

Abstract
The aryl hydrocarbon receptor (AHR) is expressed in a variety of skin cell types and contributes to skin homeostasis. Therapeutic targeting of the AHR in dermatology was first described for the treatment of inflammatory skin diseases using coal tar ointment. In addition to therapies involving active ligands like coal tar and Tapinarof, inhibition of AHR-dependent enzymatic activities in skin may be an alternative approach to resolving skin inflammation.
Introduction
Prior to 10 years ago, the aryl hydrocarbon receptor (AHR), a ligand-activated transcription factor, was best known for its role is mediating toxicity and carcinogenesis through the binding of environmental pollutants and xenobiotics. Recently, this promiscuous environmental sensor has gained tremendous attention as a modulator of immune cell development and immune tolerance in a tissue dependent manner. In the fields of skin biology and dermatology, research into AHR signaling is booming. Multiple studies describe the efficacy of exogenous ligands to induce terminal differentiation, increase expression levels of skin barrier-related and antimicrobial proteins, and thus contribute to skin barrier function. AHR activation also interferes with cytokine-induced or T-cell mediated inflammatory signaling pathways (e.g. via JAK/STAT)(Furue, 2020). These anti-inflammatory effects lead to reduced symptoms in experimental models of psoriasis and atopic dermatitis, similar to the well-known therapeutic efficacy and molecular mechanism of action of coal tar in the treatment of these chronic inflammatory skin diseases (van den Bogaard et al., 2013). It is important to keep in mind that improvement of epithelial barrier function would result in a dampening of inflammation and, thus, these two proposed functions of the AHR in the skin would be linked. The accumulating body of evidence on the role of AHR signaling in skin led to the investigation and screening of potential therapeutic AHR ligands, yielding Tapinarof, a natural AHR ligand for topical treatment of atopic dermatitis and psoriasis (currently in phase 2 and 3 trials) (Bissonnette et al., 2020).
Besides the relative straightforward ligand-mediated targeting of AHR activation, this complex signaling pathway involves multiple interaction partners (e.g. HSP90, XAP2, ARNT). In addition, there are multiple self-regulating negative feedback loops, such as AHR-dependent induction of AHRR (repressor) expression and ligand-mediated proteolytic degradation of the AHR. Degradation of ligands by cytochrome P450 enzymes (e.g. CYP1A1/1B1) that are transcriptionally induced by the AHR/ARNT complex demonstrates that multiple roads could lead to Rome (Wincent et al., 2012). In this issue of the JID, Kyoreva et al. demonstrate that inhibition of CYP1A1 enzyme activity may provide an alternative therapeutic strategy to empower AHR signaling in skin and to dampen psoriasis-like skin inflammation using murine models, analysis of human skin and in vitro studies (Kyoreva, 2020).
AHR activation in the skin: friend or foe?
The putative beneficial or detrimental effects of AHR signaling in skin are heavily debated. Studies claim pro-inflammatory effects of constitutive active AHR leading to dermatitis in conditional transgenic mouse models. Also, the higher incidence rate of dermatitis in heavily polluted regions is linked to AHR signaling, hypothetically via reactive oxygen species-mediated inflammation. In contrast, many studies using a variety of structurally diverse AHR ligands in different experimental models and target tissues demonstrate anti-inflammatory effects of AHR activation. The study by Kyoreva et al. adds to the latter, although seen from a different angle.
