

Summary
Extracellular vesicles (EVs) are released into blood from multiple organs and carry molecular cargo that facilitates inter-organ communication and an integrated response to physiological and pathological stimuli. Interrogation of the protein cargo of EVs is currently limited by the absence of optimal and reproducible approaches for purifying plasma EVs that are suitable for downstream proteomic analyses. We describe a size-exclusion chromatography (SEC)-based method to purify EVs from platelet-poor plasma (PPP) for proteomics profiling via high-resolution mass spectrometry (SEC-MS). The SEC-MS method identifies more proteins with higher precision than several conventional EV isolation approaches. We apply the SEC-MS method to identify the unique proteomic signatures of EVs released from platelets, adipocytes, muscle cells, and hepatocytes, with the goal of identifying tissue-specific EV markers. Furthermore, we apply the SEC-MS approach to evaluate the effects of a single bout of exercise on EV proteomic cargo in human plasma.
Keywords: size-exclusion chromatography, ultracentrifugation, mass spectrometry, bioinformatics, high-intensity exercise
Graphical abstract
Highlights
-
•
SEC-MS provides excellent proteomic coverage of plasma EVs
-
•
SEC-MS can precisely quantify EV proteins from plasma by using label-free proteomics
-
•
EVs derived from different cell types contain unique EV protein cargo
-
•
Distinct exosome protein cargo is released immediately after exercise
Motivation
Extracellular vesicles (EVs) are detected in most body fluids including circulation, and emerging data indicate their potential role in inter-organ communications. Currently, a variety of approaches are used to purify EVs from plasma for molecular characterization; however, important parameters such as method precision and quantitative performance have not been described. To facilitate studies that characterize the function and composition of plasma EVs, there is a critical need for optimized methods that allow for reliable, reproducible, and efficient plasma EV isolation that are compatible with downstream molecular analysis, especially for proteins, EVs’ most abundant cargo.
Vanderboom et al. describe the quantitative performance of an optimized size-exclusion-chromatography-based isolation method for proteomic analysis of plasma-derived EVs. As proof of concept, this method is applied to detect changes in EV-associated protein abundance after an acute high-intensity aerobic and low-intensity resistance exercise.
Introduction
Integrated physiology relies on proper communication between different organs, and interest in extracellular vesicles (EVs) has grown rapidly in recent years, because of their role in inter-organ communication (Colombo et al., 2014; Guay and Regazzi, 2017; van Niel et al., 2018). EVs are membranous structures released from most cells that serve as vehicles to exchange protein, nucleic acid, and lipid cargo (Korutla et al., 2019; Minciacchi et al., 2015; van Niel et al., 2018) between distant organs. To understand the role of EVs in physiological and pathological states, reliable and reproducible profiling of circulating EV molecular cargo is critically important. Currently, a variety of approaches are used by investigators to purify EVs from plasma (Wu et al., 2019), including ultracentrifugation and commercial kit-based isolations. Many of these methods suffer from poor reproducibility or incompatibility with downstream analytical approaches, highlighting the need to establish a robust and reproducible approach for purifying plasma EVs to allow in-depth characterization and accurate quantification of their molecular content.
Although EVs derived from cell culture media can be isolated with high purity and in relatively large amounts (Kowal et al., 2016), isolation of plasma-derived EVs poses several unique technical and analytical challenges. For example, proteomic characterization of plasma-derived EVs isolated with differential ultracentrifugation (UTC) has been confounded by the co-isolation of an overwhelming number of plasma protein contaminants (Bard et al., 2004; György et al., 2011). Proteomic analysis of EVs is also limited by notoriously low isolation yield due to EV loss associated with successive washing steps (Livshits et al., 2015). Alternative approaches include commercially available kits based on membrane affinity (Kuang et al., 2019), polymer precipitation (Turay et al., 2016), and immunoprecipitation (Ghosh et al., 2014). Although these methods are suitable for some types of downstream analyses, they are also incompatible with unbiased, in-depth characterization of EV proteins. Size-exclusion chromatography (SEC)-based EV isolation was reported to rapidly isolate relatively pure EVs from plasma (Böing et al., 2014; Corso et al., 2017; Kreimer and Ivanov, 2017) and was shown to produce exosomes that are suitable for downstream proteomics analysis (Jeannin et al., 2018; Karimi et al., 2018). Although this method is promising, post-isolation processing methods need to be developed to allow for in-depth characterization of plasma EV cargo. Furthermore, optimized pre-analytical methods need to be coupled with sensitive high-resolution mass spectrometry (MS) analysis to achieve precise relative quantification of proteome differences between groups. Finally, there is a need to benchmark the analytical performance of the SEC method in relation to other EV isolation approaches to serve as a resource for those interested in EV proteomics research.
Another technical challenge of investigating plasma-derived EVs is the contribution of platelets, which are believed to contribute 70%–90% of circulating EVs and are known to release EVs ex vivo when activated during sample-handling procedures. A particularly problematic issue is the several-fold increase in plasma EV concentration caused by platelets upon cooling/freezing (Artoni et al., 2012; György et al., 2012). This is especially important because cooling/freezing is a necessary pre-analytical step when analyzing a large number of plasma samples that require some interim storage. To address this issue, rigorous pre-analytical guidelines have been established by the International Society of Extracellular Vesicles (ISEV) and International Society on Thrombosis and Haemostasis (ISTH) to limit ex vivo generation of platelet EVs (Artoni et al., 2012; Ayers et al., 2011; Burnouf et al., 2014; LaCroix et al., 2012).
In the current study, we systematically evaluated an innovative approach by using SEC coupled to high-resolution MS (SEC-MS) for comprehensive proteomic characterization of human plasma EVs. We address several important factors, including sample purity, method reproducibility, and downstream MS analysis that are important for characterizing the EV protein cargo of large sample groups in both research and clinical settings. We also demonstrate the importance of following the rigorous pre-analytical guidelines established by the ISEV and ISTH to limit ex vivo generation of platelet EVs (Artoni et al., 2012; Ayers et al., 2011; Burnouf et al., 2014; LaCroix et al., 2012). Additionally, sample-processing steps downstream of EV isolation require careful attention because of the challenging nature of these samples. Finally, we address the lack of a benchmark for plasma EV proteomics method reproducibility and quantitative performance. Toward this goal, we provide an optimized and reproducible method for isolating plasma EVs from human plasma that is suitable for downstream analysis of protein cargo. To demonstrate the sensitivity of this analytical approach to detect physiological changes in exosome proteomic cargo, we evaluated the effects of acute exercise on proteome cargo of EVs isolated from human plasma samples.
Results
SEC improves plasma EV proteomics characterization
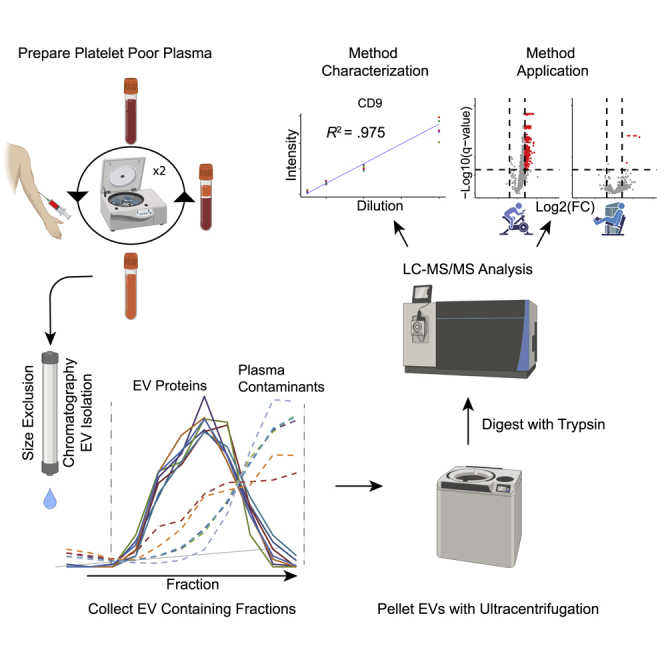
SEC is a low-resolution, size-based fractionation technique. Larger EVs do not interact with the SEC resin and pass through the column rapidly, whereas smaller particles (e.g., plasma proteins) interact with the resin and pass through at a slower rate. This separation enables improved proteomic characterization of EVs (Jeannin et al., 2018; Karimi et al., 2018), largely by increasing isolation yield and reducing plasma protein contamination (Stranska et al., 2018). We investigated the proteomic profiling compatibility of plasma EVs that were isolated with SEC compared with several conventional plasma EV isolation methods. We used platelet-poor plasma (PPP) collected from healthy donors in the resting state after overnight fast to reduce ex vivo contribution of platelets to the plasma EV population. PPP was prepared as previously described (Lacroix et al., 2013) with modifications (see STAR Methods). EVs were also isolated from PPP by differential ultracentrifugation (UTC), SEC (qEV Original), polymer precipitation (ExoQuick), and membrane affinity (ExoEasy). We show that SEC outperformed other methods, on the basis of the ability to detect greater numbers of exosome proteins (defined by the ExoCarta database; Figure S1A). Furthermore, SEC identified more canonical EV marker proteins (Kowal et al., 2016) from less sample volume than with the UTC and ExoEasy methods (Figure S1B).
