

Abstract
Mitochondrial antiviral signaling (MAVS) protein is the core signaling adaptor in the RNA signaling pathway. Thus, appropriate regulation of MAVS expression is essential for antiviral immunity against RNA virus infection. However, the regulation of MAVS expression at the mRNA level especially at the post transcriptional level is not well‐defined. Here, it is reported that the MAVS mRNA undergoes N6‐methyladenosine (m6A) modification through methyltransferase‐like protein 14 (METTL14), which leads to a fast turnover of MAVS mRNA. Knockdown or deficiency of METTL14 increases MAVS mRNA stability, and downstream phosphorylation of TBK1/IRF3 and interferon‐β production in response to RNA viruses. Compared to wild‐type mice, heterozygotes Mettl14 +/− mice better tolerate RNA virus infection. The authors' findings unveil a novel mechanism to regulate the stability of MAVS transcripts post‐transcriptionally through m6A modification.
Keywords: antiviral immunity, N6‐methyladenosine modification, methyltransferase‐like protein 14, mitochondrial antiviral signaling protein, mRNA stability
This study reveals that METTL14 plays an important role in the retinoic acid‐inducible gene‐I‐like receptors (RLR)‐mediated innate antiviral response via m6A modification of MAVS mRNA. Deficiency of METTL14 increases MAVS mRNA stability and interferon‐β production in response to RNA viruses. m6A modification on MAVS transcripts by METTL14 negatively regulates the RLR‐induced innate immunity.
1. Introduction
The innate or non‐specific immune system is the forefront of host immune defense against pathogen invasion. During viral aggression, the innate immune system recognizes microorganisms via many pattern‐recognition receptors, which detect conserved microbial components including double‐stranded, known as pathogen‐associated molecular patterns, and ultimately initiates the induction of type I interferons (IFNs), pro‐inflammatory cytokines, and other downstream IFN‐stimulated genes (ISGs).[ 1 ] Intracellular viral RNA is detected by the retinoic acid‐inducible gene‐I (RIG‐I)‐like receptors (RLRs), including retinoic‐acid‐inducible gene I (RIG‐I) and Melanoma differentiation‐associated gene 5 (MDA5). Studies have clarified that RIG‐I and MDA5 play pivotal roles in innate immune response to various types of RNA viruses, including human immunodeficiency virus (HIV), severe acute respiratory syndrome coronaviru (SARS), middle east respiratory syndrome coronavirus (MERS), Ebola virus, and SARS‐CoV‐2.[ 2 , 3 , 4 , 5 ] RIG‐I and MDA5 possess two caspase‐recruitment domains (CARDs) and a DExD/H‐box helicase domain. RIG‐I recruits a CARD‐containing adaptor mitochondrial antiviral signaling protein, mitochondrial antiviral signaling protein (MAVS, also known as IPS‐1, VISA or CARDIF), a mitochondrial integral outer‐membrane anchored protein with 540 amino acids.[ 6 ] MAVS rapidly forms very large aggregates on the mitochondrial membrane upon viral infection, activating the cytosolic kinases IKK and serine/threonine‐protein kinase (TBK1), which consequently phosphorylate the transcription factors nuclear factor NF‐kappa‐B (NF‐κB) and interferon regulatory factor 3 (IRF3), respectively. NF‐κB and IRF3 transfer into the nucleus, where they cooperate to trigger the production of type I IFNs and other antiviral molecules.[ 7 ]
As a vital adaptor that propagates signals in the innate immune response, MAVS has been shown to be regulated by various post‐translational modifications, such as phosphorylation, ubiquitination, O‐GlcNAcylation, and succinylation.[ 8 , 9 , 10 ] Upon modification, the space structure or protein quantity of MAVS could be influenced, following changes in interactions between MAVS and other molecules in RLR signaling. On the same time, control of its mRNA metabolism is also critical for managing the quantity of MAVS gene expression. For example, MiR‐3470b promotes bovine ephemeral fever virus replication via directly targeting MAVS, and miR‐27a inhibits MAVS expression, promoting the replication of vesicular stomatitis virus (VSV).[ 11 , 12 ] Recent studies demonstrated that post‐transcriptional regulation of mRNA, such as m6A or 5‐methylcytosine (m5C) modifications, help cells respond more rapidly to external signaling at the transcriptional level.[ 13 , 14 , 15 ] However, the molecular regulatory mechanism of post‐transcriptional modification on MAVS mRNA remains indistinct.
Methylation of adenosine at the N6 position (m6A) of RNA has been identified as the most common mammalian mRNA modification, which can modulate enormous genes expression and regulate extensive biological activities including metabolism, tumor progression, circadian clock, and DNA damage response.[ 16 , 17 ] The m6A modification is a dynamic and reversible change that can be controlled by proteins acting as “writers,” “erasers,” and “readers.” m6A is installed by m6A methyltransferases (writers: methyltransferase‐like protein 3‐METTL3, METTL14, Pre‐mRNA‐splicing regulator‐WTAP and others) and eliminated by m6A demethylases (erasers: fat mass and obesity‐associated protein‐FTO and alkylated DNA repair protein alkB homolog 5‐ALKBH5).[ 18 ] Reader proteins recognize m6A and participate in the degradation of downstream RNA and translation.[ 19 ] The roles of m6A modification include regulating mRNA stability, splicing, transport, localization, and translation, as well as RNA‐protein interactions.[ 16 , 17 , 18 , 19 ]
Several studies have reported that m6A modifications play an important role in innate antiviral immunity. It has been reported that m6A modification served as an inactive regulator of type I IFN response by directly guiding the fast turnover of interferon alpha (IFNΑ) and interferon beta (IFNB) mRNA.[ 20 ] But, whether other molecules in innate antiviral signaling pathway can be regulated by m6A modification is not clear. Therefore, it is of great scientific significance to explore the mechanism of m6A modification on key adaptors in the RLRs signaling pathway.
In this study, using siRNA knockdown and Mettl14 deficient mice, we demonstrated that Mettl14‐mediated m6A modification of Mavs mRNA could directly regulate Mavs mRNA stability, therefore, phosphorylation of TBK1/IRF3 and IFN‐β production in response to RNA viruses were directly regulated. Importantly, compared to wild‐type mice, heterozygotes Mettl14 +/− mice better tolerate RNA virus infection. Our findings suggested that m6A modification on MAVS transcripts by METTL14 negatively regulates RLR‐induced innate immunity.
2. Results
2.1. Methyltransferase‐Like Protein 14‐Mediated Modification Inhibits RLR‐Induced Innate Immunity Signaling
To investigate whether other molecules upstream of type I IFN were affected by m6A, we first measured the TBK1 and IRF3 phosphorylation after RNA virus infection, which are upstream molecules essential for the expression of type I IFNs.
We transfected siMETTL14 into primary peritoneal macrophages to knock down the expression of METTL14. Consistently, we found that global m6A level in peritoneal macrophages was substantially decreased in the presence of siMETTL14 compared to control siRNA (siCtrl) as measured by m6A dot blot assays (Figure S1A, Supporting Information). As reported, we found the level of Ifnb1 mRNA (encoding IFN‐β) was significantly elevated upon infection with Sendai virus (SeV) or simulation with 5′‐pppRNA in siMETTL14 transfected peritoneal macrophages compared to that in control siRNA (siCtrl) transfected cells (Figure S1B, Supporting Information). Interestingly, we found siRNA knockdown of METTL14 expression in primary peritoneal macrophages also increased the phosphorylation of IRF3 and TBK1 upon stimulation with 5′‐pppRNA (Figure S1C, Supporting Information).
