

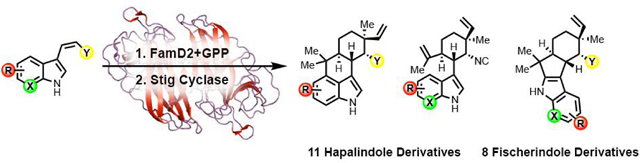
Abstract
Hapalindoles and related compounds (ambiguines, fischerindoles, welwitindolinones) are a diverse class of indole alkaloid natural products. They are typically isolated from the Stigonemataceae order of cyanobacteria and possess a broad scope of biological activities. Recently the biosynthetic pathway for assembly of these metabolites has been elucidated. In order to generate the core ring system, L-tryptophan is converted into the cis-indole isonitrile subunit before being prenylated with geranyl pyrophosphate at the C-3 position. A class of cyclases (Stig) catalyzes a three-step process including a Cope rearrangement, 6-exo-trig cyclization and electrophilic aromatic substitution to create a polycyclic core. Formation of the initial alkaloid is followed by diverse late-stage tailoring reactions mediated by additional biosynthetic enzymes to give rise to the wide array of structural variations observed in this compound class. Herein, we demonstrate the versatility and utility of the Fam prenyltransferase and Stig cyclases toward core structural diversification of this family of indole alkaloids. Through synthesis of cis-indole isonitrile subunit derivatives, and aided by protein engineering and computational analysis, we have employed cascade biocatalysis to generate a range of derivatives, and gained insights into the basis for substrate flexibility in this system.
Keywords: Stig cyclase, Fam prenyltransferase, hapalindole, fischerindole, diversification, biocatalysis
Graphical Abstract
Introduction
Hapalindoles are a large family of indole alkaloids that have been isolated from the cyanobacterial order Stigonematales.1 Along with their related compounds, fischerindoles, ambiguines and welwitindolinones, there are at least 81 members isolated from over 18 cyanobacterial strains.2 They have also been shown to have antimicrobial,3 antimycotic,3e,4 anticancer5 and immunomodulatory activity.6 Hapalindole/fischerindole metabolites possess three distinguishing features: a polycyclic ring system, diverse stereochemical variations and the late-stage introduction of a range of functional groups such as additional rings, halogens, hydroxyls, isothiocyanate, and others. Due to these unique structural features and diverse biological activities, a number of total syntheses have been devoted to this family of indole alkaloids.7 However, these efforts have been hindered due to the highly functionalized ring system and variant stereo-chemical patterns. Recently, work by several groups has explored the biogenesis of these metabolites,8 which has uncovered new prenyltransferases, cyclases, halogenases and other unique biosynthetic enzymes.
While each hapalindole subgroup has a characteristic ring connectivity and stereochemistry, current evidence indicates that they are all derived from a cis-indole isonitrile core combined with a geranyl monoterpene subunit. Through biosynthetic analysis, the mechanism for assembly of the hapalindole core was recently elucidated. l-Tryptophan is converted to cis-indole isonitrile precursor 1 by a three enzyme cascade8a followed by geranylation at the C-3 position.8f In the presence of a Stig cyclase(s), this 3-geranyl cis-indole isonitrile (3-GC) intermediate 2 undergoes a Cope rearrangement, 6-exo-trig cyclization and a terminal electrophilic aromatic substitution (EAS) at either the indole C-2 or C-4 position to afford the fischerindole or hapalindole core, respectively (Figure 1A).8f Subsequent late stage tailoring of these molecules provide further access to the ambiguines and welwitindolinones.
Figure 1:
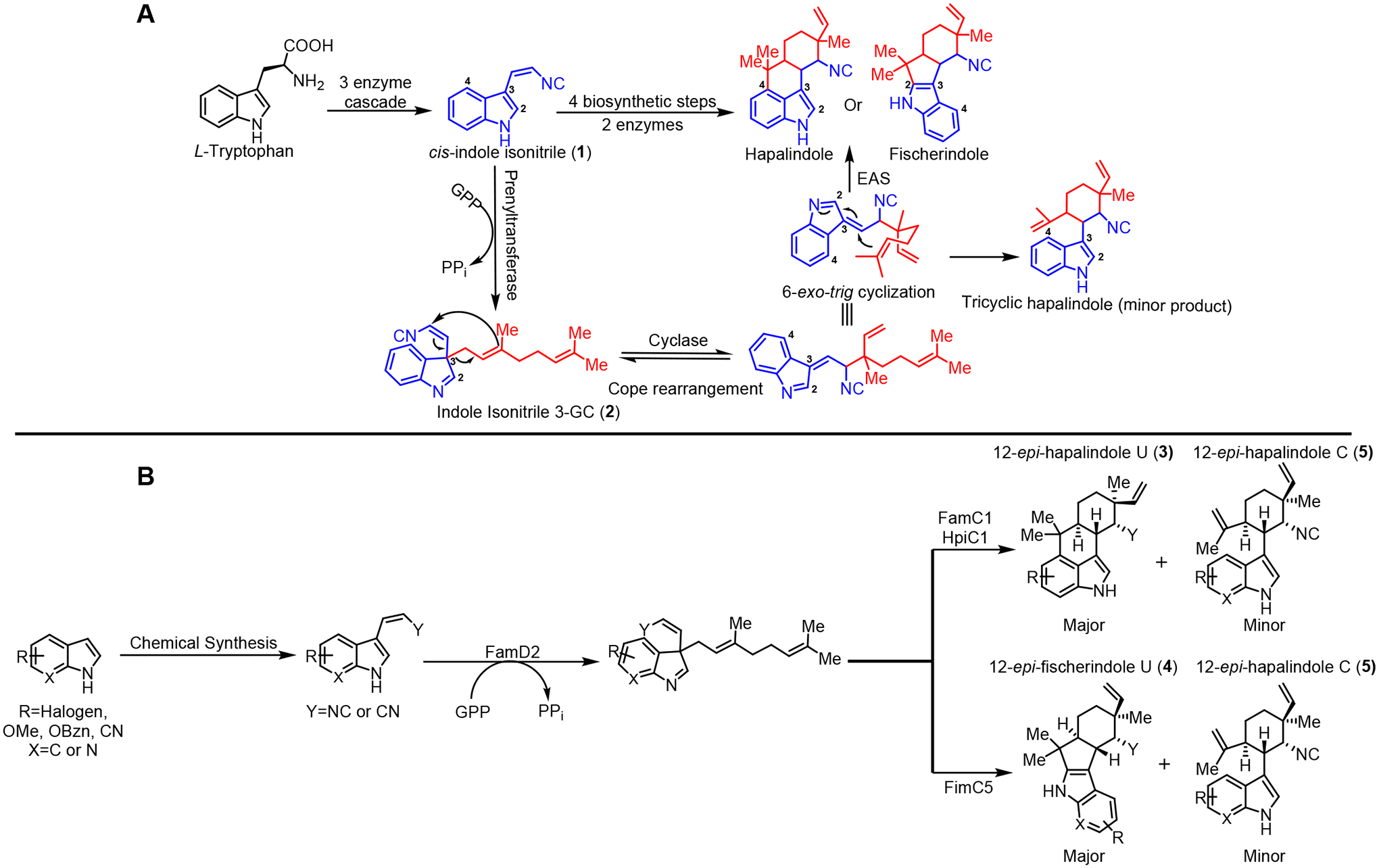
(A) Proposed biosynthesis of hapalindole type molecules. l-Tryptophan is converted into the cis-indole isonitrile core (1) (in blue) via a 3 enzyme cascade followed by geranylation (in red) at C-3 position to afford the 3-GC intermediate (2). In the presence of the cyclase(s), 2 undergoes a Cope rearrangement, 6-exo-trig cyclization and electrophilic aromatic substitution (EAS) at the C-4 or C-2 position to afford the core hapalindole or fischerindole structure, respectively. (B) The approach described herein includes chemical synthesis of unnatural cis-indole derivatives to show that the geranyltransferase (FamD2) and Stig cyclases can produce a range of unnatural 12-epi-hapalindole U (3), 12-epi-fischerindole U (4) and 12-epi-hapalindole C (5) compounds.
Recently, cyanobacterial-derived Stig cyclases have attracted interest for their ability to catalyze a multi-step core-forming cyclization cascade. Previous work has shown that the Stig cyclases exist in a dimeric state that may involve higher order oligomeric complexes to catalyze cyclization, including a terminal C-H functionalization reaction.8h,p Depending on the components of this oligomeric complex, different regio- and stereochemical outcomes have been observed.8f,g,i,s,t Structural and mutagenesis studies have revealed key residues responsible for core hapalindole and fischerindole formation alongside computational analysis that has examined the cyclase-mediated Cope rearrangement and terminal EAS.8h Although gram scale total syntheses of the hapalindoles has been achieved on a few select metabolites,7o,t further diversification is necessary for drug lead exploration. The Stig cyclases show potential as novel biocatalytic tools for developing unnatural hapalindole and fischerindole metabolites. Recently, we showed that fluorinated unnatural fischerindole and hapalindole derivatives could be produced using a microscale in vitro transcription/translation system with genes encoding prenyltransferase and Stig cyclase proteins.9 To validate and explore this approach further, we demonstrate herein that key Stig cyclases can be employed as biocatalytic tools to produce numerous unnatural hapalindole and fischerindole compounds while also providing further mechanistic insights into the unusual cyclization cascade (Figure 1A and 1B).
Results and Discussion
Substrate Synthesis
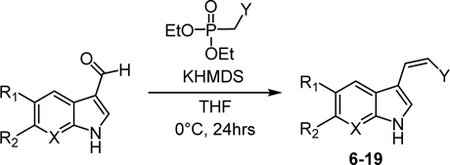
To pursue our analysis, chemical synthesis of unnatural cis-indole isonitrile derivatives was initiated. We synthesized these molecules via a Horner-Wadsworth-Emmons (HWE) olefination reaction from carboxyaldehyde indole derivatives based on the ability to produce unnatural derivatives in a facile manner (Table 1). While the HWE reaction is known to stereochemically favor trans-(E) product formation, we found that performing the reaction at 0°C yields a 50:50 ratio of the cis-(Z) and trans isomers. Using normal phase chromatography, these isomers are readily separated to obtain the desired Z-isomer product. The majority of these derivatives were designed to have functional group modification at the indole C-5 and C-6 positions, which included halogens (10–16), methoxy groups (8, 9), benzyl groups (6) and cyano (7) functionality. Two of the isomers (17, 18) were modified at the C-7 position of the indole ring by replacing with a nitrogen atom. For derivatives 16 and 18, the carboxyaldehyde indole precursor was commercially unavailable. Thus, we obtained the indole precursor and added the carboxyaldehyde functionality via a Vilsmeier-Haack reaction that offered 20 and 21 (Experimental Section), allowing us to obtain derivatives 16 and 18.