SEC-MS can reproducibly distinguish EV-associated proteins from plasma contaminants
To further characterize and optimize the SEC-MS assay, we analyzed the elution patterns of canonical EV marker proteins and plasma contaminants by using SEC. Fasting PPP (2 mL) was obtained from a donor, and plasma EVs were isolated by using a qEV2 SEC column (Izon Science, Christchurch, New Zealand). For clarity, we defined SEC fractions as each 1-mL increment eluted from the SEC column. For this experiment, individual 1-mL SEC fractions spanning the region where EVs elute from the SEC column (fractions 12–22) were collected and processed individually with a label-free proteomics workflow (see STAR Methods). The intensities of six canonical EV marker proteins measured in sequential SEC column fractions were used to generate SEC EV elution profiles (Figure 1A). The elution profiles of all six EV marker proteins show distinct peaks that co-eluted over seven distinct SEC fractions (fractions 15–21) (Figure 1A). High-molecular-weight plasma protein contaminants such as fibrinogen, apolipoprotein B, and immunoglobulin M, also eluted in this region; however, the peak profiles of these common plasma contaminant proteins were right-shifted (Figure 1A), thereby differentiating them from the well-resolved EV marker SEC peaks. These results indicate that SEC can effectively separate EVs from plasma contaminants.
Figure 1.

Size-exclusion chromatography separation of extracellular vesicles from plasma proteins
(A) Size-exclusion chromatography (SEC) fraction profiles of several extracellular vesicle (EV) markers and high-molecular-weight plasma proteins to illustrate separation of EVs from plasma proteins. Profiles are generated from protein intensities derived from label-free analysis of individually analyzed 1-mL fractions across the EV elution range as described in STAR Methods. Scaled protein abundance was obtained by adjusting protein intensity measurements from all fractions to a mean of 100.
(B) Bar plot of intensity apex versus SEC fraction for proteins in the ExoCarta top 100 (ECT100) to show where EVs are eluting from the SEC column.
(C) Schematic of a bioinformatics-based filtering process to identify potential contaminant proteins. Proteins remaining after the filtering process have low plasma concentrations and low molecular weight, indicating that they are enriched for EV-derived proteins.
Orthogonal separation of the EVs and plasma contaminants by SEC was used to refine the method and develop an algorithm to flag potential plasma contaminants in EV SEC-MS experiments. Toward this goal, we established a reference elution range for EVs by using the ExoCarta top 100 (ECT100), a list of the most commonly observed proteins in exosomes (Keerthikumar et al., 2016). Seventy-nine of 84 ECT100 proteins detected displayed Gaussian-shaped profiles, which reached their intensity apex, an indicator of maximum abundance, between fractions 17 and 19 (Figures 1B and 1C). The five ECT100 proteins that reached intensity apexes outside of fractions 17–19 exhibited chromatographically distinct peak profiles (Figures S2 and S3) that were mainly associated with the delayed elution of plasma contaminants rather than EV-derived proteins (Figure S2A). These proteins also have high molecular weights and high plasma concentrations (A2M, FN1) or the ability to form multi-molecular complexes (THBS-1 and LGALS3BP) (Sasaki et al., 1998; Tan et al., 2006). The large size and chromatographic association with plasma contaminants suggest that these proteins might be of plasma origin rather than EV derived. Consistent with this concept, serum albumin, a known plasma contaminant and ECT100 protein, exhibited an elution profile that was distinct from all other ECT100 proteins. Further review of all proteins co-eluting with albumin revealed that none of them were associated with ECT100 and those fractions contained several abundant erythrocyte proteins such as hemoglobin, band-3 anion transport protein, and peroxiredoxin-2 (Kakhniashvili et al., 2004). These observations suggest that erythrocyte fragments and large protein aggregates containing albumin elute before EVs (Figures S2A and S2B). Therefore, we determined that the reference EV SEC elution range was between fractions 17 and 19, and all proteins in the dataset with an apex falling within this fraction range were extracted and categorized as “EV-associated proteins.” Finally, high-molecular-weight proteins (>200 kDa), proteins known to form large complexes (immunoglobulin, apolipoproteins, complement), and common contaminants (keratin and hemoglobin) were filtered out (Figure 1C). After the filtering process, 454 of the original 758 identified proteins remained (Figure 1C). EV-associated proteins were less abundant than contaminants (EV-associated proteins: median = 32 ng/mL, interquartile range [IQR] = 7.0–180 ng/mL; contaminants: median = 28 μg/mL, IQR = 0.21–290 μg/mL; p = 2.2 × 10−16). Considering these characteristics, it is reasonable to hypothesize that the proteins with apex intensities within SEC fractions 17–19 are contained within a vesicle. It should be noted, however, that it might be possible for some proteins to be associated with both EVs and the EV-independent plasma proteome. In comparison with the co-migration-based filtering, 686 of the 758 proteins were contained in the ExoCarta database, suggesting that this peak filtering strategy is a more conservative but more accurate strategy to distinguish between EV and contaminant proteins.
We evaluated the reproducibility of SEC separation by using three replicate PPP samples processed by using three different SEC columns. Each replicate PPP sample was processed with each SEC column, and fractions were processed in 2-mL increments (12–13, 14–15, 16–17, 18–19, 20–21, and 22–23) for a total of 6 concatenated fractions to reduce the total number of samples. Each 2-mL fraction was subjected to proteomics analysis, and protein SEC elution profiles were constructed to determine reproducibility of EV separation. In total, 85 of the ECT100 proteins were identified in each replicate, and their elution profiles were highly correlated both within the same SEC column and across SEC columns (Figures S3AB and S3C). Furthermore, the four ECT100 proteins identified above as possible plasma contaminants (A2M, FN1, THBS1, and LGALS3BP) displayed reproducible shifts in elution, differentiating them from all other ECT100 proteins (Figure S3D). We also assessed the reproducibility of EV protein intensities by using this replicate analysis. To this end, we summed the intensity of each protein observed in each sample across all six fractions of each SEC column. The median coefficient of variation (CV) of protein intensities was 20.89 (IQR 16.75–30.54; Table S1), which is within the acceptable limits of typical label-free proteomics experiments (Al Shweiki et al., 2017; Shen et al., 2018; Tabb et al., 2010).
Collecting and analyzing individual SEC fractions is labor intensive, error prone, and restricts sample throughput. To streamline SEC-based exosome analysis, we tested the feasibility of pooling the EV-containing SEC fractions together as one sample for subsequent proteomic analysis. For this, SEC fractions 14–21 obtained from three additional replicates of the above reproducibility experiment were pooled as a single EV sample for each replicate. We identified a similar number of proteins when analyzing pooled or individual SEC EV fractions (pooled: 800 ± 46; fractions: 826 ± 23; Figure S4A), and a majority (76%) of proteins were identified by both types of sample analysis (Figure S4B). Importantly, the CV of protein intensities was slightly better when analyzing a pooled EV sample versus individual EV fractions of a single sample (Figures S4C and S4D). Taken together, these results indicate that EVs elute from SEC columns in a predictable and quantitatively reproducible manner that is amenable to downstream MS analysis of pooled SEC fractions.
SEC-MS increases EV protein identifications and precision compared with those of ultracentrifugation
The performance of SEC-MS in isolating EVs for proteomic analysis was compared with the conventional ultracentrifugation (UTC) approach. We isolated EVs from pooled PPP acquired from healthy donors (n = 2) in the rested and fasted state by using SEC and UTC. Five replicate measurements were performed. Nanoparticle tracking analysis showed similar particle size for both methods (SEC 78 ± 12 nm, UTC 82 ± 14 nm; p = 0.90). However, particle counts were higher for SEC than for UTC (SEC 1.49 × 1010 ± 3.6 × 109, UTC 2.05 × 107 ± 1.2 × 107, p = 0.0008; Figures 2A and 2B), indicating increased recovery of EVs with SEC compared with UTC. EVs isolated with SEC also appeared to be intact and have a diameter of less than 100 nm by transmission electron microscopy (Figure 2C). Proteomics analysis revealed that SEC produced significantly more protein identifications than UTC (SEC 1,268 ± 102, UTC 319 ± 45; p = 2.6 × 10−6) (Figure 2D). The CV of the protein intensities obtained from SEC-MS was significantly lower than those obtained via UTC-MS (Figure 2E). In total, 562 proteins quantified by SEC-MS had CVs less than 25% when compared with only 73 proteins reaching this threshold for UTC-MS. Moreover, CVs of canonical EV marker proteins were lower with SEC-MS than with UTC-MS (Figure 2F). A closer inspection of the canonical EV protein marker intensity data suggested that UTC-MS was either unable to detect these proteins or detected them with a lower intensity than SEC-MS, which resulted in a higher CV for these proteins (Figure 2G). This is suggestive of suboptimal EV recovery with UTC, which was noted by others (Livshits et al., 2015).
Figure 2.

Comparison of extracellular vesicle protein identifications after SEC and ultracentrifugation
(A and B) Size distribution (A) and particle count (B) characterization of extracellular vesicles (EVs) isolated from platelet-poor plasma (PPP) by using size-exclusion chromatography (SEC) or ultracentrifugation (UTC) by nanoparticle tracking. SEC resulted in a significantly higher particle count than when using UTC (p = 0.0008).
