

In this study, Shoshani et al. tested the role of aneuploidy in tumor initiation and progression, and generated mice with random aneuploidies by transient induction of polo-like kinase 4 (Plk4), a master regulator of centrosome number. Their findings show how transient CIN generates cells with random aneuploidies from which ones that acquire a karyotype with specific chromosome gains are sufficient to drive cancer formation, and that distinct CIN mechanisms can lead to similar karyotypic cancer-causing outcomes.
Keywords: aneuploidy, cancer, chromosome instability, Myc, Plk4, p53
Abstract
Abnormal numerical and structural chromosome content is frequently found in human cancer. To test the role of aneuploidy in tumor initiation and progression, we generated mice with random aneuploidies by transient induction of polo-like kinase 4 (Plk4), a master regulator of centrosome number. Short-term chromosome instability (CIN) from transient Plk4 induction resulted in formation of aggressive T-cell lymphomas in mice with heterozygous inactivation of one p53 allele and accelerated tumor development in the absence of p53. Transient CIN increased the frequency of lymphoma-initiating cells with a specific karyotype profile, including trisomy of chromosomes 4, 5, 14, and 15 occurring early in tumorigenesis. Tumor development in mice with chronic CIN induced by an independent mechanism (through inactivation of the spindle assembly checkpoint) gradually trended toward a similar karyotypic profile, as determined by single-cell whole-genome DNA sequencing. Overall, we show how transient CIN generates cells with random aneuploidies from which ones that acquire a karyotype with specific chromosome gains are sufficient to drive cancer formation, and that distinct CIN mechanisms can lead to similar karyotypic cancer-causing outcomes.
More than a century ago, chromosome instability (CIN) leading to aneuploidy was first recognized as a major hallmark in cancer (Hansemann 1890; for review, see Hanahan and Weinberg 2011). Aneuploidy is frequently found in multiple types of cancer (Taylor et al. 2018) and is correlated with poor prognosis (Stopsack et al. 2019). Although linkage of aneuploidy and tumorigenesis was proposed by Boveri in 1902 (Boveri 1902), the role of aneuploidy in cancer formation remains controversial. It has been proposed that increased CIN leading to aneuploid genotypes serves as a risk factor for carcinogenesis (Gordon et al. 2012; Naylor and van Deursen 2016). Indeed, reduced levels of the kinesin family motor protein CENP-E drives CIN from missegregation of individual whole chromosomes, and this generates spontaneous lymphomas and lung cancers (Weaver et al. 2007). Similarly, chronic CIN from weakening of the spindle assembly checkpoint (e.g., through reduction or mutation in BubR1 [Baker et al. 2009], reduction in Mps1 kinase activity [Foijer et al. 2014; Hoevenaar et al. 2020], or from deletion or overexpression of Mad2 [Sotillo et al. 2007; Foijer et al. 2017]) drives tumors in mice through loss of tumor suppressor genes. Alternatively, genome doubling yielding tetraploid cells can serve as an intermediate step toward aneuploidy and cancer (Storchova and Pellman 2004), although tetraploidy can also protect against cell transformation (Shoshani et al. 2012). Specific aneuploid backgrounds have been reported to suppress cell viability, even under oncogenic induction, thereby preventing transformation, but in cancer, aneuploidy is frequently linked to more aggressive phenotypes (Santaguida and Amon 2015; Sheltzer et al. 2017). Chronic CIN at a high level can also act as a tumor suppressor (Silk et al. 2013; Rowald et al. 2016). Together, these findings suggest that chronic CIN can influence tumorigenesis in both directions, depending on the severity of the CIN phenotype (Silk et al. 2013; Rowald et al. 2016; Hoevenaar et al. 2020). However, it is unclear whether transient CIN can lead to aneuploidy that persists in healthy cells to drive transformation leading to cancer.
Centriole duplication is controlled by Polo-like kinase 4 (Plk4) (Bettencourt-Dias et al. 2005; Habedanck et al. 2005), a self-regulatory kinase (Holland et al. 2010). Overexpression of Plk4 leads to centrosome amplification, which can persist under p53 deficiency (Holland et al. 2010), and drives CIN through merotelic attachment of chromosomes (Ganem et al. 2009). Indeed, centrosome amplification can be detected in premalignant cells in Barrett's esophagus and persists through malignant transformation (Lopes et al. 2018). The first direct evidence for a role of centrosome amplification in tumor formation came from flies in which increased SAK/Plk4 expression enabled tumorigenic growth of larval brain cells (Basto et al. 2008). Chronic induction of high levels of Plk4 in mice did not increase spontaneous tumor formation even in a p53-deficient background (Vitre et al. 2015), probably due to an excessive rate of continuous CIN. However, CIN from milder, chronic Plk4 overexpression has been reported to promote spontaneous tumorigenesis in wild-type (Levine et al. 2017) and p53-deficient mice (Coelho et al. 2015), while Plk4-dependent CIN during embryonic development in p53-deficient epidermis enhances skin cancer in adult mice (Serçin et al. 2016).
By transiently inducing Plk4 overexpression in adult mice and the use of multiple sequencing approaches, we now demonstrate that transient CIN enhances tumor formation by increasing the frequency of tumor-initiating cells and by accelerating the acquisition of a specific aneuploidy profile. This profile includes the gain of chromosomes 4, 5, 14, and 15, a tumor karyotype we demonstrate to develop early during tumorigenesis under transient CIN, and in end-stage tumors developed in an independent model with chronic CIN provoked by inactivation of the spindle assembly checkpoint (SAC). Transcriptomic analysis revealed that this aneuploidy profile drives a gene expression program that is also frequently observed in multiple human cancers and correlates with increased c-Myc expression. We conclude that transient CIN is a powerful tumor promoting mechanism driving acquisition of a specific aneuploid karyotype.
Results
Transient Plk4 overexpression leads to transient chromosome instability and aneuploidy
To determine how transient chromosome instability affects tumorigenesis, we generated a mouse model with an inducible Plk4 gene in the background of different p53 genotypes. This was achieved by two rounds of breeding (Fig. 1A) to generate mice (to be referred to as PRG5 mice) in a congenic background that carried (1) a doxycycline-regulated Plk4-EYFP gene (Levine et al. 2017), (2) a gene encoding the reverse tetracycline transactivator (rtTA) whose expression permitted doxycycline-inducible expression of Plk4 (Supplemental Fig. S1A), (3) a gene encoding centrin-GFP (Hirai et al. 2016), and (4) in which neither (PRG5+/+), one (PRG5+/−), or both (PRG5−/−) p53 alleles were inactivated (Jacks et al. 1994).
Figure 1.

Transient CIN in mice through induced Plk4 overexpression drives transient aneuploidy. (A) Breeding strategy used to obtain doxycycline-inducible Plk4 mice with centrin-GFP and under different backgrounds of p53 (PRG5 mice). P2 and R2 denote homozygosity for Plk4 and rtTA genes, respectively. (B) Overview of the experimental design using PRG5 mice. (C–E) Plk4 mRNA levels (C), measurement of centrin-GFP foci (D), and percent aneuploidy for chromosome 11 using interphase DNA-FISH (E) in thymuses from PRG5 mice before (control), immediately after (2 wk dox), and 1 mo after (4 wk later) doxycycline administration. Mean ± SD of indicated mice per group are presented. P-values determined using one-way ANOVA with Tukey's multiple comparison test. (F,H,J) Heat maps showing DNA copy number using single-cell whole-genome sequencing of cells collected from PRG5+/− mice before (F), immediately after (H), and 1 mo after (J) Plk4 induction. (G,I,K) Analysis of copy number changes of the samples at the left (shown in F,H,J). See the Materials and Methods for details about calculation of the CN (copy number) change score. Statistics below the dot plots indicate the total number and percentage of cells with at least one whole chromosome gain or loss.
Doxycycline-dependent Plk4 induction was determined to be comparable in mouse embryonic fibroblasts (MEFs) derived from PRG5 mice independent of p53 status. Plk4 RNA levels were increased fourfold within 2 d of Plk4 induction, decreasing back to basal levels within 2 d following doxycycline withdrawal (Supplemental Fig. S1B). Transient increase in Plk4 expression induced significant centrosome amplification, which upon doxycycline removal returned to basal levels within 1 wk in p53+/+ MEFs and trended toward basal levels in p53+/− and p53−/− PRG5-derived MEFs (Supplemental Fig. S1C).
Live-cell imaging revealed that centrosome amplification initiated within 6 h after Plk4 induction (Supplemental Fig. S1D), producing >50% abnormal mitoses with anaphase bridges or lagging chromosomes (Supplemental Fig. S1E). PRG5 mice fed for 2 wk with doxycycline-containing food (Fig. 1B) had increased Plk4 RNA levels in the thymus (threefold), spleen (2.5-fold), liver (twofold), colon (20-fold), and skin (20-fold), but not in the lung or kidney (Fig. 1C; Supplemental Fig. S1F). Plk4 induction was transient, as Plk4 RNA levels returned to basal levels within 1 mo after discontinuing doxycycline (Fig. 1C; Supplemental Fig. S1F).