This study builds on the current dogma that naturally occurring AHR ligands and endogenous HR signaling is tightly regulated via a concomitant negative feedback loop involving ligand degradation. Upon AHR activation, canonical signaling results in the production of cytochrome P450 enzymes, like CYP1A1 and CYP1B1. AHR ligands can be substrates for CYP1A1, which in turn metabolizes these ligands and thereby attenuates AHR activation and signaling. However, environmental pollutants or xenobiotics, like dioxins, may be resistant to CYP1A1-mediated degradation resulting in long half-lives, sustained and unbalanced AHR activation and prolonged CYP1A1 enzyme activity, compromising tissue homeostasis. The latter may be the result of CYP1A1 not only targeting the exogenous xenobiotics, but also host-derived or microbiota-derived ligands, or ligands present in the diet that undergo systemic circulation. This depletion of AHR ligands and deficiency in endogenous receptor signaling, may release a natural break on inflammatory processes. This hypothesis was experimentally addressed by Kyoreva and colleagues using R26Cyp1a1 mice, having constitutively induced CYP1A1 levels. These mice showed no apparent skin phenotype in normal conditions, however upon induction of dermatitis-like inflammation using Imiquimod (IMQ), mice were more severely affected as compared to wild type mice. The aggravated phenotype was similar to that observed in AHR−/− mice, showing increased epidermal acanthosis and elevated levels of inflammatory cytokines and chemokines. This aggravation of inflammation could be rescued by addition of a CYP1A1 inhibitor (alpha naphthoflavone) prior to IMQ application. However, this compound can exhibit weak agonist AHR activation potential. Nevertheless, this study nicely provides a proof-of-principle for targeting the AHR pathway upon skin inflammation not via conventional exogenous ligands but by modulating CYP1A1 activity. This will require replicating studies that demonstrate similar effects in other (humanized) models for inflammation considering the differences between the affinity of ligands for the human and murine AHR (Flaveny and Perdew, 2009). Additional studies of native CYP1A1 activity in human skin and specific cell types are required to put the results obtained using models of constitutive and ubiquitous expression of CYP1A1 enzymatic activity into perspective and to allow the evaluation of the biological significance of heightened enzymatic activity and resulting downstream effects for human pathology.
Whether heightened CYP1A1 levels in skin are sufficient to release the break on inflammation and cause dermatitis in normal conditions is debatable given the absence of a phenotype in R26Cyp1a1 and AHR−/− mice prior to induction of inflammation. The elevated CYP1A1 levels that may metabolize AHR ligands and phenocopy AHR deficiency in the skin seems by itself not a key trigger for inflammatory signaling. Instead of being essential, CYP1A1 activity may thus contribute to disease severity as an additional trigger. Indeed, the actual level of CYP1A1 enzymatic activity, which is also dependent on the level of NADPH-cytochrome P450 reductase, is going to be key to the importance of these observations. This notion is supported by the gene expression study on human skin biopsies in the manuscript. Here, CYP1A1 expression levels are only lower in lesional psoriasis skin (indicative of reduced AHR signaling upon active inflammation), whereas levels in non-lesional skin is similar to those of healthy volunteers. Especially considering that CYP1A1 can be repressed by inflammatory signaling, the control on AHR signaling is thus more strongly influenced by the inflammatory microenvironment, rather than by the postulated depletion of AHR ligands. Given that CYP1A1 expression levels were analyzed in bulk RNA from full thickness skin biopsies, it remains to be elucidated in which cell types aberrant AHR signaling occurs upon inflammation (e.g. epidermal keratinocytes or immune cell subsets), which inflammatory mediators are to blame, which naturally occurring ligands are depleted by CYP1A1 upon inflammation, and how this contributes to disease aggravation. Notwithstanding the above, could it be that we are on track to identify long sought environmental factors in multifactorial diseases like psoriasis and atopic dermatitis?
In a first attempt to model dysregulated AHR signaling in psoriasis, in vitro studies using Th17 skewed T-cells from psoriasis patients were performed. The higher CYP1A1 activity after FICZ exposure in blood cells from psoriasis patients is intriguing and raises several questions: are all cells that express AHR more susceptible to deregulated AHR signaling in psoriasis, or is this specific for T-cell subsets? Is this due to enhanced ligand or receptor bioavailability or is the response stronger or more rapid due to epigenetic modifications? In addition, a potential dysregulation of AHR interaction partners required for canonical signaling, like ARNT, may play a role.