(C) Transmission electron microscopy image of EVs isolated from plasma by using SEC.
(D) Experiments from five replicate analyses of pooled PPP showed that SEC produced approximately 3-fold more identifiable proteins compared with UTC. Dark-blue bars represent the total number of protein identifications for each replicate, and light-blue bars represent the number of the identified proteins that associate with the ExoCarta database.
(E) Histogram displaying the coefficient of variation (CV) calculated from protein intensity measurements of each identified protein over the five replicates. In total, 562 proteins with a CV of less than 25% were identified with SEC, compared with just 73 with UTC.
(F) CVs for several EV marker proteins identified by SEC and UTC.
(G) Protein identifications from each method plotted as ranked abundance against log10 intensity shows that SEC results in higher recovery of EV proteins than UTC.
SEC-MS measurements are linear and precise
SEC-MS outperformed the conventional UTC-MS approach for plasma-derived EV proteomic analysis. To test the quantitative performance of SEC-MS across a range of EV abundance, we serially diluted human plasma with exosome-depleted bovine serum (EDBS). For this experiment, non-platelet depleted plasma was used (i.e., platelet-rich plasma [PRP]) to extend the top end of the EV dynamic range. Five replicates of a dilution series (neat plasma, ×2, ×4, ×8, and neat EDBS) were generated, and each dilution series replicate was processed by using a separate SEC column. With this design, it was possible to simultaneously assess reproducibility across SEC columns and linearity of the dilution. More than 2,080 proteins were detected across the experiment, and 1,418 proteins remained after filtering out proteins that were indistinguishable from bovine proteins on the basis of the observed peptide evidence. Protein intensities of multiple canonical EV proteins displayed linear curves across the dilution range (Figure 3A). A dilution curve analysis of the entire dataset found 850 proteins with an R2 > 0.9 (Figure 3B), indicating high linearity across the quantitative range. Protein intensity precision of replicate measurements at each dilution point was also measured. This analysis identified 564, 564, 424, and 239 proteins with CVs less than 25% at the neat plasma, x2, ×4, and ×8 dilution points, respectively (Figure 3C). To test the potential for using total EV-associated protein intensity as a measure of overall EV abundance, we plotted the total intensity of all EV-associated proteins (i.e., proteins remaining after filtering contaminants identified by the co-migration-based filtering process described above) observed in a sample against its dilution factor (Figure 3D). Summed EV-associated protein intensity was highly correlated with the dilution factor (R2 = 0.992), indicating that this approach might provide an accurate readout of total EV abundance. Plotting summed EV plus contaminant protein intensity for each sample against the dilution factor produced a lower R2 of 0.815, reinforcing the notion that the co-migration-based filtering process is useful for identifying contaminants.
Figure 3.

Evaluation of quantification performance in biological samples
(A) Pooled human plasma was serially diluted with exosome-depleted fetal bovine serum, and EVs were isolated from five replicates of each dilution level. Five separate SEC columns were used, one for each replicate dilution curve as indicated by the experimental schematic. Dilution curves fit with a linear regression are displayed for several select EV marker proteins. Average protein abundance values at each dilution point were used to fit the curve.
(B) Histogram representing mean R2 values of the replicate dilution curves for all identified proteins showed that 850 proteins had an R2 value >0.9.
(C) Analysis of the CVs of the five replicate measurements at each dilution point showed low variation at each dilution point.
(D) Linear regression showing the high correlation between summed EV protein intensity and dilution factor. Correlation is much higher when contaminants identified by using the co-migration-based filter process are removed prior to summing protein intensity (contaminants filtered, R2 = 0.992 versus no filtering, R2 = 0.815). High correlation indicates that total EV abundance can be estimated by summed EV protein intensity after contaminant filtering.
EVs derived from different cell types contain both common and unique protein cargo
Cell-culture-based EV experiments were performed to identify EV proteins that might be unique to specific EV-producing tissues (e.g., platelets, skeletal muscle, adipose, liver, leukocytes). EVs were isolated from media collected from cultured primary human myotubes, primary human adipocytes, Huh7 hepatocytes, leukocytes, and isolated human platelets, and their protein cargo was profiled by using SEC-MS. Many identified EV proteins were common to all cell types, but a large number of putative cell-specific EV proteins were also identified (Figure 4A). A total of 118, 310, 350, 180, and 121 unique proteins were identified in platelet, leukocyte, myotube, hepatocyte, and adipocyte EVs, respectively. Gene ontology over-representation enrichment analysis of putative cell-type-specific EV proteins revealed several distinct gene sets including actin filament-based movement and muscle structure development (myotube), proteolysis (adipocyte), intracellular transport (hepatocyte), blood coagulation (platelet), and immune effector process (leukocyte) (Figure 4B). Putative cell-specific EV proteins were merged with tissue-specific proteins from the Human Protein Atlas (HPA) (Uhlén et al., 2015). Prior to merging putative cell-specific EV proteins with HPA data, proteins which were unique to the combined list of platelet plus leukocyte EVs (platelet + leukocyte-specific proteins) were determined because HPA considers “blood” as one tissue. HPA “tissue-enhanced” and “tissue-enriched” data aligned with 67 myotube (“muscle” in HPA) and 120 platelet + leukocyte (“blood” or “lymphoid” in HPA) specific proteins EV proteins, whereas only four adipocyte (“adipose” in HPA) and hepatocyte (“liver” in HPA) putative cell-specific EV proteins each agreed with HPA data (Table S2 and Figure S5). These data are a valuable resource in efforts to track the composition and origin of circulating EVs.
Figure 4.

Comparison of EV protein content from different cell types
(A) Venn diagram showing the protein composition overlap of EVs isolated from primary human myotube, primary human adipocyte, HUH7 hepatocyte, platelet, and leukocyte conditioned medium. Although many of the identified proteins were common to several cell types, some were unique to each.
(B) Over-representation enrichment analysis of proteins identified uniquely in each cell type identified gene sets associated with each respective tissue. FDR, false discovery rate.
Platelet- and leukocyte-derived EVs are abundant in plasma and can be released ex vivo
Ex vivo platelet activation is a well-characterized phenomenon with implications in transfusion medicine and blood processing. Several-fold increases in platelet-derived EVs have been observed after freeze-thaw of standard plasma (i.e., PRP) (Artoni et al., 2012; György et al., 2012). Obtaining unfrozen plasma to avoid platelet EV release prior to EV analysis is logistically difficult for human studies. Therefore, it is important to assess the effect of platelet depletion on SEC-MS toward the goal of making SEC-MS compatible with frozen plasma samples. Blood was collected from two healthy volunteers, pooled, and divided to be prepared as PPP or PRP. EVs were isolated and analyzed from six replicate samples of each preparation type by using SEC-MS. As expected, the average number of protein identifications was significantly higher in the PRP than in the PPP (PRP = 1,147 ± 20, PPP = 877 ± 32, p = 6.0 × 10−8) (Figure 5A). Furthermore, the abundance of platelet + leukocyte-specific proteins were significantly higher in the standard plasma than in PPP (Figure 5B and Table S3). In total, 308 platelet + leukocyte-specific proteins were identified in the PPP versus PRP comparison. Of these, 124 were significantly increased in PRP compared with in PPP >log2-fold change of 1.0. These results provide evidence that platelet- and leukocyte-derived EVs released ex vivo account for a majority of the increased EV proteins observed in standard plasma (i.e., PRP).
Figure 5.

Differential comparison of extracellular vesicle proteins derived from platelet-poor and platelet-rich plasma
(A) Mass spectrometry analysis of EVs isolated from six replicates of pooled platelet-poor plasma (PPP) or platelet-rich plasma (PRP) showed significantly increased protein identifications in PRP (p = 6.0 × 10−8).
(B) Volcano plot representing the relative difference in protein abundance between PPP and PRP. Proteins identified only in EVs derived from isolated platelets or leukocytes are colored in blue to illustrate the overwhelming increase in abundance of platelet- and leukocyte-derived EVs in PRP plasma. Abbreviation is as follows: FC, fold change.
(C) Table showing the number of identified and upregulated proteins form EVs derived from each cell type.
Acute exercise triggers EV release
We applied the SEC-MS method to evaluate the effect of acute exercise as a physiological stimulus on plasma EV proteome cargo in humans. The effects of exercise on EV release into circulation has been investigated by others (Frühbeis et al., 2015; Whitham et al., 2018), and we considered acute exercise as an appropriate physiological intervention to determine the ability of the approach to detect the EV proteome changes. Although the objective was not to compare and contrast with the previous reports, we wanted to determine whether the current PPP-SEC-MS approach with its methodological innovations allow the detection of similar or larger numbers of proteins from human EVs released after exercise. Moreover, we also sought to identify the specific cellular origin of many EV proteins. We determined the differential responses of EV protein abundance after one bout of acute high-intensity aerobic exercise (aerobic: n = 14) and a moderate bout of single-leg resistance exercise (resistance: n = 16) (Table S7).
