

Abstract
Surface epithelia provide a critical barrier to the outside world. Upon a barrier breach, resident epithelial and immune cells coordinate efforts to control infections and heal tissue damage. Inflammation can etch lasting marks within tissues, altering features including scope and quality of future responses. By remembering inflammatory experiences, tissues are better equipped to quickly and robustly respond to barrier breaches. Alarmingly in disease states, memory may fuel the inflammatory fire. Here, we review the cellular communication networks in barrier tissues and the integration between tissue-resident and recruited immune cells and tissue stem cells underlying tissue responses to environmental stress.
Fuchs In Brief
Barrier tissues such as skin have mechanisms to remember and adapt to insults from the environment to minimize potential future harm. Recent work sheds light on the cellular, molecular, and tissue-level mechanisms of barrier tissue memory.
INTRODUCTION
Whether invertebrates, vertebrates or plants, multicellular organisms share the ability to mount an immune response and adapt to environmental stress and infection. In mammals, these fundamental features are particularly pertinent for mammalian barrier epithelia of the skin, airway, gut, and urogenital tract which form the interface between the environment and the body. Under homeostatic conditions, epithelial barrier tissues exclude microbes and retain body fluids. To do so, these tissues constantly rejuvenate their epithelium by supporting its frequent turnover, a process required for elimination of dying or damaged cells. This is achieved through the function of tissue-specific stem cells, which reside in specialized local niches that afford relatively insulated supportive microenvironments. Positioned near barrier interfaces, epithelial stem cells must sense and respond to changes in the tissue status. When their integrity is breached and pathogens invade, the stem cells must quickly adjust and work with resident immune cells and other tissue cells to mount inflammatory and repair responses.
How epithelial barrier tissues achieve these feats differs depending upon the tissue, its principal functions, and the specific stresses it endures. Body surfaces such as the skin epidermis must contend with physical, chemical and biological factors including physical traumas, fluctuations in temperature, pH, and osmolarity, noxious substance exposure, and microbial colonization. To insulate themselves from biotic and abiotic stresses that impede their function and threaten the organism’s survival, epidermal stem cells (EpdSCs) generate a shield of overlying layers of terminally differentiating epithelial cells. The process culminates in the production of an armor of enucleated, flattened “squames” that are sealed together by lipid bilayers (Quiroz et al., 2020). These outer layers also contain ‘defensins,’ evolutionary ancient host defense peptides with direct antimicrobial and/or immune signaling activities. The dead squames are continually sloughed from the skin surface, replaced by a flux of inner differentiating cells moving outward from the innermost layer of self-renewing EpdSCs that balance growth and differentiation (Figure 1A).
Figure 1.
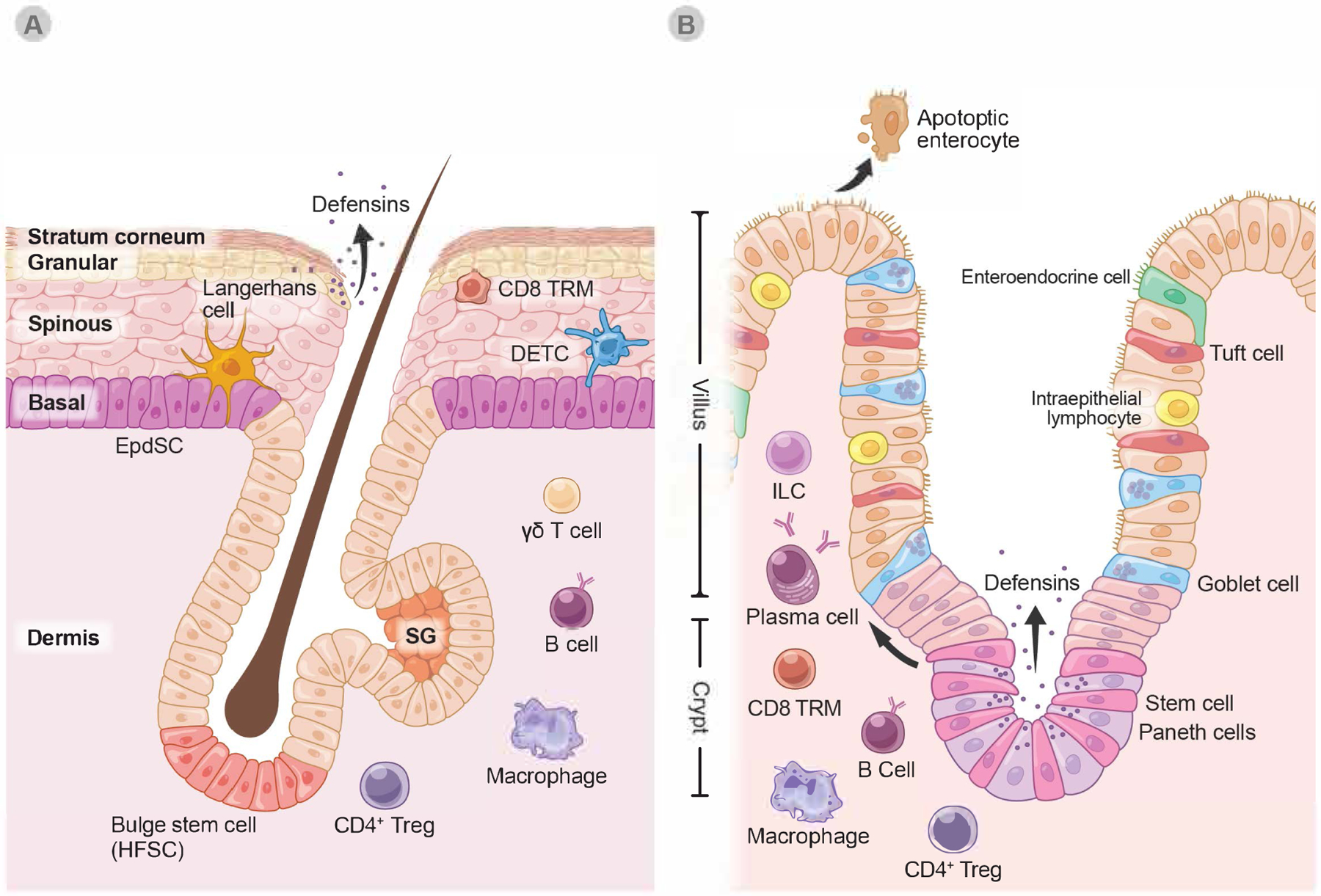
Barrier tissue composition in the skin and the small intestine
A. The skin is a multilayered barrier whose epithelium is derived from an inner layer of epidermal stem cells (EpdSC). EpdSC progeny differentiate as they move up towards the exterior surface, eventually becoming enucleated “squames” that provide the exterior coating of the body (Stratum corneum). Appendages of the epidermis, hair follicles harbor unique stem cells (HFSC) residing within an anatomical structure called the ‘bulge’. Through cyclical bouts of quiescence and activation (the ‘hair cycle’), HFSCs periodically regenerate the hair follicle and regrow hair. An appendage of the HF, sweat glands (SG) harbor their own stem cells. All skin stem cells reside within a basal layer at the interface between the epithelium and the dermis.
B. In contrast to skin, intestinal epithelium is comprised of a single layer of epithelial cells all derived from the continuous activity of intestinal stem cells (ISCs) that reside within intestinal crypts. ISCs are interspersed with Paneth cells, which are also derived from ISCs. Tuft cells, goblet cells and enteroendocrine cells are all ISC progeny that develop as ISCs commit to differentiate and move up to form the villus cells that are extruded into the lumen.
Despite these differences, barrier epithelia all display extensive involvement of immune cells, that contribute in diverse ways discussed in this review to tissue homeostasis, response to challenges, and adaptive tissue features (Figure 4). Epithelial cells in both tissues secrete antimicrobial peptides, such as defensins. CD4+ regulatory T cells (Treg), CD8 T resident memory cells (TRM), innate lymphoid cells (ILC).
Although the defensins and cellular “armor” of the epidermis provide considerable protection, they are not sufficient to meet the challenge of a severe barrier breach such as wounding. To further its defensive posture, the stem cell layer is peppered with dendritic epidermal T cells (DETCs) and a specialized type of tissue macrophages, Langerhans cells, which join forces with the EpdSCs when the barrier is breached. In the underlying dermis, additional resident and migratory immune cells are ready to relay signals to draining lymph nodes and bolster inflammatory responses to control the infection (Figure 1A). Epidermal appendages such as hair follicles and sebaceous glands also have orifices open to the body surface, which are colonized by distinct microbes and pathogens. This necessitates distinct niches of dedicated stem cell and different immune cell populations that patrol against barrier breaches at these deeper skin sites.
By contrast, simple epithelial linings of many of the internal organs have only one layer of polarized cells that distinguish the external from internal environment to form a barrier. Additionally, as exemplified by the small intestine, their stem cells multitask, producing an array of distinct cell lineages that go beyond the adsorptive villus cells that capture nutrients. At the crypt base, the niche of intestinal stem cells (ISCs) protect themselves by generating neighboring Paneth cells that produce a bevy of antimicrobial peptides. ISC progeny further up along the crypt or crypt-villus axis in the epithelium are goblet cells, which secrete mucus to protect the epithelial layer from invasive bacteria and fungi. In addition, tuft cells secrete cytokines with immunomodulatory function to communicate with resident immune cells in the nearby stroma, while enteroendocrine cells secrete gastrointestinal hormones regulating processes from digestion to defense (Figure 1B).
Despite the many intrinsic mechanisms of resistance of epithelial barrier tissues to invading pathogens, assistance of resident and migratory immune cells is required to unleash full protection. Similar to the skin, other barrier tissues harbor many of the same resident immune cell types, although their proportions, behavior and mobilization differ depending upon the circumstances. Throughout this review, we draw not only upon the specialized adaptations that contribute to epithelial barrier tissue diversity, but also the commonalities in the epithelial-immune cell communication networks that orchestrate barrier tissue dynamics.
A common facet of these networks is a combination of a sedentary and migratory behavior of their immune cell components. Many immune cells residing at epithelial barriers partake in their defense against invading microbes locally, where they assist cell-intrinsic immune function of epithelia. However, some immune cells exit the tissue to initiate protective responses within nearby draining lymph nodes. Concurrently, circulatory immune cells are recruited to the afflicted tissue in response to cues ensuing from infection, injury or other stresses. Another notable shared characteristic of stem cell niches is that blood vessels and lymphatic capillaries or sinusoids can be found in their close proximity (Gur-Cohen et al., 2019; Kiel et al., 2005; Shen et al., 2008; Tavazoie et al., 2008; Verma et al., 2018), suggesting a critical crosstalk between epithelial stem cells, the vascular and lymphatic endothelium, and resident and non-resident immune cells. Beyond immune and endothelial constituents that contribute to epithelial function, barrier tissues have sensory and motility functions controlled by neurons, which also engage in cellular communication networks [reviewed in (Hsu et al., 2014; Jacobson et al., 2021)]. Epithelia also host microbial constituents that exert potent influence on cellular composition and phenotypes within tissues [reviewed in (Honda and Littman, 2016; Kayama et al., 2020)]. Complex communications between these highly diverse cellular and microbial constituents affect barrier tissues at steady state as well as in times when tissues experience damage or infection and inform tissue responsiveness.