Table 1:
Cis-indole isonitrile derivatives generated in this study, and isolated % yield of the desired Z-isomer.
 | ||
|---|---|---|
| Compound | Substituent | % Yield of Z-isomer |
| 6 | R1=OBzn,R2=H,X=C,Y=NC | 11 |
| 7 | R1=CN,R2=H,X=C,Y=NC | 22 |
| 8 | R1=OMe,R2=H,X=C,Y=NC | 16 |
| 9 | R1=H,R2=OMe,X=C,Y=NC | 5 |
| 10 | R1=Cl,R2=H,X=C,Y=NC | 11 |
| 11 | R1=H,R2=Cl,X=C,Y=NC | 16 |
| 12 | R1=F,R2=H,X=C,Y=NC | 18 |
| 13 | R1=H,R2=F,X=C,Y=NC | 23 |
| 14 | R1=Br,R2=H,X=C,Y=NC | 18 |
| 15 | R1=H,R2=Br,X=C,Y=NC | 23 |
| 16 | R1=I,R2=H,X=C,Y=NC | 18 |
| 17 | R1=H,R2=H,X=N,Y=NC | 13 |
| 18 | R1=F,R2=H,X=N,Y=NC | 18 |
| 19 | R1=H,R2=H,X=C,Y=CN | 16 |
FamD2 prenyltransferase
With a suite of cis-indole isonitrile derivatives in hand, we assessed the flexibility of FamD2 (also known as AmbP1), the prenyltransferase from Fischerella ambigua UTEX 19038a,f for the first key transformation of 1. Through initial analytical reactions, we observed geranylation of 13 of the 14 substrates with only the 5-cyano derivative 7 failing to be geranylated. This result was rationalized based on previous structural studies of FamD2 that revealed conformational flexibility in the active site in the presence of a Mg2+ cofactor.8n In the case of 7, we hypothesized that either steric hindrance or unfavorable electronics played a role in the lack of C-3 geranylation. Because substrate 16 (containing a large 5-iodo group) was able to be geranylated, we believed that steric hinderance was less likely to be a factor. Upon examination of the crystal structure of FamD2,8n we hypothesized that the highly polar 5-cyano functionality is interacting with a specific residue or the backbone of FamD2, disrupting the derivative from a favorable position for geranylation. Because the 3-GC intermediate has been shown to undergo a 1,2-shift,8f we sought a different method to analyze the efficiency of the geranylation step. Using an HPLC assay, total turnover numbers (TTN) were calculated over a one hour reaction period at pH 10 to achieve maximum enzyme efficiency. Further cyclization reactions were conducted at pH 7 or 7.8 as cyclase activity is greater under more acidic conditions.8h,s It is possible that FamD2 turnover is attenuated at a more neutral pH, but this was not tested. At pH 10, TTN values ranged from 710 to 1902, which highlights the versatility and efficiency of FamD2 as a biocatalyst over a range of unnatural substrates (Table 2).
Table 2:
TTN values for geranylation reaction with FamD2.a
| Compound | TTN Value |
|---|---|
| 1 | 1495 |
| 6 | 1615 |
| 7 | N/Ab |
| 8 | 1481 |
| 9 | 1902 |
| 10 | 947 |
| 11 | 1547 |
| 12 | 1625 |
| 13 | 803 |
| 14 | 755 |
| 15 | 1174 |
| 16 | 710 |
| 17 | 1281 |
| 18 | 1366 |
| 19 | 1454 |
Assay conditions: 1 μM FamD2, 2 mM substrate, 1.5 mM GPP, 5 mM MgCl2, 50 mM glycine (pH 10.0), 100 μL, 37°C, 200 rpm, 1 hr.
Compound 7 showed no conversion in assay
FamC1 cyclase
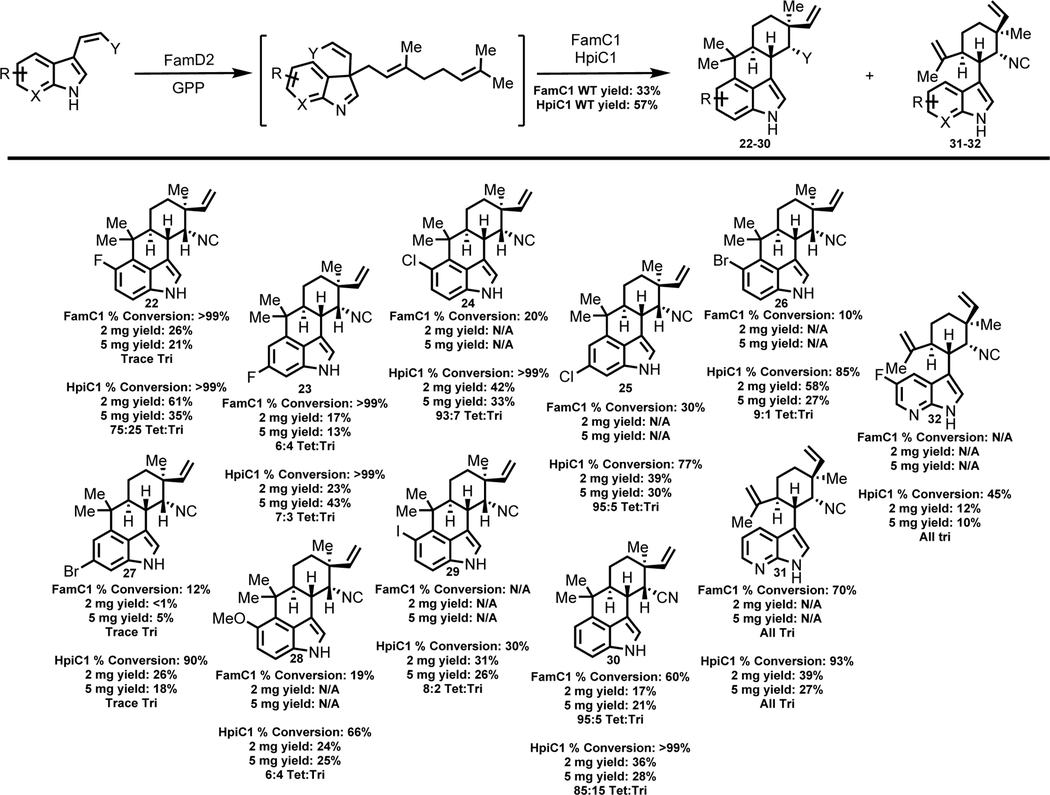
We next investigated the FamC1 cyclase from the Fischerella ambigua UTEX 1903 fam biosynthetic gene cluster. The homodimeric form of FamC1 produces 12-epi-hapalindole U8f and was previously shown to accept fluorinated cis-indole isonitrile derivatives 12 and 13 to generate new hapalindole compounds 18 and 23.9 We sought to analyze and confirm its scope beyond fluorinated derivatives. Initially, few additional substrates showed conversion to cyclized product. Lowering the pH and concentration of FamD2 did not increase cyclase activity. As Ca2+ has been shown to be an important cyclase cofactor, we supplemented the reaction mixture to test its effect on FamC1 activity.8g,h,i,s Upon the addition of 5 mM calcium chloride, we observed increased production of cyclized products in 8 of the 14 unnatural substrates. Based on HPLC analysis, we estimated the conversion values ranged from 10% to >99%. Scale up efforts of 2 mg of each substrate enabled structural characterization of these compounds. We confirmed that in all but one case, the corresponding 12-epi-hapaplindole U derivative was produced. By contrast, FamC1 catalyzed formation of tricyclic 12-epi-hapalindole C derivative 31 from azaindole compound 17. Tricyclic hapalindoles are normally a minor product observed in enzymatic reactions from previously studied Stig cyclases (Table 3). We initially reasoned that the unnatural electronegative N-7 of the azaindole inhibited the terminal EAS reaction at the C-4 position. However, C-4 tetracycle formation was observed with C-5-fluoro derivative 12, which suggests the selectivity may be guided by skeletal variation of the N-7 position instead of the electronics of the indole ring.
Table 3:
Structures of 12-epi-hapalindole U and 12-epi-hapalindole C derivatives produced by FamC1 and HpiC1 from unnatural cis-indole isonitrile or cis-indole nitrile substrates.c
|
Percent conversions, isolated yield values and tetracyclic:tricyclic ratio (ratio estimated by NMR and/or HPLC) are shown below each derivative. N/A=Derivative was either not produced by FamC1 or was not screened further in this study due to enhanced versatility of HpiC1. HPLC conversion values determined after 4 hrs in 100 μL reactions. Isolated yield values from overnight reactions.
HpiC1 cyclase
While FamC1 demonstrated the ability to convert 8 of 14 substrates, we sought a cyclase with greater flexibility. We next examined HpiC1 from Fischerella sp. ATCC 43239, which shares 84% sequence homology with FamC1. Complexes consisting of only HpiC1 have been shown to produce 12-epi-hapalindole U and trace levels of 12-epi-hapalindole C.8g,h Sequence comparison revealed only three active site sequence variations compared to FamC1.8h Analytical reactions showed an increased scope as 10 of the 14 substrates were converted to hapalindole products with conversion values ranging from 30 to >99%. As with FamC1, the majority of the compounds were confirmed to be the corresponding 12-epi-hapalindole U derivatives. Similar to reactions with FamC1, we observed production of tricyclic 12-epi-hapalindole C derivatives 31 and 32 from cis-indole isonitrile substrates 17 and 18, respectively (Table 3).
We hypothesized that the increased scope and reaction efficiency of HpiC1 could be attributed to the amino acid differences in the active site,8h resulting in less steric hindrance to accommodate larger substituents. We investigated the three amino acid differences in the active site between HpiC1 and FamC1 using site directed mutagenesis to assess their role in substrate scope, which are Val51, Phe138 and Leu147 in HpiC1. The HpiC1_V51I mutant revealed no changes in the substrate scope. Although single mutants (HpiC1_F138L and HpiC1_L147F) failed to show noticeable changes in scope, the % conversion of substrates 8, 14, and 16 were reduced (Supporting Information Table S36). The attenuated substrate scope of the double mutant (HpiC1_F138L_L147F) closely matched WT FamC1 (Supporting Information Figures S12–S22). This mutagenesis study revealed that two of the three active site residue differences (138 and 147) were responsible for the change in substrate scope and conversion values between the two cyclases. Given that FamC1 exhibits low % conversions for 14 and 15, and completely fails to convert 16, we hypothesize that the two mutations present in HpiC1 (Phe138 and Leu147) reduce steric hindrance in the area of the active site that interacts with the C-5 and C-6 substituents of the indole, resulting in higher conversion efficiencies.
