

Abstract
Understanding the bioenergetics of axon extension and maintenance has wide ranging implications for neurodevelopment and disease states. Glycolysis is a pathway consisting of 10 enzymes and separated into preparatory and payoff phases, the latter producing ATP. Using embryonic chicken sensory neurons, we report that glycolytic enzymes are found through the axon and the growth cone. Pharmacological inhibition of glycolysis in the presence of NGF impairs axon extension and growth cone dynamics within minutes without affecting axon maintenance. Experiments using microfluidic chambers show that the effect of inhibiting glycolysis on axon extension is local along distal axons and can be reversed by promoting mitochondrial respiration. Knockdown of GAPDH simplifies growth cone morphology and is rescued by shRNA-resistant GAPDH expression. Rescue of GAPDH using KillerRed fused to GAPDH followed by localized chromophore-assisted light inactivation of KillerRed-GAPDH in distal axons halts growth cone dynamics. Considering filament polymerization requires ATP, inhibition of glycolysis results in a paradoxical increase in axonal actin filament levels. The effect on actin filaments is because of enzymes before GAPDH, the first enzyme in the payoff phase. In the absence of NGF, inhibition of glycolysis along distal axons results in axon degeneration independent of cell death. These data indicate that the glycolytic pathway is operative in distal axons and contributes to the rate of axon extension and growth cone dynamics in the presence of NGF and that, in the absence of NGF, the axonal glycolytic pathway is required for axon maintenance.
SIGNIFICANCE STATEMENT Elucidation of the sources of ATP required for axon extension and maintenance has implications for understanding the mechanism of neuronal development and diseases of the nervous system. While recent work has emphasized the importance of mitochondrial oxidative phosphorylation, the role of the glycolytic pathway in axon morphogenesis and maintenance remains minimally understood. The data reveal that the glycolytic pathway is required for normal sensory axon extension in the presence of NGF, while in the absence of NGF the glycolytic pathway is required for axon maintenance. The results have implications for the understanding of the bioenergetics of axon morphogenesis and plasticity and indicate that NGF has protective effects on sensory axon maintenance in hypoglycemic states.
Keywords: ATP, axon, cytoskeleton, glycolysis, growth cone, neurotrophin
Introduction
Oxygen and glucose are used to generate ATP to power nervous system function, and the nervous system utilizes a large proportion of blood oxygen and glucose (Mergenthaler et al., 2013). Electrically active and morphologically complex cells, such as neurons, require large amounts of ATP formaintenance of their physiology. Maintenance of membrane potential, long distance axonal transport, synaptic function, and cytoskeletal remodeling underlying aspects of neuronal plasticity are all processes requiring ATP utilization (Bordone et al., 2019). Thus, understanding the contributions of ATP-generating systems in neurons is of fundamental importance to understanding neuronal function.
The extension of axons is an energetically expensive process. The growth cone is the site of axon extension, and it is characterized by a complex morphology wholly dependent on the actin filaments that underlie filopodia and lamellipodia formation and dynamics (Dent and Gertler, 2003). Actin is an ATPase and must be loaded with ATP to polymerize and subsequently hydrolyzes the ATP once incorporated into a filament. Under conditions of suppressed oxidative phosphorylation and glycolysis actin turnover in developing neurons consumes ∼50% of the available ATP (Bernstein and Bamburg, 2003), indicating that actin filament turnover is a major bioenergetic drain. As growth cones contain the highest density of dynamic actin filaments in the developing neuron, they represent a subcellular domain of high ATP consumption. Microtubule-based axonal transport of organelles and proteins represents an additional component of axon extension that consumes ATP, although its bioenergetic drain has not been assessed.
Oxidative phosphorylation in the mitochondrion and glycolysis are the two main sources of ATP generation. Recent investigations have revealed a requirement for mitochondria and oxidative phosphorylation in developmental and regenerative axon extension (for review, see Sheng, 2017; Smith and Gallo, 2018; Han et al., 2020). Some extracellular signals that promote extension and growth cone elaboration (e.g., neurotrophins) promote mitochondrial respiration and fission (Verburg and Hollenbeck, 2008; Markham et al., 2012; Su et al., 2014; Armijo-Weingart et al., 2019), while signals that inhibit extension or axon branching can decrease respiration and the targeting of mitochondria to growth cones (Pacelli et al., 2015; Sainath et al., 2017a,b). Roles for both oxidative phosphorylation and glycolysis have been described in synaptic function in established neuronal networks (for review, see Ashrafi and Ryan, 2017), and accumulation of glycolytic enzymes at synaptic sites has been reported in the context of energy stress (Jang et al., 2016). Prior work indicates that glycolysis is an important contributor to ATP levels in embryonic PNS and CNS neurons (for review, see Gallo, 2020). In cortical and retinal neurons, ATP production shifts from being largely dependent on glycolysis in embryonic neurons to depending more on oxidative phosphorylation in postnatal neurons (Steketee et al., 2012; Surin et al., 2013). Glycolytic enzymes are associated with fast axonal transport vesicles, and the payoff phase of glycolysis provides the ATP required for the transport of the vesicles, but not mitochondria motility, which instead depends on oxidative phosphorylation in embryonic CNS and PNS neurons (Spillane et al., 2013; Zala et al., 2013; Hinckelmann et al., 2016). Inhibition of glycolysis using 2-deoxyglucose treatment starting at the time of culturing prevents cortical neurons from differentiating an axon from minor processes (Agostini et al., 2016). However, whether glycolysis contributes to axon extension and growth cone dynamics, or to neuron viability and axon maintenance in embryonic neurons, has not been thoroughly explored. Herein we present evidence that the glycolytic pathway is operative in the distal axons of embryonic sensory neurons and locally contributes to axon extension and growth cone dynamics. The results also unveil that NGF has protective effects and prevents axons from degenerating when glycolysis is inhibited.
Materials and Methods
Explant and dissociated neuron culturing and reagents
Embryonic day 7 chicken embryos were used throughout. Detailed protocols for the generation of DRG explants and dissociated sensory neurons and substrata are provided in Lelkes et al. (2005) and were used accordingly. Culturing was performed on coverslips coated with polylysine (Sigma Millipore, #P-9011) and then laminin (25 mg/ml: Invitrogen, #23017-015). Microfluidic chambers were prepared and used as previously described (Sainath et al., 2017b; Armijo-Weingart et al., 2019). Unless otherwise noted, medium was supplemented with 20 ng/ml nerve growth factor (R&D Systems, #256-GF-100). For these experiments, control DMEM lacking glucose (Invitrogen, #11966025) was used and supplemented with 10 mm D-glucose (Sigma Millipore, #G7021), 10 mm HEPES (Fisher BioReagents, #BP299-100), 1% PSF (penicillin-streptomycin mixture; Thermo Fisher Scientific, #BW17745E), and 1 mm NaPyruvate (Invitrogen, #11360-070). Glycolysis inhibition medium (GIM) was made using the same formulation but replacing D-glucose with 10 mm 2-deoxyglucose (Sigma Millipore, #D8375). Antimycin-A (AA) and heptelidic acid were obtained from Sigma Millipore (#A8674) and Santa Cruz Biotechnology (#391051A), respectively. Acetyl-L-carnitine (ALC) was obtained from Sigma Millipore (#A6706).
Microscopy
Neurons were imaged using a Carl Zeiss 200 M microscope equipped with an Orca-ER camera (Hamamatsu) in series with a PC workstation running Carl Zeiss Axiovision software for image acquisition and analysis. Cultures were placed on a heated microscope stage (Carl Zeiss temperable insert P with objective heater) for 10 min at a constant 39°C before and during imaging. Imaging was performed using a Carl Zeiss Pan-Neofluar 100× and 20× objectives. For quantification of fluorescence intensity, 2 × 2 camera binning was used to obtain images. Phase-contrast time-lapse imaging was performed at 20× to measure axon extension rates and 100× to analyze growth cone dynamics. Time-lapse imaging of GFP and PercevalHR (Tantama et al., 2013) were performed at 100× using 2 × 2 binning and minimal light.
Immunocytochemistry, quantification of staining intensity, and axon and cellular metrics
Actin filaments were detected using rhodamine-phalloidin (Invitrogen, #R415) or Alexa-488-phalloidin (Invitrogen, #A12379). Tubulin was detected using anti-a-tubulin directly conjugated to Alexa-488 (DM1A; Sigma Millipore, #16-232), respectively. For actin filament and tubulin staining, cultures were fixed for 15 min using 0.5% glutaraldehyde and stained as previously described (Spillane et al., 2012, 2013). The following primary antibodies to glycolytic enzymes were used: anti-GAPDH (1:100; Thermo Fisher Scientific, #MA5-15738), anti-PKM1/2 (1:150; Cell Signaling Technology, #3190), anti-Enolase-1 (1:150; Cell Signaling Technology, #3810), anti-TPI1 (1:150; Bethyl Laboratories, #A303-755A-T), and anti-ALDOA (1:100; Bethyl Laboratories, #A304-494A-T). Secondary antibodies used were as follows: goat anti-rabbit TRITC (1:200; Sigma Millipore, #T6778) and goat anti-mouse TRITC (1:400; Sigma Millipore, #T5393). For staining of glycolytic enzymes, cultures were fixed with a final concentration of 4% PFA (EMS, #15710) and 5% sucrose (Thermo Fisher Scientific, #S5-500) for 15 min. Blocking was performed before each primary antibody application for 30 min using PBS supplemented with 10% goat serum (Sigma Millipore, #G9023) and 0.1% Triton X-100 (Sigma Millipore #9002-93-1) (GST). All antibodies were applied using GST. Omission of primary antibodies resulted in no staining along axons. All antibodies to glycolytic enzymes were determined to label a single band of the expected molecular weight by Western bot analysis.
Quantification of staining intensity along axons was performed as in our prior work (Spillane et al., 2012, 2013). Briefly, all samples were processed in parallel from the time of culturing through the fixation and staining protocols, and images for all groups within an experiment were acquired at 100× during the same microscopy section. Image acquisition parameters were set to prevent pixels from being outside the dynamic range of intensity. The total integrated staining intensity over a fixed length of axon was the main metric to address net levels of proteins/actin filaments in distal axons. Measurements of axons excluded the growth cone, defined as starting at the point when the axon shafts splays to give rise to the “neck” of the growth cones and central domain, or if the growth cone was not splayed out as the distal 10 μm of the axon shaft (excluding filopodia lengths).
Samples labeled for tubulin and actin filaments were also used to obtain measurements of average axon length from explants as described by Sainath and Gallo (2015). Scholl analysis was performed by counting axon profiles intersecting lines at 100 μm intervals starting at 100 μm from the base of explants, as in Silver et al. (2014).
Determination of the number of neurons in dissociated ganglion cultures was performed using anti-neuron-specific β-III tubulin antibodies (Biolegend, #801213; 1:100, using same fixation protocol as for glycolytic antibodies) counterstained with phalloidin. The morphology of neuronal cell bodies was also qualitatively assessed at 100× using phase contrast and the staining to determine whether signs of cell death might be evident (e.g., membrane blebbing, nuclear disruption).
GAPDH, PercevalHR, shRNA expression vectors, and transfection
The PercevalHR expression plasmid was obtained from Addgene (#49083) (Tantama et al., 2013). The vectors expressing the human sequence of GAPDH open reading frame (ORF) (pET30-2-GAPDH, Addgene #83910) was used as PCR templates to generate the cDNAs of GAPDH (GenBank accession no. NM_002046.5). The primers were engineered to include EcoRI and BamHI restriction sites in the cDNA product (full ORF, nt 1-1005). The PCR conditions were as follows: 95°C for 1 min, 58°C for 1 min, and 72°C for 1 min for 35 cycles with a final 72°C extension for 10 min. The digested PCR product was subcloned into pKillerRed-N vector (Evrogen, #FP962) to generate the construct encoding an in frame KillerRed (KR) protein fused to the C-terminus of GAPDH (pGAPDH-KR). The vector cortactin-RFP (Spillane et al., 2012) was used to replace the cortactin sequence by the GAPDH PCR product by EcoRI/BamHI restriction, generating the vector pGAPDH-RFP.
