resistance when the glucose concentration was raised (Fig. 6F–H); and 2 were GI neurons activated by a decrease in glucose concentrations (Fig. 6I–K).
Figure 6.
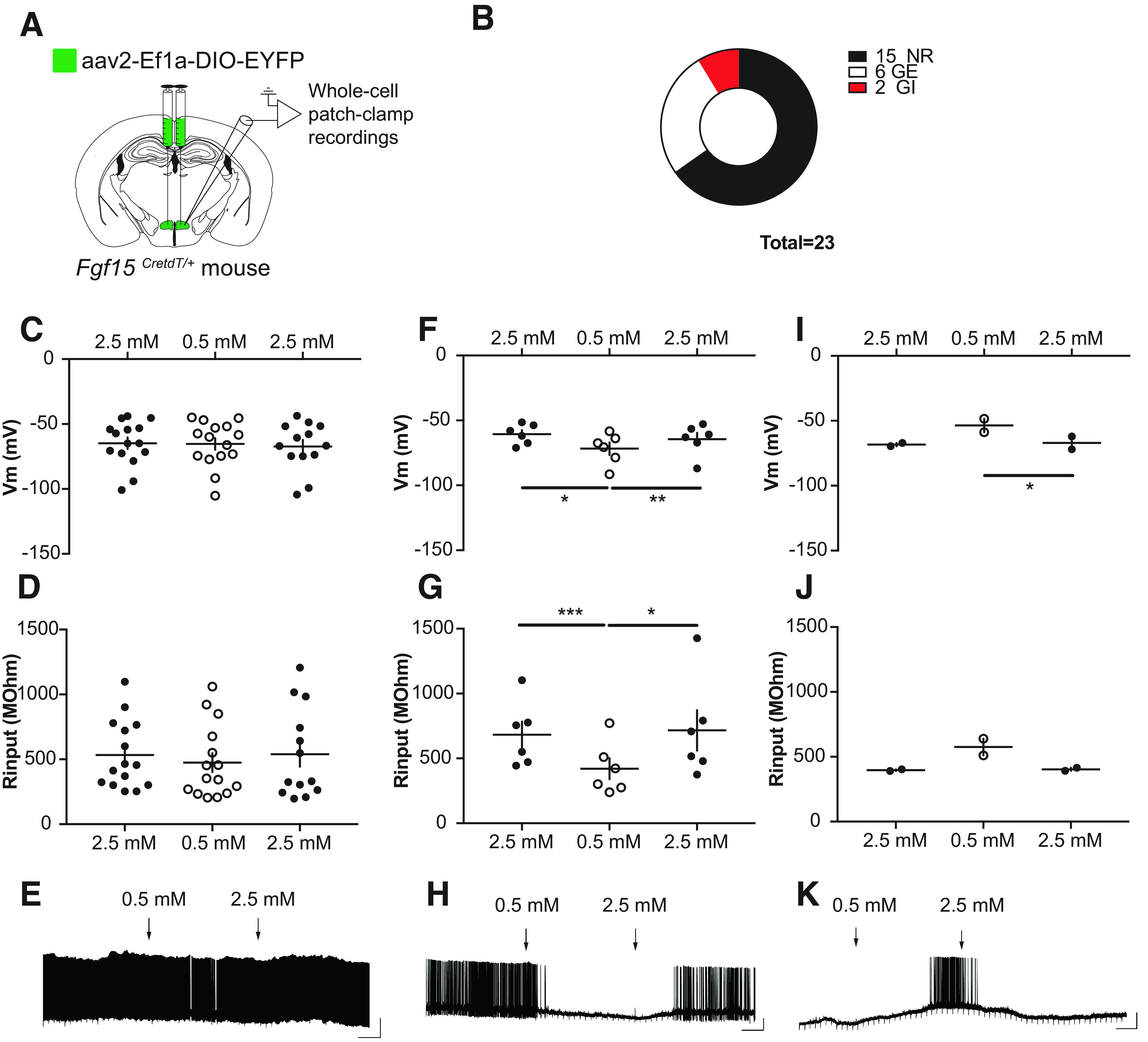
Glucose responsiveness of DMH Fgf15 neurons. A: Recombinant AAV2-EF1a-DIO-EYFP was injected in the DMH of Fgf15CreTdT/+ mice to allow fluorescence visualization of Fgf15 neurons. B: Number of recorded NR, GE, or GI Fgf15 neurons. C–E: Membrane potential (C), membrane resistance (D), and representative recording (E) of NR Fgf15 neurons. F–H: Membrane potential (F), membrane resistance (G), and representative recording (H) of GE Fgf15 neurons. I–K: Membrane potential (I), membrane resistance (J), and representative recording (K) of GI Fgf15 neurons. Data are shown as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0,001. One-way ANOVA followed by Bonferroni multiple comparisons test (F, G, and I).