FimC5 cyclase
Following analysis of two hapalindole producing cyclases, we explored FimC5, a fischerindole producing cyclase derived from Fischerella muscicola UTEX 1829. This strain produces 12-epi-fischerindole U8g and was used in our previous study to generate two fluorinated derivatives 33 and 37.9 Analytical reactions showed production of cyclized products for 8 of the 14 substrates tested with estimated conversion values ranging from 20% to >99%. The majority of the substrates were characterized as the corresponding 12-epi-fischerindole U derivatives. With substrate 17, FimC5 catalyzed formation of a 50:50 mixture of two products: the previously observed tricyclic derivative 31 and new tetracyclic derivative 36. The structural assignment of 36 was also confirmed using X-ray crystallography (Figure 2). In the case of derivative 18, we only observed production of the tricyclic derivative 32 (Table 4), with the C-5 fluorine appearing to limit reactivity at the C-2 position of the indole ring.
Figure 2:
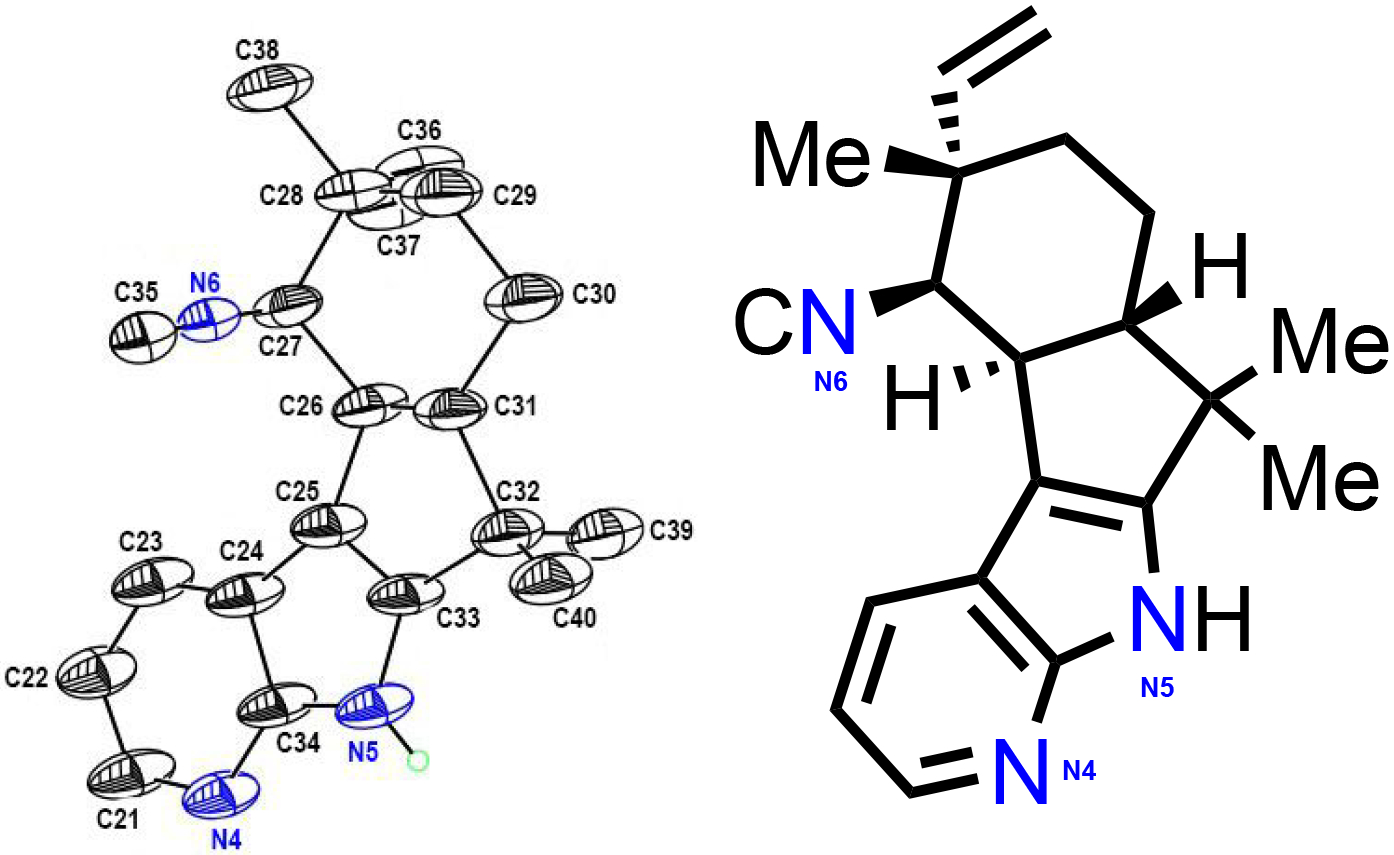
X-ray crystal structure of 36. NMR assigned structure has been added to highlight the structures similarity. Nitrogen atoms have been highlighted in blue with same numbers to orient the reader.
Table 4:
Structures of new 12-epi-fischerindole U derivatives produced by FimC5 from unnatural cis-indole isonitrile or cis-indole nitrile substrates.d
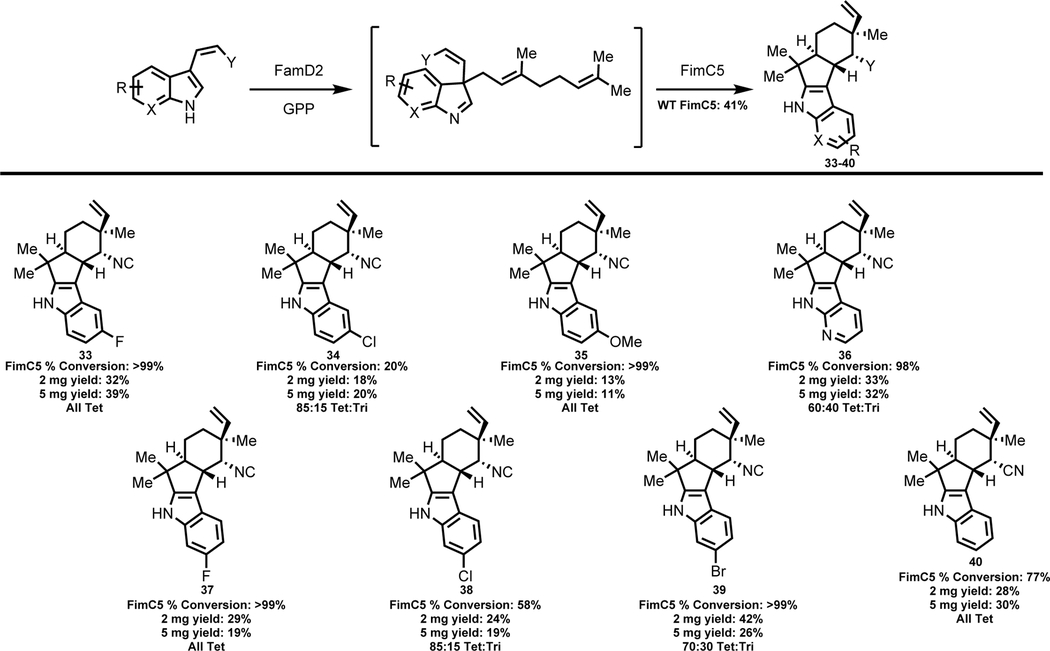
|
Percent conversions, isolated yields and tetracyclic:tricyclic ratio (ratio estimated by NMR and/or HPLC) are shown below each derivative. HPLC conversion values determined after 4 hrs in 100 μL reactions. Isolated yield values from overnight reactions.
Previous analysis of FimC5 indicated that two active site amino acid residues (Phe101 and Ser138) play a key role in selectivity towards the indole C-2 or C-4 position for the terminal EAS.8g,h Through site directed mutagenesis, we decided to explore this further with unnatural 3-GC derivatives. Three mutants of HpiC1 were produced to match the same residues identified in the FimC5 active site (Y101F, F138S and Y101F_F138S). Our results provide further support that these residues are critical for selectivity of the terminal reaction. For the Y101F and F138S mutants, we observed co-production of fischerindole and hapalindole products. However, upon screening the HpiC1 double mutant, we observed almost complete conversion to fischerindole metabolites. The F138S mutation also aided in uncovering the substrate selectivity differences between HpiC1 and FimC5 as product formation was either inhibited or completely abolished in select substrates. Exceptions to these results include compounds 13 and 17, which displayed a 50:50 ratio of products for all mutants and compound 18, which led to formation of the tricyclic hapalindole 32 for all HpiC1 mutants tested (Supporting Information Table S37 and Figures S1–S11).
Discovery and isolation of nitrile containing compounds 30 and 40
Hapalindole type-indole alkaloids are noted for containing a rare isonitrile (−NC) moiety although nitrile (−CN) containing fischerindole and ambiguine molecules have been isolated previously.1,3c,10 Derivation of the nitrile functionality from isonitrile rearrangement has been suggested previously, and we decided to examine this hypothesis by screening the Stig cyclases using cis-indole nitrile derivative 19, which was accepted by the prenyltransferase FamD2 at a TTN value comparable to the native isonitrile compound (Table 2). Cyclized product from wild-type HpiC1 was not initially observed with 19, however, generation of two new products from each of the three HpiC1→FimC5 mutant cyclases in variant ratios was observed. Scale up and structure characterization of products derived from the HpiC1 Y101F mutant led to identification of the 12-epi-hapalindole U nitrile derivative 30 (we also observed small amounts of the corresponding tricyclic hapalindole derivative). Wild-type HpiC1 and FamC1 were then rescreened with optimized conditions (Experimental Section) resulting in production of 30 for both enzymes. To our knowledge, these are the first nitrile containing hapalindole molecules reported. With this result in hand, we returned to wild type FimC5 cyclase and, under the same optimized conditions, retested substrate 19. We observed production of nitrile containing 12-epi-fischerindole U derivative 40 that was inadvertently overlooked in our initial analysis. Thus, our results demonstrated that the FamC1, HpiC1 and FimC5 cyclases have the ability to catalyze cyclization of the isonitrile and nitrile containing indole subunits. However, the nitrile substrate appeared to have lower conversion and isolated yield values (Tables 3 and 4) compared to the native isonitrile substrate, suggesting that the nitrile moiety affects turnover from the 3-GC intermediate to the terminal tetracyclic product. Regardless, this result supports that the nitrile functionality may come from early modifications of the intermediates but the source remains unknown.