The ability of epithelia to react quickly to the abiotic and biotic stresses they encounter is daunting considering the wide range of provocations. A major factor in the tissue’s ability to maintain its function while facing the barrage of ever-changing stressful environments is its ability to “learn” from its inflammatory experiences and, as a result, be poised to regain homeostasis more quickly following future exposures. Thus, rather than signifying a return to the “naïve” tissue state existing prior to a challenge, a new basal state arises in barrier tissues that have experienced inflammation. Increasing evidence indicates that the numbers, seeding density, activation state, and specific location of immune cells within tissues are influenced by environmental factors, including microbial colonization, both during normal neonatal development and also upon injury and inflammation episodes in early life and adulthood (Milutinović and Kurtz, 2016; Reimer-Michalski and Conrath, 2016; Sharrock and Sun, 2020). Highly sensitive to exposures, these immunologic features of tissue undergo iterative changes throughout life and, in a combinatorial manner, define tissue responsiveness. In this review, we discuss how barrier epithelial tissues as a whole adapt to and remember their inflammatory experiences. Their adaptive and maladaptive reactions of tissues to inflammatory processes are likely at the crux of not just their efficient healing and fitness, but also chronic inflammatory and degenerative diseases, aging and cancer.
FORMS OF CELLULAR ADAPTATIONS IN BARRIER TISSUES
Tissue adaptation is the phenomenon wherein a tissue, following an initial challenge, acquires the ability to respond differently upon future repeat or novel insults. By this definition, tissue adaptation comprises both beneficial and maladaptive responses. In this sense, tissue memory can be encoded not only by cell intrinsic epigenetic and metabolic changes that alter responsiveness, but also in lasting changes, ‘memories’, in tissue immune cell composition and feed-forward cellular circuits that impact parenchymal cell function (Figure 2).
Figure 2.
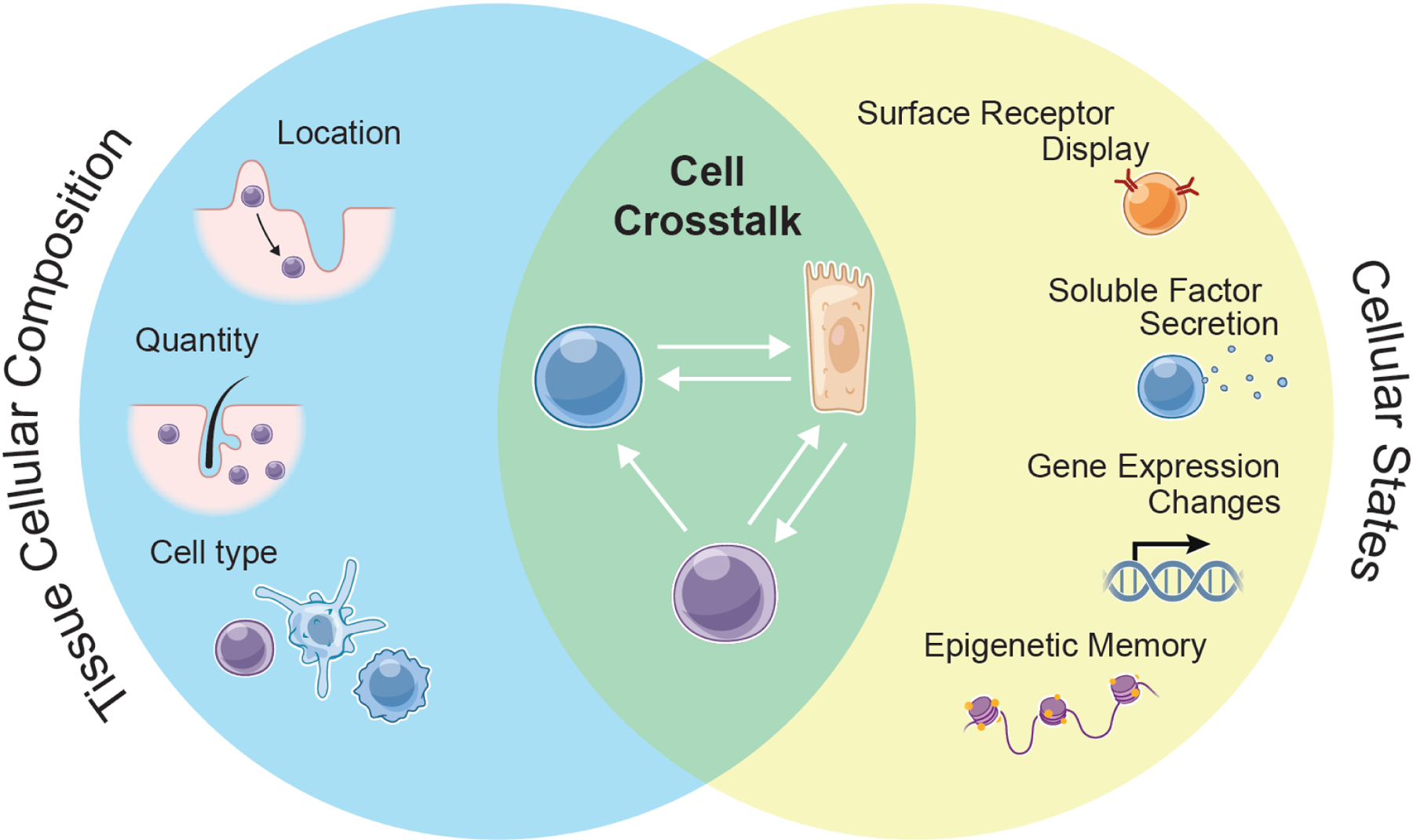
Tissue adaptation to inflammation
Adaptation in tissues comprises alterations in cellular composition (left circle) and the functional states of those cells (right circle). Interactions between numerically or phenotypically altered cellular constituents of tissues form tissue circuits (overlap) that determine the function and thresholds for reactivity. These circuits can include signals between adaptive and innate cells, adaptive and stem cells, innate and stem cells.
Concepts of memory and adaptation have emerged from studies of adaptive and innate immune cells and tissue stem cells. In vertebrates, memories of specific pathogens or harmful agents are harbored within antigen-experienced memory T and B lymphocytes. Pathogen-specific clones of adaptive immune memory cells are endowed with augmented capacity to defend the host upon re-infection, the result of (i) selection for highly sensitive antigen recognition by their receptors and (ii) enriched frequencies, sentinel location and behavior relative to other T and B cells. These cells and their characteristic features were once considered the sole mediator of vertebrate defense adaptation, hence fitting with their label as “adaptive immune cells”. This thinking evolved when it was realized that innate immune cells such as macrophages and natural killer (NK) cells can undergo stimulus-dependent epigenetic changes, some of which persist long after the inflammation has resolved (Lau and Sun, 2018; Netea et al., 2020; Sun et al., 2009).
Referred to as innate immune memory, priming, or ‘trained immunity’ (Divangahi et al., 2020), these lasting changes in response to a specific stimulus are thought to sensitize tissues to respond differently to future assaults. Through mechanisms unknown, they also prepare the tissue to grapple with multifarious secondary challenges. Importantly, these innate immune memory adaptations exist in various modes (discussed below), the diversity of which has yet to be fully revealed but with chromatin features at response genes as key determinants (Foster et al., 2007; Smale et al., 2014; West and Heagy, 2002). Dynamic and durable tolerance programs likely limit potential damage to epithelial tissues that are under frequent attack. Consequently, loss of these programs, can shift tissues towards hyperinflammatory disease states (Figure 3), while their preservation in innate immune cells may assist the transition of tissues from pro-inflammatory to anti-inflammatory states, enabling their healing and restoration of fitness.
Figure 3.
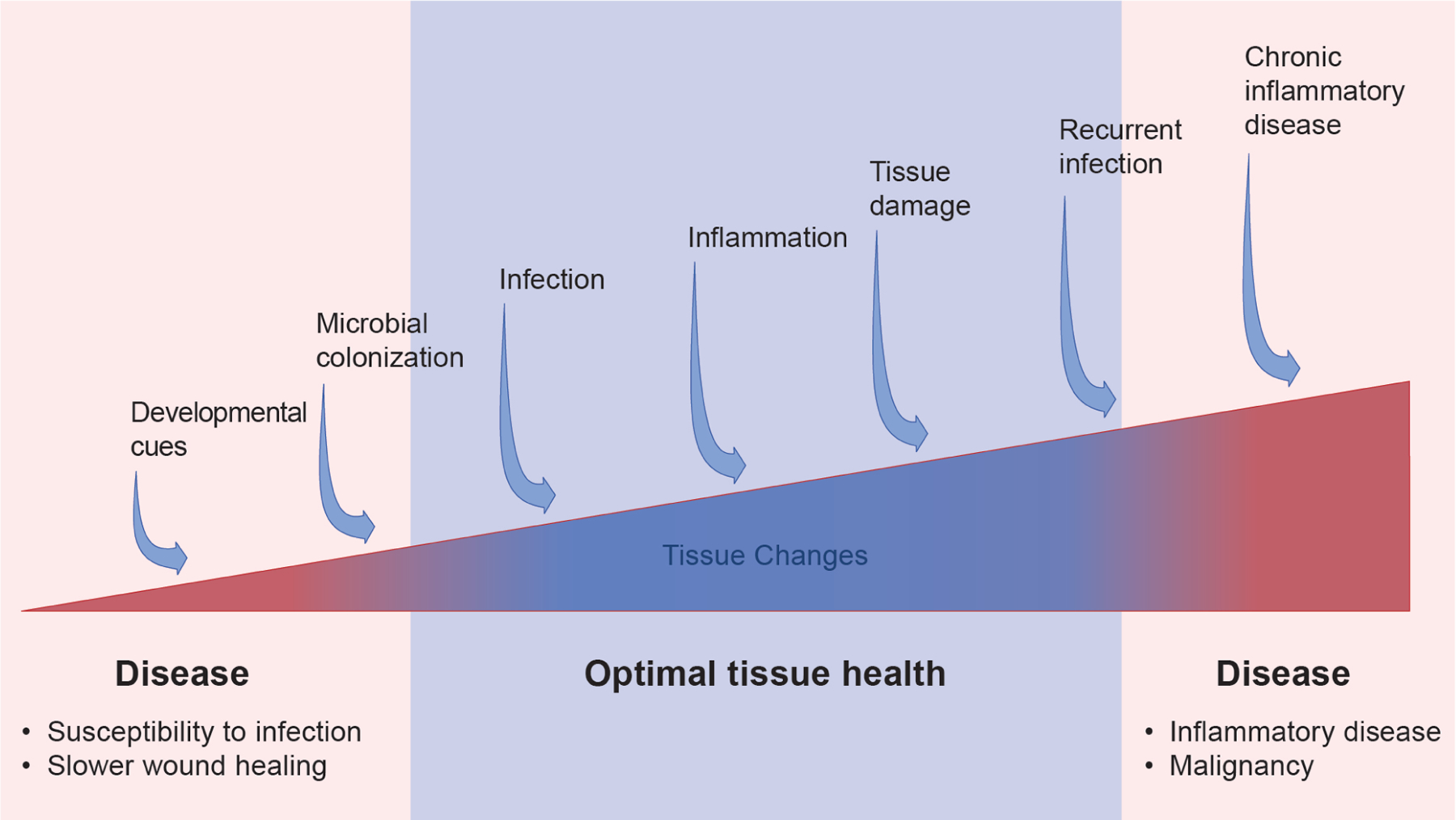
The spectrum of tissue adaptation
Cells within barrier epithelial tissues experience diverse exposures to inflammatory stimuli over a lifetime. Whether memories or adaptations might accumulate is a fascinating possibility as yet unexplored. In a model where they do so, tissue adaptation would be expected to occur on a spectrum represented by the red and blue triangle. In this model, a paucity of normal developmental cues and inflammatory exposures might render tissues vulnerable to infection, resulting in poor tissue recovery following a naïve injury. By contrast, when a tissue is perpetually exposed to excessive inflammatory signals, this may drive chronic inflammation and cancer via accumulation of tissue “memories”.