Three previously reported shRNA sequences targeting chicken GAPDH were used to generate expression vectors (Billings et al., 2010). shRNA sequences were expressed based on pSuper.puro (VEC-PBS-0007/0008, OligoEngine) with modifications. GFP sequence was subcloned PstI/NheI on the plasmid pSuper.puro generating the vector pSuper.GFP (Pacheco et al., 2020). The GAPDH silencing plasmids (pSUPERshRNA-GAPDH-GFP) were generated according to the manufacturer's instructions (OligoEngine). The control shRNA-nontarget (target sequence GCGCGATAGCGCTAATAAT) used was as in Pacheco et al. (2020). Each of the three shRNA sequences from Billings et al. (2010) were expressed for 48 h in dissociated neurons before fixation and quantification of the intensity of GAPDH levels detected through immunocytochemistry as shown in Figure 5A. Based on the efficacy of knockdown, we selected for further work the target chicken sequence GAAACTGATCTGTTTGTGTACC (nt 1233-1254, GenBank accession no. NM_204305), which yielded a 72% decrease in GAPDH median staining intensity in distal axons relative to expression of the nontarget sequence control (n = 38 and n = 37 neurons, respectively; p = 0.0006, Mann–Whitney test) without affecting the area of the distal 50 μm of axons being measured (p = 0.32, Welch t test). The shRNA sequence to chicken GAPDH used in the rest of the studies targets a sequence in the 3′UTR of the mRNA. The rescue vectors using the human GAPDH ORF do not contain the 3′UTR and as thus resistant to the shRNAs (see Fig. 5D).
Figure 5.
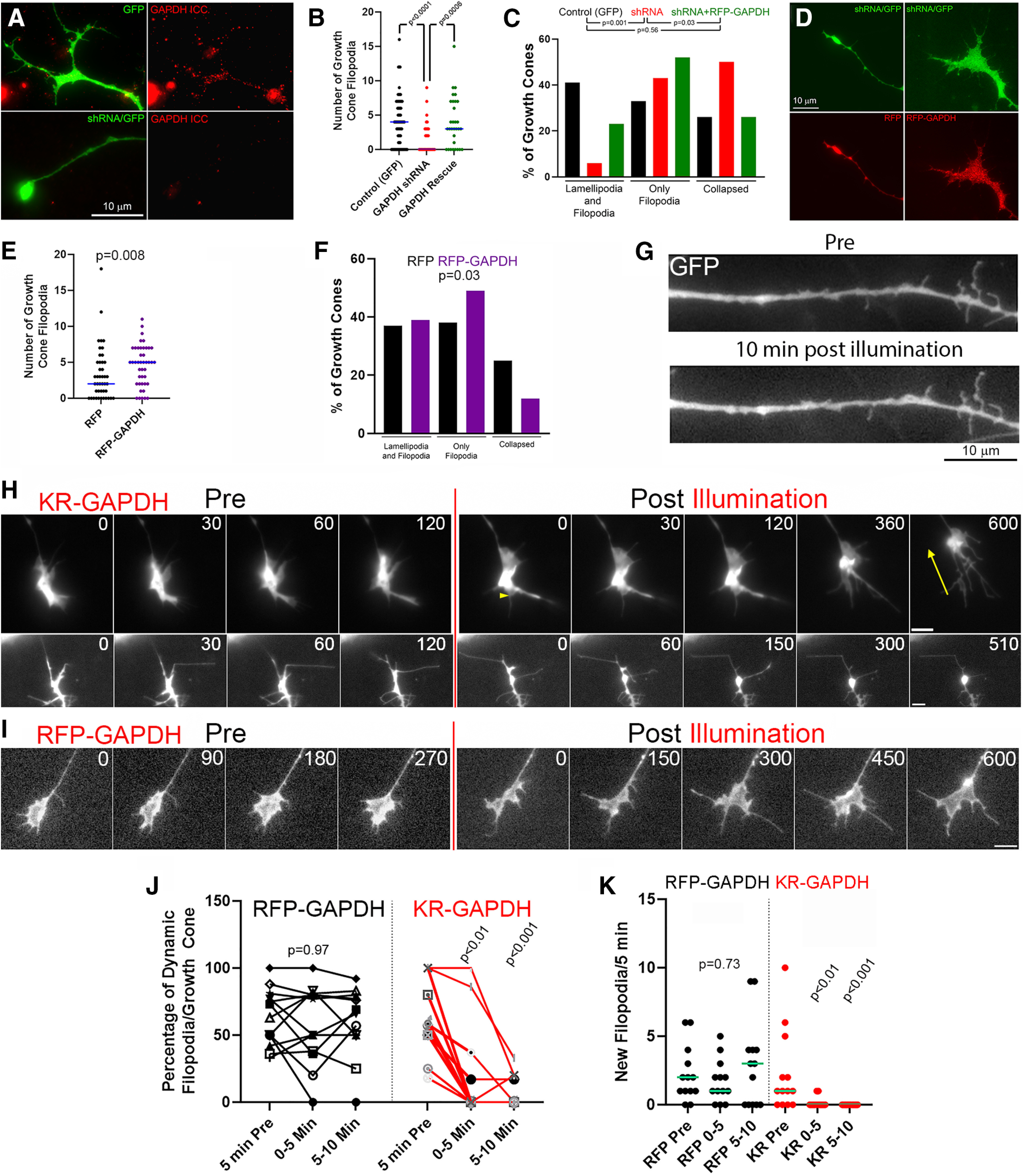
Suppression of GAPDH expression simplifies growth cones, and local ablation of GAPDH in distal axons impairs growth cone dynamics. A, Example of shRNA-mediated knockdown of GAPDH. GFP is expressed by the shRNA expression vector and used to track morphology in transfected neurons. B, Knockdown of GAPDH decreases filopodia number and is rescued by shRNA-resistant GAPDH expression. Mann–Whitney tests. Median shown. C, Knockdown of GAPDH simplifies growth cone morphology and is rescued by shRNA-resistant GAPDH expression. χ2 tests for independence. We note that, without live imaging, growth cones scored as having only filopodia may represent growth cones undergoing retraction as the filopodia cannot be strictly differentiated from the retraction fibers in static images. D, Examples of growth cones expressing GAPDH shRNA and expressing RFP (control) or rescue RFP-GAPDH. E, GAPDH overexpression increased the number of growth cone filopodia. Mann–Whitney test. Medians shown. F, GAPDH overexpression increases growth cones with filopodia and decreases the proportion of collapsed growth cone. χ2 tests for independence. G, CALI of KR-GAPDH in a RATK experimental design does not adversely affect the morphology of the axon shaft as evidenced by the absence of thinning and/or beading. H, Two examples of RATK in KR-GAPDH-expressing distal axons. Time in seconds is shown. Following illumination/CALI, the dynamic remodeling of the growth cones greatly decreases. Yellow arrow at 600 s qualitatively indicates growth cone retraction in the top sequence. The base of the yellow arrow at 600 s indicates the initial distalmost position of the leading edge of the growth cone (arrowhead at 0 s after illumination) and the tip the current position. Scale bars, 5 μm. I, Example of RATK in RFP-GAPDH-expressing distal axons. After illumination, the growth cone remains dynamic. Scale bar, 5 μm. Percentage of dynamic filopodia (J) and new filopodia (K) formed before and after illumination in RATK using RFP-GAPDH (control) and KR-GAPDH. Repeated-measures ANOVA for control. Dunn's multiple comparison tests between each time point and Pre for KR-GAPDH. Each symbol represents a growth cone. Medians are shown.
Transfection was performed as previously described (Armijo-Weingart et al., 2019). For transfection of plasmids into neurons, 40 chicken DRGs were dissociated and suspended in 100 µl nucleofector solution (Lonza Walkersville, #VPG-1002) through gentle trituration. The neuron suspension was transferred to a nucleofector cuvette containing 10 μg of plasmid DNA, and electroporated using an Amaxa Nucleofector (program G-13; Lonza Walkersville). The electroporated solution was then immediately transferred to a tube containing culturing media as described above before plating.
GAPDH rescue followed by chromophore-assisted light inactivation (CALI) of KR
CALI of KR was performed using the microscopy setup and illumination previously described for ablation of KR targeted to mitochondria (Spillane et al., 2013). Based on the expression levels of GAPDH-KR, a 25 s illumination bleached the KR signal by >95% and 25 s illumination was thus used throughout. For the CALI of KR-GAPDH and RFP-GAPDH (see Fig. 5H-K), similar lengths of distal axon were illuminated (54.8 ± 2.1 and 55.7 ± 2.5 μm, respectively, mean ± SEM and n = 13 for each group; p = 0.78, Welch t test). The number of growth cone filopodia also did not differ between the two groups before illumination (8.5 ± 0.9 and 10 ± 1.2, respectively, mean ± SEM; p = 0.33, Welch t test).
Experimental design and statistical analysis
All data were analyzed using Instat software (GraphPad Software). Determination of the normalcy of datasets was performed using the Kolmogorov–Smirnov test. Normal datasets were analyzed using the Welch t test for independent groups or the paired t test for before-after treatment experimental designs. If non-normal datasets were present, then nonparametric analysis was used (Mann–Whitney test). For multiple comparison tests within experimental designs, parametric Bonferroni or nonparametric Dunn's post hoc tests were used according to the normalcy of the datasets. For datasets representing categorical data falling into bins, the Fisher's exact test was used on the raw categorical data, although the data are expressed as percentages for visual presentation. One- or two-tailed p values are reported based on whether the hypothesis predicted the directionality of the expected difference in mean or median, respectively. Graphs were generated using Prism (GraphPad Software). Sample sizes and qualitative statistical presentation are denoted in figure legends or figures.
For experiments involving live imaging and pre-/post-treatment experimental design wherein the readout comes from a single subject/growth cone tracked over time each datum comes from a single culture. In this type of design, for any day of imaging, we sampled equally across the multiple groups in the experimental design, usually yielding 4-10 data points per day. Thus, the data are ultimately representative of multiple cultures in each group equal to the number of data points shown and over multiple separate sets of cultures derived from an equivalent number of embryos.
For experiments involving the quantification of growth cones subjected to shRNA, rescue or overexpression, population-based analysis was performed across growth cones sampled from multiple cultures derived from multiple embryos and rounds of culturing and transfection. In our transfection and culturing system, we obtain ∼5 to at most 15 growth cones that are transfected and fit criteria for sampling per transfection, each transfection representing by technical necessity cells pooled from 2 or 3 embryos generating ∼6 coverslips. In order to be sampled for morphometric analysis, growth cones cannot be contacting any other cells or be fasciculated.
For experiments collecting categorical datasets on proportions of axon degenerating, axons were sampled from a minimum of three replicate cultures per experimental condition. The data were then pooled into one population and analyzed using the Fisher's exact test. Only distal axons that were not contacting other axons or cells were counted in the analysis.
Results
Sensory axon growth cones contain glycolytic enzymes
Glycolysis is considered of consisting of two main phases: the preparatory and the payoff phases. In the preparatory phase, ATP is used to convert glucose into triose sugars. The payoff phase, starting with GAPDH, uses the triose sugars derived from the preparatory phase and results in the net gain of ATP. Immunocytochemistry to detect 5 of the 10 glycolytic enzymes representative of the preparatory and payoff phases of glycolysis revealed that enzymes are present in growth cones (Fig. 1A,A′) as well as throughout the axon. All enzymes exhibited a mostly punctate distribution, consistent with their reported targeting to membranous vesicles (Zala et al., 2013; Hinckelmann et al., 2016) and localized to the central domain and lamellipodia. Although for all the enzymes analyzed the signal was predominantly punctate, some domains of more diffuse signal could also be detected within the central and peripheral domains. The targeting of enzymes within filopodia at growth cones varied between enzymes (Fig. 1A,B). These observations indicate that the glycolytic apparatus is present in growth cones and distal axons, and that at least one of the ATP-generating enzymes (PKM) targets to both the central and peripheral domains of the growth cone. GAPDH binds actin filaments (for review, see DeWane et al., 2021); and consistently, we found a positive correlation between the mean intensity of actin filaments in growth cone filopodia and the mean intensity of GAPDH staining (Fig. 1C), indicating it may be recruited to regions of high filament content.
Figure 1.