Computational analysis for reactivity of substrates 17 and 18
To investigate why substrates 17 and 18 form tricyclic hapalindole compounds 31 and 32 with FamC1/HpiC1, respectively, while tetracyclic fischerindole 36 is formed with FimC5, we performed quantum mechanical density functional theory computations (Figure 3). For substrates 17 and 18, as well as the parent indole intermediate 2, we began with the tricyclic cationic intermediates T and considered two possible electrophilic aromatic substitutions: (1) reaction at C-4 to form the tetracyclic hapalindole scaffolds H and (2) reaction at C-2 to form the tetracyclic fischerindole scaffolds F. As shown in Figure 3, formation of hapalindole scaffolds is favored for the parent indole but disfavored for the azaindoles. The difference can be understood by consideration of resonance structures that stabilize the hapalindole scaffold. While conversion of indole cation 2-T into 2-H is exergonic by 1.9 kcal/mol and has a low free energy barrier of 7.4 kcal/mol, the conversion of azaindole cation 17-T into 17-H is endergonic by 10.0 kcal/mol and has a higher barrier of 14.7 kcal/mol. In particular, of the four resonance structures in addition to the iminium structure, the resonance structure shown places positive charge on the electronegative nitrogen, resulting in less stabilization than with the parent indole derivative where this positive charge is on a carbon. The conversion of fluorinated azaindole cation 18-T into 18-H is only slightly more favorable due to near-cancellation of resonance and inductive effects of F; this reaction is endergonic by 9.8 kcal/mol and has a barrier of 14.0 kcal/mol. Given the higher barriers to tetracyclic hapalindole formation for 17 and 18, it is not surprising that FamC1 and HpiC1 fail to catalyze this reaction; instead, deprotonation of cations 17-T and 18-T yield tricyclic compounds 31 and 32, respectively. Unlike tetracyclic hapalindole formation, conversion of azaindoles into tetracyclic fischerindole scaffolds does not exhibit differential resonance effects compared to the parent indole because the azaindole nitrogen and fluorine atoms are not on positively charged positions in any of the four additional resonance structures that stabilize each tetracyclic fischerindole scaffold. Instead, tetracyclic fischerindole formation is influenced by smaller inductive effects. Thus, conversion of indole 2-T into 2-F is exergonic by 10.1 kcal/mol, and conversion of azaindole 17-T into 17-F (with an additional inductively withdrawing nitrogen atom) is exergonic by only 8.5 kcal/mol. Conversion of fluorinated azaindole 18-T into 18-F, which contains inductively withdrawing nitrogen and fluorine atoms is exergonic by only 6.5 kcal/mol. The free energy barriers, which are 1.7, 1.9, and 2.9 kcal/mol, respectively, follow the same trend in which addition of inductively withdrawing atoms raises the transition state energy. However, because these inductive effects are smaller than resonance effects, FimC5 can efficiently catalyze the conversion of 17 into 36. Although it is important to note that these reactions take place in an enzyme active site, these computations addressing the innate reactivity of azaindoles reveal that tetracyclic hapalindole formation has a substantially higher free energy barrier than tetracyclic fischerindole formation. Accordingly, it is not surprising that FimC5 catalyzes tetracyclic fischerindole formation while FamC1 and HpiC1 form tricyclic products rather than tetracyclic hapalindoles. Evidently, the enzyme active sites do not overcome the innate difference in ease of tetracyclic product formation.
Figure 3:
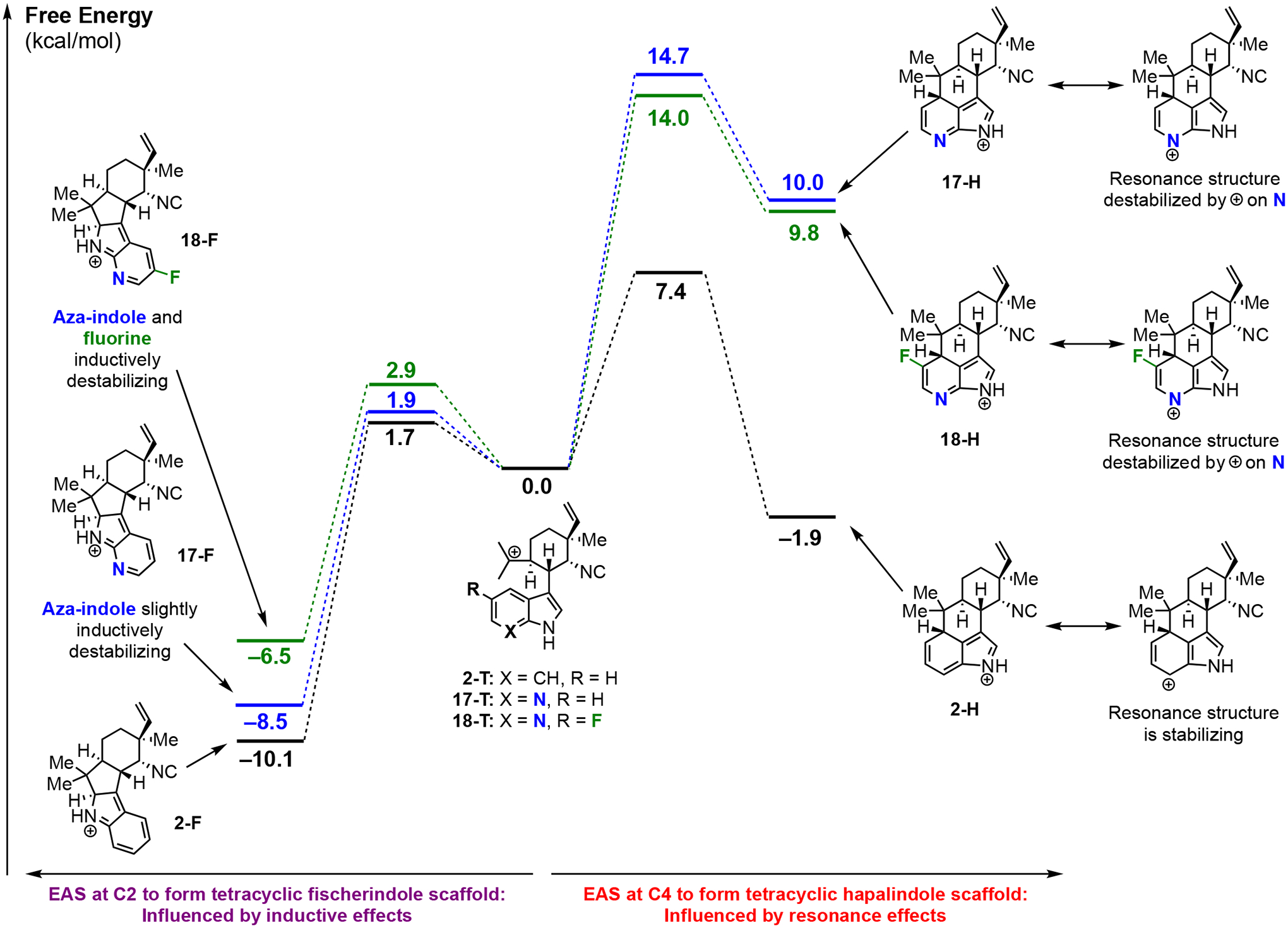
Quantum mechanical density functional theory computations comparing the energetics of tetracyclic hapalindole formation and tetracyclic fischerindole formation starting from the cationic intermediates derived from 2, 17, and 18. Tetracyclic fischerindole formation (left side) is only influenced by inductive effects, while tetracyclic hapalindole formation (right side) is also influenced by resonance effects.
Analysis of Stig cyclase biocatalytic ability
In this work, we explored the biocatalytic versatility of select Stig cyclases by assessing their substrate scope and ability to generate new derivatives. We analyzed HpiC1, select FamC1 and FimC5 reactions at both a 2 mg and 5 mg scale to assess how scalability may affect overall yield, which ranged from 10–60% (Table 3 & 4) for both reactions. The isolated yields of certain substrates (22, 26 for HpiC1 and 39 for FimC5) were coincident with, or exceeded, the levels of the native substrate, suggesting certain substituents enhance cyclase reactivity. In the majority of cases though, the isolated yields appeared to be significantly lower than the percent conversion values observed in the analytical scale reactions. We believe that through the filtration, workup and purification process, a significant amount of material was lost and optimizing isolation methods may be necessary to address this issue. At the 5 mg scale with HpiC1, we observed that the tricyclic minor product was generated in a higher ratio compared to the 2 mg or analytical reactions. While these two compounds are separable, further scalability represents an objective for protein engineering to maximize tetracycle formation. However, FamC1 did show lower production of the tricyclic product, but appears to be less efficient and flexible compared to HpiC1.
Conclusion
Through this work, we have shown the biocatalytic potential of core biosynthetic enzymes to produce unnatural hapalindole and fischerindole derivatives. The three cyclases investigated for this work, FamC1, HpiC1, and FimC5 along with select mutants and FamD2 geranyltransferase have demonstrated their ability to accept numerous unnatural cis-indole isonitrile (and nitrile) derivatives. These new compounds are poised for investigation of biological activity directly or for additional semi-synthetic or biocatalytic modifications. While HpiC1 appears to be the most versatile Stig cyclase, there are numerous additional homologs and heteromeric combinations that can be screened to further probe the full scope of Stig cyclase biocatalytic versatility. This work further supports the growing opportunity to employ natural product biosynthetic enzymes for assembly of complex, bioactive small molecules and as a complement to synthetic chemistry approaches.
Experimental Section
General
All NMR spectra were acquired on a Varian 400 and 600 MHz and Bruker 800 MHz spectrometers. Proton and carbon signals are reported in part per million (δ) using residual solvent signals as an internal standard. Analytical HPLC analysis was performed on a Shimadzu 2010 EV APCI spectrometer equipped with an LUNA C18 250 × 4.6 mm column, using a mobile phase gradient of 70–100% acetonitrile in water over 16 min. at 40°C and was monitored by UV absorption at 280 nm. LC-MS analysis was performed on a Agilent Infinity II TOF using an XBridge C18 2.1 × 150 mm column, using a mobile phase gradient of 70–100% acetonitrile in water over 12 min. Preparative-scale HPLC was performed on a Shimadzu 20-AT equipped with an LUNA C18 250 × 10 mm column for 2 mg reactions and an LUNA C8 250 × 21 mm column for 5 mg reactions, using a mobile phase gradient of 50 or 60–100% acetonitrile in water over 60 min. Optical rotations were obtained using a Jasco P2000 polarimeter at 25°C.