DMH Fgf15 Neurons Efferent Projections and Afferent Innervation
To map the anterograde projections of Fgf15 neurons, we injected bilaterally in the DMH of Fgf15CretdT/+ mice an adenoviral vector allowing for the Cre-dependent expression of a synaptophysin-mCherry fusion protein that labels synaptic vesicles (32) (Fig. 7A). We found that Fgf15 neurons send numerous intra-DMH projections (Fig. 7B). The other sites identified were the medial preoptic area (MPA) (Fig. 7C and D), the medial preoptic nucleus (MPO) (Fig. 7E and F), the arcuate nucleus of the hypothalamus (ARH) (Fig. 7G and H), and the locus coeruleus (Fig. 7I and K). The origin of presynaptic neurons contacting Fgf15 neurons was assessed with a pseudorabies retrograde labeling technique (Fig. 8A). The injection site identified by the presence of eGFP and mCherry double-positive neurons was restricted to the DMH (Fig. 8B). Most of the presynaptic neuronal populations were localized at the hypothalamic level in different nuclei of the preoptic area (Fig. 8C and G), in the paraventricular nucleus (Fig. 8D and G), in the lateral hypothalamus and PeF, and in the VMN and the ARH (Fig. 8E–G). Minor presynaptic neuronal populations were identified in the medial part of the medial mammillary nucleus, the ventral part of the premammillary nucleus, the tuberal nucleus, the retrochiasmatic area, the suprachiasmatic nucleus, and the anterior nucleus of the hypothalamus and in the anteroventral periventricular nucleus (Fig. 8G). Some neurons cells were also identified in the anterior part of the bed nucleus of the stria terminalis, the ventrolateral periaqueductal gray, and the raphe magnus nucleus (data not shown). These data are summarized in Fig. 8H.
Figure 7.

Mapping of DMH Fgf15 neuron synaptic projections. A: An Ad-CMV-synaptophysin-mCherry was injected bilaterally in the DMH of Fgf15CretdT/+ mice to map synaptic terminals of Fgf15 neurons. Immunofluorescence detection of mCherry showing positive cell bodies and fibers in the DMH (B) (scale bar = 50 μm), the MPA (C and D) (scale bar = 50 μm and 25 μm, respectively), the MPO (E and F) (scale bar = 50 μm and 25 μm), the ARH (G and H) (scale bar = 50 μm and 25 μm), and the locus coeruleus (LC) (I–K) (scale bar = 50 μm and 25 μm). 3V, third ventricle; 4V, fourth ventricle.
Discussion
In the current study, we generated a new mouse model (Fgf15CretdT/+) that allowed for the further characterization of the glucoregulatory role of Fgf15 neurons of the DMH. We found that these neurons were glutamatergic and displayed different sensitivities to changes in extracellular glucose concentrations. Physiologically, we showed that their chemogenetic activation had a dual role in the regulation of the autonomic nervous system. On the one hand, they prevented hypoglycemia-induced vagal nerve firing and glucagon secretion, while on the other hand, they increased sympathetic nervous activity leading to induction of the liver Creb1/Pck1 axis, hepatic glucose production, and glucose intolerance. Together, these data identify the role of a newly characterized subpopulation of DMH neurons, which directly promote hepatic glucose production by stimulating sympathetic nervous activity.
Fgf15 is a member of the fibroblast growth factor family that lacks a heparan sulfate proteoglycan–binding motif and, thus, diffuses in the circulation to act as a hormone (36). The major site of Fgf15 production is the intestinal endothelium, and Fgf15 was first characterized for its role in regulating bile acid synthesis (34,37). In our search for novel hypothalamic regulators of glucagon secretion, we found that Fgf15 was also expressed in neurons of the DMH and the PeF and that silencing its expression in the DMH, but not in the PeF, increased glucagon secretion in response to 2DG-induced neuroglucopenia (23), indicating that Fgf15 can also be a neurotransmitter.
