

Abstract
The deployment of molecular biomarkers that are indicative of sensitivity to tumor-targeted or immune-targeted cancer therapies improves the outcome of individual patients and increases the chances of successful drug approval. However, for many lethal malignancies, the majority of clinical trials are conducted with patients who do not have biomarkers and hence they miss the target.
In the era of precision cancer medicine, biomarkers are increasingly valuable for selecting patients in the clinical cancer research and practice therapeutic settings. Biomarkers with the potential to inform treatment, including but not limited to pathogenic genomic alterations that alter druggable signaling pathways and/or cause specific immune deregulation, are identifiable by testing approaches such as next-generation sequencing. Although molecular testing is increasingly commonplace in many clinical settings, biomarker-guided therapy is not yet the norm, and key questions remain about the contexts in which gene- or immune-targeted treatments should be given to patients with cancer without biomarker selection, and what the implications are for treatment outcomes. Meta-analysis of ~85,000 participants in clinical trials1–3 has demonstrated that patients who receive therapy selected by a biomarker have significantly better outcomes. Indeed, median objective response rates (ORRs) in phase 1 clinical trials with genomic biomarker selection were ~42%, whereas median ORRs to targeted agents in phase 1 and phase 2 trials lacking a biomarker were ~5%; cytotoxic agents without a biomarker had median ORRs of ~5–11% (refs.1–3). The use of biomarkers also correlated with greater success in the drug-development pipeline. In a recent review of reasons for failure of experimental anticancer drugs in late-stage clinical development, drug programs that reached phase 3 trials but never gained approval from the US Food and Drug Administration (FDA) were compared with anti-cancer drugs that achieved FDA approval in that time period4. Only 16% of failed drug programs adopted a biomarker-driven rationale for patient selection, versus 57% of successful drug programs (P <0.001)4. Therefore, the use of a biomarker is associated with significantly better outcomes for patients and for drug development.
Non–small-cell lung cancer (NSCLC) is now the ‘poster child’ solid tumor type for the benefit of genomic stratification, despite the fact that this cancer almost uniformly responded poorly to treatments in the past. Multiple driver mutations that are recurrent in NSCLC can be targeted with available inhibitors, and testing for these molecular alterations, including alterations in ALK, BRAF, EGFR, MET (skipping of exon 14), RET and ROS1, as well as immunohistochemistry (IHC) of the immune-checkpoint ligand PD-L1, are now approved by the FDA and/or within guidelines of the National Comprehensive Cancer Network. ORRs for drugs targeting the molecular alterations of ALK, BRAF, EGFR, MET (skipping of exon 14), RET or ROS1 have been reported in the range of ~45–85% in NSCLCs that bear the cognate alteration(s)5. However, each of these alterations occur in only ~1–10% of lung cancers5; thus, targeted drugs would be unlikely to be efficacious in a large proportion of patients. Indeed, historically, FDA approval for the EGFR inhibitor gefitinib was revoked after a post-marketing phase 3 randomized controlled trial (RCT) failed to show benefit in unselected NSCLC. However, subsequent trials of patients with NSCLC selected for EGFR alterations as a criterion for gefitinib treatment reported ORRs of ~55 to 75%, which led to re-approval of gefitinib by the FDA5. Of note, by the time the FDA approval was rescinded, gefitinib had been administered to over 20,000 unselected patients with NSCLC, the vast majority of whom derived no benefit from the treatment6. Thus, these experiences not only underscore the value of biomarker stratification in improving outcomes for patients but also point to potential consequences of excess treatments and cost incurred in absence of biomarker selection.
Critically, treatment decisions and clinical trials for many extremely difficult-to-treat cancers are for the most part still executed in absence of biomarker selection, with key examples of this being pancreatic cancer and glioblastoma multiforme (GBM). Pancreatic cancer is the fourth-most-common cause of cancer-related mortality in the USA. It is one of the few malignancies for which incidence approximates prevalence, as patients almost always succumb to the tumor. The 1-year survival for pancreatic cancer is ~20%, and the 5-year survival is about ~3% for metastatic disease, with most patients being diagnosed at advanced stages. The effort that has been invested in trials of pancreatic cancer is enormous, with inadequate return. In a review of contemporary RCTs, a total of 35 different agents or combinations had been tested in RCTs for patients with advanced pancreatic cancer in the 25 years before 2016, but only a dismal 11% had been accepted into clinical practice, and notably, no trial had biomarker stratification7. On the other hand, recent advances have provided optimism that molecularly matched treatment can make this disease more tractable. For example, olaparib, a potent inhibitor of poly (ADP-ribose) polymerase, when administered as maintenance therapy in patients with germline mutations in the tumor suppressor–encoding genes BRCA1 and BRCA2, resulted in a significantly longer median progression-free survival than that of patients who received placebo (7.4 months versus 3.8 months; P = 0.004)8. Olaparib is now FDA approved as maintenance therapy for advanced pancreatic cancer, on the condition of germline BRCA mutation. Even so, our enumeration of modern therapeutic trials for pancreatic cancer (from ClinicalTrials. gov; 1 January through 31 December 2019) showed that only 14 of 79 trials (17.7%) (expected accrual, 1,206 of 8,582 patients (14.1%)) planned for biomarker selection. While these numbers may in part reflect an ongoing need for better tailored therapies for pancreatic cancer, especially in advanced disease, the uptake of biomarker-selected trials is also hampered by the paucity of reliable biomarkers to provide guidance for current therapies. Thus, it will be imperative to focus ongoing research activities on the discovery and validation of molecular biomarkers with clinical utility.