We generated Mettl14‐deficient mice using CRISPR/Cas9 technology. Unfortunately, we could not obtain homozygous Mettl14 −/− mice through mating between heterozygous Mettl14 +/− mice, which is consistent with previous studies showing that depletion of Mettl14 resulted in embryonic lethality early in gestation.[ 21 ] However, the heterozygous Mettl14 +/− mice are viable. Western blotting and m6A dot analysis showed that the protein expression of METTL14 and the global m6A level were substantially decreased in macrophages prepared from Mettl14 +/− mice, compared to that from Mettl14 +/+ mice (Figure 1A,B). Thus, we employed Mettl14 +/+ and Mettl14 +/− mice in the following experiments. Similar to the data in siRNA knockdown cells, we observed enhanced expression of Ifnb1, Il6, and Ifna4 mRNA and production of IFN‐β and IL6 protein in peritoneal macrophages prepared from Mettl14 +/− mice upon infection with SeV, Encephalomyocarditis virus (EMCV), or simulation with 5′‐pppRNA, compared to that from wild‐type littermates (Figure 1C). Again, we found that phosphorylation of TBK1 and IRF3 induced by SeV infection or 5′‐pppRNA stimulation was remarkably increased in Mettl14 +/− primary peritoneal macrophages, compared to that in wild‐type macrophages (Figure 1D and Figure S1C, Supporting Information). And beyond that, the phosphorylation of TBK1 and IRF3 induced by EMCV infection was upregulated in Mettl14 deficient primary peritoneal macrophages (Figure 1E). However, METTL14 deficiency did not affect the phosphorylation of TBK1 and IRF3 induced by herpes simplex virus 1 (HSV‐1; Figure S1E, Supporting Information).
Figure 1.
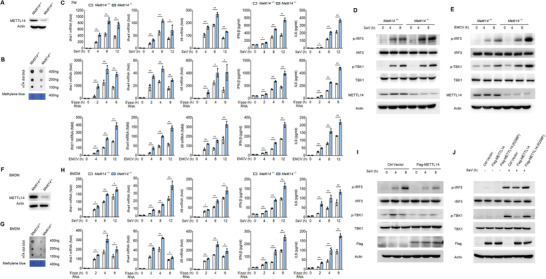
METTL14‐mediated m6A modification inhibits RLR‐induced innate immunity signaling. A) Immunoblot analysis of METTL14 in Mettl14 +/+ and Mettl14 +/− peritoneal macrophages. B) m6A dot blot assays of Mettl14 +/+ and Mettl14 +/− peritoneal macrophages, methylene blue staining (as loading control). C) qPCR analysis of Ifnb1, Il6, or Ifna4 mRNA in Mettl14 +/+ and Mettl14 +/− peritoneal macrophages, followed by infection with SeV or EMCV or stimulation with 5′‐ppp RNA for the indicated times. D) Immunoblot analysis of phosphorylated and total IRF3 and TBK1 in lysates of Mettl14 +/+ and Mettl14 +/− peritoneal macrophages infected with SeV for the indicated times. E) Immunoblot analysis of phosphorylated and total IRF3 and TBK1 in lysates of Mettl14 +/+ and Mettl14 +/− peritoneal macrophages infected with EMCV for the indicated times. F) Immunoblot analysis of METTL14 in Mettl14 +/+ and Mettl14 +/− BMDMs. G) m6A dot blot assays of Mettl14 +/+ and Mettl14 +/− BMDMs, methylene blue staining (as loading control). H) qPCR analysis of Ifnb1, Il6, or Ifna4 mRNA in Mettl14 +/+ and Mettl14 +/− BMDMs, followed by infection with SeV or stimulation with 5′‐pppRNA for the indicated times. I) Immunoblot analysis of phosphorylated and total IRF3 and TBK1 in lysates of HEK293T cells transfected with control plasmid or plasmid expressing Flag‐METTL14 (above blots) for 24 h, followed by infection with SeV for the indicated times. J) Immunoblot analysis of phosphorylated and total IRF3 and TBK1 in lysates of HEK293T cells transfected with control plasmid or plasmid expressing Flag‐METTL14 or Flag‐METTL14 (R298P) (above blots), followed by SeV infection for 8 h. Data information: Data are presented as mean ± S.D. Two‐tailed unpaired Student's t‐test; *P < 0.05; **P < 0.01 (C,H).
Similarly, we found the protein expression of METTL14 and the global m6A level were substantially decreased in bone‐marrow‐derived macrophages (BMDMs) prepared from Mettl14 +/− mice, compared to that from Mettl14 +/+ mice (Figure 1F,G). The expression of Ifnb1, Il6, and Ifna4 were increased in BMDMs from Mettl14 +/− mice after infection with SeV or treatment with 5′‐pppRNA, compared to that in wild‐type BMDMs from Mettl14 +/+ littermates (Figure 1H). We also observed that Mettl14 +/− BMDMs showed higher phosphorylation levels of IRF3 and TBK1 upon SeV infection, relative to Mettl14 +/+ BMDMs (Figure S1F, Supporting Information).
Further, we showed that SeV‐induced phosphorylation of IRF3 and TBK1 was attenuated by Flag‐METTL14 overexpression in HEK293T cells (Figure 1I). Notably, cells expressing Flag‐METTL14 mutant R298P, which has been shown to have lost the ability for METTL14 catalytic activity and mRNA substrate recognition of the methyltransferase complex,[ 21 , 22 ] could not inhibit SeV‐induced phosphorylation of IRF3 and TBK1 (Figure 1J). These data suggested that METTL14 might regulate the m6A methylation of upstream adaptors in the RLR signaling pathway, and then regulate the production of multiple cytokines including IFNs and interleukins.
2.2. Methyltransferase‐Like Protein 14 Attenuates Mitochondrial Antiviral Signaling Protein Expression
To identify molecules in the RLR signaling pathway regulated through METTL14‐mediated m6A modification, we assessed the expression of the key molecules in the RLRs signaling pathway. We prepared primary peritoneal macrophages from Mettl14 +/+ and Mettl14 +/− mice followed with SeV infection for different times. Western blotting analysis showed that SeV infection in WT macrophages increased the protein level of RIG‐I and MDA5 (Figure 2A), whose increased expression after virus infection has been reported previously.[ 23 ] While, the protein level of MAVS, TBK1, and IRF3 is not greatly changed upon virus infection in WT macrophages (Figure 2A). However, compared to wide type macrophages, knockout of Mettl14 significantly increased the MAVS protein expression in macrophages after SeV or EMCV virus infection (Figure 2A). The protein level of RIG‐I and MDA5 was similarly increased in Mettl14 +/− macrophages as that in Mettl14 +/+ macrophages (Figure 2A). Knockout of Mettl14 had no effect on the expression of TBK1 and IRF3 proteins (Figure 2A). siRNA knockdown of METTL14 expression in primary peritoneal macrophages also elevated MAVS protein level after virus infection (Figure S2A, Supporting Information). Further, we showed that knockout of Mettl14 expression in BMDMs also increased MAVS protein level, while, the protein level of RIG‐I, MDA5, TBK1 and IRF3 was not affected (Figure S2B, Supporting Information). These data suggested that METTL14 specifically regulates MAVS protein expression, which is an upstream molecule of TBK1 and IRF3 in the RLR‐induced innate signaling pathway.
Figure 2.
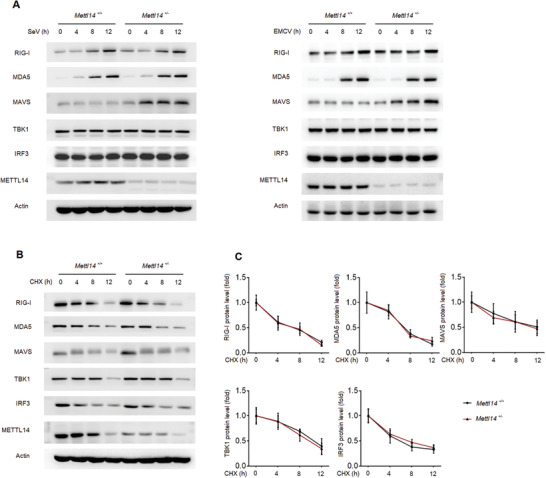
METTL14 attenuates MAVS protein expression. A) Immunoblot analysis of the main adaptors in RLRs signaling pathway in Mettl14 +/+ and Mettl14 +/− peritoneal macrophages infected with SeV or EMCV for the indicated times. B,C) Immunoblot analysis (B) and quantification (C) of innate main adaptors degradation kinetics in Mettl14 +/+ and Mettl14 +/− peritoneal macrophages treated with the protein synthesis inhibitor CHX for the indicated times.