Here, we generated Fgf15CretdT/+ reporter mice, which express the Cre recombinase and tdTomato from the Fgf15 locus. The intestinal epithelial cells of these mice, where Fgf15 is highly expressed, showed strong tdTomato fluorescence. In the brain, Fgf15 is expressed at a much lower level than in the intestine (23), and identification of Cre-expressing cells required the stereotactic delivery of a Cre-dependent eYFP viral expression system. Comparing the pattern of Fgf15 mRNA expression detected by in situ hybridization with that of eYFP expression detected by fluorescence microscopy indicated that the reporter mice allowed for specific expression of the Cre recombinase in Fgf15 neurons. In addition to expression of eYFP in the DMH and PeF, we found expression in the MHb, a bilateral epithalamic nucleus involved in the regulation of fear, anxiety, sleep, aversion, and reward (38). The availability of Fgf15CretdT/+ mice may help with investigation of the specific behavioral roles of these habenula neurons.
In the DMH, Fgf15 neurons were found to be almost exclusively glutamatergic, whereas this nucleus contains a majority of GABAergic neurons (39,40). Chemogenetic activation of Fgf15 neurons of the DMH induced glucose intolerance as assessed following an intraperitoneal glucose injection. This intolerance could not be explained by reduced insulin secretion, increased insulin resistance, or higher plasma glucagon levels. Instead, it was associated with increased hepatic glucose production, a response induced by a stimulation of sympathetic nervous activity leading to increased hepatic Creb1 phosphorylation and Pck1 expression. We showed that, at the same time as they increase liver gluconeogenesis, Fgf15 neurons negatively regulate insulin-induced vagus nerve activity and glucagon secretion. This impaired hormonal response is in line with our previous study showing that intracerebroventricular FGF19 injections reduced neuroglucopenia-induced vagal activity and glucagon secretion, whereas silencing Fgf15 expression in the DMH increased glucagon secretion (23). The fact that the Fgf15 neurons stimulate hepatic glucose production through sympathetic signaling while restricting vagally induced glucagon secretion suggests that these two counterregulatory mechanisms need to be antagonistically regulated to prevent an exaggerated hyperglycemic response. Alternatively, as glucagon has many physiological effects beyond its glucoregulatory role, in particular reducing food intake (41,42), a direct sympathetic stimulation of hepatic glucose production may avoid the anorectic effect of glucagon.
The Fgf15 neurons form a subpopulation of cells in the DMH involved in glucoregulation. Our electrophysiological analysis showed that they comprise GE, GI, and NR neurons. However, whether their glucose sensitivity is directly involved in their physiological function needs to be determined. Their activity may, however, be regulated by homeostatic signals received from the several hypothalamic nuclei identified by our retrograde-labeling experiments, including the LH, ARH, or ventromedial hypothalamic nucleus (VMH), which contain GI neurons that contribute to the counterregulatory response to hypoglycemia. On the other hand, the downstream projections of Fgf15 neurons, which control autonomic nervous activity, are not yet identified. Nevertheless, it is interesting that efferent projections were found in the locus coeruleus, a preautonomic center whose activation increases sympathetic and decreases parasympathetic activity (43), consistent with the consequences of activating Fgf15 neurons described here.
Collectively, our data provide compelling evidence that Fgf15 neurons form a subpopulation of glutamatergic DMH neurons involved in the control of glucose homeostasis. When activated, they increase the sympathetic tone to the liver to increase gluconeogenesis via the Creb1/Pepck axis, and in the presence of insulin-induced hypoglycemia, they blunt vagal nerve activation and reduce glucagon secretion. These neurons therefore contribute to the counterregulatory response to hypoglycemia by increasing hepatic glucose production. It is, however, not yet established whether they are directly activated by hypoglycemia to increase the counterregulatory response or whether they form a link in an integrated neuronal circuit that prevents hypoglycemia development and where they would function as modulators of such circuit. Nevertheless, our observation of their dual role in preventing hypoglycemia-activated vagal activity and stimulating parasympathetic nervous activity indicates that they play an important role in the regulation of autonomous nervous activity in glucose homeostasis.
Article Information
Acknowledgments. The authors thank Dr. Martin Myers Jr. (University of Michigan) for the gift of adenoviruses and the staff of the metabolic analysis platform of the Center for Integrative Genomics for the glucose turnover analysis.