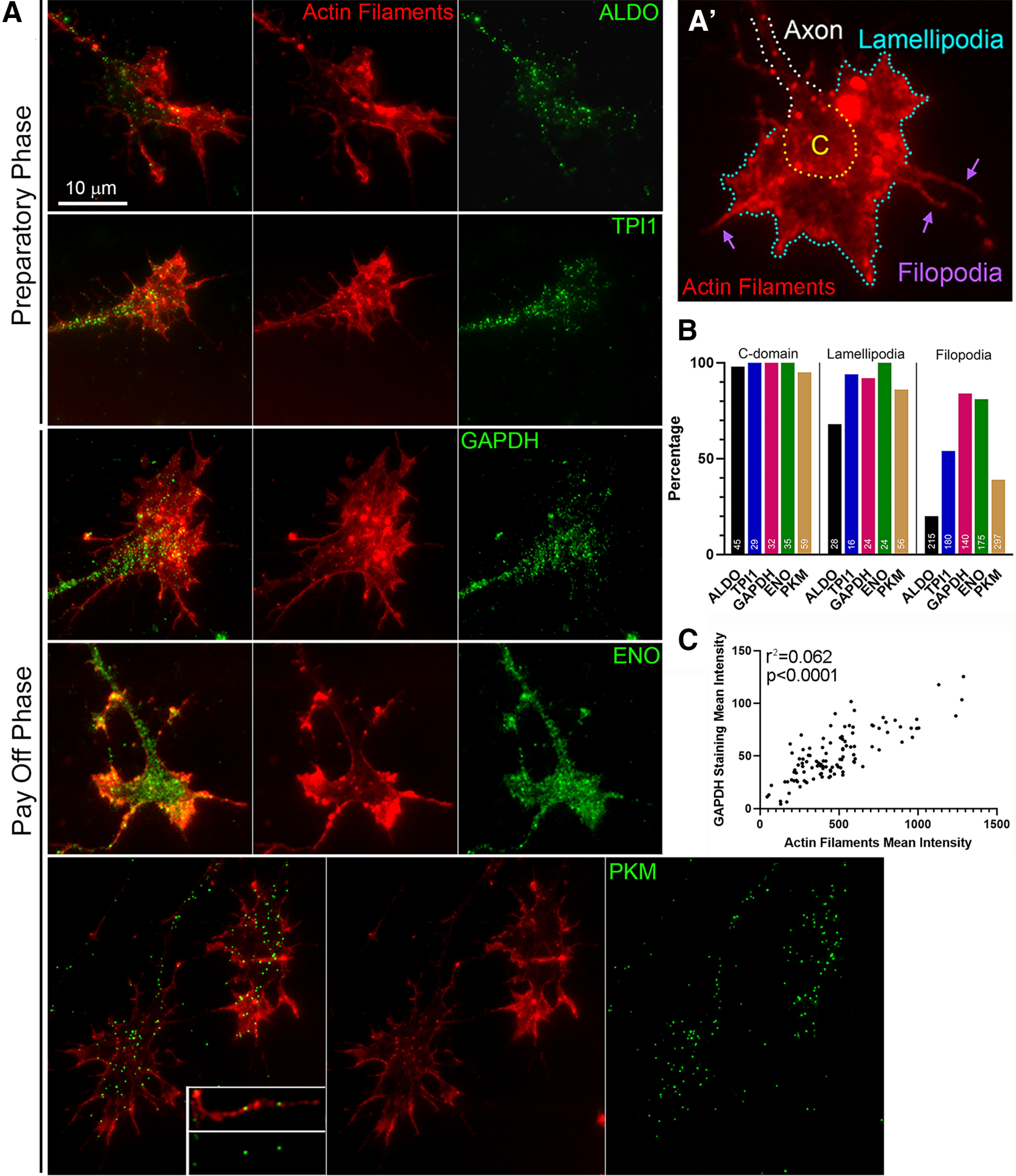
Axons and growth cones contain glycolytic enzymes. A, Both preparatory (aldolase [ALDO] and triosephosphate isomerase [TPI1]) and payoff (GAPDH, enolase [ENO], and the ATP-generating pyruvate kinase [PKM]) enzymes targeted to the central and peripheral domains of growth cones (A′). B, Percentage of growth cone domains containing glycolytic enzymes. n = total number of growth cones sampled, shown in bars for C-domain. n shown in other categories reflects the number of growth cones with lamellipodia, a subset of the total, and the total number of filopodia sampled at growth cones. C, Correlation analysis of the mean intensity of actin filaments in growth cone filopodia and the mean intensity of GAPDH staining (r = 0.79, 95% CI 0.71-0.85; n = 111 filopodia from 13 growth cones).
Acute inhibition of glycolysis rapidly halts axon extension and growth cone dynamics
To rapidly inhibit glycolysis, we exchanged the culturing medium with medium containing no glucose and the glycolysis inhibitor 2-deoxyglucose, which is hereafter referred to as GIM. 2-Deoxyglucose is phosphorylated by the first kinase in the preparatory phase (hexokinase), and its product cannot be further metabolized by phosphoglucoisomerase (second step in the preparatory phase). GIM and control medium were both supplemented with pyruvate to prevent impairment of oxidative phosphorylation downstream of glycolysis inhibition. Unless otherwise stated, all experiments were performed in the presence of NGF. Imaging the ATP/ADP ratio in the growth cones of PercevalHR-expressing neurons showed that both GIM and treatment with the oxidative phosphorylation inhibitor AA rapidly decrease the ATP/ADP ratio (Fig. 2A,B). The initial effects of GIM and AA were additive (Fig. 2A), consistent with oxidative phosphorylation not being impaired by GIM treatment. Exchange of control medium with GIM resulted in the cessation of growth cone dynamics within minutes (Fig. 2C–F) and halted axon extension (Fig. 2G). By 5 min of treatment with GIM, protrusive activity ceased and the growth cone area decreased as the edges contracted (Fig. 2C,D). Filopodia were not initially retracted but rather stabilized in place. As the edges of growth cones or filopodia underwent contraction they often generated retraction fibers (Fig. 2C, arrowheads labeled RF at 60-180 s), indicating that substratum attachment points within filopodia and lamellipodia were maintained as the rest of the structure contracted or dislocated. A general contraction of the growth cone was apparent and between 10 and 55 min of GIM treatment, and 38% of axon tips underwent transient retraction as their growth cones became simplified losing lamellipodia and filopodia (Fig. 2G). The effects of a 10 or 60 min treatment with GIM on axon extension rate were partially reversed with 55 min of exchanging GIM with control medium, although statistically different (Fig. 2G). Treatment with a heptelidic/koningic acid, a GAPDH inhibitor (Endo et al., 1985; Sakai et al., 1988; Kato et al., 1992; Kumagai et al., 2008), for 1 h decreased growth cone area and the number of filopodia (Fig. 2H–J), indicating the effects of inhibition of glycolysis on growth cones are downstream of GAPDH. Similar to GIM treatment, axon extension was stalled after acute treatment with heptelidic acid and 21% of axons underwent retraction (Fig. 2K).
Figure 2.
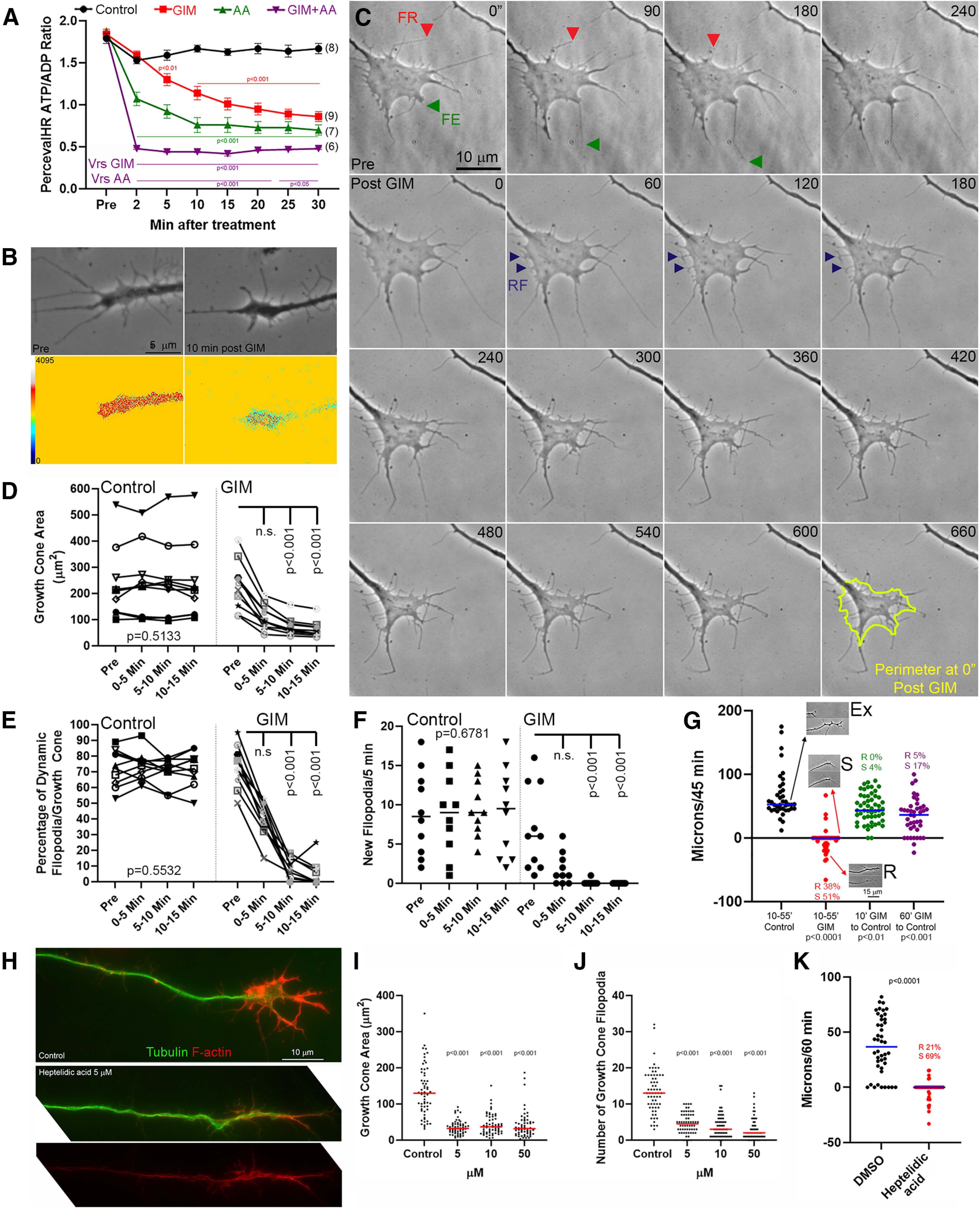
Glycolysis is required for growth cone dynamics. A, Ratiometric ATP/ADP measurements in growth cones. Treatment with either GIM and AA rapidly decreases the ATP/ADP ratio (p values color-coded for GIM [red] and AA [green] relative to control treatment, and GIM+AA relative to GIM or AA [purple]). n = growth cones. Bonferroni multiple comparison tests between groups within time point. B, Examples of ATP/ADP ratio before and after GIM treatment. C, Example of the response of growth cones to GIM. In pretreatment period (top row of panels), examples of filopodial retraction (FR) and extension (FE) are shown. Post-GIM examples of retraction fibers (RF) representing no change in the position of the stabilized filopodium are denoted at 60-180 s after treatment. At 660 s, the original outline of the growth cone is shown in yellow. D-F, Repeated-measures ANOVA for control. Dunn's multiple comparison tests between each time point and Pre for GIM treatment. Each symbol represents a growth cone. D, Growth cone area as a function of GIM treatment. E, Percentage of dynamic filopodia (i.e., filopodia that exhibited extension and/or retraction of their tips). F, Number of new filopodia formed/5 min. G, Axon extension or retraction (negative values) during a 45 min period after control (black) or GIM (red) treatment. Extension/retraction during a 45 min period after an initial 10 (green) or 60 min (purple) treatment with GIM followed by return to control medium. Each symbol is one axon. Insets, Examples of extension (Ex), stalling (S), and retraction (R) and percentage of axons in R or S categories are also shown. H, Examples of the morphology and cytoskeleton of axons treated for 1 h with DMSO (control) of heptelidic acid (5 μm). I, Effects of a 1 h treatment with varying concentrations of heptelidic acid on growth cone area. Dunn's multiple comparison tests relative to control. J, Effects of a 1 h treatment with varying concentrations of heptelidic acid on the number of growth cone filopodia. Dunn's multiple comparison tests relative to control. K, Measurements of axon extension as in G following a 1 h treatment with heptelidic acid (5 μm). Mann–Whitney test. A, Mean ± SEM is shown in A and medians are shown in F and G and I-K.