Escherichia coli strain BL21(DE3) was used for protein expression. Plasmid pET28H8T8g was used for cloning and expression of N-truncated FamC1 and FimC5. Plasmid pET28a was used for cloning and expression of FamD2, HpiC1 and HpiC1 mutants. Isopropyl β-D-thiogalactopyranoside (IPTG) was used to induce expression; DNAse and lysozyme were purchased form Sigma-Aldrich. Ni-NTA agarose from Invitrogen was used to purify His-tag proteins.
All chemicals were purchased from Sigma-Aldrich, ACROS, and Combi-Blocks. Multiplicities are abbreviated as following: singlet (s), doublet (d), triplet (t), quartet (q), doublet-doublet (dd), triplet-doublet (td), doublet-doublet-doublet (ddd), triplet-doublet-doublet (tdd), and multiplet (m). Chemical abbreviations: Ethyl Acetate (EtOAc), Dichloromethane (DCM), Tetrahydrofuran (THF), Potassium bis(trimethylsilyl)amide (KHMDS), Acetic Acid (AcOH), Sodium Sulfate (Na2SO4), Diethyl Ether (Et2O), Phosphorous Oxychloride (POCl3), Sodium Hydroxide (NaOH), Acetonitrile (CH3CN), Magnesium Chloride (MgCl2), Calcium Chloride (CaCl2), Sodium Chloride (NaCl)
Protein expression and purification
The expression and purification of proteins was performed as described.8g,9 Briefly, a single BL21(DE3) colony was inoculated in LB medium containing 50 μg/mL kanamycin and grown overnight at 37 °C shaking at 200 rpm. The main culture (1 L) was inoculated at the dilution of 1:100 in 2.8 L of Fernbach flask containing TB medium and the same concentration of antibiotic. The cells were grown (37 °C, 200 rpm) to an optical density (A600 nm) of 1.0. The culture flasks were chilled in ice, induced with IPTG (0.2 mM), and were further incubated (16 °C, 200 rpm) for 16 h. The cells were harvested (5000 rpm, 4 °C, 15 min), flash frozen, and stored at −80 °C until purification. The cell pellets were resuspended at 4 °C in the lysis buffer (10 mM HEPES, 50 mM NaCl, 0.2 mM TCEP, 10% glycerol), containing 0.5 mg/mL of lysozyme, 1 mM PMSF and 1 mL of 2 mg/mL DNAse. The mixture was stirred for 30 min and sonicated on ice for 120 s total time using 10 s pulses followed by a 50 s pause. The cellular debris was removed by centrifugation (65,000 × g, 4 °C, 35 min). The clarified lysate was loaded onto Ni-NTA agarose column equilibrated with lysis buffer. The column was washed with two column volume of wash buffer (10 mM HEPES, 300 mM NaCl, 0.2 mM TCEP, 10% glycerol, 20 mM imidazole) and the His-tagged protein was eluted with elution buffer (10 mM HEPES, 50 mM NaCl, 0.2 mM TCEP, 10% glycerol, 300 mM imidazole). The fractions were pooled and dialyzed overnight or by using a PD10-desalting column (GE Healthcare) using storage buffer (10 mM HEPES, 50 mM NaCl, 0.2 mM TCEP, 10% glycerol). The purified protein was analyzed by SDS-PAGE gel for purity, measured by Nanodrop using a calculated molar extinction coefficient for concentration, and flash-frozen in liquid nitrogen to store at −80 °C.
Analytical, TTN and scale up enzymatic reactions
Cis-indole isonitrile derivatives were synthesized as described below. For the initial assays, a 50μL reaction containing 10 μM FamD2, 15 μM cyclase, 1 mM substrate, 1 mM GPP, 5 mM MgCl2, 50 mM of Tris pH 7.8 buffer and 5 mM CaCl2, was incubated at 37°C for 4 hrs. The reaction was quenched with 3x volume of EtOAc twice. The organic layers were combined, dried and re-dissolved in 100 μL acetonitrile for LCMS and HPLC analysis. HPLC conversion values were determined by the area under the curve of the residual starting material and newly formed product. For later assays, a 100μL reaction containing 5μM FamD2, 20μM cyclase, 1 mM substrate, 1.5 mM GPP, 5 mM MgCl2, 50 mM of Tris pH 7.0 buffer and 7.5 mM CaCl2 was incubated, quenched and analyzed as previous. For FamD2 TTN assays, a 100 μL reaction containing 1 μM FamD2, 2 mM substrate, 1.5 mM GPP, 5 mM MgCl2, and 50 mM Glycine pH 10.0 buffer was incubated at 37°C for 1 hr. The reaction was quenched and analyzed as previous. TTN values were determined by standard curve analysis for the starting material (Supporting Information Tables S3–S15). For the structure analysis and isolated yield values of the enzymatic products, the reactions were scaled up to 2 mg and 5 mg starting material (10 and 25 mL respectively) and incubated at 37°C overnight or until HPLC showed consumption of starting material. Products were extracted with EtOAc and purified by preparative HPLC as described in the general methods. All products were obtained as a white solid. The purified compounds were concentrated, dissolved in C6D6 and analyzed using a Varian 600 MHz NMR and Bruker 800 MHz NMR.
Chemical synthesis of cis-indole isonitrile derivatives
All derivatives were prepared using method previously described.8f,9 Briefly, to a 50 mL two-neck round-bottom flask purged with nitrogen at −78 °C (dry ice/acetone), diethyl (isocyanomethyl) phosphonate (0.37 mL, 2.26 mmol) (diethyl cyanomethyl phosphonate was used for production of 19) was diluted with THF (5 mL). KHMDS (1 M THF, 2.60 mL, 2.60 mmol) was added dropwise, and the reaction was stirred at −78 °C for 15 min. To a separate 4 mL vial, indole-3-carboxaldehyde derivative (1.13 mmol) was dissolved in THF (5 mL), and the resulting solution was added dropwise to the KHMDS solution at −78 °C. The resulting mixture was stirred at 0 °C (cryocool) overnight or until TLC showed consumption of starting material. The resulting solution was quenched by the addition of AcOH (0.15 mL, 2.6 mmol) and concentrated. The resulting residue was diluted with EtOAc (20 mL), washed with 1 M aqueous potassium phosphate buffer (20 mL, pH 7), washed with brine, dried with Na2SO4, and concentrated to a residue. The residue was dissolved in EtOAc and purified by flash chromatography (24%–100% pentane/Et2O, SiO2) to afford the titled compound as reported below. Yields and spectral data reported below.
(Z)-5-(benzyloxy)-3-(2-isocyanovinyl)-1H-indole (6): Blue solid, 17mg, 11%
1H NMR (400 MHz, Acetone-d6) δ 5.17 (s, 2H), 5.90 (d, J = 8.9 Hz, 1H), 6.90 – 6.99 (m, 2H), 7.32 (t, J = 7.3 Hz, 1H), 7.37 – 7.47 (m, 4H), 7.51 (d, J = 7.5 Hz, 2H), 8.14 (d, J = 2.7 Hz, 1H), 10.80 (s, 1H).
13C NMR (151 MHz, acetone) δ 169.52, 154.15, 137.99, 130.84, 128.32, 127.70, 127.58, 127.55, 127.32, 124.44, 113.46, 112.60, 109.49, 101.38, 70.17.
(Z)-3-(2-isocyanovinyl)-1H-indole-5-carbonitrile (7): Tan Solid, 25mg, 22%
1H NMR (400 MHz, Acetone-d6) δ 6.06 (d, J = 8.9 Hz, 1H), 7.06 (dt, J = 9.2, 4.7 Hz, 1H), 7.53 (dd, J = 8.5, 1.6 Hz, 1H), 7.71 (dd, J = 8.4, 0.8 Hz, 1H), 8.24 – 8.29 (m, 1H), 8.32 (s, 1H), 11.38 (s, 1H).
13C NMR (101 MHz, acetone) δ 171.15, 138.20, 129.86, 127.72, 126.16, 124.72, 124.10, 120.80, 114.08, 111.07, 104.41
(Z)-3-(2-isocyanovinyl)-5-methoxy-1H-indole (8): Red Solid, 36mg, 16%
1H NMR (400 MHz, Acetone-d6) δ 3.84 (s, 3H), 5.89 (d, J = 8.8 Hz, 1H), 6.87 (dd, J = 8.8, 2.4 Hz, 1H), 6.97 (d, J = 6.0 Hz, 1H), 7.30 (d, J = 2.4 Hz, 1H), 7.41 (d, J = 8.7 Hz, 1H), 8.07 – 8.15 (m, 1H), 10.77 (s, 1H).
13C NMR (151 MHz, dmso) δ 169.82, 154.74, 130.64, 127.65, 127.49, 125.45, 124.09, 113.16, 113.00, 109.24, 100.54, 55.83.
(Z)-3-(2-isocyanovinyl)-6-methoxy-1H-indole (9): Tan Solid, 5mg, 5%
1H NMR (599 MHz, Acetone-d6) δ 3.82 (s, 4H), 5.91 (d, J = 8.8 Hz, 1H), 6.82 (dd, J = 8.7, 2.3 Hz, 1H), 6.93 (dt, J = 9.8, 4.8 Hz, 1H), 7.04 (d, J = 2.3 Hz, 1H), 7.64 (d, J = 8.7 Hz, 1H), 8.03 (d, J = 2.3 Hz, 1H), 10.67 (s, 1H).
13C NMR (151 MHz, dmso) δ 169.84, 156.73, 136.44, 125.96, 125.32, 124.46, 123.68, 119.32, 110.83, 109.34, 95.19, 55.67.
(Z)-5-chloro-3-(2-isocyanovinyl)-1H-indole (10): Tan Solid, 20mg, 11%
1H NMR (400 MHz, Acetone-d6) δ 5.98 (d, J = 8.9 Hz, 1H), 6.93 – 7.06 (m, 0H), 7.22 (dd, J = 8.6, 2.0 Hz, 1H), 7.55 (d, J = 8.6 Hz, 1H), 7.82 (d, J = 2.0 Hz, 1H), 8.22 (d, J = 2.0 Hz, 1H), 11.04 (s, 1H).