Funding. This work was supported by a European Research Council Advanced Grant (INTEGRATE, no. 694798) and a Swiss National Science Foundation grant (310030-182496) to B.T. and has received funding from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement no. 777460 (HypoRESOLVE). The JU receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA and T1D Exchange, JDRF, International Diabetes Federation, and The Leona M. and Harry B. Helmsley Charitable Trust.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. A.P. and B.T. conceived the project. A.P., G.L., and B.T. designed the experiments. A.P., S.M., D.T., W.D., X.B., S.C., and G.L. performed the experiments. A.P., G.L., and B.T. analyzed the data and wrote the manuscript. B.T. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1. Routh VH. Glucose-sensing neurons: are they physiologically relevant? Physiol Behav 2002;76:403–413 [DOI] [PubMed] [Google Scholar]
- 2. Marty N, Dallaporta M, Thorens B. Brain glucose sensing, counterregulation, and energy homeostasis. Physiology (Bethesda) 2007;22:241–251 [DOI] [PubMed] [Google Scholar]
- 3. Silver IA, Erecińska M. Glucose-induced intracellular ion changes in sugar-sensitive hypothalamic neurons. J Neurophysiol 1998;79:1733–1745 [DOI] [PubMed] [Google Scholar]
- 4. Verberne AJ, Sabetghadam A, Korim WS. Neural pathways that control the glucose counterregulatory response. Front Neurosci 2014;8:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thorens B. Neural regulation of pancreatic islet cell mass and function. Diabetes Obes Metab 2014;16(Suppl. 1):87–95 [DOI] [PubMed] [Google Scholar]
- 6. Beall C, Ashford ML, McCrimmon RJ. The physiology and pathophysiology of the neural control of the counterregulatory response. Am J Physiol Regul Integr Comp Physiol 2012;302:R215–R223 [DOI] [PubMed] [Google Scholar]
- 7. Cryer PE. Hypoglycemia: still the limiting factor in the glycemic management of diabetes. Endocr Pract 2008;14:750–756 [DOI] [PubMed] [Google Scholar]
- 8. Cryer PE, Davis SN, Shamoon H. Hypoglycemia in diabetes. Diabetes Care 2003;26:1902–1912 [DOI] [PubMed] [Google Scholar]
- 9. Dallaporta M, Himmi T, Perrin J, Orsini JC. Solitary tract nucleus sensitivity to moderate changes in glucose level. Neuroreport 1999;10:2657–2660 [DOI] [PubMed] [Google Scholar]
- 10. Lamy CM, Sanno H, Labouèbe G, et al. Hypoglycemia-activated GLUT2 neurons of the nucleus tractus solitarius stimulate vagal activity and glucagon secretion. Cell Metab 2014;19:527–538 [DOI] [PubMed] [Google Scholar]
- 11. Ritter S, Llewellyn-Smith I, Dinh TT. Subgroups of hindbrain catecholamine neurons are selectively activated by 2-deoxy-D-glucose induced metabolic challenge. Brain Res 1998;805:41–54 [DOI] [PubMed] [Google Scholar]
- 12. Chan O, Sherwin R. Influence of VMH fuel sensing on hypoglycemic responses. Trends Endocrinol Metab 2013;24:616–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou C, Teegala SB, Khan BA, Gonzalez C, Routh VH. Hypoglycemia: role of hypothalamic glucose-inhibited (GI) neurons in detection and correction. Front Physiol 2018;9:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Steinbusch LK, Picard A, Bonnet MS, Basco D, Labouèbe G, Thorens B. Sex-specific control of fat mass and counterregulation by hypothalamic glucokinase. Diabetes 2016;65:2920–2931 [DOI] [PubMed] [Google Scholar]
- 15. Quenneville S, Labouèbe G, Basco D, et al. Hypoglycemia-sensing neurons of the ventromedial hypothalamus require AMPK-induced Txn2 expression but are dispensable for physiological counterregulation. Diabetes 2020;69:2253–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meek TH, Nelson JT, Matsen ME, et al. Functional identification of a neurocircuit regulating blood glucose. Proc Natl Acad Sci U S A 2016;113:E2073–E2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lindberg D, Chen P, Li C. Conditional viral tracing reveals that steroidogenic factor 1-positive neurons of the dorsomedial subdivision of the ventromedial hypothalamus project to autonomic centers of the hypothalamus and hindbrain. J Comp Neurol 2013;521:3167–3190 [DOI] [PubMed] [Google Scholar]
- 18. Cardin S, Jackson PA, Edgerton DS, Neal DW, Coffey CS, Cherrington AD. Effect of vagal cooling on the counterregulatory response to hypoglycemia induced by a low dose of insulin in the conscious dog. Diabetes 2001;50:558–564 [DOI] [PubMed] [Google Scholar]
- 19. Hevener AL, Bergman RN, Donovan CM. Portal vein afferents are critical for the sympathoadrenal response to hypoglycemia. Diabetes 2000;49:8–12 [DOI] [PubMed] [Google Scholar]