Chronic inhibition of glycolysis reversibly halts axons extension in the absence of axon degeneration
Continuous treatment with GIM for 24 h, starting at 24 h of culturing, resulted in the persistent stalling of axon extension without further axon retraction or degeneration (Fig. 3A) as evidenced by the maintenance of a normal microtubule array, axonal morphology, the absence of axonal beading, and unaffected density of axons emanating from explants (Fig. 3A; also see Fig. 6C) and no change in the area of distal axons or tubulin content (Fig. 3B,C). The density of mitochondria in distal axons (mitochondria/unit length of axon; assayed as in Armijo-Weingart et al., 2019) was also not affected by GIM treatment (p = 0.52, Mann–Whitney test, n = 42 and n = 47 axons for control and GIM). The stalling of axons in GIM treatment conditions was verified using phase-contrast live imaging to measure extension rates. Axon tips exhibited minimal to no extension or retraction after 24 h of GIM treatment (Fig. 3D, leftmost dataset) and up to 48 h of GIM treatment (Fig. 3D, rightmost data set, and Fig. 3E) starting at 24 h after culturing as shown in Figure 3A. Consistent with prior work using rat embryonic sensory neurons (Press and Milbrandt, 2008), inhibition of glycolysis did not adversely impact the survival of dissociated neurons, even when starting at the beginning of culturing (Fig. 3F), although it prevented axon extension. Using the experimental design shown in Figure 3A, a 24 h treatment with heptelidic acid starting at 24 h of culturing similarly stalled axon extension without any signs of axon degeneration (Fig. 3G).
Figure 3.
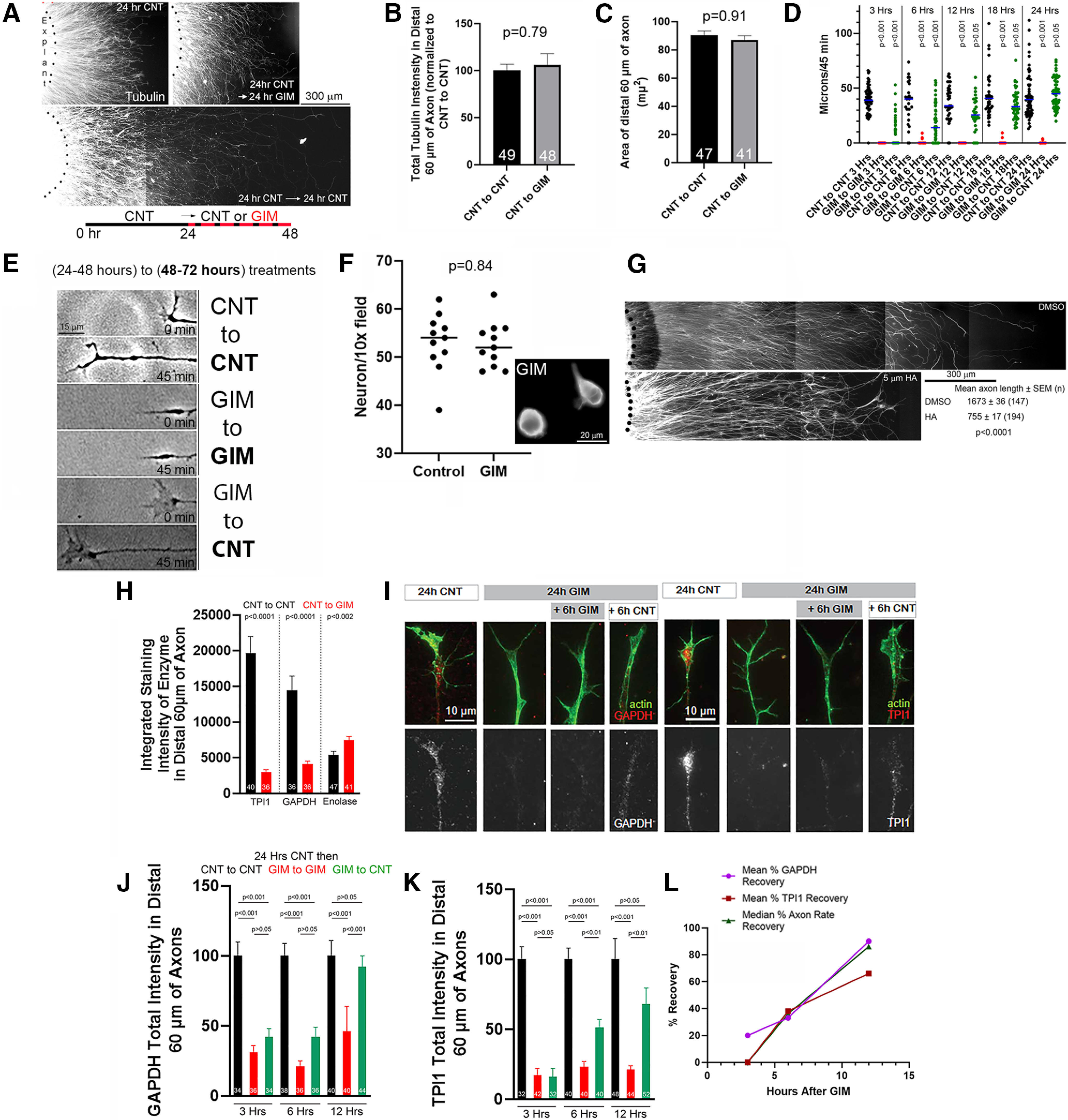
Long-term inhibition of glycolysis reversibly stalls axon extension. A, Examples of the extent of axon growth from the edges of sensory explants (black dots) cultured for varying time points under different experimental conditions (samples have been stained with anti-α-tubulin to reveal axons). Diagram below the images represents the basic experimental design used in the rest of similar experiments. After a 24 h culturing period, axons further cultured in control medium (CNT) for an additional 24 h continued to extend (p < 0.001 for 24 h compared with 48 h in control medium, Welch t test; n = 34 and n = 48, respectively). Those treated with GIM between 24 and 48 h of culturing remained the same length as at the time of initiation of GIM treatment (p = 0.54 for 24 h in control medium compared with 24 h in control medium followed by 24 h in GIM, Welch t test; n = 34 and n = 29, respectively). B, Measurement of tubulin intensity in axons cultured for 24 h, starting at 24 h of culturing as in A, in control medium or GIM. Welch t test. C, Measurement of the area of distal axons under the same conditions as in B. Welch t test. D, Measurements of axon extension using phase-contrast imaging (as in Fig. 2G,K) at various times after an initial culturing of explants following the design in A. CNT to CNT (black) means that, after the 24-48 h period in CNT, the culture was treated with CNT; GIM to GIM (red) means that, after the 24-48 h period in GIM, the culture was treated with GIM; GIM to CNT (green) means that, after the 24-48 h period in GIM, the culture was treated with CNT. Dunn's multiple comparisons relative to the CNT to CNT group in each of the 5 time categories representing 3-24 h of treatment starting at 48 h of culturing. Each data point represents one axon. E, Examples from the rightmost dataset in D (24 h treatments starting at 48 h in culture) showing axon extension and growth cone morphology in the three experimental groups at the final treatment time point (48-72 h total in culture). The treatment during the 24-48 h period is not bolded; the subsequent treatment between 48 and 72 h is shown in bold. F, Counts of dissociated neurons (Tuj-1-positive cells) per unit area cultured in control medium or GIM. Welch t test. Inset, Morphology (Tuj-1 stain) of neurons at 24 h of culturing in GIM. No evidence of membrane blebbing or nuclear disintegration is noted. G, Examples as in A of the extent of the growth of axons from explants at 48 h of culturing in control medium with 24-48 h being treated with DMSO or heptelidic acid (5 μm) treatment. The heptelidic acid-treated axons are approximately half the length of the DMSO group, indicating a stall of extension following treatment. H, Quantitative measurements of glycolytic enzymes in distal axons at 48 h as in A. Welch t tests. I, Examples of immunocytochemical staining of GAPDH and TPI1 from the datasets used in H and J, K. Counterstained to reveal actin filaments. J, Quantification, as in H, of the levels of GAPDH at 3-12 h of further GIM treatment or exchange to CNT starting at 48 h (as in D). Bonferroni multiple comparison tests within time point. K, Quantification, as in H, of the levels of TPI1 at 3-12 h of further GIM treatment or exchange to CNT starting at 48 h (as in D). Bonferroni multiple comparison tests within time point. L, Graphical representation of the relative recovery of axon extension rate and GAPDH and TPI1 levels between 3 and 12 h. Data from D, J, and K were used. The range of recovery was determined by using the GIM to GIM and CNT to CNT values to set the range (0%-100%, respectively). Mean ± SEM is shown in B,C,H,J,K and medians are shown in D,F.
Figure 6.
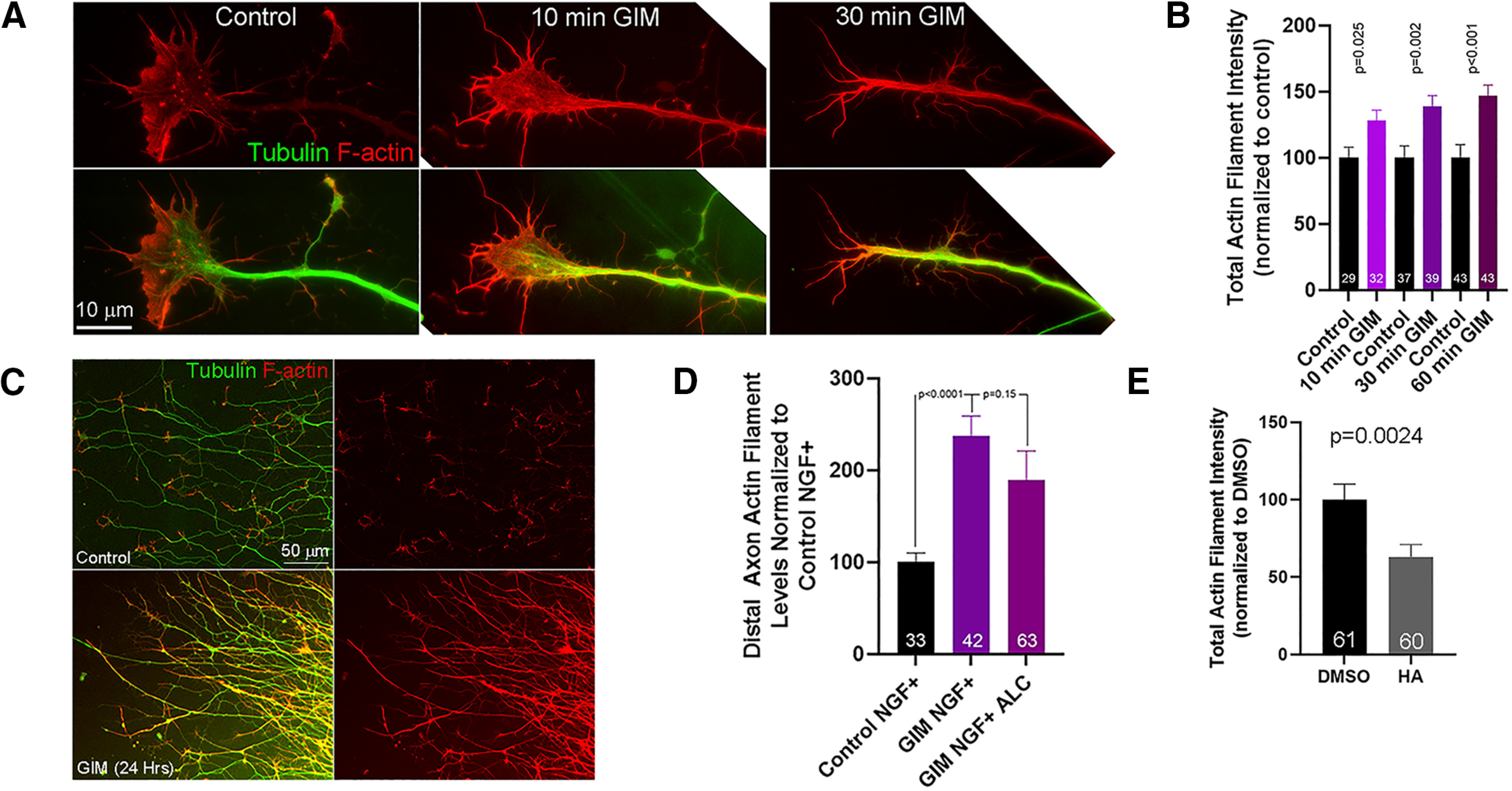
Inhibition of glycolysis impacts the axonal actin filament cytoskeleton. A, Examples of distal axons and growth cones labeled to reveal tubulin/microtubules and actin filaments (F-actin). The control growth cone exhibits the characteristic higher level of filaments in the lamellipodia relative to the central domain, and low levels along the axon shaft. In contrast, during the early response to GIM treatment (10 min), growth cones axons exhibit a relatively high level of actin filaments that are also found in the central domain. By 30 min of GIM treatment growth, cones have decreased in area. At both 10 and 30 min after GIM treatment, the actin filament content of the axon proximal to the growth cone is elevated. B, Quantification of the integrated intensity of phalloidin staining reflective of actin filaments in the distal 60 μm of axons, excluding growth cones (the distal most 10 μm if collapsed), as a function of time after GIM treatment and relative to time-matched control medium treatment groups. Welch t tests. C, Examples of distal axons emanating from explants initially cultured for 24 h in control medium and then an additional 24 h in either GIM or control medium (as in Fig. 3A) stained to revealed tubulin and actin filaments. The axons of the GIM-treated explant show high levels of actin filaments along their lengths, whereas the control axons show the expected local accumulation of filaments at growth cones (as in A). The GIM-treated axons appear more densely packed because they stopped extending on GIM treatment, whereas the control axons splayed out as they continued extending radially away from the explant. D, Quantification of actin filament levels in distal axons, as in B, for axons in the axonal chamber of microfluidic chambers (for representative examples of actin filament staining in these conditions, see Fig. 4C). Bonferroni multiple comparison tests. E, Quantification of actin filament levels in distal axons, as in B and D, following a 1 h treatment with 5 μm heptelidic acid (HA) or DMSO. Welch t test. Mean ± SEM is shown throughout.