Increased Plk4 RNA yielded transient centrosome amplification (Fig. 1D; Supplemental Fig. S1G) and CIN, leading to aneuploidy just after doxycycline removal with both gain and loss of chromosome 11 copy numbers in thymocytes (threefold increase in aneuploidy) (Fig. 1E) as determined using interphase DNA fluorescent in situ hybridization (FISH), but not in splenocytes (in which Plk4 RNA induction was only mild) (Supplemental Fig. S1H). Aneuploidy levels, determined using single-cell whole-genome sequencing (scWGS) of thymic cells from PRG5+/− mice, revealed a transient increase in aneuploidy from near zero (0%–4% before Plk4 induction) to 24%–40% after transient CIN from 2-wk Plk4 induction, which returned to near-zero within 1 mo after doxycycline withdrawal (Fig. 1F–K). Transient CIN produced heterogeneous aneuploidy without any apparent bias for particular karyotypes (Fig. 1H). Importantly, loss of p53 by itself did not promote CIN-induced aneuploidy, as determined by scWGS of cells from three independent PRG5−/− mice (Supplemental Fig. S1I).
Transient CIN accelerates thymic lymphoma formation
Survival and tumor formation in PRG5 mice were monitored for 2 yr following a 2-wk CIN induction beginning at 4 wk of age (doxycycline administered during 30–45 d of age) and in control PRG5 littermates without transient doxycycline-induced centrosome amplification (Fig. 1B). Transient induction of CIN in PRG5+/+ mice did not affect survival (median survival 837 d and 805 d in control and induced mice, respectively) (Fig. 2A) or tumor formation (17 out of 42 and 13 out of 49 control and induced mice with tumors, respectively) (Fig. 2B; Supplemental Fig. S2A) compared with noninduced PRG5+/+ mice. The tumor spectrum profile in aged PRG5+/+ mice (Fig. 2C) was not affected by Plk4 induction either, with a majority of tumors forming in spleen (∼40%), the digestive system (colorectal and intestinal tumors ∼20%), and liver (∼17%). Without Plk4 induction, PRG5+/− mice developed tumors earlier than PRG5+/+ mice, resulting in decreased mean survival from 837 to 537 d (Fig. 2A,D). Thymic tumors were rare (3.2%) in PRG5+/− (Fig. 2E).
Figure 2.

Transient CIN drives thymic lymphoma in p53-deficient mice. (A,D,G) Survival (Kaplan-Meier) plots of control (noninduced) PRG5 mice and PRG5 mice treated with doxycycline for 2 wk at the age of 30 d (dox 2 wk) with wild-type p53 (A), heterozygous p53 (D), and p53 knockout (G) backgrounds. Comparison of indicated number of mice done using log rank test. (B,E,H) Thymic and nonthymic tumor frequencies of indicated number in PRG5 mice treated with doxycycline for 2 wk at the age of 30 d with wild-type p53 (B), heterozygous p53 (E), and p53 knockout (H) backgrounds. (C,F,I) Distribution of tumor types from indicated number of tumors in PRG5 mice treated with doxycycline for 2 wk at the age of 30 d with wild-type p53 (C), heterozygous p53 (F), and p53 knockout backgrounds (I).
Two weeks of CIN induction at an early age (starting at 30 d) reduced survival of PRG5+/− mice further (median survival 341 d) (Fig. 2D) and doubled tumor frequency (from 45% to 82.9%) (Fig. 2E). Tumors in induced PRG5+/− mice developed very early (starting at 100 d of age) (Supplemental Fig. S2B), the majority (54.7%) of which were thymic lymphomas (Fig. 2E,F). Reduced survival could be largely attributed to the increased frequency of thymic lymphoma, as the median survival of mice with other tumors was unaltered (Supplemental Fig. S2C). A shorter, 1-wk induction of CIN in PRG5+/− mice had a milder effect, resulting in a much lower frequency (only one in 10 mice) of thymic lymphoma and longer survival (median survival 556 d) (Supplemental Fig. S2D).
Doubling the period of induced CIN to 4 wk resulted in reduced survival (median survival 158 d) (Supplemental Fig. S2D) but without affecting the frequency of thymic lymphomas (54% and 54.7% in 4-wk- and 2-wk-induced mice, respectively). Reduced survival of PRG5+/− mice after longer (4-wk) CIN could be attributed to nonthymic lymphoma tumors, with thymic lymphoma latency (∼150 d) nearly identical between 2-wk and 4-wk CIN (Supplemental Fig. S2E,F). Transient, 2-wk CIN induction in older (100-d-old) mice yielded indistinguishable survival and tumor formation frequencies compared with CIN induced in 30-d-old mice (Supplemental Fig. S2G–I).
All PRG5−/− mice without Plk4-dependent CIN succumbed to tumors within the first year of life (median survival of 176 d, 68.3% of mice with detected tumors) (Fig. 2G,H), with a large proportion with thymic lymphomas (44.9%) (Fig. 2H). Two weeks of doxycycline-induced CIN in PRG5−/− mice significantly decreased survival (to only 138 d) (Fig. 2G) and increased overall tumor (to 91%) and thymic lymphoma (80.4% of mice) (Fig. 2H) frequencies. Decreased survival of PRG5−/− mice correlated with thymic lymphomas developing at younger ages (with median survival with or without transient CIN of 134 and 154 d, respectively) (Supplemental Fig. S2J–L). Tumor spectrum profiles were similar, with the exception of an increased proportion of thymic tumors after transient CIN induction (Fig. 2I). In all examined cases, thymic tumors were lymphomas, many of which appeared invasive and metastatic (Supplemental Fig. S2M; Supplemental Table S1).
Transient CIN drives p53 loss and c-Myc overexpression
RNA expression profiles of tumors and normal thymuses were determined to examine the mechanism(s) underlying the conversion of normal tissue into spontaneous or doxycycline-induced thymic lymphomas. A general profile distinct from control unaffected thymus samples was identified for lymphomas (1325 differentially expressed genes; P < 0.05) (Supplemental Fig. S3A; Supplemental Table S2), with significant alterations in cell cycle and metabolic pathways (Supplemental Figs. S3B, S4). Lymphomas from transient CIN in p53+/− PRG5 mice had low expression of p53 target genes, including Bcl2l1, and activation of p19 (Supplemental Fig. S5A), indicative of loss of the wild-type p53 allele. Indeed, expression of p53 exons 2–6 (the region deleted in the p53-null allele) was completely absent (Supplemental Fig. S5B; Jacks et al. 1994). Whole-genome DNA sequencing (15× coverage) confirmed the loss of p53 gene exons 2–6 without evidence of other numerical or structural alterations in chromosome 11 (Supplemental Fig. S5C,D), consistent with whole-chromosome missegregation as the underlying mechanism in lymphomas arising in PRG5+/− mice. Several common features were identified in PRG5−/− mice (with or without induced CIN) and PRG5+/− after CIN as hallmarks of advanced thymic lymphomas, including increased centrosome numbers (Supplemental Fig. S6A) and overexpression of c-Myc RNA (Supplemental Fig. S6B) and protein (Supplemental Fig. S6C), potentially due to acquisition of additional copies of chromosome 15 (Supplemental Fig. S6D).
Transient CIN increases tumor-initiating cell frequency
We hypothesized that acceleration of tumor formation following transient CIN might occur due to CIN enhancing generation of tumor-initiating cells. To test this, we collected small biopsies (∼2%–3% of the tumor) from multiple tumors, as well as multifocal biopsies from additional tumors and sequenced the β chain of the T-cell receptors (TCRβ) expressed in the tumor T cells (Figure 3A,B). Single-biopsy analysis revealed that tumors forming after 2 wk of induced CIN in PRG5 mice had increased clone frequencies, with one out of five tumors in p53+/− mice and six out of six tumors in p53−/− mice having more than one dominant clone (>1% frequency). In contrast, no tumors (zero out of six) from noninduced PRG5−/− mice carried more than one dominant clone (Fig. 3A; Supplemental Table S3). RNA sequencing confirmed the specifically increased expression of individual variable TCRβ genes in tumors from PRG5 mice, further confirming the presence of multiple clones (Supplemental Fig. S7A).
Figure 3.

Tumors formed following transient CIN have increased tumor-initiating cell burden but share a similar transcription profile. (A,B) Results from T-cell receptor sequencing showing the top 10 T-cell receptor sequences (indicative of T-cell clones) identified in thymic T-cell lymphomas from PRG5 mice in single biopsies (A) and multiregional biopsies (B) as determined using T-cell receptor sequencing. T-cell receptor sequencing allowed the identification of T-cell lymphoma-initiating cells by using the T-cell receptor as an endogenous barcode. A control thymic sample (mouse #624) is presented in A, showing the expected low frequency of each T-cell receptor sequence, indicating there was no selection for a specific T-cell clone. (C) Heat maps showing pairwise Pearson correlation coefficients among samples (tumor single biopsies) using transcript per million (TPM) values as determined using RNA sequencing of the PRG5 mice.