Although memory and adaptation in response to an inflammatory stimulus have been traditionally been attributed to different types of immune cells, these phenomena have now been shown to extend to parenchymal cells. In the first such study of its kind, it was found that upon exposure of mouse skin to a psoriatic-like inflammatory stimulus, certain epigenetic alterations to EpdSC chromatin persist and endow the epidermis with the ability to respond more robustly to subsequent diverse challenges (Naik et al., 2017). Additionally, when human respiratory epithelial progenitors from allergen-exposed tissue are placed in culture, they are slow to restore normalcy at the chromatin level (Ordovas-Montanes et al., 2018). These findings have raised the tantalizing possibility that stem cells and perhaps other tissue cell types may be able to epigenetically adapt to inflammation and equip barrier tissues with a heightened capacity to collaborate in tackling subsequent encounters.
Two additional features of inflammatory memory are important to consider. First, although overall tissue structure is often restored following inflammation, subtle differences linger in barrier tissues. This is particularly true for resident immune cells, whose numbers and overall composition can shift during inflammation and then endure long after the initial stressor is gone. Second, within the bone marrow, early hematopoietic progenitors can respond to microbes, endotoxins and dietary changes by skewing their differentiation potential from lymphoid to myeloid fates (Christ et al., 2018; Kaufmann et al., 2018; Khan et al., 2020; Mitroulis et al., 2018). This may result in perpetuation of altered immune cell composition in circulation and within tissues.
Given the intricate cross-talk among tissue cells, persistent changes in the frequency and/or location of even a single cell type could have cascading effects on cell-to-cell communication within tissues (Adler et al., 2020; Zhou et al., 2018), thereby amplifying the imbalance in cellular compositions and behaviors and further shifting the tissue from its naïve state. Such types of changes unfold in the context of viral infection (Kadoki et al., 2017) and in inflammatory diseases such as ulcerative colitis (Smillie et al., 2019). Though the detailed parameters of most cell circuits remain poorly understood, in vivo studies using single cell approaches and defining ligand-receptor connectivity illuminate what must reflect a complex web of cellular circuits in response to inflammation.
Particular states of a tissue are predominantly defined by its cellular constituents, their function, and critically, their cooperative programs (Figures 2 and 3). In the next section, we highlight several key immune cell players with potent activity in establishing tissue status and responsiveness (Figure 4). We discuss relevant cell circuits in barrier tissues that typify an emerging paradigm of dynamic and modular inter-cellular communications in coordination of tissue function.
Figure 4.
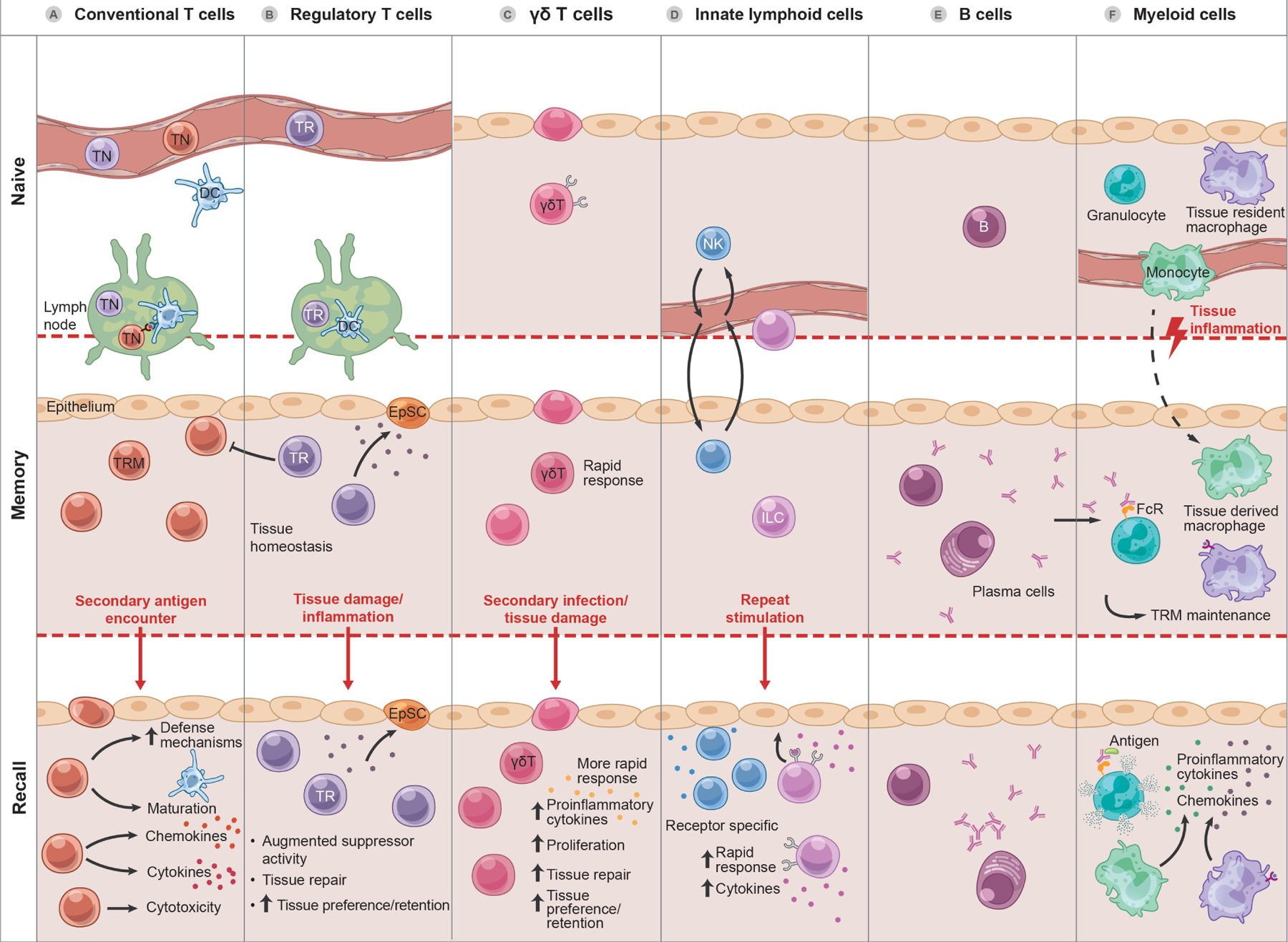
Constituents of epithelial tissue memory.
A. During an acute immune response, a subset of clonal-expanding T cells, derived from naïve T cells (TN) after selection within lymph nodes, home into tissues, where they differentiate into tissue resident memory T cells (TRM), taking up long-term residence and surveilling the tissue during the memory phase. During a recall response, these poised TRM undergo stereotypical adaptive immune responses on site with augmented cytotoxicity, cytokine and chemokine secretion, and recruitment of diverse immune cells. Accumulation and increased number of TRM in tissues occurs temporally in response to post-natal developmental exposures. Under homeostatic conditions, TRM produce factors that drive EpSC growth. Inflammation-experienced TRM may be endowed with augmented tissue repair and immunosuppressive activity
B. Accumulation and increased number of regulatory T cells (Treg, TR) in tissues represents a memory of sorts of these developmental exposures. Under homeostatic conditions, TR produce factors that drive epithelial stem cell (EpSC) growth. Inflammation-experienced TR may be endowed with augmented tissue repair and immunosuppressive activity.
C. γδT cells appear early in development in epithelial tissues. In addition to recognition of endogenous ligands, γδT cells can also respond to common microbial molecules, molecular signatures of tissue damage, and inflammatory cues to expand in tissues with capacity for long-term residence and augmented recall responses featuring cytotoxicity, cytokine and chemokine responses, and, unusual for T cells, phagocytosis and cross-presentation.
D. Natural killer cells (NK) predominantly circulate but can transit through tissues sufficiently to contribute to contact hypersensitivity reactions. “Experienced” innate lymphoid cells (ILC) can persist for months or more, endowed with cell intrinsic memory of inflammation and the ability for augmented cytokine production and proliferation in subsequent responses.
E. Upon antigen encounter and activation, tissue resident B cells can be induced to produce antibody in situ. These antibodies can also contribute to tissue adaptation by binding Fc receptors (FcR) on myeloid cells and augmenting or directing their activity.
F. Tissue-resident macrophages of embryonic origin are long-lived, self-renewing sentinels that also support tissue homeostasis. Under inflammatory conditions, newly-minted peripheral blood monocytes can be recruited into tissues where a subset may convert to adopt the phenotype and function of local tissue resident macrophages. Inflammation can also alter the phenotype of tissue resident macrophages, endowing them with long-term cell autonomous memory enabling TRM maintenance, augmented cytokine and chemokine production and immune cell recruitment upon subsequent challenge.
INFLAMMATORY ADAPTATIONS BY LYMPHOCYTES
Tissue Resident Memory T Lymphocytes
At the onset of an infection, tissue-resident dendritic cells (DCs) specialized in sensing pathogens, capture their antigens, and display them through surface major histocompatibility complexes (MHCs). DCs then traffic to local lymph nodes where they present MHC-loaded antigens to naïve T cells (TN), each unique in its expression of a particular T cell receptor (TCR). Upon recognition of the MHC-antigen complex, a T cell expressing an optimally matched TCR will proliferate and differentiate into effector and memory populations that migrate to the site of infection and exert their function (Figure 4A). By producing pro-inflammatory mediators such as IFNγ, CD8+ effector T cells are distinguished by direct potent cytolytic function while CD4+ T cells recruit and activate other immune and non-immune cells and can sometimes be cytolytic.
The role of these activated T cells is primarily to mount an attack against infectious agent following tissue injury or infection. However, once the tissue has healed, antigen-experienced CD4+ and CD8+ cells persist. These include long-lived circulating memory T cells, effector-memory T cells (capable of circulating and surveying peripheral tissues), central-memory T cells (occupying the spleen and other secondary lymphoid tissues as they await activation) and tissue resident memory T cells (TRM). Due to their strategic positioning within tissues, TRM are situated to patrol for previously encountered pathogens and react rapidly to secondary challenge (Gebhardt et al., 2009; Masopust et al., 2001, 2010; Wakim et al., 2008). In doing so, these TRM endow the tissue with a ‘memory’ of prior encounters and with a protection that is superior in its immediacy to non-resident memory T cells (Jiang et al., 2012; Park et al., 2018).
The tissue residence of pathogen-specific memory CD8+ T cells is particularly important for pathogens that can persist if not controlled at their site of entry, such as herpes simplex virus 1 (HSV1) (Gebhardt et al., 2009; Wakim et al., 2008). Beyond their direct cytotoxic activity, CD8+ TRM exert broad tissue conditioning effects to promote pathogen clearance – including upregulation of epithelial cell defense mechanisms, maturation of local dendritic cells, mobilization of natural killer (NK) cells, and recruitment of circulating T cells. Moreover, these effects can augment clearance of persistent viruses such as HSV1 and heterologous pathogens even at distant sites (Ariotti et al., 2014; Jiang et al., 2012; Schenkel et al., 2014).
In general, CD8+ TRM predominate as stable tissue-resident sentinels, while memory CD4+ T cells are more broadly distributed and show greater tendency to re-circulate and occupy regional lymph nodes (Collins et al., 2016; Gebhardt et al., 2011; Klicznik et al., 2019). That said, although typically less abundant and more transient than their CD8+ counterparts, CD4+ TRM nevertheless contribute significantly to local immune activation and tissue immune defense, as exemplified by the recent discovery of a longer-lived TRM population of CD4+ cells derived from early effector T cells and important for rapid clearance of pathogens in a secondary response (Amezcua Vesely et al., 2019; Beura et al., 2019; Glennie et al., 2015; Iijima and Iwasaki, 2014; Teijaro et al., 2011).