For a complete understanding of the role of AHR signaling in the underlying pathophysiology and a full appreciation of AHR-targeted therapeutics in inflammatory skin diseases, like psoriasis, we are in need of in-depth molecular analysis using (multiple) biologically relevant experimental models, and investigating the response to (multiple) structurally diverse AHR ligands. Furthermore, the relative small study cohorts for investigating transcriptional levels of genes related to AHR signaling, combined with bulk RNA analysis of mixed cell populations may result in an over- or underestimation of expression levels in the respective cells types that are responsible for the biological function of AHR in the skin. Cross cohort studies that combine multi-layer data on patient characteristics (e.g. epidemiology, clinical data, disease trajectories, epigenomics, transcriptomes and microbiome metagenomics), like those planned for in the recently founded consortium on biomarkers for psoriasis and atopic dermatitis (https://www.biomap-imi.eu/), could serve as a highly valuable data source for dissecting the role of AHR signaling in inflammatory skin diseases.
Identification of the natural AHR ligand pool in the skin: needles in a haystack?
The final questions related to potential CYP1A1-mediated loss of AHR activation and thus aggravation of skin inflammation revolves around the so-called natural ligand pool. Which AHR ligands are in fact present in and on our skin, which of them are CYP1A1 substrates, what is their source and do levels indeed differ between healthy skin and diseases skin? Besides a potential role for CYP1A1, ligand depletion may be the result of elevated kynureninase (KYNU) levels in psoriasis. KYNU is one of the most highly differentially regulated genes in the psoriasis transcriptome and catabolizes kynurinine, an AHR ligand generated from IDO-mediated tryptophan catabolism in an inflammatory environment. For a long time, UV radiation is been said to generate FICZ by the generation of tryptophan photoproducts in the skin, serving as a high affinity endogenous AHR ligand. Indeed, FICZ is formed from tryptophan exposed to UVB and FICZ metabolites have been found in urine. However, upon biologically relevant dosage of UVB exposure, FICZ accounts for only 0.02% of generated photoproducts rendering undetectable levels in the cytosol of UVB exposed cultured cells (Youssef et al., 2019). While FICZ has been used widely as a high affinity ligand for AHR activation studies in experimental models, other sources of AHR ligands in skin should be considered. For example, oxidized photoproducts of skin surface squalene have been shown to activate the AHR (Kostyuk et al., 2012). In addition, dietary sources of AHR ligands such as indole-3-carbinol products can influence skin barrier function (Haas et al., 2016).
The skin microbiome is a very likely source of AHR ligands considering the putative metabolites (e.g. indoles, indolo[3,2b]carbazole, quinolines) that bacteria and fungi can produce. Most lessons on this subject can be learned from the gut, where AHR is accepted to regulate host- microbe interactions by activation of AHR in gut epithelial cells and immune cells via microbial metabolites and dietary constituents, such as tryptophan metabolites, flavonoids and indole-3- carbinol condensation products. From skin microbiome studies, it seems evident that microbiome dysbiosis is associated with production of tryptophan metabolites (Chng et al., 2016), probably linked to altered abundance of bacteria or fungi that generate tryptophan metabolites, such as the commensal fungal Malassezia spp.
Figure 1. Roadmap for AHR targeting in skin inflammation.
