

Hermansky-Pudlak syndrome (HPS), a rare autosomal recessive disorder characterized by defective biogenesis of lysosome-related organelles (LROs), affects approximately 1-9 per 1,000,000 individuals worldwide (www.orpha.net) [1]. Individuals with HPS manifest with a bleeding diathesis and oculocutaneous albinism; some also develop inflammatory bowel disease, pulmonary fibrosis, and/or neutropenia [1, 2]. The diagnosis of HPS is established in a patient with a compatible clinical presentation by demonstrating absent platelet delta granules by whole mount electron microscopy (Figure 1) and/or identifying bi-allelic pathogenic variants in an HPS-associated gene.
Figure 1.
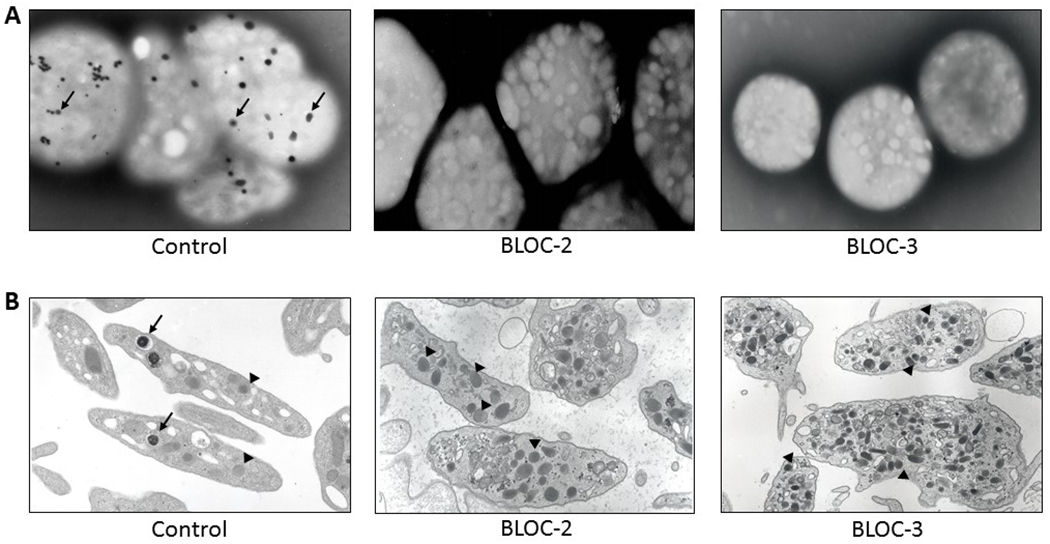
Ultrastructural imaging of platelets.
A: Whole mount electron micrographs of control platelets (×4000), each containing several delta granules (arrows) and platelets of HPS patients with defects in BLOC-2 or BLOC-3 (×6000), each showing absence of delta granules.
B: Thin section micrographs of platelets from control and HPS patients with defects in BLOC-2 or BLOC-3. Control platelets (×16000) contain delta granules (arrows), which are lacking in BLOC-2 (x20000) and BLOC-3 (x22000) defective platelets. Other cytoplasmic components include alpha granules (arrowheads), which appear similar in size, number and shape in BLOC-2 and BLOC-3 defective platelets compared to control platelets.
HPS displays locus heterogeneity; to date, ten genes associated with HPS are identified (Table I). Each HPS-associated gene encodes a protein subunit of either Biogenesis of Lysosome-related Organelles Complex (BLOC)-1, BLOC-2, BLOC-3, or the Adaptor Protein-3 (AP-3) complex [3]. These multi-subunit complexes are involved in formation of LROs, such as delta granules in platelets and melanosomes in melanocytes [3, 4]. All HPS types manifest with a bleeding diathesis and oculocutaneous albinism. Individuals with BLOC-1 (HPS types 7, 8, or 9) or BLOC-2 (HPS types 3, 5, or 6) related HPS generally exhibit mild clinical symptoms, including mild hypopigmentation. Individuals with BLOC-3 (HPS types 1 or 4) or AP-3 (HPS types 2 or 10) related HPS have more pronounced pigment defects of the skin, hair, and eyes and often have severe ancillary symptoms including progressive pulmonary fibrosis, inflammatory bowel disease, or neutropenia [2, 3].
Table I.