To further investigate how METTL14 regulates MAVS protein expression, we first measured the protein degradation rate of RIG‐I, MAVS, TBK1, and IRF3. Mettl14 +/+ and Mettl14 +/− peritoneal macrophages were first stimulated by SeV infection for 8 h, followed by treatment with cycloheximide (CHX) to inhibit protein expression for the indicated times. In these assays, we found the degradation rate of RIG‐I, MAVS, TBK1, and IRF3 was not affected by Mettl14 deficiency, indicating that METTL14 could not regulate MAVS protein stability to increase MAVS protein level in Mettl14 deficient macrophages (Figure 2B,C).
2.3. Methyltransferase‐Like Protein 14 Promotes Mitochondrial Antiviral Signaling Protein (MAVS) mRNA Decay
We then measured the steady state mRNA levels of Ddx58, Mavs, Tbk1, and Irf3 in Mettl14 +/+ and Mettl14 +/− macrophages before and after SeV infection. We found SeV infection increased the steady state mRNA level of Ddx58 in both Mettl14 +/+ and Mettl14 +/− macrophages in a similar kinetics after virus infection (Figure 3A). The steady state mRNA levels of Tbk1 and Irf3 in both Mettl14 +/+ and Mettl14 +/− macrophages were not changed after virus infection (Figure 3A). The steady state mRNA level of Mavs gradually decreased upon virus infection in WT macrophages (Figure 3A). However, the Mavs mRNA level was substantially higher in Mettl14 +/− macrophages before and after SeV infection, indicating Mettl14 regulates Mavs mRNA expression (Figure 3A). As a control, we found SeV infection‐induced Ifnb1 mRNA was further elevated in Mettl14 deficient macrophages (Figure 3A), consistent with the report that Ifnb1 mRNA is an m6A target.[ 20 ]
Figure 3.
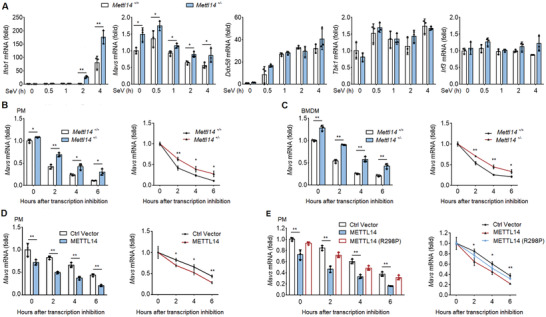
METTL14 promotes MAVS mRNA decay. A) qPCR analysis of Ifnb1, Mavs, Ddx58, Tbk1, and Irf3 mRNAs in Mettl14 +/+ and Mettl14 +/− peritoneal macrophages, followed by infection with SeV for the indicated times. B) qPCR analysis of Mavs mRNAs (left) and Mavs mRNA degradation (right) in Mettl14 +/+ and Mettl14 +/− peritoneal macrophages infected with SeV for 8 h, followed by treatment with actinomycin‐D for the indicated times. C) qPCR analysis of Mavs mRNAs (left) and Mavs mRNA degradation (right) in Mettl14 +/+ and Mettl14 +/− BMDMs infected with SeV for 8 h, followed by treatment with actinomycin‐D for the indicated times. D) qPCR analysis about the degradation of Mavs mRNA in macrophages reconstituted with empty vector and METTL14 using lentivirus followed by infection with SeV for 8 h and treatment with actinomycin‐D as indicated times. E) qPCR analysis about the degradation of Mavs mRNA in macrophages reconstituted with empty vector, METTL14, and mutant METTL14 (R298P) using lentivirus followed by infection with SeV for 8 h and treatment with actinomycin‐D as indicated times. Data information: Data are presented as mean ± S.D. Two‐tailed unpaired Student's t‐test; *P < 0.05; **P < 0.01 (A–E).
Modification of mRNA by m6A has been reported to regulate mRNA decay.[ 24 ] Thus, we measured the Mavs mRNA stability in Mettl14 +/+ and Mettl14 +/− macrophages. Mettl14 +/+ and Mettl14 +/− peritoneal macrophages were first stimulated by SeV infection for 8 h, followed by treatment with actinomycin‐D for the indicated times. Quantitative reverse transcription PCR (qPCR) analysis of Mavs mRNA showed that both Mavs mRNA stability and steady mRNA level were increased in Mettl14 +/− macrophages, relative to levels of Mavs mRNA in Mettl14 +/+ macrophages (Figure 3B). siRNA knockdown of METTL14 expression in macrophages also increased the stability of Mavs mRNA (Figure S3A, Supporting Information). We further observed that the stability and expression of Mavs mRNA were significantly increased in Mettl14 +/− BMDMs compared to that in Mettl14 +/+ BMDMs (Figure 3C). siRNA knockdown of METTL14 in THP‐1 cells also delayed MAVS mRNA degradation and increased its accumulation in THP‐1 cells (Figure S3B, Supporting Information). On the contrary, the stability and expression of Mavs mRNA were reduced in METTL14 ectopically forced expressed macrophages (Figure 3D). Similarly, the stability and expression of MAVS mRNA were decreased in HEK293T cells with the overexpression of wild‐type Flag‐tagged METTL14 (Figure S3E, Supporting Information). Notably, compared to wild‐type METTL14, the METTL14‐R298P mutant lost the ability to promote the degradation of MAVS mRNA (Figure 3E and Figure S3E, Supporting Information). To further confirm that METTL14 regulates MAVS at the post‐transcriptional level, we also measured the initiation of MAVS transcription. We extracted the nascent RNA from the cell nucleus infected with SeV and labeled with 5‐ethyluridine (EU) metabolic pulse for 30 min. We found that the newly synthesized RNA production of Ddx58, Mavs, Tbk1, Ifnb1, and Irf3 was not influenced by Mettl14 deficiency, indicating Mettl14 could not upregulate Mavs mRNA transcription to increase MAVS mRNA level in Mettl14 +/− macrophages as that in Mettl14 +/+ macrophages (Figure S3F, Supporting Information).
Taken together, these data demonstrated that METTL14, as a functionally active methyltransferase, decreases MAVS mRNA stability and its accumulation thereby reducing MAVS protein expression.
2.4. Methyltransferase‐Like Protein 14 Catalyzes m6A Modification of Mitochondrial Antiviral Signaling Protein (Mavs) mRNA
The above finding that METTL14 regulates the stability of MAVS mRNA prompted us to investigate whether MAVS mRNA was m6A‐modified by METTL14. We first performed methylated RNA immunoprecipitation (RIP) sequencing (MeRIP‐seq) to profile the transcriptome‐wide m6A modification sites in wild‐type macrophages before and after SeV infection. The data displayed that Ifnb1, Tarf3, and Traf6 mRNA were modified by m6A as previously reported (Figure S4, Supporting Information),[ 20 , 25 ] which confirmed the reliability of these assays. Interestingly, this transcriptome‐wide m6A‐seq assay showed that the m6A modification is indeed present in the Mavs mRNA before and after SeV infection, and we found a putative m6A modification site, which was adjacent to the stop codon (Figure 4A). In contrast, this analysis revealed no m6A modification on Ddx58 (Rig‐I), Ifih1 (Mda5), and Tbk1 mRNA transcripts (Figure S4, Supporting Information). If the m6A modification changes by SeV infection were mediated by METTL14, it needs a further verification.