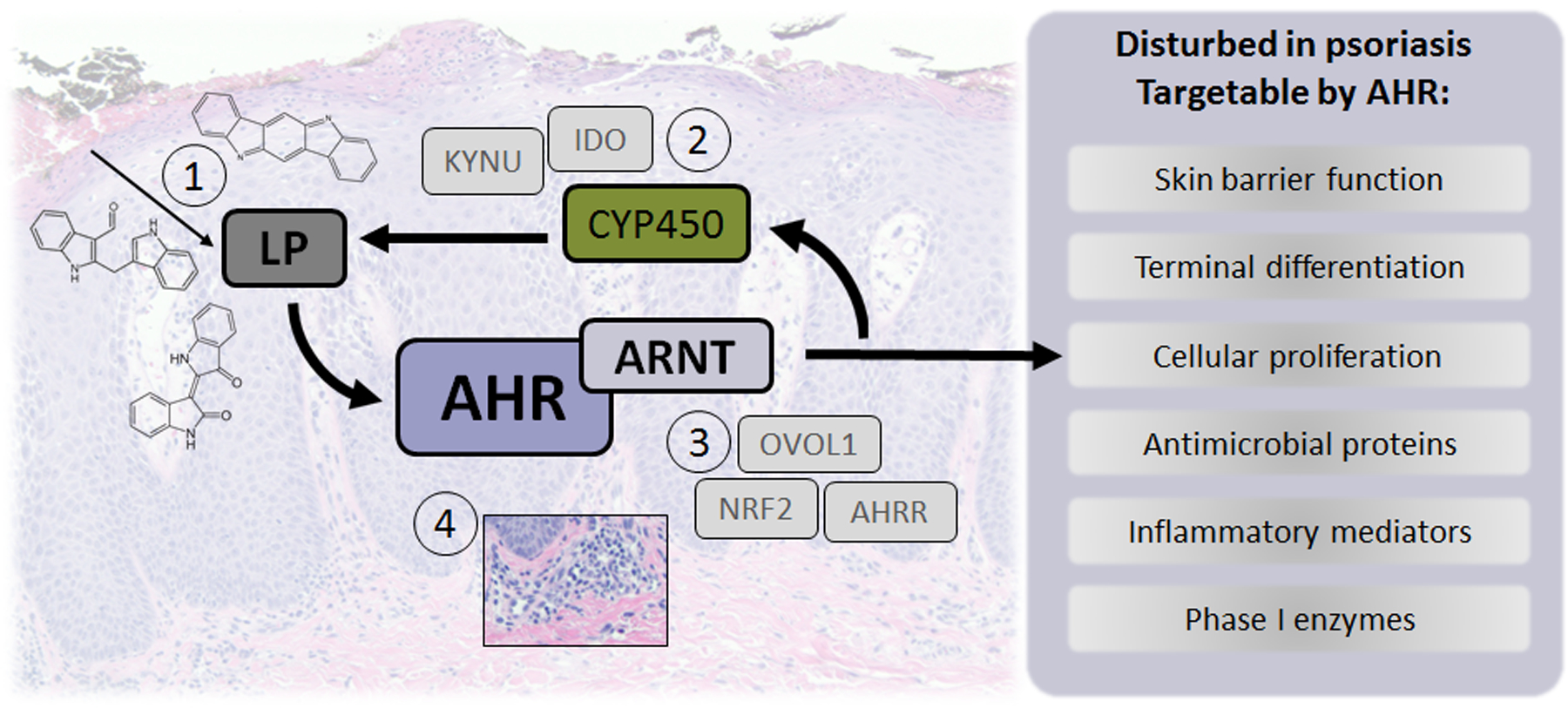
- AHR ligand supplementation: topical therapy (e.g coal tar or tapinarof), microbiome-derived (e.g. using pro- or prebiotics), or via dietary supplementation.
- Reduction of enzyme activity involved in ligand degradation, as now proposed for CYP450, but also likely for other enzymes (like KYNU, IDO).
- Identification and targeting of AHR interaction partners involved in downstream processes.
- Immune modulatory drugs changing the inflammatory milieu to normalize enzymatic levels or aberrant expression of molecules involved in deregulated AHR signaling.