GBM, as the deadliest primary brain tumor, with a 5-year survival rate of <5% (ref.9), is another example of a difficult-to-treat malignancy for which biomarker stratification has not been well addressed. Intriguingly, evidence exists for promoter methylation of the gene encoding the DNA-repair molecule MGMT as an instructive biomarker for response to a widely used GBM therapy, temozolomide. In a study that randomly assigned patients to receive concomitant temozolomide with radiotherapy versus radiotherapy alone, those who received temozolomide and had methylation of MGMT had a significant survival advantage (21.7 months versus 15.3 months; P = 0.007)9. However, temozolomide is still routinely given regardless of MGMT status, potentially due to a lack of better alternatives for the broader patient population. Similar to the scenario for pancreatic cancer, data from the website ClinicalTrials.gov indicate that only 739 of the 3,528 patients expected to accrue on therapeutic trials in 2019 (22.7%) would be selected by a biomarker in GBM trials. The broader GBM biomarker landscape often includes mutations in IDH1 and IDH2 (which encode isocitrate dehydrogenases), as well as mutations and/or amplifications of EGFR, but beyond these, biomarker discovery for GBM is again a limiting factor in the improvement of targeted therapy selections.
Experiences with other tumor types provide examples of how deploying highly targeted drugs in clinical trials in unselected populations may obscure evidence of drug efficacy in some patients. For example, in the COMET-1 and COMET-2 trials, patients with metastatic castration-resistant prostate cancer received cabozantinib, a potent multi-kinase inhibitor whose targets include MET (a receptor tyrosine kinase); however, testing for MET status or the other kinase targets was not included in the study design10. These trials found no significant impact of cabozantinib on outcome10. However, contemporaneous data have shown that the response to cabozantinib can be considerable when it is given to the patients with the relevant deleterious MET alteration11. It is conceivable that if the two previously ‘failed’ trials of prostate cancer had selected for patients’ tumors that had MET alterations, a beneficial response may have been observed, albeit in a subset of patients. It is plausible that the lack of a biomarker is also resulting in failures in drug development. For example, indoleamine-pyrrole 2,3-dioxygenase is highly elevated in <5% of cancers12. However, epacadostat, an inhibitor of indoleamine-pyrrole 2,3-dioxygenase, was tested in a RCT of unselected patients. The trial failed to show activity of epacadostat12, which led to abandonment of the drug and possibly the target. In light of this, looking forward, it may be worthwhile to re-evaluate whether existing drugs that may have failed in prior trials instead show efficacy in selected populations, and, in general, to consider the trial a success, or at least worthy of further study, even if a certain drug is relevant only for a defined subset of patients and not the unselected overall trial cohort.
Recent tissue-agnostic FDA approvals have demonstrated the wide-reaching value of a robust and reliable biomarker. For example, fusions of neurotrophic tyrosine receptor kinase (NTRK) are found in only 0.3% of adult and pediatric tumors; thus, across cancer types, in unselected patients, the response rate for drugs targeting these alterations would be predicted to be around ~0.15% (ref.13). However, after reports of striking ORRs (between 57% and 75%) for the NTRK inhibitors larotrectinib and entrectinib in patients with NTRK fusion, these drugs were granted FDA approval for any solid tumor with an NTRK fusion. In the context of immune-checkpoint inhibitors, the ORR for pembrolizumab is ~40% in patients whose tumors have high microsatellite instability, a condition that leads to a high tumor mutation burden, compared with ORRs of ~15–20% in unselected cohorts. Notably, the FDA has recently granted approval for the use of pembrolizumab in patients with high–intermediate or high tumor mutation burden (ten or more mutations per megabase) regardless of microsatellite status. Still, only 37 of 413 contemporary immunotherapeutic clinical trials listed at ClinicalTrials.gov in 2019 (~9%) (expected accrual, 5,602 of 57,853 patients (9.7%)) included biomarker for selecting patients. However, as research into correlates of response to immunotherapy is evolving at a rapid pace, there is high potential for increased adoption of biomarker-guided immunotherapy in the coming years.