Colonization with commensal bacteria and fungi in the skin can also stimulate production of TRM, equipping the tissue with commensal-sensing CD4+ and CD8+ T cells that are poised for both anti-pathogen activity and tissue homeostasis and repair (Harrison et al., 2019; Naik et al., 2015). Analogously in the intestine, resident CD8+ TRM are primed to control oral infection by Listeria monocytogenes (Sheridan et al., 2014), while virus-activated CD4+ TRM establish residence and drive local immune cell mobilization upon pathogen re-encounter (Beura et al., 2019). Their additional links to draining lymph nodes and other circulating memory T cell populations enable CD4+ and CD8+ TRM to provide an arsenal for immune surveillance and/or rapid attack upon secondary assaults against myriad fungal, parasitic, and bacterial organisms, including both pathogens and commensals.
TRM are typified by their expression of specific surface receptors that enable them to be recognized and retained by their tissue (Masopust and Soerens, 2019). Conversely, the epithelium must also employ various mechanisms to entice TRM to establish tissue residency and, subsequently create an environment conducive to retaining TRM for prolonged periods of time. Their stem cells do so by producing epithelial-derived TRM survival signals such as IL-15 and IL-7 (Adachi et al., 2015a; Mackay et al., 2013). This mutually beneficial relationship between epithelial progenitors and TRM is further augmented by cues emanating from other resident immune cells, such as macrophages (Iijima and Iwasaki, 2014). Despite the clear benefits for an epithelial tissue afforded by memory T cells, TRM can also present heightened inflammatory disease risk to tissues. CD4+ TRM-like cells, for instance, accumulate within the intestine of human inflammatory bowel disease patients, where their abundance correlates with disease activity (Zundler et al., 2019).
Taken together, the positioning of TRM at key commensal and pathogen epithelial gateways and their dependency on epithelial-derived cytokines and other factors protects barrier tissues from infection during homeostasis, but also raises the possibility of tissue dysregulation leading to hyperproliferative conditions in inflammatory disease. Striking the sweet-spot, diverse tissue constituents including epithelial cells themselves, form regulatory circuits, which can adjust the availability of maintenance factors for TRM cells, thereby modulating their numbers (Adachi et al., 2015a; Mackay et al., 2013). Because antigen specificity, localization and abundance of TRMs are key determinants of a barrier tissue’s ability to remember and adapt to an inflammatory encounter, altering TRM cell maintenance factors within barrier tissues might help not only in deciphering the cellular circuits that govern tissue adaptation, but also in therapeutically dampening down the deleterious consequences to cumulative TRM memory.
Regulatory T Lymphocytes
Regulatory T cells (Treg) represent an essential specialized immunosuppressive lineage of CD4+ T lymphocytes that is phenotypically and functionally defined by expression of transcription factor FOXP3 (Josefowicz et al., 2012). Treg have broad distribution in lymphoid tissues and diverse non-lymphoid tissues, and are especially abundant at barrier sites (Whibley et al., 2019). While Treg are best known for their critical immunomodulatory function, they also participate in homeostatic tissue maintenance and in reparative processes following injury (Figure 4B). In the skin for example, Treg have been implicated in stimulating stem cells to regenerate hair (Ali et al., 2017). In the intestine, ISCs express MHC class II, enabling them to act as unconventional antigen-presenting cells that directly stimulate resident and circulation-recruited Treg. The latter become activated and produce IL-10, a immunomodulatory cytokine that can feed back on the ISCs to support their self-renewal capacity (Biton et al., 2018). Such circuitries between barrier epithelial stem cells and immune cells must be tightly regulated to maintain the balance between proliferation and differentiation in the epithelium and at the same time to be poised to trigger an inflammatory response when a breach occurs.
In settings of tissue injury and inflammation, Treg cell numbers prominently rise in the affected tissues, as documented not only for skin and gut, but also for adipose, brain and muscle tissues. Many of these expanded Treg cells are found in the proximity or within stem cell niches, where through production of anti-inflammatory and tissue repair mediators including IL-10 and transforming growth factor β (TGFβ), they coordinate diverse processes that in addition to stem cell self-renewal, include dampening inflammation, essential for facilitating tissue repair (Arpaia et al., 2015; Burzyn et al., 2013; Cipolletta et al., 2012; Feuerer et al., 2009; Lay et al., 2018; Naik et al., 2018; Sather et al., 2007; Scharschmidt et al., 2017; Whibley et al., 2019). By secreting these factors, Treg may help to restock stem cell niches which otherwise become taxed in the course of tissue repair, and thereby aid in restoring the homeostatic balance between long-lived quiescent stem cells and their immediate proliferative short-lived progeny, which fuel parenchymal cell differentiation.
Treg can also facilitate conversion of inflammatory macrophages to a reparative state, a process again integral to tissue repair. One intriguing aspect of Treg is their accumulation in tissues following microbial colonization, which takes place early in postnatal development (Scharschmidt et al., 2015). In the intestine, specific microbes and their metabolites regulate maintenance of intestinal Treg and localized conversion of naïve CD4 T cells into immunosuppressive Treg, key in promoting intestinal homeostasis (Arpaia et al., 2013; Atarashi et al., 2013; Campbell et al., 2020; Hang et al., 2019; Ohnmacht et al., 2015; Sefik et al., 2015; Smith et al., 2013; Song et al., 2020). As such, starting at a very early age, increased Treg frequencies may already be a sign of microbial exposure, with later consequences for tissue homeostasis and immunity.
Whereas recurrent rounds of inflammatory stimulation increasingly enhance memory in TRM, activated Treg display only a transient increase in their immunomodulatory effector functions, which resolve following inflammation (van der Veeken et al., 2016). Rather, the memory of inflammation-experienced Treg is conferred through their increased preference for nonlymphoid tissues following microbial exposure. Indeed, a “memory” state in the traditional sense may be the baseline at which Treg are maintained in tissues after their neonatal exposure to commensal antigens.
In non-lymphoid tissues, Treg are transcriptionally and metabolically modified through changes in their local environment that impact combinatorial expression of tissue-restricted and inducible transcription factors (DiSpirito et al., 2018; Panduro et al., 2016; Whibley et al., 2019). The outcome of this transcriptional and metabolic tune-up is tissue-specific adaption of Treg functionality, which at a population level, supports extended tissue residency of Treg and shapes their functional outputs. Hence, regardless of the presence or absence of intrinsic Treg memory features, recruitment and incorporation of Treg facilitates their involvement in tissue-specific cellular networks. This includes their regulation of tissue stem cells, and thus can broadly impact tissue physiology and subsequent responses to inflammatory stimuli.
B Lymphocytes
While T cells are typically considered the orchestrators of tissue adaptation and defense, B lymphocytes command a spotlight through their ability to generate antibodies, which can bind to Fc receptors (FcR) on diverse tissue resident immune cells. Upon antibody-mediated FcR crosslinking, the innate immune cells become armed with antigen-specific effector function, triggering cytokine and chemokine release and/or antibody-dependent-cytotoxicity (Figure 4E). In turn, these stimuli elicit direct responses from epithelia, vasculature and smooth muscle. This antibody-dependent circuitry is a key component of the acute and memory immune responses to parasites and other extracellular pathogens, as well as venoms and noxious agents that must be expunged from the tissue and/or detoxified (Palm et al., 2012). Neatly ascribing to the spectrum from optimal defense to disease (Figure 3), these same mechanisms also define common allergic pathologies.
Beyond arming tissue resident innate immune cells with these antibody-dependent capabilities, B lymphocytes themselves can migrate through or occupy barrier tissues to mediate antigen-specific immunity and memory via antibody secretion. In mucosal tissues of the lung and gut, local plasma cells produce IgM, IgG, and dimeric IgA that are secreted and transported via immunoglobulin receptors through the epithelium to the exterior mucosa, where they mediate protective immunity (Adachi et al., 2015b; Allie et al., 2019; Iwasaki, 2007; Onodera et al., 2012). By contrast in the vaginal epithelium, protective antibody responses are conferred by memory B cells which enter the tissue upon TRM-mediated signaling (Oh et al., 2019). Though infrequent in skin, tissue resident B cells have been implicated in regulating microbial colonization, microbial defense upon barrier breach, suppressing aberrant inflammation via IL-10 secretion, and augmenting apoptotic cell clearance via IgM deposition (Debes and McGettigan, 2019).
Overall, B lymphocytes can endow resident innate immune cells with a potent and sustained antigen-specific response capacity. They can secrete antibodies in the steady-state in situ, or mount rapid migratory forays into tissues, guided by cues from CD4+ TRM. Given tissue residency and steady-state production of factors like IL-10, it is tempting to speculate that B cells may engage in tissue level circuits or adaptations, as do Treg.
Innate-like T Lymphocytes
In contrast to conventional CD4+ and CD8+ αβ TCR-expressing T cells, innate like T cells such as iNKT, MAIT and γδ T cells express TCRs with restricted diversity (Godfrey et al., 2015; Lau and Sun, 2018). These ‘innate-like’ T cells preferentially localize to barrier tissues where they rapidly generate effector molecules in response to initial antigen encounter. Beyond a robust primary response, γδ T cell function can be enhanced by a priming challenge, a feature consistent with “conventional” memory T cells (Figure 4C). For example, in the skin, psoriatic-like inflammation elicits expansion of a population of IL-17-producing γδ T cells that persist in the dermis long after the initial response. Moreover, upon re-challenge, inflammation-experienced γδ T cells proliferate more rapidly and increase their IL-17 production (Ramírez-Valle et al., 2015). Similarly, oral infections of L. monocytogenes in mice promote expansion of γδ T cells in the intestinal lamina propria, typified by classical memory features that include expansion, contraction and a more brisk response to secondary L. monocytogenes infection (Romagnoli et al., 2016; Sheridan et al., 2013). Along with their high motility, these features make γδ T cells key sentinels in surveying epithelial barrier tissues for damage.
Another intriguing facet of γδ T cells is their selective preference for particular barrier tissues and for distinct locales within. This preference depends upon their particular TCR utilization and butryophylin-like (BTNL) proteins are expressed by cells within the tissue (Hayday, 2019; Willcox and Willcox, 2019). For instance, developing thymic epithelia express BTNL protein SKINT1, which promotes the differentiation of dendritic epidermal γδ T cells (DETCs), that then home selectively to the epidermal progenitor layer (Boyden et al., 2008) [reviewed by (Nielsen et al., 2017)]. Upon injury, EpdSCs upregulate other SKINTs at the wound bed, which prompt resident DETCs to produce keratinocyte-stimulating factors that in turn promote tissue repair (Keyes et al., 2016; MacLeod et al., 2013; Naik et al., 2018).
Analogously, the intestinal preference of different γδ T cells is defined at least in part by their unique BTNL protein expression (Di Marco Barros et al., 2016; Melandri et al., 2018; Vantourout et al., 2018) [reviewed by (McCarthy and Eberl, 2018)]. Moreover, BTNL expression can change in disease states, and this can have a profound effect on tissue fitness. Such a shift occurs in patients with celiac disease, a chronic gastrointestinal inflammatory disorder, resulting in altered tissue γδ T cell populations. Intriguingly, while gluten avoidance resolves and restores normal BTNL expression, intestinal γδ T cell populations remain altered long after the inflammatory pathology resolves (Mayassi et al., 2019). Given that expression levels of BTNL molecules also correlate with inflammatory bowel disease and cancer (Lebrero-Fernández et al., 2016), memory of inflammation in tissue resident γδ T cells may not only impact subsequent protection from pathogens, but also sensitize the epithelium in adverse ways. These observations beg for future studies on the underlying mechanisms by which γδ T cells exert heightened memory-like responses and how shifts in specific populations of γδ T cells impact barrier epithelial integrity in health and disease.