Genes Associated with Hermansky-Pudlak Syndrome1
| HPS type | Gene | GeneID; mRNA2 (# exons) | Locus | Protein | Protein ID2 (# aa; Mw) | Complex | ~ # variants reported3 |
|---|---|---|---|---|---|---|---|
| HPS-1 | HPS1 | ID:3257; NM_000195 (20 exons) |
10q24.2 | HPS1 |
NP_000186 (700 aa; 79.3 kD) |
BLOC-3 | 64 |
| HPS-2 | AP3B1 | ID:8546; NM_003664 (27 exons) |
5q14.1 | AP-3 Beta 3A subunit |
NP_003655 (1094 aa; 121.3 kD) |
AP-3 | 38 |
| HPS-3 | HPS3 | ID:84343; NM_032383 (17 exons) |
3q24 | HPS3 |
NP_115759 (1004 aa; 113.7 kD) |
BLOC-2 | 23 |
| HPS-4 | HPS4 | ID:89781; NM_022081 (14 exons) |
22q12.1 | HPS4 |
NP_071364 (708 aa; 76.9 kD) |
BLOC-3 | 30 |
| HPS-5 | HPS5 | ID:11234; NM_181507 (23 exons) |
11p15.1 | HPS5 |
NP_852608 (1129 aa; 127.4kD) |
BLOC-2 | 31 |
| HPS-6 | HPS6 | ID:79803; NM_024747 (1 exon) |
10q24.32 | HPS6 |
NP_079023 (775 aa; 83.0 kD) |
BLOC-2 | 40 |
| HPS-7 | DTNBP1 | ID:84062; NM_032122 (10 exons) |
6p22.3 | Dysbindin |
NP_115498 (351 aa; 39.5 kD) |
BLOC-1 | 3 |
| HPS-8 | BLOC1S3 | ID:388552; NM_212550 (2 exons) |
19q13.32 | BLOC-1 subunit 3 |
NP_997715 (202 aa; 21.3 kD) |
BLOC-1 | 4 |
| HPS-9 | PLDN | ID:26258; NM_001311255 (5 exons) |
15q21.1 | Pallidin |
NP_001298184 (177 aa; 20.3 kD) |
BLOC-1 | 2 |
| HPS-10 | AP3D1 | ID:8943; NM_001261826 (32 exons) |
19p13.3 | AP-3 delta subunit |
NP_001248755 (1215 aa; 136.7 kD) |
AP-3 | 1 |
Abbreviations: aa, amino acids; AP, Adaptor Protein; BLOC, Biogenesis of Lysosome-related Organelles Complex; kD, kilodaltons; Mw, molecular weight.
As of July 2019
Genbank accession numbers of the mRNA and protein product encoding the longest isoform (often transcript variant 1), its number of exons, amino acids and predicted protein molecular weight.
Reported in the Human Gene Mutation Database (Professional 2019.1; http://www.hgmd.cf.ac.uk/) and additional literature reports.
HPS is a rare disorder affecting people of different ethnicities, including African American, Ashkenazi-Jewish, Chinese, Indian, Israeli Bedouin, Japanese, Latino, and Western European [1]. The prevalence of each HPS type varies due to founder mutations (Table I). HPS type 1 is the most common type, with Puerto-Rican, Swiss and Japanese genetic isolates [5–7]. In northwest Puerto Rico, the prevalence of HPS type 1 is 1 in 1,800, and approximately 80% of affected individuals from this region are homozygous for a 16-base pair duplication (c.1470_1486dup16) in HPS1 [5, 8]. Other HPS1 variants also occur in Puerto Rican patients with HPS type 1 [9]. HPS type 3 is common in central Puerto Rico, where about 1 in 4,100 individuals are affected due to a 3,904-bp deletion in HPS3 [8, 10]. Disease-causing variants in the 10 HPS genes are reported in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), Albinism Database (http://www.ifpcs.org/albinism/), and AP3B1base (http://structure.bmc.lu.se/idbase/AP3B1base/) (Table I). There may be other HPS genes that have not yet been identified in humans, and it is possible that genetic testing of patients with excessive bleeding (and minor hypopigmentation) of unknown etiology may lead to the discovery of new genes associated with HPS.
Individuals with HPS have an increased tendency to bleed which is attributed to a platelet delta storage pool deficiency. Delta granules are absent in platelets from individuals with HPS (Figure 1), which causes impairment of secondary aggregation, prolongation of bleeding times, and abnormal alpha granule secretion [4, 11,12]. Platelet delta and alpha granules arise from megakaryocyte multivesicular bodies, but ultrastructural studies show normal distribution, quantity, and size of alpha granules in patients with HPS (Figure 1) [13], suggesting that HPS gene products function in biogenesis of delta granules, and not alpha granules.