Exchange of GIM with control medium after an initial 24 h GIM treatment (as in Fig. 3A reflective of 24-48 h of culturing) resulted in restoration of the rate axon extension by 12 h (Fig. 3D,E). The delayed recovery of the rate axon extension after prolonged GIM treatment and the reported dependency of glycolytic enzymes on glycolysis for their transport along axons (Zala et al., 2013; Hinckelmann et al., 2016) led us to consider whether there may be alterations in the levels of glycolytic enzymes in axons after prolonged inhibition of glycolysis. Although not adversely impacting axonal structure (Fig. 3A–C), the initial 24-48 h treatment with GIM resulted in a decrease in the levels of GAPDH and TPI1, but not enolase, in distal axons (Fig. 3H,I). The levels of GAPDH and TPI1 in distal axons increased as a function of time after replacing GIM with control medium (Fig. 3I–K). The delayed time course of the recovery of axon extension after GIM treatment (Fig. 3D) exhibited a similar time course to the restoration of the relative levels of GAPDH and TPI1 in axons (Fig. 3L). These data indicate that the delayed resumption of axon extension after returning axons to normal glucose-containing medium is because of the relative paucity of at least two required enzymes in the glycolytic pathway (GAPDH and TPI1) after the initial 24 h treatment with GIM.
Local inhibition of glycolysis inhibits axon extension and simplifies growth cones
Microfluidic chambers were used to locally expose distal axons and growth cones to GIM. Neuronal cell bodies were located in the cell body compartment, and axons grew through microgrooves until reaching the entry point into the distal chamber (Fig. 4A) as in our prior work (Sainath et al., 2017b). The length of axon extension into the distal chamber was measured from the entry point into the distal chamber as axons exited the grooves. Axons exhibited a 52% decrease in length when challenged with extension into the distal chamber containing GIM and NGF compared with control medium and NGF (Fig. 4A,B). In the absence of NGF, GIM largely blocked the entry of axons into the distal chamber (Fig. 4A). In the absence of NGF, 81% (n = 156) of grooves containing axons exhibited axons that entered the distal chamber in control medium in contrast to 25% (n = 100) of grooves when GIM was present in the distal chamber (p < 0.001, Fisher's exact test) and the few axons extended <100 μm. The surface area of the growth cones of axons that extended into the GIM- and NGF-containing chamber was decreased relative to that in control medium and NGF chambers (Fig. 4C,D). Allowing axons to enter the distal chamber in the presence of control medium and NGF and then exchanging the medium with GIM and NGF for 30 min decreased the surface area of growth cones and filopodia number relative to exchange with control medium and NGF (Fig. 4E,F).
Figure 4.
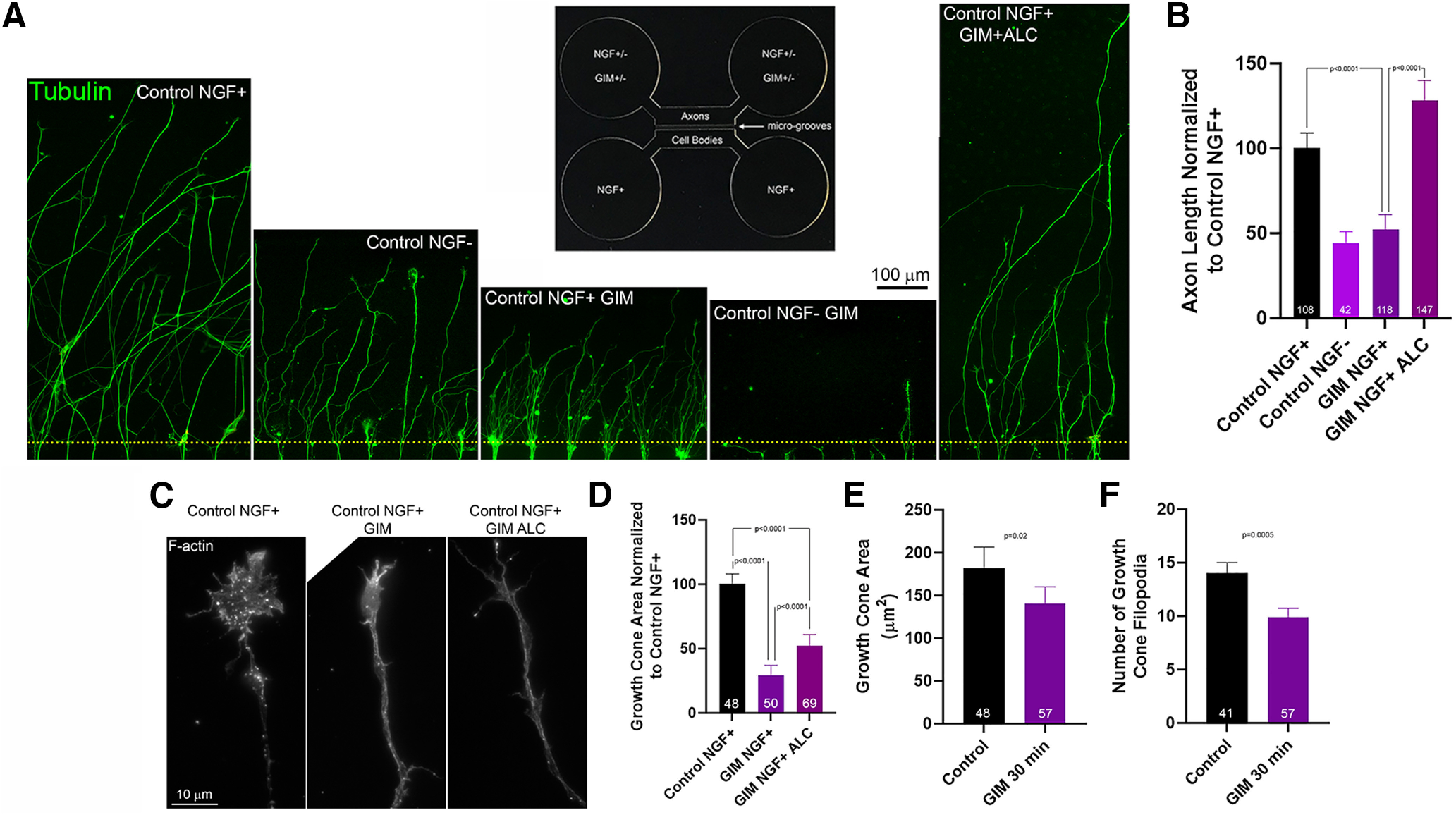
Inhibition of glycolysis in distal axons impairs axon extension in the presence of NGF and blocks it in the absence of NGF. A, Representative examples of axon extension to the axon chamber of microfluidic chambers. Inset, Design of the microfluidic chambers. Yellow dots represent the entry points into the axon chambers. B, Measurements of axon length in axon chambers. Bonferroni multiple comparisons tests. C, Examples of actin filaments (F-actin) in growth cones and distal axons in axon chambers. D, Growth cone measurement area in axon chambers as in A. Bonferroni multiple comparisons tests. Effect of a 30 min treatment with GIM or control medium in the distal chambers after axons had already grown into the chamber on growth cone area (E) and number of filopodia (F). Welch t tests. Mean ± SEM is shown throughout.
To determine whether the lower extension observed in GIM containing distal chambers relied on mitochondria respiration AA, an inhibitor of oxidative phosphorylation, was added to the GIM. The percentage of grooves in microfluidic chambers that contained axons that had arrived at the exit of the groove into the axonal compartment and extended beyond the exit point was 94% (n = 243) and 0% (n = 93) for GIM and GIM+AA chambers, respectively (p < 0.0001, Fisher's exact test). We have previously determined that inclusion of AA in the distal chamber blocks extension into the chamber in control medium with NGF (Sainath et al., 2017b) and consistently AA blocked entry into GIM and NGF containing distal chambers indicating the remaining axon extension in GIM is dependent on mitochondria respiration. Axons that entered the distal chamber in the presence of GIM did not differ in their density of mitochondria relative to those in control medium (assessed as in Armijo-Weingart et al., 2019) relative to controls (p = 0.425, Mann–Whitney test, n = 49 and n = 63 axons for control and GIM conditions, respectively). To address whether increasing mitochondrial respiration could impact the inhibitory effects of GIM, we considered the effects of including ALC, which promotes mitochondrial bioenergetic function and ATP generation (Rosca et al., 2009; Onofrj et al., 2013), in the distal chamber. We previously reported that, in control medium in the presence of NGF, as well as in the absence of NGF, ALC increases axon extension on permissive substrates and partially restores axon length on inhibitory substrates that decrease mitochondria membrane potential (Sainath et al., 2017b). ALC supplementation in GIM-containing distal chambers containing NGF restored axon extension to control levels (Fig. 4A,B), and ALC partially increased growth cone area relative to GIM alone (Fig. 4C,D).
Regulation of growth cone morphology by GAPDH knockdown and overexpression
GAPDH was knocked down through expression of shRNA using previously published sequences for chicken GAPDH (Fig. 5A; for details and efficacy, see Materials and Methods) (Billings et al., 2010). Expression of GAPDH-shRNA resulted in an increase in the number of collapsed growth cones (having 1 or less filopodia and no lamellipodia), decreased the percentage of growth cones exhibiting lamellipodial protrusions, and decreased the number of filopodia at growth cones (Fig. 5B,C). Rescue with an shRNA-resistant RFP-GAPDH construct reversed the effects of GAPDH knockdown on growth cone morphology (Fig. 5B-D). Overexpression of GAPDH decreased the percentage of collapsed growth cones, increased growth cone filopodia number and the percentage of growth cones with filopodia, but did not alter the percentage of growth cones exhibiting lamellae (Fig. 5E,F).