We next addressed tumor clonality in PRG5 mice by collecting multiregional biopsies representing different spatial regions of a tumor. TCRβ sequencing of these biopsies revealed that three out of four of CIN-induced p53+/− and three out of four of CIN-induced p53−/− -derived tumors from PRG5 mice contained more than one dominant clone, whereas none of the tumors arising in p53−/− mice without induced CIN contained more than one dominant clone (zero out of three) (Fig. 3B; Supplemental Table S3). We conclude that transient CIN increases the number of tumor-initiating clones.
Transient CIN drives tumors with defined transcriptome and aneuploidy profiles
Since CIN drives random genomic reshuffling and generates higher frequency of tumor-initiating cells (Fig. 3A,B), transient CIN could be expected to generate tumors with diverse gene expression profiles. However, this was not the case: transcriptomes of independent tumors formed in PRG5 (p53+/− or p53−/−) mice after transient CIN shared a similar gene expression profile, whereas tumors arising in PRG5−/− mice without induced CIN were more heterogeneous (Fig. 3C; Supplemental Fig. S8). Whole-genome sequencing of these tumors revealed that transcriptome changes paralleled acquisition of a specific DNA copy number profile that included recurrent gains of copy numbers for chromosomes 4, 5, 14, and 15 (Fig. 4A; Supplemental Fig. S9). RNA expression levels confirmed the apparent changes in chromosome copy numbers (Supplemental Fig. S9E).
Figure 4.
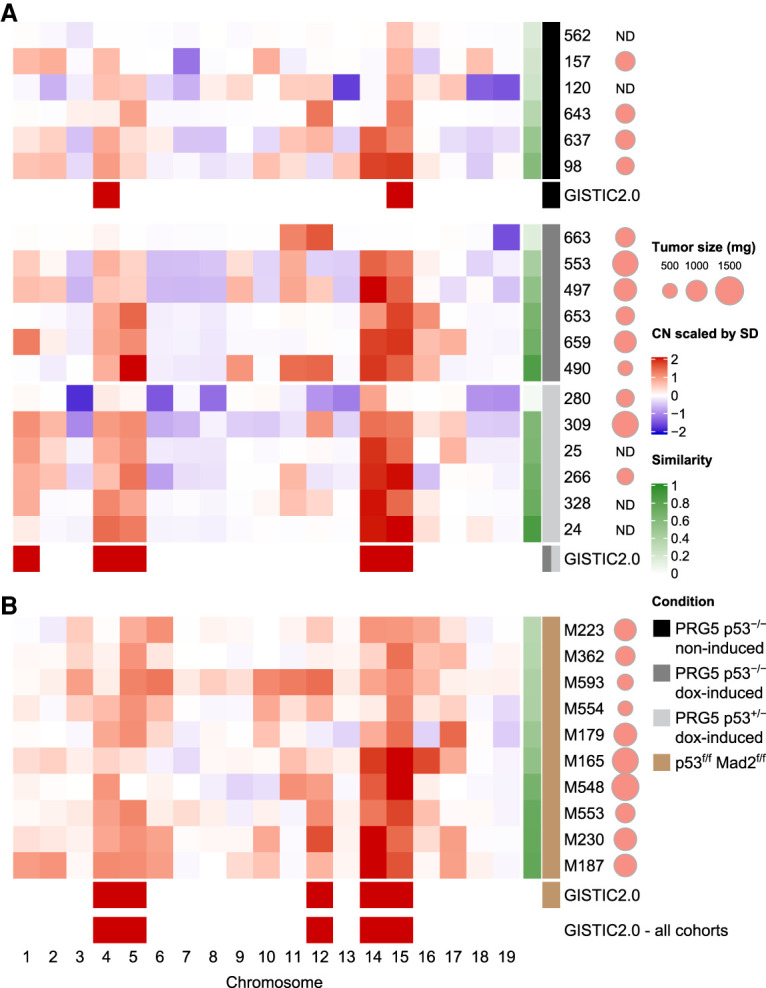
CIN accelerates the selection for a specific aneuploidy profile. (A,B) Heat maps showing averaged chromosome DNA copy number changes in late tumors (>500 mg) from noninduced p53−/− PRG5 mice (black), 2-wk doxycycline-treated (at the age of 30 d) p53−/− PRG5 mice (dark gray) and p53+/− PRG5 mice (light gray) (A), and from Lck-Cre+; Mad2f/f; p53f/f mice (brown) (B). Copy number changes of PRG5 mice were determined using whole-genome sequencing. Averaged copy number from single-cell whole-genome sequencing (as shown in Fig. 1D) is shown for tumors from Mad2 mice. Genomic identification of significant targets in cancer (GISTIC 2.0) analysis showing significant (q = 0.01) aneuploidies for each cohort and across all cohorts are presented. The similarity of individual tumors to the GISTIC 2.0 output from across all cohorts is presented in the green heat map (see Supplemental Table S4 for similarity values). Scaled tumor weights are presented. (ND) Not determined.
The most significant chromosomal events in the different tumor groups were determined by use of the genomic identification of significant targets in cancer (GISTIC) algorithm (Mermel et al. 2011), which allows the identification of significant events in DNA copy number across multiple samples. GISTIC analysis revealed that tumors from noninduced PRG5−/− mice displayed significantly increased copy numbers of chromosomes 4 and 15. However, tumors induced by transient CIN in PRG5−/− as well as PRG5+/− mice showed a more complex karyotype, encompassing increased copy numbers of chromosomes 1, 4, 5, 14, and 15 (q-value = 0.01) (Fig. 4A).
Independent CIN mechanisms drive tumors with similar aneuploidy profiles
We previously showed that Lck-Cre; Mad2f/f; p53f/f mice succumb to thymic lymphomas within ∼4 mo with recurrent aneusomies involving chromosomes 4, 5, 14, 15, and 17 (Foijer et al. 2017). The karyotype landscapes in these lymphomas are remarkably similar to those observed in induced PRG5 mice despite a different underlying CIN-driving mechanism. In the Lck-Cre; Mad2f/f; p53f/f model, CIN is chronic, suggesting that murine lymphomas exhibiting transient or chronic CIN select for (nearly) identical karyotypes.
Since the type and driver of CIN between these models are so different, we compared the dynamics of karyotype evolution during tumor development driven by transient CIN in PRG5 mice or chronic CIN in Lck-Cre Mad2f/f; p53f/f mice. To quantify karyotype evolution at various stages of tumorigenesis, we used scWGS (Supplemental Fig. S10A). We first assessed the impact of Mad2 inactivation, which leads to spindle checkpoint inactivation and thus CIN (Foijer et al. 2017) in the p53 wild-type mouse thymus. scWGS analysis of two Mad2f/f; Lck-Cre thymuses from 8-wk-old mice revealed that 48% and 46% of the cells (n = 46 from each mouse) displayed aneuploidy for one or multiple chromosomes without selection for specific chromosome copy number changes (Supplemental Fig. S10B). Thus, Mad2 loss drives a random CIN phenotype in nontransformed thymocytes, similar to what is seen after Plk4 overexpression (Fig. 1H).
Tumors from Lck-Cre; Mad2f/f; p53f/f mice, which developed with an average latency of 16 wk in Foijer et al. (2017), were harvested between 8 and 16 wk and represented a spectrum of tumor development from early (100–500 mg) to advanced (>500 mg) thymic lymphomas (Supplemental Fig. S10A,C,D). scWGS revealed that developing (Supplemental Fig. S10C) or advanced (Supplemental Fig. S10D) thymic lymphomas in these mice had frequent whole-chromosome changes, with an overwhelming majority of tumor cells presenting chromosome gains accompanied by few or no losses. Gains of chromosomes 4, 5, 14, 15, and 17 (Supplemental Fig. S10E) were frequent in advanced tumors, indicating progressive karyotype evolution with increasing tumor size (Supplemental Fig. S10F,G). Lck-Cre; Mad2f/f; p53f/f tumors displayed a chronic CIN phenotype, as (1) cultured primary Lck-Cre; Mad2f/f; p53f/f lymphoma cells were spindle assembly checkpoint-deficient, as evidenced by their lack of mitotic arrest when exposed to the spindle poison nocodazole (Supplemental Fig. S11A); (2) these lymphoma cells displayed frequent missegregation events (seen with time-lapse microscopy) (Supplemental Fig. S11B); and (3) cells from primary lymphomas showed significant cell-to-cell karyotype heterogeneity (as quantified by scWGS) (Supplemental Fig. S10C,D).