Conclusions and future directions
The advent of next-generation sequencing and transcriptomic sequencing has enabled the rapid identification of potentially pathogenic molecular abnormalities. In addition, many new agents with striking ability to modify the action of specific aberrant proteins have been developed. However, both a deep understanding of the mechanism of action of a drug as well as exploitation of molecular biomarkers to choose the appropriate patients for treatment are essential for propelling the field of precision cancer medicine forward (Fig. 1). As highlighted in the examples provided above, progress toward widespread adoption of biomarker-guided treatment has been inconsistent and calls both for improved biomarker-discovery efforts and for changes to trial design and policy to increase implementation of biomarker selection. In addition to rapidly advancing genomic technologies, vast potential lies in transcriptomic, proteomic and immunomic data as minable resources for biomarker discovery, both preclinically and via correlative studies in clinical trials14. However, continued progress will need to overcome key challenges to discovery, such as the availability of suitable datasets for analysis and validation of a proposed biomarker. The need for improved patient selection extends to non-targeted therapy such as chemotherapy and immunotherapy, and to the need to define biomarkers for response to combination therapies as well; patient-to-patient variability and independent drug action may be sufficient to explain the superiority of many FDA-approved drug combinations, even in the absence of drug additivity or synergy15,16. A large volume of literature now suggests that giving targeted drugs to unselected patients is associated with paltry ORRs of <5% (refs.1–3). While trials even of unselected patients are occasionally successful just by chance alone or because a large enough fraction of the unselected population has the appropriate biological underpinnings, it is becoming increasingly clear that moving away from unselected treatment will stand to benefit patients and facilitate advances in the field of oncology research in general. Increasing the implementation of biomarker-guided trials will require large-scale efforts from key opinion leaders and a departure from the treatment dogma that some cancers do not require a biomarker for treatment. This is a formidable but surmountable task that can be accomplished through adaptation and standardization of the treatment paradigm in oncology.
Fig. 1 |. Example of responders versus non-responders among hypothetical patients with cancer of unknown primary and lung metastases.
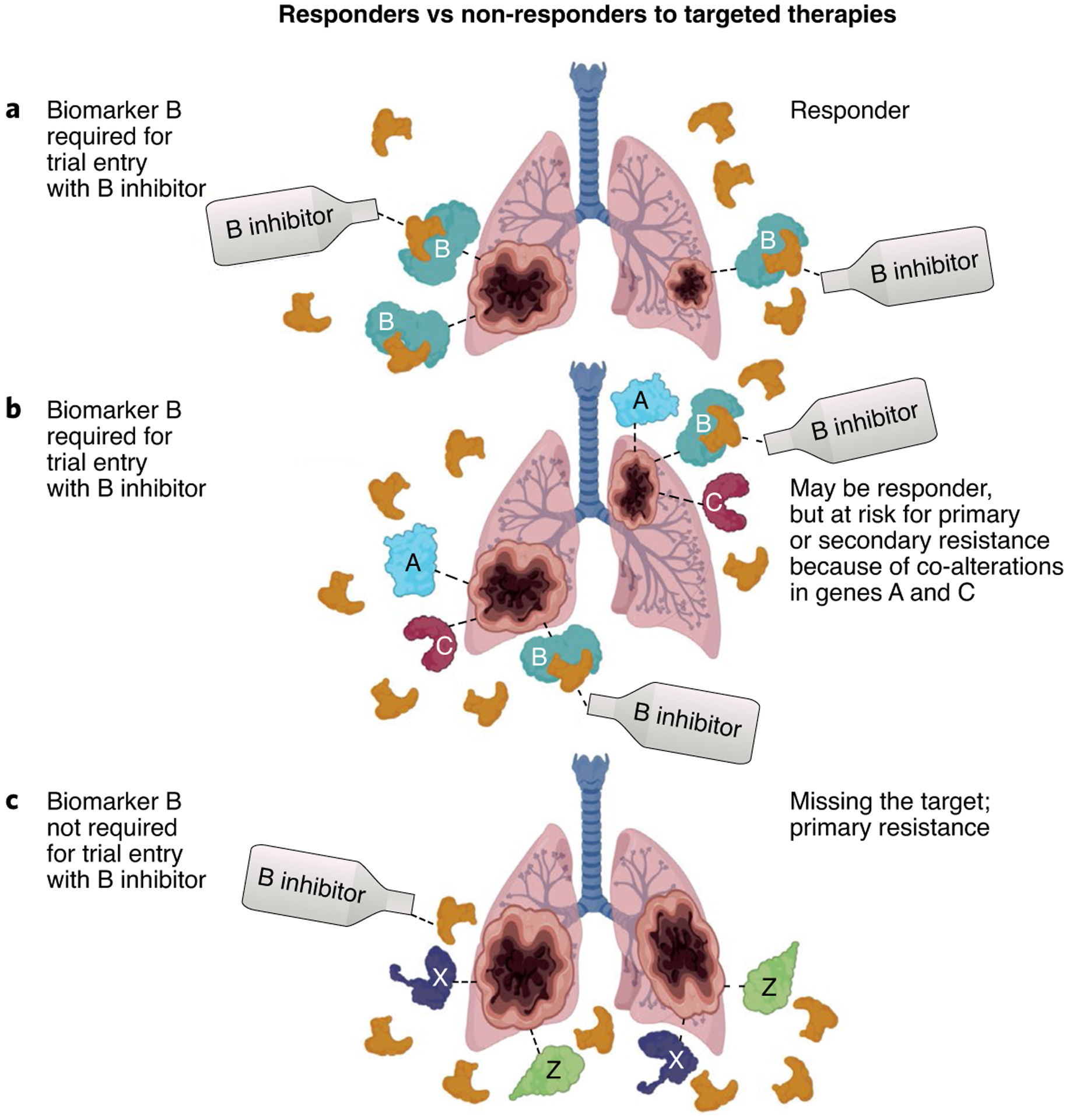