Fourteen healthy individuals performed a single bout of aerobic exercise at ~90% of VO2 peak (Robinson et al., 2017), and samples were collected before exercise, then within 10 min and at 1 h and 3 h after exercise. Sixteen additional individuals performed a single bout of unilateral knee extensor resistance exercise consisting of three sets of ten repetitions at 70% of their 1-repetition maximum (1RM) (Robinson et al., 2017). Each participant's 1RM was determined at least 7 days, but no more than 30 days, prior to the study visit. Samples were collected before, then within 10 min and at 30 min and 60 min after exercise. All samples were analyzed by PPP-SEC-MS. We identified 2,736 proteins in the aerobic exercise group (Figure 6A). Time-course analysis of the response to aerobic exercise utilizing summed EV-associated protein intensity showed a significant increase in circulating EV protein abundance immediately after aerobic exercise (p = 0.0006), which returned to pre-training levels by 3 h after exercise (Figures 6B, S6A, and S6B). Notably, a similar trend was observed in ADAM10-positive particles measured by nano-flow cytometry (nFLC) immediately after training (p = 0.055; Figure S6D). Conventional nanoparticle tracking analysis (Figure S6E) did not reveal the same post-exercise increase in particle count as summed EV protein intensity of nFLC. This suggests that SEC-MS and nFLC are more sensitive methods for detecting shifts in EV abundance with exercise compared with particle counting. The immediately after (10 min) aerobic exercise time point revealed an increase in the abundance of 321 EV-associated proteins (Figures 6A and S7; Table S4). Functional enrichment analysis of these proteins indicated that they are central to CDC42-mediated cell migration, integrin signaling, and immune function as indicated by enrichment of lymphocyte adhesion molecules, monocyte surface molecules, and regulation of T cell activation (Table S5). Similarly, Ingenuity Pathway Analysis (IPA) showed that these proteins were enriched for additional immune regulation processes such as leukocyte extravasation and CXCR4 signaling. Additional pathways including glycolysis and signaling via Rho GTPase, interleukin-8, and integrin were also differentially regulated by exercise (Table S6). We next overlaid these data with the cell-type-specific EV data we generated previously (Figure 4A) to assess the source of circulating EV proteins. When using cell-type-specific EV proteins, we found proteins specific to myotubes (n = 96), hepatocytes (n = 58), adipocytes (n = 59), leukocytes (n = 189), and platelets (n = 93) (Figures 7A and 7B). Interestingly, at the immediate post-exercise time point we found significantly upregulated proteins that were specific to each cell type profiled, including 30 leukocyte EV-specific and 5 myotube EV-specific proteins, of which 19 and 3 were supported by HPA tissue-specific data, respectively, demonstrating that EVs from different tissues can be measured in the plasma circulation. Collectively, these data highlight that one session of high-intensity aerobic exercise leads to robust changes in circulating EV protein abundance, which might play an important role in regulating the metabolic and immune responses of training.
Figure 6.

Aerobic exercise triggers an increase in EV protein abundance
(A) Volcano plots showing the increase in protein abundance immediately after aerobic exercise and resistance exercise. Abbreviation is as follows: FC, fold change.
(B) Summed EV-associated protein intensity normalized to pre-exercise is displayed for aerobic and resistance exercise to illustrate temporal changes in EV abundance. Summed EV-associated protein is significantly increased after exercise (∗∗∗p = 0.0006) and returns to pre-exercise levels by 3 h.
(C) Venn diagram showing common and uniquely regulated proteins for aerobic and resistance exercise.
Figure 7.

Cell-specific EV proteins are upregulated after aerobic exercise
(A) Volcano plots illustrating the increase in EV protein abundance after aerobic exercise. Proteins identified uniquely from each cell type (Figure 5) are highlighted in individual volcano plots and significantly upregulated proteins (log2 fold change [FC] > 0.5) are labeled.
(B) Table showing the number of identified and upregulated proteins from each cell type. The highest number of cell-type-specific proteins were found to be from leukocytes and platelets.
The samples from the single-leg resistance exercise analysis identified a total of 3,138 proteins. EV-associated protein intensity displayed an upward trend after exercise; however, this increase failed to reach significance (p = 0.17) (Figure 6B). Analysis of the post-resistance exercise time point found only seven proteins that were significantly upregulated and one protein that was downregulated, in stark contrast to the robust increase of EV proteins triggered by the acute bout of aerobic exercise (Figure 6A). Of the seven upregulated proteins, three were commonly upregulated by the aerobic exercise bout (Figure 6C). Interestingly, two of the three commonly upregulated proteins were found to be specific to leukocytes in our cell-type-specific experiments, further implicating immune modulation in response to different modes and intensities of exercise. Among the upregulated proteins common to both exercise responses was MMP9 (matrix metallopeptidase 9), which is involved in functions that include neutrophil action, angiogenesis, and wound healing.
Discussion
Here, we describe an optimized SEC-MS method for proteomic analysis of plasma EVs that offers superior quantitative performance and reproducibility compared with conventional EV analytical approaches. Importantly, this SEC-MS method exhibited linearity over a biologically relevant dynamic range and is sensitive to small differences in protein abundance. Additionally, the orthogonal separation of EVs and plasma protein contaminants provided by the SEC column was leveraged to filter contaminants from plasma proteins within the EV proteome. Using SEC-MS, we demonstrate the importance of using PPP to avoid the confounding effects of ex vivo release of platelet EVs. After benchmarking quantitative performance of SEC-MS, we used the method to demonstrate the time-related effects of two different modes and intensities of acute exercise bouts on changes in plasma EV protein abundance.
The proteomic depth of plasma EVs isolated with UTC or commercially available kits is limited by factors including plasma protein contamination and low EV recovery (Figure S1). We show that SEC purification of EVs followed by proteomic analysis substantially mitigates plasma protein contamination. Particle counts, EV protein intensities, and EV protein identifications were markedly higher when plasma EVs were isolated with SEC in comparison with UTC (Figures 2A, 2B, and 2D). Furthermore, the precision of replicate SEC-MS measures is far superior to that based on UTC-MS, capable of quantifying >500 proteins with CV less than 25% (Figure 2E).
Beyond demonstrating the utility of SEC for isolating EVs, we also present a strategy, based on the fractionation properties of SEC, to distinguish EV-associated proteins from plasma protein contaminants on the basis of their elution patterns (Figure 1C). This process relies on identification of proteins that co-elute with known EV proteins, allowing us to reproducibly identify four proteins in the ECT100 as likely plasma protein contaminants rather than EV-derived proteins (Figure S3D). However, we cannot categorically exclude the possibility that some proteins might be commonly present in both EVs and plasma proteome. Nevertheless, the SEC-MS method can discern whether the majority of the protein abundance in a sample is derived from plasma contaminants or EVs. This is particularly useful because accurate EV abundance measurements are obtained by filtering contaminants and summing EV-associated protein intensities (Figure 3C). Additionally, SEC-MS can be used to validate whether a candidate protein is derived from EV, with an added advantage of being applicable for any EV protein and not limited to EV membrane proteins. There are previous reports (Jeannin et al., 2018; Whitham et al., 2018) of the relative quantification of plasma EV proteome. However, the quantitative performance of those methods has not been described. We benchmarked the quantitative performance of SEC-MS to validate it for relative quantification of EV-associated proteins in biological samples. Using a dilution series experiment, we showed that over 850 EV proteins can be measured from human plasma with an R2 > 0.9. We also observed low quantitative CVs at multiple dilution points, indicating high precision. Ex vivo activation of platelets is known to cause a massive release of EVs, resulting in sample contamination and confounding results. To avoid this issue, ISEV and ISTH guidelines recommend including a platelet-depletion step prior to refrigerating plasma. Although this step is strongly recommended and widely recognized by the EV research community, several recent studies examining the plasma EV proteome have utilized frozen serum or plasma without platelet depletion (Chen et al., 2017; Jeannin et al., 2018; Whitham et al., 2018; Zhang et al., 2018), which facilitates analysis of samples from previous well-characterized studies. To assess the extent of platelet-derived EVs released during sample processing, we performed a differential analysis on pooled plasma with and without platelet depletion by using SEC-MS. The detected proteins were also cross-referenced with platelet + leukocyte-specific EVs derived from the cell-culture-based EV experiment. This analysis showed large increases in platelet- and leukocyte-specific EV protein identifications and abundances in non-platelet-depleted plasma in relation to PPP. These results highlight the importance of the platelet-depletion step in avoiding sample contamination and confounding results. This study demonstrates that the SEC-MS offers an opportunity to analyze the PPP EV proteome, which contains fewer EVs but more accurately reflects the composition of circulating EVs.