Figure 4.
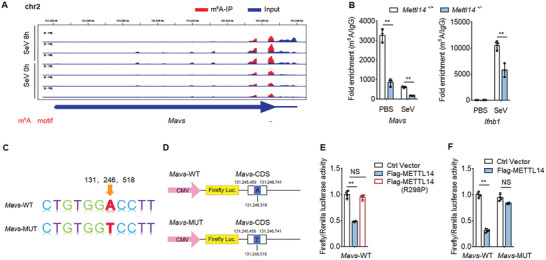
METTL14 catalyzes m6A modification of Mavs mRNA. A) RNA‐seq of Mavs mRNA in input RNA and m6A immunoprecipitated RNA from peritoneal macrophages infected with SeV for 8 h; m6A motif sequences that correspond to an immunoprecipitate‐enriched region are marked in red. B) The enrichment of MAVS mRNA in Mettl14 +/+ and Mettl14 +/− macrophages with anti‐m6A antibody, followed by m6A‐RIP‐qPCR analysis, with IgG as a negative control. C) Schematic plot of the Mavs‐WT or Mavs‐MUT mRNA nucleotide sequence. D) Schematic plot of the luciferase reporter of Mavs‐WT or Mavs‐MUT. E) Relative luciferase activity of Mavs‐WT was measured after cotransfection with Flag‐METTL14, Flag‐METTL14‐R298P, or control vector in HEK293T cells. Cell lysis was quantified for firefly luciferase activity, with the normalization to Renilla luciferase activity as an inner control. F) Relative luciferase activity of Mavs‐WT and a mutant vector carrying mutation in the m6A site were measured after cotransfection with Flag‐METTL14 or control vector in HEK293T cells. Cell lysis was quantified for firefly luciferase activity, with the normalization to Renilla luciferase activity as an internal control. Data information: Data are presented as mean ± S.D. Two‐tailed unpaired Student's t‐test; *P < 0.05; **P < 0.01; NS: no significance (B,E,F).
We next utilized RIP followed by qPCR (RIP‐qPCR) to verify the transcriptome‐wide m6A‐seq data. We observed that Mavs mRNA was m6A‐modified in macrophages before SeV infection, and SeV infection decreased m6A modification on Mavs mRNA. Importantly, m6A‐modified Mavs mRNA levels were substantially weakened in the absence of Mettl14 (Figure 4B, left). Again, we found m6A modification was present in Ifnb1 mRNA especially after SeV infection, and the increase of m6A modification decreased in Mettl14 deficient macrophages (Figure 4B, right). To evaluate the m6A modifications site on Mavs mRNA by METTL14 and if this modification directly regulates Mavs mRNA stability, we designed a series of experiments. Combining the transcriptome‐wide m6A‐seq data and a software prediction, we found the putative m6A modification site (131246518 in Chromosome 2), which was adjacent to the stop codon (Figure 4C). We then generated a luciferase reporter that included the wild‐type Mavs CDS truncation sequence (nucleic acid from 131245450 to 131246741 in Chromosome 2, named Mavs‐WT) or a mutant luciferase reporter CDS truncation sequence (named Mavs‐MUT), in which we replaced the adenine into thymine at the 131246518 site of Mavs (Figure 4D). Luciferase reporter assays showed that expressing wild‐type Flag‐METTL14 markedly weakened Mavs‐WT luciferase activity, which was not observed with mutated METTL14‐R298P (Figure 4E). Importantly, ectopical expression of Flag‐METTL14 dampened luciferase activity of reporter constructs with wild‐type Mavs, whereas the restraint abolished when the m6A site mutation (Mavs‐MUT) was made (Figure 4F). Collectively, these findings prove that the regulation of MAVS stability and innate immunity signaling mediated by METTL14 depends on the methyltransferase activation and putative m6A site on the Mavs mRNA.
2.5. Methyltransferase‐Like Protein 14 Inhibits Cellular Antiviral Response to RNA Virus
RLRs‐mediated IFN‐β production plays essential roles in the innate immune responses against RNA viral infection. To investigate the function of METTL14 on antiviral responses in vivo, VSV was used as an RNA virus. Similar to SeV infection, VSV infection induced elevated level of Ifnb1 mRNA expression in primary peritoneal macrophages transfected with siMETTL14, compared to that in macrophages transfected with control siRNA (siCtrl) (Figure S5A, Supporting Information). Accordingly, VSV mRNA measured by qPCR and VSV titers measured by plaque assay substantially decreased in siMETTL14 transfected peritoneal macrophages compared to siCtrl transfected cells (Figure S5A, Supporting Information). Vesicular stomatitis virus G protein (VSV‐G) also decreased in siMETTL14 transfected macrophages compared to that in the control siRNA transfected cells (Figure S5B, Supporting Information). Similarly, knockdown of METTL14 expression using siRNA in THP‐1 cells increased IFNB1 expression and decreased VSV replication after VSV infection (Figure S5C, Supporting Information). VSV‐G also decreased in METTL14‐knockdown THP‐1 cells (Figure S5D, Supporting Information).
We also prepared primary peritoneal macrophages from Mettl14 +/+ mice and Mettl14 +/− mice followed infection with VSV. We found Ifnb1 mRNA expression was upregulated in peritoneal macrophages prepared from Mettl14 +/− mice relative to that from Mettl14 +/+ mice after VSV infection (Figure 5A). Accordingly, VSV replication was potently inhibited in Mettl14 +/− peritoneal macrophages (Figure 5B).
Figure 5.
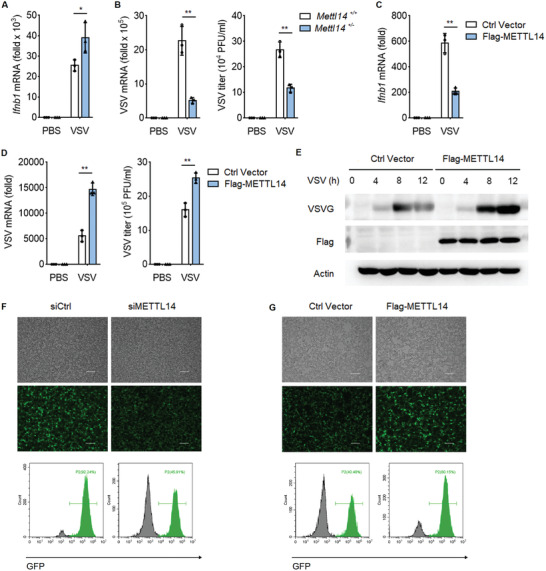
METTL14 inhibits cellular antiviral response to RNA virus. A) qPCR analysis of Ifnb1 mRNA in Mettl14 +/+ and Mettl14 +/− peritoneal macrophages infected with VSV (MOI, 0.1) for 12 h. B) qPCR analysis of VSV mRNA (left) and plaque assay of VSV titers (right) in Mettl14 +/+ and Mettl14 +/− peritoneal macrophages infected with VSV (MOI, 0.1) for 12 h. C) qPCR analysis of Ifnb1 mRNA in HEK293T cells transfected with indicated plasmids for 24 h, followed by infection with VSV (MOI, 0.1) for 12 h. D) qPCR analysis of VSV mRNA (left) and plaque assay of VSV titers (right) in HEK293T cells transfected as in (C), followed by infection with VSV (MOI, 0.1) for 12 h. E) Immunoblot analysis of VSV glycoprotein (VSV‐G) in HEK293T cells transfected as in (C), followed by infection with VSV for the indicated times. F) Fluorescence microscopy (above) and flow cytometry analysis (bottom) of VSV‐GFP replication in HEK293T cells transfected with control siRNA (siCtrl) or siRNA targeting METTL14 (siMETTL14) for 48 h, followed by infection with VSV‐GFP (MOI, 0.1) for 12 h (bright‐field, upper; fluorescence, bottom). Scale bars, 100 µm. G) Fluorescence microscopy (above) and flow cytometry analysis (bottom) of VSV‐GFP replication in HEK293T cells transfected as in (C), followed by infection with VSV‐GFP (MOI, 0.05) for 12 h (bright‐field, upper; fluorescence, bottom). Scale bars, 100 µm. Data information: Data are presented as mean ± S.D. Two‐tailed unpaired Student's t‐test; *P < 0.05; **P < 0.01 (A–D).