Furthermore, analysis of copy number (CN) and karyotype heterogeneity revealed an increase in CN changes but not heterogeneity in progressing lymphomas (see the Materials and Methods for detailed definitions; Supplemental Fig. S11C,D), indicating that, with constant CIN, karyotypes became more complex during tumor progression. Some of the early lymphomas, most notably M503, M522, and M559, showed aneuploidy landscapes closer to Lck-Cre; Mad2f/f untransformed thymocytes (Supplemental Fig. S10, cf. B and C), with similar CN scores, heterogeneity, and percentage of aneuploid cells (Supplemental Fig. S11C,D), indicating that, in these early malignancies, selection for chromosome aberrations had not (yet) occurred. In contrast, other early lymphomas showed clear signs of aneuploidy karyotype selection biased toward the recurrent clonal chromosome copy number changes that became more apparent in endpoint lymphomas (Supplemental Fig. S10F,G). The most advanced lymphomas also displayed an increase in copy number state transitions within chromosomes, indicating that structural chromosomal damage occurred in lymphomas with chronic CIN, but only at advanced stages (Supplemental Fig. S10D). Finally, RNA sequencing of Lck-Cre; Mad2f/f; p53f/f lymphomas revealed that chronic CIN induced by SAC inactivation also increased the clonal frequency (quantified by the number of T-cell receptor variants present in a single tumor) (Supplemental Fig. S7B), similar to tumors arising from transient CIN induced by Plk4 overexpression.
Systematic comparison with GISTIC 2.0 of karyotype makeup of end-point tumors confirmed that the most significant chromosomal events in tumors from mice with chronic CIN were increases in chromosomes 4, 5, 12, 14, and 15 (q-value = 0.01). A similarity test (see the Materials and Methods) of individual tumors to the overall GISTIC profile (calculated from all cohorts in Fig. 4) revealed that tumors forming following transient CIN had a higher similarity value to the GISTIC profile than the tumors spontaneously occurring in noninduced PRG5−/− mice (P = 0.065, two-tailed t-test) (Fig. 4; Supplemental Table S4). While all chronic CIN tumors displayed key aspects of the GISTIC profile, they typically displayed additional chromosome copy number changes, which varied between individual tumors.
Aneuploidy profile selection occurs early in lymphoma development driven by transient CIN
We used scWGS to determine the karyotypes of individual tumor cells in lymphomas developing in PRG5 mice. Comparing single-cell karyotypes of early tumors (<500 mg, with mice lacking any clinical signs such as weight loss or dyspnea) with late tumors (Fig. 5A; Supplemental Fig. S12) revealed that gains of chromosomes 4, 5, 14, and 15 were already present in almost every cell in early tumors from induced p53+/− and p53−/− PRG5 mice. Indeed, both early and late tumors with these genotypes displayed karyotypes that had near-identity to the GISTIC profile, consistent with early formation of tumor-initiating cells under transient CIN. The most striking difference in the aneuploidy landscapes of tumors developed under chronic versus transient CIN was that the latter had more efficiently acquired extra copies of chromosomes 4, 5, 14, and 15 early in tumor development.
Figure 5.

Transient CIN selects for a specific aneuploidy profile early during tumor development. (A) Heat map showing averaged DNA copy number changes in early tumors (<500 mg) from noninduced p53−/− PRG5 mice (black), 2-wk doxycycline-treated (at the age of 30 d) p53−/− PRG5 mice (dark gray) and p53+/− PRG5 mice (light gray), and Lck-Cre+; Mad2f/f; p53f/f mice (brown) as determined using single-cell whole-genome sequencing. Single-cell data of tumors from PRG5 mice are in Supplemental Figure S12, and of Mad2 mice in Supplemental Figure S10. Genomic identification of significant targets in cancer (GISTIC) 2.0 analysis showing significant aneuploidies across all tumors is shown at the bottom. The similarity of individual tumors to the GISTIC 2.0 output is presented in the green heat map. (B, left) Uniform manifold approximation and projection (UMAP) analysis of DNA copy number changes in control (gray shades), p53−/− noninduced PRG5 tumors (red shades), doxycycline-induced PRG5 tumors (blue shades), and Mad2 tumors (green shades). (Right) Embedding of the GISTIC 2.0 similarity score for each sample shown in the UMAP plot at the left. Control samples were not used for the GISTIC 2.0 analysis and appear in gray shades.
Compared with early chronic CIN-driven tumors, early tumors formed as a result of transient CIN showed increased aneuploidy but with lower copy number variability (i.e., cell-to-cell karyotype heterogeneity) (Supplemental Fig. S13). In contrast, early tumors from PRG5−/− mice without CIN yielded a different result: One tumor (PRG5-693) showed high similarity to the GISTIC profile; a second tumor (PRG5-926) underwent genome doubling (gaining chromosomes 4, 5, 14, and 15 in the process), an event observed also in two out of three late tumors (PRG5-98 and PRG5-120); and a third tumor did not acquire any similarity to the GISTIC profile (Fig. 5A; Supplemental Fig. S12).
To determine how different individual tumor cells of all cohorts (early and late tumors from all genotypes) varied based on their genomic profiles, we performed a uniform manifold approximation and projection (UMAP) analysis (Fig. 5B). UMAP separated cell populations according to their tumor of origin (sample cell types were not given as an input), indicating that cells within a single tumor had highest similarity to cells from the same tumor. Cells from tumors formed under chronic CIN, as well as spontaneous tumors from PRG5−/− mice without CIN, were not localized to a specific cluster (Fig 5B, left panel). In contrast, cells from both early and terminal tumors of mice experiencing transient CIN at an early age were localized in a region opposite to euploid noncancer cells, with most cells from other tumors (e.g., spontaneous tumors from p53−/− PRG5 mice and tumors forming under chronic CIN) spread in between. In agreement with this, when labeling the individual cells according to their respective GISTIC similarity score (Fig. 5B, right panel), UMAP successfully separated single cells according to their similarity to the GISTIC score, with a visible gradient from euploid cells to a cluster of cells with high similarity where cells from both early and terminal induced PRG5 tumors localized. Thus, a very specific selection for particular karyotypes occurs already in early tumors following transient CIN, which is counteracted in tumors experiencing chronic CIN. We therefore conclude that ongoing CIN delays the evolutionary path toward an optimal GISTIC karyotype.
Aneuploidy selection generates a gene expression profile found in human cancers
To assess the relevance of the CIN-driven lymphomas to human cancer, we first determined which RNA expression signature correlated best with the genomic GISTIC score that defined the optimal karyotype as determined above. To determine this GISTIC-correlated transcriptome signature, we used the similarity indices of individual tumors as continuous covariates in the RNA sequencing analysis to identify genes whose expression (RNA) significantly correlated with the similarity index (similarity to the DNA copy number profile as determined by GISTIC). This analysis revealed significant enrichment of genes related to cell cycle progression and pathways allowing cells to overcome oncogene-induced senescence and replication stress (Fig. 6A; Supplemental Fig. S14; Supplemental Table S5). This outcome raised the possibility that aneuploidy selection might provide higher tolerance to oncogene stress due to overexpression of c-Myc, for example.
Figure 6.

Acquisition of the aneuploidy-specific profile reshapes gene expression and correlates with increasing c-Myc expression in human cancers. (A) Heat map showing genes from the indicated chromosomes whose expression (RNA) significantly correlated with the similarity index (similarity to the DNA copy number profile as determined by GISTIC from Fig. 4) of terminal tumors from the indicated PRG5 and Mad2 mice. Gene names are color-coded according to chromosome numbers and are sorted from top left to bottom right. See Supplemental Table S5 for a list of genes. (B) Enrichment of the CIN-induced gene expression profile (as shown in A and in Supplemental Table S5) in TCGA tumor cohorts gene lists of significantly up-regulated genes relative to their nontumor controls. (C) Enrichment of the CIN-induced gene expression profile (as shown in A and in Supplemental Table S5) in TCGA cohorts in correlation with MYC expression. A Spearman correlation coefficient between the gene's expression and MYC expression (among all samples) and the posterior probability that the gene is correlated with MYC was calculated. The ranked gene lists were then tested for enrichment with Bioconductor fgsea on the CIN-induced gene expression profile (Supplemental Table S5).
We next examined the relevance of this gene expression signature to RNA expression signatures in human cancers in the TCGA database (Fig. 6B,C). This revealed that 11 out of 24 of the TGCA cancer types in this cohort acquired a gene expression profile highly similar to that occurring in tumors from transient and chronic CIN (Fig. 6A). This gene profile was highly correlated with increased c-Myc expression in 13 out of 34 of the TCGA cancers (Fig. 6C) and was highly significant in tumor types with known involvement of c-MYC dysregulation such as diffuse large B-cell lymphoma (DLBC), uterine corpus endometrial carcinoma (UCEC), prostate adenocarcinoma (PRAD), and lung adenocarcinoma (LUAD) (Koh et al. 2010; Iwakawa et al. 2011; The Cancer Genome Atlas Research 2013; Nguyen et al. 2017). We conclude that transient and chronic CIN drive cancer cell evolution in our lymphoma models to acquire karyotypes producing a transcriptome signature that includes up-regulated c-Myc expression, and that is common in many human cancers.