We applied SEC-MS to characterize the acute effects of a single bout of high-intensity aerobic exercise or resistance exercise on plasma EV proteome. We identified 3,409 proteins in this experiment by using a short 1-h MS method, substantially shorter than UTC-MS methods described previously (Whitham et al., 2018). In the present study, >300 proteins were significantly increased after a single bout of aerobic exercise. We also noted that an acute single-leg resistance exercise bout increased only seven EV proteins, of which three of them are common to both exercise modes. Increased cardiac output and more dynamic nature and high intensity (90% of VO2peak) of aerobic exercise are likely reasons for the substantially more robust response to aerobic exercise in comparison with the response to the modest one-leg resistance exercise. Although the goal of this exercise was not to contrast the effects of exercise modality on circulating EV cargo, it is interesting to note that some modest changes in EV proteomic cargo could be detected even after a low-intensity, short-duration bout of single-leg resistance exercise. We also performed cell-culture-based EV profiling experiments and identified candidate tissue-specific proteins that are released immediately after an aerobic exercise bout. Although skeletal muscle contributed to the circulating EV pool, it is of great interest that EV proteins specific to other tissues including leukocytes were dominant after a bout of aerobic exercise and linked these proteins with important functions including signaling, immunological response, repair, and angiogenesis (Table S6). How aerobic exercise interacts with other tissues, especially leukocytes, remains to be determined. It has been shown that exercise-induced muscle tissue damage attracts many circulating cells, including those involved in immunological reactions that remove degraded proteins and stimulate protein synthesis (Pedersen and Hoffman-Goetz, 2000). It appears that acute exercise stimulates these circulating cells to release proteins via EVs, which might enable the transport of important signals to multiple organs. It remains to be determined whether these signals contribute to the exercise responses in remote organs such as liver and brain. The exact mechanism of exercise-mediated EV release from liver, adipocytes, and platelets also remain to be determined.
In summary, our study describes the development and quantitative performance of a reproducible SEC-MS assay for plasma EV protein analysis. We demonstrate that this assay yields deeper proteomic coverage and higher precision when compared with more commonly used approaches, especially UTC-MS. Our study makes an important advance in the quest to determine the tissues of origin of EVs found in circulation. We demonstrate the importance of PPP in avoiding ex vivo contamination from platelet-derived EVs. We also describe an approach to filter plasma protein contamination even after SEC-based exosome isolation. We benchmarked the quantitative performance of SEC-MS and demonstrated that it can be usefully employed in human clinical studies. Finally, we demonstrated the EV proteome response to acute bouts of exercise. Future studies will be needed to determine the effects of exercise intensity, duration, and modality on the molecular cargo of circulating EVs in humans.
Limitations of the study
Isolating EVs from plasma for proteomics analysis is technically challenging, and the currently presented method has several limitations. In the present study we demonstrated the reproducibility of EV elution from the SEC column with up to three replicate samples on three different columns (Figures S3A–S3C). We also report the ability of the method to measure known differences in biological samples (Figure 3), reusing the column up to four times. However, we have not extensively characterized column performance beyond a reuse of four times, and investigators should take this into consideration when designing experiments. We also have not assessed the EV elution range for each new lot of SEC columns. Regarding the exercise study, the endurance and resistance exercise protocols were not intensity balanced and we did not intend to compare differences in exosomal protein composition or abundance between two different exercise modalities. Rather, our aim was to demonstrate the ability to measure physiological changes with the approach.
In conclusion, we believe that SEC-MS is a powerful tool for investigators interested in studying the composition and function of plasma EVs in biological systems.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified anti-human ADAM10 Antibody | BioLegend | Cat# 352702; RRID: AB_10897813 |
| Chemicals, peptides, and recombinant proteins | ||
| Sequencing Grade Modified Trypsin | Promega | V5073 |
| TCEP HCl (tris(2-carboxyethyl)phosphine hydrochloride) | Sigma | 646547 |
| Methanol (Optima™ LC/MS) | Fisher Scientific | Cat# A454SK-4 |
| Water (Optima™ LC/MS) | Fisher Scientific | Cat# W6-4 |
| Acetonitrile (Optima™ LC/MS) | Fisher Scientific | Cat# A9554 |
| Honeywell Fluka™ Formic acid, for mass spectrometry | Fisher Scientific | Cat# 60-006-17 |
| Phosphate Buffered Saline (PBS) | BioRad | Cat# 1610780 |
| ProteaseMAX™ Surfactant | Promega | Cat# V2072 |
| Dithiothreitol (DTT) | Millipore Sigma | Cat# 10708984001 |
| Iodoacetamide (IAA) | Millipore Sigma | Cat# I1149 |
| Triflouroacetic Acid (TFA) | Millipore Sigma | Cat# 302031 |
| ZWITTERGENT® 3-16 Detergent | Millipore Sigma | Cat# 693023 |
| Isopropanol (Optima™ LC/MS) | Fisher Scientific | Cat# A461-1 |
| Deposited data | ||
| Mass Spectrometry Raw and Analyzed Data | This Study | PRIDE: PXD026483 |
| Experimental models: Cell lines | ||
| Human Skeletal Muscle Myoblasts (HSMM) | Lonza | Cat# CC-2580 |
| Isolated Human Platelets | Mayo Clinic Blood Bank | NA |
| Isolated Human Leukocytes | Donor | NA |
| Software and algorithms | ||
| MaxQuant Quantitative Software (v1.6.7.0) | Cox and Mann, 2008 | https://www.maxquant.org/ |
| Perseus (v1.6.7.0) | Tyanova et al., 2016 | https://maxquant.net/perseus/ |
| FlowJo (v10.5) | FlowJo | https://www.flowjo.com/ |
| Other | ||
| qEV 2.0 Size Exclusion Chormatography Column (70nm Pore Size) | Izon | Cat# qEV 2.0 |
| Self-Pack PicoFrit Columns 360x75x10 | New Objective | Part# PF-360-75-10-N-5 |
| EXP®2 Stem Trap | Optimize Technologies | Part# 04003HNC18 |
| Skeletal Muscle Cell Growth Medium | Cell Applications | Cat# 151K-500 |
| Gibco™ Dulbecco's Modified Eagle Medium/F-12 (DMEM/F-12) | Fisher Scientific | Cat# 11320 |
| Gibco™ Fetal Bovine Serum, exosome-depleted | Fisher Scientific | Cat# A2720801 |
| Preadipocyte Growth Medium-2 (PGM-2 Bullekit) | Lonza | cat# PT-8002 |
| Gibco™ Fetal Bovine Serum (FBS) | Fisher Scientific | Cat# A31605 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: K Sreekumaran Nair (nair@mayo.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD026483.
Experimental models and subject details
Study overview
This study was approved by the Mayo Clinic Institutional Review Board and conducted in accordance with the Declaration of Helsinki. For method development and analytical validation, blood was collected from a group of volunteers composed of both males and females. For the high intensity aerobic exercise study seven male and seven female subjects were recruited. For the resistance exercise study eight male and eight female subjects were recruited. Subject characteristics are summarized in Table S7.
Cell culture
Human skeletal muscle myoblasts (HSMM) were purchased from Lonza and grown in skeletal muscle cell growth medium (Cell Applications, cat# 151K-500). Media was changed three times a week until he cells reached 70% confluency at which point they were differentiated to myotubes in Dulbecco's Modified Eagle Medium/F-12 (DMEM/F-12) containing penicillin(100 U/mL), streptomycin(100 ug/mL) and 2% horse serum. After differentiation the cells washed twice with PBS and were placed back into growth medium supplemented with 2% exosome depleted bovine serum (EDBS,Gibco,cat# A2720801). After 24hours the media was harvested and frozen.
Human pre adipocytes were cultured in preadipocyte growth medium-2 (PGM-2 Bullekit, Lonza cat# PT-8002) and the media was changed 2-3 times a week until the cells reached 80-90% confluency. The preadipocytes were differentiated to adipocytes according to the manufacturer instructions. Once differentiated, the cells were washed twice with PBS and the media was replaced with PGM-2 prepared with 10% EDBS (Gibco,cat# A2720801 ) and cultured for an additional 24hrs before the media was collected and frozen.
Isolated platelets were obtained from the Mayo Clinic blood bank. Platelet EVs were prepared as previously described (Prokopi et al., 2009). Briefly, the isolated platelets were washed with 3x in Tyrode-HEPES buffer (145mM NaCl, 2.9 mM KCl, 10mM HEPES, 1mM MgCl2, 5mM glucose, pH 7.3) and re-suspended at a concentration of 6X108/ml. Platelets were activated with thrombin (0.1U/ml) and incubated at 37°C for 30min with rotation every 10min. Platelets were then pelleted by centrifugation at 3200x g for 10min and the supernatant was collected and frozen at -80°C.
Huh7 hepatocytes were cultured in DMEM containing glucose(4.5 g/L) penicillin(100 U/mL), streptomycin(100 ug/mL) and 10% fetal bovine serum (FBS). Cells were grown to 90% confluency on 150mm tissue culture dishes. Cells were washed twice with PBS and then cultured for an additional 24hr in growth medium supplemented with 5% extracellular vesicle-depleted FBS, which was prepared by overnight centrifugation at 100,000g at 4C and 1% bovine serum albumin.
Human leukocytes were isolated from blood donated by two donors. Whole blood was drawn into 10-ml ETDA tubes and placed on the benchtop for 15minutes. After 15minutes each tube was centrifuged at 2,000xg for 10min at room temperature (RT) to pellet red blood cells. The buffy coat was isolated from 10 blood collection tubes and transferred to a 50ml Falcon tube. Cells were then re-suspended in 25ml of PBS, pelleted at 2,000xg and transferred to a new tube 3x to remove as many red blood cells as possible. The isolated leukocytes were then re-suspended in 25ml of PBS and pelleted them at 800rpm. The supernatant was then removed and this process was repeated 3x to remove platelets. Isolated leukocytes were then re-suspended in 10ml of PBS and placed on ice for 1.5hr. The supernatant containing the EVs was then collected for further processing.