“Rescue and then kill” (RATK) of GAPDH in growth cones suppresses growth cone motility
KR is a fluorescent protein that on illumination generates reactive oxygen species that can inactivate immediately adjacent proteins in CALI experimental designs (Bulina et al., 2006). In order to manipulate glycolysis in a spatiotemporal manner, we reasoned that, if neurons with shRNA-mediated knockdown of GAPDH were rescued through expression of KR-GAPDH, we could then locally impair glycolysis through CALI of KR-GAPDH specifically in distal axons. This strategy was inspired by the prior use of KR-β1 integrin to rescue the effects of β1 loss in β1 KO cells followed by CALI of KR-β1 (Destaing et al., 2010). As in our prior work using mitochondrially targeted KR to ablate mitochondria in axon segments, and as indicated by the standard protocol for the use of KR (Bulina et al., 2006), we used rescue with RFP-GAPDH as a control for CALI. Based on preliminary studies, we found that 25 s illumination was sufficient to bleach >90% of the KR-GAPDH signal in distal axons, and this duration of illumination in the absence of KR or RFP expression did not adversely affect growth cone filopodial dynamics or area during the subsequent 10 min period (not shown). In all rescue experiments, GFP was expressed by the shRNA plasmid and used to track axon morphology and growth cone behavior. CALI of KR-GAPDH did not adversely affect the integrity of axonal shafts (Fig. 5G), indicating no widespread damage to microtubules as axons would then be expected to thin and undergo beading. CALI of KR-GAPDH, but not RFP-GAPDH (control), resulted in the cessation of filopodial and lamellipodial dynamics within 0-10 min after illumination (Fig. 5H–J) and the termination of the formation of new filopodia (Fig. 5K). Growth cones that exhibited lamellipodia before CALI of KR-GAPDH similarly lost protrusive activity as the lamellipodia contracted (Fig. 5H), and 31% of growth cones underwent retraction (Fig. 5H), similar to the effects of acute GIM and heptelidic acid treatment (Fig. 2G,K). CALI of RFP-GAPDH did not result in retraction by any of the growth cones.
Inhibition of glycolysis alters the axonal actin filament cytoskeleton
Growth cones exhibit higher levels of actin filaments in lamellipodia and lower levels in the central domain (Figs. 1A′, 6A). The initial termination of growth cone dynamics in response to GIM correlated with the loss of preferentially distally located lamellipodial meshworks of filaments and the development of a network of actin filaments throughout growth cones (Fig. 6A). Although growth cones subsequently decreased in size and lost protrusive actin filament based structures (lamellipodia and filopodia) in response to GIM treatment (Fig. 2; Fig. 6A, 30 min GIM), the axonal actin filament content proximal to and excluding the growth cone (see Materials and Methods) was increased as early as 10 min after GIM treatment (Fig. 6A,B) and up to 24 h of continuous GIM treatment starting at 24 h of culturing as in Figure 3A (Fig. 6C; also see actin filament stains in Fig. 3I). Sensory axons normally exhibit sparse actin filament levels characterized by focal puncta of actin filaments that serve as platforms for the formation of axonal filopodia (Gallo, 2011) (Fig. 6A) and submembranous actin filament rings (Unsain et al., 2018; G. Wang et al., 2019), the latter not detectable without the use of super resolution microscopy. In contrast, GIM-treated axons exhibited what appeared to be longitudinal bundles of actin filaments (Fig. 6A) qualitatively similar to those we previously described forming in response to semaphorin-3A treatment (Gallo, 2006). Axons growing into the axonal compartment of microfluidic chambers containing NGF with GIM similarly showed increased axonal actin filament content compared with axons in control medium with NGF (Fig. 6D; for examples of actin filament staining, see also Fig. 4C), indicating the effect is local. Supplementation of GIM with ALC, as in Figure 4, did not impact the increase in axonal actin levels induced by GIM treatment (Fig. 6D; for examples of actin filament staining, see also Fig. 4C). Treatment with heptelidic acid did not increase the axonal actin filament content (Fig. 6E; for an example of actin filaments staining pattern, see also Fig. 2H), indicating the effect of GIM is before GAPDH in the glycolytic pathway. In contrast, and as expected for partial ATP depletion because of inhibition of the first enzyme in the payoff phase of glycolysis, inhibition of GAPDH partially decreased axonal actin filament content.
Inhibition of glycolysis in the absence of NGF results in the degeneration of axons but not cell death
As described previously, inhibition of glycolysis using GIM for up to 24 h in the presence of NGF did not adversely affect axons or result in cell death, consistent with a prior report also noting the absence of cell death in response to inhibition of glycolysis using sensory neurons in the presence of NGF (Press and Milbrandt, 2008). The observation that in microfluidic chambers axons failed to enter the axon chamber when GIM was present in the absence of NGF (Fig. 4A) led us to consider whether inhibition of glycolysis in the absence of NGF might differently affect axons. Using the same experimental protocol as in Figure 3A, in the absence of NGF a 24 h GIM treatment starting at 24 h of culturing resulted in the degeneration of axons (Fig. 7A). Degenerated axons were evident as “beads on a string” profiles when stained with tubulin antibodies (Fig. 7A,A′). Scholl analysis of the number of axons that did not have a degenerative morphology revealed that as expected between 24 and 48 h of culturing in control medium axon continued to extend and also increased in number as more emanated from the explants relative to the 24 h time point (Fig. 7B), although branching may also contribute to this metric. In contrast, GIM treatment between 24 and 48 h resulted in a decrease in the number of healthy axons relative to the 24 h time point, consistent with the degeneration of the axon population initially present at 24 h (Fig. 7B). An increase in the proportion of degenerating axons became detectable following the exchange to GIM after an initial 24 h period of culturing, although at minor levels, as early as after 30 min of GIM treatment (Fig. 7C). A concentration response curve for the protective effect of NGF against GIM treatment induced axon degeneration showed protective effects starting at 0.05 ng/ml NGF and saturating at 10 ng/ml (Fig. 7D), consistent with the involvement of the high-affinity TrkA receptor (Kaplan et al., 1991). In contrast to the ensuing axon degeneration, in the absence of NGF GIM unexpectedly increased the number of surviving dissociated neurons after 24 h of culturing relative to control medium without affecting the numbers of non-neuronal cells in the cultures (reflective of mixture of fibroblasts and Schwann cells) (Fig. 7E).
Figure 7.
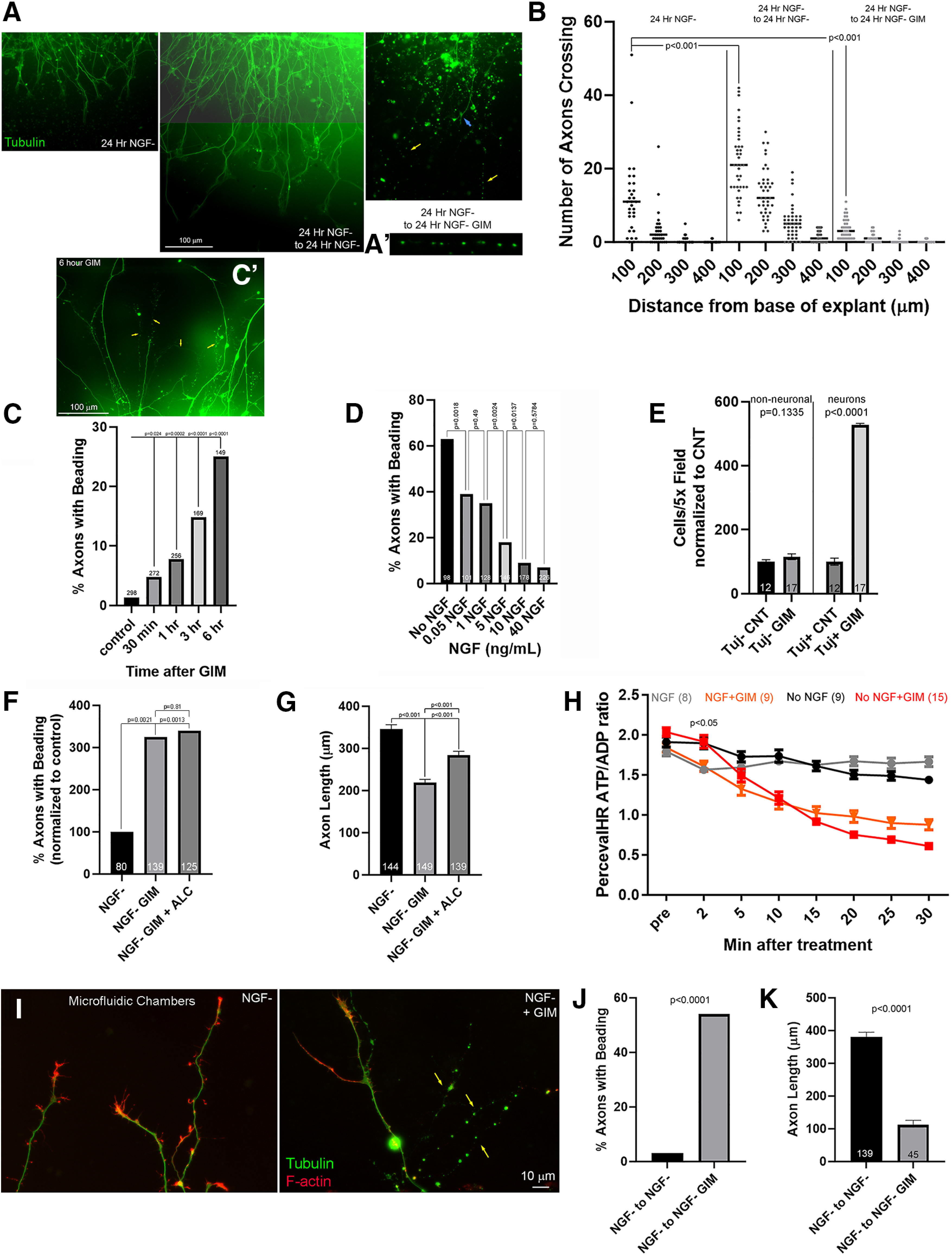
In the absence of NGF, inhibition of glycolysis results in axon degeneration independent of cell death. A, Examples of axons growing from explants fixed and stained after 24 h in no NGF control medium, 24 h in no NGF control medium followed by another 24 h in no NGF control medium, and 24 h in no NGF control medium followed by an additional 24 h in no NGF GIM. Yellow arrows in the latter group point to degenerated axons characterized by sequences of beads. A′, Higher-magnification example of the degenerated axon labeled by the rightmost arrow in the panel above. B, Scholl analysis of the number of healthy, not beaded, axons crossing lines at 100-400 μm from the base of the explants. The groups are in the same left to right order as in A. Dunn multiple comparison tests for analysis of the most proximal (100 μm) crossing to the base of the axons. C, Quantification of the percentage of axons exhibiting beading following a 30 min to 6 h treatment with GIM. Control reflects a 3 h treatment with control medium. Fisher's exact tests. C′, Example of 6 h treatment with GIM, annotated as in A. D, Quantification of the percentage of axons exhibiting beading following a 24 h treatment with GIM starting at 24 h in culture (as in A) but raised initially in the various concentrations of NGF and maintained at such concentrations during the 24-48 h treatment with GIM. Fisher's exact tests. E, Counts of neurons (Tuj+) and non-neuronal cells (Tuj–) in dissociated sensory ganglion cultures raised overnight in no NGF in control or GIM medium. Welch t tests. F, Quantification of the percentage of degenerating axons in no NGF conditions with GIM treatment with or without ALC supplementation. In all groups, explants were cultured for 24 h in control medium with no NGF before exchange with control medium, GIM, or GIM and ALC for an additional 24 h. Fisher's exact tests. G, Same experimental design as in F, but measuring the lengths of nondegenerating axons. Bonferroni multiple comparisons tests. H, Measurements of ATP/ADP ratio using PercevalHR, as in Figure 2A, in NGF and no NGF culturing conditions. The ratio after treatment with GIM was analyzed within time point across the NGF and no NGF groups using Bonferroni post hoc tests. A difference at p < 0.05 was detected only at 2 min after GIM treatment. The NGF condition group is replicated from Figure 2A. I, Examples of axons in axon chambers of microfluidic chambers. All were initially cultured in no NGF and then treated for 24 h with no NGF GIM or no NGF control medium before fixing and staining. Yellow arrows point to degenerating axons exhibiting beading. J, Quantification of the percentage of axons exhibiting beading following from the experiments shown in G. Fisher's exact test. K, Length of axons not exhibiting beading under the same conditions as in G and H. Welch t tests. Medians are shown in B and Mean ± SEM is shown in E,G,H,K.
Treatment with GIM containing ALC in the absence of NGF, for 24 h after an initial 24 h of culturing in control medium lacking NGF, did not decrease the percentage of degenerating axons (Fig. 7F). However, ALC treatment increased the length of nondegenerating axons albeit to ∼50% of the length increase observed in the control group (Fig. 7G). These observations indicate that promoting mitochondrial oxidative phosphorylation is not sufficient to decrease axon degeneration induced by GIM in the absence of NGF, although it partially restores the extension of nondegenerating axons consistent with the extension promoting effects of ALC in NGF and GIM treatment conditions (Fig. 4A,B) and our prior demonstration that ALC increases axon extension in the presence or absence of NGF (Sainath et al., 2017a).