Innate Lymphoid Cells
Innate lymphoid cells (ILCs) comprise NK cells, ILC1, ILC2, ILC3 and lymphocyte tissue inducer cells (LTi) (Vivier et al., 2018). Although they derive from common lymphoid progenitors, they differ from T and B cells in that they do not express antigen-specific receptors that undergo genomic receptor rearrangement. ILCs are largely confined to tissues where their homeostatic and effector functions rely upon cytokine and innate immune receptors such as TLRs. In these tissues, ILCs participate in tissue homeostasis, repair and immunity (Figure 4D).
The field of innate lymphocyte memory began with the discovery that NK cells can form antigen-specific, cell intrinsic memory replete with all the features of adaptive immune memory in response to systemic viral infection (Sun et al., 2009). While NK cells reside primarily in the liver and spleen, their contribution to barrier tissue inflammation is evident in contact hypersensitivity models (a stereotypical “memory” response) that lack T and B cells (O’Leary et al., 2006; Paust et al., 2010; Peng et al., 2013). By contrast, ILCs are prominent in barrier tissues, where they too are capable of inflammatory memory (Gasteiger et al., 2015).
ILC1 function in responses to viruses and bacteria (Cortez and Colonna, 2016; Fuchs et al., 2013) where following an infection, they form a stable pool of memory cells that are typified by persistent transcriptional and epigenetic alterations. This endows them with the ability to enhance protective effector responses upon a secondary challenge (Weizman et al., 2017). By contrast, ILC2 function in epithelial repair, parasitic expulsion, and type 2 inflammatory reactions (Vivier et al., 2018). In response to lipid mediators and alarmins (IL-25, IL-33 and thymic stromal lymphopoietin or TSLP), ILC2 produce and secrete type 2 cytokines (IL-4, IL-5, IL-9 and IL-13), which can either have host-protective or pathogenic activity. In the lung and lymph nodes, exposure to IL-33 results in robust expansion of ILC2. Although these numbers wane when the allergen-response subsides, a subpopulation of “allergen-experienced” ILC2 persists for days to months with heightened capacity for proliferation and type 2 cytokine production upon re-exposure to IL-33 elicited by the original or unrelated stimulus (Martinez-Gonzalez et al., 2016).
While ILC2 appear capable of intrinsic inflammatory memory, their role in tissue memory may take several forms. For example, in the intestinal epithelium, tuft cells constitutively express IL-25, which supports ILC2 under physiologic conditions (Gerbe et al., 2016; Howitt et al., 2016; von Moltke et al., 2016). Following helminth infection, however, microbial metabolites stimulate tuft cells to activate ILC2s, which in turn, act on the ISCs to favor their differentiation into goblet and tuft lineages over other ISC fate options. This creates a feed-forward inflammatory circuit, triggered by helminths but fueled by ILC2s, ISCs and tuft/goblet cells (von Moltke et al., 2016; Nadjsombati et al., 2018). Indeed, goblet cell hyperplasia is a characteristic of chronic inflammatory disease in the intestine, suggesting that these circuits may become entrenched in maladaptive epithelial responses.
Another ILC-epithelial stem cell circuit involves ILC3, whose members are typified by their expression of nuclear retinoid orphan receptor RORγ, and by their ability to express and secrete IL-17 and IL-22, when provoked by pathogens. IL-17 triggers a potent inflammatory response through its ability to mobilize resident immune cell populations and recruit neutrophils to tissues. IL-22 is thought to fuel repair, tissue reorganization, and/or hypertrophy through its ability to stimulate barrier epithelial stem cells to induce phosphorylation and activity of the transcription factor STAT3, which then increases stem cell proliferation and suppresses tissue differentiation (Ekman et al., 2019; Lindemans et al., 2015).
Although it remains unclear how ILC3 generate cell intrinsic memory of inflammation, by instructing durable intrinsic reprogramming in ILCs and epithelial stem cells, lasting feed-forward cellular circuits can be considered a form of memory within barrier tissues. In addition, as judged by the effects of their cytokines on inflammation and EpdSC behavior, activity of ILC3 subpopulations may invoke long-lasting epigenetic memory features in the stem cells in barrier tissues (Naik et al., 2017).
In summary, this section has highlighted the diversity of immune cell types and their complex communication circuitry involved in regulating barrier tissue homeostasis and adaptation. We next highlight concepts of cell-intrinsic adaption in two especially influential tissue-resident cell types, macrophages and epithelial stem cells. The combination of these cell-intrinsic adaptations and alterations in tissue cellular composition and cell circuit composition combine to orchestrate overall tissue behavior (Figure 2).
INFLAMMATORY MEMORY IN NON-LYMPHOID BARRIER TISSUE RESIDENTS
Macrophages
Macrophages and other mature myeloid lineage cells diverge from lymphoid cells early in hematopoiesis and many establish long-term residence in barrier tissues. Although lacking somatically rearranged antigen receptors altogether (Artis and Spits, 2015), these cells respond to local cues to adopt distinct transcriptional and phenotypic profiles to perform their roles in tissue maintenance and responsiveness. This tailors tissue resident macrophages to receive microbial signals, stimulate adaptive immune cells and coordinate temporal tissue inflammatory and healing responses.
In times of duress, macrophages can also derive from blood monocytes, which typically circulate for one to two weeks as sentinels that are poised to enter barrier tissues when called upon, where they can differentiate to augment local inflammatory responses. Fueled by stimulation of innate sensors like TLRs or inflammasomes and metabolic perturbations, monocyte-derived tissue macrophages undergo epigenetic and transcriptional remodeling. Although these changes resolve following withdrawal of the causative insult, they do so to varying degrees and rates with some epigenetic features (histone modifications and chromatin accessibility) persisting for days to months afterward (Netea et al., 2020; Saeed et al., 2014). Such chromatin remodeling bestows the associated genes with the ability to become activated quickly should the stimulus return to the tissue (Figure 4F).
Additional intrigue comes from the recent discoveries of epigenetic memory in longer-lived inflammation-experienced tissue-resident macrophages. Following respiratory viral infection of the airway epithelium, tissue resident alveolar macrophages can display augmented responsiveness for several weeks, including increased expression of MHCII, elevated glycolysis and neutrophil recruitment potential (Yao et al., 2018). CD8+ T cell-derived interferon-γ was required to initiate heightened macrophage responsiveness, but dispensable for its maintenance. Moreover, the memory appeared to be autonomous, as upon transplantation into naïve mice, it was propagated, augmenting the response upon rechallenge. Similar findings were made following helminth infection, initiating a long-term memory that endowed airway macrophages with enhanced parasite clearance capabilities for at least 45 days (Chen et al., 2014).
Further supporting the concept of tissue resident macrophages as a repository for inflammatory memory, brain microglia remain changed for at least six months following systemic inflammation (Wendeln et al., 2018). In this study, the authors used different challenge conditions to induce either microglia “tolerance” or “training” phenotypes, which persisted and either promoted or helped reverse diseased state. While these states might be more accurately described as ‘tissue maintenance and repair (reparative)’ and ‘chronic inflammation promoting (inflamed)’ states, it was interesting that in an experimental model of Alzheimer’s disease, the “reparative” state of microglia was linked to protection against neurodegeneration and cognitive decline, while “inflamed” conditions exacerbated disease (Wendeln et al., 2018). Additionally, “reparative” microglia reduced neuronal damage following induced focal ischemia (Wendeln et al., 2018). These findings further underscore the exciting potential for tissue-resident macrophage memory across tissues in health and disease.
Barrier Epithelial Stem Cells
Until recently, tissue memory was thought to be the exclusive purview of immune cells. The discovery that epidermal stem cells also harbor memories of their inflammatory experiences upended this view and also unleashed the concept that long-lived, often quiescent stem cells harbor ideal conditions for establishing a record of inflammatory experiences(Naik et al., 2017).
In a mouse model of psoriasis, cutaneous application of a synthetic TLR7/8 ligand, imiquimod, sparked a chain of signaling events that first prompted dendritic cells to produce IL-23, which then stimulated ILCs and RORγt+ T cells to generate IL-22 and IL-17 (van der Fits et al., 2009; Pantelyushin et al., 2012). In turn, IL-22 and IL-17 induced phosphorylation of STAT3 in EpdSCs, dramatically altering their chromatin landscape and triggering hyperproliferation (Naik et al., 2017). Upon resolution of inflammation, most changes in chromatin reverted, but some chromatin domains retained enhanced accessibility for months afterwards. These open chromatin domains harbored binding motifs for both STATs and epidermal stem cell TFs, and although few were transcribed after pathology resolved, the genes associating with these domains were more rapidly induced upon a subsequent assault. Additionally, as judged by transgenic reporter assays, these persistent open chromatin domains harbored inflammation-sensing activity (Naik et al., 2017). By these criteria, EpdSCs retain an epigenetic memory of their inflammatory experiences, in much the same way as first described for macrophages.
Recent studies extended the concept of tissue progenitor memory to early hematopoietic progenitors in the bone marrow. While underlying mechanisms are still unfolding, there are a few intriguing parallels between inflammatory memory of EpdSCs and hematopoietic progenitors that invite further investigation. Both require IL1β for manifesting inflammatory memory, and in both populations, memories can persist for months and confer more rapid response upon secondary stimulation (Naik et al., 2017; Mitroulis et al., 2018). Another possible parallel is that Western diet, recently found to enhance inflammatory memory in hematopoietic progenitors, results in increased expression of cholesterol biosynthesis genes (Christ et al., 2018). Although the effects of Western diet and/or metabolism on EpdSCs have not yet been examined, it is curious that shifts in cholesterol biosynthesis seem to underlie the heightened memory in both of these progenitor populations (Naik et al., 2017).
Further insights await deeper probing into the underlying process of inflammation-associated epigenetic memory. Like EpdSCs, long-term hematopoietic stem cells (HSCs) also possess epigenetic memories of inflammation (de Laval et al., 2020), and it is tempting to speculate that different stem cell populations may employ converging mechanisms to achieve similar shifts in rapid-response employment of gene expression. Intriguingly, both stem cell types appear to use memory to achieve optimal performance in fighting infections and tissue damage. However, HSCs exposed to repetitive inflammatory stimulants skew their fate towards the myeloid lineages, which armors the organism with more macrophages for the next inflammatory assault (Bekkering et al., 2018). By contrast, inflammatory memory in EpdSCs enables a more rapid reaction to a secondary assault such as wounding that enhances the reparative phase to seal wounds and restore the skin barrier (Naik et al., 2017).
The realization that inflammatory memory extends beyond immune cells also opens the door for addressing other non-immune cell populations that might also retain epigenetic memory. To this end, it was recently reported that basal cells in the human respiratory epithelium in human chronic allergic disease develop distinct epigenetic signatures, some of which were still detectable even after 5 weeks in the same culture media (Ordovas-Montanes et al., 2018). Moreover, polyp-derived basal keratinocytes still responded to IL-4 and IL-13 in a fashion distinct from their control counterparts. Although mutational and intrinsic differences may at least partially account for these differences, it will be exciting in the future to learn whether these long-lived epigenetic changes impact downstream tissue defense in normal nasal progenitors and in disease.