(Z)-6-chloro-3-(2-isocyanovinyl)-1H-indole (11): Yellow solid, 36mg, 16%
1H NMR (599 MHz, Acetone-d6) δ 5.96 (d, J = 8.9 Hz, 1H), 6.94 (dt, J = 9.5, 4.9 Hz, 1H), 7.16 (dd, J = 8.5, 1.9 Hz, 1H), 7.57 (d, J = 1.8 Hz, 1H), 7.75 (d, J = 8.5 Hz, 1H), 8.17 – 8.20 (m, 1H), 11.02 (s, 1H).
13C NMR (151 MHz, acetone) δ 170.89, 137.02, 128.93, 128.64, 128.47, 126.68, 124.71, 121.72, 120.25, 112.65, 110.61.
(Z)-5-fluoro-3-(2-isocyanovinyl)-1H-indole (12): Tan Solid, 36.7mg, 18%
1H NMR (400 MHz, Acetone-d6) δ 5.95 (d, J = 8.9 Hz, 1H), 6.94 (p, J = 4.8 Hz, 1H), 7.03 (td, J = 9.1, 2.5 Hz, 1H), 7.52 (td, J = 9.9, 3.5 Hz, 2H), 8.23 (s, 1H), 10.97 (s, 1H).
(Z)-6-fluoro-3-(2-isocyanovinyl)-1H-indole (13): Tan Solid, 48mg, 23%
1H NMR (400 MHz, Acetone-d6) δ 5.97 (d, J = 8.9 Hz, 1H), 6.93 – 7.01 (m, 2H), 7.27 (dd, J = 9.7, 2.3 Hz, 1H), 7.77 (dd, J = 8.7, 5.2 Hz, 1H), 8.17 (s, 1H), 10.93 (s, 1H).
(Z)-5-bromo-3-(2-isocyanovinyl)-1H-indole (14): Red Solid, 51mg, 18%
1H NMR (400 MHz, Acetone-d6) δ 5.96 (d, J = 8.9 Hz, 1H), 6.97 (dt, J = 9.2, 4.7 Hz, 1H), 7.33 (dd, J = 8.6, 1.9 Hz, 1H), 7.49 (d, J = 8.6 Hz, 1H), 7.95 (d, J = 1.8 Hz, 1H), 8.20 (d, J = 2.2 Hz, 1H), 11.08 (s, 1H).
13C NMR (101 MHz, acetone) δ 170.77, 135.27, 129.69, 128.97, 128.80, 126.21, 124.63, 121.61, 114.62, 114.27, 110.11
(Z)-6-bromo-3-(2-isocyanovinyl)-1H-indole (15): Red Solid, 63mg, 23%
1H NMR (400 MHz, Acetone-d6) δ 5.98 (d, J = 8.9 Hz, 1H), 6.85 – 7.00 (m, 1H), 7.29 (dd, J = 8.5, 1.8 Hz, 1H), 7.67 – 7.76 (m, 2H), 8.18 (d, J = 1.9 Hz, 1H), 10.99 (s, 1H).
13C NMR (151 MHz, acetone) δ 169.98, 136.50, 127.61, 126.03, 123.70, 123.38, 119.69, 115.57, 114.74, 109.70, 104.77.
(Z)-5-iodo-3-(2-isocyanovinyl)-1H-indole (16): Red Solid, 60mg, 18%
1H NMR (400 MHz, Acetone-d6) δ 5.98 (d, J = 8.9 Hz, 1H), 7.00 (d, J = 9.0 Hz, 1H), 7.40 (d, J = 8.5 Hz, 1H), 7.51 (dd, J = 8.6, 1.7 Hz, 1H), 8.16 (d, J = 3.0 Hz, 2H), 11.03 (s, 1H).
13C NMR (151 MHz, acetone) δ 169.93, 152.01, 134.82, 130.90, 129.54, 127.63, 127.06, 123.71, 114.15, 108.91, 83.50.
(Z)-3-(2-isocyanovinyl)-1H-pyrrolo[2,3-b]pyridine (17): White Solid, 26mg, 13%
1H NMR (599 MHz, Acetone-d6) δ 1.70 – 1.84 (m, 0H), 3.56 – 3.66 (m, 0H), 6.01 (d, J = 8.9 Hz, 1H), 6.97 (dt, J = 9.8, 5.1 Hz, 1H), 7.20 (dd, J = 7.9, 4.7 Hz, 1H), 8.20 (dd, J = 7.9, 1.6 Hz, 1H), 8.26 (s, 1H), 8.35 (dd, J = 4.6, 1.6 Hz, 1H), 11.38 (s, 1H).
13C NMR (151 MHz, acetone) δ 170.07, 148.22, 144.26, 127.05, 126.63, 123.64, 118.94, 116.61, 108.38, 105.03.
(Z)-5-fluoro-3-(2-isocyanovinyl)-1H-pyrrolo[2,3-b]pyridine (18): White Solid, 38mg, 18%
1H NMR (400 MHz, Acetone-d6) δ 6.04 (d, J = 8.9 Hz, 1H), 6.95 (dt, J = 9.5, 5.0 Hz, 1H), 8.04 (dd, J = 9.3, 2.7 Hz, 1H), 8.26 (t, J = 2.2 Hz, 1H), 8.33 (d, J = 2.1 Hz, 1H), 11.44 (s, 1H).
13C NMR (151 MHz, acetone) δ 170.20, 156.91, 155.31, 144.88, 132.67, 132.48, 129.23, 123.38, 119.29, 119.25, 112.50, 112.35, 108.61.
(Z)-3-(1H-indol-3-yl)acrylonitrile (19): Yellow solid, 30mg, 16%
1H NMR (400 MHz, Acetone-d6) δ 5.37 (dd, J = 11.8, 1.1 Hz, 1H), 7.14 – 7.31 (m, 2H), 7.50 – 7.60 (m, 1H), 7.67 (d, J = 11.7 Hz, 1H), 7.77 – 7.86 (m, 1H), 8.37 (s, 1H), 11.02 (s, 1H).
Chemical synthesis of Indole-3-carboxaldehyde derivatives
All derivatives were prepared using the same method. Briefly, in a 25mL round-bottom flask purged with nitrogen at 0 °C (ice-water bath), POCl3 (1.38 mL, 14.7 mmol) was stirred in dry DMF (4 mL) for 20 minutes. To a separate 4 mL vial, the reactant indole compound (2.94 mmol) was dissolved in dry DMF (4 mL) and added to the POCl3 solution at 0 °C. The reaction was slowly brought to room temperature and allowed to stir for 1 h or until TLC showed consumption of starting material. The reaction mixture was cooled to 0°C and quenched with ice-water and 1M NaOH (5 mL each). The reaction mixture was allowed to stir at room temperature for 1 h or until TLC showed consumption of intermediate. The resulting solution was extracted with EtOAc (2×10 mL), washed with brine and dried with Na2SO4 and concentrated to a residue. The residue was dissolved in EtOAc and purified by flash chromatography (16%−100% Hexanes/EtOAc, SiO2) to afford the titled compound as reported below. Yields and spectral data reported below.
5-iodo-1H-indole-3-carbaldehyde (20): Off-white solid, 259mg, 46%
1H NMR (400 MHz, Acetone-d6) δ 7.42 (dd, J = 8.5, 0.6 Hz, 1H), 7.58 (dd, J = 8.6, 1.8 Hz, 1H), 8.22 (s, 1H), 8.56 – 8.66 (m, 1H), 10.01 (s, 1H).
5-fluoro-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde (21): White Solid, 88mg, 18%
1H NMR (400 MHz, Acetone-d6) δ 8.20 (dd, J = 8.8, 2.8 Hz, 1H), 8.31 (dd, J = 2.8, 1.8 Hz, 1H), 8.48 (s, 1H), 10.01 (s, 1H), 11.69 (s, 1H).
Chemical Synthesis of Geranyl diphosphate, tri ammonium
Geranyl diphosphate was synthesized as described previously.11 To a 10 mL round bottom flask purged with nitrogen, tris (tetrabutylammonium) hydrogen pyrophosphate (1.0 g, 1.04 mmol) was dissolved in CH3CN (1.0 mL). Geranyl chloride (0.09 mL, 0.475 mmol) was added and the reaction mixture was stirred at room temperature for 2 h.
Dowex 50WX8 resin preparation:
Dowex 50WX8 resin (20 g, hydrogen form) was washed with half saturated aqueous ammonium chloride (5×50 mL) and water (5×50 mL) until the pH of the supernatant equaled 5. The slurry was rinsed twice with ion exchange buffer (2% isopropanol in 25 mM aqueous ammonium bicarbonate) and loaded into a flash column and equilibrated with ion exchange buffer.
Purification:
The reaction mixture was concentrated to afford an orange residue which was diluted with ion exchange buffer. The crude mixture was chromatographed with two column volumes of ion exchange buffer (75 mL). The fractions were combined and concentrated by rotary evaporation, flash frozen and lyophilized for 2 d. The resulting white powder was diluted with 0.1 M ammonium bicarbonate (4 mL) and 50% isopropanol/CH3CN (10 mL), vortexed for 30 seconds and centrifuged (2,000 rpm, rt, 5 min). The organic layer was extracted and the residual 0.5 mL of yellow liquid was diluted with 50% isopropanol/CH3CN and the dilution/vortex/centrifugation process was repeated twice. The combined organic layers were concentrated to afford a while solid. The white solid was taken up in 50% isopropanol:25% CH3CN:25% 0.1 M aqueous ammonium bicarbonate and chromatographed with cellulose. The resulting fractions were combined and lyophilized affording the title compound as a white powder (138 mg, 80.3%).
1H NMR (400 MHz, D2O/ND4OD) δ 1.92 (d, J = 1.3 Hz, 3H), 1.98 (s, 3H), 2.01 (d, J = 1.3 Hz, 3H), 2.39 (d, J = 6.5 Hz, 2H), 2.41 – 2.49 (m, 2H), 5.45 – 5.53 (m, 1H), 5.74 (dt, J = 6.1, 3.9 Hz, 1H).
13C NMR (151 MHz, D2O/ND4OD) δ 142.46, 133.67, 124.40, 120.46, 62.61, 39.10, 25.92, 25.16, 17.26, 15.89.
31P NMR (162 MHz, D2O/ND4OD) δ −9.93 (d, J = 21.6 Hz), −6.08 (d, J = 21.6 Hz).