Discussion
In his prophetic monograph, Boveri (1914) laid the foundations for cancer research in years to come. In the present study, we investigated Boveri's (1914) hypothesis and tested whether a transient period of CIN is sufficient to drive cancer. We indeed found that a very short time frame of CIN is sufficient to trigger tumor initiation and acceleration in p53 heterozygous and null mice, respectively. The fact that tumor latency in p53 heterozygous mice experiencing transient CIN is near-identical to tumor latency in p53null mice suggests that malignant transformation in p53null mice occurs within the same time frame in which we induced transient CIN in p53 heterozygous mice. Since p53 heterozygous mice that do not experience CIN rarely develop malignancies in their first year (Fig. 2D), we conclude that transient CIN is a powerful driver of cancer in a genetically predisposed setting (i.e., p53 heterozygosity).
We found that all lymphomas arising in induced p53+/− PRG5 mice have completely lost p53. This is consistent with whole-chromosome missegregation as the underlying mechanism for the early loss of the wild-type p53-containing chromosome (and acquisition of a second copy of chromosome 11 with the mutant p53 allele), although gene conversion cannot be excluded. Therefore, transient CIN drives tumorigenesis, at least partly, by facilitating loss of the p53 wild-type allele in this model, in line with our own earlier findings and those of others (Baker et al. 2009; Foijer et al. 2014). We also found that CIN increases the cancer-initiating clonal burden, as we found more tumor clones in PRG5 mice with CIN compared with the number of clones we observed in lymphomas arising in mice without CIN. While it has been reported that thymic lymphomas in p53−/− mice can also form from more than one tumor-initiating cell (Dudgeon et al. 2014), our study shows that transient or chronic CIN drives this further.
Tumors from both p53+/− and p53−/− mice experiencing transient CIN at an early age show similar genomic landscapes in which chromosomes 4, 5, 14, and 15 are gained. This genomic profile shows close similarities with previous reports in which thymic lymphomas in mice were provoked by chronic CIN induced through completely different mechanisms (Foijer et al. 2014, 2017). However, our DNA and RNA analyses of the multifocal tumor biopsies suggest that, in lymphomas arising following transient CIN, single cells that acquired the optimal genomic landscape are selected for and originate very early during tumor formation, as different regions in the tumor contain almost identical genome landscapes.
Single-cell DNA sequencing of early and late tumors provides further evidence for early, major aneuploidy-generating events involving missegregation of multiple chromosomes occurring during the 2-wk induction of CIN, consistent with the idea of the “Big Bang” model proposed for human breast and colon cancers (Sottoriva et al. 2015; Gao et al. 2016). We propose that within this time frame multiple optimal and suboptimal tumorigenic cells form, with optimal clones acquiring the genomic landscape that is also detected in advanced tumors. The same aneuploidy events emerge in Lck-Cre; Mad2f/f; p53f/f tumors, although these take much longer to reach the optimal karyotype. This is well in line with the “Big Bang” model, as both in case of chronic CIN and as well as transient CIN optimal and suboptimal clones form, but in case of chronic CIN, cells will frequently lose a recently-acquired more optimal karyotype, due to ongoing chromosome missegregation events, thus slowing down overall tumor growth. Overall, transient CIN generates rapid genomic chaos that provides an increased opportunity for formation of a tumor initiating cell in a short time frame, as seen also in catastrophic chromosome structural events such as chromothripsis (Stephens et al. 2011; Shoshani et al. 2021).
While we found that when CIN is chronic, thymocytes accumulate random aneuploidies, we also found that when we remove CIN (in our model for transient CIN), aneuploid cells rapidly disappear, indicating that aneuploid cells are selected against in vivo in agreement with earlier evidence that aneuploidy is not well-tolerated in normal cells (Santaguida and Amon 2015; Sheltzer et al. 2017). Further work is required to better understand the impact of aneuploidy on thymocytes in a chronic CIN background. Importantly, the two CIN models used in this work are unrelated (nonisogenic), therefore making it difficult to draw conclusions from a direct comparison beyond the analysis of karyotypic outcomes as we performed. Future work should address this by performing comparisons in isogenic models in which different levels and periods of CIN are induced.
Our data further show that p53 inactivation alone does not lead to a CIN phenotype (Supplemental Fig. S1I). However, once additional perturbations occur (such as c-Myc overexpression), p53−/− tumors acquire an aneuploid karyotype that frequently involves genome doubling. Further work is required to identify the drivers of CIN in the PRG5−/− lymphomas without Plk4 induction.
Altogether, we present evidence for a tissue-specific aneuploidy profile that optimizes malignant transformation. We propose that such optimal genomic landscapes exist for each tissue and cell type, with evidence for this already seen through our knowledge of recurrent chromosome abnormalities and of the involvement of chromosome instability (Foijer et al. 2017; Sansregret and Swanton 2017; Ben-David and Amon 2020). Interestingly, thymic lymphomas forming in developmentally arrested RAG2-deficient cells present a completely different genomic landscape lacking gains of chromosomes 1/4/5/14/15 but containing a unique rearrangement of chromosome 9qA4-5.3 (Bianchi et al. 2019), suggesting that even within the same cell type, cancer genome evolution is dependent on the initial genetic changes. Our work shows that clonal evolution underlies the development of thymic lymphoma and is accelerated both under transient and chronic CIN, with transient CIN being a more efficient driver of optimal karyotypes. Future work is now needed to reveal how tumors displaying transient and chronic CIN compare in their ability to adapt to new circumstances, such as exposure to anticancer drugs.
Materials and methods
Cell culture
Mouse embryonic fibroblasts (MEFs) were derived as previously described (Putkey et al. 2002). Briefly, E13.5 embryos were washed with PBS, and head, liver, and tail were removed. Embryos were minced in 0.05% trypsin (Gibco) and incubated for 15 min at 37°C. Dissociated cells were plated in DMEM media (Gibco) supplemented with 15% fetal bovine serum (Omega Scientific), 50 µg/mL penicillin and streptomycin (Gibco), 2 mM L-glutamine (Gibco), 1 µM 2-mercaptoethanol (Sigma), 1 mM sodium pyruvate (Gibco), and 0.1 mM nonessential amino acids (Gibco). Cells were maintained at 37°C, 5% CO2, and 3% O2. Doxycycline (Sigma) was dissolved in water and used at a final concentration of 1 μg/mL. Thymic lymphoma cells were derived as previously described (Jinadasa et al. 2011). Briefly, dissected tumors were dissociated using bent needles, and cells were plated in RPMI 1640 with 25 mM HEPES and 200 mM L-glutamine (Lonza), and supplemented with 10% fetal bovine serum (Hyclone), 1% penicillin and streptomycin (Gibco), 1% nonessential amino acids (Gibco), and 55 mM 2-mercaptoethanol (Sigma).
Mice
Generation of doxycycline-inducible Plk4 mouse with the tetracycline-responsive Plk4-YFP gene inserted downstream from the Col1a1 locus and the M2-rtTA gene inserted into the ROSA locus was previously described (Levine et al. 2017). To generate the PRG5 mice, Plk4-inducible mice were crossed with centrin-GFP mice (Hirai et al. 2016) and with mice carrying a knockout allele of p53 (Jacks et al. 1994). Mice homozygous for the Plk4 transgene and for the M2-rtTA gene and heterozygous for p53 were crossed with mice homozygous for centrin-GFP and heterozygous for p53 to generate cohort mice heterozygous for Plk4, M2-rtTA, and centrin-GFP, and homozygous or heterozygous for p53. Doxycycline was administered for indicated times through mouse diet containing 0.625 g/kg doxycycline hyclate (Envigo, TD.08541). Genotyping performed on tail DNA was done using the following primers: Plk4-YFP (CACAGGAACAGGCGTCTCTTCAAGTC and GTGCAGATGAACTTCAGGGTCAGCTTG), rtTA (AGGAGCATCAAGTAGCAAAAGAG and AAGAGCGTCAGCAGGCAGCA), centrin-GFP (GACAAGCAGAAGAACGGCATCAAGGTG and CTTGCTTCTGATCCTCAGTGAGCTC), p53 (ACAGCGTGGTGGTACCTTAT, TATACTCAGAGCCGGCCT, and TCCTCGTGCTTTACGGTATC), Rosa (AAAGTCGCTGAGTTGTTAT, GCGAAGAGTTTGTCCTCAACC, and GGAGCGGGAGAAATGGATATG), and Col1a1 (CCAGCTTCACCAGTTCAATCATCC and CAGTCCCTGTTTCTGCTGCTTGAATC). Mice were housed and cared for in an Association for the Assessment and Accreditation of Laboratory Animal Care-accredited facility, and all animal experiments were conducted in accordance with Institutional Animal Care and Use Committee-approved protocols. Lck-Cre; Mad2f/f; p53f/f mice were previously described (Foijer et al. 2017). Genotyping of Lck-Cre; Mad2f/f; p53f/f mice was performed as described previously (Foijer et al. 2017). Mice were housed and experiments were conducted according to Dutch law and approved by the Central Committee of Animal Experiments (CCD, permit AVD105002016465). Tissue sections from formalin-fixed paraffin embedded (FFPE) tissues and from cryopreserved tissues were collected as previously described (Vitre et al. 2015).