To further investigate the role of METTL14 on virus replications, we transfected METTL14 expression plasmid into HEK293T cells and then the cells were infected with VSV. Overexpression of Flag‐METTL14 decreased IFNB1 mRNA level upon infection with VSV (Figure 5C). Accordingly, the replication of VSV as measured by VSV‐specific mRNA, VSV titers, and VSV‐G, was substantially increased in METTL14 transfected cells (Figure 5D,E). We further used VSV‐GFP virus to infect HEK293T cells and measured the function of METTL14 on VSV‐GFP infection. Fluorescence microscopy showed that siRNA knockdown of METTL14 expression in HEK293T cells inhibited VSV‐GFP levels relative to control siRNA transfected cells (Figure 5F). In contrast, transfection of METTL14 expression plasmid increased replication of VSV‐GFP in HEK293T cells (Figure 5G). All together, these data suggested that METTL14 negatively regulates RLRs‐mediated IFN‐β production and innate antiviral immune responses.
2.6. Methyltransferase‐Like Protein 14 Negatively Regulates Antiviral Response to RNA Virus In Vivo
To investigate the role of METTL14 on antiviral immunity in physiological condition, we challenged Mettl14 +/+ and Mettl14 +/− mice with VSV. Mettl14 +/− mice showed improved survival than their WT littermates during viral infection (Figure 6A). Hematoxylin and‐eosin staining showed less damage in the lungs of Mettl14 +/− mice compared to that of Mettl14 +/+ mice (Figure 6B). ELISA assays showed increased IFN‐β production in the sera from Mettl14 +/− mice compared to that from WT mice (Figure 6C). qPCR analysis also showed that Mettl14 +/− mice had substantially increased Ifnb1 mRNA in the lung, liver, and spleen (Figure 6D). Accordingly, the replication of VSV in the lung, liver, and spleen was attenuated as indicated by VSV mRNA measured by qPCR and viral titers measured by plaque assays (Figure 6E,F). In order to validate the role of macrophages in our viral infection model, clodronate liposomes were treated for 3 days to delete macrophages before viral infection. Flow cytometry analysis showed the successful deletion of macrophages in the blood and spleen (Figure S6A, Supporting Information). In the absence of macrophages, Mettl14 +/+ and Mettl14 +/− mice showed the same antiviral ability to VSV challenge, which was evaluated based on survival rate, IFN‐β secretion in serum, and viral burden in lung, liver, and spleen (Figure S6B–D, Supporting Information). This indicates that macrophages are the key to the antiviral response against viral infections in METL14‐deficient mice. Taken together, these data demonstrated that Mettl14 facilitates VSV replication in vivo.
Figure 6.
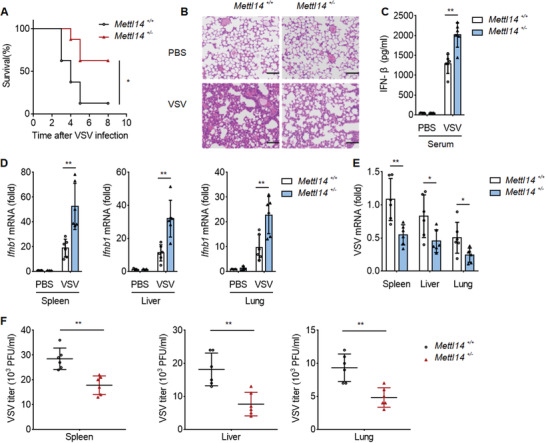
METTL14 negatively regulates antiviral response to RNA virus in vivo. A) Survival of Mettl14 +/+ and Mettl14 +/− mice infected intravenously with VSV (1 × 108 PFU per mouse). B) Hematoxylin‐and‐eosin‐stained images of lung sections of Mettl14 +/+ and Mettl14 +/− mice infected by intraperitoneal injection of VSV (1 × 108 PFU per mouse) for 48 h, Scale bars, 100 µm. C) ELISA analysis of IFN‐β of Serum from Mettl14 +/+ and Mettl14 +/− mice infected by intraperitoneal injection of VSV (1 × 108 PFU per mouse) for 8 h. D) qPCR analysis of Ifnb1 mRNA (middle right) of the spleen (left), liver (middle), and lungs (right) from Mettl14 +/+ and Mettl14 +/− (six per group) mice infected by intraperitoneal injection of VSV (1 × 108 PFU per mouse) for 48 h. E) qPCR analysis of VSV RNA of the spleen (left), liver (middle), and lungs (right) from Mettl14 +/+ and Mettl14 +/− mice infected by intraperitoneal injection of VSV (1 × 108 PFU per mouse) for 48 h. F) Plaque assay of VSV titers of the spleen, liver, and lungs from Mettl14 +/+ and Mettl14 +/− mice infected by intraperitoneal injection of VSV (1 × 108 PFU per mouse) for 48 h. Each symbol represents an individual mouse; small horizontal lines indicate the mean. Data information: Data are presented as mean ± S.D. Log‐rank test (A); Two‐tailed unpaired Student's t‐test; *P < 0.05; **P < 0.01 (C–F).
3. Discussion
Type I IFN production is the hallmark of immunity against a variety of viral infections, and it results in the production of autocrine and paracrine antiviral factors. Type I IFN response can be regulated by various enhancement and inhibitory signals that induce robust and powerful antiviral responses. The effect of m6A modification in the innate antivirus signaling pathway is not fully understood.
One study showed that attenuating m6A writer METTL3 and reader YTHDF2 could directly reduce the m6A modification of mouse Ifnb1 mRNA, thereby accelerating its mRNA degradation, which decreased ISGs expression and weakened antiviral effect during viral infection.[ 20 ] It was also reported that knockdown of METTL14 using siRNA increased the production and stability of nascent Ifnb1 mRNA, consequently inhibiting the propagation of DNA virus, and increasing the yield of Ifnb1 mRNA induced by dsDNA or HCMV. By comparison, knockout of ALKBH5 reduced the output of primary Ifnb1 mRNA, but did not significantly affect the degradation rate of Ifnb1 mRNA.[ 26 ] It was shown that HMPV RNAs were m6A methylated and that m6A methylation restrained the binding efficiently of viral RNA to RIG‐I, which inhibited IFN expression and promoted HMPV replication.[ 27 ] It was also reported that m6A modification of viral transcripts inhibited viral RNA recognition by RIG‐I and regulated host innate immunity against hepatitis B and C viral infections by inducing.[ 28 ]
As mentioned above, m6A modification play important roles on IFNB1 production, the main mechanisms focused on m6A modification on IFNB1 mRNA or virus RNA directly. Whether the vital participants in the RLR‐induced innate signaling pathway, like RIG‐1, MDA5, MAVS, TBK1, and IRF3, could be modified by RNA modification m6A is still unclear. It was reported that DDX46 RNA helicase interacted with m6A “eraser” ALKBH5 directly but not the writers like METTL3 or METTL14, which then demethylate m6A‐modified Mavs, TNF receptor‐associated factor 3 (Traf3), and TNF receptor associated factor 6 (Traf6) transcripts. The m6A demethylation inhibited the mRNA translocation of those adaptors from nucleus to cytoplasm.[ 25 ] However, there were still some unsolved problems in the paper. For example, ALKBH5 could demethylase m6A modification of Mavs, Traf3, and Traf6 mRNA, that means the demethylation of m6A lacks specificity, and the m6A modification sites or the sequences similarity on the adaptors mRNA were not clearly identified. Moreover, the demethylation of m6A modification on Mavs, Traf3, and Traf6 mRNA by AKBH5 depended on the existence of DDX46; the role of m6A “writers” was not elaborated. Thereby, different “writers” might play distinct roles on those adaptors mRNA. Discovering new m6A targets and mechanisms in RLR‐induced innate signaling pathway is still a challenge in the field.