Structure Determination
Colorless plates of 36 were grown from a diethyl ether/hexanes solution of the compound at 4 °C. A crystal of dimensions 0.06 × 0.04 × 0.02 mm was mounted on a Rigaku AFC10K Saturn 944+ CCD-based X-ray diffractometer equipped with a low temperature device and Micromax-007HF Cu-target micro-focus rotating anode (λ = 1.54187 A) operated at 1.2 kW power (40 kV, 30 mA). The X-ray intensities were measured at 85(1) K with the detector placed at a distance 42.00 mm from the crystal. A total of 2028 images were collected with an oscillation width of 1.0° in ω. The exposure times were 5 sec. for the low angle images, 45 sec. for high angle. Rigaku d*trek images were exported to CrysAlisPro for processing and corrected for absorption. The integration of the data yielded a total of 61153 reflections to a maximum 2θ value of 139.83° of which 7384 were independent and 5502 were greater than 2σ(I). The final cell constants (Table S36) were based on the xyz centroids of 8462 reflections above 10σ(I). Analysis of the data showed negligible decay during data collection. The structure was solved and refined with the Bruker SHELXTL (version 2018/3) software package, using the space group P2(1)2(1)2(1) with Z = 4 for the formula C44H56N6O. All non-hydrogen atoms were refined anisotropically with the hydrogen atoms placed in a combination of idealized and refined positions. Full matrix least-squares refinement based on F2 converged at R1 = 0.0885 and wR2 = 0.2349 [based on I > 2σ(I)], R1 = 0.1102 and wR2 = 0.2582 for all data. Additional details are presented in Tables S36–S41 and are given as Supporting Information in a CIF file.
Supplementary Material
Acknowledgements
We are grateful to the National Science Foundation under the CCI Center for Selective C−H Functionalization (CHE-1700982), National Institutes of Health (R35 GM118101), and the Hans W. Vahlteich Professorship (to D.H.S.) and ACS MEDI Pre-Doctoral Fellowship (to R.M.H) for financial support. J. N. S. acknowledges support from the National Institute of General Medical Sciences of the National Institutes of Health (F32 GM122218). The authors thank Dr. Jeffery Kampf (University of Michigan Department of Chemistry) for X-ray crystallographic work and Dr. Pavel Nagorny for access to their polarimeter. Funding from NSF grant CHE-0840456 helped defray costs of small molecule X-ray instrumentation. Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE) and by the San Diego Supercomputing Center (SDSC) through XSEDE (ACI-1548562).
Footnotes
Supporting Information: This information is available free of charge on the ACS Publications website
Full amino acid sequences, mutagenic primers, TTN tables, compound characterization tables, mutagenesis percent conversions tables, mutagenesis HPLC traces, NMR spectra and MS traces, crystallographic data and parameters, computational methods
The authors declare no competing financial interest.
References
- (1).Walton K; Berry JP Indole Alkaloids of the Stigonematales (Cyanophyta): Chemical Diversity, Biosynthesis and Biological Activity. Mar. Drugs 2016, 14, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bhat V; Dave A; MacKay JA; Rawal VH Chapter Two- The Chemistry of Hapalindoles, Fischerindoles, Ambiguines, and Welwitindolinones. In The Alkaloids: Chemistry and Biology; Academic Press: New York, 2014; Vol. 73, p 65–160. [DOI] [PubMed] [Google Scholar]
- (3).(a) Shunyan M; Krunic A; Chlipala G; Orjala J Antimicrobial Ambiguine Isonitriles from the Cyanobacterium Fischerella ambigua. J. Nat. Prod 2009, 72, 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shunyan M; Krunic A; Santarsiero BD; Orjala J Hapalindole-related alkaloids from the cultured cyanobacterium Fischerella ambigua. Phytochemistry 2010, 71, 2116–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim H; Lantivit D; Hwang CH; Kroll DJ; Swanson SM; Franzblau SG; Orjala J Indole alkaloids from two cultured cyanobacteria, Westiellopsis sp. and Fischerella muscicola. Bioorg. Med. Chem 2012, 20, 5290–5295. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Raveh A; Carmeli S Antimicrobial Ambiguines from the Cyanobacterium Fischerella sp. Collected in Israel. J. Nat. Prod 2007, 70, 196–201. [DOI] [PubMed] [Google Scholar]; (e) Moore RE; Cheuk C; Yang XQG; Patterson GML; Bonjouklian R; Smitka TA; Mynderse JS; Foster RS; Jones ND; Swartzendruber JK; Deeter JB Hapalindoles, antibacterial and antimycotic alkaloids from the cyanophyte Hapalosiphon fontinalis. J. Org. Chem 1987, 52, 1036–1043. [Google Scholar]; (f) Asthana RK; Srivastava A; Singh AP; Deepali; Singh SP; Nath G; Srivastava R; Srivastava BS Identification of an antimicrobial entity from the cyanobacterium Fischerella sp. isolated from bark of Azadirachta indica (Neem) tree. J. App. Phycol 2006, 18, 33–39. [Google Scholar]
- (4).Smitka TA; Bonjouklian R; Doolin L; Jones ND; Deeter JB; Yoshida WY; Prinsep MR; Moore RE; Patterson GML Ambiguine isonitriles, fungicidal hapalindole-type alkaloids from three genera of blue-green algae belonging to the Stigonemataceae. J. Org. Chem 1992, 57, 857–861. [Google Scholar]
- (5).(a) Stratmann K; Moore RE; Bonjouklian R; Deeter JB; Patterson GML; Shaffer S; Smith CD; Smitka TA Welwitindolinones, Unusual Alkaloids from the Blue-Green Algae Hapalosiphon welwitschii and Westiella intricata. Relationship to Fischerindoles and Hapalinodoles. J. Am. Chem. Soc 1994, 116, 9935–9942. [Google Scholar]; (b) Smith CD; Zilfou JT; Stratmann K, Patterson GML; Moore RE Welwitindolinone analogues that reverse P-glycoprotein-mediated multiple drug resistance. Mol. Pharmacol 1995, 47, 241–247. [PubMed] [Google Scholar]; (c) Zhang X; Smith CD Microtubule effects of welwistatin, a cyanobacterial indolinone that circumvents multiple drug resistance. Mol. Pharmacol 1996, 49, 288–294. [PubMed] [Google Scholar]
- (6).Chilczuk T; Steinborn C; Breinlinger S; Zimmermann-Klemd AM; Huber R; Enke H; Enke D; Niedermeyer THJ; Grundemann C Hapalindoles from the Cyanobacterium Hapalosiphon sp. Inhibit T Cell Proliferation. Planta Med. 2020, 86, 96–103. [DOI] [PubMed] [Google Scholar]
- (7).(a) Dethe DH; Das S; Kumar VB; Mir NA Enantiospecific Total Syntheses of (+)- Hapalindole H and (−)-12-epi-Hapalindole U. Eur. J. Chem 2018, 24, 8980–8984. [DOI] [PubMed] [Google Scholar]; (b) Richter JM; Ishihara Y; Masuda T; Whitefield BW; Llamas T; Pohjakallio A; Baran PS Enantiospecific Total Synthesis of the Hapalindoles, Fischerindoles, and Welwitindolinones via a Redox Economic Approach. J. Am. Chem. Soc 2008, 130, 17938–17954. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rafferty RJ; Williams RM Formal Synthesis of Hapalindole O and Synthetic Efforts towards Hapalindole K and Ambiguine A. Heterocycles 2012, 86, 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Connon R; Guiry PJ Recent advances in the development of one-pot/multistep syntheses of 3,4-annulated indoles. Tetrahedron Lett. 2020, 151696. [Google Scholar]; (e) Maimone TJ; Ishihara Y; Baran PS Scalable total syntheses of (−)-hapalindole U and (+)-ambiguine H. Tetrahedron 2015, 71, 3652–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Fukuyama T; Chen X Stereocontrolled Synthesis of (−)-Hapalindole G. J. Am. Chem. Soc 1994, 116, 3125–3126. [Google Scholar]; (g) Sahu S; Das B; Maji MS Stereodivergent Total Synthesis of Hapalindoles, Fischerindoles, Hapalonamide H, and Ambiguine H Alkaloids by Developing a Biomimetic, Redox-Neutral, Cascade Prins-Type Cyclization. Org. Lett 2018, 20, 6485–6489. [DOI] [PubMed] [Google Scholar]; (h) Vaillancourt V; Albizati KF Synthesis and absolute configuration of (+)-hapalindole Q. J. Am. Chem. Soc 1993, 115, 3499–3502. [Google Scholar]; (i) Kinsman AC; Kerr MA The Total Synthesis of (+)-Hapalindole Q by an Organomediated Diels−Alder Reaction. J. Am. Chem. Soc 2003, 125, 14120–14125. [DOI] [PubMed] [Google Scholar]; (j) Kinsman AC; Kerr MA Total Synthesis of (±)-Hapalindole Q. Org. Lett 2001, 3, 3189–3191. [DOI] [PubMed] [Google Scholar]; (k) Bhat V; Allan KM; Rawal VH Total Synthesis of N - Methylwelwitindolinone D Isonitrile. J. Am. Chem. Soc 2011, 133, 5798–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Gademann K; Bonazzi S Total Synthesis of Complex Cyanobacterial Alkaloids without Using Protecting Groups. Angew. Chem. Int. Ed 2007, 46, 5656–5658. [DOI] [PubMed] [Google Scholar]; (m) Lu Z; Yang M; Chen P; Xiong X; Li A Total Synthesis of Hapalindole-Type Natural Products. Angew. Chem. Int. Ed 2014, 53, 13840–13844. [DOI] [PubMed] [Google Scholar]; (n) Rafferty RJ; Williams RM Total Synthesis of Hapalindoles J and U. J. Org. Chem 2012, 77, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Baran PS; Maimone TJ; Richter JM Total synthesis of marine natural products without using protecting groups. Nature 2007, 446, 404–408. [DOI] [PubMed] [Google Scholar]; (p) Johnson RE; Ree H; Hartmann M; Lang L; Sawano S; Sarpong R Total Synthesis of Pentacyclic (−)-Ambiguine P Using Sequential Indole Functionalizations. J. Am. Chem. Soc 2019, 141, 2233–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Xu J; Rawal VH Total Synthesis of (−)-Ambiguine P. J. Am. Chem. Soc 2019, 141, 4820–4823. [DOI] [PubMed] [Google Scholar]; (r) Chandra A; Johnston JN Total Synthesis of the Chlorine-Containing Hapalindoles K, A, and G. Angew. Chem. Int. Ed 2011, 50, 7641–7644. [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Reisman SE; Ready JM; Hasuoka A; Smith CJ; Wood JL Total Synthesis of (±)- Welwitindolinone A Isonitrile. J. Am. Chem. Soc 2006, 128, 1448–1449. [DOI] [PubMed] [Google Scholar]; (t) Baran PS; Richter JM Direct Coupling of Indoles with Carbonyl Compounds: Short, Enantioselective, Gram-Scale Synthetic Entry into the Hapalindole and Fischerindole Alkaloid Families. J. Am. Chem. Soc 2004, 126, 7450–7451. [DOI] [PubMed] [Google Scholar]; (u) Muratake H; Natsume M Total synthesis of marine alkaloids (±)-hapalindoles J and M. Tetrahedron Lett. 1989, 30, 1815–1818. [Google Scholar]; (v) Muratake H; Natsume M Synthetic studies of marine alkaloids hapalindoles. Part 2. Lithium aluminum hydride reduction of the electron-rich carbon-carbon double bond conjugated with the indole nucleus. Tetrahedron. 