Centrosome enumeration
Centrosomes were visualized by the centrin-GFP marker in PRG5-derived mouse embryonic fibroblasts grown in chamber slides (ibidi) and were imaged (1 µM, 10×, 40×/1.35 NA) using the DeltaVision elite system (Applied Precision). Centrosomes visualized by the centrin-GFP marker in tissue cryosections were imaged (at 0.2-μm Z-sections with a Nikon 100× APO TIRF 1.49 NA objective) using the Nikon A1 scanning confocal microscope operated with NIS-Elements (Nikon). Additional quantification was done using an Olympus bx43 microscope equipped with a X-Cite series 120Q fluorescence lamp (60× magnification) and using Thermo Scientific Menzel-Gläser Deckgläser Coverslips ø12 mm #1.
Live-cell imaging
PRG5-derived mouse embryonic fibroblasts were seeded in CELLSTAR μClear 96-well plates (Greiner bio-one). Cells were stained with DNA SiR (Spirochrome) and 3 h later were imaged using a CQ1 spinning disk confocal system (Yokogawa Electric Corporation) with a 40× magnification at 37°C and 5% CO2. To induce Plk4 overexpression, doxycycline (Sigma, 1 µg/mL) was added 1 h prior to imaging. Live imaging of 8 × 3-µm z-sections was conducted for 24 h. Image acquisition and data analysis were performed using CQ1 software and ImageJ, respectively. For time-lapse imaging of Lck-Cre Mad2f/f p53f/f T-ALL cells, primary T-ALL-derived cell lines were transduced with H2B-Cherry using retroviral transduction as described previously (Foijer et al. 2005), cultured in LabTek imaging chambers (Nunc), and imaged on a DeltaVision Elite microscope (Applied Precision). Mitotic abnormalities were quantified by manual inspection of the movies.
Quantitative real-time PCR
RNA from PRG5-derived mouse embryonic fibroblasts was extracted using the Nucleospin RNA kit (Macherey Nagel). Mouse tissues were homogenized using a mechanical tissue homogenizer in Trizol reagent (Invitrogen), and RNA was extracted according to the manufacturer's guidelines. cDNA was prepared from 1 μg of total RNA using the high-capacity reverse transcription kit (ABI) according to the manufacturer's instructions. Quantitative real-time PCR was done in triplicate, using iTaq Universal SYBR green (Bio-Rad) and a CFX384 real-time PCR machine. For detection of Plk4, the following primers were used: GGAGAGGATCGAGGACTTTAAGG and CCAGTGTGTATGGACTCAGCT. The following primers were used as housekeeping control genes: Rsp9 (GACCAGGAGCTAAAGTTGATTGGA and GCGTCAACAGCTCCCGGGC) and Actg1 (TGGATCAGCAAGCAGGAGTATG and CCTGCTCAGTCCATCTAGAAGCA).
Protein analysis
Total protein from PRG5 mouse tissues (10-mg biopsies) was extracted in 500 µL of 2× Laemmli sample buffer, and 10 µL from each sample was loaded into a 10% acrylamide gel for SDS/PAGE separation. Proteins were transferred to a nitrocellulose membrane, blocked with 5% milk in Tris-buffered saline and 0.1% Tween-20 (TBS-T), and incubated overnight with the following primary antibodies: anti-GAPDH (1:10,000; Cell Signaling 2118), Myc (1:1000; Abcam ab32072). Immunoblots were washed with TBS-T and incubated with HRP-conjugated secondary antibodies (1:5,000; GE Healthcare) for 1 h at room temperature before development on films. For quantification of mitotic cells, cultured T-ALL cells were exposed to nocodazole (Sigma) or DMSO (Sigma) for 6 h, fixated in 70% ethanol, washed with PBS, and blocked in blocking buffer (0.05% Tween [Sigma]/2% BSA [Sigma] in PBS) and stained with FITC-conjugated MPM2 antibody (Upstate) for 2 h. Cells were washed in PBS and resuspended in staining buffer to label DNA, which was stained using FACS staining buffer (20 μg/mL propidium iodide [Sigma], 0.2 mg/mL RNaseA [Sigma] in PBS). Cells were analyzed on a FACSCanto analyzer (BD) and quantified using FlowJo software (BD).
Single-cell whole-genome DNA sequencing
Single cells from mouse thymuses and thymic tumors were isolated using flow cytometry sorting and prepared for sequencing as previously described (Foijer et al. 2017). Sequencing was performed using a NextSeq 500 machine (Illumina), up to 51, 77, or 84 cycles; single end. The generated data were subsequently demultiplexed using sample-specific barcodes and changed into fastq files using bcl2fastq (Illumina; version 1.8.4). Reads were afterward aligned to the mouse reference genome (GRCm38/mm10) using Bowtie2 (version 2.2.4) (Langmead and Salzberg 2012). Duplicate reads were marked with BamUtil (version 1.0.3) (Jun et al. 2015). The aligned read data (bam files) were analyzed with AneuFinder (Version 1.14.0) (Bakker et al. 2016). Following GC correction and blacklisting of artifact-prone regions (extreme low or high coverage in control samples), libraries were analyzed using the dnacopy and edivisive copy number calling algorithms with variable width bins (binsize: 1 Mb; stepsize: 500 kb) and breakpoint refinement (R = 20, confint = 0.95; other settings as default). Results were afterward curated by requiring a minimum concordance of 95% between the results of the two algorithms. Libraries with less than five reads per bin per chromosome copy (∼ 25,000 reads for a diploid genome) were discarded. AneuFinder gave unexpected results for sample P309. About half of the libraries showed an average copy number of 1.5 and the other half an average copy number of 3 (twice as high). Examination of the model results showed poor fits for the first group of libraries. Sample P309 was therefore reanalyzed with the developer version of AneuFinder (version 1.7.4; from GitHub) using a minimum ground ploidy of 2.5 (min.groun.plody = 2.5) and a maximum ground ploidy of 3.5 (max.ground.ploidy = 3.5). Results were subsequently curated as described above. As a final step, breakpoints that were located within 5 Mb of each other (across libraries) were grouped and centered using custom made R functions (R version 3.6.3, https://www.R-project.org) to prevent the heterogeneity scores from reaching unlikely high values just because breakpoints are off by a few bins.
Genome-wide karyotype measures
Copy number change score (CN change score)
For each cell, the CN change score was calculated as the average absolute difference between the observed copy number of each bin and the expected copy number of each bin (euploid genome). Bins had variable width and therefore a weighted average was used. The score of the sample was calculated as the average score of all cells.
Heterogeneity score
For each bin, the heterogeneity score was calculated as the proportion of all pairwise comparisons (cell 1 vs. cell 2, cell 1 vs. cell 3, etc.; total number = n × [n − 1]/2) that showed differences in copy number. The score of the sample was calculated as the average score of all bins.
Mean number of transitions per megabase (structural score)
For each cell, the mean number of transitions per megabase (structural score) was calculated as the total number of transitions (or breakpoints) divided by the total genome length (sum of bin widths). The score of the sample was calculated as the average score of all cells.
Uniform manifold approximation and projection (UMAP)
The UMAP function of the R package umap (McInnes et al. 2020) was used to compute the manifold approximation and projection of all single-cell DNA sequencing samples described here (arguments: random_state = 1, n_neighbors = 50, min_dist = 0.9999) (Fig. 1F,H,J; Supplemental Figs. S10B–D; S12). A matrix containing the copy number calls of the 1-Mb bins of the autosomes of all samples was used as input. Chromosome X was excluded due to differences in gender.
DNA fluorescent in situ hybridization (FISH)
Dissociated splenic and thymic cells were fixed using methanol/acetic acid (3:1), washed with fixative three times, and kept in fixative at −20°C until use. To make custom Trp53 BAC probes, the following BACs were ordered from bacpac (https://bacpacresources.org): RP24-285L20 and RP23-243M15. BACs were isolated from 50-mL bacterial cultures using the BACMAX Bac extraction kit (Epicentre). Isolated BACs were sonicated (10 cycles of 15 sec “on” 45 sec “off” at constant intensity with power set to “3”; Branson Sonifier 450) and labeled with TM-rhodamine Label-IT (Mirus). Labeled BAC probes were suspended in commercial chromosome paint probe for chromosome 11 (Metasystems). DNA-FISH was performed by applying probes onto samples and covering with a glass coverslip. Genomic DNA and probes were codenatured for 2 min at 75°C by placing the slide on a preheated metal plate. Samples were hybridized overnight at 37°C in a dark humidified chamber. Slides were subsequently washed with 0.4× SSC for 2 min at 72°C and rinsed in 2× SSC, 0.05% Tween-20 for 30 sec at room temperature. Slides were then rinsed in PBS, counterstained with DAPI, and mounted using Prolong Gold (Invitrogen). FISH images were acquired on a DeltaVision elite system (Applied Precision) at 40× magnification (10 × 0.5-μm z-sections). Maximum intensity projections were generated using the softWoRx program.