In this study, we found an evolutionarily conserved mechanism for regulating type I IFN response, in which METTL14 mediated m6A modification on MAVS mRNA specifically. Importantly, both knockdown and knockout of METTL14 could increase the stability and expression of MAVS mRNA, but not affect the nuclear translocation of MAVS mRNA, which is a different and new mechanism compared to previous reports. Using MeRIP‐seq and MeRIP‐PCR, we also confirmed that the Mavs mRNA was m6A‐modified by Mettl14. We used a luciferase reporter that included the wild‐type CDS truncation sequence (Mavs‐WT) and demonstrated that expressing wild‐type Flag‐METTL14 markedly weakened MAVS response, but not that of Mavs‐MUT, compared with empty vector. Thereby, we identified that the 131246518 site on Mavs mRNA was the modification targets of Mettl14. These data proved that Mavs mRNA was regulated by METTL14 through m6A modification and therefore affects its stability. Our study found an interesting phenomenon that m6A methylation of Mavs mRNA is decreased in virus‐induced macrophages (Figure 4B). We measured the expression of methyltransferases (METTL3, METTL14) and demethylases (FTO, ALKBH5), and found that the expression of FTO and ALKBH5 rather than METTL3 and METTL14 were gradually increased upon virus infection (Figure S4B, Supporting Information), which may account for that the m6A methylation of Mavs mRNA is decreased in virus‐induced macrophages (Figure 4B).
In conclusion, we found that METTL14 plays an important role in the RLRs‐mediated phosphorylation of IRF3, as well as transcription of IFNs and inflammatory cytokines. The m6A‐modified 131, 246, and 518 sites on Mavs mRNA by MTTL14 impaired its mRNA stability and antivirus innate immunology response. Our study suggested that m6A modification on MAVS transcripts by METTL14 negatively regulates the RLR‐induced innate immunity. Deficiency of METTL14 increased MAVS protein stability and IFN‐β production in response to RNA viruses (Figure 7 ).
Figure 7.
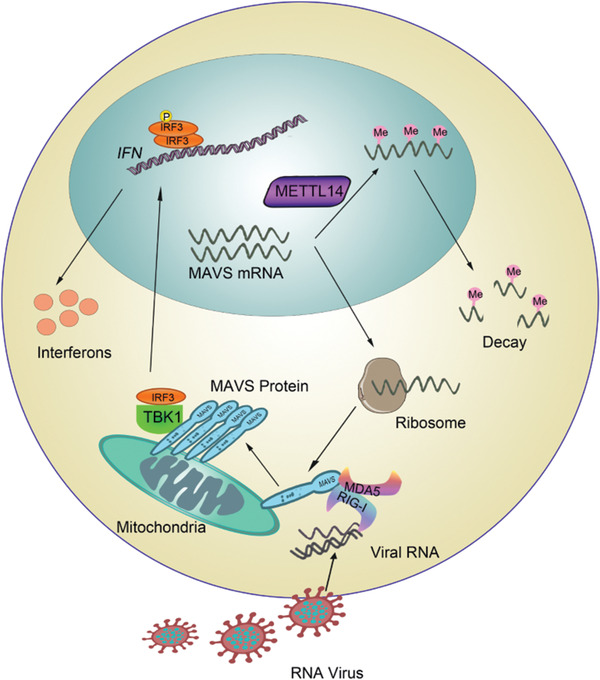
The proposed schematic model of METTL14 in RLR‐mediated innate immunity.
4. Experimental Section
Cells and Viruses
HEK293T, THP‐1, and Hela cells were obtained from the American Type Culture Collection. The cells were cultured in DMEM (HyClone) supplemented with 10% FBS, 100 U mL−1 penicillin and 100 µg mL−1l streptomycin at 37 °C with 5% CO2. BMDMs were prepared as previously described.[ 8 ] SeV was purchased from the China Center for Type Culture Collection (Wuhan University, China). VSV and VSV‐GFP were provided by H. Meng (Institute of Basic Medicine, Shandong Academy of Medical Sciences, China). The corresponding empty vectors were used as negative controls in all transfection experiments.
Plasmids and Transfection
METTL14 cDNA was amplified from THP‐1 cells by standard PCR and cloned in pCMV2‐Flag plasmids. For dual‐luciferase reporter assay, the DNA fragments of wild‐type MAVS (Mavs‐WT) CDS truncation sequence (from 131245450 to 131246741) was amplified from macrophage and inserted downstream a firefly luciferase gene in pMIR‐REPORT vector (Luciferase miRNA Expression Reporter Vector, Ambion). Mutant plasmids for METTL14 R298P or Mavs‐MUT with an analogous sequence in which its putative m6A site was abolished by A to T mutations at 131246518 were made using the KOD‐Plus‐Mutagenesis kit (Toyobo, Osaka, Japan). Phorbol 12‐myristate 13‐acetate (PMA) was purchased from Invitrogen. Actinomycin‐D was from MedChemExpress. For transfection of plasmids into HEK293 cells, lipofectamine 3000 reagents were used (Invitrogen). For transfection of siRNA knockdown, THP‐1, BMDMs, or primary peritoneal macrophages were cultured in 24‐ or 6‐well plates, and siRNA transfection was performed using RNAIMAX (Invitrogen) by following the manufacturer's instruction. siRNAs were transfected into cells in each well. 48 h after transfection, knockdown efficiency was monitored by qPCR, or incubation continued to the subsequent indicated treatment. Small interfering sequences targeting METTL14 were as follows: mouse: 5′‐GCACCTCGGTCATTTATAT‐3′; human: 5′‐GGACUUCAUUCAUGCUAAUTT‐3′.
Antibodies and Reagents
Antibodies used in this study were summarized at the dilutions listed: anti‐METTL14, 1:1000, (IB; Cell Signaling Technology, #: 51104S), anti‐N6‐methyladenosine modifications of RNA and DNA (m6A), 1:2000, (dot blot; Synaptic Systems, #: 202 003); anti‐IRF3, 1:1000, (IB; Cell Signaling Technology, #: 4302S); anti‐pIRF3, 1:1000, (IB; Cell Signaling Technology, #: 4947S) anti‐TBK1, 1:1000, (IB; Cell Signaling Technology, #: 3504S); anti‐pTBK1, 1:1000, (IB; Cell Signaling Technology, #: 5483S); anti‐RIG‐I, 1:1000, (IB; Cell Signaling Technology, #: D14G6); anti‐MDA5, 1:1000, (IB; Cell Signaling Technology, #: D74E4); anti‐FTO, 1:1000 (IB; Cell Signaling Technology, #: D6Z8W); anti‐METTL3, 1:1000 (IB; Cell Signaling Technology, #: E3F2A); anti‐ALKBH5, 1:1000 (IB; Abcam, #: ab195377); anti‐MAVS, 1:500, (IB; Santa Cruz Biotechnology, #: sc‐365333); anti‐FLAG (M2), 1:1000, (IB; Sigma‐Aldrich, #: F1804); anti‐β‐actin, 1:2000, (IB; ZSGB‐BIO, #:TA‐09). CHX (HY‐12320), actinomycin‐D, (HY‐17559), were obtained from MedChemExpress (MCE, NJ, USA); PMA (P1585) and Dynabeads mRNA Purification Kit (#: 61006) were purchased from Invitrogen; Click‐iT nascent RNA capture kit (#: C10365) was purchased from Life Technologies; EpiMark N6‐Methyladenosine Enrichment Kit (E1610S) was purchased from New England Biolabs.