The overall effects of treatment with GIM on the decline in the growth cone's ATP/ADP ratio were not different as a function of NGF (Fig. 7H). GIM treatment decreased the ratio equally during the first 30 min of treatment in both NGF and no NGF culturing conditions, with the only difference observed at 2 min after treatment at which point the ratio declined more in the presence of NGF than its absence. These data indicate that the ensuing axon degeneration is not dependent on a different effect of GIM treatment on the ATP/ADP ratio and are generally consistent with the failure to rescue the degeneration via ALC supplementation.
We next used microfluidic chambers and allowed axons to enter the axon compartment before exchanging medium with no NGF medium or no NGF medium and GIM for 24 h. Under these conditions, GIM treatment resulted in the presence of degenerating axons profiles (Fig. 7I,J). The length of axons that did not degenerate in the axon chamber following GIM treatment was 76% shorter than controls in no NGF medium that had continued extending during the 24 h treatment period (Fig. 7K), indicating a stall of extension. Collectively, the data indicate that axonal glycolysis is required locally for the maintenance of axons in the absence of NGF.
Discussion
The data presented herein unveil a requirement for glycolysis in distal axons in the regulation of embryonic growth cone dynamics and axon extension. In the presence of NGF, inhibition of glycolysis throughout the whole neuron did not exhibit cytotoxicity or axonal toxicity for a period of up to 48 h but stalled axon extension, and local inhibition of glycolysis in distal axons attenuated axon extension by ∼50%. In the absence of NGF, inhibition of glycolysis in distal axons resulted in axon degeneration without however impacting neuron survival.
Glycolysis powers the fast axonal transport of some vesicular cargoes but not mitochondria (Spillane et al., 2013; Zala et al., 2013; Hinckelmann et al., 2016). The difference between spatially homogeneous and local inhibition of glycolysis in distal axons alone on axon extension (full and partial, respectively) may reflect the absence of an effect on fast transport until the axons reach the distal chambers in microfluidic chambers. Under homogeneous inhibition, we observed a decline in the levels of some axonal glycolytic enzymes, which may reflect a failure to maintain transport of the glycolytic apparatus in axons with ensuing degradation of the enzymes in situ. The mechanism for the decline in enzyme levels will require further consideration. The restoration of axon extension after prolonged inhibition of glycolysis correlated with the return of glycolytic enzymes in axons, consistent with the notion that active glycolysis is required for axon extension even if glucose is readily available.
In hippocampal neurons, there is a developmental transition between the embryonic to postnatal period from glycolysis to mitochondrial respiration as the main source of ATP production (Surin et al., 2013), and glycolysis has been reported to contribute to synapse function in established networks (Ashrafi and Ryan, 2017). Although mitochondria respiration is an important aspect of embryonic axon extension and adult regeneration (Sheng, 2017; Smith and Gallo, 2018), whether glycolysis may contribute to regenerative axon extension in adult neurons remains to be considered. In this context, it will be of interest to investigate the expression and function of glycolytic enzymes in regenerating neurons given that injured neurons can undergo transcriptional changes indicative of an attempt to revert to an embryonic mode of axon extension (Hilton and Bradke, 2017; Lee and Cho, 2020).
Actin is an ATPase and requires ATP loading for polymerization. It is thus surprising that inhibition of glycolysis using GIM to deprive of glucose and inhibit glycolysis with 2-deoxyglucose results in an increase in axonal actin filaments while growth cones undergo simplification and contraction and eventually exhibit decreased filament content. Although ALC treatment restored axon extension in GIM, it did not impact the increase in axonal actin filament levels. This observation indicates that the effect of glycolysis inhibition on axonal actin levels cannot be rescued by increased mitochondrial ATP production. The increase in axonal actin filaments was also not observed in response to pharmacological inhibition of GAPDH, the first enzyme in the ATP-generating payoff phase. In contrast, inhibition of GAPDH resulted in the expected decrease in actin filament content because of decreased ATP availability. Collectively, the data indicate that the mechanism driving the formation of axonal actin filaments is upstream of GAPDH and does not involve changes in ATP availability, suggesting that glycolytic flux in the preparatory phase regulates axonal actin filaments. Glycolytic enzymes can have roles other than the generation of ATP, including binding to and regulating the cytoskeleton (Snaebjornsson and Schulze, 2018), possibly underlying the observed effects on axonal actin filaments. We have previously described that the formation of axonal actin bundles, which qualitatively are not different from those detailed herein, is downstream of RhoA-ROCK activation (Gallo, 2006; Orlova et al., 2007). Whether inhibition of glycolysis may be activating the RhoA-ROCK signaling axis and promoting myosin II activity remains to be determined, but such a hypothetical mechanism is consistent with the inhibition of axon extension, simplification of growth cones, and the formation of axonal actin bundles at the expense of growth cone dynamics and axon formation. Regardless, the initial formation of the filament bundles may persist for as long as determined (at least 24 h) because of decreased ATP levels as a consequence of conjoined suppression of the payoff phase that may in turn decrease filament turnover.
In the presence of NGF, the restoration of axon extension by increasing mitochondrial respiration through treatment of distal axons with ALC under conditions of GIM treatment in microfluidic chambers argues that the decrease in axon extension is a consequence of decreases in net axonal ATP levels. Consistent with this idea, inhibition of glycolysis and oxidative phosphorylation each partially decreased the ATP/ADP ratio in distal axons, and GIM treatment of isolated distal axons partially decreased axon extension that was fully blocked by additional inhibition of oxidative phosphorylation. The results of the RATK experiments and pharmacological inhibition of GAPDH indicate that growth cone dynamics and overall morphology are supported by elements of the glycolytic pathway at the level of GAPDH or downstream thereof. As GAPDH is the first step in the ATP-generating payoff phase of glycolysis, ATP production may be the mechanistic contribution of glycolysis to growth cone dynamics. This interpretation is also supported by the partial restoration of growth cone morphology observed in the microfluidic chamber experiments involving ALC supplementation to promote mitochondrial oxidative phosphorylation. In conclusion, this report identifies the payoff phase of the glycolytic pathway as operative in distal axons and indicates that it contributes to the rate of axon extension and growth cone dynamics. The specific roles of ATP derived from the payoff phase in the various aspects of the mechanism of axon extension remain to be determined.
Consistent with a prior report showing that, in NGF, embryonic sensory axons do not undergo degeneration on inhibition of glycolysis using 2-deoxyglucose (Press and Milbrandt, 2008), in our studies neither GIM nor pharmacological inhibition of GAPDH resulted in axon degeneration in the presence of NGF. However, in the absence of NGF, inhibition of glycolysis resulted in axon degeneration. The mechanism responsible for the ensuing degeneration remains to be addressed. Intra-axonal protein synthesis may contribute to axon health; and, in its absence, axons could become more susceptible to degeneration (Kim and Jung, 2020). NGF drives intra-axonal protein synthesis in the sensory axon population used in this study (Spillane et al., 2012, 2013), and it will be of interest to address whether the protective effect of NGF is mediated by local protein synthesis. At face value, the form of axon degeneration noted here is similar to the Wallerian degeneration of distal axon segments after axotomy. ATP levels are known to decline during Wallerian degeneration, although a causative role for the decline in the degeneration is not clear (Conforti et al., 2014). Both glycolysis and oxidative phosphorylation are decreased during Wallerian degeneration, and rescue of degeneration by Sarm1 KO more strongly attenuated the decrease in glycolytic ATP output (Godzik and Coleman, 2015). It remains to be addressed whether degeneration in response to inhibition of glycolysis in the absence of NGF operates through the same mechanism as Wallerian degeneration. The susceptibility to inhibition of glycolysis in the absence of NGF may be linked to lower axonal mitochondrial function in the absence of NGF (Verburg and Hollenbeck, 2008). Finally, sensory axons also exhibit a submembranous actin filament ring cytoskeleton considered to maintain axonal integrity through its association with spectrin and ankyrin that is also dependent for its maintenance on NGF signaling (Unsain et al., 2018; G. Wang et al., 2019). Disruption of this submembranous cytoskeleton disposes axons to degeneration (Hammarlund et al., 2007; C. Y. Huang et al., 2017; Unsain et al., 2018; G. Wang et al., 2019), suggesting that NGF may at least in part protect axons from degeneration through the maintenance of the submembranous cytoskeleton. Insulin acting through its tyrosine receptor kinase increases the activity of the first kinase in glycolysis (hexokinase) through the PI3K pathway (T. J. Huang et al., 2005; Arboleda et al., 2007; Z. Wang et al., 2008). This raises the possibility that NGF could also promote glycolytic activity through Trk-mediated PI3K signaling, but this issue remains to be addressed, and such an effect of NGF would not impact the response to GIM, which blocks glycolysis through glucose deprivation and 2-deoxyglucose.
In conclusion, this study paves the way for further analysis of the role of glycolysis in axon morphogenesis, branching, guidance, and the neuronal cytoskeleton. Whether glycolysis may have a role in axon regeneration, be it in developmental or adult contexts, remains to be determined. The cytoskeletal and signaling mechanisms underlying axon extension that are impacted by glycolysis will need specific analysis. Considering the similarities in the effects of, and signaling pathways used by, the multiple members of the neurotrophin family, it seems likely that the axon degeneration protective effects of NGF described here are not specific to NGF. The detection of this form of axon degeneration may be of significance for the understanding of axonopathies in hypoglycemic states (Mohseni, 2014) and in diabetic neuropathy because of decreased expression of NGF in the skin (Tang et al., 2019).
Footnotes
This work was supported by National Institutes of Health Grant R21NS118000 to G.G. We thank Dr. Almudena Pacheco for technical assistance.
The authors declare no competing financial interests.