Individuals with HPS exhibit easy bruisability and increased bleeding. Excessive blood loss can be seen with medical procedures, dental extractions, or surgery, and trauma can cause significant hemorrhage [2]. Ecchymoses may be unusual in size or location, and they may be in various stages of resolution [2]. Venipuncture sites may bleed for prolonged periods. Children with HPS may experience severe epistaxis or pronounced bleeding with loss of deciduous teeth [2]. Individuals with inflammatory bowel disease may bleed excessively per rectum [2, 14, 15]. The inflammatory bowel disease of HPS affects some pediatric and adult patients [2, 15]. Clinical manifestations and therapy are generally similar to those for Crohn’s disease, including treatment with anti-tumor necrosis factor alpha drugs for severe disease and surgical bowel resection for cases refractory to medical treatment [15]. Menorrhagia, menometrorrhagia, and post-partum hemorrhage requiring transfusion of platelets or packed red blood cells can occur in female patients [2, 16, 17].
A bleeding diathesis is found in HPS patients irrespective of genetic type, other phenotypic features are dependent upon HPS type. Recurrent infections due to neutropenia and/or immunodeficiency may develop in patients with HPS types 2 or 10, and neutropenia generally responds to treatment with G-CSF [18, 19]. Pulmonary fibrosis is diagnosed in patients with HPS types 1, 2, or 4 [2, 3, 20]. Adults with HPS type 1 may present with progressive fibrotic lung disease in middle age, and children and young adults with HPS type 2 may develop pulmonary fibrosis [2, 3, 20]. Results of clinical trials investigating pirfenidone as treatment for HPS pulmonary fibrosis were inconclusive [21, 22], and thus medical therapy approved as treatment for HPS pulmonary fibrosis is not available. Single or double lung transplantation has been successfully performed on several patients with HPS, and it remains an option for certain candidates with severe HPS pulmonary fibrosis [23].
Desmopressin, pro-coagulants (e.g., aminocaproic acid, tranexamic acid), or platelet transfusion may be used as prophylaxis or treatment for excessive bleeding in patients with HPS [24, 25]. Response to desmopressin can be variable between and within patients with HPS, and some patients may not respond to desmopressin [16, 25]. Platelets are effective in preventing or treating bleeding in patients with HPS. However, platelets should be transfused judiciously. Donor organs were not found for some lung transplant candidates with severe HPS pulmonary fibrosis who were sensitized from prior blood product transfusions [23]. Thus, interventions such as transfusion of leukoreduced single donor platelets or blood products to limit alloimmunization should be implemented in patients with HPS who are at risk of developing pulmonary fibrosis.
Overall, HPS is a rare disorder associated with several genetic variants and phenotypic heterogeneity. Despite the spectrum of molecular and clinical features in HPS, genotype-phenotype associations have not been reported. Progress in genetic analyses including use of next-generation sequencing or panel-based platforms may improve the ability to diagnose patients with HPS and facilitate the identification of novel genes associated with HPS. Ultimately, these scientific advances may result in an expansion of the current understanding of HPS.
Acknowledgments
We thank our patients who participate in our studies and Dr. James G. White for providing platelet images.
Funding
This research was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
Footnotes
Declaration of interest
The authors report no conflicts of interest.