The ability of epithelial progenitors to retain epigenetic marks of their inflammatory encounters make the links between chronic inflammation, dysplasia and oncogenesis all the more thought provoking. In the intestinal epithelium, a high fat diet results in increased proliferation and “stemness” in ISCs and their progeny as well as alterations to the resident immune cell composition and a balance between inflammatory vs. regenerative factors in this milieu (Beyaz et al., 2016). Acting in epithelial stem and progenitor cells, the transcription factor PPARγ, which serves as a fatty acid sensor, acted at the helm to drive these effects. Intriguingly, intestinal crypts retained enhanced growth 1 week after mice were returned to a normal chow diet. Although the proliferative effects subsided by 4 weeks, the sustained high-fat diet nevertheless increased susceptibility of ISCs to spontaneous intestinal dysplasia and carcinomas. The possibility that a diet-induced or microbial dysbiosis-driven inflammatory “overdrive” could result in heightened stem cell activity could come with a downside of potential hypertrophy and persistent pathophysiology.
Taken together, these studies highlight two features of epithelial stem cells that have major implications for their participation in tissue adaptation to inflammation. First, under both homeostatic conditions and upon challenge, epithelial stem cells are recipients of immune cell-derived cues that directs their growth and differentiation. Second, reception of these signals can imprint durable memory of inflammation on epithelial stem cells enabling augmented responses to future challenge or dysregulation of growth and disease.
Memory Mechanics
How inflammatory memory works at a molecular level is a major question whose answer has been elusive. In response to inflammation, otherwise closed chromatin has to be opened. Often, this activity is attributed to ‘pioneer factors’-- transcription factors that initiate new regulatory chromatin events by de novo binding and opening of nucleosomal DNA, an activity sometimes enabled by linker histones mimicry and scanning function through repressed or inactive chromatin (Zaret, 2020). Even if inflammation-driven signal activated transcription factors, e.g. STATs or NF-kB, could bind to nucleosomes, their activity is only transient, implying that there must be some other means to keep chromatin open once accessible.
Most histone modifications are expected to be reset or “diluted out” with each round of DNA replication, making post-mitotic and/or infrequently dividing stem cells optimal candidates to retain memory. That said, certain histone modifications can recruit DNA methyltransferases to specific target sites in the genome (Baubec et al., 2015; Weinberg et al., 2019). This opens the possibility that inflammation-induced memory might be propagated through cytosine methylation, which can be stably inherited over years, even on evolutionary time scales (Catania et al., 2020). The notion that offspring might inherit the inflammatory adaptations of their parents is both enticing and unsettling.
Another clue to inflammatory memory, however, is that DNA domains harboring epigenetic memory are typically in more accessible states and can contain sequence motifs for the binding of both inflammation induced transcription factors and lineage defining transcription factors, defining the particular cell specific memory features (Naik et al., 2017). In support of an appealing potential model for propagating memory, studies on enhancers have shown that key regulatory DNA elements can act as dynamic binding platforms for both lineage-specific and signal-activated transcription factors (Adam et al., 2015, 2018; Ciofani et al., 2012; Josefowicz, 2013; Long et al., 2016; Samstein et al., 2012; Vahedi et al., 2012). If such a mechanism is operative in inflammatory memory, as has been proposed (de Laval et al., 2020; Naik et al., 2017; Ostuni et al., 2013; Saeed et al., 2014), the initial inflammatory transcription factor would not need to remain bound to maintain the open chromatin state (Figure 5).
Figure 5.
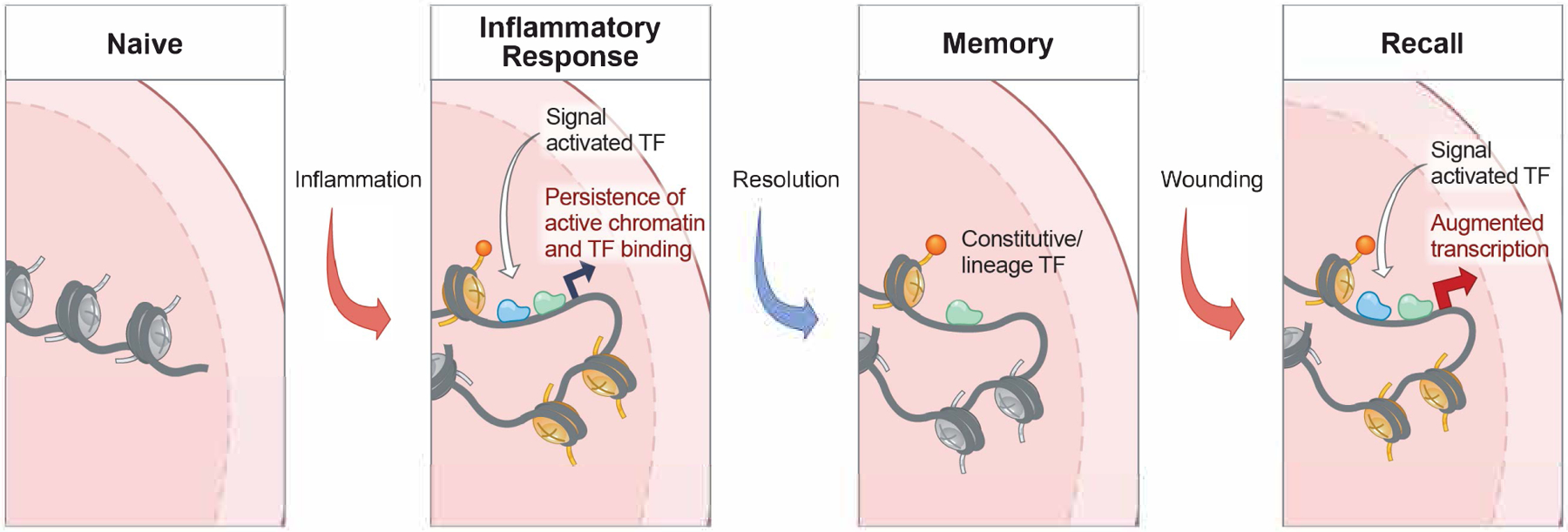
Model of the acquisition of cell intrinsic memory and augmented function in barrier tissue cells.
During exposure to inflammation, a barrier tissue cell experiences the transient activity of signal activated transcription factors such as pSTAT3 and NFkB. In a model still untested, these transcription factors open inflammation response gene chromatin, possibly providing accessibility for the binding of native transcription factors and/or histone marks. Following resolution of the inflammatory state, these homeostatic chromatin marks could then leave behind a durable epigenetic memory of inflammation. In barrier epithelial stem cells such as EpdSCs, most of these genes exist in a poised (accessible) but transcriptionally dormant state (Naik et al., 2017). Upon rechallenging, this poise state then becomes rapidly re-activated, presumably though easier accessibility of inflammation-activated transcription factors, rapidly fueling transcription and augmented growth and tissue repair. How memory endows cells with the ability to respond more rapidly to different inflammatory experiences remains a mystery, even in current models such as this one.
As attractive as this model may be, it still leaves a number of questions unaddressed. What are the specific transcription factors that initiate and/or sustain heritable memory over time and through cell divisions? What are the epigenetic mechanisms initiated by these transcription factors, for example mitotic bookmarking-- an ability for transcription factors to remain bound through mitosis-- or establishment of heritable DNA or histone modifications? Finally, and perhaps most importantly, how can epigenetic memory begin with a specific inflammatory stimulus, but later lend itself to recall by diverse stimuli?
CONCLUSIONS AND PERSPECTIVES
It is now clear that within tissues, diverse cell types ranging from cells of the adaptive and innate immune system to tissue stem cells, are subject to inflammation-induced modulation of their numbers and cell-intrinsic characteristics and along with other long-lived cells within barrier tissues, including fibroblasts, endothelial cells, and neurons enable collective retention of inflammatory memory. Rather than acting individually, these diverse tissue residents engage in interconnected circuits, which can encode memory through positive feedback loops or through long-term cell-intrinsic memory.
While still in its infancy, a conserved mechanism of cell intrinsic memory across cell types is beginning to emerge that includes temporary cytokine receptor or inflammatory signaling and results in durable changes in cellular metabolism and epigenetic landscape. As they accumulate, tissue memories or adapted tissue states are likely to define the level of readiness to challenge but also bring with it the risk of disease resulting from chronic inflammation of tissue hypertrophy or diminished adaptability to divergent insults. Although the environmental cues and memory mechanisms still remain poorly understood, it is reasonable to hypothesize that changes accumulate in a progressive and iterative manner. This “buildup” may even begin during development and then continue throughout lifespan, with “normal” homeostatic fluctuations, microbial colonization, and stress likely being major contributors. As future studies are conducted, the extent to which adaptations persist as well as the relevance of these tissue states to susceptibility to infection, inflammatory disease, and transitions from inflamed to neoplastic tissue will most assuredly continue to unfold.
Acknowledgments
We thank SciStories LLC, www.scistories.com for their expert preparation of the artwork associated with this review. We are grateful to the many friends and colleagues in the immunology and stem cell biology whose work inspired this review. We apologize to those whose work could not be cited in this Review due to space constraints. We thank the anonymous reviewers of this manuscript for their thoughtful insights and suggestions. We thank Steven Josefowicz and members of the Fuchs and Rudensky labs for helpful discussion. E.F. and S. R. are Investigators of the Howard Hughes Medical Institute. R. N. is a Clinical Scholar at the Rockefeller University and receives support in part by the National Center for Advancing Translational Sciences, NIH, through The Rockefeller University, Grant # UL1 TR001866 and Grant # KL2TR001865. R. N. is also a fellow in the Department of Gastroenterology and Hepatology at Weill Cornell Medicine. This work was supported by grants from the NIH (R01-AR31737, R01-AR27883, R01-AR050452 (E.F.), the Starr Foundation (E.F.) and NYSTEM (C32585GG, E.F.), NCI Cancer Center Support Grant P30 CA08748 (A.Y.R.), R01-AI034206 (A.Y.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
The authors declare no competing financial interests. E.F. is on the scientific advisory boards of L’Oreal and Arsenal Biosciences. A.Y.R. is a co-founder and SAB member of Sonoma Biotherapeutics.