Animal Experiments
All animal experiments were carried out in agreement with the regulations of the National Institute of Health Guide for the Care and Use of Laboratory Animals, and approved by the Ethics Committee on Scientific Research of Shandong University Qilu Hospital, Jinan, Shandong Province, China. Permission‐numbers: KYLL‐2017(KS)‐361. All mice were housed in individually ventilated cages under specific pathogen‐free conditions. METTL14‐deficient mice were generated by Cyagen Biosciences Inc. (Guangzhou, China) using CRISPR/Cas system. Briefly, Cas9/sgRNA expression plasmid and targeting vector inserted with the upstream and downstream of the exon 7–10 of METTL14 were constructed. Linearized Cas9/sgRNA and targeting vector were micro‐injected into mouse zygote. Injected zygotes were implanted into C57BL/6 female mice. Genotyping was performed by PCR using the following primers: F1: 5′‐GCCTTCAGAGATGACGATGACTTC‐3′; F2: 5′‐CTGCCTAAAAGTCCTCCCTACTC‐3′; R1: 5′‐GTTACAAGAGGCCAGGTAAGAGTG‐3′.
All mice used were 6–8 weeks old. The mouse experiments were carried out following the general guidelines published by the Association for Assessment and Accreditation of Laboratory Animal Care. All of the mice were on the C57BL/6 background and were fed under specific‐pathogen free conditions with the approval of the Scientific Investigation Board of the Medical School of Shandong University.
Viral Infection and Plaque Assay
HEK293 (2 × 105), THP‐1 (5 × 105), BMDMs (5 × 105), or primary peritoneal macrophages (5 × 105) were plated 24 h before infection. VSV (MOI, 0.1) or SeV were transduced into cell for the indicated times. VSV plaque assay and VSV replication were performed in HEK293 cells as previously described.[ 29 ]
mRNA Isolation and Quantitative Reverse Transcription PCR
RNA was extracted from whole cell lysates using EASYspin Plus tissue/cell rapid RNA exaction kit (Aidlab) and 0.5 µg of total RNA was reverse transcribed with a PrimeScript RT reagent Kit (Takara). qPCR analysis was performed in triplicate wells with an iCycler IQ thermal cycler and detection system (Bio‐Rad) using the SYBR RT‐PCR kit (Roche) according to the manufacturer's instructions. The data were normalized to the expression of the actin housekeeping gene in each individual sample. The 2−∆∆Ct method was utilized to calculate relative expression variations. Specific primers used for RT‐PCR assays are list as Table S1, Supporting Information.
Virus Infection In Vivo
Littermate mice of Mettl14 +/+ and Mettl14 +/− mice were allocated into groups according to age and sex and intraperitoneally (i.p.) injected with VSV (1 × 108 PFU per mouse). Mice were sacrificed and serum IFN‐beta levels were measured using ELISA. The VSV titers in the spleen, lung, or liver were detected by standard plaque assays. Lungs from mice were dissected, fixed in 10% phosphate‐buffered formalin, embedded into paraffin, sectioned, stained with hematoxylin–eosin solution, and inspected by light microscopy for histological changes. For survival experiments, mice were evaluated for survival after VSV infection. To deplete the macrophages in vivo, 200 vL of clodronate liposome (FormuMax Scientific Inc.) or 200 vL of control liposome suspension (FormuMax Scientific Inc.) was intravenously administered to mice for 3 days and i.p. injected with VSV (5 × 107 PFU per mouse) and then the next experiments were performed.
Lentivirus Production and Infection
METTL14 and METTL14 (R298P) were cloned into the lentiviral expression vector pLVX‐IRES‐Puro. The viral particles were produced by transfecting HEK293T cells with METTL14‐ or METTL14 (R298P)‐expressing, or control plasmids in combination with pLVX‐IRES‐Puro, pMD2.G, and psPAX2 using Lipofectamine 3000 (Thermo Fisher Scientific). Viral supernatant was collected after 48 h. Macrophages were infected with lentivirus for at least 3 days and then the efficiency of expression was assessed by western blot 24 h after infection.
Analysis of Nascent RNA Synthesis
To detect the transcription of newly born RNA, a Click‐iT nascent RNA capture kit (Life Technologies) was used. Mettl14 +/+ and Mettl14 +/− peritoneal macrophages were exposed to a 30‐min EU pulse after SeV infection at the indicated times. Following EU exposure, cells were washed with PBS and were harvested, and RNA samples were extracted for qPCR analysis.
RNA Decay Assays
Cells were seeded in 12‐well plates and cultured overnight at 37 °C. The next day, the authors performed the corresponding transfection operation. Cells were then infected for 8 h with SeV, followed by treatment with actinomycin‐D at a final concentration of 5 µg mL−1 for the indicated times. Cells were harvested and RNA samples were extracted for qPCR analysis to detect MAVS mRNA levels. The data were normalized to the t = 0 time point.
m6A Dot Blot Assays
Total RNA was isolated from different cells according to the manufacturer's instructions. The m6A dot blot assay was executed following a published protocol[ 30 ] with appropriate modifications. In brief, diluted RNA was heated at 95 °C for 3 min to disrupt secondary structure and then cooled down. The cold RNA samples were loaded on Amersham Hybond‐N+ membrane (GE Healthcare), dried, and fixed by UV cross‐linking. The membrane was blocked with 5% nonfat dry milk (in 1× PBST) for 1–2 h and incubated with a specific anti‐m6A antibody (1:2000 dilution, Synaptic Systems, 202003) overnight at 4 °C. After washing, the membrane was incubated with HRP‐conjugated goat anti‐rabbit IgG (1: 5000 dilution) for 1 h at room temperature. Blots were exposed with Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific).
Luciferase Reporter Assay
For dual‐luciferase reporter assay, HEK‐293T cells (0.5 × 105) were seeded in 24‐well plate overnight and 200 ng Mavs‐WT or Mavs‐MUT, 200 ng Flag‐METTL14‐WT or Flag‐METTL14‐R298P plasmid, and 20 ng pRL‐TK (Renilla luciferase plasmid) were cotransfected for 48 h, followed by SeV infection for 8 h. The relative luciferase activities were determined using Dual‐Luciferase Reporter Assay System (Promega).
m6A RNA Immunoprecipitation
Over 100 µg total RNA from Mettl14 KO and WT peritoneal macrophages were extracted with TRIzol reagent (Invitrogen) and subjected to Poly(A)+ mRNA purification via Dynabeads mRNA Purification Kit (Invitrogen) according to the manufacturer's instructions. For m6A RNA, immunoprecipitation was performed using an EpiMark N6‐Methyladenosine Enrichment Kit (NEB). Briefly, 25 µL of Protein G Magnetic Beads (NEB #S1430) were washed twice with 200 µL reaction buffer (150 mm NaCl, 10 mm Tris‐HCl, pH 7.5, 0.1% NP‐40 in nuclease free H2O), and resuspended completely in 250 µL reaction buffer. 1 µL N6‐methyladenosine antibody or IgG antibody was added to the beads and incubated with orbital rotation for 30 min at 4 °C. Beads were washed twice with Reaction Buffer and resuspended in 250 µL reaction buffer. Poly(A) selected purified polyadenylated RNA was added to the resuspended beads and incubated with orbital rotation for 1 h at 4 °C. Beads were washed twice with Reaction Buffer, twice with Low Salt Reaction Buffer (50 mm NaCl, 10 mm Tris‐HCl, pH 7.5, 0.1% NP‐40 in nuclease free H2O), twice with High Salt Reaction Buffer (500 mm NaCl, 10 mm Tris‐HCl, pH 7.5, 0.1% NP‐40 in nuclease free H2O), and resuspended completely in 30 µL of Buffer RLT (Qiagen, 20 µL of Dynabeads MyOne Silane (Life Technologies,) was washed with 100 µL of Buffer RLT, resuspended in 30 µL of Buffer RLT, and added to the eluted RNA. Subsequently, 60 µL of 100% ethanol was added to the RNA and Dynabeads mixture. RNA‐bound beads were washed twice with 200 µL 70% ethanol and incubated with 16 µL nuclease‐free water for 1 min at room temperature to elute the RNA, followed immediately by cDNA synthesis.