Method details
Measurement of VO2 max
VO2max was measured with indirect calorimetry (Medgraphics Diagnostics) using an electronically braked cycle ergometer (Lode Medical Technologies). VO2max was defined as reaching a perceived exertion > 17 on the Borg RPE scale with an RER > 1.1 (mean ± SD [Range]: 1.17 ± 0.02 [1.15–1.19]), and achieving a heart rate within 10% of the age-predicted maximal heart rate.
High intensity aerobic exercise
Subjects completed one bout of high intensity cycling (4 bouts of 4 min at > 90% of VO2max with 3 min pedaling at no load between bouts), as previously described (Robinson et al., 2017).
Acute resistance exercise
Subjects completed one bout of one-legged knee extension exercise (3 sets of 10 repetitions at 70% of 1-repetition-maximum, with 1min rest in-between sets).
EV isolation from cell culture media
Myotube, adipocyte, hepatocyte, platelet and leukocyte conditioned media were centrifuged at 2,000xg for 10 minutes to pellet cell debris. The media was then transferred to a new tube and centrifuged at 10,000xg for 20min to pellet large vesicles. The media was then filtered through a 0.2μm filter. Myotube, adipocyte, hepatocyte and platelet media were transferred to a 70-ml 100kD Centricon® Plus-70m filter unit to concentrate the media as previously described (Lobb et al., 2015). The concentrated media was then diluted up to 2ml at which point the EVs were isolated using a qEV2.0 (Izon) size exclusion chromatography column and processed as described for plasma EVs. Leukocyte and red blood cell EVs were processed by ultracentrifugation (UTC). Briefly, samples were pelleted at 10,000 x g to pellet large vesicles. The supernatant was transferred to a new tube and centrifuged at 100,000 x g for 1.5hr and digested as described for plasma EVs.
Blood collection and plasma preparation
Whole blood was drawn into ETDA tubes and placed on the benchtop for 15minutes. After 15minutes each tube was centrifuged at 2,000xg for 10min at room temperature (RT) to pellet red blood cells. The upper plasma fraction was then collected, transferred to a new sterile tube, and centrifuged again at RT for 10min at 2000xg. Platelet poor plasma was isolated by carefully transferring the top layer of plasma to a new tube and then frozen at -80°C in 1ml aliquots.
Isolation of plasma extracellular vesicles by size exclusion chromatography (SEC)
Frozen platelet poor plasma was removed from the freezer, thawed on ice and centrifuged at 2,000xg for 10 min to pellet debris. The plasma was then transferred to a new microcentrifuge tube and centrifuged at 10,000xg for 10min to pellet larger vesicles. Following centrifugation the plasma was transferred to a new tube and placed on ice until analysis. Extracellular vesicle isolation was performed using a qEV 2.0 size exclusion chromatography column (Izon Science, Christchurch, New Zealand) following the manufactures protocol with minor modifications. Briefly, SEC columns were equilibrated to room temperature and flushed with 90ml of phosphate buffered saline (PBS) before use. Prior to use, each column was passivated with 2ml of 5% BSA in PBS and flushed with 90ml of PBS. Platelet poor plasma was transferred into the sample loading reservoir of the SEC column. All experiments utilized 2.0ml of plasma except for exercise study, where limited sample volume was available and 1.2ml was used for 6 or the 14 aerobic exercise subjects. The column cap was then removed and the plasma sample was allowed to completely enter the column, at which point the sample loading reservoir was filled with PBS. The first 13ml of PBS fraction was discarded and the following 8ml’s of EV containing fraction were collected for each sample. All samples were processed using pooled fractions (fractions 14-21) except for the fractionation experiments where the individual fractions were processed as described in the text. The SEC columns were then flushed with 90ml of PBS before the addition of the next sample. The 8ml of EV containing fraction for each sample was then pelleted by ultracentrifugation at 100,000xg for 1.5hr at 4°C in a swing bucket rotor with a conical bottom tube. Following ultracentrifugation the supernatant was discarded and all residual PBS was removed before tryptic digestion.
Nano-particle tracking analysis (NTA) and transmission electron microscopy (TEM)
For the SEC versus UTC comparison, EVs isolated by SEC were measured by NTA using a 10 fold dilution of the pooled SEC column eluate (Fractions 14-21). EVs isolated by UTC were re-suspended in PBS prior to measurement. For the exercise study, NTA measurements were obtained using a 10-fold dilution of the pooled SEC column eluate (fractions 14-21). All NTA measurements were obtained using a Nano-sight LM10-HS with a 532nm laser running on NTA software v3.2. Samples were diluted until they reached the manufacturers recommended concentration of 20-60 particles per frame and data was acquired in 3 x 60sec videos with a viscosity of 0.985-0.897cP, camera level 13, detection threshold 2 and temperature of 25°C. For TEM, a 1ml volume of the pooled SEC column eluate (fractions 14-21) was fixed with 16% paraformaldehyde (4% final). The sample (5μl) was deposited on formvar-carbon-coated nickel grid (150 mes), air dried for 15min, washed 6x with PBS, fixed in 1% glutaraldehyde/PBS for 5min and then washed 6x for 2min with stilled water drops. The grid was then transferred to a drop mixture of 4% uranyl acetate and 2% methylcellulose (in a 1:9 ratio) for 5min. A paper filter was then used to remove excess solution and the sample was air dried for 1hr prior to observation with a JEOL (Peabody, MA) 1400 electron microscope at 80 KV.
Nanoscale flow cytometry (NFLC)
Plasma samples were diluted 10-fold in particle-free PBS and incubated with ADAM10 antibody conjugated with PE (SHM14 clone, Biolegend) for 30 minutes at 4°C. To control for non-specific binding and autofluorescence, samples were also incubated with antibody-matched isotype control. Samples were further diluted in particle-free PBS and analyzed using the A60-MicroPlus nanoscale flow cytometer (Apogee Flow Systems Inc.). Samples were run at 0.75 μl/min for 1 minute. All samples were run in triplicates and data were analyzed using FlowJo v10.5 software.
Protein digestion and label free mass spectrometry analysis
Samples were processed in three separate batches. For the first batch, EV pellets were lysed with the addition of 50μl 0.04% ProteaseMax Surfactant (Promega, Madison WI) in 50mM Tris buffer pH 8.2 and vortexed for 1min. 5ul of 110mM DTT (5mM final) was then added to reduce disulfide bonds and the samples were heated at 70°C for 10min. Samples were then equilibrated back to room temperature over 20 minutes before alkylation with 5μl 120mM IAA (10mM final) and incubated for 30min protected from light. Following alkylation an additional 0.675μl of 1.0% ProteaseMax was added prior to the addition of 0.1μg of Trypsin LysC mix. The samples were then vortexed and incubated at 37°C for 16hr in the capped conical bottom ultracentrifugation tube. Finally, the digestion was terminated by adding 5μl of 5.25% TFA. Sample batches two and three were processed with an optimized method to yield deeper proteome coverage. For these samples, EV pellets were precipitated with the addition of 50μl of methanol. The methanol was then evaporated in a SpeedVac (Thermo Scientific, Waltham MA) and protein was reconstituted with 50μl of 50mM Tris pH 8.2 containing 0.002% zwittergent Z3-16 (EMD Millipore, Burlington MA) and subsequently heated at 95°C for 10min. All additional steps are the same as described above except ProteaseMax wan not added before the digestion.
LC-MS conditions
Digested samples (15μl) were loaded onto a 0.33μl OptiPak trap column (Optimize Technologies) packed with Halo C18 peptide ES stationary phase. The trap was then washed with an aqueous loading buffer composed of 0.2% FA and 0.05% TFA for 4 minutes at 10μl/min. After the wash, the 10-port valve was switched and peptides were flushed off of the trap onto a 25cm x 75μm PicoFrit (New Objective) analytical column packed with Waters BEH 1.7μm stationary phase using a Dionex UltiMate 3000 RSLC liquid chromatography (LC) system (Thermo Scientific). The analytical gradient for peptide separation began at 2% mobile phase B (MPB) and 98% mobile phase A (MPA) for 4min, MPB was then increased to 30% over 40min, raised to 40% MPB over 20min, increased to 95% over 10min, held for 2min, returned to 2% B in one minute and equilibrated for 15min. MPA was composed of 98:2 (water/acetonitrile) with 0.2% FA and MPB was composed of 80:10:10 (acetonitrile/ isopropyl alcohol/water) with 0.2% FA. Analysis of the eluting peptides was performed using an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific) operated in data dependent mode. Survey scans were acquired from 300-1400m/z with 120,000 resolving power and an AGC of 4e5 and max fill time of 50ms. MS/MS scans of selected precursor ions were performed for a maximum of 3s or until the list was exhausted and dynamic exclusion was set to 45s. Quadrupole isolation for MS/MS scans was set at 0.7 m/z followed by fragmentation in the ion trap with “Rapid” scan speed and an ion target value of 5e4 and maximum injection time of 22ms using normalized collision energy of 28% from 200-1200 m/z. The monoisotopic precursor selection was set to peptide and charge states of 1, greater than 5, or unknown were excluded.