- 20. Burcelin R, Dolci W, Thorens B. Glucose sensing by the hepatoportal sensor is GLUT2-dependent: in vivo analysis in GLUT2-null mice. Diabetes 2000;49:1643–1648 [DOI] [PubMed] [Google Scholar]
- 21. Thorens B. Brain glucose sensing and neural regulation of insulin and glucagon secretion. Diabetes Obes Metab 2011;13(Suppl. 1):82–88 [DOI] [PubMed] [Google Scholar]
- 22. Peirce JL, Lu L, Gu J, Silver LM, Williams RW. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet 2004;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Picard A, Soyer J, Berney X, et al. A genetic screen identifies hypothalamic Fgf15 as a regulator of glucagon secretion. Cell Rep 2016;17:1795–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koentgen F, Lin J, Katidou M, et al. Exclusive transmission of the embryonic stem cell-derived genome through the mouse germline. Genesis 2016;54:326–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates, Cambridge, Massachusetts: Academic Press; 1982 [Google Scholar]
- 26. Minehira K, Young SG, Villanueva CJ, et al. Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J Lipid Res 2008;49:2038–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kamohara S, Burcelin R, Halaas JL, Friedman JM, Charron MJ. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature 1997;389:374–377 [DOI] [PubMed] [Google Scholar]
- 28. Mounien L, Marty N, Tarussio D, et al. Glut2-dependent glucose-sensing controls thermoregulation by enhancing the leptin sensitivity of NPY and POMC neurons. FASEB J 2010;24:1747–1758 [DOI] [PubMed] [Google Scholar]
- 29. Barbier M, González JA, Houdayer C, Burdakov D, Risold PY, Croizier S. Projections from the dorsomedial division of the bed nucleus of the stria terminalis to hypothalamic nuclei in the mouse. J Comp Neurol 2021;529:929–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Magnan C, Collins S, Berthault MF, et al. Lipid infusion lowers sympathetic nervous activity and leads to increased beta-cell responsiveness to glucose. J Clin Invest 1999;103:413–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tarussio D, Metref S, Seyer P, et al. Nervous glucose sensing regulates postnatal β cell proliferation and glucose homeostasis. J Clin Invest 2014;124:413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Opland D, Sutton A, Woodworth H, et al. Loss of neurotensin receptor-1 disrupts the control of the mesolimbic dopamine system by leptin and promotes hedonic feeding and obesity. Mol Metab 2013;2:423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu K, Kim J, Kim DW, et al. Lhx6-positive GABA-releasing neurons of the zona incerta promote sleep. Nature 2017;548:582–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005;2:217–225 [DOI] [PubMed] [Google Scholar]
- 35. Liu JS, Park EA, Gurney AL, Roesler WJ, Hanson RW. Cyclic AMP induction of phosphoenolpyruvate carboxykinase (GTP) gene transcription is mediated by multiple promoter elements. J Biol Chem 1991;266:19095–19102 [PubMed] [Google Scholar]
- 36. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov 2009;8:235–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jung D, Inagaki T, Gerard RD, et al. FXR agonists and FGF15 reduce fecal bile acid excretion in a mouse model of bile acid malabsorption. J Lipid Res 2007;48:2693–2700 [DOI] [PubMed] [Google Scholar]
- 38. Roberson S, Halpern ME. Development and connectivity of the habenular nuclei. Semin Cell Dev Biol 2018;78:107–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liao GY, Kinney CE, An JJ, Xu B. TrkB-expressing neurons in the dorsomedial hypothalamus are necessary and sufficient to suppress homeostatic feeding. Proc Natl Acad Sci U S A 2019;116:3256–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vong L, Ye C, Yang Z, Choi B, Chua S Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 2011;71:142–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Habegger KM, Heppner KM, Geary N, Bartness TJ, DiMarchi R, Tschöp MH. The metabolic actions of glucagon revisited. Nat Rev Endocrinol 2010;6:689–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Castro JM, Paullin SK, DeLugas GM. Insulin and glucagon as determinants of body weight set point and microregulation in rats. J Comp Physiol Psychol 1978;92:571–579 [DOI] [PubMed] [Google Scholar]
- 43. Samuels ER, Szabadi E. Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part II: physiological and pharmacological manipulations and pathological alterations of locus coeruleus activity in humans. Curr Neuropharmacol 2008;6:254–285 [DOI] [PMC free article] [PubMed] [Google Scholar]