References
- Adachi T, Kobayashi T, Sugihara E, Yamada T, Ikuta K, Pittaluga S, Saya H, Amagai M, and Nagao K (2015a). Hair follicle–derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat. Med 21, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi Y, Onodera T, Yamada Y, Daio R, Tsuiji M, Inoue T, Kobayashi K, Kurosaki T, Ato M, and Takahashi Y (2015b). Distinct germinal center selection at local sites shapes memory B cell response to viral escape. J. Exp. Med 212, 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam RC, Yang H, Rockowitz S, Larsen SB, Nikolova M, Oristian DS, Polak L, Kadaja M, Asare A, Zheng D, et al. (2015). Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 521, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam RC, Yang H, Ge Y, Lien W-H, Wang P, Zhao Y, Polak L, Levorse J, Baksh SC, Zheng D, et al. (2018). Temporal Layering of Signaling Effectors Drives Chromatin Remodeling during Hair Follicle Stem Cell Lineage Progression. Cell Stem Cell 22, 398–413.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler M, Mayo A, Zhou X, Franklin RA, Meizlish ML, Medzhitov R, Kallenberger SM, and Alon U (2020). Principles of Cell Circuits for Tissue Repair and Fibrosis. IScience 23, 100841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong H-A, Lai K, Ahn R, Corbin K, Lowe MM, Scharschmidt TC, et al. (2017). Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell 169, 1119–1129.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allie SR, Bradley JE, Mudunuru U, Schultz MD, Graf BA, Lund FE, and Randall TD (2019). The establishment of resident memory B cells in the lung requires local antigen encounter. Nat. Immunol 20, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amezcua Vesely MC, Pallis P, Bielecki P, Low JS, Zhao J, Harman CCD, Kroehling L, Jackson R, Bailis W, Licona-Limón P, et al. (2019). Effector TH17 Cells Give Rise to Long-Lived TRM Cells that Are Essential for an Immediate Response against Bacterial Infection. Cell 178, 1176–1188.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song J-Y, Jacobs H, Haanen JB, and Schumacher TN (2014). T cell memory. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 346, 101–105. [DOI] [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, Van Der Veeken J, Deroos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting PM, and Rudensky AY (2015). A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 162, 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artis D, and Spits H (2015). The biology of innate lymphoid cells. Nature 517, 293–301. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. [DOI] [PubMed] [Google Scholar]
- Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, Akalin A, and Schübeler D (2015). Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243–247. [DOI] [PubMed] [Google Scholar]
- Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, Popa CD, ter Horst R, van Tuijl J, Netea-Maier RT, et al. (2018). Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 172, 135–146.e9. [DOI] [PubMed] [Google Scholar]
- Beura LK, Fares-Frederickson NJ, Steinert EM, Scott MC, Thompson EA, Fraser KA, Schenkel JM, Vezys V, and Masopust D (2019). CD4+ resident memory T cells dominate immunosurveillance and orchestrate local recall responses. J. Exp. Med 216, 1214–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, Hong S-J, Bauer-Rowe KE, Xifaras ME, Akkad A, Arias E, et al. (2016). High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A, Ashenberg O, Su C-W, Smillie C, Shekhar K, et al. (2018). T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell 175, 1307–1320.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden LM, Lewis JM, Barbee SD, Bas A, Girardi M, Hayday AC, Tigelaar RE, and Lifton RP (2008). Skint1, the prototype of a newly identified immunoglobulin superfamily gene cluster, positively selects epidermal γδ T cells. Nat. Genet 40, 656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, et al. (2013). A Special Population of regulatory T Cells Potentiates muscle repair. Cell 155, 1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, Mai C, Jin WB, Guo CJ, Violante S, et al. (2020). Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catania S, Dumesic PA, Pimentel H, Nasif A, Stoddard CI, Burke JE, Diedrich JK, Cook S, Shea T, Geinger E, et al. (2020). Evolutionary Persistence of DNA Methylation for Millions of Years after Ancient Loss of a De Novo Methyltransferase. Cell 180, 263–277.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Wu W, Millman A, Craft JF, Chen E, Patel N, Boucher JL, Urban JF, Kim CC, and Gause WC (2014). Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat. Immunol 15, 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, Scholz CJ, Oosting M, Haendler K, Baßler K, et al. (2018). Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 172, 162–175.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkhurst CN, Muratet M, et al. (2012). A validated regulatory network for Th17 cell specification. Cell 151, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, and Mathis D (2012). PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue T reg cells. Nature 486, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins N, Jiang X, Zaid A, Macleod BL, Li J, Park CO, Haque A, Bedoui S, Heath WR, Mueller SN, et al. (2016). Skin CD4+memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat. Commun 7, 11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez VS, and Colonna M (2016). Diversity and function of group 1 innate lymphoid cells. Immunol. Lett 179, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debes GF, and McGettigan SE (2019). Skin-Associated B Cells in Health and Inflammation. J. Immunol 202, 1659–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiSpirito JR, Zemmour D, Ramanan D, Cho J, Zilionis R, Klein AM, Benoist C, and Mathis D (2018). Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci. Immunol 3, eaat5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divangahi M, Aaby P, Khader SA, Barreiro LB, Bekkering S, Chavakis T, van Crevel R, Curtis N, DiNardo AR, Dominguez-Andres J, et al. (2020). Trained immunity, tolerance, priming and differentiation: distinct immunological processes. Nat. Immunol 22, 2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekman AK, Bivik Eding C, Rundquist I, and Enerbäck C (2019). IL-17 and IL-22 Promote Keratinocyte Stemness in the Germinative Compartment in Psoriasis. J. Invest. Dermatol 139, 1564–1573.e8. [DOI] [PubMed] [Google Scholar]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, et al. (2009). Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med 15, 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Fits L, Mourits S, Voerman JSA, Kant M, Boon L, Laman JD, Cornelissen F, Mus A-M, Florencia E, Prens EP, et al. (2009). Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J. Immunol 182, 5836–5845. [DOI] [PubMed] [Google Scholar]
- Foster SL, Hargreaves DC, and Medzhitov R (2007). Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447, 972–978. [DOI] [PubMed] [Google Scholar]
- Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, and Colonna M (2013). Intraepithelial Type 1 Innate Lymphoid Cells Are a Unique Subset of IL-12- and IL-15-Responsive IFN-γ-Producing Cells. Immunity 38, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger G, Fan X, Dikiy S, Lee SY, and Rudensky AY (2015). Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350, 981–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, and Carbone FR (2009). Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol 10, 524–530. [DOI] [PubMed] [Google Scholar]
- Gebhardt T, Whitney PG, Zaid A, MacKay LK, Brooks AG, Heath WR, Carbone FR, and Mueller SN (2011). Different patterns of peripheral migration by memory CD4+and CD8+T cells. Nature 477, 216–219. [DOI] [PubMed] [Google Scholar]
- Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V, Cesses P, Garnier L, Pouzolles M, Brulin B, et al. (2016). Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glennie ND, Yeramilli V. a, Beiting DP, Volk SW, Weaver CT, and Scott P (2015). Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J. Exp. Med 212, 1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, and Moody DB (2015). The burgeoning family of unconventional T cells. Nat. Immunol 16, 1114–1123. [DOI] [PubMed] [Google Scholar]
- Gur-Cohen S, Yang H, Baksh SC, Miao Y, Levorse J, Kataru RP, Liu X, de la Cruz-Racelis J, Mehrara BJ, and Fuchs E (2019). Stem cell-driven lymphatic remodeling coordinates tissue regeneration. Science 366, 1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang S, Paik D, Yao L, Kim E, Jamma T, Lu J, Ha S, Nelson BN, Kelly SP, Wu L, et al. (2019). Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison OJ, Linehan JL, Shih H-Y, Bouladoux N, Han S-J, Smelkinson M, Sen SK, Byrd AL, Enamorado M, Yao C, et al. (2019). Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science (80-.) 363, eaat6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayday AC (2019). γδ T Cell Update: Adaptate Orchestrators of Immune Surveillance. J. Immunol 203, 311–320. [DOI] [PubMed] [Google Scholar]
- Honda K, and Littman DR (2016). The microbiota in adaptive immune homeostasis and disease. Nature 535, 75–84. [DOI] [PubMed] [Google Scholar]
- Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, Gallini CA, Redding K, Margolskee RF, Osborne LC, et al. (2016). Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science (80-.) 351, 1329–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y-C, Li L, and Fuchs E (2014). Emerging interactions between skin stem cells and their niches. Nat. Med 20, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima N, and Iwasaki A (2014). A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science (80-.) 346, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A (2007). Mucosal Dendritic Cells. Annu. Rev. Immunol 25, 381–418. [DOI] [PubMed] [Google Scholar]
- Jacobson A, Yang D, Vella M, and Chiu IM (2021). The intestinal neuro-immune axis: crosstalk between neurons, immune cells, and microbes. Mucosal Immunol. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, and Kupper TS (2012). Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature 483, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ (2013). Regulators of chromatin state and transcription in CD4 T-cell polarization. Immunology 139, 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, and Rudensky AY (2012). Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol 30, 531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoki M, Patil A, Thaiss CC, Brooks DJ, Pandey S, Deep D, Alvarez D, von Andrian UH, Wagers AJ, Nakai K, et al. (2017). Organism-Level Analysis of Vaccination Reveals Networks of Protection across Tissues. Cell 171, 398–413.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, Tzelepis F, Pernet E, Dumaine A, Grenier J-C, et al. (2018). BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 172, 176–190.e19. [DOI] [PubMed] [Google Scholar]
- Kayama H, Okumura R, and Takeda K (2020). Interaction between the Microbiota, Epithelia, and Immune Cells in the Intestine. Annu. Rev. Immunol 38, 23–48. [DOI] [PubMed] [Google Scholar]
- Keyes BE, Liu S, Asare A, Naik S, Levorse J, Polak L, Lu CP, Nikolova M, Pasolli HA, and Fuchs E (2016). Impaired Epidermal to Dendritic T Cell Signaling Slows Wound Repair in Aged Skin. Cell 167, 1323–1338.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N, Downey J, Sanz J, Kaufmann E, Blankenhaus B, Pacis A, Pernet E, Ahmed E, Cardoso S, Nijnik A, et al. (2020). M. tuberculosis Reprograms Hematopoietic Stem Cells to Limit Myelopoiesis and Impair Trained Immunity. Cell 183, 752–770.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz ÖH, Iwashita T, Yilmaz OH, Terhorst C, and Morrison SJ (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121. [DOI] [PubMed] [Google Scholar]
- Klicznik MM, Morawski PA, Höllbacher B, Varkhande SR, Motley SJ, Kuri-Cervantes L, Goodwin E, Rosenblum MD, Alice Long S, Brachtl G, et al. (2019). Human CD4+CD103+ cutaneous resident memory T cells are found in the circulation of healthy individuals. Sci. Immunol 4, 8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CM, and Sun JC (2018). The widening spectrum of immunological memory. Curr. Opin. Immunol 54, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laval B, Maurizio J, Kandalla PK, Brisou G, Simonnet L, Huber C, Gimenez G, Matcovitch-Natan O, Reinhardt S, David E, et al. (2020). C/EBPβ-Dependent Epigenetic Memory Induces Trained Immuniy in Hematopoietic Stem Cells. Cell Stem Cell 26, 793. [DOI] [PubMed] [Google Scholar]
- Lay K, Yuan S, Gur-Cohen S, Miao Y, Han T, Naik S, Pasolli HA, Larsen SB, and Fuchs E (2018). Stem cells repurpose proliferation to contain a breach in their niche barrier. Elife 7, e41661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrero-Fernández C, Wenzel UA, Akeus P, Wang Y, Strid H, Simrén M, Gustavsson B, Börjesson LG, Cardell SL, Öhman L, et al. (2016). Altered expression of Butyrophilin (BTN) and BTN-like (BTNL) genes in intestinal inflammation and colon cancer. Immunity, Inflamm. Dis 4, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, elardi E, Young LF, Smith OM, Lawrence G, et al. (2015). Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long HK, Prescott SL, and Wysocka J (2016). Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 167, 1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon M-L, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, et al. (2013). The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol 14, 1294–1301. [DOI] [PubMed] [Google Scholar]