Quantification and statistical analysis
MS raw files were processed in MaxQuant software version 1.6.7.0 (Cox and Mann, 2008). Peptides were searched using the Andromeda search engine against the human Uniprot FASTA database downloaded July 24th, 2019. Cysteine carbamidomethylation was set as a fixed modification and N-terminal acetylation and methionine oxidations were set as variable modifications. Searches were performed with a false discovery rate of 1% for both peptides and proteins using a target-decoy approach. A minimum of two peptides were required, peptide length was at least 7 amino acids long and MS2 match tolerance was set to 0.5 Da. Match between runs was enabled with a retention time window of 0.7min. Enzyme specificity was set to trypsin and a maximum of 2 missed cleavages were allowed. Protein data was extracted from the “proteinGroups.txt” file and differential quantitation was carried out in Perseus version 1.6.0.7 (Tyanova et al., 2016). Aerobic exercise samples were processed in three separate batches and protein quantification results were merged by gene name prior to statistical analysis. All other mass spectrometry data was processed using custom R scripts.
For all exercise data, a minimum of 60% non-zero values were required in the post exercise group and the protein intensities were log2 transformed. The data was assessed for normality and the Pre versus Post exercise data were analyzed with a paired, two sample T test with the S0 parameter set to 0.1 and a permutation based FDR threshold of P < 0.05 was applied to the data set.
MS raw files from the cell culture experiments and PPP versus PRP comparison were searched as described above. Method comparison and SEC fractionation experiment MS raw files searched against the human SwissProt FASTA database downloaded February 2017, all other parameters are as described above, Proteomics, nFLC and NTA time course analysis measurements of total EV abundance were analyzed using a one tailed two sample T test and data is displayed as mean ± SEM. Plasma concentrations obtained from the plasma proteome database for EV enriched proteins and contaminants were compared using the Mann-Whitney U test. All other comparisons were performed using a two tailed two sample T test.
Pathway and functional enrichment analysis
Functional annotation enrichment analysis was carried out using DAVID Bioinformatics Resources functional annotation tool (v6.8). Enrichment p values were FDR corrected using Benjamini-Hochberg procedure and gene sets with a p value ≤ 0.05 were reported. Proteins that were statistically up or down regulated after exercise were subjected to Ingenuity Pathway Analysis (IPA) and regulated canonical pathways with a -log(p-value) ≥ 1.3 and a non-zero z-score were reported. For cell culture experiments, proteins identified uniquely in each cell type were subjected to an overrepresentation enrichment analyzed (ORA) using WEBGESTALT software. Redundancy of enriched gene sets was then reduced with the affinity propagation method using the R package apcluster and gene sets passing FDR (Benjamini-Hochberg procedure ≤ 0.05) were reported.
Acknowledgments
Support for this work was provided by National Institutes of Health grants MoTrPAC Chemical Analysis Center grant U24-DK-112326, CTSA grant number UL1 TR002377 from the National Center for Advancing Translational Sciences, Mayo Clinic Metabolomics Resource Core grants U24-DK-100469, T32-AG-49672, R01 AG062859, and the Dr. Richard F. Emslander Professorship in Endocrinology and Nutrition Research (K.S.N.). The authors thank Dawn Morse, Katherine Klaus, and F.L.-Matteoni for their skillful assistance.
Author contributions
Conceptualization, K.S.N., I.R.L., P.V., and S.D.; methodology, P.V.; formal analysis, P.V.; investigation, P.V. and G.N.R.; data curation, P.V. and S.D.; writing – original draft, P.V., I.R.L., and K.S.N.; writing – review & editing, P.V., I.R.L., S.D., K.S.N., and G.N.R.; visualization, P.V.; supervision, I.R.L. and K.S.N.; project administration, I.R.L. and K.S.N.; funding acquisition, I.R.L. and K.S.N.
Declaration of interests
The authors declare no competing interests.
Published: July 26, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100055.
Supplemental information
Proteins identified uniquely in adipocyte, Huh7 hepatocyte, myotube, leukocyte, and platelet EVs are listed with their corresponding cell-type specificity in the “Cell Line Specificity (EVs)” column. Proteins identified commonly from each cell type are also listed. These data are merged with tissue- and cell-type-specific proteins from the Human Protein Atlas (Uhlén et al., 2015).
Proteins that were significantly regulated greater than a log2 fold change of ±1.0 and adjusted p value of 0.05 are included in this table. Putative cell-specific EV proteins determined from the cell-line experiment are labeled in the “Cell Line Specificity (EVs)” column. Tissue-specific data from the Human Proteome Atlas is also included (columns that begin with HPA).
Proteins with an adjusted p value of less than 0.05 and a log2 fold change greater than ±0.5 are listed with their corresponding cell-line specificity as determined in the cell-line experiment (Figure 5) and the Human Protein Atlas.
References
- Al Shweiki M.H.D.R., Mönchgesang S., Majovsky P., Thieme D., Trutschel D., Hoehenwarter W. Assessment of label-free quantification in discovery proteomics and impact of technological factors and natural variability of protein abundance. J. Proteome Res. 2017;16:1410–1424. doi: 10.1021/acs.jproteome.6b00645. [DOI] [PubMed] [Google Scholar]
- Artoni A., Merati G., Padovan L., Scalambrino E., Chantarangkul V., Tripodi A. Residual platelets are the main determinants of microparticles count in frozen-thawed plasma. Thromb. Res. 2012;130:561–562. doi: 10.1016/j.thromres.2012.04.012. [DOI] [PubMed] [Google Scholar]
- Ayers L., Kohler M., Harrison P., Sargent I., Dragovic R., Schaap M., Nieuwland R., Brooks S.A., Ferry B. Measurement of circulating cell-derived microparticles by flow cytometry: sources of variability within the assay. Thromb. Res. 2011;127:370–377. doi: 10.1016/j.thromres.2010.12.014. [DOI] [PubMed] [Google Scholar]
- Bard M.P., Hegmans J.P., Hemmes A., Luider T.M., Willemsen R., Severijnen L.-A.A., van Meerbeeck J.P., Burgers S.A., Hoogsteden H.C., Lambrecht B.N. Proteomic analysis of exosomes isolated from human malignant pleural effusions. Am. J. Respir. Cell Mol. Biol. 2004;31:114–121. doi: 10.1165/rcmb.2003-0238OC. [DOI] [PubMed] [Google Scholar]
- Böing A.N., van der Pol E., Grootemaat A.E., Coumans F.A.W., Sturk A., Nieuwland R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracellular Vesicles. 2014;3 doi: 10.3402/jev.v3403.23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnouf T., Goubran H.A., Chou M.-L., Devos D., Radosevic M. Platelet microparticles: detection and assessment of their paradoxical functional roles in disease and regenerative medicine. Blood Rev. 2014;28:155–166. doi: 10.1016/j.blre.2014.04.002. [DOI] [PubMed] [Google Scholar]
- Chen I.-H., Xue L., Hsu C.-C., Paez J.S.P., Pan L., Andaluz H., Wendt M.K., Iliuk A.B., Zhu J.-K., Tao W.A. Phosphoproteins in extracellular vesicles as candidate markers for breast cancer. Proc. Natl. Acad. Sci. U S A. 2017;114:3175–3180. doi: 10.1073/pnas.1618088114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M., Raposo G., Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014;30:255–289. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- Corso G., Mäger I., Lee Y., Görgens A., Bultema J., Giebel B., Wood M.J.A., Nordin J.Z., Andaloussi S.E. Reproducible and scalable purification of extracellular vesicles using combined bind-elute and size exclusion chromatography. Sci. Rep. 2017;7:11561. doi: 10.1038/s41598-017-10646-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Frühbeis C., Helmig S., Tug S., Simon P., Krämer-Albers E.-M. Physical exercise induces rapid release of small extracellular vesicles into the circulation. J. Extracellular Vesicles. 2015;4:28239. doi: 10.3402/jev.v4.28239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A., Davey M., Chute I.C., Griffiths S.G., Lewis S., Chacko S., Barnett D., Crapoulet N., Fournier S., Joy A., et al. Rapid isolation of extracellular vesicles from cell culture and biological fluids using a synthetic peptide with specific affinity for heat shock proteins. PLoS One. 2014;9:e110443. doi: 10.1371/journal.pone.0110443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guay C., Regazzi R. Exosomes as new players in metabolic organ cross-talk. Diabetes Obes. Metab. 2017;19:137–146. doi: 10.1111/dom.13027. [DOI] [PubMed] [Google Scholar]
- György B., Módos K., Pállinger É., Pálóczi K., Pásztói M., Misják P., Deli M.A., Sipos Á., Szalai A., Voszka I., et al. Detection and isolation of cell-derived microparticles are compromised by protein complexes resulting from shared biophysical parameters. Blood. 2011;117:e39–e48. doi: 10.1182/blood-2010-09-307595. [DOI] [PubMed] [Google Scholar]