1990, 46, 6343–6350. [Google Scholar]; (w) Muratake H; Kumagami H; Natsume M Synthetic studies of marine alkaloids hapalindoles. Part 3 Total synthesis of (±)-hapalindoles H and U. Tetrahedron. 1990, 46, 6351–6360. [Google Scholar]; (x) Bhat V; Allan KM; Rawal VH Total Synthesis of N-Methylwelwitindolinone D Isonitrile. J. Am. Chem. Soc 2011, 133, 5798–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]; (y) Weires NA; Styduhar ED; Baker EL; Garg NK Total Synthesis of (−)-N-Methylwelwitindolinone B Isothiocyanate via a Chlorinative Oxabicycle Ring-Opening Strategy. J. Am. Chem. Soc 2014, 136, 14710–14713. [DOI] [PMC free article] [PubMed] [Google Scholar]; (z) Styduhar ED; Huters AD; Weires NA; Garg NK Enantiospecific Total Synthesis of N-Methylwelwitindolinone D Isonitrile. Angew. Chem. Int. Ed 2013, 52, 12422–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]; (aa) Quasdorf KW; Huters AD; Lodewyk MW; Tantillo DJ; Garg NK Total Synthesis of Oxidized Welwitindolinones and (−)-N-Methylwelwitindolinone C Isonitrile. J. Am. Chem. Soc 2012, 134, 1396–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ab) Huters AD; Styduhar ED; Garg NK Total Syntheses of the Elusive Welwitindolinones with Bicyclo[4.3.1] Cores. Angew. Chem. Int. Ed 2012, 51, 3758–3765. [DOI] [PubMed] [Google Scholar]; (ac) MacKay JA; Bishop RL; Rawal VH Rapid Synthesis of the N-Methylwelwitindolinone Skeleton. Org. Lett 2005, 7, 3421–3424. [DOI] [PubMed] [Google Scholar]; (ad) Bhat V; Rawal VH Stereocontrolled Synthesis of 20,21-Dihydro N-Methylwelwitindolinone B Isothiocyanate. Chem. Commun 2011, 47, 9705–9707. [DOI] [PubMed] [Google Scholar]; (ae) Bhat V; MacKay JA; Rawal VH Lessons Learned While Traversing the Welwitindolinone Alkaloids Obstacle Course. Tetrahedron 2011, 67, 10097–10104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (af) Bhat V; MacKay JA; Rawal VH Directed Oxidative Cyclizations to C2- or C4-Positions of Indole: Efficient Construction of the Bicyclo[4.3.1]Decane Core of Welwitindolinones. Org. Lett 2011, 13, 3214–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ag) Allan KM; Kobayashi K; Rawal VH A Unified Route to the Welwitindolinone Alkaloids: Total Syntheses of (−)-N-Methylwelwitindolinone C Isothiocyanate, (−)-N-Methylwelwitindolinone C Isonitrile, and (−)-3-Hydroxy-N-Methylwelwitindolinone C Isothiocyanate. J. Am. Chem. Soc 2012, 134, 1392–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Hillwig ML; Zhu Q; Liu X Biosynthesis of Ambiguine Indole Alkaloids in Cyanobacterium Fischerella ambigua. ACS Chem. Biol 2014, 9, 372–377. [DOI] [PubMed] [Google Scholar]; (b) Chang WC; Sanyal D; Huang JL; Ittiamornkul K; Zhu Q; Liu X In Vitro Stepwise Reconstitution of Amino Acid Derived Vinyl Isocyanide Biosynthesis: Detection of an Elusive Intermediate. Org. Lett 2017, 19, 1208–1211. [DOI] [PubMed] [Google Scholar]; (c) Ittiamornkul K; Zhu Q; Gkotsi DS; Smith DMR; Hillwig ML; Nightingale N; Goss RJM; Liu X Promiscuous indolyl vinyl isonitrile synthases in the biogenesis and diversification of hapalindole-type alkaloids. Chem. Sci 2015, 6, 6836–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu X; Hillwig ML; Koharudin LMI; Gronenborn AM Unified biogenesis of ambiguine, fischerindole, hapalindole and welwitindolinone: identification of a monogeranylated indolenine as a cryptic common biosynthetic intermediate by an unusual magnesium-dependent aromatic prenyltransferase. Chem. Comm 2016, 52, 1737–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hillwig ML; Fuhrman HA; Ittiamornkul K; Sevco TJ; Kwak DH; Liu X Identification and Characterization of a Welwitindolinone Alkaloid Biosynthetic Gene Cluster in the Stigonematalean Cyanobacterium Hapalosiphon welwitschii. ChemBioChem 2014, 15, 665–669. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Li S; Lowell AN; Yu F; Raveh A; Newmister SA; Bair N; Schaub JM; Williams RM; Sherman DH Hapalindole/Ambiguine Biogenesis Is Mediated by a Cope Rearrangement, C–C Bond-Forming Cascade. J. Am. Chem. Soc 2015, 49, 15366–15369. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Li S; Lowell AN; Newmister SA; Yu F; Williams RM; Sherman DH Decoding cyclase-dependent assembly of hapalindole and fischerindole alkaloids. Nat. Chem. Biol 2017, 13, 467–469. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Newmister SA; Li S; Garcia-Borras M; Sanders JN; Yang S; Lowell AN; Yu F; Smith JL; Williams RM; Houk KN; Sherman DH Structural basis of the Cope rearrangement and cyclization in hapalindole biogenesis. Nat. Chem. Biol 2018, 14, 345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Li S; Newmister SA; Lowell AN; Zi J; Chappell CR; Yu F; Hohlman RM; Orjala J; Williams RM; Sherman DH Control of Stereoselectivity in Diverse Hapalindole Metabolites is Mediated by Cofactor-Induced Combinatorial Pairing of Stig Cyclases. Angew. Chem. Int. Ed 2020, 59, 8166–8172. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Micallef ML; Sharma D; Bunn BM; Gerwick L; Viswanathan R; Moffitt MC Comparative analysis of hapalindole, ambiguine and welwitindolinone gene clusters and reconstitution of indole-isonitrile biosynthesis from cyanobacteria. BMC Microbiology 2014, 14, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Yu CP; Tang Y; Cha L; Milikisiyants S; Smirnova TI; Smirnov AI; Guo Y; Chang WC Elucidating the Reaction Pathway of Decarboxylation-Assisted Olefination Catalyzed by a Mononuclear Non-Heme Iron Enzyme. J. Am. Chem. Soc 2018, 140, 15190–15193. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Bornemann V; Patterson GML; Moore RE Isonitrile biosynthesis in the cyanophyte Hapalosiphon fontinalis. J. Am. Chem. Soc 1988, 110, 2339–2340. [Google Scholar]; (m) Harris NC; Born DA; Cai W; Huang Y; Martin J; Khalaf R; Drennan CL; Zhang W Isonitrile Formation by a Non-Heme Iron(II)-Dependent Oxidase/Decarboxylase. Angew. Chem. Int. Ed 2018, 130, 9855–9858. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Awakawa T; Mori T; Nakashima Y; Zhai R; Wong CP; Hillwig ML; Liu X; Abe I Molecular Insight into the Mg2+-Dependent Allosteric Control of Indole Prenylation by Aromatic Prenyltransferase AmbP1. Angew. Chem. Int. Ed 2018, 57, 6810–6813. [DOI] [PubMed] [Google Scholar]; (o) Wang J; Chen CC; Yang Y; Liu W; Ko TP; Shang N; Hu X; Xie Y; Huang JW; Zhang Y; Guo RT Structural insight into a novel indole prenyltransferase in hapalindole-type alkaloid biosynthesis. Biochem. Biophys. Res. Commun 2018, 495, 1782–1788. [DOI] [PubMed] [Google Scholar]; (p) Chen CC; Hu X; Tang X; Yang Y; Ko TP; Gao J; Zheng Y; Huang JW; Yu Z; Li L; Han S; Cai N; Zhang Y; Liu W; Guo RT The Crystal Structure of a Class of Cyclases that Catalyze the Cope Rearrangement. Angew. Chem. Int. Ed 2018, 57, 15060–15064. [DOI] [PubMed] [Google Scholar]; (q) Wong CP; Awakawa T; Nakashima Y; Mori T; Zhu Q; Liu X; Abe I Two Distinct Substrate Binding Modes for the Normal and Reverse Prenylation of Hapalindoles by the Prenyltransferase AmbP3. Angew. Chem. Int. Ed 2018, 57, 560–563. [DOI] [PubMed] [Google Scholar]; (r) Hillwig ML; Liu X A new family of iron-dependent halogenases act on freestanding substrates. Nat. Chem. Bio 2014, 10, 921–923. [DOI] [PubMed] [Google Scholar]; (s) Zhu Q; Liu X Discovery of a Calcium-Dependent Enzymatic Cascade for the Selective Assembly of Hapalindole-Type Alkaloids: On the Biosynthetic Origin of Hapalindole U. Angew. Chem. Int. Ed 2017, 56, 9062–9066. [DOI] [PubMed] [Google Scholar]; (t) Zhu Q; Liu X Molecular and genetic basis for early stage structural diversifications in hapalindole-type alkaloid biogenesis. Chem. Comm 2017, 53, 2826–2829. [DOI] [PubMed] [Google Scholar]
- (9).Khatri Y; Hohlman RM; Mendoza J; Li S; Lowell AN; Asahara H; Sherman DH Multicomponent Microscale Biosynthesis of Unnatural Cyanobacterial Indole Alkaloids. ACS Syn. Biol 2020, 9, 1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Huber U; Moore RE; Patterson GML Isolation of a Nitrile-Containing Indole Alkaloid from the Terrestrial Blue-Green Alga Hapalosiphon delicatulus. J. Nat. Prod 1998, 61, 1304–1306. [DOI] [PubMed] [Google Scholar]
- (11).Davisson VJ; Woodside AB; Neal TR; Stremler KE; Muehlbacher M; Poulter CD Phosphorylation of isoprenoid alcohols. J. Org. Chem 1986, 51, 4768–4779. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.