Preparation of DNA/RNA from tissue biopsies for sequencing
Tissues were snap-frozen in liquid nitrogen and kept at −80°C. Biopsies were taken from frozen tissues placed on a −80°C chilled metallic stage. Stage and tools were cleaned between different tissues and biopsies to minimize cross-contamination of DNA and RNA. Biopsies were homogenized using a QIAshresdder (Qiagen), and DNA/RNA were prepared using the AllPrep DNA/RNA mini kit (Qiagen) according to the manufacturer's guidelines.
TCR sequencing
TCR libraries were prepared using an immunoSEQ kit mmTCRB according to the user manual (Adaptive Biotechnologies). TCR libraries were sequenced using illumina Nextseq 500 (DNA Link Facility), and TCR sequences were analyzed using the immunoSEQ Analyzer platform (Adaptive Biotechnologies). Rare sequences (found at <1%) with 100% identity to dominant sequences were filtered and are marked in Supplemental Table S3.
Whole-genome DNA sequencing (WGS)
Library preparation: 450 ng of genomic DNA from each sample was fragmented by adaptive focused acoustics (E220 Focused Ultrasonicator, Covaris) to produce an average fragment size of 350 bp. Fragmented DNA was purified using Agencourt AMPure XP beads (Beckman Coulter), and sequencing libraries were generating using the KAPA Hyper preparation kit (KAPA Biosystems) following the manufacturer's instructions using four cycles of amplification. The quality of the library was assessed using a High Sensitivity D1000 kit on a 2200 TapeStation instrument (Agilent Technologies). Sequencing was performed using the NovaSeq 6000 sequencing system (Illumina), generating 150-bp paired-end reads to obtain 10× coverage.
RNA sequencing
cDNA libraries were prepared using the TruSeq stranded mRNA sample preparation kit (Illumina) according to the manufacturer's guidelines. Then, cDNA libraries were sequenced on an Illumina HiSeq 4000 using single-read, 50-cycle runs. FASTQ files were processed to assess quality by determining general sequencing bias, clonality, and adapter sequence contamination. RNA sequencing reads were aligned to the mm10 mouse reference genome using STAR (Dobin et al. 2013). Gene expression levels TPM and raw counts were calculated by using RSEM (Li and Dewey 2011). The log2(TPM) values of selected TRBV transcripts were shown in a heat map where the blue color stands for low expression and red for high expression. The heat map was generated by the “pheatmap” package for the R program (https://www.R-project.org).
GISTIC 2.0 similarity index
A CNVkit was used to determine DNA copy number from tumor WGS. We executed GISTIC 2.0 broad-level analysis (Mermel et al. 2011) using CNVkit segment files as input. Amplifications and deletions with q < 0.01 were kept, leaving chromosomes 4, 5, 12, 14, and 15 as significant amps. Segments were binned into 100,000-bp bins, each keeping their respective segment mean. Bin value z-scores were applied to the data set, followed by an ecdf function. The resulting measure was then subtracted from 1 to create a “similarity index,” indicating a similarity to the “optimal clone,” which is described by significantly amplified regions.
CNV heat maps
R Bioconductor package ComplexHeatmap was used to plot the broad-level gistic results from the file “broad_values_by_arm.txt.”
RNA-seq vs. normal thymus
The R Bioconductor packages edgeR and limma were used to implement the limma-voom method for differential expression analysis. Lowly expressed genes were removed, and then a trimmed mean of M-values (TMM) normalization was applied. The experimental design was modeled upon the experimental treatment of tumors (∼0 + treatment). The Voom method was employed to model the mean-variance relationship in the log cpm values, after which lmFit was used to fit per-gene linear models and empirical Bayes moderation was applied with the eBayes function.
RNA-seq using GISTIC 2.0 index as covariate
Samples’ similarity indices were used as continuous covariates in the RNA-seq gene expression analysis using the limma-voom method to determine genes whose expression significantly correlates with the similarity index. The experimental design was modeled upon the gistic index and batch (∼gistic + batch).
Data availability
RNA sequencing has been deposited in the Gene Expression Omnibus (GEO) database, accession number GSE161728. Whole-genome DNA sequencing has been deposited in the Sequence Read Archive (SRA) database, accession number PRJNA678822. Single-cell whole-genome sequencing has been deposited in the European Nucleotide Archive (ENA) under accession number PRJEB41176.
Supplementary Material
Acknowledgments
This work was funded by a grant from the National Institutes of Health (R35 GM122476 to D.W.C.). D.W.C. receives salary support from the Ludwig Institute for Cancer Research. This work was further funded by a Dutch Cancer Society Grant (2017-RUG-11457) to F.F. This work was partially supported by the National Institutes of Health (NIH), grant UL1TR001442 of Clinical and Translational Science Awards. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. L.d.H. received support from the Nora Baart Foundation, the K.F. Hein Foundation, and the Jo Kolk Foundation. We thank Andrew Shiau for providing access to the CQ1 spinning-disk confocal system. We thank Dr. Nissi Varki (University of California at San Diego Pathology Core) for histology sample analysis. We thank Kristen Jepsen from the University of California at San Diego Institute for Genomic Medicine Genomics Center for help with whole-genome DNA sequencing (NIH Shared Instrument Grant S10 OD026929). We thank Stefan Kessler for help with uploading the raw sequencing data to public databases.
Author contributions: O.S. and D.W.C. conceived the project and wrote the manuscript. O.S. designed, performed, and analyzed the experiments. B.B. performed and analyzed the experiments related to samples from the Mad2 mouse cohort. Y.W. assisted in maintaining the Plk4 mouse cohort and helped with breeding and samples collections. D.H.K. assisted with live-cell imaging. M.M. helped with tumor sample processing and imaging. M.A.D. helped with sample processing for RNA expression analysis. J.A. helped with maintenance of the Plk4 mouse colony and with RNA library preparation for RNA sequencing. O.Z. helped with analysis of Plk4 cohort RNA sequencing analysis. L.d.H. assisted with MEF experiments. A.M., R.W., R.S., and K.F. helped with analyzing RNA sequencing, whole-genome DNA sequencing, and single-cell whole-genome DNA sequencing analysis. D.C.J.S. and A.E.T. helped prepare samples for single-cell whole-genome DNA sequencing. B.V. helped generate the Plk4 mouse cohort. All authors provided input on the manuscript, and D.W.C., F.F., and O.S. supervised all aspects of the work.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348319.121.
Competing interest statement
The authors declare no competing interests.
References
- Baker DJ, Jin F, Jeganathan KB, van Deursen JM. 2009. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell 16: 475–486. 10.1016/j.ccr.2009.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker B, Taudt A, Belderbos ME, Porubsky D, Spierings DC, de Jong TV, Halsema N, Kazemier HG, Hoekstra-Wakker K, Bradley A, et al. 2016. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol 17: 115. 10.1186/s13059-016-0971-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW. 2008. Centrosome amplification can initiate tumorigenesis in flies. Cell 133: 1032–1042. 10.1016/j.cell.2008.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David U, Amon A. 2020. Context is everything: aneuploidy in cancer. Nat Rev Genet 21: 44–62. 10.1038/s41576-019-0171-x [DOI] [PubMed] [Google Scholar]
- Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L, Riparbelli M, Lehmann L, Gatt MK, Carmo N, Balloux F, Callaini G, Glover DM. 2005. SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol 15: 2199–2207. 10.1016/j.cub.2005.11.042 [DOI] [PubMed] [Google Scholar]
- Bianchi JJ, Murigneux V, Bedora-Faure M, Lescale C, Deriano L. 2019. Breakage-fusion-bridge events trigger complex genome rearrangements and amplifications in developmentally arrested T cell lymphomas. Cell Rep 27: 2847–2858.e4. 10.1016/j.celrep.2019.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveri T. 1902. über mehrpolige mitosen als mittel zur analyse des zellkerns. Verhandl Phys Med Ges 35: 67–90. (English translation by: Gluecksohn-Waelsch S. 1974. On multipolar mitosis as a means of analysis of the cell nucleus. In Foundations of experimental embryology, 2nd ed. [ed. Willier BH, Oppenheimer JM], pp. 74–97. Prentice-Hall, Englewood Cliffs, NJ.). [Google Scholar]
- Boveri T. 1914. Zur frage der entstehung maligner tumoren. Verlag von Gustav Fischer, Jena, Germany. (English translation and annotation by: Harris H. 2008. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci 121: 1–84. 10.1242/jcs.025742 [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2013. Integrated genomic characterization of endometrial carcinoma. Nature 497: 67–73. 10.1038/nature12113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho PA, Bury L, Shahbazi MN, Liakath-Ali K, Tate PH, Wormald S, Hindley CJ, Huch M, Archer J, Skarnes WC, et al. 2015. Over-expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol 5: 150209. 10.1098/rsob.150209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudgeon C, Chan C, Kang W, Sun Y, Emerson R, Robins H, Levine AJ. 2014. The evolution of thymic lymphomas in p53 knockout mice. Genes Dev 28: 2613–2620. 10.1101/gad.252148.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foijer F, Wolthuis RM, Doodeman V, Medema RH, te Riele H. 2005. Mitogen requirement for cell cycle progression in the absence of pocket protein activity. Cancer Cell 8: 455–466. 10.1016/j.ccr.2005.10.021 [DOI] [PubMed] [Google Scholar]