Statistical Analysis
For statistical analysis, GraphPad Prism 7.0 was used for all analyses. Unpaired Student's t‐test was applied to compare the differences between the two groups. All data were presented as mean ± standard deviation (SD) of one representative experiment. For the mouse survival study, Kaplan–Meier survival curves were generated and analyzed by the log‐rank Mantel–Cox test; All statistical tests were two‐tailed; P < 0.05 was considered statistically significant, *P < 0.05, **P < 0.01, NS: no significance
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
C.G. conceived and designed research; B.L. and F.Q. performed research; L.Z. and Y.Z. provided discussions; B.L., F.Q., and C.G. analyzed data; C.G. and B.L. wrote the paper.
Supporting information
Supporting Information
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31730026, 81930039, 81525012 to C.G. and 31900680 to B.L.). This work was also supported by the National Postdoctoral Program for Innovative Talents (BX201700146 to B.L.) and Shandong Provincial Natural Science Foundation (ZR2018BC021 to B.L.).
Qin F., Cai B., Zhao J., Zhang L., Zheng Y., Liu B., Gao C., Methyltransferase‐Like Protein 14 Attenuates Mitochondrial Antiviral Signaling Protein Expression to Negatively Regulate Antiviral Immunity via N6‐methyladenosine Modification. Adv. Sci. 2021, 8, 2100606. 10.1002/advs.202100606
Contributor Information
Bingyu Liu, Email: liubingyu@sdu.edu.cn.
Chengjiang Gao, Email: cgao@sdu.edu.cn.
Data Availability Statement
Research data are not shared.
References
- 1. Akira S., Uematsu O., Takeuchi, Cell 2006, 124, 783. [DOI] [PubMed] [Google Scholar]
- 2. Solis M., Nakhaei P., Jalalirad M., Lacoste J., Douville R., Arguello M., Zhao T., Laughrea M., Wainberg M. A., Hiscott, J. Virol. 2011, 85, 1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lu X., Pan J., Tao J., Guo D., Virus Genes 2011, 42, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao X., Chu H., Wong B. H., Chiu M. C., Wang D., Li C., Liu X., Yang D., Poon V. K., Cai J., Chan J. F., To K. K., Zhou J., Yuen K. Y., J. Infect. Dis. 2020, 221, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ramanan P., Edwards M. R., Shabman R. S., Leung D. W., Endlich‐Frazier A. C., Borek D. M., Otwinowski Z., Liu G., Huh J., Basler C. F., Amarasinghe G. K., Proc. Natl. Acad. Sci. USA. 2012, 109, 20661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu B., Hur S., Curr. Opin. Virol. 2015, 12, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu S., Cai X., Wu J., Cong Q., Chen X., Li T., Du F., Ren J., Wu Y. T., Grishin N. V., Chen Z. J., Science 2015, 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- 8. Liu B., Zhang M., Chu H., Zhang H., Wu H., Song G., Wang P., Zhao K., Hou J., Wang X., Zhang L., Gao C., Nat. Immunol. 2017, 18, 214. [DOI] [PubMed] [Google Scholar]
- 9. Li T., Li X., Attri K. S., Liu C., Li L., Herring L. E., Asara J. M., Lei Y. L., Singh P. K., Gao C., Wen H., Cell Host Microbe 2018, 24, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu X., Zhu C., Zha H., Tang J., Rong F., Chen X., Fan S., Xu C., Du J., Zhu J., Wang J., Ouyang G., Yu G., Cai X., Chen Z., Xiao W., EMBO J. 2020, 39, 103285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hou P., Wang H., Zhao G., Hu G., Xia X., He H., BMC Microbiol. 2018, 18, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu L., Peng L., Gu T., Yu D., Yao Y. G., Biochim. Biophys. Acta, Gene Regul. Mech. 2019, 1862, 47. [DOI] [PubMed] [Google Scholar]
- 13. Zhao B. S., Roundtree I. A., He C., Nat. Rev. Mol. Cell Biol. 2017, 18, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roundtree I. A., Evans M. E., Pan T., He C., Cell 2017, 169, 1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frye M., Harada B. T., Behm M., He C., Science 2018, 361, 1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee M., Kim B., Kim V. N., Cell 2014, 158, 980. [DOI] [PubMed] [Google Scholar]
- 17. Zhang C., Chen Y., Sun B., Wang L., Yang Y., Ma D., Lv J., Heng J., Ding Y., Xue Y., Lu X., Xiao W., Yang Y. G., Liu F., Nature 2017, 549, 273. [DOI] [PubMed] [Google Scholar]
- 18. Yu R., Li Q., Feng Z., Cai L., Xu Q., Int. J. Mol. Sci. 2019, 20, 1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luo S., Tong L., Proc. Natl. Acad. Sci. USA 2014, 111, 13834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Winkler R., Gillis E., Lasman L., Safra M., Geula S., Soyris C., Nachshon A., Tai‐Schmiedel J., Friedman N., Le‐Trilling V., Trilling M., Mandelboim M., Hanna J. H., Schwartz S., Stern‐Ginossar N., Nat. Immunol. 2019, 20, 17. [DOI] [PubMed] [Google Scholar]
- 21. Weng H., Huang H., Wu H., Qin X., Zhao B. S., Dong L., Shi H., Skibbe J., Shen C., Hu C., Sheng Y., Wang Y., Wunderlich M., Zhang B., Dore L. C., Su R., Deng X., Ferchen K., Li C., Sun M., Lu Z., Jiang X., Marcucci G., Mulloy J. C., Yang J., Qian Z., Wei M., He C., Chen J., Cell Stem Cell 2018, 22, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang P., Doxtader K. A., Nam Y., Mol. Cell 2016, 63, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao C., Jia M., Song H., Yu Z., Wang W., Li Q., Zhang L., Zhao W., Cao X., Cell Rep. 2017, 21, 1613. [DOI] [PubMed] [Google Scholar]
- 24. Liu Y., You Y., Lu Z., Yang J., Li P., Liu L., Xu H., Niu Y., Cao X., Science 2019, 365, 1171. [DOI] [PubMed] [Google Scholar]
- 25. Zheng Q., Hou J., Zhou Y., Li Z., Cao X., Nat. Immunol. 2017, 18, 1094. [DOI] [PubMed] [Google Scholar]
- 26. Rubio R. M., Depledge D. P., Bianco C., Thompson L., Mohr I., Genes Dev. 2018, 32, 1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu M., Zhang Z., Xue M., Zhao B. S., Harder O., Li A., Liang X., Gao T. Z., Xu Y., Zhou J., Feng Z., Niewiesk S., Peeples M. E., He C., Li J., Nat. Microbiol. 2020, 5, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim G. W., Imam H., Khan M., Siddiqui A., J. Biol. Chem. 2020, 295, 13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang P., Zhao W., Zhao K., Zhang L., Gao C., PLoS Pathog. 2015, 11, e1004726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nagarajan A., Janostiak R., Wajapeyee N., in Epitranscriptomics (Eds: Wajapeyee N., Gupta R.), Humana Press, New York: 2019, p. 263. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.