References
- 1.Huizing M, Malicdan MCV, Gochuico BR, Gahl WA. Hermansky-Pudlak Syndrome. 2000. July 24 [Updated 2017 Oct 26]. In: Adam MP, Ardinger HH, Pagon RA, et al. , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1287/ [Google Scholar]
- 2.Gahl WA, Brantly M, Kaiser-Kupfer MI, et al. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome). N Engl J Med. 1998;338:1258–1264. [DOI] [PubMed] [Google Scholar]
- 3.Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 2008;9:359–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ambrosio AL, Di Pietro SM. Storage pool diseases illuminate platelet dense granule biogenesis. Platelets. 2017;28:138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Witkop CJ, Nuñez Babcock M, Rao GH, et al. Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Bol Asoc Med P R. 1990;82:333–339. [PubMed] [Google Scholar]
- 6.Schallreuter KU, Frenk E, Wolfe LS, Witkop CJ, Wood JM. Hermansky-Pudlak syndrome in a Swiss population. Dermatology. 1993;187:248–256. [DOI] [PubMed] [Google Scholar]
- 7.Ito S, Suzuki T, Inagaki K, et al. High frequency of Hermansky-Pudlak syndrome type 1 (HPS1) among Japanese albinism patients and functional analysis of HPS1 mutant protein. J Invest Dermatol. 2005;125:715–720. [DOI] [PubMed] [Google Scholar]
- 8.Santiago Borrero PJ, Rodríguez-Pérez Y, Renta JY, et al. Genetic testing for oculocutaneous albinism type 1 and 2 and Hermansky-Pudlak syndrome type 1 and 3 mutations in Puerto Rico. J Invest Dermatol. 2006;126:85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carmona-Rivera C, Hess RA, O’Brien K, et al. Novel mutations in the HPS1 gene among Puerto Rican patients. Clin Genet. 2011;79:561–567. [DOI] [PubMed] [Google Scholar]
- 10.Anikster Y, Huizing M, White J, et al. Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet. 2001;28:376–380. [DOI] [PubMed] [Google Scholar]
- 11.Meng R, Wu J, Harper DC, et al. Defective release of α granule and lysosome contents from platelets in mouse Hermansky-Pudlak syndrome models. Blood. 2015;125:1623–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharda A, Kim SH, Jasuja R, et al. Defective PDI release from platelets and endothelial cells impairs thrombus formation in Hermansky-Pudlak syndrome. Blood. 2015;125:1633–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huizing M, Parkes JM, Helip-Wooley A, White JG, Gahl WA. Platelet alpha granules in BLOC-2 and BLOC-3 subtypes of Hermansky-Pudlak syndrome. Platelets. 2007;18:150–157. [DOI] [PubMed] [Google Scholar]
- 14.Hussain N, Quezado M, Huizing M, et al. Intestinal disease in Hermansky-Pudlak syndrome: occurrence of colitis and relation to genotype. Clin Gastroenterol Hepatol. 2006;4:73–80. [DOI] [PubMed] [Google Scholar]
- 15.Seward SL Jr, Gahl WA. Hermansky-Pudlak syndrome: health care throughout life. Pediatrics. 2013;132:153–160. [DOI] [PubMed] [Google Scholar]
- 16.Van Avermaete F, Muys J, Jacquemyn Y. Management of Hermansky-Pudlak syndrome in pregnancy and review of literature. BMJ Case Rep. 2016;2016. pii:bcr2016217719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivera-Concepción J, Acevedo-Canabal J, Burés A, Vargas G, Cadilla C, Izquierdo NJ. Bleeding assessment in female patients with the Hermansky-Pudlak syndrome-A case series. Eur J Haematol. 2019;102:432–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huizing M, Scher CD, Strovel E, et al. Nonsense mutations in ADTB3A cause complete deficiency of the beta3A subunit of adaptor complex-3 and severe Hermansky-Pudlak syndrome type 2. Pediatr Res. 2002;51:150–158. [DOI] [PubMed] [Google Scholar]
- 19.Ammann S, Schulz A, Krägeloh-Mann I, et al. Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome. Blood. 2016;127:997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gochuico BR, Huizing M, Golas GA, et al. Interstitial lung disease and pulmonary fibrosis in Hermansky-Pudlak syndrome-2, an AP-3 complex disease. Mol Med. 2012;18:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gahl WA, Brantly M, Troendle J, et al. Effect of pirfenidone on the pulmonary fibrosis of Hermansky-Pudlak syndrome. Mol Genet Metab. 2002;76:234–242. [DOI] [PubMed] [Google Scholar]
- 22.O’Brien K, Troendle J, Gochuico BR, et al. Pirfenidone for the treatment of Hermansky-Pudlak syndrome pulmonary fibrosis. Mol Genet Metab. 2011;103:128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Chemaly S, O’Brien KJ, Nathan SD, et al. Clinical management and outcomes of patients with Hermansky-Pudlak syndrome pulmonary fibrosis evaluated for lung transplantation. PLoS One. 2018;13:e0194193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nisal M, Pavord S, Oppenheimer CA, Francis S, Khare M. Hermansky-Pudlak syndrome: management of a rare bleeding disorder in a twin pregnancy. J Obstet Gynaecol. 2012;32:185–186. [DOI] [PubMed] [Google Scholar]
- 25.Cordova A, Barrios NJ, Ortiz I, Rivera E, Cadilla C, Santiago-Borrero PJ. Poor response to desmopressin acetate (DDAVP) in children with Hermansky-Pudlak syndrome. Pediatr Blood Cancer. 2005;44:51–54. [DOI] [PubMed] [Google Scholar]