a, A patient with cancer and mutations in the gene encoding B who is enrolled in a clinical trial of a B inhibitor that required an alteration in the gene encoding B for trial entry. The patient would be expected to respond to the B inhibitor. b, A patient with cancer and mutations in genes encoding A, B and C who is enrolled in a clinical trial of a B inhibitor that required an alteration in the gene encoding B for trial entry. The patient may respond or may show primary or secondary resistance to the B inhibitor because of the concomitant presence of mutations in the genes encoding A and C. c, A patient with cancer with mutations in genes encoding X and Z who is enrolled in a clinical trial of a B inhibitor (because the trial has no biomarker selection criteria). It would be expected that the tumor would be resistant.
Acknowledgements
Funded in part by the Joan and Irwin Jacobs Fund, and by the National Cancer Institute grant P30 CA023100 (R.K.).
Competing interests
S.K. consults for Foundation Medicine, has received a speaker’s fee from Roche, and has research grants from ACT Genomics, Sysmex, Konica Minolta and OmniSeq. R.K. has research funding from Incyte, Genentech, Merck Serono, Pfizer, Sequenom, Foundation Medicine, Guardant Health, Grifols, Boehringer Ingelheim and Konica Minolta; has consultant and/or speaker fees from LOXO, X-Biotech, Actuate Therapeutics, Genentech, Pfizer, Roche and NeoMed; has an equity interest in IDbyDNA and Curematch; and is a board member of CureMatch and CureMetrix and co-founder of CureMatch.
References
- 1.Jardim DL et al. J. Natl. Cancer Inst 107, djv253 (2015).26378224 [Google Scholar]
- 2.Schwaederle M et al. J. Clin. Oncol 33, 3817–3825 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwaederle M et al. JAMA Oncol. 2, 1452–1459 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Jardim DL, Groves ES, Breitfeld PP & Kurzrock R Cancer Treat. Rev 52, 12–21 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Blumenthal GM et al. J. Clin. Oncol 33, 1008–1014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herbst RS et al. J. Clin. Oncol 22, 785–794 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Goel N & Reddy SS Surg. Oncol. Clin. N. Am 26, 767–790 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Golan T et al. N. Engl. J. Med 381, 317–327 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegi ME et al. N. Engl. J. Med 352, 997–1003 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Pond GR et al. J. Clin. Oncol 36, 225–225 (2018).29148892 [Google Scholar]
- 11.Adashek JJ, Salgia M & Pal SK Urol. Case Rep 19, 60–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long GV et al. Lancet Oncol. 20, 1083–1097 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Okamura R et al. JCO Precis. Oncol 10.1200/PO.18.00183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kato S et al. Nat. Commun 11, 4965 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adashek JJ, Subbiah V & Kurzrock R Trends Cancer 10.1016/j.trecan.2020.08.009 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Palmer AC & Sorger PK Cell 171, 1678–1691.e1613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]