Future perspectives
To move the field forward, studies on the identification and quantification of the AHR ligand pool in human skin in health and disease; and on the existence and biological relevance of ligand-dependent downstream effects in the various tissue contexts (healthy vs. inflammatory state) are key. For this, humanized mice with human AHR to faithfully mimic the ligand-receptor affinity and magnitude of signaling events are important, combined with multiple AHR ligands of various stability and dosage in multiple experimental (human) disease models. Besides examining AHR and target gene expression, assessment of AHR protein localization and functional levels among different cell types in target tissues is crucial to identify the cells responsible for mediating biological events and highlighting targets for intervention. From the study by Kyoreva, we can conclude that when exploring the avenues for the therapeutic targeting of the AHR in dermatology, AHR-targeted therapies using exogenous ligands that drive canonical AHR signaling and thus also induce CYP1A1 enzymatic activity in skin cells may result in metabolizing naturally occurring AHR ligands in the skin that normally contribute to controlling inflammatory processes (Figure 1). Thus, AHR ligands that minimally induce CYP1A1 enzymatic activity may yield more effective drugs for the treatment inflammatory skin diseases.
Clinical relevance.
Monitoring (dys)regulation of AHR signaling in patient cohorts can provide key insights into disease pathophysiology.
Selective inhibitors for CYP1A1 enzyme activity may have therapeutic potential in the treatment of inflammatory skin diseases.
Lead optimization of AHR targeting ligands to minimize induction of CYP1A1 enzymatic activity could improve therapeutic efficacy.
Acknowledgements
This work was supported by the National Institutes of Health Grant ES028244 (GHP) and NWO-VENI grant 19616054 (EB)
Footnotes
Conflict of interest
EVDB and GHP report no conflicts of interest
References
- Bissonnette R, Gold LS, Rubenstein DS, Tallman AM, Armstrong A. Tapinarof in the treatment of psoriasis: A review of the unique mechanism of action of a novel therapeutic AhR modulating agent (TAMA). J Am Acad Dermatol 2020. [DOI] [PubMed] [Google Scholar]
- Chng KR, Tay AS, Li C, Ng AH, Wang J, Suri BK, et al. Whole metagenome profiling reveals skin microbiome-dependent susceptibility to atopic dermatitis flare. Nat Microbiol 2016;1(9):16106. [DOI] [PubMed] [Google Scholar]
- Flaveny CA, Perdew GH. Transgenic Humanized AHR Mouse Reveals Differences between Human and Mouse AHR Ligand Selectivity. Mol Cell Pharmacol 2009;1(3):119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furue M Regulation of Skin Barrier Function via Competition between AHR Axis versus IL-13/IL-4JAKSTAT6/STAT3 Axis: Pathogenic and Therapeutic Implications in Atopic Dermatitis. J Clin Med 2020;9(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas K, Weighardt H, Deenen R, Kohrer K, Clausen B, Zahner S, et al. Aryl Hydrocarbon Receptor in Keratinocytes Is Essential for Murine Skin Barrier Integrity. J Invest Dermatol 2016;136(11):2260–9. [DOI] [PubMed] [Google Scholar]
- Kostyuk V, Potapovich A, Stancato A, De Luca C, Lulli D, Pastore S, et al. Photo-oxidation products of skin surface squalene mediate metabolic and inflammatory responses to solar UV in human keratinocytes. PLoS One 2012;7(8):e44472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyoreva. CYP1A1 enzymatic activity influences skin inflammation via regulation of the AHR pathway. Journal of Investigative Dermatology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaard EH, Bergboer JG, Vonk-Bergers M, van Vlijmen-Willems IM, Hato SV, van der Valk PG, et al. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J Clin Invest 2013;123(2):917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wincent E, Bengtsson J, Mohammadi Bardbori A, Alsberg T, Luecke S, Rannug U, et al. Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A 2012;109(12):4479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef A, von Koschembahr A, Caillat S, Corre S, Galibert MD, Douki T. 6-Formylindolo[3,2-b]carbazole (FICZ) is a Very Minor Photoproduct of Tryptophan at Biologically Relevant Doses of UVB and Simulated Sunlight. Photochem Photobiol 2019;95(1):237–43. [DOI] [PubMed] [Google Scholar]