- Foijer F, Xie SZ, Simon JE, Bakker PL, Conte N, Davis SH, Kregel E, Jonkers J, Bradley A, Sorger PK. 2014. Chromosome instability induced by Mps1 and p53 mutation generates aggressive lymphomas exhibiting aneuploidy-induced stress. Proc Natl Acad Sci 111: 13427–13432. 10.1073/pnas.1400892111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foijer F, Albacker LA, Bakker B, Spierings DC, Yue Y, Xie SZ, Davis S, Lutum-Jehle A, Takemoto D, Hare B, et al. 2017. Deletion of the MAD2L1 spindle assembly checkpoint gene is tolerated in mouse models of acute T-cell lymphoma and hepatocellular carcinoma. Elife 6: e20873. 10.7554/eLife.20873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. 2009. A mechanism linking extra centrosomes to chromosomal instability. Nature 460: 278–282. 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Davis A, McDonald TO, Sei E, Shi X, Wang Y, Tsai PC, Casasent A, Waters J, Zhang H, et al. 2016. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet 48: 1119–1130. 10.1038/ng.3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DJ, Resio B, Pellman D. 2012. Causes and consequences of aneuploidy in cancer. Nat Rev Genet 13: 189–203. 10.1038/nrg3123 [DOI] [PubMed] [Google Scholar]
- Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. 2005. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol 7: 1140–1146. 10.1038/ncb1320 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hansemann D. 1890. Ueber asymmetrische zelltheilung in epithelkrebsen und deren biologische bedeutung (On the asymmetrical cell division in epithelial cancers and its biological significance). Archiv f pathol Anat 119: 299–326. 10.1007/BF01882039 [DOI] [Google Scholar]
- Hirai M, Chen J, Evans SM. 2016. Generation and characterization of a tissue-specific centrosome indicator mouse line. Genesis 54: 286–296. 10.1002/dvg.22937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoevenaar WHM, Janssen A, Quirindongo AI, Ma H, Klaasen SJ, Teixeira A, van Gerwen B, Lansu N, Morsink FHM, Offerhaus GJA, et al. 2020. Degree and site of chromosomal instability define its oncogenic potential. Nat Commun 11: 1501. 10.1038/s41467-020-15279-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Lan W, Niessen S, Hoover H, Cleveland DW. 2010. Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J Cell Biol 188: 191–198. 10.1083/jcb.200911102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakawa R, Kohno T, Kato M, Shiraishi K, Tsuta K, Noguchi M, Ogawa S, Yokota J. 2011. MYC amplification as a prognostic marker of early-stage lung adenocarcinoma identified by whole genome copy number analysis. Clin Cancer Res 17: 1481–1489. 10.1158/1078-0432.CCR-10-2484 [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. 1994. Tumor spectrum analysis in p53-mutant mice. Curr Biol 4: 1–7. 10.1016/S0960-9822(00)00002-6 [DOI] [PubMed] [Google Scholar]
- Jinadasa R, Balmus G, Gerwitz L, Roden J, Weiss R, Duhamel G. 2011. Derivation of thymic lymphoma T-cell lines from Atm−/− and p53−/− mice. J Vis Exp 2598. 10.3791/2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun G, Wing MK, Abecasis GR, Kang HM. 2015. An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data. Genome Res 25: 918–925. 10.1101/gr.176552.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM, Bieberich CJ, Dang CV, Nelson WG, Yegnasubramanian S, De Marzo AM. 2010. MYC and prostate cancer. Genes Cancer 1: 617–628. 10.1177/1947601910379132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MS, Bakker B, Boeckx B, Moyett J, Lu J, Vitre B, Spierings DC, Lansdorp PM, Cleveland DW, Lambrechts D, et al. 2017. Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev Cell 40: 313–322.e5. 10.1016/j.devcel.2016.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323. 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes CAM, Mesquita M, Cunha AI, Cardoso J, Carapeta S, Laranjeira C, Pinto AE, Pereira-Leal JB, Dias-Pereira A, Bettencourt-Dias M, et al. 2018. Centrosome amplification arises before neoplasia and increases upon p53 loss in tumorigenesis. J Cell Biol 217: 2353–2363. 10.1083/jcb.201711191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, Melville J. 2020. UMAP: uniform manifold approximation and projection for dimension reduction. arXiv 1802.03426v3 [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. 2011. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12: R41. 10.1186/gb-2011-12-4-r41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor RM, van Deursen JM. 2016. Aneuploidy in cancer and aging. Annu Rev Genet 50: 45–66. 10.1146/annurev-genet-120215-035303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, Papenhausen P, Shao H. 2017. The role of c-MYC in B-cell lymphomas: diagnostic and molecular aspects. Genes (Basel) 8: 116. 10.3390/genes8040116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putkey FR, Cramer T, Morphew MK, Silk AD, Johnson RS, McIntosh JR, Cleveland DW. 2002. Unstable kinetochore–microtubule capture and chromosomal instability following deletion of CENP-E. Dev Cell 3: 351–365. 10.1016/S1534-5807(02)00255-1 [DOI] [PubMed] [Google Scholar]
- Rowald K, Mantovan M, Passos J, Buccitelli C, Mardin BR, Korbel JO, Jechlinger M, Sotillo R. 2016. Negative selection and chromosome instability induced by Mad2 overexpression delay breast cancer but facilitate oncogene-independent outgrowth. Cell Rep 15: 2679–2691. 10.1016/j.celrep.2016.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansregret L, Swanton C. 2017. The role of aneuploidy in cancer evolution. Cold Spring Harb Perspect Med 7: a028373. 10.1101/cshperspect.a028373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Amon A. 2015. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol 16: 473–485. 10.1038/nrm4025 [DOI] [PubMed] [Google Scholar]
- Serçin O, Larsimont JC, Karambelas AE, Marthiens V, Moers V, Boeckx B, Le Mercier M, Lambrechts D, Basto R, Blanpain C. 2016. Transient PLK4 overexpression accelerates tumorigenesis in p53-deficient epidermis. Nat Cell Biol 18: 100–110. 10.1038/ncb3270 [DOI] [PubMed] [Google Scholar]
- Sheltzer JM, Ko JH, Replogle JM, Habibe Burgos NC, Chung ES, Meehl CM, Sayles NM, Passerini V, Storchova Z, Amon A. 2017. Single-chromosome gains commonly function as tumor suppressors. Cancer Cell 31: 240–255. 10.1016/j.ccell.2016.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshani O, Massalha H, Shani N, Kagan S, Ravid O, Madar S, Trakhtenbrot L, Leshkowitz D, Rechavi G, Zipori D. 2012. Polyploidization of murine mesenchymal cells is associated with suppression of the long noncoding RNA H19 and reduced tumorigenicity. Cancer Res 72: 6403–6413. 10.1158/0008-5472.CAN-12-1155 [DOI] [PubMed] [Google Scholar]
- Shoshani O, Brunner SF, Yaeger R, Ly P, Nechemia-Arbely Y, Kim DH, Fang R, Castillon GA, Yu M, Li JSZ, et al. 2021. Chromothripsis drives the evolution of gene amplification in cancer. Nature 591: 137–141. 10.1038/s41586-020-03064-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, Weaver BA. 2013. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci 110: E4134–E4141. 10.1073/pnas.1317042110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Hernando E, Díaz-Rodríguez E, Teruya-Feldstein J, Cordón-Cardo C, Lowe SW, Benezra R. 2007. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 11: 9–23. 10.1016/j.ccr.2006.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata D, et al. 2015. A Big Bang model of human colorectal tumor growth. Nat Genet 47: 209–216. 10.1038/ng.3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. 2011. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144: 27–40. 10.1016/j.cell.2010.11.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopsack KH, Whittaker CA, Gerke TA, Loda M, Kantoff PW, Mucci LA, Amon A. 2019. Aneuploidy drives lethal progression in prostate cancer. Proc Natl Acad Sci 116: 11390–11395. 10.1073/pnas.1902645116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchova Z, Pellman D. 2004. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 5: 45–54. 10.1038/nrm1276 [DOI] [PubMed] [Google Scholar]
- Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, et al. 2018. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell 33: 676–689.e3. 10.1016/j.ccell.2018.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitre B, Holland AJ, Kulukian A, Shoshani O, Hirai M, Wang Y, Maldonado M, Cho T, Boubaker J, Swing DA, et al. 2015. Chronic centrosome amplification without tumorigenesis. Proc Natl Acad Sci 112: E6321–E6330. 10.1073/pnas.1519388112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. 2007. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11: 25–36. 10.1016/j.ccr.2006.12.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing has been deposited in the Gene Expression Omnibus (GEO) database, accession number GSE161728. Whole-genome DNA sequencing has been deposited in the Sequence Read Archive (SRA) database, accession number PRJNA678822. Single-cell whole-genome sequencing has been deposited in the European Nucleotide Archive (ENA) under accession number PRJEB41176.