References
- Agostini M, Romeo F, Inoue S, Niklison-Chirou MV, Elia AJ, Dinsdale D, Morone N, Knight RA, Mak TW, Melino G (2016) Metabolic reprogramming during neuronal differentiation. Cell Death Differ 23:1502–1514. 10.1038/cdd.2016.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arboleda G, Huang TJ, Waters C, Verkhratsky A, Fernyhough P, Gibson RM (2007) Insulin-like growth factor-1-dependent maintenance of neuronal metabolism through the phosphatidylinositol 3-kinase-Akt pathway is inhibited by C2-ceramide in CAD cells. Eur J Neurosci 25:3030–3038. 10.1111/j.1460-9568.2007.05557.x [DOI] [PubMed] [Google Scholar]
- Armijo-Weingart L, Ketschek A, Sainath R, Pacheco A, Smith GM, Gallo G (2019) Neurotrophins induce fission of mitochondria along embryonic sensory axons. Elife 8:e49494. 10.7554/eLife.49494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Ryan TA (2017) Glucose metabolism in nerve terminals. Curr Opin Neurobiol 45:156–161. 10.1016/j.conb.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BW, Bamburg JR (2003) Actin-ATP hydrolysis is a major energy drain for neurons. J Neurosci 23:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings NA, Emerson MM, Cepko CL (2010) Analysis of thyroid response element activity during retinal development. PLoS One 5:e13739. 10.1371/journal.pone.0013739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordone MP, Salman MM, Titus HE, Amini E, Andersen JV, Chakraborti B, Diuba AV, Dubouskaya TG, Ehrke E, Espindola de Freitas A, Braga de Freitas G, Gonçalves RA, Gupta D, Gupta R, Ha SR, Hemming IA, Jaggar M, Jakobsen E, Kumari P, Lakkappa N, et al. (2019) The energetic brain: a review from students to students. J Neurochem 151:139–165. 10.1111/jnc.14829 [DOI] [PubMed] [Google Scholar]
- Bulina ME, Lukyanov KA, Britanova OV, Onichtchouk D, Lukyanov S, Chudakov DM (2006) Chromophore-assisted light inactivation (CALI) using the phototoxic fluorescent protein KillerRed. Nat Protoc 1:947–953. 10.1038/nprot.2006.89 [DOI] [PubMed] [Google Scholar]
- Conforti L, Gilley J, Coleman MP (2014) Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci 15:394–409. 10.1038/nrn3680 [DOI] [PubMed] [Google Scholar]
- Dent EW, Gertler FB (2003) Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron 40:209–227. 10.1016/s0896-6273(03)00633-0 [DOI] [PubMed] [Google Scholar]
- Destaing O, Planus E, Bouvard D, Oddou C, Badowski C, Bossy V, Raducanu A, Fourcade B, Albiges-Rizo C, Block MR (2010) β1A integrin is a master regulator of invadosome organization and function. Mol Biol Cell 21:4108–4119. 10.1091/mbc.E10-07-0580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWane G, Salvi AM, DeMali KA (2021) Fueling the cytoskeleton: links between cell metabolism and actin remodeling. J Cell Sci 134:jcs248385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo A, Hasumi K, Sakai K, Kanbe T (1985) Specific inhibition of glyceraldehyde-3-phosphate dehydrogenase by koningic acid (heptelidic acid). J Antibiot (Tokyo) 38:920–925. 10.7164/antibiotics.38.920 [DOI] [PubMed] [Google Scholar]
- Gallo G (2006) RhoA-kinase coordinates F-actin organization and myosin II activity during semaphorin-3A-induced axon retraction. J Cell Sci 119:3413–3423. 10.1242/jcs.03084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo G (2011) The cytoskeletal and signaling mechanisms of axon collateral branching. Dev Neurobiol 71:201–220. 10.1002/dneu.20852 [DOI] [PubMed] [Google Scholar]
- Gallo G (2020) The bioenergetics of neuronal morphogenesis and regeneration: frontiers beyond the mitochondrion. Dev Neurobiol 80:263–276. 10.1002/dneu.22776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godzik K, Coleman MP (2015) The axon-protective WLD(S) protein partially rescues mitochondrial respiration and glycolysis after axonal injury. J Mol Neurosci 55:865–871. 10.1007/s12031-014-0440-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Jorgensen EM, Bastiani MJ (2007) Axons break in animals lacking beta-spectrin. J Cell Biol 176:269–275. 10.1083/jcb.200611117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q, Xie Y, Ordaz JD, Huh AJ, Huang N, Wu W, Liu N, Chamberlain KA, Sheng ZH, Xu XM (2020) Restoring cellular energetics promotes axonal regeneration and functional recovery after spinal cord injury. Cell Metab 31:623–641. 10.1016/j.cmet.2020.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton BJ, Bradke F (2017) Can injured adult CNS axons regenerate by recapitulating development? Development 144:3417–3429. 10.1242/dev.148312 [DOI] [PubMed] [Google Scholar]
- Hinckelmann MV, Virlogeux A, Niehage C, Poujol C, Choquet D, Hoflack B, Zala D, Saudou F (2016) Self-propelling vesicles define glycolysis as the minimal energy machinery for neuronal transport. Nat Commun 7:13233. 10.1038/ncomms13233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Zhang C, Zollinger DR, Leterrier C, Rasband MN (2017) An alphaII spectrin-based cytoskeleton protects large-diameter myelinated axons from degeneration. J Neurosci 37:11323–11334. 10.1523/JNEUROSCI.2113-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TJ, Verkhratsky A, Fernyhough P (2005) Insulin enhances mitochondrial inner membrane potential and increases ATP levels through phosphoinositide 3-kinase in adult sensory neurons. Mol Cell Neurosci 28:42–54. 10.1016/j.mcn.2004.08.009 [DOI] [PubMed] [Google Scholar]
- Jang S, Nelson JC, Bend EG, Rodríguez-Laureano L, Tueros FG, Cartagenova L, Underwood K, Jorgensen EM, Colón-Ramos DA (2016) Glycolytic enzymes localize to synapses under energy stress to support synaptic function. Neuron 90:278–291. 10.1016/j.neuron.2016.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Martin-Zanca D, Parada LF (1991) Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature 350:158–160. 10.1038/350158a0 [DOI] [PubMed] [Google Scholar]
- Kato M, Sakai K, Endo A (1992) Koningic acid (heptelidic acid) inhibition of glyceraldehyde-3-phosphate dehydrogenases from various sources. Biochim Biophys Acta 1120:113–116. 10.1016/0167-4838(92)90431-c [DOI] [PubMed] [Google Scholar]
- Kim E, Jung H (2020) Local mRNA translation in long-term maintenance of axon health and function. Curr Opin Neurobiol 63:15–22. 10.1016/j.conb.2020.01.006 [DOI] [PubMed] [Google Scholar]
- Kumagai S, Narasaki R, Hasumi K (2008) Glucose-dependent active ATP depletion by koningic acid kills high-glycolytic cells. Biochem Biophys Res Commun 365:362–368. 10.1016/j.bbrc.2007.10.199 [DOI] [PubMed] [Google Scholar]
- Lee J, Cho Y (2020) Comparative gene expression profiling reveals the mechanisms of axon regeneration. FEBS J. Advance online publication. Retrieved November 28, 2020. doi: 10.1111/febs.15646. [DOI] [PubMed] [Google Scholar]
- Lelkes PI, Unsworth BR, Saporta S, Cameron DF, Gallo G (2005) Culture of neuroendocrine and neuronal cells for tissue engineering. In: Culture of cells of tissue engineering (Freshney RI, ed), Chapter 14. Hoboken, NJ: Wiley. [Google Scholar]
- Markham A, Cameron I, Bains R, Franklin P, Kiss JP, Schwendimann L, Gressens P, Spedding M (2012) Brain-derived neurotrophic factor-mediated effects on mitochondrial respiratory coupling and neuroprotection share the same molecular signalling pathways. Eur J Neurosci 35:366–374. 10.1111/j.1460-9568.2011.07965.x [DOI] [PubMed] [Google Scholar]
- Mergenthaler P, Lindauer U, Dienel GA, Meisel A (2013) Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 36:587–597. 10.1016/j.tins.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohseni S (2014) Neurologic damage in hypoglycemia. Handb Clin Neurol 126:513–532. 10.1016/B978-0-444-53480-4.00036-9 [DOI] [PubMed] [Google Scholar]
- Onofrj M, Ciccocioppo F, Varanese S, di Muzio A, Calvani M, Chiechio S, Osio M, Thomas A (2013) Acetyl-L-carnitine: from a biological curiosity to a drug for the peripheral nervous system and beyond. Expert Rev Neurother 13:925–936. 10.1586/14737175.2013.814930 [DOI] [PubMed] [Google Scholar]
- Orlova I, Silver L, Gallo G (2007) Regulation of actomyosin contractility by PI3K in sensory axons. Dev Neurobiol 67:1843–1851. 10.1002/dneu.20558 [DOI] [PubMed] [Google Scholar]
- Pacelli C, Giguère N, Bourque MJ, Lévesque M, Slack RS, Trudeau LÉ (2015) Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr Biol 25:2349–2360. 10.1016/j.cub.2015.07.050 [DOI] [PubMed] [Google Scholar]
- Pacheco A, Merianda TT, Twiss JL, Gallo G (2020) Mechanism and role of the intra-axonal calreticulin translation in response to axonal injury. Exp Neurol 323:113072. 10.1016/j.expneurol.2019.113072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press C, Milbrandt J (2008) Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J Neurosci 28:4861–4871. 10.1523/JNEUROSCI.0525-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca MG, Lemieux H, Hoppel CL (2009) Mitochondria in the elderly: is acetylcarnitine a rejuvenator? Adv Drug Deliv Rev 61:1332–1342. 10.1016/j.addr.2009.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainath R, Gallo G (2015) The dynein inhibitor Ciliobrevin D inhibits the bidirectional transport of organelles along sensory axons and impairs NGF-mediated regulation of growth cones and axon branches. Dev Neurobiol 75:757–777. 10.1002/dneu.22246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainath R, Ketschek A, Grandi L, Gallo G (2017a) CSPGs inhibit axon branching by impairing mitochondria-dependent regulation of actin dynamics and axonal translation. Dev Neurobiol 77:454–473. 10.1002/dneu.22420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainath R, Armijo-Weingart L, Ketscheck A, Xu Z, Li S, Gallo G (2017b) Chondroitin sulfate proteoglycans negatively regulate the positioning of mitochondria and endoplasmic reticulum to distal axons. Dev Neurobiol 77:1351–1370. 10.1002/dneu.22535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Hasumi K, Endo A (1988) Inactivation of rabbit muscle glyceraldehyde-3-phosphate dehydrogenase by koningic acid. Biochim Biophys Acta 952:297–303. 10.1016/0167-4838(88)90130-6 [DOI] [PubMed] [Google Scholar]
- Sheng ZH (2017) The interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol 27:403–416. 10.1016/j.tcb.2017.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver L, Michael JV, Goldfinger LE, Gallo G (2014) Activation of PI3K and R-Ras signaling promotes the extension of sensory axons on inhibitory chondroitin sulfate proteoglycans. Dev Neurobiol 74:918–933. 10.1002/dneu.22174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GM, Gallo G (2018) The role of mitochondria in axon development and regeneration. Dev Neurobiol 78:221–237. 10.1002/dneu.22546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snaebjornsson MT, Schulze A (2018) Non-canonical functions of enzymes facilitate cross-talk between cell metabolic and regulatory pathways. Exp Mol Med 50:1–16. 10.1038/s12276-018-0065-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillane M, Ketschek A, Donnelly CJ, Pacheco A, Twiss JL, Gallo G (2012) Nerve growth factor-induced formation of axonal filopodia and collateral branches involves the intra-axonal synthesis of regulators of the actin-nucleating Arp2/3 complex. J Neurosci 32:17671–17689. 10.1523/JNEUROSCI.1079-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillane M, Ketschek A, Merianda TT, Twiss JL, Gallo G (2013) Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep 5:1564–1575. 10.1016/j.celrep.2013.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steketee MB, Moysidis SN, Weinstein JE, Kreymerman A, Silva JP, Iqbal S, Goldberg JL (2012) Mitochondrial dynamics regulate growth cone motility, guidance, and neurite growth rate in perinatal retinal ganglion cells in vitro. Invest Ophthalmol Vis Sci 53:7402–7411. 10.1167/iovs.12-10298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B, Ji YS, Sun XL, Liu XH, Chen ZY (2014) Brain-derived neurotrophic factor (BDNF)-induced mitochondrial motility arrest and presynaptic docking contribute to BDNF-enhanced synaptic transmission. J Biol Chem 289:1213–1226. 10.1074/jbc.M113.526129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surin AM, Khiroug S, Gorbacheva LR, Khodorov BI, Pinelis VG, Khiroug L (2013) Comparative analysis of cytosolic and mitochondrial ATP synthesis in embryonic and postnatal hippocampal neuronal cultures. Front Mol Neurosci 5:102. 10.3389/fnmol.2012.00102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HY, Jiang AJ, Ma JL, Wang FJ, Shen GM (2019) Understanding the signaling pathways related to the mechanism and treatment of diabetic peripheral neuropathy. Endocrinology 160:2119–2127. 10.1210/en.2019-00311 [DOI] [PubMed] [Google Scholar]
- Tantama M, Martínez-François JR, Mongeon R, Yellen G (2013) Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun 4:2550. 10.1038/ncomms3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsain N, Bordenave MD, Martinez GF, Jalil S, von Bilderling C, Barabas FM, Masullo LA, Johnstone AD, Barker PA, Bisbal M, Stefani FD, Cáceres AO (2018) Remodeling of the actin/spectrin membrane-associated periodic skeleton, growth cone collapse and F-actin decrease during axonal degeneration. Sci Rep 8:3007. 10.1038/s41598-018-21232-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verburg J, Hollenbeck PJ (2008) Mitochondrial membrane potential in axons increases with local nerve growth factor or semaphorin signaling. J Neurosci 28:8306–8315. 10.1523/JNEUROSCI.2614-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Simon DJ, Wu Z, Belsky DM, Heller E, O'Rourke MK, Hertz NT, Molina H, Zhong G, Tessier-Lavigne M, Zhuang X (2019) Structural plasticity of actin-spectrin membrane skeleton and functional role of actin and spectrin in axon degeneration. Elife 8:e38730. 10.7554/eLife.38730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gardiner NJ, Fernyhough P (2008) Blockade of hexokinase activity and binding to mitochondria inhibits neurite outgrowth in cultured adult rat sensory neurons. Neurosci Lett 434:6–11. 10.1016/j.neulet.2008.01.057 [DOI] [PubMed] [Google Scholar]
- Zala D, Hinckelmann MV, Yu H, Lyra da Cunha MM, Liot G, Cordelières FP, Marco S, Saudou F (2013) Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 152:479–491. 10.1016/j.cell.2012.12.029 [DOI] [PubMed] [Google Scholar]