- MacLeod AS, Hemmers S, Garijo O, Chabod M, Mowen K, Witherden DA, and Havran WL (2013). Dendritic epidermal T cells regulate skin antimicrobial barrier function. J. Clin. Invest 123, 4364–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marco Barros R, Roberts NA, Dart RJ, Vantourout P, Jandke A, Nussbaumer O, Deban L, Cipolat S, Hart R, Iannitto ML, et al. (2016). Epithelia Use Butyrophilin-like Molecules to Shape Organ-Specific γδ T Cell Compartments. Cell 167, 203–218.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Gonzalez I, Mathä L, Steer CA, Ghaedi M, Poon GFT, and Takei F (2016). Allergen-Experienced Group 2 Innate Lymphoid Cells Acquire Memory-like Properties and Enhance Allergic Lung Inflammation. Immunity 45, 198–208. [DOI] [PubMed] [Google Scholar]
- Masopust D, and Soerens AG (2019). Tissue-Resident T Cells and Other Resident Leukocytes. Annu. Rev. Immunol 37, 521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masopust D, Vezys V, Marzo AL, and Lefrançois L (2001). Preferential localization of effector memory cells in nonlymphoid tissue. Science 291, 2413–2417. [DOI] [PubMed] [Google Scholar]
- Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, et al. (2010). Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med 207, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayassi T, Ladell K, Gudjonson H, McLaren JE, Shaw DG, Tran MT, Rokicka JJ, Lawrence I, Grenier J-C, van Unen V, et al. (2019). Chronic Inflammation Permanently Reshapes Tissue-Resident Immunity in Celiac Disease. Cell 176, 967–981.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy NE, and Eberl M (2018). Human γδ T-cell control of mucosal immunity and inflammation. Front. Immunol 9, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melandri D, Zlatareva I, Chaleil RAG, Dart RJ, Chancellor A, Nussbaumer O, Polyakova O, Roberts NA, Wesch D, Kabelitz D, et al. (2018). The γδTCR combines innate immunity with adaptive immunity by utilizing spatially distinct regions for agonist selection and antigen responsiveness. Nat. Immunol 19, 1352–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milutinović B, and Kurtz J (2016). Immune memory in invertebrates. Semin. Immunol 28, 328–342. [DOI] [PubMed] [Google Scholar]
- Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, et al. (2018). Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 172, 147–161.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, Ji M, Liang H-E, and Locksley RM (2016). Tuft-cell-derived IL-25 regulates an intestinal ILC2–epithelial response circuit. Nature 529, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadjsombati MS, McGinty JW, Lyons-Cohen MR, Jaffe JB, DiPeso L, Schneider C, Miller CN, Pollack JL, Nagana Gowda GA, Fontana MF, et al. (2018). Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immunity 49, 33–41.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Linehan JL, Han S-J, Harrison OJ, Wilhelm C, Conlan S, Himmelfarb S, Byrd AL, Deming C, et al. (2015). Commensal–dendritic-cell interaction specifies a unique protective skin immune signature. Nature 520, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S, Polak L, Kulukian A, Chai S, and Fuchs E (2017). Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550, 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Larsen SB, Cowley CJ, and Fuchs E (2018). Two to Tango: Dialog between Immunity and Stem Cells in Health and Disease. Cell 175, 908–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, Joosten LAB, van der Meer JWM, Mhlanga MM, Mulder WJM, et al. (2020). Defining trained immunity and its role in health and disease. Nat. Rev. Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MM, Witherden DA, and Havran WL (2017). γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat. Rev. Immunol 17, 733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary JG, Goodarzi M, Drayton DL, and von Andrian UH (2006). T cell– and B cell–independent adaptive immunity mediated by natural killer cells. Nat. Immunol 7, 507–516. [DOI] [PubMed] [Google Scholar]
- Oh JE, Iijima N, Song E, Lu P, Klein J, Jiang R, Kleinstein SH, and Iwasaki A (2019). Migrant memory B cells secrete luminal antibody in the vagina. Nature 571, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, Gaboriau-Routhiau V, Marques R, Dulauroy S, Fedoseeva M, et al. (2015). The microbiota regulates type 2 immunity through RORγt+ T cells. Science (80-.) 349, 989–993. [DOI] [PubMed] [Google Scholar]
- Onodera T, Takahashi Y, Yokoi Y, Ato M, Kodama Y, Hachimura S, Kurosaki T, and Kobayashi K (2012). Memory B cells in the lung participate in protective humoral immune responses to pulmonary influenza virus reinfection. Proc. Natl. Acad. Sci. U. S. A 109, 2485–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, Wadsworth MH, Hughes TK, Kazer SW, Yoshimoto E, et al. (2018). Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature 560, 649–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, and Natoli G (2013). Latent Enhancers Activated by Stimulation in Differentiated Cells. Cell 152, 157–171. [DOI] [PubMed] [Google Scholar]
- Palm NW, Rosenstein RK, and Medzhitov R (2012). Allergic host defences. Nature 484, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panduro M, Benoist C, and Mathis D (2016). Tissue Tregs. Annu. Rev. Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, and Becher B (2012). Rorγt+ innate lymphocytes and γδ T cells initiate psoriasiform plaque formation in mice. J. Clin. Invest 122, 2252–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, Alexandre YO, Gregory JL, Russell TA, Gebhardt T, et al. (2018). Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol 19, 183–191. [DOI] [PubMed] [Google Scholar]
- Paust S, Gill HS, Wang B-Z, Flynn MP, Moseman EA, Senman B, Szczepanik M, Telenti A, Askenase PW, Compans RW, et al. (2010). Critical role for the chemokine receptor CXCR6 in NK cell–mediated antigen-specific memory of haptens and viruses. Nat. Immunol 11, 1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Jiang X, Chen Y, Sojka DK, Wei H, Gao X, Sun R, Yokoyama WM, and Tian Z (2013). Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J. Clin. Invest 123, 1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz FG, Fiore VF, Levorse J, Polak L, Wong E, Pasolli HA, and Fuchs E (2020). Liquid-liquid phase separation drives skin barrier formation. Science 367, eaax9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez-Valle F, Gray EE, and Cyster JG (2015). Inflammation induces dermal Vγ4+ γδT17 memory-like cells that travel to distant skin and accelerate secondary IL-17-driven responses. Proc. Natl. Acad. Sci. U. S. A 112, 8046–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer-Michalski E-M, and Conrath U (2016). Innate immune memory in plants. Semin. Immunol 28, 319–327. [DOI] [PubMed] [Google Scholar]
- Romagnoli PA, Sheridan BS, Pham Q-M, Lefrançois L, and Khanna KM (2016). IL-17A-producing resident memory γδ T cells orchestrate the innate immune response to secondary oral Listeria monocytogenes infection. Proc. Natl. Acad. Sci. U. S. A 113, 8502–8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F, Cheng SC, Ratter J, Berentsem K, Van Der Ent MA, et al. (2014). Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science (80-.) 345, 1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, et al. (2012). Foxp3 Exploits a Pre-Existent Enhancer Landscape for Regulatory T Cell Lineage Specification. Cell 151, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, and Campbell DJ (2007). Altering the distribution of Foxp3 + regulatory T cells results in tissue-specific inflammatory disease. J. Exp. Med 204, 1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharschmidt TC, Vasquez KS, Truong H-A, Gearty SV, Pauli ML, Nosbaum A, Gratz IK, Otto M, Moon JJ, Liese J, et al. (2015). A Wave of Regulatory T Cells into Neonatal Skin Mediates Tolerance to Commensal Microbes. Immunity 43, 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharschmidt TC, Vasquez KS, Pauli ML, Leitner EG, Chu K, Truong H-A, Lowe MM, Sanchez Rodriguez R, Ali N, Laszik ZG, et al. (2017). Commensal Microbes and Hair Follicle Morphogenesis Coordinately Drive Treg Migration into Neonatal Skin. Cell Host Microbe 21, 467–477.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, and Masopust D (2014). Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science (80-.) 346, 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, Burzyn D, Ortiz-Lopez A, Lobera M, Yang J, et al. (2015). Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science (80-.) 349, 993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharrock J, and Sun JC (2020). Innate immunological memory: from plants to animals. Curr. Opin. Immunol 62, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Wang Y, Kokovay E, Lin G, Chuang SM, Goderie SK, Roysam B, and Temple S (2008). Adult SVZ Stem Cells Lie in a Vascular Niche: A Quantitative Analysis of Niche Cell-Cell Interactions. Cell Stem Cell 3, 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan BS, Romagnoli PA, Pham Q-M, Fu H-H, Alonzo F, Schubert W-D, Freitag NE, and Lefrançois L (2013). γδ T Cells Exhibit Multifunctional and Protective Memory in Intestinal Tissues. Immunity 39, 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan BS, Pham Q-M, Lee Y-T, Cauley LS, Puddington L, and Lefrançois L (2014). Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity 40, 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale ST, Tarakhovsky A, and Natoli G (2014). Chromatin Contributions to the Regulation of Innate Immunity. Annu. Rev. Immunol 32, 489–511. [DOI] [PubMed] [Google Scholar]
- Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, et al. (2019). Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 178, 714–730.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, and Garrett WS (2013). The microbial metabolites, short-chain fatty acids, regulate colonic T reg cell homeostasis. Science (80-.) 341, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, Geva-Zatorsky N, Jupp R, Mathis D, Benoist C, et al. (2020). Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 577, 410–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Beilke JN, and Lanier LL (2009). Adaptive immune features of natural killer cells. Nature 457, 557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, Garcia-Verdugo JM, and Doetsch F (2008). A Specialized Vascular Niche for Adult Neural Stem Cells. Cell Stem Cell 3, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrançois L, and Farber DL (2011). Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J. Immunol 187, 5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahedi G, Takahashi H, Nakayamada S, Sun H-W, Sartorelli V, Kanno Y, and O’Shea JJ (2012). STATs shape the active enhancer landscape of T cell populations. Cell 151, 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vantourout P, Laing A, Woodward MJ, Zlatareva I, Apolonia L, Jones AW, Snijders AP, Malim MH, and Hayday AC (2018). Heteromeric interactions regulate butyrophilin (BTN) and BTN-like molecules governing γδ T cell biology. Proc. Natl. Acad. Sci. U. S. A 115, 1039–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veeken J, Gonzalez AJ, Cho H, Arvey A, Hemmers S, Leslie CS, and Rudensky AY (2016). Memory of Inflammation in Regulatory T Cells. Cell 166, 977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma M, Asakura Y, Murakonda BSR, Pengo T, Latroche C, Chazaud B, McLoon LK, and Asakura A (2018). Muscle Satellite Cell Cross-Talk with a Vascular Niche Maintains Quiescence via VEGF and Notch Signaling. Cell Stem Cell 23, 530–543.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, et al. (2018). Innate Lymphoid Cells: 10 Years On. Cell 174, 1054–1066. [DOI] [PubMed] [Google Scholar]
- Wakim LM, Waithman J, van Rooijen N, Heath WR, and Carbone FR (2008). Dendritic Cell-Induced Memory T Cell Activation in Nonlymphoid Tissues. Science (80-.) 319, 198–202. [DOI] [PubMed] [Google Scholar]
- Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, Horth C, McGuire JT, Xu X, Nikbakht H, et al. (2019). The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weizman O-E, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, Degli-Esposti MA, Leslie CS, Sun JC, and O’Sullivan TE (2017). ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell 171, 795–808.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendeln A-C, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, Wagner J, Häsler LM, Wild K, Skodras A, et al. (2018). Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MA, and Heagy W (2002). Endotoxin tolerance: a review. Crit. Care Med 30, S64–73. [PubMed] [Google Scholar]
- Whibley N, Tucci A, and Powrie F (2019). Regulatory T cell adaptation in the intestine and skin. Nat. Immunol 20, 386–396. [DOI] [PubMed] [Google Scholar]
- Willcox BE, and Willcox CR (2019). γδ TCR ligands: the quest to solve a 500-million-year-old mystery. Nat. Immunol 20, 121–128. [DOI] [PubMed] [Google Scholar]
- Yao Y, Jeyanathan M, Haddadi S, Barra NG, Vaseghi-Shanjani M, Damjanovic D, Lai R, Afkhami S, Chen Y, Dvorkin-Gheva A, et al. (2018). Induction of Autonomous Memory Alveolar Macrophages Requires T Cell Help and Is Critical to Trained Immunity. Cell 175, 1634–1650.e17. [DOI] [PubMed] [Google Scholar]
- Zaret KS (2020). Pioneer Transcription Factors Initiating Gene Network Changes. Annu. Rev. Genet 54, 367–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Franklin RA, Adler M, Jacox JB, Bailis W, Shyer JA, Flavell RA, Mayo A, Alon U, and Medzhitov R (2018). Circuit Design Features of a Stable Two-Cell System. Cell 172, 744–757.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zundler S, Becker E, Spocinska M, Slawik M, Parga-Vidal L, Stark R, Wiendl M, Atreya R, Rath T, Leppkes M, et al. (2019). Hobit- and Blimp-1-driven CD4+ tissue-resident memory T cells control chronic intestinal inflammation. Nat. Immunol 20, 288–300. [DOI] [PubMed] [Google Scholar]