- György B., Szabó T.G., Turiák L., Wright M., Herczeg P., Lédeczi Z., Kittel Á., Polgár A., Tóth K., Dérfalvi B., et al. Improved flow cytometric assessment reveals distinct microvesicle (cell-derived microparticle) signatures in joint diseases. PLoS One. 2012;7:e49726. doi: 10.1371/journal.pone.0049726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeannin P., Chaze T., Giai Gianetto Q., Matondo M., Gout O., Gessain A., Afonso P.V. Proteomic analysis of plasma extracellular vesicles reveals mitochondrial stress upon HTLV-1 infection. Sci. Rep. 2018;8:5170. doi: 10.1038/s41598-018-23505-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakhniashvili D.G., Bulla L.A., Goodman S.R. The human erythrocyte proteome. Mol. Cell. Proteomics. 2004;3:501. doi: 10.1074/mcp.M300132-MCP200. [DOI] [PubMed] [Google Scholar]
- Karimi N., Cvjetkovic A., Jang S.C., Crescitelli R., Hosseinpour Feizi M.A., Nieuwland R., Lötvall J., Lässer C. Detailed analysis of the plasma extracellular vesicle proteome after separation from lipoproteins. Cell Mol. Life Sci. 2018;75:2873–2886. doi: 10.1007/s00018-018-2773-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keerthikumar S., Chisanga D., Ariyaratne D., Al Saffar H., Anand S., Zhao K., Samuel M., Pathan M., Jois M., Chilamkurti N., et al. ExoCarta: a web-based compendium of exosomal cargo. J. Mol. Biol. 2016;428:688–692. doi: 10.1016/j.jmb.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korutla L., Rickels M.R., Hu R.W., Freas A., Reddy S., Habertheuer A., Harmon J., Korutla V., Ram C., Naji A., et al. Noninvasive diagnosis of recurrent autoimmune type 1 diabetes after islet cell transplantation. Am. J. Transplant. 2019;19:1852–1858. doi: 10.1111/ajt.15322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal J., Arras G., Colombo M., Jouve M., Morath J.P., Primdal-Bengtson B., Dingli F., Loew D., Tkach M., Théry C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. U S A. 2016;113:E968–E977. doi: 10.1073/pnas.1521230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreimer S., Ivanov A.R. Rapid isolation of extracellular vesicles from blood plasma with size-exclusion chromatography followed by mass spectrometry-based proteomic profiling. Methods Mol. Biol. 2017;1660:295–302. doi: 10.1007/978-1-4939-7253-1_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang M., Tao X., Peng Y., Zhang W., Pan Y., Cheng L., Yuan C., Zhao Y., Mao H., Zhuge L., et al. Proteomic analysis of plasma exosomes to differentiate malignant from benign pulmonary nodules. Clin. Proteomics. 2019;16:5. doi: 10.1186/s12014-019-9225-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix R., Judicone C., Mooberry M., Boucekine M., Key N.S., Dignat-George F., The ISTH SSC Workshop Standardization of pre-analytical variables in plasma microparticle determination: results of the International Society on Thrombosis and Haemostasis SSC Collaborative workshop. J. Thromb. Haemost. 2013 doi: 10.1111/jth.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCroix R., Judicone C., Poncelet P., Robert S., Arnaud L., Sampol J., Dignat-George F. Impact of pre-analytical parameters on the measurement of circulating microparticles: towards standardization of protocol. J. Thromb. Haemost. 2012;10:437–446. doi: 10.1111/j.1538-7836.2011.04610.x. [DOI] [PubMed] [Google Scholar]
- Livshits M.A., Khomyakova E., Evtushenko E.G., Lazarev V.N., Kulemin N.A., Semina S.E., Generozov E.V., Govorun V.M. Isolation of exosomes by differential centrifugation: theoretical analysis of a commonly used protocol. Sci. Rep. 2015;5:17319. doi: 10.1038/srep17319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobb R.J., Becker M., Wen Wen S., Wong C.S.F., Wiegmans A.P., Leimgruber A., Möller A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracellular Vesicles. 2015;4:27031. doi: 10.3402/jev.v4.27031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minciacchi V.R., Freeman M.R., Di Vizio D. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin. Cell Dev. Biol. 2015;40:41–51. doi: 10.1016/j.semcdb.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen B.K., Hoffman-Goetz L. Exercise and the immune system: regulation, integration, and adaptation. Physiol. Rev. 2000;80:1055–1081. doi: 10.1152/physrev.2000.80.3.1055. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M., et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokopi M., Pula G., Mayr U., Devue C., Gallagher J., Xiao Q., Boulanger C.M., Westwood N., Urbich C., Willeit J., et al. Proteomic analysis reveals presence of platelet microparticles in endothelial progenitor cell cultures. Blood. 2009;114:723–732. doi: 10.1182/blood-2009-02-205930. [DOI] [PubMed] [Google Scholar]
- Robinson M.M., Dasari S., Konopka A.R., Johnson M.L., Manjunatha S., Esponda R.R., Carter R.E., Lanza I.R., Nair K.S. Enhanced protein translation underlies improved metabolic and physical adaptations to different exercise training modes in young and old humans. Cell Metab. 2017;25:581–592. doi: 10.1016/j.cmet.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T., Brakebusch C., Engel J., Timpl R. Mac-2 binding protein is a cell-adhesive protein of the extracellular matrix which self-assembles into ring-like structures and binds beta1 integrins, collagens and fibronectin. EMBO J. 1998;17:1606–1613. doi: 10.1093/emboj/17.6.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X., Shen S., Li J., Hu Q., Nie L., Tu C., Wang X., Poulsen D.J., Orsburn B.C., Wang J., et al. IonStar enables high-precision, low-missing-data proteomics quantification in large biological cohorts. Proc. Natl. Acad. Sci. U S A. 2018;115:E4767–E4776. doi: 10.1073/pnas.1800541115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranska R., Gysbrechts L., Wouters J., Vermeersch P., Bloch K., Dierickx D., Andrei G., Snoeck R. Comparison of membrane affinity-based method with size-exclusion chromatography for isolation of exosome-like vesicles from human plasma. J. Transl. Med. 2018;16:1. doi: 10.1186/s12967-017-1374-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb D.L., Vega-Montoto L., Rudnick P.A., Variyath A.M., Ham A.-J.L., Bunk D.M., Kilpatrick L.E., Billheimer D.D., Blackman R.K., Cardasis H.L., et al. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2010;9:761–776. doi: 10.1021/pr9006365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan K., Duquette M., Liu J.-H., Zhang R., Joachimiak A., Wang J.-h., Lawler J. The structures of the thrombospondin-1 N-terminal domain and its complex with a synthetic pentameric heparin. Structure. 2006;14:33–42. doi: 10.1016/j.str.2005.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turay D., Khan S., Diaz Osterman C.J., Curtis M.P., Khaira B., Neidigh J.W., Mirshahidi S., Casiano C.A., Wall N.R. Proteomic profiling of serum-derived exosomes from ethnically diverse prostate cancer patients. Cancer Invest. 2016;34:1–11. doi: 10.3109/07357907.2015.1081921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- van Niel G., D'Angelo G., Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018;19:213. doi: 10.1038/nrm.2017.125. [DOI] [PubMed] [Google Scholar]
- Whitham M., Parker B.L., Friedrichsen M., Hingst J.R., Hjorth M., Hughes W.E., Egan C.L., Cron L., Watt K.I., Kuchel R.P., et al. Extracellular vesicles provide a means for tissue crosstalk during exercise. Cell Metab. 2018;27:237–251.e4. doi: 10.1016/j.cmet.2017.12.001. [DOI] [PubMed] [Google Scholar]
- Wu A.Y.-T., Ueda K., Lai C.P.-K. Proteomic analysis of extracellular vesicles for cancer diagnostics. Proteomics. 2019;19:1800162. doi: 10.1002/pmic.201800162. [DOI] [PubMed] [Google Scholar]
- Zhang H., Liu J., Qu D., Wang L., Wong C.M., Lau C.-W., Huang Y., Wang Y.F., Huang H., Xia Y., et al. Serum exosomes mediate delivery of arginase 1 as a novel mechanism for endothelial dysfunction in diabetes. Proc. Natl. Acad. Sci. U S A. 2018;115:E6927–E6936. doi: 10.1073/pnas.1721521115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Proteins identified uniquely in adipocyte, Huh7 hepatocyte, myotube, leukocyte, and platelet EVs are listed with their corresponding cell-type specificity in the “Cell Line Specificity (EVs)” column. Proteins identified commonly from each cell type are also listed. These data are merged with tissue- and cell-type-specific proteins from the Human Protein Atlas (Uhlén et al., 2015).
Proteins that were significantly regulated greater than a log2 fold change of ±1.0 and adjusted p value of 0.05 are included in this table. Putative cell-specific EV proteins determined from the cell-line experiment are labeled in the “Cell Line Specificity (EVs)” column. Tissue-specific data from the Human Proteome Atlas is also included (columns that begin with HPA).
Proteins with an adjusted p value of less than 0.05 and a log2 fold change greater than ±0.5 are listed with their corresponding cell-line specificity as determined in the cell-line experiment (Figure 5) and the Human Protein Atlas.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD026483.